 Krupa Naran
Krupa Naran Trishana Nundalall
Trishana Nundalall Shivan Chetty
Shivan Chetty Stefan Barth1,2*
Stefan Barth1,2*- 1Medical Biotechnology and Immunotherapy Unit, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
- 2South African Research Chair in Cancer Biotechnology, Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
The advances in cancer biology and pathogenesis during the past two decades, have resulted in immunotherapeutic strategies that have revolutionized the treatment of malignancies, from relatively non-selective toxic agents to specific, mechanism-based therapies. Despite extensive global efforts, infectious diseases remain a leading cause of morbidity and mortality worldwide, necessitating novel, innovative therapeutics that address the current challenges of increasing antimicrobial resistance. Similar to cancer pathogenesis, infectious pathogens successfully fashion a hospitable environment within the host and modulate host metabolic functions to support their nutritional requirements, while suppressing host defenses by altering regulatory mechanisms. These parallels, and the advances made in targeted therapy in cancer, may inform the rational development of therapeutic interventions for infectious diseases. Although “immunotherapy” is habitually associated with the treatment of cancer, this review accentuates the evolving role of key targeted immune interventions that are approved, as well as those in development, for various cancers and infectious diseases. The general features of adoptive therapies, those that enhance T cell effector function, and ligand-based therapies, that neutralize or eliminate diseased cells, are discussed in the context of specific diseases that, to date, lack appropriate remedial treatment; cancer, HIV, TB, and drug-resistant bacterial and fungal infections. The remarkable diversity and versatility that distinguishes immunotherapy is emphasized, consequently establishing this approach within the armory of curative therapeutics, applicable across the disease spectrum.
Introduction
Numerous host factors which constitute the immune system influence treatment outcomes and are accountable for disease progression or regression. Immunotherapy is collectively defined as a therapeutic approach that targets or manipulates the immune system (Papaioannou et al., 2016). Ultimately, immunotherapy aims to harness the host’s adaptive and innate immune response to effectuate long-lived elimination of diseased cells and can be categorized broadly into passive (including adoptive and antibody-based) and active (including vaccine therapy and allergen-specific) approaches. Passive-mediated immunotherapy involves the administration of ex vivo-generated immune elements (antibodies, immune cells) to patients and does not stimulate the host immune response, while active immunotherapy induces the patient’s immune response and results in the development of specific immune effectors (antibodies and T cells) (Tur and Barth, 2014).
Consequently, these targeted therapies advance host cellular responses to disease by stimulating immune responses, targeting disease-causing virulence factors and improving immunological memory (Zumla et al., 2016b). As such, several types of targeted therapies have been approved for cancer treatment and act by blocking regulatory biochemical pathways or mutant proteins, essential to tumor growth and maintenance and have reduced toxic side effects as they act with precision on diseased cells with little collateral tissue damage (Wykes and Lewin, 2018). As such, “immunotherapy” is almost exclusively, and somewhat biasedly, associated with the treatment of cancer. In this review, clinically relevant examples of adoptive therapies are provided to emphasize the versatility of their corresponding general principles of action for both non-communicable and infectious diseases.
T Cell Therapies
T cells initiate potent and specific immune responses against foreign antigens. Strategies that activate these immune responses, broadly define T cell therapies and are often developed in combination with monoclonal antibodies (mAbs) (Figure 1).
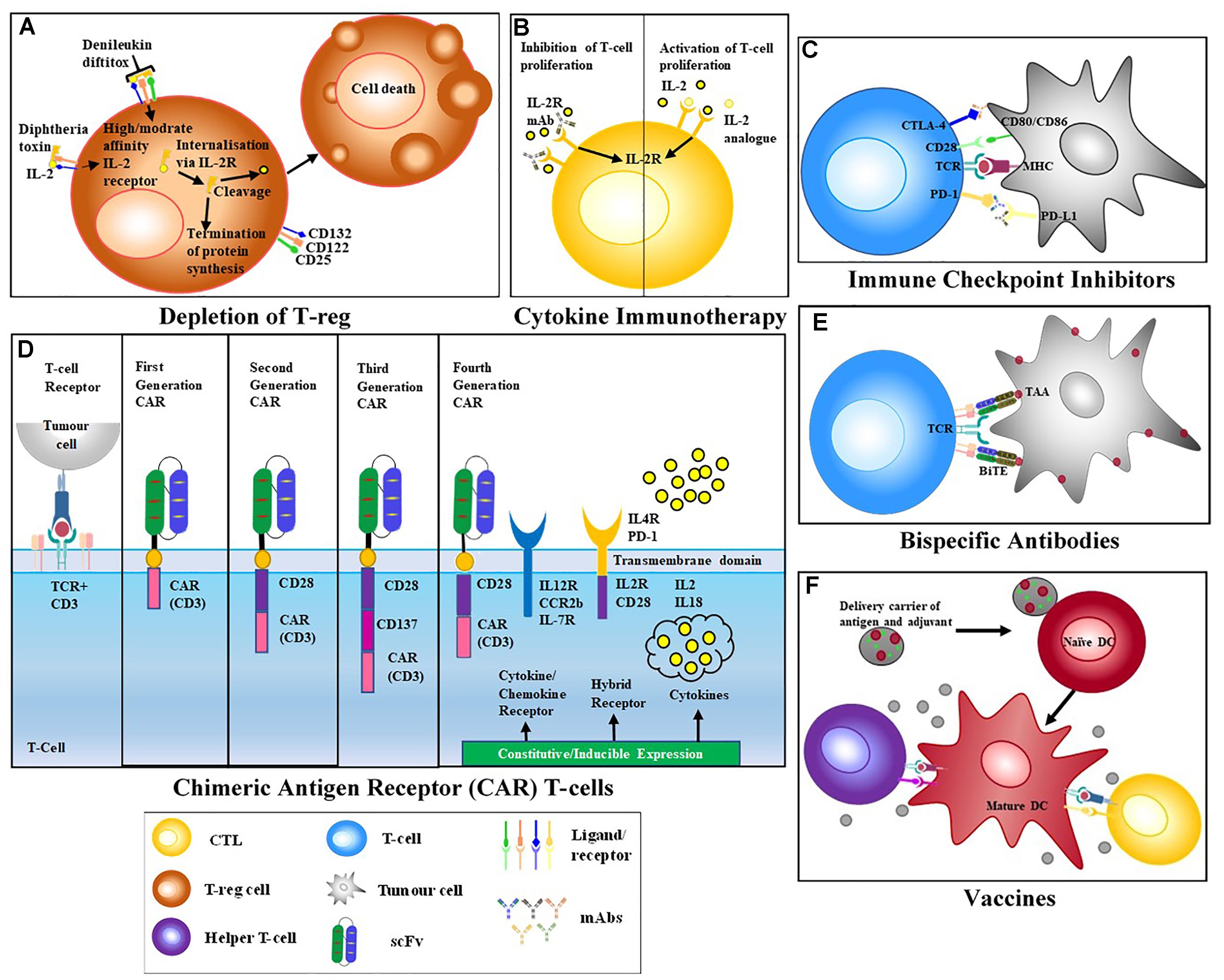
Figure 1. T cell-activating Therapeutic Strategies. (A) Treg depletion – biologics such Denileukin diftitox (DD) bind to target receptors on suppressor cells and initiate apoptosis via down-stream signaling. (B) Cytokine therapy – addition of pro-inflammatory cytokines increases immune activation while the addition of anti-inflammatory cytokines reduces immune activation. MAbs specific for cytokine receptors may also be used to block cytokine stimulation of the immune system. (C) Immune checkpoint blockade – mAbs block the interaction of inhibitory receptors CTLA-4 and PD-1, resulting in the activation of effector T cells (QYResearch) (D) Chimeric antigen receptors (CARs) T cells are modified T cells with a recombinant receptor; usually a scFv that redirects the specificity of effector T cells. First generation CARs that only comprised an activation domain were prone to anergy. Due to this signaling failure, second and third generation CARs, incorporating a CD3 chain and cytoplasmic domain of a co-stimulatory receptor, like CD28 were generated. Fourth generation CARs also included constitutive or inducible expression of co-receptors or soluble cytokines together with T cell activating CAR (Golubovskaya and Wu, 2016). (E) Bispecific antibodies containing two binding arms one specific for a target antigen and a second arm specific for CD3, thereby bringing T cells into close proximity to target cells and activating T cells while bypassing the need for MHC restricted engagement. (F) Vaccines – Introduction of non-infectious component to stimulate activation of T cells and development of memory immune cells.
Vaccines
Vaccination represents the first form of host-directed immunotherapy to be introduced and includes various categories (Table 1). Most vaccines work by introducing a non-infectious version of a disease-causing microbe into an individual, thereby providing a better stimulus for the activation of disease-specific T cells and the development of immunological memory. Memory immune cells are able to rapidly kill microbes and prevent infection. While this type targeted therapy has been able to eradicate smallpox and drastically reduce the disease burden of multiple infectious agents such as rabies, typhoid, cholera, hepatitis and more, it has been far less effective against cancer and chronic infectious diseases such as human immunodeficiency virus (HIV). The combination of therapeutic vaccines and other immune-based therapies may provide improved efficacy (Waldmann, 2003).
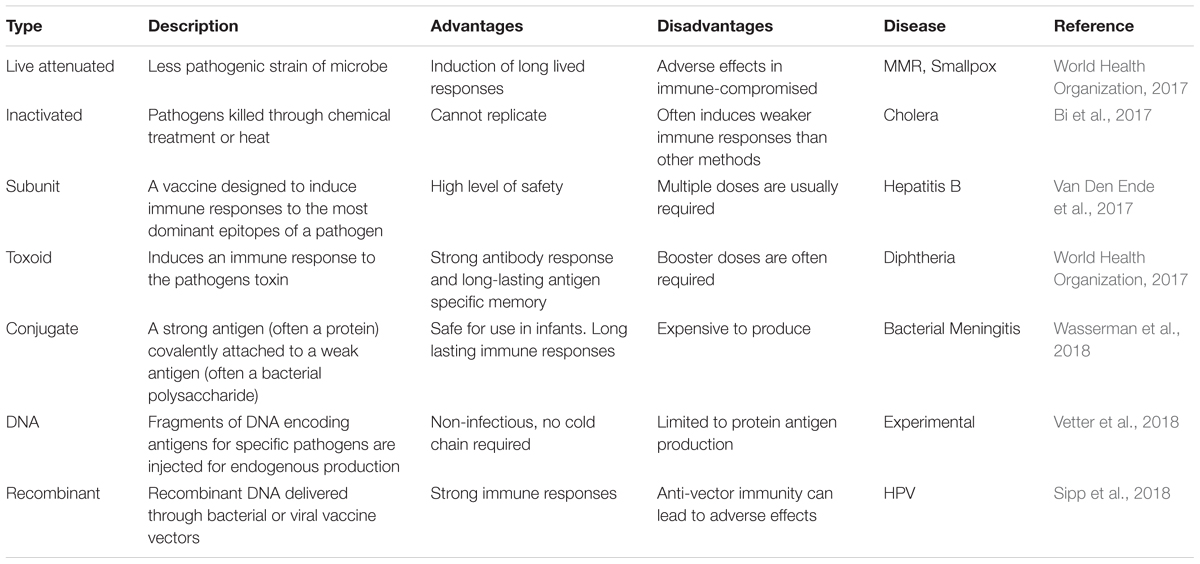
Table 1. Vaccine types and examples.
Following the discovery of Edward Jenner’s smallpox vaccine in 1796 (Nabel, 2013) vaccination, especially against viral infections, has been the leading practice of infection prevention. Developed in 1971, an example of a modern viral vaccine success is the MMR (mumps, measles and rubella) vaccine (Larson et al., 2018). This live-attenuated vaccine contains variants of all three pathogens. MMR can be transported in lyophilized form and is usually administered subcutaneously. Developing vaccines for other viral pathogens remains challenging despite enormous technological advancements. This pertains particularly to HIV, where in spite of more than 30 years of research, no reports of a completely effective prophylactic or therapeutic vaccine exist (Dangeti, 2014). The lack of efficacy of candidate HIV vaccines has been attributed to HIV escape from vaccine specific CD8+ T cells, lack of CD8+ T cells replenishment and inability to target latently infected cells (reviewed by Perreau et al., 2017). Presently, it is accepted that a successful HIV vaccine would comprise of an optimized vector and/or immunogen proficient in stimulating broad, long-lived and safe immune responses (Esparza and Van Regenmortel, 2014). Up until 2016 only 6 HIV-vaccine candidates reached clinical trials and most of these failed to produce protective broadly neutralizing antibodies thus changing the focus of vaccine strategies to elicit CTL responses (Stephenson et al., 2016). Effective T cell-based vaccine can limit the initial viral replication and lower viral load set point, thereby preventing the initial infection or/and slow disease progression (Haynes, 2015). The first CTL based vaccine trial was the step study (HVTN502) which also did not show any protection and actually induced higher infection rates in a subset of participants (Cao et al., 1995; Duerr et al., 2012). Eventually, the RV144 trial, which used a prime-boost strategy with a canarypox viral vector and envelope protein gp120 demonstrated a 31% efficacy rate (Rerks-Ngarm et al., 2009). Results from the RV144 trail demonstrated that even non-neutralizing antibodies which match the infecting strain also have the ability to prevent infection (NCT01435135) primarily via ADCC and Fc mediated responses. The major concern with such a vaccine strategy is the enormous diversity of HIV-1 strains (Stephenson et al., 2016). To improve on the RV144 results, current strategies are focused on understanding immune correlates of protection that would increase the breadth of ADCC and FcR activities (Haynes, 2015). Although these trials have been mostly disappointing, they are informative and highlight the remarkable struggle associated in developing effective HIV vaccines and provide essential information to support pre-clinical development of other novel T cell vaccine strategies.
Unlike viral-targeted vaccines, cancer vaccines do not prevent disease but rather stimulate the immune system to attack an already existing disease. Many cancer vaccines consist of cancer cells, parts of cells, or pure antigens. Often, a patient’s immune cells are isolated and exposed to cancer antigens, and once activated, these immune cells are re-introduced into the patient’s body and are better able to suppress cancer cells. Sipuleucel-T (Provenge®) (Sip-T) is the first FDA (United States Food and Drug Administration)-approved therapeutic cancer vaccine. It is an autologous vaccine used for the treatment of patients with asymptomatic or minimally symptomatic castration-resistant metastatic prostate cancer (mCRPC) and who are not responsive to hormone therapy (Cheever and Higano, 2011). Sip-T elicit anti-tumor activity via activation of T cells that are specific for prostatic acid phosphatase (PAP), an enzyme found on the surface of 95% of prostate cancer cells (Alteri et al., 2018). The vaccine is formulated to comprise of a patient’s mature antigen-presenting cells (APCs) expressing PAP after ex vivo exposure to a granulocyte macrophage colony stimulating factor (GM-CSF)-and PAP fusion protein (Gardner et al., 2012).
There is still no clinically approved vaccine for fungal infections; however, there are a growing number of candidates in pre-clinical development and at various phases of clinical trials (Health, 2012). Fungal vaccine strategies have mainly prioritized CD4+ T cell and B cell stimulation, thereby enhancing protection mediated by these defense mechanisms (Nanjappa and Klein, 2014). This involves targeting common antigens that are shared among a variety of medically relevant fungi. One example is the β-1,3-D-glucan, a key component of the fungal cell wall (Armstrong-James et al., 2017). Mice immunized with this glucan, conjugated to diphtheria toxin, elicit strong antibody responses that are protective against models of aspergillosis, candidiasis and cryptococcosis. Moreover, immunizing mice with antigen encapsulated in glucan, also stimulate antigen-specific antibody and T cell responses. Preclinical studies involving the vaccination of mice with an attenuated strain of Blastomyces dermatitidis showed protection against subsequent challenge from virulent strains (Wüthrich et al., 2003). Even upon CD4+ T cell depletion, protection was seen due to the emergence of protective CD8+ T cells. More recently, the focus of fungal vaccines has been on subunit vaccines and the two containing Candida albicans-derived proteins, currently in phase I/II clinical trials, were found to confer immunogenicity (De Bernardis et al., 2012; Schmidt et al., 2012).
Majority of FDA approved vaccines work by stimulating a CD4+ T cell dependent antibody response. However, for cancer and intracellular infections a CD8+ cytotoxic T cell (CTL) response is required for protection. Thus far, the primary limitation of such vaccines has been a lack of ability to prime and activate disease specific-CD8+ T cells necessary for protection. This is attributed to the inherent mechanism by which external foreign antigens administered in a vaccine are presented to the immune system via MHCII, thus stimulating CD4+ based responses. Recently, these challenges have been addressed by photochemical internalization (PCI) which has been shown to deliver antigenic peptides to the cytosol of antigen presenting cells by disruption of the endosomal membranes using a co-endocytosed photosensitizer (Haug et al., 2018). The 30-fold increase in MHCI presentation and resulting 30–100-fold increase in disease specific CD8+ activation in comparison to antigen stimulation alone, is promising. Furthermore, preclinical in vivo experiments involving the induction of antigen-specific CTL responses against cancer antigens in mice confirmed the efficacy of PCI as a peptide-based vaccine. Strategies such as these are not only applicable to cancer by have great potential to improve various peptide vaccines especially for diseases like HIV where an appropriate CTL response is required for protection.
Enhancing T Cell Activation
Successful T cell activation requires two signals: T cell receptor (TCR) binding to peptide-MHC complex and binding of T cell co-receptors with counter-receptors on APCs. T cell exhaustion is a state of T cell dysfunction that arises during persistent antigen exposure and/or inflammation and is associated with many chronic infections and cancer. It is characterized by persistent expression and diversity of inhibitory receptors, progressive and hierarchical loss of effector cytokines, metabolic imbalances, altered expression and function of transcription factors, failure to convert to quiescence and inability to acquire antigen-independent memory T cell homeostasis (Wherry, 2011; Schietinger and Greenberg, 2014). Thus, T cell exhaustion is a mechanism of immune evasion essentially leading to the inefficient control of infection and tumors. Importantly, exhausted T cells are not inert but sustain suboptimal, essential functions that encumber ongoing pathogen infection or tumor progression (Wherry and Kurachi, 2015).
This state of T cell dysfunction was initially described in the murine lymphocytic choriomeningitis virus (LCMV) model (Zajac et al., 1998), and has since been observed in animal and human models during chronic viral infections such as HIV (Kaufmann et al., 2007), Hepatitis C virus (HCV), Hepatitis B virus (HBV) (Guidotti and Chisari, 2006), simian immunodeficiency virus SIV (Zeng et al., 2011), along with various cancers (Lee et al., 1999), malaria infections (Illingworth et al., 2013) and Mycobacterium tuberculosis infection (Khan et al., 2017). Major advances have been made in three significant areas including inhibitory receptors and negative regulatory pathways, the absence of canonical memory T cell properties and maintenance, and the origin and homeostasis of exhausted T cells (Kim and Ahmed, 2010; Paley et al., 2012; Crawford et al., 2014). As such, there has been considerable interest in avoiding or reversing this dysfunctional state of exhaustion, and consequently, restoring or augmenting immune responses to effectively control infection or malignancies (Pauken and Wherry, 2015).
Modulating Intrinsic Inhibitory Receptors
To minimize tissue damage, negative pathways of immunoregulation such as those used in immune checkpoint inhibition offer a plethora of inhibitory pathways that are essential for preserving self-tolerance and moderating the duration and magnitude of immune responses in peripheral tissues (Pardoll, 2012). These pathways are inherently involved in T cell exhaustion and involve cell surface inhibitory receptors that modulate autoreactivity and immunopathology (Sharpe et al., 2007). Inhibitory receptors are transiently expressed in functional effector T cells, however, increased and prolonged expression can be considered a hallmark of exhausted T cells (Wherry, 2011). Pathogens and tumors promote immune checkpoint-mediated inhibitory interactions to escape immune control. Many of these checkpoint proteins have already been studied extensively and will be addressed in relation to cancer, HIV, TB, and malaria infections.
Within cancer immunotherapy, cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death protein 1 (PD-1) are the canonical immune-checkpoint receptors that illustrate ligand–receptor interactions between T cells and APCs that modulate the T cell response to antigen (MHC- mediated molecule complexes that are recognized by the TCR). CTLA-4 is a co-inhibitory receptor expressed on T cells that competes with CD28, a T cell co-stimulatory receptor, for binding on shared identical ligands CD80 (B7.1) and CD86 (B7.2) (Pardoll, 2012). Binding of CTLA-4 inhibits the activation of T cells, whereas binding of CD28 to CD80/CD86 activates the T cell responses (Dyck and Mills, 2017). CTLA-4 expression on activated T cells reduces CD28 co-stimulation by; outcompeting CD28 for binding to B.7 ligands as it has a greater affinity for the B7 ligands and, possibly also by depletion of CD80/CD86 via trans-endocytosis (Qureshi et al., 2011). Subsequently, CTLA-4 controls the activation state of effector T cells and initiates tolerance by inhibiting cytokine production and activating transcription factor STAT5 (Srahna et al., 2006). Furthermore, CTLA-4 plays a physiological role in the negative modulation of helper T cell activity, stimulating the suppressive function of T regulatory (Treg) cells (Topalian et al., 2015).
PD-1 has also been extensively studied and predominantly modulates effector T cell activity within tissue and tumors and is therefore more widely expressed than CTLA-4; it is induced on activated T cells, B cells, macrophages, dendritic cells and Tregs. The potent inhibitory signal is provided through its interaction with programmed death-ligand 1 (PD-L1) and/or PD-L2, whereby PD-1 inhibits kinases involved in T cell activation (Latchman et al., 2001; Francisco et al., 2009; Araki et al., 2013).
CTLA-4 is constitutively expressed on all cancer cells but found to be highly expressed on breast tumor cells and is implicated in immune dysregulation and associated with poor prognosis (Mao et al., 2010; Yu et al., 2015; Yuan et al., 2015). PD-L1 is expressed on a majority of tumor cells, and tumor-derived APCs, signifying the principal role of the PD-1 pathway in tumor immune evasion associated with poor prognosis (Hirano et al., 2005; Currie et al., 2009). There is gathering evidence that inhibitory receptors have a significant role in modulating T cell responses during chronic viral infections, including HIV, as well as during acute phase malarial infections (Trautmann et al., 2006; Kaufmann et al., 2007; Crawford and Wherry, 2009; Gonçalves-Lopes et al., 2016). Furthermore, CTLA-4 is expressed on Tregs and CD8+ T cells, while PD-1 is expressed on neutrophils, Tregs and natural killer (NK) cells of patients infected with M. tuberculosis, however, over a course of standard anti-TB therapy, this expression declines as responsiveness of effector T cells to antigenic stimulation improves (Merlo et al., 2001; Li et al., 2015; Zumla et al., 2016a).
Lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin-3 (TIM-3), band T lymphocyte attenuator (BTLA) and T-cell immunoglobulin and ITIM domain (TIGIT) are highly expressed on exhausted T-cells and represent the next generation of immune checkpoints that are under evaluation for the treatment of chronic diseases including cancer and chronic viral infections such as HIV (Topalian et al., 2015; Chew et al., 2016; Dyck and Mills, 2017; Grabmeier-Pfistershammer et al., 2017; Manieri et al., 2017). Interestingly, Anderson et al. (2016) recently proposed a model describing LAG-3, TIM-3, and TGIT as second tier co-inhibitory molecules that play a specific role in regulating immune responses at sites of tissue inflammation, distinct to first tier co-inhibitory receptors CTLA-4 ad PD-1 which are primarily involved in maintaining self-tolerance. This hierarchal ranking of co-inhibitory receptors provides a basis for selecting synergistic receptor blockade combinations that will result in improved T cell and NK cell effector function (Anderson et al., 2016).
Immune checkpoint inhibitors
As immune checkpoints are mediated by ligand–receptor connections, these pathways can be easily blocked by monoclonal antibodies (mAbs) or regulated by recombinant forms of ligands or receptors (Pardoll, 2012). As such, PD-1 and CTLA-4 present ideal targets for immune checkpoint inhibition therapies, one of the most noteworthy immunomodulatory therapies of current times. In contrast to vaccines which activate immune cells to attack specific targets, this type of therapy removes the natural disruptions of the immune system (Postow et al., 2015).
Subsequent to the clinical success of Ipilimumab, an anti-CTLA4 mAb for the treatment of advanced melanoma which acts by blocking the binding of CTLA-4 expressed on T cells to CD80/86 ligands expressed on tumor cells (Kyi and Postow, 2014), an onslaught of immune checkpoint inhibitors have received accelerated FDA-approval. This includes Pembrolizumab and Nivolumab, both recently approved mAbs which bind the inhibitory receptor PD-1, blocking its interaction with ligands PDL-1/PDL-2, thus releasing T cells for cytotoxic action (Nguyen and Ohashi, 2015). These immune-checkpoint inhibitors are effective for the treatment of melanoma, renal cell carcinoma, and non-small lung cancer (Robert et al., 2015a,b). In addition to targeting inhibitory receptors, blockade of the ligand PD-L1, by Atezolizumab, Durvalumab, and Avelumab, has demonstrated remarkable efficacy in lung, bladder, urothelial carcinoma and other cancers (Massard et al., 2016; Balar et al., 2017; Hsu et al., 2017; Rittmeyer et al., 2017).
However, most patients do not respond to immune checkpoint monotherapy; attributed to the absence of tumor-infiltrating effector T cells and tumor microenvironment properties. As checkpoint inhibitors target ligands/receptors of immune regulation, expression of these targets on target cell membranes is an essential prerequisite for the treatment to be effective. More than 50% of cancers induce a quiescent immune tumor microenvironment which is naturally depleted of immune effector cells and thus, lacks sufficient targets for checkpoint immunotherapy to be effective. Recently, the high levels of circulating exosomal PD-L1 was shown to correlate with immune dysfunction, suggests a role as a predictive biomarker for patient responses to anti-PD-1 therapy (Chen et al., 2018). Furthermore, combining checkpoint therapies with cancer vaccine strategies like PCI has the potential to significantly enhance the efficacy of both therapies. Cancer vaccines stimulate CD8+ T cells which express checkpoint targets; consequently the normal inhibitory effects of checkpoints would be reduced by checkpoint therapy, thereby further enhancing CD8+ activation. The synergistic effects of cancer vaccines and checkpoint combination therapy has been demonstrated (Duraiswamy et al., 2013; Karyampudi et al., 2014; Fu et al., 2015; Soares et al., 2015). The positive pre-clinical results of some of these studies has initiated 2 clinical trials assessing various vaccines in combination with nivolumab for the treatment of pancreatic cancer (Kleponis et al., 2015).
In addition to cancer vaccines, combinations of other immunotherapies with checkpoint inhibitors, holds promise for the rational development of curative therapies. The simultaneous blocking of the CTLA-4 and PD-1 pathways, given their distinct and non-overlapping mechanisms of action, has been the most successful combination tested thus far, resulting in tumor regression in patients with advanced melanoma, demonstrating the synergistic effects of combining checkpoint inhibitors (Wolchok et al., 2013; Hodi et al., 2016; Weber et al., 2016). The safety and efficacy of various combinations of immune checkpoint inhibitors with cytotoxic chemotherapy (referred to as immunochemotherapy), with small-molecule inhibitors or with radiation therapy are also under intense clinical investigation1 (Mahoney et al., 2015; Li et al., 2017). Recently, prolonged progression-free survival of patients with metastatic triple-negative breast cancer treated with a combination of the chemotherapy drug nab-paclitaxel and the PDL-1 inhibitor Atezolizumab, was demonstrated in a Phase 3 trial (Schmid et al., 2018).
The development of primary or acquired resistance remains clinically pertinent. Recent studies suggest that inactivation of JAK1 and JAK2 by cancer cells results in resistance to interferon gamma which contributes to checkpoint resistance and immune escape. In addition, resistance is likely driven by multiple other mechanisms including loss of function of CD8+ T cells due to a lack of mutational or shared antigens required for T cell recognition, reduced antigen presenting components, tumor cell induced T-cell signaling inhibition and depressed sensitivity to apoptotic inducting molecules (Shin et al., 2016; Zaretsky et al., 2016). Methods of reducing these types of resistances would need to be considered for the development of new checkpoint therapies.
In addition, as with other therapies, immune checkpoint therapies are associated with a high rate of toxicities, characterized as immune-related adverse events (IRAEs) and include diarrhea/colitis and hepatic/dermatologic/endocrine toxicities. These adverse effects, although severe, are manageable with immunosuppressive agents such as corticosteroids, which in turn, may also be associated with other short-term side effects (Michot et al., 2016; Spain et al., 2016). In chronic infection, the preclinical development of checkpoint inhibitors has had mixed results, suggesting intrinsic differences in the mechanisms of T cell dysfunction between chronic infection and cancer. In in vitro settings, CTLA-4 blockade enhanced HIV-specific CD4+ T cell function, PD-1/PD-L1 blockade improved HIV-, HCV-, and HBV-specific CD8+ T cell function, while CTLA-4/PD-1 combination blockade reversed HCV-specific CD8+ T cell exhaustion (Kim and Ahmed, 2010). However, in chronic LCMV-infected mice, neither CTLA-4 nor LAG-3 blockade improved T cell function or viral load, whereas PD-1 blockade alone and in combination with LAG-3 blockade effectively salvaged T cells from exhaustion and decreased the virus load (Barber et al., 2006). In the SIV macaque model, CTLA-4 blockade did not enhance viral-specific T cell responses, but rather amplified viral replication at mucosal sites, whereas a recent study demonstrated anti-PD-1 treatment delayed viral rebound after combined antiretroviral therapy (cART) was withdrawn (Cecchinato et al., 2008; Gill et al., 2016). The ex vivo dual blockade of co-inhibitory receptor TIGIT and ligand PD-L1 restored HIV-specific CD8+ T cell effector responses (Chew et al., 2016). In HIV-infected patients, treatment with Ipilimumab resulted in reactivation of latently virus-infected CD4+ T-cells, rendering these cells discernable to the immune system (Wightman et al., 2015) (Figure 2). Furthermore, a recent Phase I trial demonstrated enhanced HIV-specific CD8+ T cell responses following treatment with an anti-PD-L1 antibody (Gay et al., 2017). Overall, these studies suggest a role for immune checkpoints in limiting T cell responses during HIV infection and the subsequent potential of immune checkpoint inhibitors. Although the anti-PD-1 therapy Nivolumab effectively reduced viral load in HCV-infected individuals, it has been proposed that established virus-specific T cells in the liver is crucial for enhanced T cell responses by PD-1 blockade (Fuller et al., 2013; Gardiner et al., 2013). In patients with pulmonary TB, PD-L1 is highly expressed on M. tuberculosis-infected macrophages, and in vitro blockade with mAbs induces IFN-γ-initiated killing of infected cells by autologous peripheral blood T cells (Zumla et al., 2016a).
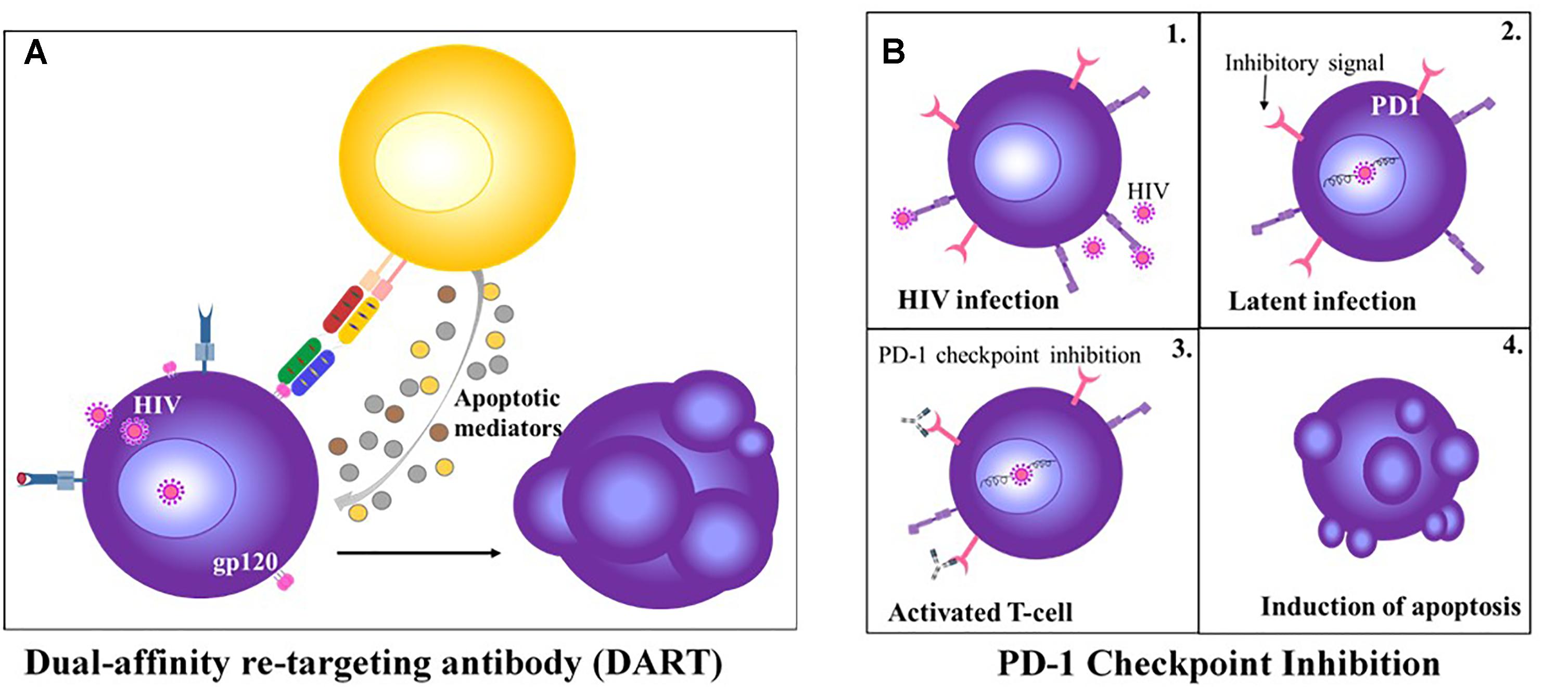
Figure 2. HIV-targeting T cell Therapies. (A) Anti-CD3 and anti-gp120 DART treatment redirects CD8+ T cell to kill HIV infected CD4+ T cells (Perreau et al., 2017). (B) PD-1 check point inhibition of latently infected CD4+ T cell results in re-activation of the T cell and induction of apoptosis (Wykes and Lewin, 2018).
While checkpoint inhibitors have revolutionized cancer immunotherapy there are clearly also many drawbacks associated with this therapy at this stage. The many combination immunotherapy studies involving checkpoint inhibitors will provide much needed information on the molecular mechanisms of resistances and toxicity associated with checkpoint therapy and subsequently assist in improving efficacy. Despite the many issues that still require addressing there have been overwhelming positive results associated with PD1/PDL-1 and CTLA-4 checkpoint inhibition studies demonstrating the potential of checkpoint inhibitors to greatly improve treatment outcomes not only in cancer patients but with other diseases as well, especially in combination with other immunotherapies.
Cytokine Therapy
Cytokines are protein mediators involved in essentially all important biological processes including immunity, cell proliferation and inflammation, wound healing and repair, cell migration, fibrosis and angiogenesis. Cytokines provide crucial signals for fundamental processes involved in a plethora of diseases thus it is intuitive that manipulation of cytokines may alter diseased states both positively and negatively. With the field of cancer, multiple mice studies have demonstrated the anti-tumor effects of cytokines leading to cytokine-based therapies for cancer treatment. Cytokines such as IL-1, IL-12, IL-15, IL-18, IL-21, and GM-CSF are just some examples of cytokines that have entered clinical trials for treatment of patients with advanced cancer (Feldmann, 2008). Recently, administration of pro-inflammatory cytokines IL-12 and IL-18 demonstrated a reversal in MHC-1 deficient tumor induced NK cell anergy. Treatment increase NK cell activity which mediated antitumor responses resulting in improved survival of mice challenged with MHC-1 deficient tumors (Ardolino and Raulet, 2016).
Neutralization of suppressive cytokines like TGF-β and IL-10 have shown promising results and will the remain the focus of this section. IL-10 is expressed by diverse cell types such as dendritic cells (DCs), monocytes and/or CD4+ T cells, and its upregulation is associated with disease progression during chronic infections as well as poor prognosis in cancer patients (Said et al., 2010; Richter K. et al., 2013). Furthermore, IL-10 has been implicated in promoting human visceral leishmaniasis, by restricting Th1 cell-type responses and/or neutralizing parasitized tissue macrophages and, in parallel, compromising responsiveness to chemotherapy (Murray et al., 2002). During in vivo M. tuberculosis infection, IL-10 has been shown to be a negative regulator of the immune response, contributing to chronic infection (Pitt et al., 2012). In addition, fungal clearance is impaired by IL-10, as demonstrated by Cryptococcus neoformans infections, the leading cause of fatal mycosis in HIV+ individuals (Murdock et al., 2014).
Blockade of the IL-10 receptor (IL-10R) with anti-IL-10 mAbs enhances control of LCMV infection and restores T cell responses (Brooks et al., 2006; Ejrnaes et al., 2006), while simultaneous blockade of IL-10 and PD-1 reverses CD8+ T cell exhaustion, improving HIV control (Wherry, 2011). Furthermore, simultaneous IL-10 blockade during therapeutic DNA vaccination enhanced clearance of chronic LCMV infection (Ni et al., 2015). In the mouse model, the use of antimony, the conventional chemotherapy for visceral leishmaniasis, in conjunction with anti-IL-10R treatment permitted a considerable reduction in duration and dosage of treatment (Weiner et al., 2012). During M. tuberculosis infection, antibody blockade of IL-10R specifically during BCG vaccination improves Th1 and Th17 responses, and IFN-γ and IL-17A expression in vivo, resulting in significantly enhanced protection against aerogenic challenge with M. tuberculosis, compared to BCG vaccination alone (Pitt et al., 2012). In the murine model for C. neoformans infection, IL-10 blockade amplified various effector mechanisms, including improved accumulation of CD4+ T cells, B cells, Th1 and Th17 cells, but not CD8+ T cells, and, furthermore, effectively abrogated dissemination of infection to the brain (Murdock et al., 2014).
TGFβ is often over-expressed in several tumors and has been implicated in promoting growth, progression, and metastatic potential of advanced cancers (Jakowlew, 2006; Tian et al., 2011; Drabsch and Ten Dijke, 2012). Various TGFβ pathway inhibitors including small molecules, anti-TGFβ antibodies and synthetic small peptides have been reported to restore immune response and increase the efficacy of combination immunotherapy (Neuzillet et al., 2015). During chronic viral infection, both murine and human, TGFβ signaling has been implicated in T cell exhaustion (Wherry and Kurachi, 2015). However, in contrast to cancer therapy, the systemic administration of antibody- and inhibitor-based strategies to prevent TGFβ signaling during chronic viral infection have proven futile, at least in the murine model, the reasons for which remain unclear (Boettler et al., 2012; Garidou et al., 2012).
Given that cytokines play such a multifunctional role in different biological systems, systemic inhibition of any given cytokine will have wanted and unwanted effects, necessitating the need to make this therapy more targeted. Cytokines have differing signaling cascades that may be exploited for selecting a target to inhibit their functions. TNF is an ideal example; its two forms, membrane-bound or secreted, each interact with distinct receptors, TNFR1 or TNFR2, differentially expressed on multiple cell types (Drutskaya et al., 2018). Depending on the source of cells secreting TNF, and the neighboring cytokines, it may act as pro-inflammatory or anti-inflammatory. Thus, targeting selected downstream pathways may provide a more controlled treatment option than systemic TFN inhibition. TNFR1 is responsible for majority of TNF pro-inflammatory functions which signals via NF-κB and AP-1 resulting in expression of other pro-inflammatory cytokines like IL-1 and IL-6 (Simmonds and Foxwell, 2008). A TNFR1-specific antibody, ASTROSAB has demonstrated effective inhibition of TNF induced IL-6 and IL-8 secretion in cell culture (Richter F. et al., 2013). A second study in chimeric humanized mice demonstrated that the inhibition if TNFR1 by a human TFNR1 silencer (TROS) lowered TNFR1-induced proinflammation reactions but did not alter TNFR2 signaling (Steeland et al., 2015). Studies looking at the source of TNF found that TNF produced by myeloid cells play a role in pathogenesis of experimental arthritis and autoimmune encephalomyelitis (Grivennikov et al., 2005; Kruglov et al., 2011). Contrastingly TNF secreted by T cells demonstrated protection against M. tuberculosis infections (Allie et al., 2013). TNF perfectly demonstrates the pleiotropic functionality typical of pro-inflammatory cytokines, balancing regulatory functions and pro-inflammatory signaling to maintain homeostasis of the immune system. Thus, systemic knock-out of TNF while beneficial in controlling certain inflammatory and autoimmune diseases, has major drawbacks like increased risk of severe infections, cardiovascular, neurological and immunological complications. Currently antibody-based immunotherapies with dual affinities for TNF and specific cell types are in development; one example is a myeloid-specific TFN inhibitor (MYSTI) that binds to both myeloid cells and TNF thus only restricting myeloid-secreted TNF activity. MYSTI demonstrated better protection against LPS/D-gal hepatotoxicity in humanized mice compared to anti-TNF treatment (Efimov et al., 2016).
Multiple other recombinant cytokines have already been approved for treatment of patients; IL-2 for cancer (Baron and Narula, 1990; Dutcher, 2002), several IFNα derivatives for cancer and viral infections (Syed and Ahmadpour, 1998; Melian and Plosker, 2001; Marcellin et al., 2004), IL-11 thrombocytopenia induced by chemotherapy (Isaacs et al., 1997), IFNβ for multiple sclerosis and IFNγ for osteoporosis and cancer (Cutler and Brombacher, 2005). Understanding the molecular mechanisms of action of different cytokines in the context of a specific disease will be an essential prerequisite for developing more targeted approaches to anti-cytokine/cytokine therapy. Targeting specialized processes rather than systemic cytokines has the added benefit of restricting toxicity and reducing interference with non-related biological processes. With the exponential development in antibody technologies it is likely that cytokine therapies in the near future might all be targeted to specific functions rather than cytokines overall.
T Cell Engineering
Recruitment and Activation of Cytotoxic T Cells
Bispecific antibodies (BsAbs) have two binding sites, each with a unique antigen specificity. Multiple formats of bsAbs exist including bi-specific T cell engagers (BiTEs), dual-affinity re-targeting antibodies (DARTs) and Tandem Diabodies. The most commonly used format is a tandem scFv, of which BiTEs are the most clinically advanced in oncology and are distinguished for their binding specificity to the TCR complex. These antibodies comprise both an antigen-specific arm and a T cell activating arm via generic CD3-interaction, which bypasses the requirement for MHC-I-restricted engagement. The activated T cells are redirected to target cells through the recognition of a pathogen/tumor-specific antigen by BiTE antibodies (Wolf et al., 2005; Bargou et al., 2008). BsAb mechanism of action is via creation of a temporary cytolytic synapse between the T cell and the target cell and the ensuing activation and proliferation of T cells elicits target cell lysis (Offner et al., 2006). The immunological synapses created by BiTEs are indistinguishable from those induced during natural CTL responses. The size of the synapse is determined by the distance between anti-CD3 and anti-antigen moieties and is a vital measure of cytotoxic potency; the smaller the synapse the closer the T cell membrane is to the target cell and since delivery of apoptotic mediators, such as granzyme proteases and perforin, is conducted by passive diffusion, this results in more efficient target-cell lysis (Moore et al., 2011).
BsAbs have been posited for the treatment of malignancies and inflammatory conditions for decades but have only recently begun to yield results (Fan et al., 2015). As such, catumaxomab (Removab) was the first bispecific trifunctional drug approved for the treatment of malignant ascites (Linke et al., 2010; Sedykh et al., 2018). Thereafter, blinatumomab (Blincyto®), a CD19 × CD3 BiTE, gained accelerated FDA-approval for the treatment of relapsed and/or refractory (R/R) acute lymphoblastic leukemia (ALL), and numerous other BiTE candidates are currently in phase I clinical trials for the treatment of various cancers (Przepiorka et al., 2015). Furthermore, as T cell exhaustion due to high antigen load has been suggested, concurrent PD-1 or PD-L1/2 blockade is under investigation (Köhnke et al., 2015). Studies are now focused on the efficacy of BiTEs for solid tumor malignancies, in which penetration of the tumor by BiTEs and T cells are at the forefront (Yuraszeck et al., 2017).
Given the success of bsAbs in cancer immunotherapy, similar strategies to eliminate pathogen-infected cells have been adopted. Following early studies on natural HIV receptor CD4 and an anti-CD3 binding moiety, no further development of bsAbs ensued for more than 20 years (Berg et al., 1991). Recently, DARTs, an alternative tandem ligand format, have been exploited for T cell–mediated cytolysis of latently HIV-infected cells (Figure 2). These bsAbs comprise anti-Env and anti-CD3 arms which simultaneously bind to HIV-infected cells and CD3-expressing polyclonal T cells, subsequently inducing CD8+ T cell elimination of HIV-infected CD4+ T cells in vitro, reducing virion levels ex vivo and mediating clearance of latent HIV reservoirs from resting CD4+ T cells (Sloan et al., 2015; Sung et al., 2015). The efficacy of DARTs on the HIV-1 reservoir in vivo remains undetermined. Apart from DARTs, BiTEs that target the HIV-1 envelope protein gp120 and CD4 have been recently described with potent antiviral activity in vitro and ex vivo (Brozy et al., 2018). In addition to HIV, bsABs have been developed to redirect effector T cells to HBV- and human cytomegalovirus (HCMV)-infected cells, in vitro (Meng et al., 2018).
The appeal of BsAbs lies in their synergistic effects attributed to their ability to simultaneously target several antigens (Sedykh et al., 2018). Although a promising therapeutic strategy, BsAbs are not without their challenges. In clinical settings, continuous delivery is necessary due to the rapid clearance of these agents, related to their small size and consequently, short half-life. Furthermore, blinatumomab is associated with severe adverse events including neurotoxicity and cytokine release syndrome, which although reversible and manageable by treatment with corticosteroids and/or Tocilizumab, an interleukin-6 receptor antagonist, discontinuation of use by patients is still common (Thakur et al., 2018). In addition, clinical success in solid tumors remains to be demonstrated, which may provide insights into the applicability of these agents against intracellular pathogens.
Reprogramming T Cells
Chimeric antigen receptor (CAR) T cells are modified T cells with a recombinant receptor, comprising a scFv that redirects the specificity of effector T-lymphocytes, fused to a transmembrane and signaling domain that mediates T cell activation without MHC engagement. By bypassing the standard kinetics of active immunization, CARs rapidly generate tumor-targeting T cells and modulate T cell proliferation and persistence in the tumor microenvironment (Ho et al., 2003; Sadelain et al., 2003). The therapy involves the ex vivo expansion of the patient’s peripheral blood T cells, genetically modifying the cells to express the CAR and re-infusing the engineered T cells back into the patient (Gattinoni et al., 2006).
First generation CAR T cells comprising a targeting scFv and chimeric CD3/FcεRIγ have undergone phase I clinical trials for the treatments of ovarian cancer (scFv-FcεRIγ) (Sadelain et al., 2003), refractory follicular lymphoma (scFv-CD3ζ) (Ho et al., 2003), neuroblastoma and melanoma (scFv-CD3ζ) (NCT00085930) (Zhao et al., 2006; Birkholz et al., 2009). In theory, any cell surface molecule, even those that elicit tolerance due to self-reactivity, may be targeted by CAR modified T cells, thereby increasing the natural T cell repertoire. Transgenic mice studies with first generation CARs recognized that these CAR T cells were prone to anergy (Brocker and Karjalainen, 1995). Thus, second and third generation CARs comprising an activating CD3 domain and cytoplasmic domain co-stimulatory receptors like CD28, 4-1BB, DAP10, ICOS or OX40 were generated. These CARs demonstrated superior activity as confirmed by a comparative study in patients treated with CD28/CD3ζ or CD3ζ-only CAR (Savoldo et al., 2011).
Chimeric antigen receptor T cell immunotherapy has achieved impressive clinical outcomes for the treatment of hematological malignancies, specifically B cell acute lymphoblastic leukemia (ALL) and lymphoma (Kochenderfer and Rosenberg, 2013; Maude et al., 2014). As such, two CD19-targeted CAR T cell therapies, Tisagenlecleucel (2nd generation) and Axicabtagene Ciloleucel (3rd generation) were recently FDA-approved (Neelapu et al., 2017; Schuster et al., 2017). Majority of cancer-related morbidity and mortality is associated to solid malignancies, however, attempts to apply CAR T cell therapy to solid tumors have been less effective compared to hematologic cancers. This has been attributed to a number of obstacles; the lack of suitable tumor-specific antigens and the subsequent toxicity to normal tissue, inefficient CAR T cell infiltration of tumor barriers, such as the extracellular matrix (ECM), as well as the immunosuppressive tumor microenvironment (Zhukovsky et al., 2016; Srivastava and Riddell, 2018).
As such, genetically modified fourth generation CARs also known as TRUCKs (T cell redirected for universal cytokine-mediated killing) with the ability to secret pro-inflammatory and pro-proliferative cytokines have been developed (Golubovskaya and Wu, 2016). This ability to induce cytokine expression is highly advantageous in the setting of solid tumors where T-cell penetration is poor and due to high phenotypic diversity, many cancer cells remain invisible to penetrating T-cells. The expression of cytokines such as IL-12 induced by TRUCKs provides a cytokine gradient and initiates universal cytokine mediated killing of cancer cells which eradicates cancer cells lacking antigen at the tumor site (Li et al., 2018). Recently, a TRUCK system utilizing the synNotch receptor was developed to express a variety of payloads in response to target antigens (Roybal et al., 2016; Roybal and Lim, 2017). In addition to inducible cytokines this synNotch system can induce adjuvants, checkpoint, bispecific and pro-tumor cytokine antibodies. With this system more anti-tumor cytokines and factors with the potential to remodel the suppressive tumor environment may be co-administered.
The unique obstacles imposed by solid tumors are also associated with brain tumors, although further exacerbated by additional biological complexities. With regards to glioblastoma, the most common malignant brain tumor in adults, recent developments in CAR T cells have addressed these challenges which include penetration through the blood–brain barrier (BBB), inadequate neoantigen presentation and the presence of glioma stem cells (GSCs) and their intrinsic role in drug and radiation resistance [reviewed by (Bagley et al., 2018)]. The poor homing of CAR T cells to target sites (Ager et al., 2016) has recently been addressed by the rational reengineering of CD6 to generate cytotoxic homing system (HS) T cells that readily infiltrate the otherwise restrictive BBB (Samaha et al., 2018). Studies such as these may provide significant insight into engineering T cells to target elusive infectious agents.
While most CAR T cell studies conducted to date are in the context of cancer immunotherapy, Kumaresan and colleagues developed a fungal-specific Dectin-1 C-type lectin receptor. The resulting D-CAR T cell specific for β-1.3-D-glucan showed effective killing of Aspergillus fumigatus in vitro and in vivo (Kumaresan et al., 2014). This proof-of-principle study shows that CAR technology can encompass C-type lectins, a key innate pattern recognition receptor with broad repertoires against fungal pathogens and thus, may be applied to other infectious diseases (Armstrong-James et al., 2017).
As with other immunotherapies, adverse effects are associated with CAR T cells. These include the on-target toxic effects of cytokine release syndrome (CRS) and neurotoxicity which can be managed by treatment with corticosteroids and/or Tocilizumab (Gauthier and Turtle, 2018). In a recent phase I/II clinical trial, CRS severity was found to be associated with infectious complications (Hill et al., 2018). Reducing the adverse effects of treatment, improved models to evaluate pre-clinical efficacy and importantly, the ultimate cost implications to patients, will determine the extent to which CAR T cell therapy can be applied (Srivastava and Riddell, 2018). The advances in CAR T cell therapy for solid tumors offers promise for intracellular infectious agents, such as M. tuberculosis, for which the inherent heterogeneity of granulomas accounts for poor antimicrobial drug penetration and accumulation (Kiran et al., 2016).
Depletion of Suppressor Cell Populations
Tregs are a subpopulation of T cells that modulate the immune system by reducing activation of effector T cell populations during prolonged inflammatory responses and inducing tolerance to benign molecules and self-antigens. Modern cancer immunotherapy approaches include biologics that selectively deplete suppressor cell populations, like Tregs. One such agent, a diphtheria toxin (DT) (Liedtke et al., 2008)-related interleukin 2 (IL-2) fusion protein, known as DD depletes cells with high expression levels of CD25 (Kiyokawa et al., 1991). DD is comprised of 3 domains; the DT catalytic domain, the DT membrane translocation domain and a substituted human IL-2 instead of the native DT-receptor. The IL-2 moiety allows for selective targeting of IL-2R bearing cells. Upon IL-2 and IL-2R binding, the fusion protein is internalized and transported to the cell cytosol where the catalytic domain inhibits protein synthesis resulting in downstream alterations that ultimately lead to apoptosis (Zahaf and Schmidt, 2017). DD has been approved for treatment of cutaneous T cell lymphoma and subsequent human studies. Results from these studies indicate that in addition to killing CD25+ lymphoma T cells, DD also transiently depleted Tregs (Litzinger et al., 2007). More recent studies assessing DD treatment in Leishmanian-infected mice verified a reduction of lesional Tregs, better lesion resolution and reduced parasite burden (Divanovic et al., 2011).
During M. tuberculosis infection, bacilli are engulfed by dendritic cells in the lung and transported to lymph nodes where CD4+ and CD8+ T cells are activated. Tregs have an opposing effect that limit potential protective responses and facilitate bacterial replication (Kursar et al., 2007). Additional studies using specific antibodies to deplete Tregs in M. tuberculosis-infected mice showed decreased bacterial burden (Gupta et al., 2017). Similarly to Tregs, another suppressor cell type, myeloid-derived suppressor cell (MDSC) populations also increase during M. tuberculosis infection contributing to T cell dysfunction and exuberant inflammation (Knaul et al., 2014). As with Tregs, specific antibody depletion of MDSCs ameliorated pathology and reduced bacterial replication in vivo. In addition to the suppressive effect of these cell populations, both Tregs and MDSCs express IL-2R, rendering these cell populations susceptible to DD treatment. M. tuberculosis-infected mice treated with DD monotherapy, or in conjunction with standard anti-TB treatment of indicated the effective reduction of Treg and MDSC populations resulting in reduced bacterial burdens (Gupta et al., 2017). While no studies have yet been conducted in the patients with TB, these studies suggest that IL-2 receptor targeting might be a viable treatment option in the future and should be validated in clinical trials.
Although the role of Tregs in the pathogenesis of HIV and HCV infections remains controversial, several studies have indicated Treg-mediated suppression of effector T cell responses in these diseases (Li et al., 2008). As such, the Treg depletion mouse model (DEREG), which expresses the diphtheria toxin (DT) receptor under the control of the Foxp3 promoter, has shown the reactivation of virus-specific CD8+ T cells following the transient depletion of Tregs during chronic viral infection (Kim et al., 2007; Lahl et al., 2007; Dietze et al., 2011; Berod et al., 2012). The limitations to DEREG, including DT-mediated toxicity, warrants further studies to investigate the role of Tregs during chronic viral infection (Christiaansen et al., 2014).
Antibody/Ligand-Based Therapies
Selecting the Ideal Ligand
The variety of mechanisms of action that exist to accomplish ligand-mediated control of disease suppression designates this approach an attractive one. The distinct structure of mAbs/natural ligands define their highly efficient function in immunity and underpins antibody/ligand engineering and design. The aim of ligand-targeted therapeutics (LTT) is to neutralize or eliminate pathogenic infections and/or malignant cells (Figure 3). This is done by three major mechanisms of action: (a) inhibiting the function of certain molecules, (b) targeting particular cells or (c) acting as signaling molecules (Brekke and Sandlie, 2003).
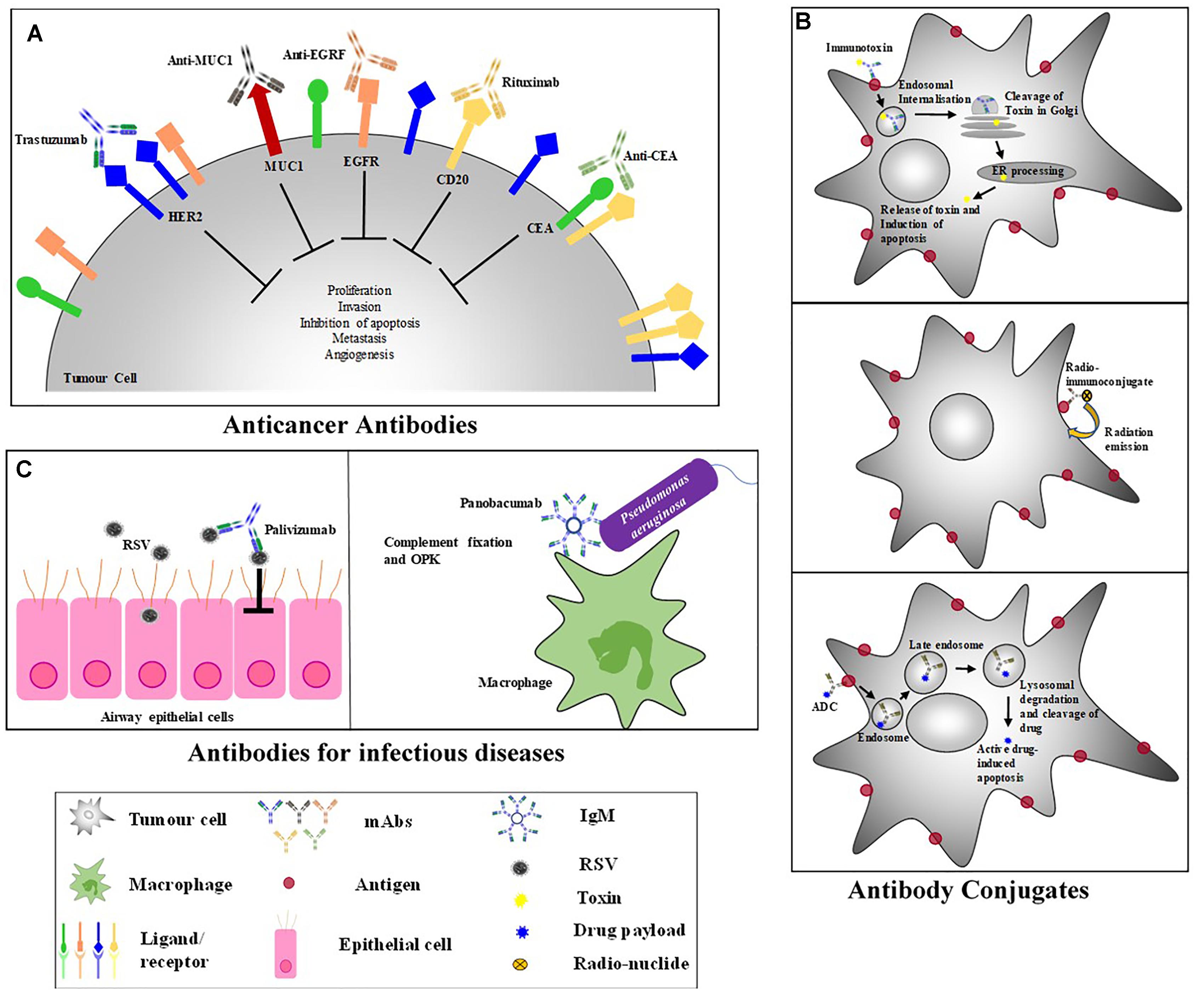
Figure 3. Antibody-Based Therapeutic Strategies. (A) Anticancer antibodies eliminate cancer cells and cause tumor destruction by targeting cancer antigens. (B) Antibody-conjugates – (i) Immunotoxins: bind to a surface receptor of an infected cell, undergo endocytosis and intracellular trafficking to the cytosol where most toxins induce cell death; (Becker and Benhar, 2012) (ii) ADCs: combine the specificity of mAbs with the cytotoxic potential of drugs and binds to internalizing receptors on target cell and are taken up by endocytosis; Once in the cell, ADCs undergo cellular trafficking to a lysosome where lysosomal degradation results in the cleavage and release of the active drug into the cellular cytoplasm where the drug induces apoptosis; (Scotti et al., 2015) (iii) Radioimmunoconjugates: antibodies attached to a radioactive molecule, once the antibody binds the target cell, the radio-particle’s radiation interacts with target cells, resulting in cell death. (C) Anti-viral antibodies – to eliminate a viral inhibition of cell infection, viral replication, cell-cell transmission, viral release as well as mediated killing of infected cells needs to occur; Palivizumab is a neutralizing antibody that binds to RSV preventing virus-host cell interactions (Groothuis and Nishida, 2002). Most antibacterial therapeutic mAbs function by inducing complement fixation and opsonophagocytic killing (OPK) of target bacteria; Panobacumab induces macrophage OPK of Pseudomonas aeruginosa (Que et al., 2014).
The complex set of interaction that occur between antibodies/ligands and their cognate receptors, marks choosing an appropriate targeting ligand a crucial step in developing successful targeting applications. Multiple variables should be accounted; the level of receptor expression, the binding affinity of the targeting molecule (Damelin et al., 2015), a role for receptor-mediated internalization, the use of non-antibody based ligands and full antibody or antibody-fragment (Kovtun et al., 2006). For most therapies, the target antigen/receptor should ideally be expressed in high density on the target cell surface to permit accurate selectivity (Beck et al., 2017). In addition, avoidance of a high degree of target heterogenicity may limit off-target effects. Importantly, targets which are shed are not favored as free targets may compete with cell-bound targets thus reducing the therapeutic effects of a treatment (Allen, 2002).
Non-antibody ligands such as Fas, tumor necrosis factor (TNF) and TFN-related apoptosis-inducing ligand (TRIAL) are often easily available, relatively cheap to manufacture and easy to handle (Mathew and Verma, 2009). The main disadvantage is their relatively non-selective expression on diseased cells, making antibodies the preferred targeted delivery agent (Allen, 2002). Furthermore, recent advances in antibody engineering and phage display have improved the identification and selection of antibodies/antibody derivatives with a high degree of specificity and wide range of binding affinity for target tissue/cells (Zhao et al., 2016).
The use of whole antibodies is advantageous, allowing increased binding avidity that occurs through multiple binding sites on one molecule. Additionally, antibodies possess an Fc domain which engages with other immune cells that potentiate the natural immune response by antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Whole antibodies may also be more stable that antibody fragments over prolonged storage periods (Allen, 2002). Despite the advantages, whole antibodies also have their limitations; they commonly bind to normal tissues via Fc receptors, specifically on macrophages resulting in high liver and spleen uptake that can cause increased immunogenicity. Fab and scFv (single-chain variable fragments) lack both an Fc domain and complement-activating region thus reducing the potential for immunogenicity. Their smaller size also permits better cellular penetration which is a desirable effect of certain treatments. Furthermore, scFvs are attractive choices due to ease of identification and production, for example using phage display or Escherichia coli fermentation, as well as their reduced immunogenicity (Ahmad et al., 2012).
Therapeutic Monoclonal Antibodies
Monoclonal Antibody (mAb) therapy has been studied and developed for the treatment of cancer, more so than for any other disease area. Subsequently, the trials and errors and ensuing solutions encountered in the cancer field, have paved the way and advanced antibody-based therapies. These include the initial complications associated with administering mouse hybridoma-derived antibodies to humans and the human anti-mouse antibody (HAMA) response which was subsequently resolved by chimerization and humanization, using recombinant DNA technology (Morrison et al., 1984; Jones et al., 1986). Thus, after years of research, a variety of mAb-based approaches that mediate their antitumor effects through diverse mechanisms have been developed (Weiner, 2015). Attesting to the success of these strategies, numerous mAbs have received clinical approval for the treatment of various cancers, as reviewed by (Kim and Kim, 2015).
The inhibitory activity of mAbs is conducted by preventing soluble mediators like growth factors and cytokines from reaching their target receptors. This is achieved by binding to the mediator or its cognate receptor. Targeting involves directing antibodies to a particular cell population where Fc receptor functions are elicited or to which effector moieties are transported. Subsequently, antibody signaling is conducted by cross linking of receptors related to programmed cell death or by specific receptors that function as agonists for activation of specific cell types (Brekke and Sandlie, 2003).
Antibody therapy for infectious diseases can be traced back more than 100 years ago to serum therapy for the neutralization of bacterial toxins (Graham and Ambrosino, 2015). In comparison to neutralization of toxins, prevention of viral diseases with the use of mAbs is more complex. To eliminate a viral infection a number of events need to occur, these include inhibition of: cell infection, viral replication, cell–cell transmission, viral release as well as mediated killing of infected cells (Parren and Burton, 2001; Burton, 2002). A typical immune response to such an infection results in the generation of specific polyclonal antibodies. Some of these antibodies are neutralizing others are not. The overall outcome, with a combination of blocking, neutralizing and eliminating antibodies might indeed be effective protection. The anti-RSV mAb Palivizumab is approved for high risk infants, and is clinically effective prophylactically, but not therapeutically (Hu and Robinson, 2010). As such, super-antibodies are a new generation of highly potent and/or broadly cross-reactive mAbs that offer prophylactic and therapeutic possibilities for viral infections. This includes highly antigenically variable viruses such as HIV, influenza virus, and Ebola virus, the treatments of which by super-antibodies, are under clinical evaluation (Walker and Burton, 2018).
As with most antibodies, surface antigens are attractive targets for antibacterial mAbs. The key features of surface-specific antibacterial mAbs are complement fixation for engagement of the host immune system and opsonophagocytic killing (OPK). Panobacumab, one of the most clinically advanced antibacterial mAbs, is a human anti-Pseudomonas aeruginosa LPS serotype O11 (Horn et al., 2010; Que et al., 2014). This mAb showed OPK activity in vitro and exhibited decreased bacterial burden and improved survival in infection models (Horn et al., 2010). In a phase II clinical trial, Panobacumab was used adjunctively with standard of care antibiotics resulting in shortened duration to disease resolution in P. aeruginosa 011 pneumonia patients (Que et al., 2014). While strain-specific mAbs have shown promising results, its activity is limited to strains that express its target receptors. Contrastingly, a protective mAb targeting P. aeruginosa type III secretion protein, PcrV plays an important role in transporting virulence factors into host cells (Frank et al., 2002). This led to the development of KB001, a phase II mAb for the prevention of P. aeruginosa ventilator-associated pneumonia (Baer et al., 2009).
In addition, the secreted virulence factor, alpha toxin (AT) which causes cellular death by the formation permeable pores in the plasma membrane, is targeted by two anti-staphylococcal mAbs currently undergoing efficacy trials (Berube and Wardenburg, 2013; Oganesyan et al., 2014). This prospective therapy has demonstrated prophylactic protection in Staphylococcus aureus pneumonia mouse models (Hua et al., 2014; Sause et al., 2016).
Furthermore, scFvs to the spirochete Borrelia have been found to have direct bactericidal effects (Larocca et al., 2008). Similarly, an anti-idiotype antibody that mimics yeast killer toxin has also been described to have direct cell-killing effects against an array of broad spectrum bacteria along with yeast, fungi and mycobacteria (Conti et al., 2000). Such antibodies may overcome the necessity for a competent host immune response for efficacy. More often, however, antibacterial antibodies rely on recruitment of varying antibody-directed immune effector functions such as; initiating antibody-directed cellular cytotoxicity (ADCC), activating the complement cascade, and inducing oxidative bursts. Moreover, all antibody variable regions are able to catalyze redox reactions resulting in the generation of potent oxidizing agents that directly harm bacteria (Wentworth et al., 2003). This process causes inflammation which further enhances the immune response by recruitment of immune system elements (Babior et al., 2003).
Antibody-Conjugates
Different antibody formats consisting of an antibody binding fragment conjugated to varying effector molecules like radionuclides (radioimmunotherapy/RITs), drugs (antibody-drug conjugates/ADCs), toxins (immunotoxins) and enzymes (antibody-directed enzyme prodrug therapy/ADEPT) are referred to as antibody conjugates. Conjugated mAbs are used to deliver one of these effector molecules specifically to infected cells while limiting the side effects of off-target drug delivery (Weiner et al., 2012).
Radioimmunotherapy/radioimmunoconjugates
The first effective form of targeted cancer therapy was131I, a β-particle emitter that also emits small amounts of γ-radiation. It was used for treatment of thyroid cancer because of its specificity for elemental iodine in the thyroid. The success of this treatment led researchers to explore various carrier molecules including mAbs which could be utilized for targeted delivery of radioisotopes to infected cells (Weiner, 2015). The first 2 FDA approved conjugated antibody therapies, were radioimmunotherapies, both for the treatment of hematological malignancies. These RITs were 90Y-ibritumomab tiuxetan and 131I-tositumomab, both targeting CD20 for treatment of relapsed and/or rituximab-refractory follicular or low-grade lymphomas. There are a number of other RITs in development, several of which target solid tumors (Weiner, 2015). Surface receptors (non-internalizing) are sufficient for treatment with RITs as internalization is not required by all RITs to induce killing of target cells (Illidge and Johnson, 2000). In addition, while most antibody-based therapies rely on targets with high binding affinity, RITs may be more effective against diseased cells with lower binding affinity targets; therapeutic proteins with high binding affinity will likely bind the first target receptor it contacts, predicted to be on the periphery of the tumor, and given its high affinity, will not penetrate deeper into the tumor thus resulting in radiation exposure to a higher number of healthy cells circulating around the tumor. However, lower binding affinity targets may permit better tumor penetration by allowing the therapeutic protein to move from one target expressing cell to the next, within the tumor microenvironment (Allen, 2011).
Whilst the use of mAbs has significantly improved specificity of radiotherapies many challenges persist limiting its widespread clinical application. Radioisotopes are in a continual state of decay harming healthy tissue during circulation or taken up non-specifically (Pouget et al., 2011). Bone marrow in particular is highly sensitive to radiation and is therefore affected by circulating RITs. In addition, the kidney and liver are exposed to high doses of radiation due to their roles in clearing RITs and free radioisotopes, resulting in a limited amount of radiation being delivered to target sites, even with the use of very specific targeting agents (Pouget et al., 2011). Further, the actual production of RITs is highly complex and requires experienced nuclear-medicine physicians due to the dangers of radiation poisoning. To overcome these challenges, many ongoing studies are assessing novel radioisotopes like α-emitters which deliver ionizing radiation adjacent, within up to a few cell diameters, to diseased cells consequently restricting radiation exposure to target cells and reducing exposure to neighboring benign cells (Allen, 2011). Current clinical and preclinical studies on such α-particles are ongoing and have the potential to significantly enhance RITs (Weiner, 2015).
There are presently no clinically approved RITs for treatment of infectious diseases, but studies have demonstrated promising progress in this field. The potential efficacy of RIT against C. neoformans (CN) has been of particular focus (Dadachova et al., 2003). Due to the current HIV pandemic, CN has emerged as a major fungal disease-causing life-threatening meningoencephalitis in 6–8% of AIDS patients. Further, CN infections are usually incurable in immunocompromised patients as current drug treatments are unable to eradicate the infection in this population (Casadevall et al., 1993; Currie and Casadevall, 1994). The α-particle emitter Bismuth-213 (213Bi) has been proposed for use in certain solid cancers and single-cell disorders and is currently in phase I/II clinical trials for treatment of leukemia patients (Boll et al., 1997; Sgouros et al., 1999; Adams et al., 2000; Mcdevitt et al., 2000). Consequently, a RIT comprising either of two radioisotopes, a high-energy β-emitter Rhenium-188 (188Re) or 213Bi and the capsule-specific mAb 18B7, were assessed with the CN mouse models. 18B7 has no inherent fungicidal activity by itself, but when conjugated to either 188Re or 213Bi, resulted in decreased fungal burden and prolonged survival in RIT treated mice (Dadachova et al., 2003).
The positive results of the CN study has prompted researchers to conducted a second investigation assessing the RIT approach in animal models of Streptococcus pneumonia (Dadachova et al., 2004). In this study the 213Bi radioisotope was conjugated to mAb D11, which has specific binding of pneumococcal capsular polysaccharide 8 (PPS8) and is protective against this serotype in multiple strains of mice and infection models (Zhong et al., 1999; Burns et al., 2003). 213Bi was specifically chosen over β-emitters for its short half-life that allows delivery of substantial amounts of radiation in a shorter time which is imperative when targeting rapidly dividing cells like pneumococci. Treatment of S. pneumonia with 213Bi-D11 RIT demonstrated bacterial killing in a dose-dependent manner and increased survival in S. pneumonia-infected C57BL/6 mice (Dadachova et al., 2004).
Furthermore, RIT has been reported to significantly decrease the number of HIV-infected cells in vivo, via conjugation of 213Bi and 188Re radioisotopes to gp41 (HIV-envelope protein)-specific mAb 246-D (Dadachova et al., 2006). A major advantage of using RIT as a treatment for viruses is that the RIT itself does not neutralize the virus, consequently reducing the probability of applying selection pressure that may result resistant strains forming (Dadachova et al., 2006). Recently, the potential therapeutic utility of RIT in combination with antiretroviral drugs (ARVs) has been demonstrated as a novel treatment for the eradication of HIV (Dadachova and Casadevall, 2014).
The morphological similarities observed between granuloma formation and that of a solid tumor, a highly fibrotic and hypoxic microenvironment, suggests this type of treatment may prove useful in TB patients (Datta et al., 2015). Furthermore, granulomas and solid tumors have functionally abnormal vasculature resulting in impaired small molecule distribution, exhibiting a wide range of variation in spatial drug distribution with majority of drugs accumulating in the periphery of the tumor/granuloma and not penetrating into the central diseased tissue. There are currently no RITs approved for the treatment of TB but given that granuloma penetration is a major challenge, RITs with a wider radiation field may be able to bind to the cells on the outside of the granuloma and still elicit killing of bacilli within the granuloma. Additionally, other forms of immunotherapy used for treatment of solid tumors may be applicable to TB granulomas; one important example is bevacizumab, an anti-VEGF antibody used to normalize tumor vasculature in solid tumors. A study conducted in rabbit TB models demonstrated the ability of bevacizumab to normalize the abnormal vasculature of granulomas, resulting in reduced hypoxia and increased small molecule drug penetration into the central granuloma (Datta et al., 2015).
Antibody-drug conjugates (ADCs)
Antibody-drug conjugates combine the specificity of mAbs with the cytotoxic potential of drugs (Weiner, 2015). Currently there are two FDA- approved chemotherapeutic ADCs, Ado-rastuzumab, a HER2+-targeting conjugate that exerts cytotoxicity via inhibition of microtubule assembly, and Brentuximab Vedotin, a CD30-targeting mAb conjugated to an antimitotic drug for the treatment of Hodgkin (NCT00848926) and anaplastic large cell lymphoma (NCT00866047) (Amiri-Kordestani et al., 2014; Herrera et al., 2018). This CD30-specific ADC, although approved for cancer treatment, has also been tested as an anti-viral ADC for the treatment of HIV (Hogan et al., 2018). CD30 is part of the TFN receptor superfamily, expressed on tumor cells and on a small population of lymphocytes in healthy individuals (Stein et al., 1985; Falini et al., 1995). Additionally, CD30 has been associated with HIV disease progression, with cell-associated HIV-1 RNA being considerably enriched in CD30 expressing CD4+ T cells. The significant reduction in HIV-1 DNA in PBMCs from HIV-infected donors by the ADC, suggests CD30 maybe a potential therapeutic target for HIV (Hogan et al., 2018).
Unlike RITs all ADC’s require internalizing targets to elicit their effector functions thus only targets which undergo receptor-mediated endocytosis may be used for ADC treatment (Trail et al., 2017). The distribution of receptors on diseased cells and tissue is also an important consideration when choosing a target for ADC’s. For diseases where penetration is not an issue, such as hematological cancers and other blood borne diseases, targeted therapeutics may easily bind to their target cells thus even a moderately expressed target may be sufficient for effective killing. However, for solid tumors, TB granulomas and other difficult to penetrate diseases it is important that the therapeutic protein have an increased serum half-life to allow sufficient time for uptake into diseased tissue (Allen, 2002). Furthermore, the frequency of target receptor internalization/recycling may also affect the binding of the therapeutic protein. For targets with fast turnover rates, changes in surface receptor expression have little impact on the accumulation of the ADC in target cells whereas targets with slow turnover rates show reduced efficacy with decrease surface receptor expression (Sadekar et al., 2015).
Recently, an antibody-antibiotic conjugate (AAC), Thiomab combining essential characteristics of a mAB and an antibiotic has been shown to be an effective therapy against S. aureus (Lehar et al., 2015). This ACC works similarly to ADC’s in that it uses and antibody to deliver an antibiotic payload to bacteria (Mariathasan and Tan, 2017). As with ADC’s the antibody, linker and payload used in the design of the ACC play critical roles in the binding, stability, and effector functions, respectively. A mAb, directed against β-N-acetylglucosamine cell-wall teichoic acid (β-GlcNAc) residues on the cell wall of S. aureus was chosen as this target is highly expressed on S. aureus but not on mammalian cells. For this ACC, a dimethyl DNA31, referred to as dmDNA31, was the chosen antibiotic as it possesses a functional group amenable to conjugation, it is active against S. aureus in both its dormant and active state and has potent bactericidal activity. Finally, the linker selected for the ACC was a MC-ValCit-PABQ linker consisting of caproic acid and maleimide for attachment to the antibody, a protease-cleavable dipeptide, valine citrulline and a novel salt, PABQ for attachment of the antibiotic. Thiomab effector function comprises killing of intracellular bacteria via intracellular release of dmDNA31 and internalization of extracellular bacteria through rapid opsonization. Given the rapid emergence of antibiotic resistance and lack of discovery of new antibiotics the ACC avenue provides opportunity for developing novel therapeutics to treat infectious diseases (Mariathasan and Tan, 2017).
Immunotoxins
A classical IT is comprised of a binding domain and a toxic protein domain originally attached by chemical conjugation and later recombinantly fused. The binding domain of an IT may be natural ligands (growth factors, surface receptors), an antibody, or a recombinant derivative (Barth, 2009). To be effective, an IT needs to bind to a surface receptor of an infected cell, undergo endocytosis and intracellular trafficking of its catalytically active subunit to the cytosol where it induces cell death. Additionally, the natural binding domains of the toxins need to be removed so that cellular uptake may be controlled by the specificity of the mAb alone. Historically, bacterial- or plant-derived toxins have first been used to generate recombinant ITs (rITs). Their recognition as foreign antigens by the human immune system, has resulted in induction of neutralizing antibody responses in patients. To reduce immunogenicity, (a) B and T cell epitopes have been removed from the protein toxins, and (b) the bacterial/plant toxins have been replaced by human apoptosis-inducing enzymes which are non-toxic until delivered intracellularly to the target cell (Jordaan et al., 2018). As such, targeted human cytolytic fusion proteins represent the 4th and most recent generation of anticancer immunotherapeutics, with a plethora of candidates such as granzyme B, angiogenin, DAP-K and Map-tau currently under preclinical investigation (Hristodorov et al., 2013; Amoury et al., 2016; Lilienthal et al., 2016; Akinrinmade et al., 2017).
Majority of the focus on developing ITs has been for treatment of cancer, however, there are also those that target viruses (Spiess et al., 2016). Anti-viral ITs interfere with the infection cycle of viruses by targeting virus entry, intracellular replication, viral particle formation and cellular extrusion or by modulating the anti-viral cellular immune responses (Zhu et al., 2015). In addition, anti-viral ITs have a capacity to kill viral infected cells (Spiess et al., 2015; Zhu et al., 2015) and because target molecules are often encoded by the virus itself, off-target killing of cells expressing low levels of the target receptor is greatly reduced compared to anti-cancer immunotoxins. Furthermore, interference of protein synthesis in infected cells prevents viral dissemination and destroys reservoirs of latently infected cells (Geoghegan et al., 2015) which pertains to diseases such as HIV, where current therapies only target active viral infections and the lack of eradication of latently infected cells results in patients needing lifelong therapy (Kennedy et al., 2006). ITs comprising anti-HIV capsid protein gp120 mAb linked to a truncated form of Pseudomonas exotoxin A (PE) display specific and potent cytotoxicity against HIV replication in monocyte-derived macrophages and PBMC’s (Ashorn et al., 1991; Berger and Pastan, 2010). Mice studies utilizing gp120-specific ITs in combination with standard therapy showed significant improvement over standard therapy alone but was unable to eradicate the disease (Goldstein et al., 2000), probably as a result of immunogenicity-related neutralization. A major complication of immunotoxin-based treatment of HIV is the limited expression of HIV-proteins in latently infected cells, engendering this cell population invisible to the immunotoxin. Combination therapy of ADC’s with drugs such as latency-revering agents, may increase the target receptor expression on latently infected cells enhancing susceptibility to ADC-induced toxicity (Perreau et al., 2017). Furthermore, two PE-based ITs YC15-PE38 and 2014-PE38, have been developed for the specific killing of Kaposi’s sarcoma-associated herpesvirus (KSHV)-infected cells and target KSHV lytic glycoproteins. Both ITs impede the construction of infectious KSHV particles and specifically kill KSHV-infected cells (Cai and Berger, 2011; Chatterjee et al., 2012).
In recent times, antibodies isolated from members of the camelid family have been used to develop ITs directed against herpes-simplex virus 2 (HSV-2) (Geoghegan et al., 2015). These antibodies are termed VHH as they consist of the variable domains of antibody heavy-chains and lack light chain and CH1 domains (Hamers-Casterman et al., 1993). VHH share a large degree of homology with mammalian variable domains and have enhanced tissue penetration due their smaller size (Cortez-Retamozo et al., 2002). A VHH, R33 antibody with specific binding to viral cell surface glycoprotein D on HSV-2 was identified using phage display. R33 alone did not have any neutralizing effect on HSV-2, however, in combination with the cytotoxic domain of PE, this IT demonstrated extremely efficient killing of HSV-2-infected cells in vitro.
In addition to anti-viral ITs, the targeted delivery of granzyme B (Araki et al., 2013) to Plasmodium falciparum provided the first evidence of granzyme B’s anti-parasitic effects on malaria (Kapelski et al., 2015). The merozoite surface protein 4 (MSP4)-targeting IT induced enhanced inhibition of parasite growth in infected erythrocytes and although the exact mechanism of action is unknown the evidence strongly suggests that malaria-specific ITs are valuable drug candidates for treatment of multidrug resistant P. falciparum strains.
Antibody drug conjugates has significantly improved in efficacy over the last few years, however, its therapeutic application is still limited and require further improvements. All ADC’s currently approved for therapy target tumor associated antigens not tumor specific antigens thus resulting in undesired toxicity that limits its therapeutic application. New generation ADC’s should focus on disease-specific targets and will most likely require payload that do not interact with cells unless conjugated to an antibody. The major challengers in developing ADC’s are identifying appropriate targets, designing chemically stable linkers and choosing an appropriately cytotoxic drug (Lin et al., 2018). All of these factors require extensive optimization to fully improve the efficacy and tolerability of ADC’s thus understanding each of these components is key to developing new generation ADCs.
Conclusion
In spite of the conventional association of the term “immunotherapy” with the treatment of cancer, indeed publication records are indicative of this bias, this approach is fast-establishing itself within the armory of therapeutics applicable across the disease spectrum. Herewith, the versatility of the strategies that distinguish the immunotherapeutic approach have been described, with an emphasis on therapies that enhance T cell effector function as well as ligand-based therapies that neutralize or eliminate diseased cells. Although not within the scope of this review, natural killer (NK) cell immunotherapies, particularly CAR-modified NK cells and bispecific killer cell engagers (BiKEs), are rapidly gaining attention for application in cancer and HIV (Gleason et al., 2014; Rezvani and Rouce, 2015; Schmohl et al., 2016; Liu et al., 2017). Although the development of vaccines, whether therapeutic or prophylactic, still remains a major focus for infectious diseases such as HIV, based on the number and progress of preclinical studies, the development of immune checkpoint inhibitors and antibody-based therapies are emerging as promising approaches. T cell engineering has transformed adoptive therapies; CAR T cells in particular have gained massive recognition in the cancer field and although not initially successful in clinical trials, second-generation anti-HIV CAR T cells are being explored for their potential to provide long-term protection (Zhen et al., 2017; Wagner, 2018). In addition, it is evident that advances in antibody-conjugates against cancer have paved the way for similar strategies in infectious diseases, especially HIV and various bacterial and fungal infections. In this regard, an onslaught of novel agents and a significant increase in pre-clinical studies in various disease models can be expected.
Despite these encouraging results, ultimately, the high cost of these agents will necessitate comprehensive evaluations of the economic sustainability on healthcare systems, particularly in low- and middle-income countries where the burden of diseases such as HIV and TB remain significant. In addition, the common adverse effects amongst T cell-based therapies, specifically neurotoxicity and CRS, although manageable, remains a major challenge during patient care. Importantly, not all patients respond to immunotherapy, and in cancer, this is attributed to various resistance mechanisms. Combination immunotherapy may provide a solution; a multipronged approach to a curative treatment that has been heralded as the next wave of therapeutic strategies for cancer, proving successful with the recent case of complete durable regression of metastatic breast cancer (Morrissey et al., 2016; Zacharakis et al., 2018). Combinatorial strategies that promote an immunogenic tumor microenvironment by introducing existing chemotherapeutic agents with immune checkpoint inhibitors are fast gaining clinical recognition (Sharma and Allison, 2015). Recently, a mathematical model was developed to predict the efficacy of the combination of a cancer vaccine with an immune checkpoint inhibitor (Lai and Friedman, 2017). The establishment of such tools will be essential to provide rational insight to the advancement of combination immunotherapy. Clinical studies evaluating the configuration of these combinatorial regimens will be equally important (Schmid et al., 2018). Certainly, a combinatorial approach is central to achieving an HIV cure (Perreau et al., 2017). Of note, despite the incredibly diverse application of mAbs for infectious diseases, its relative underutilization in comparison to cancer may be attributable to the high cost compared with small molecule drugs or vaccines. Consequently, it is possible that immunotherapies may be relegated to adjunct or interim intervention strategies.
The age of immunotherapies has revolutionized cancer treatment. However, there is still much room left for improvement and efforts are ongoing to address better patient outcomes (Mohindra, 2018). With the growing understanding of the biological etiology of diseases, the current portfolio of immunotherapies is expected to expand significantly and may be, just as groundbreaking in combating infectious diseases.
Author Contributions
KN and TN contributed equally to the development and writing of the paper, reviewing relevant literature, and preparation of figures in the paper. SC contributed to the writing of the paper and provided the table on vaccines. SB provided substantial, direct, and intellectual contribution to the work. All authors consented for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Adams, G., Shaller, C., Chappell, L., Wu, C., Horak, E., Simmons, H., et al. (2000). Delivery of the α-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl. Med. Biol. 27, 339–346. doi: 10.1016/S0969-8051(00)00103-7
Ager, A., Watson, H. A., Wehenkel, S. C., and Mohammed, R. N. (2016). Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem. Soc. Trans. 44, 377–385. doi: 10.1042/BST20150254
Ahmad, Z. A., Yeap, S. K., Ali, A. M., Ho, W. Y., Alitheen, N. B. M., and Hamid, M. (2012). scFv antibody: principles and clinical application. Clin. Dev. Immunol. 2012:980250. doi: 10.1155/2012/980250
Akinrinmade, O. A., Jordaan, S., Hristodorov, D., Mladenov, R., Mungra, N., Chetty, S., et al. (2017). Human MAP tau based targeted cytolytic fusion proteins. Biomedicines 5:E36. doi: 10.3390/biomedicines5030036
Allen, B. J. (2011). Can α-radioimmunotherapy increase efficacy for the systemic control of cancer? Immunotherapy 3, 455–458. doi: 10.2217/imt.11.13
Allen, T. M. (2002). Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2, 750–763. doi: 10.1038/nrc903
Allie, N., Grivennikov, S. I., Keeton, R., Hsu, N.-J., Bourigault, M.-L., Court, N., et al. (2013). Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci. Rep. 3:1809. doi: 10.1038/srep01809
Alteri, R., Kalidas, M., Gadd, L., and Wyant, T. (2018). Cancer Immunotherapy. Atlanta: American Cancer Society.
Amiri-Kordestani, L., Blumenthal, G. M., Xu, Q. C., Zhang, L., Tang, S. W., Ha, L., et al. (2014). FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 20, 4436–4441. doi: 10.1158/1078-0432.CCR-14-0012
Amoury, M., Kolberg, K., Pham, A.-T., Hristodorov, D., Mladenov, R., Di Fiore, S., et al. (2016). Granzyme B-based cytolytic fusion protein targeting EpCAM specifically kills triple negative breast cancer cells in vitro and inhibits tumor growth in a subcutaneous mouse tumor model. Cancer Lett. 372, 201–209. doi: 10.1016/j.canlet.2016.01.027
Anderson, A. C., Joller, N., and Kuchroo, V. K. (2016). Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004. doi: 10.1016/j.immuni.2016.05.001
Araki, K., Youngblood, B., and Ahmed, R. (2013). Programmed cell death 1-directed immunotherapy for enhancing T-cell function. Cold Spring Harb. Symp. Quant. Biol. 78, 239–247. doi: 10.1101/sqb.2013.78.019869
Ardolino, M., and Raulet, D. H. (2016). Cytokine therapy restores antitumor responses of NK cells rendered anergic in MHC I-deficient tumors. Oncoimmunology 5:e1002725. doi: 10.1080/2162402X.2014.1002725
Armstrong-James, D., Brown, G. D., Netea, M. G., Zelante, T., Gresnigt, M. S., Van De Veerdonk, F. L., et al. (2017). Immunotherapeutic approaches to treatment of fungal diseases. Lancet Infect. Dis. 17, e393–e402. doi: 10.1016/S1473-3099(17)30442-5
Ashorn, P., Englund, G., Martin, M. A., Moss, B., and Berger, E. A. (1991). Anti-HIV activity of CD4- Pseudomonas exotoxin on infected primary human lymphocytes and monocyte/macrophages. J. Infect. Dis. 163, 703–709. doi: 10.1093/infdis/163.4.703
Babior, B. M., Takeuchi, C., Ruedi, J., Gutierrez, A., and Wentworth, P. (2003). Investigating antibody-catalyzed ozone generation by human neutrophils. Proc. Natl. Acad. Sci. U.S.A. 100, 3031–3034. doi: 10.1073/pnas.0530251100
Baer, M., Sawa, T., Flynn, P., Luehrsen, K., Martinez, D., Wiener-Kronish, J. P., et al. (2009). An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 77, 1083–1090. doi: 10.1128/IAI.00815-08
Bagley, S. J., Desai, A. S., Linette, G. P., June, C. H., and O’rourke, D. M. (2018). CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neurooncology 20, 1429–1438. doi: 10.1093/neuonc/noy032
Balar, A. V., Galsky, M. D., Rosenberg, J. E., Powles, T., Petrylak, D. P., Bellmunt, J., et al. (2017). Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 389, 67–76. doi: 10.1016/S0140-6736(16)32455-2
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., et al. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. doi: 10.1038/nature04444
Bargou, R., Leo, E., Zugmaier, G., Klinger, M., Goebeler, M., Knop, S., et al. (2008). Tumor regression in cancer patients by very low doses of a T cell–engaging antibody. Science 321, 974–977. doi: 10.1126/science.1158545
Baron, E., and Narula, S. (1990). From cloning to a commercial realization: human alpha interferon. Crit. Rev. Biotechnol. 10, 179–190. doi: 10.3109/07388559009038206
Barth, S. (2009). Editorial [Hot Topic: recombinant immunotoxins-The Next Generation (Executive Editor: Stefan Barth)]. Curr. Pharm. Des. 15, 2650–2651. doi: 10.2174/138161209788923912
Beck, A., Goetsch, L., Dumontet, C., and Corvaïa, N. (2017). Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 16, 315–317. doi: 10.1038/nrd.2016.268
Becker, N., and Benhar, I. (2012). Antibody-based immunotoxins for the treatment of cancer. Antibodies 1, 39–69. doi: 10.3390/antib1010039
Berg, J., Lötscher, E., Steimer, K. S., Capon, D. J., Baenziger, J., Jäck, H., et al. (1991). Bispecific antibodies that mediate killing of cells infected with human immunodeficiency virus of any strain. Proc. Natl. Acad. Sci. U.S.A. 88, 4723–4727. doi: 10.1073/pnas.88.11.4723
Berger, E. A., and Pastan, I. (2010). Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog. 6:e1000803. doi: 10.1371/journal.ppat.1000803
Berod, L., Puttur, F., Huehn, J., and Sparwasser, T. (2012). Tregs in infection and vaccinology: heroes or traitors? Microbial. Biotechnol. 5, 260–269. doi: 10.1111/j.1751-7915.2011.00299.x
Berube, B. J., and Wardenburg, J. B. (2013). Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins 5, 1140–1166. doi: 10.3390/toxins5061140
Bi, Q., Ferreras, E., Pezzoli, L., Legros, D., Ivers, L. C., Date, K., et al. (2017). Protection against cholera from killed whole-cell oral cholera vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 17, 1080–1088. doi: 10.1016/S1473-3099(17)30359-6
Birkholz, K., Hombach, A., Krug, C., Reuter, S., Kershaw, M., Kämpgen, E., et al. (2009). Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 16, 596–604. doi: 10.1038/gt.2008.189
Boettler, T., Cheng, Y., Ehrhardt, K., and Von Herrath, M. (2012). TGF-β blockade does not improve control of an established persistent viral infection. Viral. Immunol. 25, 232–238. doi: 10.1089/vim.2011.0079
Boll, R. A., Mirzadeh, S., and Kennel, S. J. (1997). Optimizations of radiolabeling of immunoproteins with 213Bi. Radiochim. Acta 79, 145–150. doi: 10.1524/ract.1997.79.2.145
Brekke, O. H., and Sandlie, I. (2003). Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2, 52–62. doi: 10.1038/nrd984
Brocker, T., and Karjalainen, K. (1995). Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 181, 1653–1659. doi: 10.1084/jem.181.5.1653
Brooks, D. G., Trifilo, M. J., Edelmann, K. H., Teyton, L., Mcgavern, D. B., and Oldstone, M. B. (2006). Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12, 1301–1309. doi: 10.1038/nm1492
Brozy, J., Schlaepfer, E., Mueller, C. K., Rochat, M.-A., Rampini, S. K., Myburgh, R., et al. (2018). Antiviral Activity of HIV gp120 Targeting Bispecific T Cell Engager (BiTE®) Antibody Constructs. J. Virol. 92:JVI.00491-18.
Burns, T., Zhong, Z., Steinitz, M., and Pirofski, L.-A. (2003). Modulation of polymorphonuclear cell interleukin-8 secretion by human monoclonal antibodies to type 8 pneumococcal capsular polysaccharide. Infect. Immun. 71, 6775–6783. doi: 10.1128/IAI.71.12.6775-6783.2003
Burton, D. R. (2002). Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2, 706–713. doi: 10.1038/nri891
Cai, Y., and Berger, E. A. (2011). An immunotoxin targeting the gH glycoprotein of KSHV for selective killing of cells in the lytic phase of infection. Antiviral. Res. 90, 143–150. doi: 10.1016/j.antiviral.2011.03.175
Cao, Y., Qin, L., Zhang, L., Safrit, J., and Ho, D. D. (1995). Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. New Engl. J. Med. 332, 201–208. doi: 10.1056/NEJM199501263320401
Casadevall, A., Spitzer, E., Webb, D., and Rinaldi, M. (1993). Susceptibilities of serial Cryptococcus neoformans isolates from patients with recurrent cryptococcal meningitis to amphotericin B and fluconazole. Antimicrob. Agents Chemother. 37, 1383–1386. doi: 10.1128/AAC.37.6.1383
Cecchinato, V., Tryniszewska, E., Ma, Z. M., Vaccari, M., Boasso, A., Tsai, W.-P., et al. (2008). Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 180, 5439–5447. doi: 10.4049/jimmunol.180.8.5439
Chatterjee, D., Chandran, B., and Berger, E. A. (2012). Selective killing of Kaposi’s sarcoma-associated herpesvirus lytically infected cells with a recombinant immunotoxin targeting the viral gpK8. 1A envelope glycoprotein. MAbs 4, 233–242. doi: 10.4161/mabs.4.2.19262
Cheever, M. A., and Higano, C. S. (2011). PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 17, 3520–3526. doi: 10.1158/1078-0432.CCR-10-3126
Chen, G., Huang, A. C., Zhang, W., Zhang, G., Wu, M., Xu, W., et al. (2018). Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560, 382–386. doi: 10.1038/s41586-018-0392-8
Chew, G. M., Fujita, T., Webb, G. M., Burwitz, B. J., Wu, H. L., Reed, J. S., et al. (2016). TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog. 12:e1005349. doi: 10.1371/journal.ppat.1005349
Christiaansen, A. F., Boggiatto, P. M., and Varga, S. M. (2014). Limitations of Foxp3+ Treg depletion following viral infection in DEREG mice. J. Immunol. Methods 406, 58–65. doi: 10.1016/j.jim.2014.03.005
Conti, S., Magliani, W., Arseni, S., Dieci, E., Frazzi, R., Salati, A., et al. (2000). In vitro activity of monoclonal and recombinant yeast killer toxin-like antibodies against antibiotic-resistant gram-positive cocci. Mol. Med. 6, 613–619. doi: 10.1007/BF03401799
Cortez-Retamozo, V., Lauwereys, M., Hassanzadeh Gh, G., Gobert, M., Conrath, K., Muyldermans, S., et al. (2002). Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 98, 456–462. doi: 10.1002/ijc.10212
Crawford, A., Angelosanto, J. M., Kao, C., Doering, T. A., Odorizzi, P. M., Barnett, B. E., et al. (2014). Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity 40, 289–302. doi: 10.1016/j.immuni.2014.01.005
Crawford, A., and Wherry, E. J. (2009). The diversity of costimulatory and inhibitory receptor pathways and the regulation of antiviral T cell responses. Curr. Opin. Immunol. 21, 179–186. doi: 10.1016/j.coi.2009.01.010
Currie, A. J., Prosser, A., Mcdonnell, A., Cleaver, A. L., Robinson, B. W., Freeman, G. J., et al. (2009). Dual control of antitumor CD8 T cells through the programmed death-1/programmed death-ligand 1 pathway and immunosuppressive CD4 T cells: regulation and counterregulation. J. Immunol. 183, 7898–7908. doi: 10.4049/jimmunol.0901060
Currie, B. P., and Casadevall, A. (1994). Estimation of the prevalence of cryptococcal infection among patients infected with the human immunodeficiency virus in New York City. Clin. Infect. Dis. 19, 1029–1033. doi: 10.1093/clinids/19.6.1029
Cutler, A., and Brombacher, F. (2005). Cytokine therapy. Ann. N. Y. Acad. Sci. 1056, 16–29. doi: 10.1196/annals.1352.002
Dadachova, E., Burns, T., Bryan, R., Apostolidis, C., Brechbiel, M., Nosanchuk, J., et al. (2004). Feasibility of radioimmunotherapy of experimental pneumococcal infection. Antimicrob. Agents Chemother. 48, 1624–1629. doi: 10.1128/AAC.48.5.1624-1629.2004
Dadachova, E., and Casadevall, A. (2014). Radiolabeled antibodies for therapy of infectious diseases. Microbiol. Spectr. 2:0023. doi: 10.1128/microbiolspec.AID-0023-2014
Dadachova, E., Nakouzi, A., Bryan, R. A., and Casadevall, A. (2003). Ionizing radiation delivered by specific antibody is therapeutic against a fungal infection. Proc. Natl. Acad. Sci. U.S.A. 100, 10942–10947. doi: 10.1073/pnas.1731272100
Dadachova, E., Patel, M. C., Toussi, S., Apostolidis, C., Morgenstern, A., Brechbiel, M. W., et al. (2006). Targeted killing of virally infected cells by radiolabeled antibodies to viral proteins. PLoS Med. 3:e427. doi: 10.1371/journal.pmed.0030427
Damelin, M., Zhong, W., Myers, J., and Sapra, P. (2015). Evolving strategies for target selection for antibody-drug conjugates. Pharm. Res. 32, 3494–3507. doi: 10.1007/s11095-015-1624-3
Dangeti, S. R. (2014). Distinct advancements and challenges in HIV 1 vaccine development and cure—A review. HIV AIDS Rev. 13, 1–5. doi: 10.1016/j.hivar.2013.10.001
Datta, M., Via, L. E., Kamoun, W. S., Liu, C., Chen, W., Seano, G., et al. (2015). Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc. Natl. Acad. Sci. U.S.A. 112, 1827–1832. doi: 10.1073/pnas.1424563112
De Bernardis, F., Amacker, M., Arancia, S., Sandini, S., Gremion, C., Zurbriggen, R., et al. (2012). A virosomal vaccine against candidal vaginitis: immunogenicity, efficacy and safety profile in animal models. Vaccine 30, 4490–4498. doi: 10.1016/j.vaccine.2012.04.069
Dietze, K. K., Zelinskyy, G., Gibbert, K., Schimmer, S., Francois, S., Myers, L., et al. (2011). Transient depletion of regulatory T cells in transgenic mice reactivates virus-specific CD8+ T cells and reduces chronic retroviral set points. Proc. Natl. Acad. Sci. U.S.A. 108, 2420–2425. doi: 10.1073/pnas.1015148108
Divanovic, S., Trompette, A., Ashworth, J. I., Rao, M. B., and Karp, C. L. (2011). Therapeutic enhancement of protective immunity during experimental leishmaniasis. PLoS Negl. Trop. Dis. 5:e1316. doi: 10.1371/journal.pntd.0001316
Drabsch, Y., and Ten Dijke, P. (2012). TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 31, 553–568. doi: 10.1007/s10555-012-9375-7
Drutskaya, M. S., Efimov, G. A., Astrakhantseva, I. V., Kruglov, A. A., and Nedospasov, S. A. (2018). Making anti-cytokine therapy more selective: studies in mice. Cytokine 101, 33–38. doi: 10.1016/j.cyto.2016.08.022
Duerr, A., Huang, Y., Buchbinder, S., Coombs, R. W., Sanchez, J., Del Rio, C., et al. (2012). Extended follow-up confirms early vaccine-enhanced risk of HIV acquisition and demonstrates waning effect over time among participants in a randomized trial of recombinant adenovirus HIV vaccine (Step Study). J. Infect. Dis. 206, 258–266. doi: 10.1093/infdis/jis342
Duraiswamy, J., Kaluza, K. M., Freeman, G. J., and Coukos, G. (2013). Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T cell rejection function in tumors. Cancer Res. 73, 3591–3603. doi: 10.1158/0008-5472.CAN-12-4100
Dutcher, J. (2002). Current status of interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma. Oncology 16, 4–10.
Dyck, L., and Mills, K. H. (2017). Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 47, 765–779. doi: 10.1002/eji.201646875
Efimov, G. A., Kruglov, A. A., Khlopchatnikova, Z. V., Rozov, F. N., Mokhonov, V. V., Rose-John, S., et al. (2016). Cell-type–restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. U.S.A. 113, 3006–3011. doi: 10.1073/pnas.1520175113
Ejrnaes, M., Filippi, C. M., Martinic, M. M., Ling, E. M., Togher, L. M., Crotty, S., et al. (2006). Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 203, 2461–2472. doi: 10.1084/jem.20061462
Esparza, J., and Van Regenmortel, M. H. (2014). More surprises in the development of an HIV vaccine. Front. Immunol. 5:329. doi: 10.3389/fimmu.2014.00329
Falini, B., Pileri, S., Pizzolo, G., Durkop, H., Flenghi, L., Stirpe, F., et al. (1995). CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 85, 1–14.
Fan, G., Wang, Z., Hao, M., and Li, J. (2015). Bispecific antibodies and their applications. J. Hematol. Oncol. 8:130. doi: 10.1186/s13045-015-0227-0
Feldmann, M. (2008). Many cytokines are very useful therapeutic targets in disease. J. Clin. Invest. 118, 3533–3536. doi: 10.1172/JCI37346
Francisco, L. M., Salinas, V. H., Brown, K. E., Vanguri, V. K., Freeman, G. J., Kuchroo, V. K., et al. (2009). PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029. doi: 10.1084/jem.20090847
Frank, D. W., Vallis, A., Wiener-Kronish, J. P., Roy-Burman, A., Spack, E. G., Mullaney, B. P., et al. (2002). Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J. Infect. Dis. 186, 64–73. doi: 10.1086/341069
Fu, J., Kanne, D. B., Leong, M., Glickman, L. H., Mcwhirter, S. M., Lemmens, E., et al. (2015). STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 7:283ra252. doi: 10.1126/scitranslmed.aaa4306
Fuller, M. J., Callendret, B., Zhu, B., Freeman, G. J., Hasselschwert, D. L., Satterfield, W., et al. (2013). Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. U.S.A. 110, 15001–15006. doi: 10.1073/pnas.1312772110
Gardiner, D., Lalezari, J., Lawitz, E., Dimicco, M., Ghalib, R., Reddy, K. R., et al. (2013). A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One 8:e63818. doi: 10.1371/journal.pone.0063818
Gardner, T., Elzey, B., and Hahn, N. M. (2012). Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum. Vaccin. Immunother. 8, 534–539. doi: 10.4161/hv.19795
Garidou, L., Heydari, S., Gossa, S., and Mcgavern, D. B. (2012). Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J. Virol. 86, 7060–7071. doi: 10.1128/JVI.00164-12
Gattinoni, L., Powell, D. J. Jr., Rosenberg, S. A., and Restifo, N. P. (2006). Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 6, 383–393. doi: 10.1038/nri1842
Gauthier, J., and Turtle, C. J. (2018). Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr. Res. Transl. Med. 66, 50–52. doi: 10.1016/j.retram.2018.03.003
Gay, C. L., Bosch, R. J., Ritz, J., Hataye, J. M., Aga, E., Tressler, R. L., et al. (2017). Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J. Infect. Dis. 215, 1725–1733. doi: 10.1093/infdis/jix191
Geoghegan, E. M., Zhang, H., Desai, P. J., Biragyn, A., and Markham, R. B. (2015). Antiviral activity of a single-domain antibody immunotoxin binding to glycoprotein D of herpes simplex virus 2. Antimicrob. Agents Chemother. 59, 527–535. doi: 10.1128/AAC.03818-14
Gill, A. L., Green, S. A., Abdullah, S., Le Saout, C., Pittaluga, S., Chen, H., et al. (2016). Programed death-1/programed death-ligand 1 expression in lymph nodes of HIV infected patients: results of a pilot safety study in rhesus macaques using anti–programed death-ligand 1 (Avelumab). AIDS 30, 2487–2493. doi: 10.1097/QAD.0000000000001217
Gleason, M. K., Ross, J. A., Warlick, E. D., Lund, T. C., Verneris, M. R., Wiernik, A., et al. (2014). CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 123, 3016–3026. doi: 10.1182/blood-2013-10-533398
Goldstein, H., Pettoello-Mantovani, M., Bera, T. K., Pastan, I. H., and Berger, E. A. (2000). Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-SCID-Hu mice. J. Infect. Dis. 181, 921–926. doi: 10.1086/315351
Golubovskaya, V., and Wu, L. (2016). Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers 8:E36. doi: 10.3390/cancers8030036
Gonçalves-Lopes, R. M., Lima, N. F., Carvalho, K. I., Scopel, K. K., Kallás, E. G., and Ferreira, M. U. (2016). Surface expression of inhibitory (CTLA-4) and stimulatory (OX40) receptors by CD4+ regulatory T cell subsets circulating in human malaria. Microbes Infect. 18, 639–648. doi: 10.1016/j.micinf.2016.06.003
Grabmeier-Pfistershammer, K., Stecher, C., Zettl, M., Rosskopf, S., Rieger, A., Zlabinger, G. J., et al. (2017). Antibodies targeting BTLA or TIM-3 enhance HIV-1 specific T cell responses in combination with PD-1 blockade. Clin. Immunol. 183, 167–173. doi: 10.1016/j.clim.2017.09.002
Graham, B. S., and Ambrosino, D. M. (2015). History of passive antibody administration for prevention and treatment of infectious diseases. Curr. Opin. HIV AIDS 10, 129–134. doi: 10.1097/COH.0000000000000154
Grivennikov, S. I., Tumanov, A. V., Liepinsh, D. J., Kruglov, A. A., Marakusha, B. I., Shakhov, A. N., et al. (2005). Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity 22, 93–104. doi: 10.1016/j.immuni.2004.11.016
Groothuis, J. R., and Nishida, H. (2002). Prevention of respiratory syncytial virus infections in high-risk infantsby monoclonal antibody (palivizumab). Pediatr. Int. 44, 235–241. doi: 10.1046/j.1442-200X.2002.01558.x
Guidotti, L. G., and Chisari, F. V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. Mech. Dis. 1, 23–61. doi: 10.1146/annurev.pathol.1.110304.100230
Gupta, S., Cheung, L., Pokkali, S., Winglee, K., Guo, H., Murphy, J. R., et al. (2017). Suppressor cell–depleting immunotherapy with denileukin diftitox is an effective host-directed therapy for Tuberculosis. J. Infect. Dis. 215, 1883–1887. doi: 10.1093/infdis/jix208
Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hammers, C., Songa, E. B., et al. (1993). Naturally occurring antibodies devoid of light chains. Nature 363, 446–448. doi: 10.1038/363446a0
Haug, M., Brede, G., Håkerud, M., Nedberg, A. G., Gederaas, O. A., Flo, T. H., et al. (2018). Photochemical internalization of Peptide antigens Provides a novel strategy to realize Therapeutic cancer Vaccination. Front. Immunol. 9:650. doi: 10.3389/fimmu.2018.00650
Haynes, B. F. (2015). New approaches to HIV vaccine development. Curr. Opin. Immunol. 35, 39–47. doi: 10.1016/j.coi.2015.05.007
Health, U. N. I. O. (2012). ClinicalTrials.gov is a Database of Privately and Publicly Funded Clinical Studies Conducted Around the World. Available at: https://clinicaltrials.gov/
Herrera, A. F., Moskowitz, A. J., Bartlett, N. L., Vose, J. M., Ramchandren, R., Feldman, T. A., et al. (2018). Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 131, 1183–1194. doi: 10.1182/blood-2017-10-811224
Hill, J. A., Li, D., Hay, K. A., Green, M. L., Cherian, S., Chen, X., et al. (2018). Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 131, 121–130. doi: 10.1182/blood-2017-07-793760
Hirano, F., Kaneko, K., Tamura, H., Dong, H., Wang, S., Ichikawa, M., et al. (2005). Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096.
Ho, W. Y., Blattman, J. N., Dossett, M. L., Yee, C., and Greenberg, P. D. (2003). Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell 3, 431–437. doi: 10.1016/S1535-6108(03)00113-2
Hodi, F. S., Chesney, J., Pavlick, A. C., Robert, C., Grossmann, K. F., Mcdermott, D. F., et al. (2016). Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 17, 1558–1568. doi: 10.1016/S1470-2045(16)30366-7
Hogan, L. E., Vasquez, J., Hobbs, K. S., Hanhauser, E., Aguilar-Rodriguez, B., Hussien, R., et al. (2018). Increased HIV-1 transcriptional activity and infectious burden in peripheral blood and gut-associated CD4+ T cells expressing CD30. PLoS Pathog. 14:e1006856. doi: 10.1371/journal.ppat.1006856
Horn, M. P., Zuercher, A. W., Imboden, M. A., Rudolf, M. P., Lazar, H., Wu, H., et al. (2010). Preclinical in vitro and in vivo characterization of the fully human monoclonal IgM antibody KBPA101 specific for Pseudomonas aeruginosa serotype IATS-O11. Antimicrob. Agents Chemother. 2338–2344. doi: 10.1128/AAC.01142-09
Hristodorov, D., Mladenov, R., Pardo, A., Pham, A., Huhn, M., Fischer, R., et al. (2013). Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br. J. Cancer 109, 1570. doi: 10.1038/bjc.2013.457
Hsu, F.-S., Su, C.-H., and Huang, K.-H. (2017). A comprehensive review of US FDA-approved immune checkpoint inhibitors in urothelial carcinoma. J. Immunol. Res. 2017:6940546. doi: 10.1155/2017/6940546
Hu, J., and Robinson, J. L. (2010). Treatment of respiratory syncytial virus with palivizumab: a systematic review. World J. Pediatrics 6, 296–300. doi: 10.1007/s12519-010-0230-z
Hua, L., Hilliard, J., Shi, Y., Tkaczyk, C., Cheng, L., Yu, X., et al. (2014). Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob. Agents Chemother. 58, 1108–1117. doi: 10.1128/AAC.02190-13
Illidge, T., and Johnson, P. (2000). The emerging role of radioimmunotherapy in haematological malignancies. Br. J. Haematol. 108, 679–688. doi: 10.1046/j.1365-2141.2000.01926.x
Illingworth, J., Butler, N. S., Roetynck, S., Mwacharo, J., Pierce, S. K., Bejon, P., et al. (2013). Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J. Immunol. 190, 1038–1047. doi: 10.4049/jimmunol.1202438
Isaacs, C., Robert, N. J., Bailey, F. A., Schuster, M. W., Overmoyer, B., Graham, M., et al. (1997). Randomized placebo-controlled study of recombinant human interleukin-11 to prevent chemotherapy-induced thrombocytopenia in patients with breast cancer receiving dose-intensive cyclophosphamide and doxorubicin. J. Clin. Oncol. 15, 3368–3377. doi: 10.1200/JCO.1997.15.11.3368
Jakowlew, S. B. (2006). Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 25, 435–457. doi: 10.1007/s10555-006-9006-2
Jones, P. T., Dear, P. H., Foote, J., Neuberger, M. S., and Winter, G. (1986). Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321, 522–525. doi: 10.1038/321522a0
Jordaan, S., Akinrinmade, O. A., Nachreiner, T., Cremer, C., Naran, K., Chetty, S., et al. (2018). Updates in the development of immunornases for the selective killing of tumor cells. Biomedicines 6:28. doi: 10.3390/biomedicines6010028
Kapelski, S., De Almeida, M., Fischer, R., Barth, S., and Fendel, R. (2015). Antimalarial Activity of Granzyme B and Its Targeted Delivery by a Granzyme B–Single-Chain Fv Fusion Protein. Antimicrob. Agents Chemother. 59, 669–672. doi: 10.1128/AAC.04190-14
Karyampudi, L., Lamichhane, P., Scheid, A. D., Kalli, K. R., Shreeder, B., Krempski, J. W., et al. (2014). Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 74, 2974–2984. doi: 10.1158/0008-5472.CAN-13-2564
Kaufmann, D. E., Kavanagh, D. G., Pereyra, F., Zaunders, J. J., Mackey, E. W., Miura, T., et al. (2007). Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 8, 1246–1254. doi: 10.1038/ni1515
Kennedy, P. E., Bera, T. K., Wang, Q. C., Gallo, M., Wagner, W., Lewis, M. G., et al. (2006). Anti-HIV-1 immunotoxin 3B3 (Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J. Leukoc. Biol. 80, 1175–1182. doi: 10.1189/jlb.0306139
Khan, N., Vidyarthi, A., Amir, M., Mushtaq, K., and Agrewala, J. N. (2017). T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit. Rev. Microbiol 43, 133–141. doi: 10.1080/1040841X.2016.1185603
Kim, E. G., and Kim, K. M. (2015). Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol. Ther. 23, 493–509. doi: 10.4062/biomolther.2015.116
Kim, J. M., Rasmussen, J. P., and Rudensky, A. Y. (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197. doi: 10.1038/ni1428
Kim, P. S., and Ahmed, R. (2010). Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol 22, 223–230. doi: 10.1016/j.coi.2010.02.005
Kiran, D., Podell, B. K., Chambers, M., and Basaraba, R. J. (2016). Host-directed therapy targeting the Mycobacterium tuberculosis granuloma: a review. Semin. Immunopathol. 38, 167–183. doi: 10.1007/s00281-015-0537-x
Kiyokawa, T., Williams, D. P., Snider, C. E., Strom, T. B., and Murphy, J. R. (1991). Protein engineering of diphtheria-toxin-related interleukin-2 fusion toxins to increase cytotoxic potency for high-affinity IL-2-receptor-bearing target cells. Protein Eng. Design Select. 4, 463–468. doi: 10.1093/protein/4.4.463
Kleponis, J., Skelton, R., and Zheng, L. (2015). Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol. Med. 12, 201–208. doi: 10.7497/j.issn.2095-3941.2015.0046
Knaul, J. K., Jörg, S., Oberbeck-Mueller, D., Heinemann, E., Scheuermann, L., Brinkmann, V., et al. (2014). Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 190, 1053–1066. doi: 10.1164/rccm.201405-0828OC
Kochenderfer, J. N., and Rosenberg, S. A. (2013). Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 10, 267–276. doi: 10.1038/nrclinonc.2013.46
Köhnke, T., Krupka, C., Tischer, J., Knösel, T., and Subklewe, M. (2015). Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 8:111. doi: 10.1186/s13045-015-0213-6
Kovtun, Y. V., Audette, C. A., Ye, Y., Xie, H., Ruberti, M. F., Phinney, S. J., et al. (2006). Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 66, 3214–3221. doi: 10.1158/0008-5472.CAN-05-3973
Kruglov, A. A., Lampropoulou, V., Fillatreau, S., and Nedospasov, S. A. (2011). Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J. Immunol. 187, 5660–5670. doi: 10.4049/jimmunol.1100663
Kumaresan, P. R., Manuri, P. R., Albert, N. D., Maiti, S., Singh, H., Mi, T., et al. (2014). Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. U.S.A. 111, 10660–10665. doi: 10.1073/pnas.1312789111
Kursar, M., Koch, M., Mittrücker, H.-W., Nouailles, G., Bonhagen, K., Kamradt, T., et al. (2007). Cutting edge: regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J. Immunol. 178, 2661–2665. doi: 10.4049/jimmunol.178.5.2661
Kyi, C., and Postow, M. A. (2014). Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett. 588, 368–376. doi: 10.1016/j.febslet.2013.10.015
Lahl, K., Loddenkemper, C., Drouin, C., Freyer, J., Arnason, J., Eberl, G., et al. (2007). Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204, 57–63. doi: 10.1084/jem.20061852
Lai, X., and Friedman, A. (2017). Combination therapy of cancer with cancer vaccine and immune checkpoint inhibitors: a mathematical model. PLoS One 12:e0178479. doi: 10.1371/journal.pone.0178479
Larocca, T. J., Katona, L. I., Thanassi, D. G., and Benach, J. L. (2008). Bactericidal action of a complement-independent antibody against relapsing fever Borrelia resides in its variable region. J. Immunol. 180, 6222–6228. doi: 10.4049/jimmunol.180.9.6222
Larson, H. J., Clarke, R. M., Jarrett, C., Eckersberger, E., Levine, Z., Schulz, W. S., et al. (2018). Measuring trust in vaccination: a systematic review. Hum. Vaccin. Immunother. 14, 1599–1609. doi: 10.1080/21645515.2018.1459252
Latchman, Y., Wood, C. R., Chernova, T., Chaudhary, D., Borde, M., Chernova, I., et al. (2001). PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261. doi: 10.1038/85330
Lee, P. P., Yee, C., Savage, P. A., Fong, L., Brockstedt, D., Weber, J. S., et al. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685. doi: 10.1038/9525
Lehar, S. M., Pillow, T., Xu, M., Staben, L., Kajihara, K. K., Vandlen, R., et al. (2015). Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 527, 323–328. doi: 10.1038/nature16057
Li, J., Li, W., Huang, K., Zhang, Y., Kupfer, G., and Zhao, Q. (2018). Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J. Hematol. Oncol. 11:22. doi: 10.1186/s13045-018-0568-6
Li, N., Xie, W., Kong, H., Min, R., Hu, C., Zhou, X., et al. (2015). Enrichment of regulatory T-cells in blood of patients with multidrug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 19, 1230–1238. doi: 10.5588/ijtld.15.0148
Li, S., Gowans, E. J., Chougnet, C., Plebanski, M., and Dittmer, U. (2008). Natural regulatory T cells and persistent viral infection. J. Virol. 82, 21–30. doi: 10.1128/JVI.01768-07
Li, X., Xu, P., Wang, C., Xu, N., Xu, A., Xu, Y., et al. (2017). Synergistic effects of the immune checkpoint inhibitor CTLA-4 combined with the growth inhibitor lycorine in a mouse model of renal cell carcinoma. Oncotarget 8, 21177–21186. doi: 10.18632/oncotarget.15505
Liedtke, C., Mazouni, C., Hess, K. R., André, F., Tordai, A., Mejia, J. A., et al. (2008). Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–1281. doi: 10.1200/JCO.2007.14.4147
Lilienthal, N., Lohmann, G., Crispatzu, G., Vasyutina, E., Zittrich, S., Mayer, P., et al. (2016). A novel recombinant anti-CD22 immunokinase delivers proapoptotic activity of death-associated protein kinase (DAPK) and mediates cytotoxicity in neoplastic B cells. Mol. Cancer Ther. 15, 971–984. doi: 10.1158/1535-7163.MCT-15-0685
Lin, J. H., Guo, Y., and Wang, W. (2018). Challenges of antibody drug conjugates in cancer therapy: current understanding of mechanisms and future strategies. Curr. Pharmacol. Rep. 4, 10–26. doi: 10.1007/s40495-018-0122-9
Linke, R., Klein, A., and Seimetz, D. (2010). Catumaxomab: clinical development and future directions. MAbs 2, 129–136. doi: 10.4161/mabs.2.2.11221
Litzinger, M. T., Fernando, R., Curiel, T. J., Grosenbach, D. W., Schlom, J., and Palena, C. (2007). IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood 110, 3192–3201. doi: 10.1182/blood-2007-06-094615
Liu, D., Tian, S., Zhang, K., Xiong, W., Lubaki, N. M., Chen, Z., et al. (2017). Chimeric antigen receptor (CAR)-modified natural killer cell-based immunotherapy and immunological synapse formation in cancer and HIV. Protein Cell 8, 861–877. doi: 10.1007/s13238-017-0415-5
Mahoney, K. M., Rennert, P. D., and Freeman, G. J. (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14, 561–584. doi: 10.1038/nrd4591
Manieri, N. A., Chiang, E. Y., and Grogan, J. L. (2017). TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. 38, 20–28. doi: 10.1016/j.it.2016.10.002
Mao, H., Zhang, L., Yang, Y., Zuo, W., Bi, Y., Gao, W., et al. (2010). New insights of CTLA-4 into its biological function in breast cancer. Curr. Cancer Drug. Targets 10, 728–736. doi: 10.2174/156800910793605811
Marcellin, P., Lau, G. K., Bonino, F., Farci, P., Hadziyannis, S., Jin, R., et al. (2004). Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 351, 1206–1217. doi: 10.1056/NEJMoa040431
Mariathasan, S., and Tan, M.-W. (2017). Antibody–antibiotic conjugates: a novel therapeutic platform against bacterial infections. Trends Mol. Med. 23, 135–149. doi: 10.1016/j.molmed.2016.12.008
Massard, C., Gordon, M. S., Sharma, S., Rafii, S., Wainberg, Z. A., Luke, J., et al. (2016). Safety and efficacy of durvalumab (MEDI4736), an anti–programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J. Clin. Oncol. 34, 3119–3125. doi: 10.1200/JCO.2016.67.9761
Mathew, M., and Verma, R. S. (2009). Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 100, 1359–1365. doi: 10.1111/j.1349-7006.2009.01192.x
Maude, S. L., Frey, N., Shaw, P. A., Aplenc, R., Barrett, D. M., Bunin, N. J., et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517. doi: 10.1056/NEJMoa1407222
Mcdevitt, M. R., Barendswaard, E., Ma, D., Lai, L., Curcio, M. J., Sgouros, G., et al. (2000). An α-particle emitting antibody ([213Bi] J591) for radioimmunotherapy of prostate cancer. Cancer Res. 60, 6095–6100.
Melian, E. B., and Plosker, G. L. (2001). Interferon alfacon-1. Drugs 61, 1661–1691. doi: 10.2165/00003495-200161110-00009
Meng, W., Tang, A., Ye, X., Gui, X., Li, L., Fan, X., et al. (2018). Targeting human-cytomegalovirus-infected cells by redirecting T cells using an anti-CD3/anti-glycoprotein B bispecific antibody. Antimicrob. Agents Chemother. 62:e01719-17. doi: 10.1128/AAC.01719-17
Merlo, A., Saverino, D., Tenca, C., Grossi, C. E., Bruno, S., and Ciccone, E. (2001). CD85/LIR-1/ILT2 and CD152 (Cytotoxic T Lymphocyte Antigen 4) inhibitory molecules down-regulate the cytolytic activity of human CD4+ T-Cell Clones Specific for Mycobacterium tuberculosis. Infect. Immun. 69, 6022–6029. doi: 10.1128/IAI.69.10.6022-6029.2001
Michot, J., Bigenwald, C., Champiat, S., Collins, M., Carbonnel, F., Postel-Vinay, S., et al. (2016). Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54, 139–148. doi: 10.1016/j.ejca.2015.11.016
Mohindra, N. (2018). Current state of immunotherapy: chipping away at the tip of the iceberg. J. Cancer Immunol. Ther. 1, 1–2.
Moore, G. L., Bautista, C., Pong, E., Nguyen, D.-H. T., Jacinto, J., Eivazi, A., et al. (2011). A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 3, 546–557. doi: 10.4161/mabs.3.6.18123
Morrison, S. L., Johnson, M. J., Herzenberg, L. A., and Oi, V. T. (1984). Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. U.S.A. 81, 6851–6855. doi: 10.1073/pnas.81.21.6851
Morrissey, K. M., Yuraszeck, T., Li, C. C., Zhang, Y., and Kasichayanula, S. (2016). Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin. Transl. Sci. 9, 89–104. doi: 10.1111/cts.12391
Murdock, B. J., Teitz-Tennenbaum, S., Chen, G.-H., Dils, A. J., Malachowski, A. N., Curtis, J. L., et al. (2014). Early or late IL-10 blockade enhances Th1 and Th17 effector responses and promotes fungal clearance in mice with cryptococcal lung infection. J. Immunol. 193, 4107–4116. doi: 10.4049/jimmunol.1400650
Murray, H. W., Lu, C. M., Mauze, S., Freeman, S., Moreira, A. L., Kaplan, G., et al. (2002). Interleukin-10 (IL-10) in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect. Immun. 70, 6284–6293. doi: 10.1128/IAI.70.11.6284-6293.2002
Nabel, G. J. (2013). Designing tomorrow’s vaccines. N. Engl. J. Med. 368, 551–560. doi: 10.1056/NEJMra1204186
Nanjappa, S. G., and Klein, B. S. (2014). Vaccine immunity against fungal infections. Curr. Opin. Immunol 28, 27–33. doi: 10.1016/j.coi.2014.01.014
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544. doi: 10.1056/NEJMoa1707447
Neuzillet, C., Tijeras-Raballand, A., Cohen, R., Cros, J., Faivre, S., Raymond, E., et al. (2015). Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 147, 22–31. doi: 10.1016/j.pharmthera.2014.11.001
Nguyen, L. T., and Ohashi, P. S. (2015). Clinical blockade of PD1 and LAG3—potential mechanisms of action. Nat. Rev. Immunol. 15, 45–56. doi: 10.1038/nri3790
Ni, G., Wang, T., Walton, S., Zhu, B., Chen, S., Wu, X., et al. (2015). Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. 293, 126–129. doi: 10.1016/j.cellimm.2014.12.012
Offner, S., Hofmeister, R., Romaniuk, A., Kufer, P., and Baeuerle, P. A. (2006). Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 43, 763–771. doi: 10.1016/j.molimm.2005.03.007
Oganesyan, V., Peng, L., Damschroder, M. M., Cheng, L., Sadowska, A., Tkaczyk, C., et al. (2014). Mechanisms of neutralization of a human anti-α-toxin antibody. J. Biol. Chem. 289, 29874–29880. doi: 10.1074/jbc.M114.601328
World Health Organization (2017). Measles vaccines: WHO position paper, April 2017–Recommendations. Vaccine doi: 10.1016/j.vaccine.2017.07.066 [Epub ahead of print].
World Health Organization (2018). Diphtheria vaccine: WHO position paper, August 2017–Recommendations. Vaccine 36, 199–201. doi: 10.1016/j.vaccine.2017.08.024
Paley, M. A., Kroy, D. C., Odorizzi, P. M., Johnnidis, J. B., Dolfi, D. V., Barnett, B. E., et al. (2012). Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225. doi: 10.1126/science.1229620
Papaioannou, N. E., Beniata, O. V., Vitsos, P., Tsitsilonis, O., and Samara, P. (2016). Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 36, 199–201. doi: 10.21037/atm.2016.04.01
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. doi: 10.1038/nrc3239
Parren, P. W., and Burton, D. R. (2001). The antiviral activity of antibodies in vitro and in vivo. Adv. Immunol. 77, 195–262. doi: 10.1016/S0065-2776(01)77018-6
Pauken, K. E., and Wherry, E. J. (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276. doi: 10.1016/j.it.2015.02.008
Perreau, M., Banga, R., and Pantaleo, G. (2017). Targeted immune interventions for an HIV-1 Cure. Trends Mol. Med. 23, 945–961. doi: 10.1016/j.molmed.2017.08.006
Pitt, J. M., Stavropoulos, E., Redford, P. S., Beebe, A. M., Bancroft, G. J., Young, D. B., et al. (2012). Blockade of IL-10 signaling during BCG vaccination enhances and sustains Th1, Th17, and innate lymphoid IFN-γ and IL-17 responses and increases protection to Mycobacterium tuberculosis infection. J. Immunol. 189, 4079–4087. doi: 10.4049/jimmunol.1201061
Postow, M. A., Callahan, M. K., and Wolchok, J. D. (2015). Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 33, 1974–1982. doi: 10.1200/JCO.2014.59.4358
Pouget, J.-P., Navarro-Teulon, I., Bardiès, M., Chouin, N., Cartron, G., Pèlegrin, A., et al. (2011). Clinical radioimmunotherapy—the role of radiobiology. Nat. Rev. Clin. Oncol. 8, 720–734. doi: 10.1038/nrclinonc.2011.160
Przepiorka, D., Ko, C.-W., Deisseroth, A., Yancey, C. L., Candau-Chacon, R., Chiu, H.-J., et al. (2015). FDA approval: blinatumomab. Clin. Cancer Res. 21, 4035–4039. doi: 10.1158/1078-0432.CCR-15-0612
Que, Y.-A., Lazar, H., Wolff, M., François, B., Laterre, P.-F., Mercier, E., et al. (2014). Assessment of panobacumab as adjunctive immunotherapy for the treatment of nosocomial Pseudomonas aeruginosa pneumonia. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1861–1867. doi: 10.1007/s10096-014-2156-1
Qureshi, O. S., Zheng, Y., Nakamura, K., Attridge, K., Manzotti, C., Schmidt, E. M., et al. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. doi: 10.1126/science.1202947
∗Qyresearch, G., and Global Immune Checkpoint Inhibitors Market Size (2017). Revenue and Forecast 2022 [Online]. Available: Global Immune Checkpoint Inhibitors Market Size 2017. Revenue and Forecast 2022 [Accessed].
Rerks-Ngarm, S., Pitisuttithum, P., Nitayaphan, S., Kaewkungwal, J., Chiu, J., Paris, R., et al. (2009). Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220. doi: 10.1056/NEJMoa0908492
Rezvani, K., and Rouce, R. H. (2015). The application of natural killer cell immunotherapy for the treatment of cancer. Front. Immunol. 6:578. doi: 10.3389/fimmu.2015.00578
Richter, F., Liebig, T., Guenzi, E., Herrmann, A., Scheurich, P., Pfizenmaier, K., et al. (2013). Antagonistic TNF receptor one-specific antibody (ATROSAB): receptor binding and in vitro bioactivity. PLoS One 8:e72156. doi: 10.1371/journal.pone.0072156
Richter, K., Perriard, G., Behrendt, R., Schwendener, R. A., Sexl, V., Dunn, R., et al. (2013). Macrophage and T cell produced IL-10 promotes viral chronicity. PLoS Pathog. 9:e1003735. doi: 10.1371/journal.ppat.1003735
Rittmeyer, A., Barlesi, F., Waterkamp, D., Park, K., Ciardiello, F., Von Pawel, J., et al. (2017). Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265. doi: 10.1016/S0140-6736(16)32517-X
Robert, C., Long, G. V., Brady, B., Dutriaux, C., Maio, M., Mortier, L., et al. (2015a). Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330. doi: 10.1056/NEJMoa1412082
Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., et al. (2015b). Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532. doi: 10.1056/NEJMoa1503093
Roybal, K. T., and Lim, W. A. (2017). Synthetic immunology: hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol. 35, 229–253. doi: 10.1146/annurev-immunol-051116-052302
Roybal, K. T., Williams, J. Z., Morsut, L., Rupp, L. J., Kolinko, I., Choe, J. H., et al. (2016). Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432.e16. doi: 10.1016/j.cell.2016.09.011
Sadekar, S., Figueroa, I., and Tabrizi, M. (2015). Antibody drug conjugates: application of quantitative pharmacology in modality design and target selection. AAPS J. 17, 828–836. doi: 10.1208/s12248-015-9766-0
Sadelain, M., Rivière, I., and Brentjens, R. (2003). Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 3, 35–45. doi: 10.1038/nrc971
Said, E. A., Dupuy, F. P., Trautmann, L., Zhang, Y., Shi, Y., El-Far, M., et al. (2010). Programmed death-1–induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 16, 452–459. doi: 10.1038/nm.2106
Samaha, H., Pignata, A., Fousek, K., Ren, J., Lam, F. W., Stossi, F., et al. (2018). A homing system targets therapeutic T cells to brain cancer. Nature 561, 331–337. doi: 10.1038/s41586-018-0499-y
Sause, W. E., Buckley, P. T., Strohl, W. R., Lynch, A. S., and Torres, V. J. (2016). Antibody-based biologics and their promise to combat Staphylococcus aureus infections. Trends Pharmacol. Sci. 37, 231–241. doi: 10.1016/j.tips.2015.11.008
Savoldo, B., Ramos, C. A., Liu, E., Mims, M. P., Keating, M. J., Carrum, G., et al. (2011). CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J. Clin. Invest. 121, 1822–1826. doi: 10.1172/JCI46110
Schietinger, A., and Greenberg, P. D. (2014). Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 35, 51–60. doi: 10.1016/j.it.2013.10.001
Schmid, P., Adams, S., Rugo, H. S., Schneeweiss, A., Barrios, C. H., Iwata, H., et al. (2018). Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121. doi: 10.1056/NEJMoa1809615
Schmidt, C. S., White, C. J., Ibrahim, A. S., Filler, S. G., Fu, Y., Yeaman, M. R., et al. (2012). NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine 30, 7594–7600. doi: 10.1016/j.vaccine.2012.10.038
Schmohl, J., Gleason, M., Dougherty, P., Miller, J., and Vallera, D. (2016). Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target. Oncol. 11, 353–361. doi: 10.1007/s11523-015-0391-8
Schuster, S. J., Svoboda, J., Chong, E. A., Nasta, S. D., Mato, A. R., Anak, Ö, et al. (2017). Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554. doi: 10.1056/NEJMoa1708566
Scotti, C., Iamele, L., and Vecchia, L. (2015). Antibody–drug conjugates: targeted weapons against cancer. Antibody Technol. J. 5, 1–13. doi: 10.2147/ANTI.S52914
Sedykh, S. E., Prinz, V. V., Buneva, V. N., and Nevinsky, G. A. (2018). Bispecific antibodies: design, therapy, perspectives. Drug Des. Dev. Ther. 12, 195–208. doi: 10.2147/DDDT.S151282
Sgouros, G., Ballangrud, A. M., Jurcic, J. G., and Mcdevitt, M. R. (1999). Phrmacokinetics and dosimetry of an (alpha-particle) emitter labeled antibody: 213Bi-HuM195 (Anti-CD33) in patients with leukemia. J. Nucl. Med. 40, 1935–1946.
Sharma, P., and Allison, J. P. (2015). The future of immune checkpoint therapy. Science 348, 56–61. doi: 10.1126/science.aaa8172
Sharpe, A. H., Wherry, E. J., Ahmed, R., and Freeman, G. J. (2007). The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8, 239–245. doi: 10.1038/ni1443
Shin, D. S., Zaretsky, J. M., Escuin-Ordinas, H., Garcia-Diaz, A., Hu-Lieskovan, S., Kalbasi, A., et al. (2016). Primary resistance to PD-1 blockade mediated by JAK½ mutations. Cancer Discov. 17, 188–201.
Simmonds, R. E., and Foxwell, B. M. (2008). Signalling, inflammation and arthritis: NF-κ B and its relevance to arthritis and inflammation. Rheumatology 47, 584–590. doi: 10.1093/rheumatology/kem298
Sipp, D., Frazer, I. H., and Rasko, J. E. (2018). No Vacillation on HPV Vaccination. Cell 172, 1163–1167. doi: 10.1016/j.cell.2018.02.045
Sloan, D. D., Lam, C.-Y. K., Irrinki, A., Liu, L., Tsai, A., Pace, C. S., et al. (2015). Targeting HIV reservoir in infected CD4 T cells by dual-affinity re-targeting molecules (DARTs) that bind HIV envelope and recruit cytotoxic T cells. PLoS Pathog. 11:e1005233. doi: 10.1371/journal.ppat.1005233
Soares, K. C., Rucki, A. A., Wu, A. A., Olino, K., Xiao, Q., Chai, Y., et al. (2015). PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T cell infiltration into pancreatic tumors. J. Immunother. 38, 1–11. doi: 10.1097/CJI.0000000000000062
Spain, L., Diem, S., and Larkin, J. (2016). Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 44, 51–60. doi: 10.1016/j.ctrv.2016.02.001
Spiess, K., Jakobsen, M. H., Kledal, T. N., and Rosenkilde, M. M. (2016). The future of antiviral immunotoxins. J. Leukoc. Biol. 99, 911–925. doi: 10.1189/jlb.2MR1015-468R
Spiess, K., Jeppesen, M. G., Malmgaard-Clausen, M., Krzywkowski, K., Dulal, K., Cheng, T., et al. (2015). Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc. Natl. Acad. Sci. U.S.A. 112, 8427–8432. doi: 10.1073/pnas.1509392112
Srahna, M., Grunsven, L., Remacle, J., and Vandenberghe, P. (2006). CTLA-4 interacts with STAT5 and inhibits STAT5-mediated transcription. Immunology 117, 396–401. doi: 10.1111/j.1365-2567.2005.02313.x
Srivastava, S., and Riddell, S. R. (2018). Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J. Immunol. 200, 459–468. doi: 10.4049/jimmunol.1701155
Steeland, S., Puimège, L., Vandenbroucke, R. E., Van Hauwermeiren, F., Haustraete, J., Devoogdt, N., et al. (2015). Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 290, 4022–4037. doi: 10.1074/jbc.M114.617787
Stein, H., Mason, D., Gerdes, J., O’connor, N., Wainscoat, J., Pallesen, G., et al. (1985). The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 66, 848–858.
Stephenson, K. E., T D’couto, H., and Barouch, D. H. (2016). New concepts in HIV-1 vaccine development. Curr. Opin. Immunol 41, 39–46. doi: 10.1016/j.coi.2016.05.011
Sung, J. A., Pickeral, J., Liu, L., Stanfield-Oakley, S. A., Lam, C.-Y. K., Garrido, C., et al. (2015). Dual-Affinity Re-Targeting proteins direct T cell–mediated cytolysis of latently HIV-infected cells. J. Clin. Invest. 125, 4077–4090. doi: 10.1172/JCI82314
Syed, T. A., and Ahmadpour, O. A. (1998). Human leukocyte derived interferon-alpha in a hydrophilic gel for the treatment of intravaginal warts in women: a placebo-controlled, double-blind study. Int. J. STD AIDS 9, 769–772. doi: 10.1258/0956462981921396
Thakur, A., Huang, M., and Lum, L. G. (2018). Bispecific antibody based therapeutics: strengths and challenges. Blood Rev. 32, 339–347. doi: 10.1016/j.blre.2018.02.004
Tian, M., Neil, J. R., and Schiemann, W. P. (2011). Transforming growth factor-β and the hallmarks of cancer. Cell. Signal. 23, 951–962. doi: 10.1016/j.cellsig.2010.10.015
Topalian, S. L., Drake, C. G., and Pardoll, D. M. (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. doi: 10.1016/j.ccell.2015.03.001
Trail, P. A., Dubowchik, G. M., and Lowinger, T. B. (2017). Antibody drug conjugates for treatment of breast cancer: novel targets and diverse approaches in ADC design. Pharmacol. Ther. 181, 126–142. doi: 10.1016/j.pharmthera.2017.07.013
Trautmann, L., Janbazian, L., Chomont, N., Said, E. A., Gimmig, S., Bessette, B., et al. (2006). Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12, 1198–1202. doi: 10.1038/nm1482
Tur, M. K., and Barth, S. (2014). “Immunotherapy”, in Encyclopedia of Cancer, ed. M. Schwab (Berlin: Springer).
Van Den Ende, C., Marano, C., Van Ahee, A., Bunge, E. M., and De Moerlooze, L. (2017). The immunogenicity and safety of GSK’s recombinant hepatitis B vaccine in adults: a systematic review of 30 years of experience. Expert Rev. Vaccin. 16, 811–832. doi: 10.1080/14760584.2017.1338568
Vetter, V., Denizer, G., Friedland, L. R., Krishnan, J., and Shapiro, M. (2018). Understanding modern-day vaccines: what you need to know. Ann. Med. 50, 110–120. doi: 10.1080/07853890.2017.1407035
Wagner, T. A. (2018). Quarter Century of Anti-HIV CAR T Cells. Curr. HIV AIDS Rep. 15, 147–154. doi: 10.1007/s11904-018-0388-x
Waldmann, T. A. (2003). Immunotherapy: past, present and future. Nat. Med. 9, 269–277. doi: 10.1038/nm0303-269
Walker, L. M., and Burton, D. R. (2018). Passive immunotherapy of viral infections:’super-antibodies’ enter the fray. Nat. Rev. Immunol. 18, 297–308. doi: 10.1038/nri.2017.148
Wasserman, M., Sings, H. L., Jones, D., Pugh, S., Moffatt, M., and Farkouh, R. (2018). Review of vaccine effectiveness assumptions used in economic evaluations of infant pneumococcal conjugate vaccine. Expert Rev. Vaccin. 17, 71–78. doi: 10.1080/14760584.2018.1409116
Weber, J. S., Gibney, G., Sullivan, R. J., Sosman, J. A., Slingluff, C. L. Jr., Lawrence, D. P., et al. (2016). Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. 17, 943–955. doi: 10.1016/S1470-2045(16)30126-7
Weiner, G. J. (2015). Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370. doi: 10.1038/nrc3930
Weiner, L. M., Murray, J. C., and Shuptrine, C. W. (2012). Antibody-based immunotherapy of cancer. Cell 148, 1081–1084. doi: 10.1016/j.cell.2012.02.034
Wentworth, P., Wentworth, A. D., Zhu, X., Wilson, I. A., Janda, K. D., Eschenmoser, A., et al. (2003). Evidence for the production of trioxygen species during antibody-catalyzed chemical modification of antigens. Proc. Natl. Acad. Sci. U.S.A. 100, 1490–1493. doi: 10.1073/pnas.0437831100
Wherry, E. J., and Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. doi: 10.1038/nri3862
Wightman, F., Solomon, A., Kumar, S. S., Urriola, N., Gallagher, K., Hiener, B., et al. (2015). Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS 29, 504–506. doi: 10.1097/QAD.0000000000000562
Wolchok, J. D., Kluger, H., Callahan, M. K., Postow, M. A., Rizvi, N. A., Lesokhin, A. M., et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133. doi: 10.1056/NEJMoa1302369
Wolf, E., Hofmeister, R., Kufer, P., Schlereth, B., and Baeuerle, P. A. (2005). BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 10, 1237–1244. doi: 10.1016/S1359-6446(05)03554-3
Wüthrich, M., Filutowicz, H. I., Warner, T., Deepe, G. S., and Klein, B. S. (2003). Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune-deficient hosts. J. Exp. Med. 197, 1405–1416. doi: 10.1084/jem.20030109
Wykes, M. N., and Lewin, S. R. (2018). Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 18, 91–104. doi: 10.1038/nri.2017.112
Yu, H., Yang, J., Jiao, S., Li, Y., Zhang, W., and Wang, J. (2015). Cytotoxic T lymphocyte antigen 4 expression in human breast cancer: implications for prognosis. Cancer Immunol. Immunother. 64, 853–860. doi: 10.1007/s00262-015-1696-2
Yuan, P., Zhang, H., Cai, C., Zhu, S., Zhou, Y., Yang, X., et al. (2015). Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 25, 157–168. doi: 10.1038/cr.2014.169
Yuraszeck, T., Kasichayanula, S., and Benjamin, J. E. (2017). Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin. Pharmacol. Ther. 101, 634–645. doi: 10.1002/cpt.651
Zacharakis, N., Chinnasamy, H., Black, M., Xu, H., Lu, Y.-C., Zheng, Z., et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730. doi: 10.1038/s41591-018-0040-8
Zahaf, N.-I., and Schmidt, G. (2017). Bacterial toxins for cancer therapy. Toxins 9:236. doi: 10.3390/toxins9080236
Zajac, A. J., Blattman, J. N., Murali-Krishna, K., Sourdive, D. J., Suresh, M., Altman, J. D., et al. (1998). Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213. doi: 10.1084/jem.188.12.2205
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829. doi: 10.1056/NEJMoa1604958
Zeng, M., Smith, A. J., Wietgrefe, S. W., Southern, P. J., Schacker, T. W., Reilly, C. S., et al. (2011). Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J. Clin. Invest. 121, 998–1008. doi: 10.1172/JCI45157
Zhao, A., Tohidkia, M. R., Siegel, D. L., Coukos, G., and Omidi, Y. (2016). Phage antibody display libraries: a powerful antibody discovery platform for immunotherapy. Crit. Rev. Biotechnol 36, 276–289. doi: 10.3109/07388551.2014.958978
Zhao, Y., Zheng, Z., Cohen, C. J., Gattinoni, L., Palmer, D. C., Restifo, N. P., et al. (2006). High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 13, 151–159. doi: 10.1016/j.ymthe.2005.07.688
Zhen, A., Peterson, C. W., Carrillo, M. A., Reddy, S. S., Youn, C. S., Lam, B. B., et al. (2017). Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 13:e1006753. doi: 10.1371/journal.ppat.1006753
Zhong, Z., Burns, T., Chang, Q., Carroll, M., and Pirofski, L. (1999). Molecular and functional characteristics of a protective human monoclonal antibody to serotype 8 Streptococcus pneumoniae capsular polysaccharide. Infect. Immun. 67, 4119–4127.
Zhu, J.-D., Meng, W., Wang, X.-J., and Wang, H.-C. R. (2015). Broad-spectrum antiviral agents. Front. Microbiol. 6:517. doi: 10.3389/fmicb.2015.00517
Zhukovsky, E. A., Morse, R. J., and Maus, M. V. (2016). Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr. Opin. Immunol. 40, 24–35. doi: 10.1016/j.coi.2016.02.006
Zumla, A., Rao, M., Dodoo, E., and Maeurer, M. (2016a). Potential of immunomodulatory agents as adjunct host-directed therapies for multidrug-resistant tuberculosis. BMC Med. 14:89. doi: 10.1186/s12916-016-0635-1
Keywords: immunotherapy, T cell therapy, antibody therapy, cancer, infectious diseases
Citation: Naran K, Nundalall T, Chetty S and Barth S (2018) Principles of Immunotherapy: Implications for Treatment Strategies in Cancer and Infectious Diseases. Front. Microbiol. 9:3158. doi: 10.3389/fmicb.2018.03158
Received: 06 June 2018; Accepted: 05 December 2018;
Published: 21 December 2018.
Edited by:
Alessandro Marcello, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Luiz Zerbini, International Centre for Genetic Engineering and Biotechnology (ICGEB), South AfricaFederica Benvenuti, International Centre for Genetic Engineering and Biotechnology, Italy
Copyright © 2018 Naran, Nundalall, Chetty and Barth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krupa Naran, a3J1cGEubmFyYW5AdWN0LmFjLnph Stefan Barth, c3RlZmFuLmJhcnRoQHVjdC5hYy56YQ==
†These authors have contributed equally to this work