 Yongjing Guan
Yongjing Guan Zaizhao Wang
Zaizhao Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 21 December 2018
Sec. Aquatic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.03191
This article is part of the Research TopicContribution of Wastewaters and Wastewater Treatment Plants on Antibiotic Resistance Situation: Health Risk Characterization, Intervention Technologies, and Surveillance Concepts View all articles
The microbial communities in freshwater have raised concerns about the ecosystem and human health. Many ecological environmental problems have been found in urban river because of the unreasonable use and long-term wastewater discharge. In this study, we explored the bacterial community composition, abundance of 14 antibiotics and 21 antibiotic resistance genes (ARGs), and water environment features in seven water samples and seven sediment samples from Ba River in Xi’an, China. Results showed Proteobacteria and Bacteroidetes were the dominant phyla in all samples, and sediment samples had a higher bacterial diversity and richness than it in water. Bacterial communities of site 5 and 6 were clustered in discrepant patterns compared to those at remaining sites from other samples. It might be influenced by nutrients, heavy metals and antibiotics. Antibiotics concentrations ranged from 1.26 to 1.61 × 103 ng L-1 in water samples and 1.55 to 4.05 × 102 μg kg-1 in sediment samples. Sulfamerazine (SM1) and erythromycin (ERY) were the chief antibiotics in water samples, while the level of oxytetracycline (OTC) and cefazolin (CFZ) were higher in sediment samples. Canonical correspondence analysis showed that trimethoprim (TMP) was significantly related to Acinetobacter in W6, and that SM1 and OTC had positive correlation with Arcobacter in W5. The tetC, blaTEM, ermF and sul1 had higher pollution abundance ranging from 10-4 to 100 copies/16S rRNA gene copies in all samples. Significant correlations were observed between ARGs and matching antibiotics, suggesting that antibiotics can pose the selective pressure on ARGs in this river. In summary, these finding might provide some new data to the limited information available on the bacterial community characteristics, abundance of antibiotics and ARGs in urban river of China.
As an important ingredient of the ecosystems for terrestrial freshwater and sediment, bacterial community play a crucial role in microbial food webs, biogeochemical cycles, energy flows and the decomposition of pollutants in the aquatic environment. As a result, bacterial community has raised concerns about the ecosystem and human health (Ruiz-González et al., 2015; Monard et al., 2016; Miao et al., 2017). The changes of bacterial community are reliable signals for pollution in water or sediment (Garrido et al., 2014; Su et al., 2017). Antibiotics, as an emerging contaminant in aquatic environments, have posed potential adverse effects on humans, animals and microorganisms (Milic et al., 2013; Petrie et al., 2015). In addition, the antibiotic-resistant bacteria and antibiotic-resistant genes (ARGs) in water increased the risks for aquatic ecological balance and human health (Michael-Kordatou et al., 2017; Zhu et al., 2017). Thus, it is indispensable to explore the bacterial community, antibiotics and ARGs in freshwater ecosystems.
As the major ingredient of freshwater ecosystems, urban river ecosystem is used for agricultural irrigation, entertainment, and it plays a key role in the source of water, traffic channel and pollution purification. However, with increasing human activities, urban rivers are polluted by wastewaters from communities, hospitals, pharmaceutical industries and animal husbandry. In previous studies, large amounts of antibiotics and high levels of ARGs have been identified in urban rivers (Sim et al., 2011; Rico et al., 2014; Rodriguez-Mozaz et al., 2015). Many reports have suggest that the abundance of ARGs may be correlated with heavy metal (He et al., 2017; Zhao et al., 2017). Besides, the structure of bacterial community was notably affected (Subirats et al., 2017). Pathogenic bacteria such as Clostridium difficile, Arcobacter butzleri, Escherichia coli and Kluyvera georgiana were detected in polluted rivers (Jia et al., 2017; Yang et al., 2017), which could cause potential health risks to residents nearby. Additionally, a recent study has proved that antibiotics can affect microbiological compositions in water (Xi et al., 2015). Therefore, it is worthwhile to explore the relationships among antibiotic, ARGs and bacterial community in urban river, which plays a major role in providing a stable and healthy environment for human beings and guaranteeing the city sustainable development (Zhou et al., 2017).
Ba River, one of the main water resources in Xi’an, China, has many ecological problems caused by unreasonable exploitation and long-term wastewater discharge. A survey of primary health care settings of Xi’an showed 40.5% was prescribed antibacterial in 780 outpatient prescriptions from April to May in 2013. And the rate of combined use was 8.2% (Ye et al., 2016). In 2015, the defined daily dose of antibacterial drugs was 4.83 × 106 in 13 tertiary hospitals in Xi’an (Li and Hu, 2016). Our laboratory studies indicated that significant gradient pollution exist along Ba River, including high concentrations of phenolic and steroidal endocrine disrupting compounds in the middle and lower reaches, and antibiotic pollution in the downstream (Jia et al., 2018; Wang et al., 2018). So far, there are no studies on the bacterial community structure in response to the gradient pollution in river system. The previous studies on microbial composition or environmental quality mainly focused on water bodies (Abia et al., 2017). Therefore, in this study, we examined bacterial community, abundance of antibiotics and ARGs and environment features in both water and sediment samples from different sites along the pollution gradient.
In this study, surface water and sediment were sampled in May 2017 at 14 locations in triplicate along the Ba River in Xi’an (Figure 1). Seven of these samples, labeled as W1, W2, W3, W4, W5, W6, and W7, were collected from water. Sediment samples were marked with S1, S2, S3, S4, S5, S6, and S7. The sampling sites for W1 and S1 were located at the upper reach of the Ecological Wetland Park of Ba River Bridge. Sites for W2 and S2 were located in the downstream of the Riverside Park. Sites for W3 and S3 were located at the hundred meters upstream from W4. Sites for W4 and S4 were located at the mouth of Chan River, which flows into Ba River. The sites for W5 and S5 were located at a resident domestic sewage outlet. The site for W6 was located at the wastewater treatment plant (WWTP) discharge port. The site for S6 was located at the two hundred meters downstream from W6. The site for W7 was located close to Jing Wei Wetland, and S7 was located at several hundred meters upstream from Chan Ba National Wetland Park. Water samples (2.0 L) from the top 0.5 m of the water surface were collected. Sediment samples (50 g) were collected from the top 5 cm layer using a bottom sampler. All samples were collected using sterile containers and transported on ice to the laboratory for analysis.
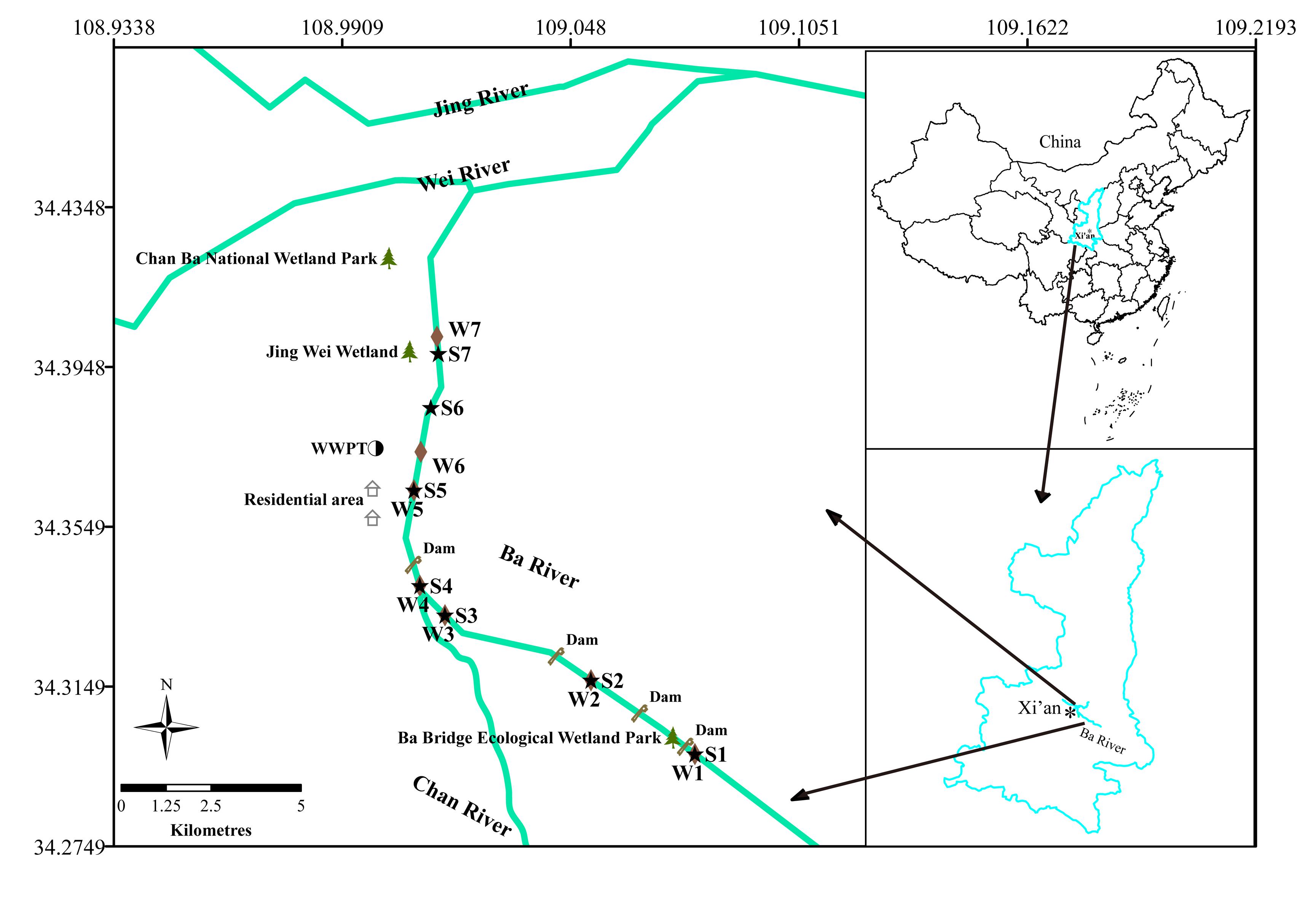
Figure 1. Map of the study area and the sampling sites.
Ultra-high performance liquid chromatography (UHPLC, Agilent 1290 Infinity, United States) coupled with tandem mass spectrometry (MS/MS, Agilent 6460 Triple Quadrupole, United States) and isotope dilution were used to determine the concentrations of fourteen antibiotics belonging to the sulfonamides, quinolones, tetracyclines, macrolides, β-lactams and amphenicols including sulfadiazine (SDZ), sulfamerazine (SM1), trimethoprim (TMP); norfloxacin (NOR), ciprofloxacin (CIP); oxytetracycline (OTC), tetracycline (TC), chlortetracycline (CTC); erythromycin (ERY), roxithromycin (ROX); cefotaxime (CTX), cefazolin (CFZ), penicillin G (PEN G); chloramphenicol (CHP). Trimethyl-13C3 caffeine was used as standards (Sigma-Aldrich Co., United States). Samples were pretreated and solid phase extracted according to the previous study (Luo et al., 2010). Chromatographic analyses were performed by injecting 2 μL extract into a ZORBAX Eclipse Plus C18 column (3.0 × 50 mm, 1.8 μm, Agilent Technologies, United States) at 35°C. The total mobile phase flow rate was 0.45 mL min-1. The washing gradient program of flowing phase was described in Supplementary Tables S1, S2 (CHP). The tandem MS analyses were performed in the positive electron spray ionization (ESI) and negative ESI (CHP ionization) modes in this study. The optimum MS conditions were: the source temperature, 325°C; dry gas temperature, 350°C; dry gas flow, 12 L min-1; nebulizer pressure, 45 psi; sheath gas heater temperature, 300°C; sheath gas flow, 6 L min-1; capillary voltage, 3000 V; Function, multiple reaction monitoring (MRM). Fourteen compounds and Trimethyl-13C3 caffeine used as standards (Sigma-Aldrich Co., United States) were described in Supplementary Table S3. The recovery rates varied between 63.4 and 107.9% (Supplementary Table S4).
Temperature (T), pH, specific conductivity (S), and dissolved oxygen (DO) levels of water samples were measured with the Multi 3410 Set KS1 (WTW, Germany). Water transparency was measured using a secchi disk (SD). The permanganate index [chemical oxygen demand (COD)] and the total contents of nitrogen (TN) and phosphate (TP) were measured according to the national standard method (Jia et al., 2018). The concentrations of six heavy metals (Cr, Cd, Cu, As, Pb, Hg) in water samples were analyzed by the inductively coupled plasma mass spectrometer (ICP-MS, Thermo Fisher Scientific, iCAP Q, United States). All measurements were conducted in triplicate.
Water samples (1 L) were vacuum filtered through 0.22-μm filter. The filters were stored at -20°C before DNA extraction. Water DNA was extracted with the Water DNA kit (Omega Bio-tek, United States) according to the instructions of the manufacturer. For sediment, DNA from 0.5 g lyophilized samples were extracted with the Soil DNA kit (Omega Bio-tek, United States). The DNA quality was detected with 1% agarose gel electrophoresis, and the concentration was measured by a NanoDrop Spectrophotometer (Thermo Fisher Scientific Inc., United States). DNA samples were stored at -20°C for further analysis.
The abundance of target genes were analyzed by qPCR including 16S rRNA and 21 antibiotic resistance genes (ARGs) including three sulfanilamide resistance genes (sul1, sul2, sul3), three quinolone resistance genes (gyrA, qnrB, qnrS), six tetracycline resistance genes (tetA, tetB, tetC, tetM, tetW, tetZ), three macrolide resistance genes (ermB, ermC, ermF), three β-lactam resistance genes (blaIMP4, blaNDM1, blaTEM), three amphenicol resistance genes (cat1, cmlA, floR). The 16S rRNA gene was quantified to normalize the abundance of the ARGs and to assess the total bacterial population. The primers used were designed in our laboratory (Supplementary Table S5; Pei et al., 2006; Xi et al., 2009; Jia et al., 2018). All qPCR assays were performed using CFX96TM Real-Time PCR Detection System (Bio-Rad, United States). The qPCR reactions were carried out in a final volume of 25 μL, using 1 × SYBR Premix Ex TaqTM, 0.4 μM of each gene specific primer, and 2.5 μL RT reaction solution. Each individual sample was run in triplicate using the following protocol: 95°C/30 s, 40 cycles of 95°C/5 s, 62°C/30 s. To ensure the specificity of each amplicon, a melting curve analysis was performed after amplification. CFX Manager software (Bio-Rad) was used to analyze the density of SYBR green I and to determine the threshold cycle (Ct) value. The qPCR efficiency (E) of each PCR reaction was calculated, and all the E values were between 90 and 110%. Plasmids containing ARGs and 16S rDNA were created to produce the standard curves as described previously (Pei et al., 2006). R2 values were more than 0.99 for all calibration curves.
The V3-V4 regions within the 16S rRNA gene were amplified from the DNA extracts using the forward primer 341F (CCTACGGGNGGCWGCAG) and reverse primer 805R (GACTACHVGGGTATCTAATCC). Each primer was labeled with an Illumina adaptor sequence and a unique multiplex identifier code. The standard 50 μL polymerase chain reaction (PCR) system included 2x Phanta Max Master Mix (Vazyme, China), 10 μM forward and reverse primers, 5 μL of DNA template and 16 μL of ddH2O. Thermal cycling program of 3 min initial denaturation at 95°C, followed by 8 cycles at 95°C for 30 s, 55°C for 30 s, 72°C for 45 s, and a final elongation step at 72°C for 5 min. The amplicons were quantified using Quant-It Pico Green kit (Invitrogen, United States) with Qubit Spectrophotometer (Invitrogen, United States), and samples were pooled together for library preparation. The library concentration was measured with Agilent 2100 Bioanalyzer (Agilent Technologies, United States), and diluted to 4 nM using Tris pH 8.5. After denaturation, 6 pM of the combined sample library and PhiX control was loaded on a MiSeq Platform (Illumina; United States) using 600 cycles MiSeq Reagent Kit PE300 v3 (Illumina; United States), and 300 bps paired-end reads were cluster generated.
The sequence date generated in this study have been deposited in the NCBI Short Read Archive under accession number PRJNA504751. Raw FASTQ files were processed using the QIIME 1.80 (Caporaso et al., 2010), and the paired reads were joined with a combination of the FLASH 1.2.7 using the default setting and the PANDASEQ 2.9 (Magoc and Salzberg, 2011; Masella et al., 2012), and then joined sequences were quality filtered and analyzed with QIIME. The remaining sequences were chimeras detected and removed using UCHIME 4.2.40 (Edgar et al., 2011). Then they were clustered into operational taxonomic units (OTUs) using UPARSE 7.0, with 97% sequence similarity. Ultimately, they were identified down to different taxonomic levels using Ribosomal Database Project (RDP) classifier 2.2 at a bootstrap cutoff of 80% (Cole et al., 2014). A heat map was generated from the relative abundance of OTUs with R 2.15.3 to classify provincial patterns in bacterial community composition. Venn diagrams were performed using R 2.15.3. A phylogenic tree was generated from the filtered alignment using FASTTREE. Based on the OTUs information, rarefaction curves and alpha diversity referring to community diversity were also calculated by MOTHUR. The phylogenetic beta diversity, including principal coordinate analysis (PCA) and hierarchical clustering analysis was evaluated with Bray-Curtis distance created by QIIME.
The linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed on the OTU table using the online Galaxy interface (Segata et al., 2011). LEfSe method uses the Kruskal-Wallis’s test to identify taxa with significant differences and performs LDA to evaluate the effect size of each feature. The LDA threshold score of 2.0 and a significant p of 0.05 were used to detect biomarkers. Canonical correspondence analysis (CCA) or redundancy analysis (RDA) was performed to find out the probable links among bacterial community structure, antibiotics/ARGs distribution and sampling sites, the associations among environmental parameters of water samples, bacterial community and sampling sites, and the correlations among antibiotics abundance, ARGs distribution and sampling sites using R software. In the analysis process, the significant variables were selection to examine the correction between these variables and bacterial community. Maps of sampling sites and geostatistical analysis were performed in ARCGIS 10.1. All the data were shown as mean ± standard error of the mean (SEM).
The sequence libraries from 42 samples contained 342 OTUs (smallest) in water samples and 2767 OTUs (largest) in sediment (Supplementary Table S6). The rarefaction curves of OTUs were saturated for all samples (Supplementary Figure S1), indicating that the sequencing depth was enough. Simultaneously, the rarefaction curves revealed that the community richness of sediment samples was higher than that of water samples. Chao and ACE indexes suggest that higher OTU richness was captured in sediment samples than water samples apart from site 6 (Figures 2A,B). This is also supported by Venn diagrams, demonstrating the water samples of the seven sites shared a lower number of OTUs compared with the sediment samples (Supplementary Figure S2). Shannon (species richness) and Simpson (species evenness) indexes illustrated diversity of bacterial community was higher in sediment sample than water sample (Figures 2C,D). Besides, the diversity index showed similar values for W1, W2, W3, W6, and W7 and lower values for W4 and W5. There was a clear difference between S5 and other sites of sediment samples (Figures 2C,D). The PCA showed that the bacterial community structure was significantly different between water and sediment samples (Figure 2E). Also, the bacterial community structures in site 5 and 6 were different from other sites (Figure 2E). These were also supported by the Bray-Curtis cluster tree of the water and sediment samples (Figure 2F).
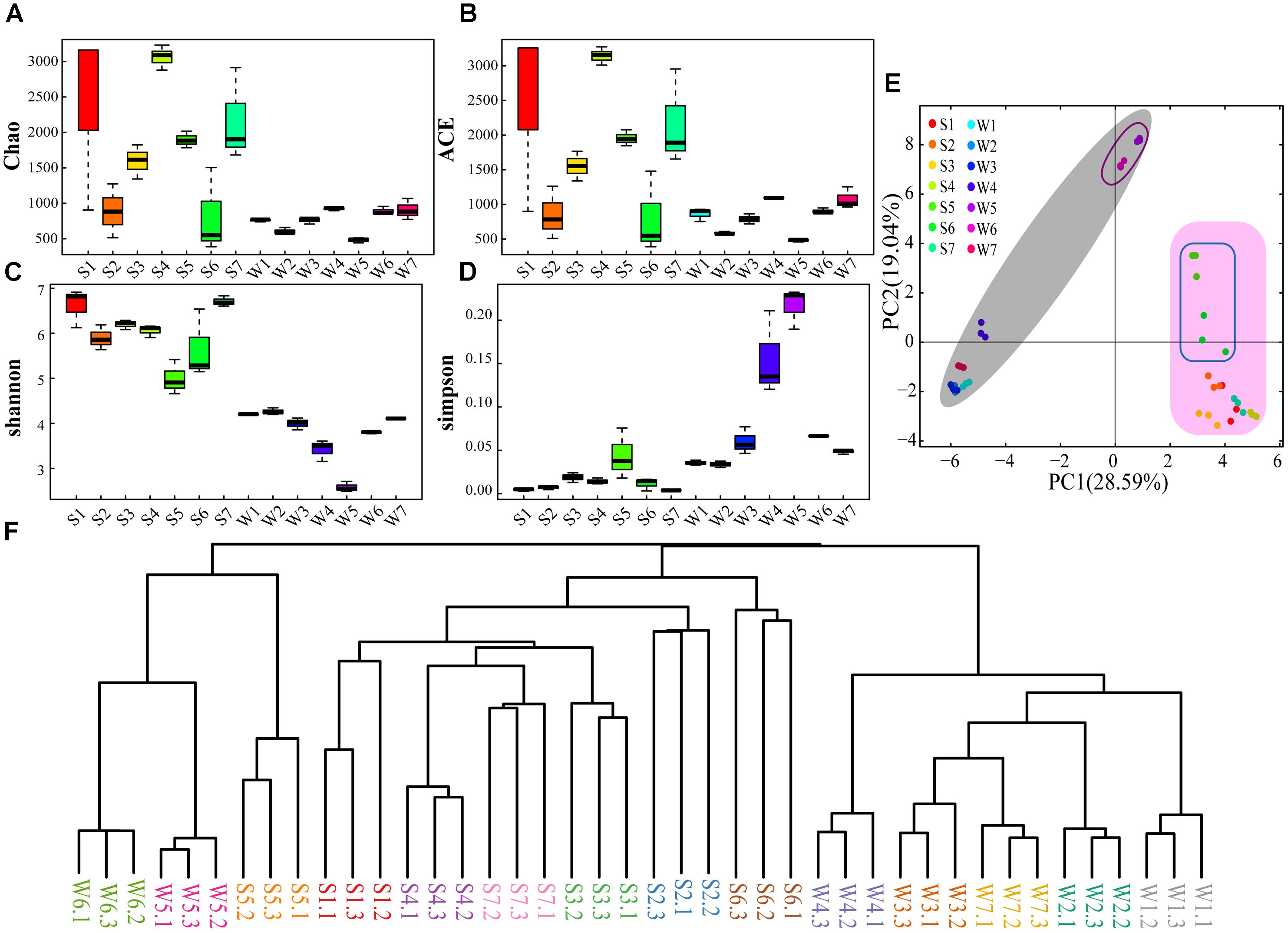
Figure 2. The diversity and phylogenetic structure of community composition. Sampling was performed at different sites for three times. W, Water samples; S, sediment samples. (A) Chao index. (B) ACE index. (C) Shannon index. (D) Simpson index. (E) PCA of the water and sediment samples based on the analysis of OTUs. (F) Bray-Curtis cluster tree of the water and sediment samples.
The 16S rRNA gene libraries detected 37 bacterial phyla of 42 samples. The community composition in the sediment and surface water samples is different (Figure 3). Two phyla (Proteobacteria and Bacteroidetes) had the high percentage in all water and sediment samples, accounting for 43.77–81.02% of the total reads in sediment and 42.23–95.09% in water using RDP Classifier at a confidence threshold of 80% (Supplementary Figure S3). In sediment samples, the two phyla were followed by a few other major (relative abundance > 1%) phyla, including Verrucomicrobia, Acidobacteria, Firmicutes, Chloroflexi, Actinobacteria, Cyanobacteria (except for S5 and S6), Spirochaetes (except for S5), Parcubacteria (only in S1, S3 and S7), Chlamydiae (only in S7), Aminicenantes and Latescibacteria (only in S6). However, Acidobacteria, Chloroflexi, Spirochaetes, Chlamydiae and Aminicenantes were minor abundance (<1%) in water samples. The two phyla were followed by Actinobacteria (except for W5 and W6), Cyanobacteria (except for W5 and W6), Verrucomicrobia (except for W5 and W6), Firmicutes (except for W3, W4, and W7), Planctomycetes (only in W2, W3, and W7), Gemmatimonadetes (only in W2), Parcubacteria (only in W6) and Fusobacteria (only in W5 and W6).
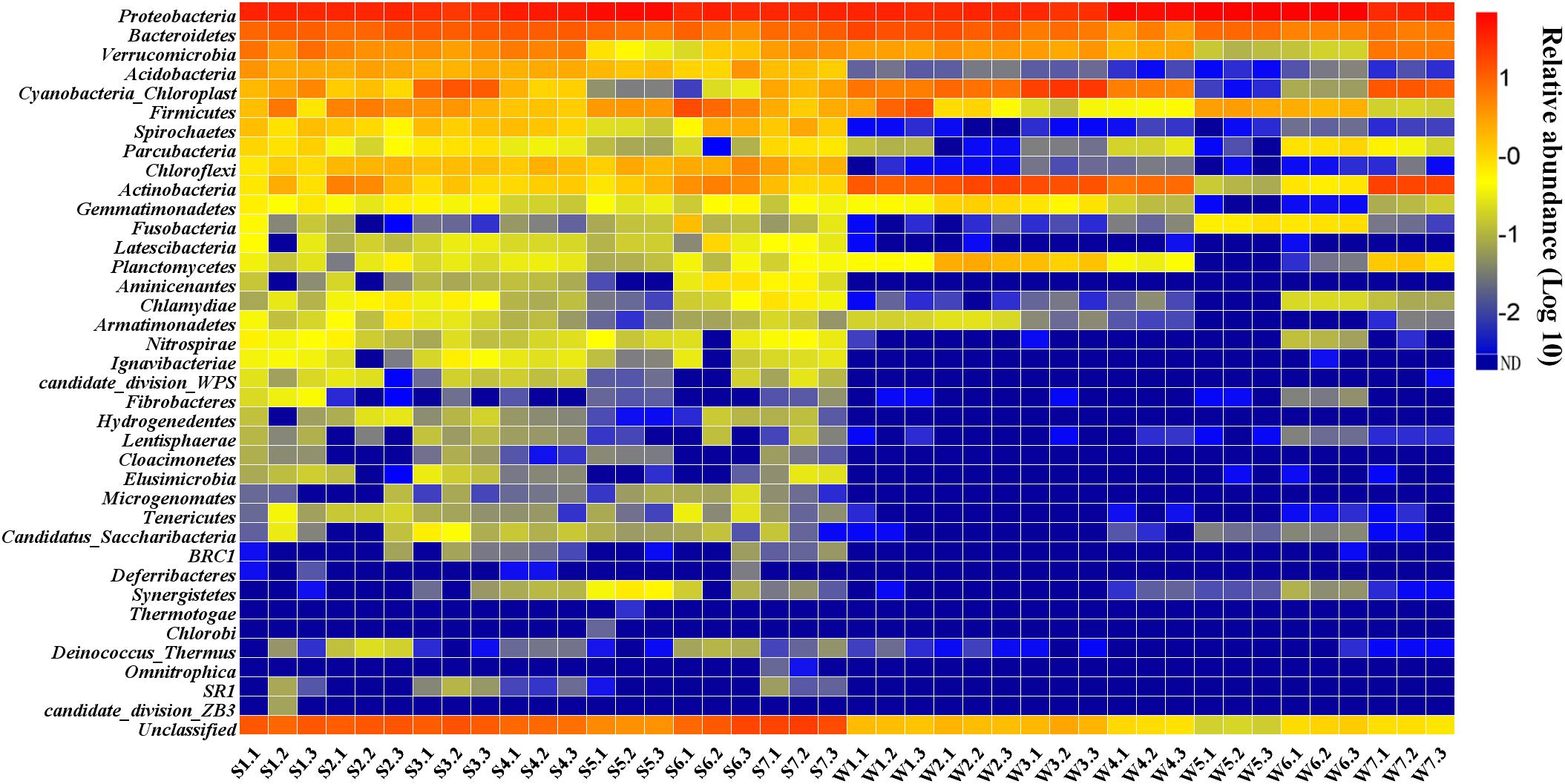
Figure 3. Percentage of different phyla in the water and sediment.
The significant differences of specific bacteria in phylum, class, order and family levels among samples were shown in cladogram for LEfSe analysis (Figure 4). W1 was mainly enriched in Firmicutes and Bacteroidetes phyla. Also, Lactobacillales, Bacillales, Flavobacteriales, Cytophagaceae, Coriobacteriaceae, and Caulobacterales order were significantly enriched in W1 compared with other water samples (Figure 4A). Some bacterial taxonomy levels showed consistent abundance advantage from the phylum or order to genus in water samples, including W2 (Gemmatimonadetes, Acidobacteria, Rhodobacterales, Methylophilales, and Opitutales), W3 (Acidimicrobiales) and W7 (Verrucomicrobiae and Methylococcales, Figure 4A). Compared with other water samples, W5 was significantly enriched in Clostridia, Bacteroidia, and Epsilonproteobacteria (from class to family). Within the Proteobacteria phylum, Alphaproteobacteria and Betaproteobacteria (class) were significantly enriched in W4 while Gammaproteobacteria was mainly enriched in W6 (Figure 4A). Some bacterial taxonomy levels had consistent abundance advantage from phylum or class or order to genus in sediment samples, including S3 (Lgnavibacteriae, Elusimicrobia, and Flavobacteriia), S1 (Opitutae, Holophagae, and Cytophagia), S4 (Verrucomicrobia, Methylococcales, and Rhodocyciaceae), S6 (Hydrogenophilales, Figure 4B). S2 was mainly enriched in Tenericutes, Armatimonadetes and Deinococcus-Thermus (phylum), Mollicutes (class) and Deinococcales (order, Figure 4B). The four orders within Proteobacteria phylum and Synergistetes (from phylum to family) were significantly enriched in S5. Firmicutes (phylum) and Clostridia and Bacilli (class) showed abundance advantage in S6 (Figure 4B).
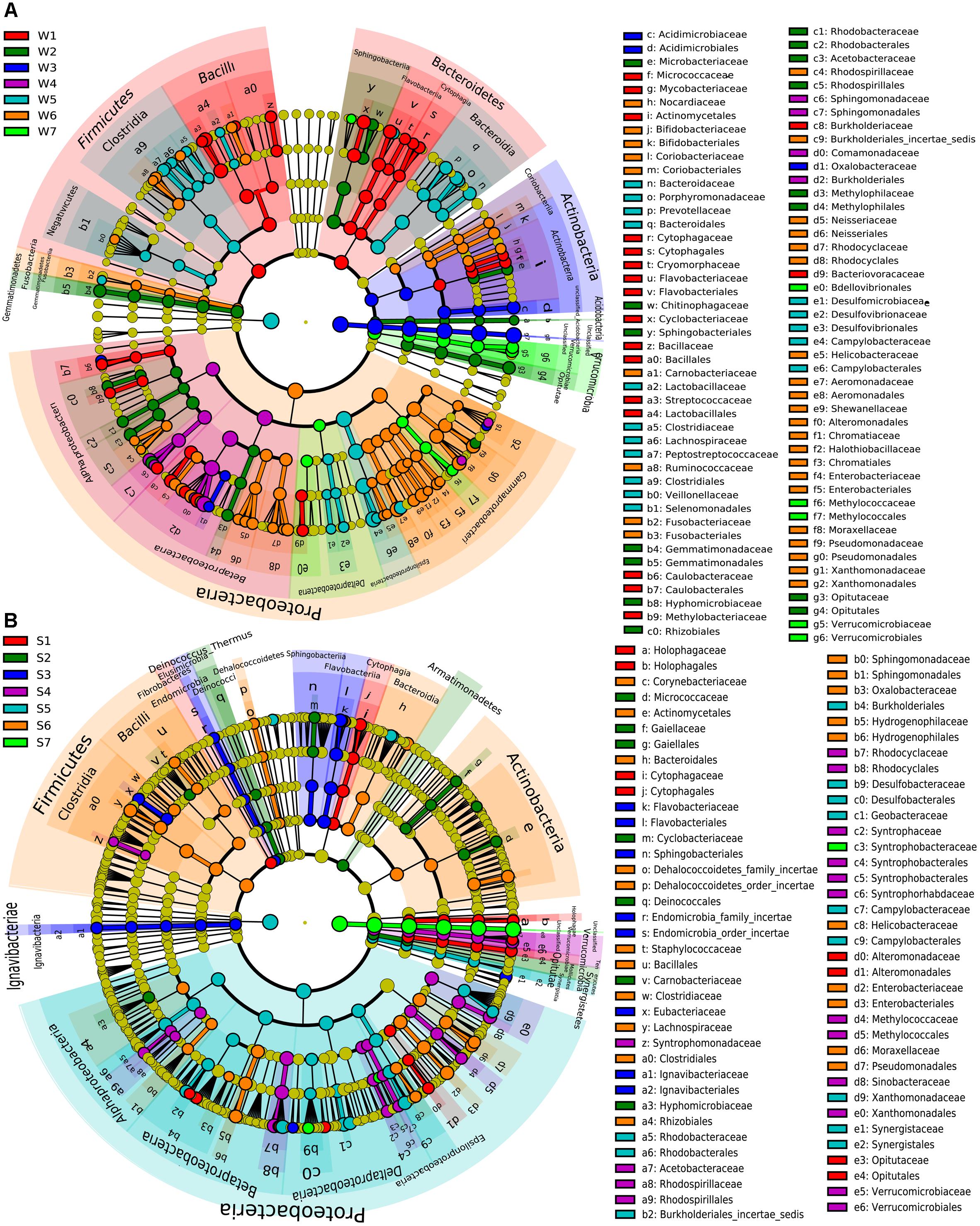
Figure 4. LEfSe analysis of bacteria abundance among different water samples (A) or sediment samples (B). The phylum, class, order, family levels are listed in order from inside to outside of the cladogram and the labels for levels of order and family are abbreviated by a single letter. The red, green, blue, purple, light blue, orange and emerald green circles represent the bacteria enriched in the site 1, 2, 3, 4, 5, 6, and 7, respectively, whereas the yellow circles represent the taxa with no significant differences between 7 sites of the water (A) or sediment (B).
Concentrations of heavy metals, nutrients and physicochemical properties of water samples showed in Supplementary Figure S4. In general, SD, pH, Cd, and Hg had normal variations among all water samples. However, the abnormal high level of T, S, Cr, Cu, As, COD, TN, and TP and the low level of DO were found in W5 and W6, whose sampling sites are close to a resident domestic sewage outlet or a WWTP discharge port. CCA was performed to examine the potential relationships between environmental parameters and bacterial community composition. Forward selection was used to identify the most influential gradients, which represented the drivers of bacterial community composition changes. The significant environmental parameters (p < 0.05) were added to improve the model’s explanatory power. Results indicated that TP, TN, DO, SD, S, Cu, and Pb exhibited significant correlations with bacterial community. The biplot score showed that 63.38% of the variations were explained by CCA1 while 13.21% were explained by CCA2. In W4, Betaproteobacteria showed a positive association with SD and DO. In addition, Epsilonproteobacteria and Bacteroidia appeared to be linked with TN and Cu in W5, while Gammaproteobacteria was associated with TN and S in W6.
Fourteen antibiotics were detected at concentrations ranging from 1.26 to 1.61 × 103 ng L-1 in water samples and 1.55 to 4.05 × 102 μg kg-1 in sediment samples (Figure 5). SM1, TMP, ERY, and ROX were detected in all samples, while PEN G was not found for all samples. Besides, SDZ, NOR, OTC, TC, and CHP were found for all sediment samples. The relatively low concentrations of the total antibiotics were detected in site 1 and 2 including water and sediment samples, where are the upstream of the Ba River city section. The concentrations of all the detected antibiotics consistently increased for water samples from site 1 to 5. The high concentrations of antibiotics were found in W5, and they consistently decreased from site 5 to 7. In sediment samples, the total concentrations of antibiotics apart from S3 were increased continuously along the river flow, while S3 had the highest concentrations.
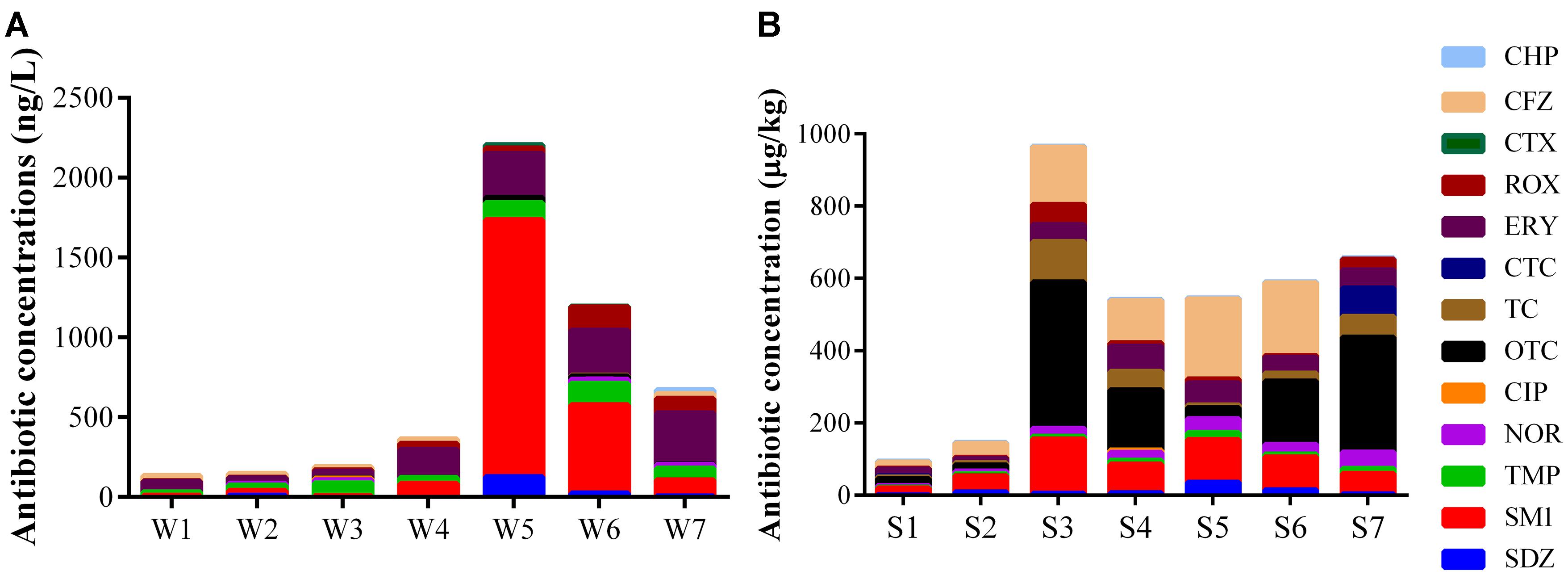
Figure 5. Concentration of antibiotics in water (A) and sediment (B) samples.
Canonical correspondence analysis was performed to examine the potential relationships between antibiotics and bacterial community. Results showed that water bacterial community (family) was significantly shaped by several key antibiotics, including TC, ERY, TMP, OTC, CHP, CIP, and CFZ (Figure 6A). OTC and CFZ were kept separate from each other on the CCA1 (explaining 54.16% of the variations), and Campylobacteraceae, Gplla, Sphingomonadaceae, and Rhodobacteraceae were scattered on the CCA1 compared to other bacteria. In sediment samples (Figure 6B), the overall pattern of the bacterial community (class) was significantly related to TC, ROX, CHP, ERY, NOR, and SDZ. SDZ was positively correlated with the CCA1 (explaining 41.21% of the variations), whereas Actimobacteria was on the CCA2 (explaining 31.35% of the variations). Cyanobacteria showed a positive association with TC and ROX. In addition, Deltaproteobacteria appeared to be linked with ERY and NOR.
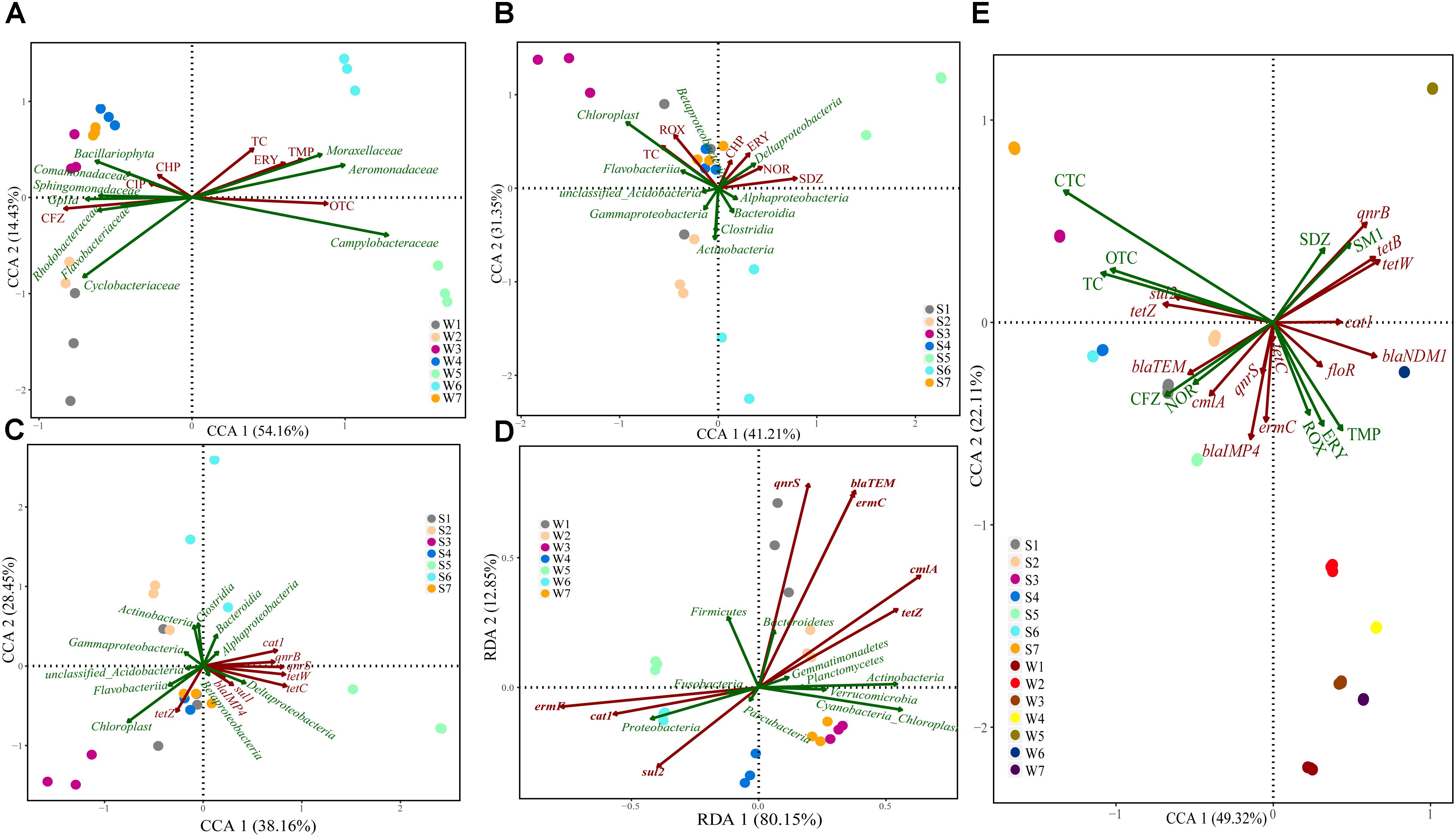
Figure 6. Canonical correspondence or Redundancy analysis of the relationship between bacterial communities and antibiotics/ARGs distribution (A–D), and the correlations among antibiotics abundance and ARGs distribution (E). Samples are performed at different sites for three times in different color solid circles separately.
The total bacteria load (16S rRNA gene copy numbers) were in the range of 1.34 × 106 to 4.64 × 108 copy numbers per mL (copies/mL) in water samples (Figure 7A) and 2.78 × 108 to 1.03 × 109 copy numbers per g (copies g-1) in sediment samples (Figure 7B), and the absolute abundance of ARGs in sediment was 1–3 orders of magnitudes higher than that in water, illustrating that the sediment was a natural reservoir of ARGs. Relative abundance of ARGs (the absolute abundance of ARGs normalized to the absolute abundance of 16S rRNA) in all samples was shown in Figure 7C. Overall, the tetC, blaTEM, ermF, sul1, cmlA, and gyrA were the predominant ARGs with the abundance ranged from 10-4 to 100 copies/16S rRNA. The relative abundance of ARGs was not obviously different between the sites of upstream (site 1 and 2) and downstream. The six tetracycline resistance genes were detected in all samples (except tetW in S3 and tetZ in S6). The tetC had the highest relative abundance with values ranging from 2.23 × 10-2 to 9.92 × 10-1 in water and 4.99 × 10-2 to 4.09 × 100 in sediment. For three β-lactam resistance genes, the blaTEM was the most abundant with values ranging from 5.14 × 10-3 to 6.68 × 10-1 in water and 1.99 × 10-1 to 1.16 × 100 in sediment, while the blaIMP4 was the lower abundance than other detected ARGs in water samples. Among the three macrolide resistance genes, the ermF had the highest relative abundance ranging from 1.26 × 10-4 to 9.38 × 10-3 in all samples, and it was higher in sediment than matching water samples. The relative abundance of three sulfanilamide resistance genes ranged from 4.12 × 10-5 to 2.47 × 10-2 in all samples, with higher abundance in sediment than in water.
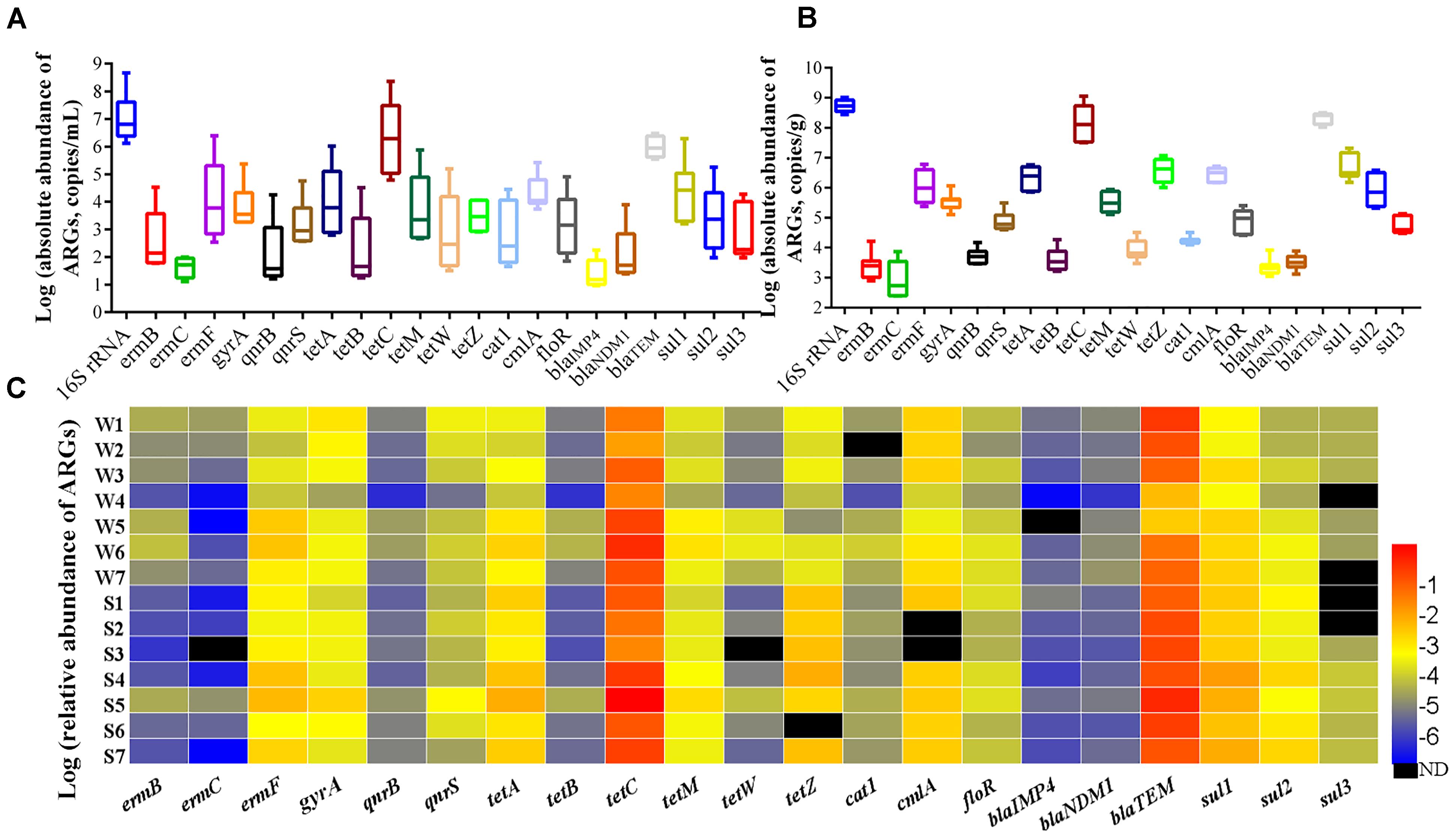
Figure 7. Absolute abundance of ARGs in water (A) and sediment (B) samples, and relative abundance of ARGs (C).
CCA and RDA were performed to identify the potential relationships between the most significant ARGs and bacterial community structure, which were based on their relative abundance. RDA showed that the bacterial community (phylum) was significantly related to the dominant ARGs in water samples (Figure 6D), such as cat1, ermF, sul2, qnrS, blaTEM, ermC, cmlA, and tetZ, with 80.15% of the constrained variance explained by RDA1 and 12.85% by RDA2. In sediment samples, the variation, of which 38.16% could be explained by CCA1 and 28.45% by CCA2. CCA showed that the bacterial community (class) was significantly related to cat1, qnrB, qnrS, tetW, tetC, sul1, blaIMP4 and tetZ (Figure 6C).
In our study, 16S rRNA sequencing techniques were employed to identify bacterial community structures from the Ba River. There was a clear differentiation of bacterial communities between sediment and surface water. For example, relative abundance of Acidobacteria, Chloroflexi, Spirochaetes, Chlamydiae, and Aminicenantes in sediment were higher than 1%, while those were minor abundance (<1%) in water samples. Additionally, the results showed that bacterial diversity and richness in sediment were relatively higher than those in water, suggesting that sediment in river environments may serve as a reservoir of microorganisms and a mainly environmental monitor. These results are consistent with previous studies demonstrating a higher concentration of microorganisms in sediment than that in water (Zeng et al., 2016).
Additionally, the bacterial communities in W5 and W6 were clustered away from the other water samples, and interestingly similar results were founded in S5 and S6. It should be noted that site 5 and 6 were close to domestic sewage outlet and WWTP discharge port. Thus, bacterial communities in site 5 and 6 might be affected by wastewater from residential area and WWTP. The previous findings also indicated that the sediment bacterial community in Tai Lake were affected by four different pollution sources (Wang et al., 2016). Proteobacteria and Bacteroidetes were proved to be the dominant components in this study. Similar results in sediment samples were also found in WWTPs in Colombia and China (Silva-Bedoya et al., 2016; Guo et al., 2017). Within the Proteobacteria phylum, Alphaproteobacteria and Betaproteobacteria class were significantly enriched in W4, whose sampling site is close to the confluence of Ba River and Chan River which is also received wastewater from WWTPs (Zhang et al., 2015). LEfse also showed that the four classes within Proteobacteria phylum were significantly enriched in S5 polluted by domestic sewage, which was consistent with previous investigation that Proteobacteria had the highest percentage in all river water samples contaminated by livestock breeding wastewater (Yang et al., 2017). In this study, Bacteroidia was significantly enriched from class to family in W5. These results suggest that Proteobacteria and Bacteroidia could survive in the wastewater environment, and their high abundance and diversity might be a reflection of human feces contamination. Some previous studies have also demonstrated that Bacteroidales order was enriched in the gut microbiota of many mammals and that specific species within this order have been proposed as fecal indicators (Fremaux et al., 2009; Liang et al., 2012). It was reported that Bacilli was the most active bacteria in the de novo DNA synthesis of the spore germination in dried river sediment, suggesting that Bacilli could become primary microbial colonizers and then passively release planktonic bacteria into the freshwater (Fazi et al., 2008). Besides, previous study indicated the wide distribution of Clostridia DNA in sediment samples more than surface water (Marcheggiani et al., 2008). Similarly, in the Firmicutes phylum, Clostridia and Bacilli class showed abundance advantage in S6 sediment sample in this study. The Firmicutes phylum was associated with wastewater samples with high antibiotics and extreme environmental conditions (Novo et al., 2013; Meng et al., 2016). Also, CCA showed that Epsilonproteobacteria and Bacteroidia were linked with TP, Cu, SM1, and OTC in W5, and Gammaproteobacteria were associated with TN, S, TMP, and TC in W6, which suggested that the changed abundance for these bacterial groups might result from both the antibiotics and the co-existing pollutants in water (Li et al., 2011). Based on the above, we speculate that the complicated impacts of the possible pollutants such as nutrients, heavy metals and antibiotics might prompt to select the appropriate bacterial groups and subsequently change the microbial diversity and composition.
Antibiotics analysis showed that high concentrations of SM1 and ERY were detected in water samples and high concentrations of OTC and CFZ were detected in sediment samples. The tetracyclines were mainly accumulated in sediment compared to water samples, whereas the concentration of TMP in the water was an order of magnitude higher than the corresponding sediment apart from site 4 and 7. The possible explanation was that the different distribution of antibiotics should be dependent on antibiotic physicochemical stability and geographical areas characteristics. Along the river flow from site 5 to 7, the concentration of OTC decreased in water samples and increased in sediment samples, supporting that tetracyclines were highly absorbed in organic matter, sediment and soil (Christou et al., 2017). The jump of ERY level at site 4 and its consistent increase in water samples from site 4 to 7 indicated that ERY was from Chan River, resident domestic sewage and the WWTP near by the river. There was no substantial increase in sediment ERY from site 4 to 7 with only somewhat elevation in S4 and S5. The shorter distance from the confluence of Chan River to the downstream rubber dam (less than 400 m) may facilitate the precipitation of suspended particles containing abundant ERY from Chan River, which could account for the relatively higher level of ERY in S4. The direct discharge of untreated wastewater at site 5 might prompt more ERY into sediment. In the present study, the high concentrations of SDZ and SM1 were detected in site 5, indicating its main source was residential domestic sewage. Compared to site 5, the concentrations of SDZ and SM1 were distinctly decreased in site 6 and 7, suggesting that the SDZ and SM1 might be removed by the WWTPs before their discharge into river near site 6. It might be attributed to the high removal efficiency for sulfonamides (up to 99.3%) in WWTPs (Zhang et al., 2017). CCA results showed that TMP was significantly related to Acinetobacter in W6 (Supplementary Figure S5). Although some species of Acinetobacter are environmental commensal bacteria, other species might cause serious nosocomial infections and community-acquired infections. Furthermore the treatment of Acinetobacter infections was difficult due to intrinsic resistance to multiple antimicrobial agents (Dijkshoorn et al., 2007; Clark et al., 2016). A previous study has also described that several Arcobacter carrying multidrug resistance was related to human and animal disease (Ferreira et al., 2016). Arcobacter, known as fecal contamination indicators, has gained visibility as opportunistic pathogens (Novo et al., 2013). In this study, SM1 and OTC were positively related to Arcobacter in W5. Therefore, it indicated that antibiotics posed selective pressure on some opportunistic pathogens, which was contributed to the increase of resistant prevalence and had adverse impact on water quality and human health. The concentrations of antibiotics including CHP, ERY, NOR, and SDZ were positively correlated with the abundance of Deltaproteobacteria and negatively with Actinobacteria and Clostridia among sediment samples. In addition, Cyanobacteria showed a positive association with TC and ROX. Thus, we speculated that different responses of bacterial communities to antibiotic pollution probably due to antibiotics pose selective pressure on indigenous bacterial communities. These were consistent with a previous study, in which showed that Deltaproteobacteria, Bacilli, Clostridia, and Epsilonproteobacteria might be specifically associated with antibiotic (PEN G and OTC, respectively) polluted rivers (Xiong et al., 2015).
To investigate how the bacterial composition changes under antibiotic selective pressure, the abundance of 21 ARGs were determined in water and sediment samples. In the present study, the tetC, blaTEM, ermF and sul1 had a higher relative abundance ranging from 10-4 to 100 copies/16S rRNA gene copies in all samples. Previous studies have indicated that the relative abundance of ARGs were higher than 10-4 in contaminated sites (Graham et al., 2011). Therefore, it was reasonable to assume that our samples had relatively higher pollution levels of ARGs. The concentrations of antibiotics in freshwater play an important role in the maintenance and enrichment of ARGs and antibiotic resistance (Xiong et al., 2015). In this study, significant correlations were observed between several ARGs and corresponding antibiotics. For examples, the tetC and blaTEM showed a similar tendency for the residue of OTC and CFZ, suggesting that antibiotics might pose the selective pressure on ARGs. Whereas, the relative abundance of sul2 was negatively correlated with SDZ and SM1 (Figure 6E), Huang et al. (2017) indicated that sul2 was significant correlated to intI1 in water and sediment samples, which meant that sul2 gene were associated with the mobile genetic elements (Cheng et al., 2013). Researchers also found that sul2 and sul3 located on plasmids are transferable and stable even in the absence of antibiotics (Suhartono et al., 2016). Thus, this negative correlation between sul2 and residue of sulfonamides might be attributed to mobility of sul2 located on mobile genetic elements. RDA showed that the relative abundance of qnrS, blaTEM, ermC, cmlA, and tetZ displayed significant positive correlations with Bacteroidetes and Actinobacteria, as well as cat1, ermF, and sul2 showed significant positive correlations with Proteobacteria in water samples. Researchers have found that the transfer of ARGs between bacterial species occurred mainly among Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, and mobile ARGs were most enriched in Proteobacteria (Hu et al., 2016). Similarly, in sediment samples, CCA showed that Deltaproteobacteria was significantly related to cat1, qnrB, qnrS, tetW, tetC, sul1, and blaIMP4. Based on the above, it indicated that ARGs might transfer among bacterial community by MGEs in this riverine system.
The present study aimed to analyze the respond of microbial composition to the gradient pollution in river system. Wastewater from residential area and WWTP discharge may have contributed to the differences among bacterial communities in this riverine system. Sediment in river environments may serve as a reservoir of microorganisms. SM1 and ERY were the predominant antibiotics in water samples while high concentrations of OTC and CFZ were detected in sediments. TMP was significantly related to Acinetobacter in W6, and SM1 and OTC had positive correlation with Arcobacter in W5. Some antibiotics posed selective pressure on some opportunistic pathogens or indigenous bacterial communities. The tetC, blaTEM, ermF and sul1 had the higher pollution abundance ranging from 10-4 to 100 copies/16S rRNA gene copies in all samples. And significant correlations were observed between ARGs and matching antibiotics. These finding provide new data to the limited information available on the bacterial community characteristics, abundance of antibiotics and ARGs in urban river of China.
YG and ZW contributed conception and design of the study. LW and GZ organized the database. JJ and XX performed the statistical analysis. YG wrote the first draft of the manuscript. JJ and ZW wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This study was supported by grant from the National Natural Science Foundation of China (31870487).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Xuan Chen Biological Technology Co., Ltd. (Shaanxi, China) for the help in data analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03191/full#supplementary-material
Abia, A. L. K., Alisoltani, A., Keshri, J., and Ubomba-Jaswa, E. (2017). Metagenomic analysis of the bacterial communities and their functional profiles in water and sediments of the Apies River, South Africa, as a function of land use. Sci. Total Environ. 616–617, 326–334. doi: 10.1016/j.scitotenv.2017.10.322
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cheng, W., Chen, H., Su, C., and Yan, S. (2013). Abundance and persistence of antibiotic resistance genes in livestock farms: a comprehensive investigation in eastern China. Environ. Int. 61, 1–7. doi: 10.1016/j.envint.2013.08.023
Christou, A., Aguera, A., Bayona, J. M., Cytryn, E., Fotopoulos, V., Lambropoulou, D., et al. (2017). The potential implications of reclaimed wastewater reuse for irrigation on the agricultural environment: the knowns and unknowns of the fate of antibiotics and antibiotic resistant bacteria and resistance genes - A review. Water Res. 123, 448–467. doi: 10.1016/j.watres.2017.07.004
Clark, N. M., Zhanel, G. G., and Lynch, J. P. III (2016). Emergence of antimicrobial resistance among Acinetobacter species: a global threat. Curr. Opin. Crit. Care 22, 491–499. doi: 10.1097/MCC.0000000000000337
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., Mcgarrell, D. M., Sun, Y., et al. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Dijkshoorn, L., Nemec, A., and Seifert, H. (2007). An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5, 939–951. doi: 10.1038/nrmicro1789
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fazi, S., Amalfitano, S., Piccini, C., Zoppini, A., Puddu, A., and Pernthaler, J. (2008). Colonization of overlaying water by bacteria from dry river sediments. Environ. Microbiol. 10, 2760–2772. doi: 10.1111/j.1462-2920.2008.01695.x
Ferreira, S., Queiroz, J. A., Oleastro, M., and Domingues, F. C. (2016). Insights in the pathogenesis and resistance of Arcobacter: a review. Crit. Rev. Microbiol. 42, 364–383. doi: 10.3109/1040841X.2014.954523
Fremaux, B., Gritzfeld, J., Boa, T., and Yost, C. K. (2009). Evaluation of host-specific Bacteroidales 16S rRNA gene markers as a complementary tool for detecting fecal pollution in a prairie watershed. Water Res. 43, 4838–4849. doi: 10.1016/j.watres.2009.06.045
Garrido, L., Sanchez, O., Ferrera, I., Tomas, N., and Mas, J. (2014). Dynamics of microbial diversity profiles in waters of different qualities. Approximation to an ecological quality indicator. Sci. Total Environ. 468–469, 1154–1161. doi: 10.1016/j.scitotenv.2013.08.065
Graham, D. W., Olivares-Rieumont, S., Knapp, C. W., Lima, L., Werner, D., and Bowen, E. (2011). Antibiotic resistance gene abundances associated with waste discharges to the Almendares River near Havana, Cuba. Environ. Sci. Technol. 45, 418–424. doi: 10.1021/es102473z
Guo, J., Li, J., Chen, H., Bond, P. L., and Yuan, Z. (2017). Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 123, 468–478. doi: 10.1016/j.watres.2017.07.002
He, X., Xu, Y., Chen, J., Ling, J., Li, Y., Huang, L., et al. (2017). Evolution of corresponding resistance genes in the water of fish tanks with multiple stresses of antibiotics and heavy metals. Water Res. 124, 39–48. doi: 10.1016/j.watres.2017.07.048
Hu, Y., Yang, X., Li, J., Lv, N., Liu, F., Wu, J., et al. (2016). The bacterial mobile resistome transfer network connecting the animal and human microbiomes. Appl. Environ. Microbiol. 82, 6672–6681. doi: 10.1128/AEM.01802-16
Huang, L., Xu, Y. B., Xu, J. X., Ling, J. Y., Chen, J. L., Zhou, J. L., et al. (2017). Antibiotic resistance genes (ARGs) in duck and fish production ponds with integrated or non-integrated mode. Chemosphere 168, 1107–1114. doi: 10.1016/j.chemosphere.2016.10.096
Jia, J., Guan, Y., Cheng, M., Chen, H., He, J., Wang, S., et al. (2018). Occurrence and distribution of antibiotics and antibiotic resistance genes in Ba River, China. Sci. Total Environ. 642, 1136–1144. doi: 10.1016/j.scitotenv.2018.06.149
Jia, S., Zhang, X. X., Miao, Y., Zhao, Y., Ye, L., Li, B., et al. (2017). Fate of antibiotic resistance genes and their associations with bacterial community in livestock breeding wastewater and its receiving river water. Water Res. 124, 259–268. doi: 10.1016/j.watres.2017.07.061
Li, D., Qi, R., Yang, M., Zhang, Y., and Yu, T. (2011). Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 45, 6063–6073. doi: 10.1016/j.watres.2011.09.002
Li, H., and Hu, B. (2016). Investigation in clinical application of antibacterial drugs from 13 tertiary hospitals in Xi’an City from 2013 to 2015. China Med. Her. 13, 176–180.
Liang, Z., He, Z., Zhou, X., Powell, C. A., Yang, Y., Roberts, M. G., et al. (2012). High diversity and differential persistence of fecal Bacteroidales population spiked into freshwater microcosm. Water Res. 46, 247–257. doi: 10.1016/j.watres.2011.11.004
Luo, Y., Mao, D., Rysz, M., Zhou, Q., Zhang, H., Xu, L., et al. (2010). Trends in antibiotic resistance genes occurrence in the Haihe River, China. Environ. Sci. Technol. 44, 7220–7225. doi: 10.1021/es100233w
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Marcheggiani, S., Iaconelli, M., D’angelo, A., Pierdominici, E., La Rosa, G., Muscillo, M., et al. (2008). Microbiological and 16S rRNA analysis of sulphite-reducing clostridia from river sediments in central Italy. BMC Microbiol. 8:171. doi: 10.1186/1471-2180-8-171
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31
Meng, L., Liu, H., Bao, M., and Sun, P. (2016). Microbial community structure shifts are associated with temperature, dispersants and nutrients in crude oil-contaminated seawaters. Mar. Pollut. Bull. 111, 203–212. doi: 10.1016/j.marpolbul.2016.07.010
Miao, L., Wang, C., Hou, J., Wang, P., Ao, Y., Li, Y., et al. (2017). Influence of silver nanoparticles on benthic oxygen consumption of microbial communities in freshwater sediments determined by microelectrodes. Environ. Pollut. 224, 771–778. doi: 10.1016/j.envpol.2017.01.017
Michael-Kordatou, I., Karaolia, P., and Fatta-Kassinos, D. (2017). The role of operating parameters and oxidative damage mechanisms of advanced chemical oxidation processes in the combat against antibiotic-resistant bacteria and resistance genes present in urban wastewater. Water Res. 129, 208–230. doi: 10.1016/j.watres.2017.10.007
Milic, N., Milanovic, M., Letic, N. G., Sekulic, M. T., Radonic, J., Mihajlovic, I., et al. (2013). Occurrence of antibiotics as emerging contaminant substances in aquatic environment. Int. J. Environ. Health Res. 23, 296–310. doi: 10.1080/09603123.2012.733934
Monard, C., Gantner, S., Bertilsson, S., Hallin, S., and Stenlid, J. (2016). Habitat generalists and specialists in microbial communities across a terrestrial-freshwater gradient. Sci. Rep. 6:37719. doi: 10.1038/srep37719
Novo, A., Andre, S., Viana, P., Nunes, O. C., and Manaia, C. M. (2013). Antibiotic resistance, antimicrobial residues and bacterial community composition in urban wastewater. Water Res. 47, 1875–1887. doi: 10.1016/j.watres.2013.01.010
Pei, R., Kim, S. C., Carlson, K. H., and Pruden, A. (2006). Effect of river landscape on the sediment concentrations of antibiotics and corresponding antibiotic resistance genes (ARG). Water Res. 40, 2427–2435. doi: 10.1016/j.watres.2006.04.017
Petrie, B., Barden, R., and Kasprzyk-Hordern, B. (2015). A review on emerging contaminants in wastewaters and the environment: current knowledge, understudied areas and recommendations for future monitoring. Water Res. 72, 3–27. doi: 10.1016/j.watres.2014.08.053
Rico, A., Oliveira, R., Mcdonough, S., Matser, A., Khatikarn, J., Satapornvanit, K., et al. (2014). Use, fate and ecological risks of antibiotics applied in tilapia cage farming in Thailand. Environ. Pollut. 191, 8–16. doi: 10.1016/j.envpol.2014.04.002
Rodriguez-Mozaz, S., Chamorro, S., Marti, E., Huerta, B., Gros, M., Sanchez-Melsio, A., et al. (2015). Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 69, 234–242. doi: 10.1016/j.watres.2014.11.021
Ruiz-González, C., Niño-García, J. P., and Giorgio, P. A. (2015). Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol. Lett. 18, 1198–1206. doi: 10.1111/ele.12499
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Silva-Bedoya, L. M., Sanchez-Pinzon, M. S., Cadavid-Restrepo, G. E., and Moreno-Herrera, C. X. (2016). Bacterial community analysis of an industrial wastewater treatment plant in Colombia with screening for lipid-degrading microorganisms. Microbiol. Res. 192, 313–325. doi: 10.1016/j.micres.2016.08.006
Sim, W. J., Lee, J. W., Lee, E. S., Shin, S. K., Hwang, S. R., and Oh, J. E. (2011). Occurrence and distribution of pharmaceuticals in wastewater from households, livestock farms, hospitals and pharmaceutical manufactures. Chemosphere 82, 179–186. doi: 10.1016/j.chemosphere.2010.10.026
Su, L., Cai, H., Kolandhasamy, P., Wu, C., Rochman, C. M., and Shi, H. (2017). Using the Asian clam as an indicator of microplastic pollution in freshwater ecosystems. Environ. Pollut. 234, 347–355. doi: 10.1016/j.envpol.2017.11.075
Subirats, J., Triado-Margarit, X., Mandaric, L., Acuna, V., Balcazar, J. L., Sabater, S., et al. (2017). Wastewater pollution differently affects the antibiotic resistance gene pool and biofilm bacterial communities across streambed compartments. Mol. Ecol. 26, 5567–5581. doi: 10.1111/mec.14288
Suhartono, S., Savin, M., and Gbur, E. E. (2016). Genetic redundancy and persistence of plasmid-mediated trimethoprim/sulfamethoxazole resistant effluent and stream water Escherichia coli. Water Res. 103, 197–204. doi: 10.1016/j.watres.2016.07.035
Wang, J., Li, Y., Wang, P., Niu, L., Zhang, W., and Wang, C. (2016). Response of bacterial community compositions to different sources of pollutants in sediments of a tributary of Taihu Lake, China. Environ. Sci. Pollut. Res. Int. 23, 13886–13894. doi: 10.1007/s11356-016-6573-9
Wang, S., Zhu, Z., He, J., Yue, X., Pan, J., and Wang, Z. (2018). Steroidal and phenolic endocrine disrupting chemicals (EDCs) in surface water of Bahe River, China: distribution, bioaccumulation, risk assessment and estrogenic effect on Hemiculter leucisculus. Environ. Pollut. 243, 103–114. doi: 10.1016/j.envpol.2018.08.063
Xi, C., Zhang, Y., Marrs, C. F., Ye, W., Simon, C., Foxman, B., et al. (2009). Prevalence of antibiotic resistance in drinking water treatment and distribution systems. Appl. Environ. Microbiol. 75, 5714–5718. doi: 10.1128/AEM.00382-09
Xi, X., Wang, M., Chen, Y., Yu, S., Hong, Y., Ma, J., et al. (2015). Adaption of the microbial community to continuous exposures of multiple residual antibiotics in sediments from a salt-water aquacultural farm. J. Hazard. Mater. 290, 96–105. doi: 10.1016/j.jhazmat.2015.02.059
Xiong, W., Sun, Y., Ding, X., Wang, M., and Zeng, Z. (2015). Selective pressure of antibiotics on ARGs and bacterial communities in manure-polluted freshwater-sediment microcosms. Front. Microbiol. 6:194. doi: 10.3389/fmicb.2015.00194
Yang, F., Huang, L., Li, L., Yang, Y., Mao, D., and Luo, Y. (2017). Discharge of KPC-2 genes from the WWTPs contributed to their enriched abundance in the receiving river. Sci. Total Environ. 581–582, 136–143. doi: 10.1016/j.scitotenv.2016.12.063
Ye, D., Chang, J., Ji, Y., Zhu, S., Yan, K., Lu, B., et al. (2016). Analysis of the use of prescribed antibacterial in primary health care facilities in Shaanxi province. Chin. Pharm. Aff. 30, 215–220.
Zeng, J., Zhao, D., Li, H., Huang, R., Wang, J., and Wu, Q. L. (2016). A monotonically declining elevational pattern of bacterial diversity in freshwater lake sediments. Environ. Microbiol. 18, 5175–5186. doi: 10.1111/1462-2920.13526
Zhang, C., Wang, Y., and Du, C. (2015). Antibiotic resistance of heterotrophic bacteria and characteristics of microbial community structure in urban rivers in China. Res. Environ. Sci. 28, 713–719.
Zhang, X., Zhao, H., Du, J., Qu, Y., Shen, C., Tan, F., et al. (2017). Occurrence, removal, and risk assessment of antibiotics in 12 wastewater treatment plants from Dalian, China. Environ. Sci. Pollut. Res. Int. 24, 16478–16487. doi: 10.1007/s11356-017-9296-7
Zhao, Z., Wang, J., Han, Y., Chen, J., Liu, G., Lu, H., et al. (2017). Nutrients, heavy metals and microbial communities co-driven distribution of antibiotic resistance genes in adjacent environment of mariculture. Environ. Pollut. 220, 909–918. doi: 10.1016/j.envpol.2016.10.075
Zhou, Z. C., Zheng, J., Wei, Y. Y., Chen, T., Dahlgren, R. A., Shang, X., et al. (2017). Antibiotic resistance genes in an urban river as impacted by bacterial community and physicochemical parameters. Environ. Sci. Pollut. Res. Int. 24, 23753–23762. doi: 10.1007/s11356-017-0032-0
Keywords: antibiotics, antibiotic resistance genes, bacterial community, wastewater treatment plant, resident domestic sewage, urban river
Citation: Guan Y, Jia J, Wu L, Xue X, Zhang G and Wang Z (2018) Analysis of Bacterial Community Characteristics, Abundance of Antibiotics and Antibiotic Resistance Genes Along a Pollution Gradient of Ba River in Xi’an, China. Front. Microbiol. 9:3191. doi: 10.3389/fmicb.2018.03191
Received: 21 July 2018; Accepted: 10 December 2018;
Published: 21 December 2018.
Edited by:
Thomas Schwartz, Karlsruhe Institute of Technology (KIT), GermanyReviewed by:
Jian-Qiang Su, Institute of Urban Environment (CAS), ChinaCopyright © 2018 Guan, Jia, Wu, Xue, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zaizhao Wang, enp3YW5nQG53c3VhZi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.