
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 21 December 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.03170
This article is part of the Research TopicHerpesvirus LatencyView all 14 articles
 Océane Sorel
Océane Sorel Ilhem Messaoudi*
Ilhem Messaoudi*Varicella zoster virus (VZV) is a neurotropic alphaherpesvirus and the causative agent of varicella (chickenpox) in humans. Following primary infection, VZV establishes latency in the sensory ganglia and can reactivate to cause herpes zoster, more commonly known as shingles, which causes significant morbidity, and on rare occasions mortality, in the elderly. Because VZV infection is highly restricted to humans, the development of a reliable animal model has been challenging, and our understanding of VZV pathogenesis remains incomplete. As an alternative, infection of rhesus macaques with the homologous simian varicella virus (SVV) recapitulates the hallmarks of VZV infection and thus constitutes a robust animal model to provide critical insights into VZV pathogenesis and the host antiviral response. In this model, SVV infection results in the development of varicella during primary infection, generation of an adaptive immune response, establishment of latency in the sensory ganglia, and viral reactivation upon immune suppression. In this review, we discuss our current knowledge about host and viral factors involved in the establishment of SVV latency and reactivation as well as the important role played by T cells in SVV pathogenesis and antiviral immunity.
Varicella zoster virus (VZV) is one of the nine human herpesviruses. Primary VZV infection results in varicella (also known as chickenpox), a disease characterized by a vesicular rash, fever, headache, and loss of appetite (Heininger and Seward, 2006). Like other alphaherpesviruses, VZV exhibits neurotropism and establishes latency in sensory ganglia neurons. VZV transmission is thought to occur through either inhalation of saliva droplets containing infectious particles and by direct contact with virus in varicella or zoster skin lesions (Leclair et al., 1980; Sawyer et al., 1994; Suzuki et al., 2004). Subsequently, VZV is presumed to undergo initial replication in the upper respiratory tract and tonsillar lymph nodes before viremia and dissemination to the skin leading to the development of varicella (Zerboni et al., 2014). Although primary VZV infection in immunocompetent individuals usually results in a benign disease, serious complications can occur in immune compromised individuals, including pneumonia, secondary bacterial infection, and stroke (Gnann, 2002; Chiner et al., 2010; Wiegering et al., 2011). Two hypotheses are proposed to explain how VZV reaches the ganglia: (Heininger and Seward, 2006) VZV infects sensory neurons via retrograde axonal transport from the infected skin, and (Suzuki et al., 2004) VZV is carried by infected T cells to the ganglia through the hematogenous route (Depledge et al., 2018b). During reactivation, VZV travels from the ganglia to the skin via anterograde axonal transport to cause herpes zoster (HZ, also known as shingles), a painful and debilitating disease that primarily affects the elderly and immunocompromised. HZ is characterized by severe prodromal pain followed by a rash restricted to the dermatome innervated by the ganglia from which the virus reactivated (Wareham and Breuer, 2007). The incidence of HZ is estimated to be 3 per 1000 adults between the age of 40 and 50 years old and increases to 11 cases per 1000 adults above the age of 80 years old (Keating, 2016). VZV reactivation can also cause other complications such as HZ ophthalmicus, vasculitis, stroke, as well as pain without development of a rash, referred as zoster sine herpete (Dayan and Peleg, 2017). Routine vaccination of children against chickenpox was implemented in several countries including Japan (1988), the United States (1995), and Canada (1999) using the live attenuated VZV vaccine that was derived from the Oka strain (Gershon, 2017). There are currently two available vaccines to prevent HZ in the elderly: a live-attenuated (Zostavax®, licensed 2005, ∼55% efficacious) and a recombinant (Shingrix®, licensed 2018, 97% efficacious) vaccine (Arnold and Messaoudi, 2017a; James et al., 2018).
Despite extensive studies, our understanding of VZV pathogenesis remains incomplete. First, the mechanisms by which VZV disseminates from the initial site of infection to the skin and ganglia are poorly understood. The prevailing model proposes that VZV initially replicates within mucosal epithelial cells at the sites of entry, followed by spread to tonsils and other regional lymphoid tissues, where VZV gains access to T cells that deliver the virus to cutaneous sites of replication and sensory ganglia (Zerboni et al., 2014). However, this model was constructed primarily using data obtained from in vitro studies carried out using the attenuated Oka vaccine strain and in vivo studies utilizing a severe-combined immunodeficient (SCID) mouse model implanted with human fetal tissues (SCID-hu) (Moffat et al., 1995; Ku et al., 2004). Moreover, the exact timeline as well as the mechanisms through which the latency is established and maintained following primary infection still remains unclear. In order to address these questions, a reliable animal model that recapitulates the key hallmarks of VZV infection is necessary.
Numerous attempts have been made to develop a reliable animal model that recapitulates the hallmarks of VZV infection. However, the success of these models remains limited due to the strict human specificity of VZV. Although seroconversion was observed following VZV inoculation in different rodent models including guinea pigs, mice, and rats; no virus was detected in circulation in these models (Haberthur and Messaoudi, 2013). Infection of guinea pigs was rendered possible through the derivation of a guinea pig-adapted VZV strain (by passaging the virus multiple times in fetal guinea pig cells) and injection of peripheral blood mononuclear cells (PBMCs) that are first infected in vitro (Gan et al., 2014). Although VZV was shown to establish latency in enteric neurons in vivo, the inconsistent development of both viremia and rash in addition to the inability to induce VZV reactivation in vivo limits the use of this small animal model (Haberthur and Messaoudi, 2013). Reactivation can be induced in vitro through overexpression of VZV ORF61 in latently infected guinea pigs enteric neurons (Gershon et al., 2008). Subcutaneous injection of VZV-infected cells in rats was reported to lead to establishment of a latency-like quiescent state in sensory ganglia although the virus was not shown to be able to reactivate (Annunziato et al., 1998; Sadzot-Delvaux et al., 1990). In addition, footpad inoculation of VZV-infected cells in the rat model has been used to study post-herpetic neuralgia (PHN), long-term chronic pain associated with zoster (Dalziel et al., 2004). Inoculation of non-human primates with VZV also resulted in latency and the development of immunity in the absence of viremia or varicella, suggestive of abortive infection (Felsenfeld and Schmidt, 1979; Meyer et al., 2015a; Myers et al., 1987, Provost et al., 1987; Cohen et al., 1996; Willer et al., 2012). Intradermal inoculation of chimpanzees resulted in a local rash, however, several restrictions have been placed on the use of apes for biomedical research (Myers et al., 1987; Cohen et al., 1996).
In order to overcome the host specificity restriction of VZV, a humanized SCID mouse model was developed using human tissue xenografts. The engraftment of different human fetal tissues (thymus/liver, skin, ganglia, and lung) in this model allowed direct inoculation of VZV and resulted in several important insights into VZV pathogenesis (Moffat et al., 1995; Ku et al., 2004; Zerboni et al., 2005; Reichelt et al., 2008; Wang et al., 2017). However, this model also presents several limitations including: (1) direct inoculation into the human xenografts tissues does not mimic natural route of transmission; (2) the lack of adaptive immunity, which is critical to control viral infection; and (3) the possibility that the strict human host specificity of VZV may alter virus behavior in this model; (4) the use of the attenuated Oka vaccine strain in some of these studies, which compared to the parent wild type strain contains numerous nucleotide substitutions found in multiple open reading frames (ORFs) and may therefore not accurately model the behavior of wild type virus strains (Jones and Arvin, 2003; Yamanishi, 2008; Sen et al., 2015).
To overcome these limitations an alternative animal model was developed where non-human primates are inoculated with Simian varicella virus (SVV), an alphaherpesvirus that causes a vesicular rash in Old World monkeys. SVV and VZV virions have a diameter of 170–200 nm and 80–120 nm, respectively, and are composed of a nucleocapsid of icosahedral symmetry surrounded by a viral envelope (Gray, 2010). The nucleocapsid of both SVV and VZV contains a linear double-stranded DNA genome of 124,138 and 124,884 bp, respectively. The viral genomes of SVV and VZV include a unique long sequence of 104.1 and 104.8 kb, respectively, and a unique short region that comprises a 4.9 and 5.2 kb sequence for SVV and VZV, respectively, as well as internal repeat and terminal repeat regions (Clarke et al., 1992). SVV and VZV genomes share 70–75% DNA homology (Gray and Oakes, 1984) and an amino acid identity ranging from 27 to 75% (Gray et al., 2001). Both VZV and SVV encode 74 ORFs of which 71 are distinct and 3 (ORFs 69, 70, and 71) are duplicated within the repeat regions (Mahalingam and Gilden, 2007; Zerboni et al., 2014). Despite exhibiting co-linearity with respect to gene organization, SVV ORFA is absent in VZV genome while SVV does not include a gene homolog of VZV ORF2 (Gray et al., 2001).
The first outbreak of a varicella-like disease in non-human primates was reported in 1967 followed by several epizootics in primates facilities worldwide (Clarkson et al., 1967; Gray, 2008). Depending on the non-human species, SVV infection can cause disease that ranges from a mild varicella (in rhesus macaques also called Macaca mulata) to a severe and life-threatening disease associated with high morbidity and mortality rates [cynomolgus monkeys (Macaca fascicularis) and African green monkeys (Chlorocebus sabaeus)] (Gray, 2008). This spectrum of disease outcomes is a hallmark of herpesvirus infection in species that are closely related to the natural host, e.g., Macacine herpesvirus 1 (also known as herpes simian B virus) infection in humans (Elmore and Eberle, 2008), Elephant endotheliotropic herpesvirus infection in Asian elephants (Long et al., 2016), and Alcelaphine herpesvirus 1 in cattle (Sorel et al., 2017). Moreover, SVV infection in cynomolgus monkeys and African green monkeys results in persistent viremia which limits the use of these models to study the adaptive immune response against SVV (White et al., 2002; Mahalingam et al., 2007). In contrast, intra-bronchial inoculation of rhesus macaques with SVV faithfully recapitulates the hallmarks of VZV pathogenesis including: viremia, development of varicella, generation of robust cellular and humoral immune responses, establishment of latency in the sensory ganglia, and viral reactivation following immune suppression (Messaoudi et al., 2009; Meyer et al., 2011; Haberthur et al., 2013, 2014; Arnold et al., 2017; Figure 1).
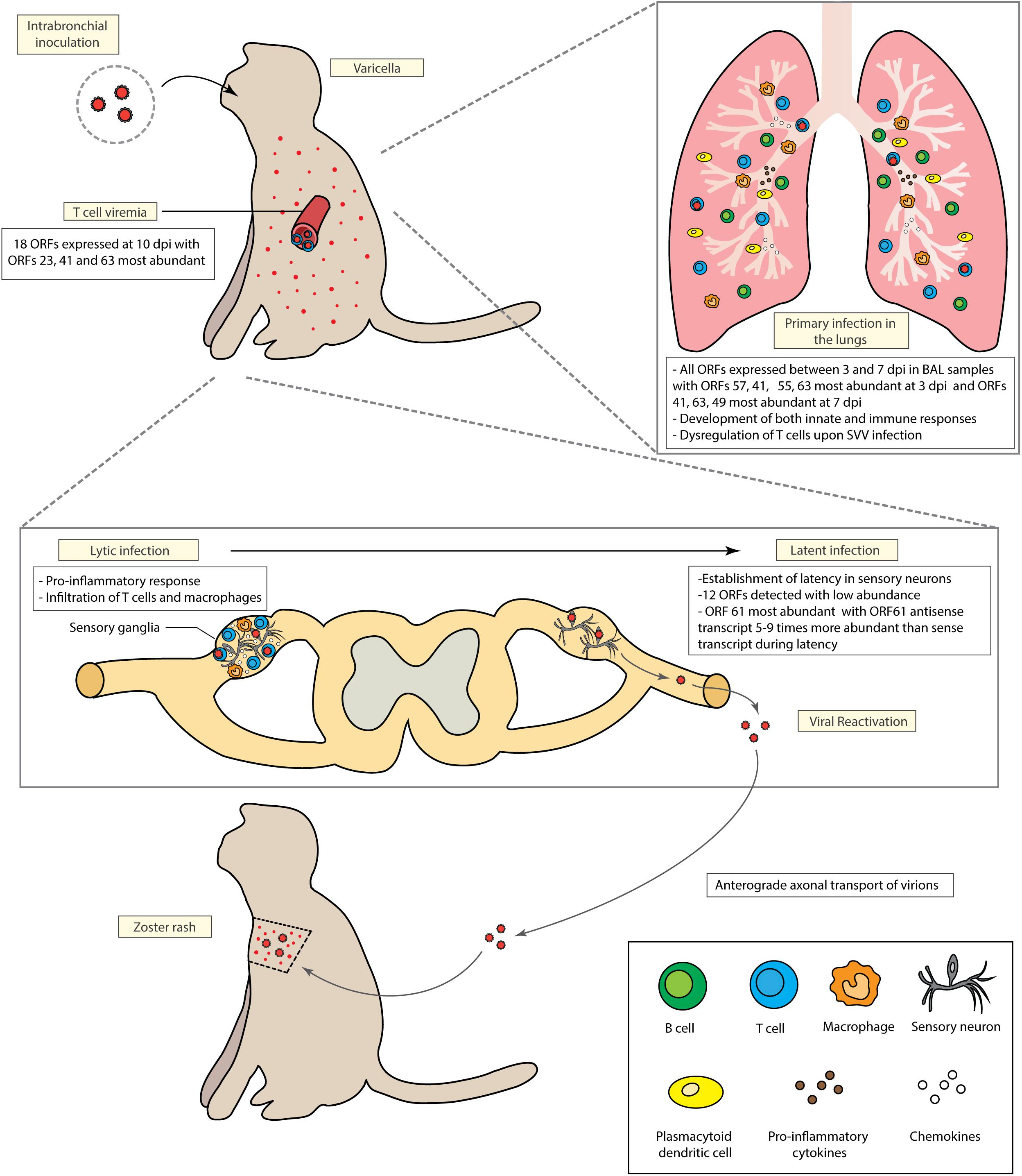
Figure 1. Model of Simian Varicella Virus (SVV) pathogenesis in rhesus macaques following intrabronchial inoculation. Intrabronchial inoculation of rhesus macaques with SVV results in primary infection associated with viral replication in the lung, T cell viremia and the development of varicella. SVV reaches sensory ganglia as early as 3 days post infection. Initial viral replication is followed by the establishment of latency in the sensory ganglia. SVV can reactivate upon immune suppression. Both primary infection and viral reactivation induce robust cellular and humoral immune responses.
Data from several studies carried out using the hu-SCID mouse model strongly suggest a critical role for T cells in VZV dissemination. First, direct inoculation of VZV-infected fibroblasts into human fetal thymus and liver xenografts placed under the kidney capsule of SCID mice revealed that T cells support VZV replication (Moffat et al., 1995). Furthermore, injection of VZV-infected T cells into human skin xenografts implanted in the SCID model demonstrated the importance of the type I interferon response in restricting VZV replication in the skin (Ku et al., 2004). More importantly, intravenous injection of VZV-infected T cells but not fibroblasts resulted in vesicular rash of human skin implants suggesting that T cells can traffic the virus to the skin (Ku et al., 2004). These observations gave rise to the current model which stipulates that VZV gains access to T cells that deliver the virus to cutaneous sites of replication (Zerboni et al., 2014). However, given the limitations of this mouse model, findings from these studies cannot be extrapolated to decisively uncover the mechanisms by which VZV hijacks T cells to disseminate to the ganglia in the human host in vivo.
In both rhesus macaques and African green monkeys, different subsets of immune cells, including T cells, were shown to reach the ganglia as early as 3 days post intrabronchial inoculation (dpi) during acute infection (Ouwendijk et al., 2013b; Arnold et al., 2016a), prior to the detection of anti-SVV specific T cell immunity, these results suggest that T cells play an important role in SVV dissemination to sensory ganglia. Interestingly, CD8 memory T cells were the most abundant subset of immune cells infiltrating the ganglia of SVV-infected African green monkeys and rhesus macaques (Arnold et al., 2016a; Ouwendijk et al., 2016). In support of the potential role of T cells as Trojan horse for SVV, T cells isolated from bronchial alveolar lavage (BAL) samples during primary infection supported viral replication (Arnold et al., 2016a). Similarly, T cells infiltrate the enteric nervous system during acute SVV infection in African green monkeys (Ouwendijk et al., 2018). These results obtained in the SVV infection model along with the studies reporting that VZV infected T cells can traffic the virus to human xenografts in the SCID-hu mouse model further emphasize the importance played by T cells in varicella viruses dissemination and pathogenesis (Ku et al., 2004; Zerboni et al., 2005).
In order to improve our understanding of the mechanisms by which SVV alters T cells migratory behavior and to in SVV trafficking, a recent study analyzed the transcriptional profile of T cells in BAL samples following SVV infection in rhesus macaques (Arnold and Messaoudi, 2017b). This study reported that multiple cellular processes were dysregulated in T cells upon SVV infection, including genes involved in chromatin assembly, immune response, cell cycle, and cellular metabolism. These results suggest that SVV might alter T cell functions in order to achieve efficient viral replication and allow the virus to spread in the host while evading the immune system. Interestingly, in line with this hypothesis, previous in vitro studies using single-cell mass-spectrometry analysis (CyTOF) have reported that VZV infection of human tonsil T cells leads to upregulation of several immune genes including components of the TCR signaling machinery (Sen et al., 2014, 2015).
Transcriptomic analysis of BAL cells collected from rhesus macaques during SVV primary infection showed that all ORFs were expressed between 3 and 7 dpi with increasing intensity that correlated with viremia (Meyer et al., 2011). The most highly expressed SVV ORFs detected at 3 dpi in BAL samples were ORF 57 (unknown function), ORF 41 (capsid protein), ORF 55 (component of the DNA helicase-primase complex), and ORF 63 (transactional activator) (Meyer et al., 2011). At 7 dpi, ORF63, ORF41 as well as ORF49 (structural protein) were the most abundant transcripts expressed in BAL cells (Meyer et al., 2011). Parallel analysis of the viral transcriptome profiles of PBMCs derived from rhesus macaques infected with SVV revealed that only 18 SVV ORFs were expressed at 10 dpi in these samples, consistent with significantly lower viremia (Meyer et al., 2011). However, similarly to the BAL samples, the most highly expressed viral genes included ORFs 23, 41, and 63 (Meyer et al., 2011).
Latency is a state that is characterized by a restricted viral gene expression pattern. In line with that expectation, only 12 SVV ORFs were detected sporadically during SVV latent infection in the sensory ganglia. Importantly, ORF61 was the most abundant and consistently detected transcript in the ganglia during latent infection (Meyer et al., 2011). More specifically, the antisense transcript of ORF61 was found to be 5–9 times more abundant than sense transcripts (Messaoudi et al., 2009). Similar findings were reported for latently infected ganglia collected from Vervet and African green monkeys (Ou et al., 2007; Ouwendijk et al., 2013a). Recently, the presence of an ORF61 anti-sense transcript was reported for VZV latent infection (Depledge et al., 2018a). Although multiple isoforms of VZV ORF61 antisense transcripts were detected during lytic infection, only one isoform was predominant during latency and was shown to suppress VZV ORF61 expression (Depledge et al., 2018a). Taken together, these results suggest that cessation of ORF61 expression by the anti-sense transcript may be critical in the establishment and maintenance of latency. Interestingly, establishment of latency was not impaired in animals infected with an SVV mutant deleted of ORF61 (SVVΔORF61) (Meyer et al., 2013c). Since ORF61 is thought to be shut off by the anti-sense transcript in order to prevent reactivation during latency, the lack of the ORF61 anti-sense transcript following infection with SVVΔORF61 would explain why establishment and maintenance of latency are not affected by the deletion.
SVV ORF61 is an immediate early gene that encodes a protein with a RING finger motif at the amino terminus, which is important for potential E3 ubiquitin ligase activity as well as a nuclear localization signal at the N terminus (Gray et al., 2007). Previous in vitro studies showed that ORF61 protein can transactivate its own promoter as well as promoters of SVV genes of all kinetic classes (Gray et al., 2007). Although SVV ORF61 is non-essential for SVV lytic cycle in vitro, SVVΔORF61 replicates 2- to 5-fold less efficiently compared to the wild-type (WT) virus (Gray et al., 2007). Similarly, in vivo infection with SVVΔORF61 was associated to a decreased expression of all viral transcripts and decreased viral loads in rhesus macaques (Meyer et al., 2013c). Infection with SVVΔORF61 also led to increased infiltration of plasmacytoid dendritic cells (pDC) into the lungs and expression of interferon stimulated genes in vivo suggesting a potential role of ORF61 in evasion of the host innate immune response (Meyer et al., 2013c). Indeed, both SVV and VZV ORF61 were shown to interfere with NF-κB signaling in vitro (Whitmer et al., 2015).
Intrabronchial infection of rhesus macaques with SVV results in the development of both innate and adaptive immune responses in the lungs concomitant with a decrease in the SVV viral loads observed (Arnold et al., 2016b). The mucosal innate immune response is characterized by a significant production of pro-inflammatory cytokines, chemokines (including T cell chemoattractants) and IFNα into the alveolar space that correlates with increased frequency of pDCs (Haberthur et al., 2014; Arnold et al., 2016b). This initial response is followed by a robust proliferation and infiltration of B and T cells in the lungs (Haberthur et al., 2014). Although CD8 T cells were found to be more abundant, a higher proportion of CD4 T cells were specific to SVV in the BAL (Haberthur et al., 2014). This observation is in line with several studies that reported a critical role for CD4 T cells in controlling both SVV and VZV acute infection (Haberthur et al., 2011; Duncan and Hambleton, 2015; Sen et al., 2015; Sen and Arvin, 2016). Indeed, whereas depletion of B cells and CD8 T cells showed no or limited effect on disease severity, CD4 depletion led to higher viral loads, prolonged viremia, and disseminated varicella (Haberthur et al., 2011). These results explain why children with T cell deficiencies are more prone to developing serious complications following VZV infection whereas children with B cell deficiencies have uncomplicated disease (Arvin et al., 1978; Wilson et al., 1992; Nader et al., 1995; Redman et al., 1997; Zerboni et al., 1998).
The anti-SVV T cell responses during acute infection in rhesus macaques is broad with CD8 T cell responses directed mainly against immediate-early (IE) and early (E) viral proteins whereas CD4 T cell responses were mostly specific to late (L) proteins (Haberthur et al., 2013). During latency, the magnitude of the T cell response decreases dramatically and becomes more restricted (Haberthur et al., 2013). Specifically, T cell responses directed against only 5 ORFs (ORF 4, 11, 19, 31, and 37) were maintained during latency whereas specific T cell responses to ORFs 10, 20, 29, 31, 62, 63, 68 showed a significant decrease compared to primary infection (Haberthur et al., 2013). Amongst these viral antigens, ORF68 (gE) is the most abundant glycoprotein, a critical determinant of VZV pathogenesis (Moffat et al., 2004; Berarducci et al., 2009; Zerboni et al., 2011), and a highly immunogenic viral antigen (Vizoso Pinto et al., 2010). These data suggest that boosting T cell responses against these viral antigens that are highly immunogenic during acute infection but poorly recognized during latency may be a promising direction for HZ vaccine. Indeed, the highly efficacious new recombinant subunit HZ vaccine (Shingrix®) contains an adjuvanted form of VZV gE that was shown to elicit a robust humoral and cell-mediated immunity (Mo et al., 2002; James et al., 2018; Syed, 2018). In contrast, ZostavaxTM induces a lower VZV-specific cell-mediated immunity including a reduced gE-specific memory T cell responses compared to Shingrix®(Levin et al., 2018; Weinberg et al., 2018).
The importance of T cell responses during acute infection in the establishment of latency is evidenced by the detection of high level of viral transcription in ganglia of animals depleted of CD4 T cells during acute infection (Meyer et al., 2013b). It should be noted that at the time of ganglia analysis, the animals were no longer viremic. These results strongly suggest that loss of CD4 T cell immunity during acute infection impaired the establishment of a latency in sensory ganglia of infected macaques (Meyer et al., 2013b). In accordance with this observation, SVV infection of aged rhesus macaques was also characterized by dampened T cell responses and high levels of viral transcription inconsistent with latent infection (Meyer et al., 2013b). More recently, direct inoculation of VZV-infected fibroblasts into human fetal dorsal root ganglia (DRG) implanted under the kidney capsule as well as intravenous transfer of VZV-infected CD4 T cells showed persistent viral replication in the ganglia tissue followed eventually by latency (Zerboni et al., 2005; Reichelt et al., 2008). These data from the hu-SCID mouse suggest that adaptive immune responses may not be critical for the establishment of latency. However, the CD4 T cells were most likely obtained from VZV-seropositive individuals and therefore the fact that they may harbor VZV-specific T cells cannot be dismissed. Similarly to VZV, stress and immune suppression can induce SVV reactivation leading to anterograde axonal transport of virions to the skin causing HZ lesions (Soike et al., 1984; Mahalingam et al., 2007, 2010; Traina-Dorge et al., 2014). Because T cells were shown to be critical in the establishment of latency, a recent study investigated the specific role of T cell immunity in preventing SVV reactivation (Arnold et al., 2017). This study showed that depletion of either CD4 or CD8 T cells in latently infected animals led to subclinical reactivation (defined as viremia detected in the absence of zoster rash) and an increase in the viral loads in the ganglia (Arnold et al., 2017). Moreover, large transcriptional changes of genes involved in inflammation and neuronal functions were reported in the ganglia obtained from animals that experienced subclinical reactivation (Arnold et al., 2017). Taken together, these results support the critical role of T cell immunity in maintaining SVV latency.
Other studies have attempted to induce reactivation in SVV-latently infected rhesus macaques using a combination of total body irradiation (2–8 Gy) and immune suppressant regimens (cyclosporine and tacrolimus). In some of these studies, cynomolgus and rhesus macaques were irradiated before receiving tacrolimus and prednisone, resulting in clinical reactivation in 25 and 100% of animals, respectively (Mahalingam et al., 2007; Traina-Dorge et al., 2014). In another study, treatment with only immune suppressants resulted in 75% reactivation (Ouwendijk et al., 2013a). The incidence of HZ obtained following these experimental treatments is significantly higher than the reactivation rate reported in humans. Another perplexing outcome of these studies includes the very high incidence of reactivation in the non-treated controls which is often ∼100%. Other groups failed to reproduce these findings using the same approaches (Meyer et al., 2015b), potentially due to a higher level of stress induced by a longer transportation to the irradiation site for the animals in the compared to those housed in the Oregon National Primate Center.
Following reactivation, SVV antigens were detected in multiple tissues, including skin and lymph nodes in rhesus macaques despite the lack of viremia at the time of HZ (Traina-Dorge et al., 2015). In skin tissues, SVV antigens were found mainly in sweat glands, whereas in lymph nodes, they were detected in macrophages, dendritic cells (DCs), and T cells. It is possible that DCs containing SVV antigens are activating T cells in the peripheral lymph nodes or that infected DCs are transferring SVV to T cells as previously described for VZV (Abendroth et al., 2001). Additionally, SVV reactivation in rhesus macaques induces the development of a strong systemic pro-inflammatory response (Traina-Dorge et al., 2014) associated with an overall increased in the number of total T cells compared to latency (James et al., 2014). T cell infiltration was detected in the sensory ganglia of cynomolgus macaques experiencing reactivation where neurons were found to be surrounded mainly by CD8 rather than CD4 T cells (Ouwendijk et al., 2013a). Moreover, as previously reported for post-mortem human sensory ganglia derived from patients who suffered from HZ at the time of death (Steain et al., 2011), the authors detected elevated levels of CXCL10, a chemokine involved in T cell migration (Ouwendijk et al., 2013a). Taken together these results suggest that the pro-inflammatory response play an important role in initiating T cell recruitment to the site of SVV/VZV reactivation. However, the high reactivation rates in the control animals raises concerns about the clinical significance of these findings.
Although studies over the last few decades have led to significant advances in our understanding of VZV pathogenesis, several questions remain unanswered. Specifically, although it is now well established that that T cells play a critical in the pathogenesis of both VZV and SVV, the exact mechanisms by which VZV/SVV modulate T cell functions to alter their migratory properties and confer ability to access into the central nervous system are not known. Furthermore, the viral and cellular factors that control establishment and maintenance of SVV/VZV latency in sensory ganglia remain poorly understood. Notably, the role of the ORF61 anti-sense transcript during the transition from lytic to latent phases has yet to be investigated. Similarly, the role of epigenetic modifications (such as histone/DNA methylation or histone acetylation) in the maintenance of latency remains to be studied. Future studies should uncover the impact of persistent transcriptional changes within the ganglia on neuronal function. SVV infection in rhesus macaques provides a model well-suited to further our knowledge of varicella viruses’ pathogenesis. The availability of this model together with a versatile bacterial artificial chromosome (Gray et al., 2011; Meyer et al., 2013a) that facilitates manipulation of the viral genome will play a critical role in addressing these remaining gaps in our knowledge.
OS and IM wrote the manuscript.
This work was supported by NIH award 1UM01AR065705.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abendroth, A., Morrow, G., Cunningham, A. L., and Slobedman, B. (2001). Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J. Virol. 75, 6183–6192. doi: 10.1128/JVI.75.13.6183-6192.2001
Annunziato, P., LaRussa, P., Lee, P., Steinberg, S., Lungu, O., Gershon, A. A., et al. (1998). Evidence of latent Varicella-zoster virus in rat dorsal root ganglia. J. Infect. Dis. 178(Suppl. 1)), S48–S51. doi: 10.1086/514261
Arnold, N., Girke, T., Sureshchandra, S., and Messaoudi, I. (2016a). Acute Simian varicella virus infection causes robust and sustained changes in gene expression in the sensory ganglia. J. Virol. 90, 10823–10843. doi: 10.1128/JVI.01272-16
Arnold, N., Girke, T., Sureshchandra, S., Nguyen, C., Rais, M., and Messaoudi, I. (2016b). Genomic and functional analysis of the host response to acute simian varicella infection in the lung. Sci. Rep. 28:34164. doi: 10.1038/srep34164
Arnold, N., and Messaoudi, I. (2017a). Herpes zoster and the search for an effective vaccine. Clin. Exp. Immunol. 187, 82–92. doi: 10.1111/cei.12809
Arnold, N., and Messaoudi, I. (2017b). Simian varicella virus causes robust transcriptional changes in T cells that support viral replication. Virus Res. 15, 226–235. doi: 10.1016/j.virusres.2017.07.004
Arnold, N., Meyer, C., Engelmann, F., and Messaoudi, I. (2017). Robust gene expression changes in the ganglia following subclinical reactivation in rhesus macaques infected with Simian varicella virus. J. Neurovirol. 23, 520–538. doi: 10.1007/s13365-017-0522-3
Arvin, A. M., Pollard, R. B., Rasmussen, L. E., and Merigan, T. C. (1978). Selective impairment of lymphocyte reactivity to Varicella-zoster virus antigen among untreated patients with lymphoma. J. Infect. Dis. 137, 531–540. doi: 10.1093/infdis/137.5.531
Berarducci, B., Rajamani, J., Reichelt, M., Sommer, M., Zerboni, L., and Arvin, A. M. (2009). Deletion of the first cysteine-rich region of the Varicella-zoster virus glycoprotein E ectodomain abolishes the gE and gI interaction and differentially affects cell-cell spread and viral entry. J. Virol. 83, 228–240. doi: 10.1128/JVI.00913-08
Chiner, E., Ballester, I., Betlloch, I., Blanquer, J., Aguar, M. C., Blanquer, R., et al. (2010). Varicella-zoster virus pneumonia in an adult population: has mortality decreased? Scand. J. Infect. Dis. 42, 215–221. doi: 10.3109/00365540903428166
Clarke, P., Rabkin, S. D., Inman, M. V., Mahalingam, R., Cohrs, R., Wellish, M., et al. (1992). Molecular analysis of Simian varicella virus DNA. Virology 190, 597–605. doi: 10.1016/0042-6822(92)90897-X
Clarkson, M. J., Thorpe, E., and McCarthy, K. (1967). A virus disease of captive vervet monkeys (Cercopithecus aethiops) caused by a new herpesvirus. Archiv. Gesamte Virusforschung. 22, 219–234. doi: 10.1007/BF01240517
Cohen, J. I., Moskal, T., Shapiro, M., and Purcell, R. H. (1996). Varicella in Chimpanzees. J. Med. Virol. 50, 289–292. doi: 10.1002/(SICI)1096-9071(199612)50:4<289::AID-JMV2>3.0.CO;2-4
Dalziel, R. G., Bingham, S., Sutton, D., Grant, D., Champion, J. M., Dennis, S. A., et al. (2004). Allodynia in rats infected with Varicella zoster virus–a small animal model for post-herpetic neuralgia. Brain Res. Brain Res. Rev. 46, 234–242. doi: 10.1016/j.brainresrev.2004.07.008
Dayan, R. R., and Peleg, R. (2017). Herpes zoster - typical and atypical presentations. Postgraduate Med. 129, 567–571. doi: 10.1080/00325481.2017.1335574
Depledge, D. P., Ouwendijk, W. J. D., Sadaoka, T., Braspenning, S. E., Mori, Y., Cohrs, R. J., et al. (2018a). A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 9:1167. doi: 10.1038/s41467-018-03569-2
Depledge, D. P., Sadaoka, T., and Ouwendijk, W. J. D. (2018b). Molecular aspects of Varicella-zoster virus latency. Viruses 10:E349. doi: 10.3390/v10070349
Duncan, C. J., and Hambleton, S. (2015). Varicella zoster virus immunity: a primer. J. Infect. 71(Suppl. 1), S47–S53. doi: 10.1016/j.jinf.2015.04.015
Elmore, D., and Eberle, R. (2008). Monkey B virus (Cercopithecine herpesvirus 1). Comp. Med. 58, 11–21.
Felsenfeld, A. D., and Schmidt, N. J. (1979). Varicella-zoster virus immunizes patas monkeys against simian varicella-like disease. J. Gen. Virol. 42, 171–178. doi: 10.1099/0022-1317-42-1-171
Gan, L., Wang, M., Chen, J. J., Gershon, M. D., and Gershon, A. A. (2014). Infected peripheral blood mononuclear cells transmit latent Varicella zoster virus infection to the guinea pig enteric nervous system. J. Neurovirol. 20, 442–456. doi: 10.1007/s13365-014-0259-1
Gershon, A. A. (2017). Is chickenpox so bad, what do we know about immunity to Varicella zoster virus, and what does it tell us about the future? J. Infect. 74(Suppl. 1), S27–S33. doi: 10.1016/S0163-4453(17)30188-3
Gershon, A. A., Chen, J., and Gershon, M. D. (2008). A model of lytic, latent, and reactivating Varicella-zoster virus infections in isolated enteric neurons. J. Infect. Dis. 197(Suppl. 2), S61–S65. doi: 10.1086/522149
Gnann, J. W. Jr. (2002). Varicella-zoster virus: atypical presentations and unusual complications. J. Infect. Dis. 186(Suppl. 1), S91–S98. doi: 10.1086/342963
Gray, W. L. (2010). Simian varicella virus: molecular virology. Curr. Top. Microbiol. Immunol. 342, 291–308. doi: 10.1007/82_2010_27
Gray, W. L., Davis, K., Ou, Y., Ashburn, C., and Ward, T. M. (2007). Simian varicella virus gene 61 encodes a viral transactivator but is non-essential for in vitro replication. Arch. Virol. 152, 553–563. doi: 10.1007/s00705-006-0866-0
Gray, W. L., and Oakes, J. E. (1984). Simian varicella virus DNA shares homology with human varicella-zoster virus DNA. Virology 136, 241–246. doi: 10.1016/0042-6822(84)90263-0
Gray, W. L., Starnes, B., White, M. W., and Mahalingam, R. (2001). The DNA sequence of the Simian varicella virus genome. Virology 284, 123–130. doi: 10.1006/viro.2001.0912
Gray, W. L., Zhou, F., Noffke, J., and Tischer, B. K. (2011). Cloning the Simian varicella virus genome in E. coli as an infectious bacterial artificial chromosome. Arch. Virol. 156, 739–746. doi: 10.1007/s00705-010-0889-4
Haberthur, K., Engelmann, F., Park, B., Barron, A., Legasse, A., Dewane, J., et al. (2011). CD4 T cell immunity is critical for the control of Simian varicella virus infection in a nonhuman primate model of VZV infection. PLoS Pathog. 7:e1002367. doi: 10.1371/journal.ppat.1002367
Haberthur, K., Kraft, A., Arnold, N., Park, B., Meyer, C., Asquith, M., et al. (2013). Genome-wide analysis of T cell responses during acute and latent Simian varicella virus infections in rhesus macaques. J. Virol. 87, 11751–11761. doi: 10.1128/JVI.01809-13
Haberthur, K., and Messaoudi, I. (2013). Animal models of varicella zoster virus infection. Pathogens 2, 364–382. doi: 10.3390/pathogens2020364
Haberthur, K., Meyer, C., Arnold, N., Engelmann, F., Jeske, D. R., and Messaoudi, I. (2014). Intrabronchial infection of rhesus macaques with Simian varicella virus results in a robust immune response in the lungs. J. Virol. 88, 12777–12792. doi: 10.1128/JVI.01814-14
Heininger, U., and Seward, J. F. (2006). Varicella. Lancet 368, 1365–1376. doi: 10.1016/S0140-6736(06)69561-5
James, S. F., Chahine, E. B., Sucher, A. J., and Hanna, C. (2018). Shingrix: the new adjuvanted recombinant herpes zoster vaccine. Ann. Pharmacother. 52, 673–680. doi: 10.1177/1060028018758431
James, S. F., Traina-Dorge, V., Deharo, E., Wellish, M., Palmer, B. E., Gilden, D., et al. (2014). T cells increase before zoster and PD-1 expression increases at the time of zoster in immunosuppressed nonhuman primates latently infected with Simian varicella virus. J. Neurovirol. 20, 309–313. doi: 10.1007/s13365-014-0237-7
Jones, J. O., and Arvin, A. M. (2003). Microarray analysis of host cell gene transcription in response to Varicella-zoster virus infection of human T cells and fibroblasts in vitro and SCIDhu skin xenografts in vivo. J. Virol. 77, 1268–1280. doi: 10.1128/JVI.77.2.1268-1280.2003
Keating, G. M. (2016). Shingles (Herpes Zoster) Vaccine (Zostavax((R))): a review in the prevention of herpes zoster and postherpetic neuralgia. BioDrugs 30, 243–254. doi: 10.1007/s40259-016-0180-7
Ku, C.-C., Zerboni, L., Ito, H., Graham, B. S., Wallace, M., and Arvin, A. M. (2004). Varicella-zoster virus transfer to skin by t cells and modulation of viral replication by epidermal cell interferon-{alpha}. J. Exp. Med. 200, 917–925. doi: 10.1084/jem.20040634
Leclair, J. M., Zaia, J. A., Levin, M. J., Congdon, R. G., and Goldmann, D. A. (1980). Airborne transmission of chickenpox in a hospital. N. Engl. J. Med. 302, 450–453. doi: 10.1056/NEJM198002213020807
Levin, M. J., Kroehl, M. E., Johnson, M. J., Hammes, A., Reinhold, D., Lang, N., et al. (2018). Th1 memory differentiates recombinant from live herpes zoster vaccines. J. Clin. Invest. 128, 4429–4440. doi: 10.1172/JCI121484
Long, S. Y., Latimer, E. M., and Hayward, G. S. (2016). Review of elephant endotheliotropic herpesviruses and acute hemorrhagic disease. ILAR J. 56, 283–296. doi: 10.1093/ilar/ilv041
Mahalingam, R., and Gilden, D. H. (2007). “Simian varicella virus,” in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, eds A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, et al. (Cambridge: Cambridge University Press), 2007.
Mahalingam, R., Traina-Dorge, V., Wellish, M., Deharo, E., Singletary, M. L., Ribka, E. P., et al. (2010). Latent Simian varicella virus reactivates in monkeys treated with tacrolimus with or without exposure to irradiation. J. Neurovirol. 16, 342–354. doi: 10.3109/13550284.2010.513031
Mahalingam, R., Traina-Dorge, V., Wellish, M., Lorino, R., Sanford, R., Ribka, E. P., et al. (2007). Simian varicella virus reactivation in cynomolgus monkeys. Virology 368, 50–59. doi: 10.1016/j.virol.2007.06.025
Messaoudi, I., Barron, A., Wellish, M., Engelmann, F., Legasse, A., Planer, S., et al. (2009). Simian varicella virus infection of rhesus macaques recapitulates essential features of Varicella zoster virus infection in humans. PLoS Pathog. 5:e1000657. doi: 10.1371/journal.ppat.1000657
Meyer, C., Dewane, J., Haberthur, K., Engelmann, F., Arnold, N., Gray, W., et al. (2013a). Bacterial artificial chromosome derived Simian varicella virus is pathogenic in vivo. Virol. J. 8:278. doi: 10.1186/1743-422X-10-278
Meyer, C., Dewane, J., Kerns, A., Haberthur, K., Barron, A., Park, B., et al. (2013b). Age and immune status of rhesus macaques impact Simian varicella virus gene expression in sensory ganglia. J. Virol. 87, 8294–8306. doi: 10.1128/JVI.01112-13
Meyer, C., Engelmann, F., Arnold, N., Krah, D. L., ter Meulen, J., Haberthur, K., et al. (2015a). Abortive intrabronchial infection of rhesus macaques with Varicella-zoster virus provides partial protection against Simian varicella virus challenge. J. Virol. 89, 1781–1793. doi: 10.1128/JVI.03124-14
Meyer, C., Kerns, A., Barron, A., Kreklywich, C., Streblow, D. N., and Messaoudi, I. (2011). Simian varicella virus gene expression during acute and latent infection of rhesus macaques. J. Neurovirol. 17, 600–612. doi: 10.1007/s13365-011-0057-y
Meyer, C., Kerns, A., Haberthur, K., Dewane, J., Walker, J., Gray, W., et al. (2013c). Attenuation of the adaptive immune response in rhesus macaques infected with Simian varicella virus lacking open reading frame 61. J. Virol. 87, 2151–2163. doi: 10.1128/JVI.02369-12
Meyer, C., Walker, J., Dewane, J., Engelmann, F., Laub, W., Pillai, S., et al. (2015b). Impact of irradiation and immunosuppressive agents on immune system homeostasis in rhesus macaques. Clin. Exp. Immunol. 181, 491–510. doi: 10.1111/cei.12646
Mo, C., Lee, J., Sommer, M., Grose, C., and Arvin, A. M. (2002). The requirement of Varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology 304, 176–186. doi: 10.1006/viro.2002.1556
Moffat, J., Mo, C., Cheng, J. J., Sommer, M., Zerboni, L., Stamatis, S., et al. (2004). Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J. Virol. 78, 12406–12415. doi: 10.1128/JVI.78.22.12406-12415.2004
Moffat, J. F., Stein, M. D., Kaneshima, H., and Arvin, A. M. (1995). Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69, 5236–5242.
Myers, M. G., Kramer, L. W., and Stanberry, L. R. (1987). Varicella in a gorilla. J. Med. Virol. 23, 317–322. doi: 10.1002/jmv.1890230403
Nader, S., Bergen, R., Sharp, M., and Arvin, A. M. (1995). Age-related differences in cell-mediated immunity to Varicella-zoster virus among children and adults immunized with live attenuated varicella vaccine. J. Infect. Dis. 171, 13–17. doi: 10.1093/infdis/171.1.13
Ou, Y., Davis, K. A., Traina-Dorge, V., and Gray, W. L. (2007). Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 81, 8149–8156. doi: 10.1128/JVI.00407-07
Ouwendijk, W. J., Abendroth, A., Traina-Dorge, V., Getu, S., Steain, M., Wellish, M., et al. (2013a). T-Cell Infiltration Correlates with CXCL10 expression in ganglia of cynomolgus macaques with reactivated Simian varicella virus. J. Virol. 87, 2979–2982. doi: 10.1128/JVI.03181-12
Ouwendijk, W. J., Getu, S., Mahalingam, R., Gilden, D., Osterhaus, A. D., and Verjans, G. M. (2016). Characterization of the immune response in ganglia after primary Simian varicella virus infection. J. Neurovirol. 22, 376–388. doi: 10.1007/s13365-015-0408-1
Ouwendijk, W. J., Mahalingam, R., de Swart, R. L., Haagmans, B. L., van Amerongen, G., Getu, S., et al. (2013b). T-Cell tropism of Simian varicella virus during primary infection. PLoS Pathog. 9:e1003368. doi: 10.1371/journal.ppat.1003368
Ouwendijk, W. J. D., van Veen, S., Mehraban, T., Mahalingam, R., and Verjans, G. (2018). Simian varicella virus infects enteric neurons and alpha4beta7 integrin-expressing gut-tropic t-cells in nonhuman primates. Viruses 10:156. doi: 10.3390/v10040156
Provost, P. J., Keller, P. M., Banker, F. S., Keech, B. J., Klein, H. J., Lowe, R. S., et al. (1987). Successful infection of the common marmoset (Callithrix jacchus) with human varicella-zoster virus. J. Virol. 61, 2951–2955.
Redman, R. L., Nader, S., Zerboni, L., Liu, C., Wong, R. M., Brown, B. W., et al. (1997). Early reconstitution of immunity and decreased severity of herpes zoster in bone marrow transplant recipients immunized with inactivated varicella vaccine. J. Infect. Dis. 176, 578–585. doi: 10.1086/514077
Reichelt, M., Zerboni, L., and Arvin, A. M. (2008). Mechanisms of Varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J. Virol. 82, 3971–3983. doi: 10.1128/JVI.02592-07
Sadzot-Delvaux, C., Merville-Louis, M. P., Delree, P., Marc, P., Piette, J., Moonen, G., et al. (1990). An in vivo model of Varicella-zoster virus latent infection of dorsal root ganglia. J. Neurosci. Res. 26, 83–89. doi: 10.1002/jnr.490260110
Sawyer, M. H., Chamberlin, C. J., Wu, Y. N., Aintablian, N., and Wallace, M. R. (1994). Detection of Varicella-zoster virus DNA in air samples from hospital rooms. J Infect Dis. 169, 91–94. doi: 10.1093/infdis/169.1.91
Sen, N., and Arvin, A. M. (2016). Dissecting the molecular mechanisms of the tropism of Varicella-zoster virus for human T Cells. J. Virol. 90, 3284–3287. doi: 10.1128/JVI.03375-14
Sen, N., Mukherjee, G., and Arvin, A. M. (2015). Single cell mass cytometry reveals remodeling of human T cell phenotypes by Varicella zoster virus. Methods 15, 85–94. doi: 10.1016/j.ymeth.2015.07.008
Sen, N., Mukherjee, G., Sen, A., Bendall, S. C., Sung, P., Nolan, G. P., et al. (2014). Single-cell mass cytometry analysis of human tonsil T cell remodeling by Varicella zoster virus. Cell Rep. 8, 633–645. doi: 10.1016/j.celrep.2014.06.024
Soike, K. F., Rangan, S. R., and Gerone, P. J. (1984). Viral disease models in primates. Adv. Vet. Sci. Comp. Med. 28, 151–199. doi: 10.1016/B978-0-12-039228-5.50011-5
Sorel, O., Chen, T., Myster, F., Javaux, J., Vanderplasschen, A., and Dewals, B. G. (2017). Macavirus latency-associated protein evades immune detection through regulation of protein synthesis in cis depending upon its glycin/glutamate-rich domain. PLoS Pathog. 13:e1006691. doi: 10.1371/journal.ppat.1006691
Steain, M., Gowrishankar, K., Rodriguez, M., Slobedman, B., and Abendroth, A. (2011). Upregulation of CXCL10 in human dorsal root ganglia during experimental and natural Varicella-zoster virus infection. J. Virol. 85, 626–631. doi: 10.1128/JVI.01816-10
Suzuki, K., Yoshikawa, T., Tomitaka, A., Matsunaga, K., and Asano, Y. (2004). Detection of aerosolized Varicella-zoster virus DNA in patients with localized herpes zoster. J. Infect. Dis. 189, 1009–1012. doi: 10.1086/382029
Syed, Y. Y. (2018). Recombinant Zoster Vaccine (Shingrix((R))): a review in herpes zoster. Drugs Aging 35, 1031–1040. doi: 10.1007/s40266-018-0603-x
Traina-Dorge, V., Doyle-Meyers, L. A., Sanford, R., Manfredo, J., Blackmon, A., Wellish, M., et al. (2015). Simian varicella virus is present in macrophages, dendritic cells, and T cells in lymph nodes of rhesus macaques after experimental reactivation. J. Virol. 89, 9817–9824. doi: 10.1128/JVI.01324-15
Traina-Dorge, V., Sanford, R., James, S., Doyle-Meyers, L. A., de Haro, E., Wellish, M., et al. (2014). Robust pro-inflammatory and lesser anti-inflammatory immune responses during primary Simian varicella virus infection and reactivation in rhesus macaques. J. Neurovirol. 20, 526–530. doi: 10.1007/s13365-014-0274-2
Vizoso Pinto, M. G., Pfrepper, K. I., Janke, T., Noelting, C., Sander, M., Lueking, A., et al. (2010). A systematic approach for the identification of novel, serologically reactive recombinant Varicella-Zoster Virus (VZV) antigens. Virol. J. 20:165. doi: 10.1186/1743-422X-7-165
Wang, W., Pan, D., Fu, W., Cai, L., Ye, J., Liu, J., et al. (2017). A SCID mouse-human lung xenograft model of Varicella-zoster virus infection. Antiviral Res. 146, 45–53. doi: 10.1016/j.antiviral.2017.08.012
Wareham, D. W., and Breuer, J. (2007). Herpes zoster. BMJ 334, 1211–1215. doi: 10.1136/bmj.39206.571042.AE
Weinberg, A., Kroehl, M. E., Johnson, M. J., Hammes, A., Reinhold, D., Lang, N., et al. (2018). Comparative immune responses to licensed herpes zoster vaccines. J. Infect. Dis. 218(Suppl._2), S81–S87.
White, T. M., Mahalingam, R., Traina-Dorge, V., and Gilden, D. H. (2002). Persistence of Simian varicella virus DNA in CD4(+) and CD8(+) blood mononuclear cells for years after intratracheal inoculation of African green monkeys. Virology 303, 192–198. doi: 10.1006/viro.2002.1664
Whitmer, T., Malouli, D., Uebelhoer, L. S., DeFilippis, V. R., Fruh, K., and Verweij, M. C. (2015). The ORF61 protein encoded by Simian varicella virus and Varicella-zoster virus inhibits NF-kappaB Signaling by Interfering with IkappaBalpha Degradation. J. Virol. 89, 8687–8700. doi: 10.1128/JVI.01149-15
Wiegering, V., Schick, J., Beer, M., Weissbrich, B., Gattenlohner, S., Girschick, H. J., et al. (2011). Varicella-zoster virus infections in immunocompromised patients - a single centre 6-years analysis. BMC Pediatr. 10:31. doi: 10.1186/1471-2431-11-31
Willer, D. O., Ambagala, A. P., Pilon, R., Chan, J. K., Fournier, J., Brooks, J., et al. (2012). Experimental infection of Cynomolgus Macaques (Macaca fascicularis) with human Varicella-zoster virus. J. Virol. 86, 3626–3634. doi: 10.1128/JVI.06264-11
Wilson, A., Sharp, M., Koropchak, C. M., Ting, S. F., and Arvin, A. M. (1992). Subclinical Varicella-zoster virus viremia, herpes zoster, and T lymphocyte immunity to Varicella-zoster viral antigens after bone marrow transplantation. J. Infect. Dis. 165, 119–126. doi: 10.1093/infdis/165.1.119
Yamanishi, K. (2008). Molecular analysis of the Oka vaccine strain of Varicella-zoster virus. J. Infect. Dis. 197(Suppl. 2), S45–S48. doi: 10.1086/522122
Zerboni, L., Berarducci, B., Rajamani, J., Jones, C. D., Zehnder, J. L., and Arvin, A. (2011). Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J. Virol. 85, 98–111. doi: 10.1128/JVI.01902-10
Zerboni, L., Ku, C.-C., Jones, C. D., Zehnder, J. L., and Arvin, A. M. (2005). Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6490–6495. doi: 10.1073/pnas.0501045102
Zerboni, L., Nader, S., Aoki, K., and Arvin, A. M. (1998). Analysis of the persistence of humoral and cellular immunity in children and adults immunized with varicella vaccine. J. Infect. Dis. 177, 1701–1704. doi: 10.1086/517426
Keywords: herpesvirus, viral latency, varicella zoster virus, simian varicella virus, non-human primates, viral reactivation, shingles
Citation: Sorel O and Messaoudi I (2018) Varicella Virus-Host Interactions During Latency and Reactivation: Lessons From Simian Varicella Virus. Front. Microbiol. 9:3170. doi: 10.3389/fmicb.2018.03170
Received: 02 October 2018; Accepted: 07 December 2018;
Published: 21 December 2018.
Edited by:
Benedikt B. Kaufer, Freie Universität Berlin, GermanyReviewed by:
Donald Scott Schmid, Centers for Disease Control and Prevention (CDC), United StatesCopyright © 2018 Sorel and Messaoudi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilhem Messaoudi, aW1lc3Nhb3VAdWNpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.