
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol., 12 December 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.03052
 Rudolf O. Schlechter1,2
Rudolf O. Schlechter1,2 Hyunwoo Jun1†Michał Bernach1,2†Simisola Oso1Erica Boyd1Dian A. Muñoz-Lintz1
Hyunwoo Jun1†Michał Bernach1,2†Simisola Oso1Erica Boyd1Dian A. Muñoz-Lintz1 Renwick C. J. Dobson1,2,3Daniela M. Remus1,2,4
Renwick C. J. Dobson1,2,3Daniela M. Remus1,2,4 Mitja N. P. Remus-Emsermann1,2*
Mitja N. P. Remus-Emsermann1,2*Differential fluorescent labeling of bacteria has become instrumental for many aspects of microbiological research, such as the study of biofilm formation, bacterial individuality, evolution, and bacterial behavior in complex environments. We designed a variety of plasmids, each bearing one of eight unique, constitutively expressed fluorescent protein genes in conjunction with one of four different antibiotic resistance combinations. The fluorophores mTagBFP2, mTurquoise2, sGFP2, mClover3, sYFP2, mOrange2, mScarlet-I, and mCardinal, encoding for blue, cyan, green, green–yellow, yellow, orange, red, and far-red fluorescent proteins, respectively, were combined with selectable markers conferring tetracycline, gentamicin, kanamycin, and/or chloramphenicol resistance. These constructs were cloned into three different plasmid backbones: a broad host-range plasmid, a Tn5 transposon delivery plasmid, and a Tn7 transposon delivery plasmid. The utility of the plasmids and transposons was tested in bacteria from the phyla Actinobacteria, Proteobacteria, and Bacteroidetes. We were able to tag representatives from the phylum Proteobacteria at least via our Tn5 transposon delivery system. The present study enables labeling bacteria with a set of plasmids available to the community. One potential application of fluorescently-tagged bacterial species is the study of bacteria–bacteria, bacteria–host, and bacteria–environment interactions.
Labeling bacterial strains using fluorescent proteins has been used in manifold investigations, such as the study of biofilm formation, bacterial individuality and evolution, and bacterial behavior in complex environments (e.g., plant surfaces, soil, or the mammalian gut) (Tolker-Nielsen and Molin, 2000; Monier and Lindow, 2005; Kroupitski et al., 2009; Tecon and Leveau, 2012; Diard et al., 2013; Remus-Emsermann et al., 2013b; Whitaker et al., 2017; Remus-Emsermann and Schlechter, 2018). Such studies require the equipment of bacteria with bright, stable, fast maturing, and highly abundant fluorescent proteins, which ideally offer unique spectral properties to unambiguously distinguish between differently labeled fluorescent bacteria. This is especially true during non-invasive in situ studies without additional staining procedures, such as fluorescence in situ hybridization, and where autofluorescence and low signal-to-noise ratio might hinder observations (Remus-Emsermann and Schlechter, 2018).
To date, several mechanisms have been described by which fluorescent protein genes can be delivered into bacterial cells and/or integrated into their chromosomes (Andersen et al., 1998; Bloemberg et al., 2000; Miller et al., 2000; Lambertsen et al., 2004; Choi and Schweizer, 2006; Lagendijk et al., 2010; Ledermann et al., 2015; Schada von Borzyskowski et al., 2015; Barbier and Heath Damron, 2016; Remus-Emsermann et al., 2016a). Plasmids are attractive tools for fluorescent protein expression in bacteria, as they can be easily delivered into host cells and lead to high fluorescent protein production due to their presence in multiple copies (Million-Weaver et al., 2012). Plasmid stability usually requires a continuous selection (e.g., antibiotics; Andersson and Hughes, 2011) and, while only some plasmids were found to be maintained in bacterial populations without the pressure exerted by antibiotics (Bloemberg et al., 2000; Rodriguez et al., 2017), the absence of a selective pressure was reported to lead to plasmid loss in growing bacterial populations (Summers, 1991; Smith and Bidochka, 1998; Lau et al., 2013). However, some experimental systems are not compatible with the use of antibiotics as a selective pressure. In natural environments, bacteria often occur as biofilms, where cells are fixed in space and enclosed in an extracellular polymeric substance matrix. Within a biofilm, individual cells experience heterogeneous environments, and owing to their location, they might either be exposed to or protected from antibiotic pressure (Stewart and Costerton, 2001).
To overcome the disadvantage of plasmid loss or their inability to replicate in some hosts, chromosomal insertion of fluorescent markers is advantageous. Chromosomal insertions cannot be lost in the same fashion as plasmids during division, and the probability of mutations that would disrupt the functionality of fluorescent protein genes is low. Several molecular tools and mechanisms exist to integrate chromosomal insertions into bacterial genomes, such as homologous recombination (Ledermann et al., 2015), CRISPR-Cas9 (Jinek et al., 2012), Zinc-finger nucleases (Carroll, 2011), or transposase-based systems (Reznikoff, 2008; Liu et al., 2013; Peters, 2015).
Transposon systems can differ widely in their host specificity and mode of integration. For instance, the Tn5 transposon has been shown to be functional in a wide range of Gram-negative bacteria and to randomly insert into their genomes with high efficiency (Reznikoff, 2008). This ability has been exploited to generate random mutation libraries (de Lorenzo et al., 1990; Christen et al., 2011), but also to integrate fluorescent protein genes into bacterial genomes (Andersen et al., 1998; Schada von Borzyskowski et al., 2015). In contrast to Tn5 transposons, Tn7 transposons only integrate into specific regions, such as the attTn7 site, in the host chromosome. Integration at attTn7 is mediated by four transposases, TnsA, B, C, and D and appears to be prevalent in phylogenetically diverse species (Choi and Schweizer, 2006; McKenzie and Craig, 2006; Parks and Peters, 2007). Even though the host range of Tn7 transposons is limited compared to the Tn5 transposon, it has the tremendous advantage that the insertion does not disrupt any genes and has been argued to have little effect on bacterial fitness (Enne et al., 2005).
Here, we developed three sets of plasmids, which contain genes encoding for the latest generation of fluorescent proteins, i.e., that have blue, cyan, green, yellow, orange, red, and far-red fluorescence emissions, with different degrees of spectral overlap. The fluorescent protein genes mTagBFP2 (mTB2), mTurquoise2 (mTq2), sGFP2, mClover3 (mCl3), sYFP2, mOrange2 (mO2), mScarlet-I (mSc), and mCardinal (mCa) were combined with four combinations of antibiotic resistance genes, i.e., gentamicin, kanamycin, tetracycline and/or chloramphenicol, on three different plasmid backbones: a broad host-range plasmid, a Tn5 transposon delivery plasmid, and a Tn7 transposon delivery plasmid (Figure 1). The broad host-range plasmid contains the pBBR1 origin of replication (Miller et al., 2000). The Tn5 transposon plasmid pAG408 is based on an R6K origin of replication. It is a suicide plasmid that only replicates in presence of the π factor (Suarez et al., 1997). The Tn7 transposon plasmid pGRG36 is based on the thermo-unstable pSC101 origin of replication and is therefore a conditional suicide plasmid for species closely related to Escherichia coli and a suicide plasmid in all other bacteria (McKenzie and Craig, 2006). The expression of the fluorescence protein genes is driven by the nptII promoter of the neomycin phosphotransferase gene (i.e., kanamycin resistance gene). The nptII promoter is considered constitutive, strong and was previously used to drive fluorescent protein expression from chromosomal insertions (Ledermann et al., 2015; Ramirez-Mata et al., 2018). We demonstrate the delivery of plasmids and transposons in a broad phylogenetic background of recently isolated environmental bacteria. Furthermore, we show the heterologous expression of fluorescent proteins at the single-cell resolution in those environmental bacterial strains using fluorescence microscopy.
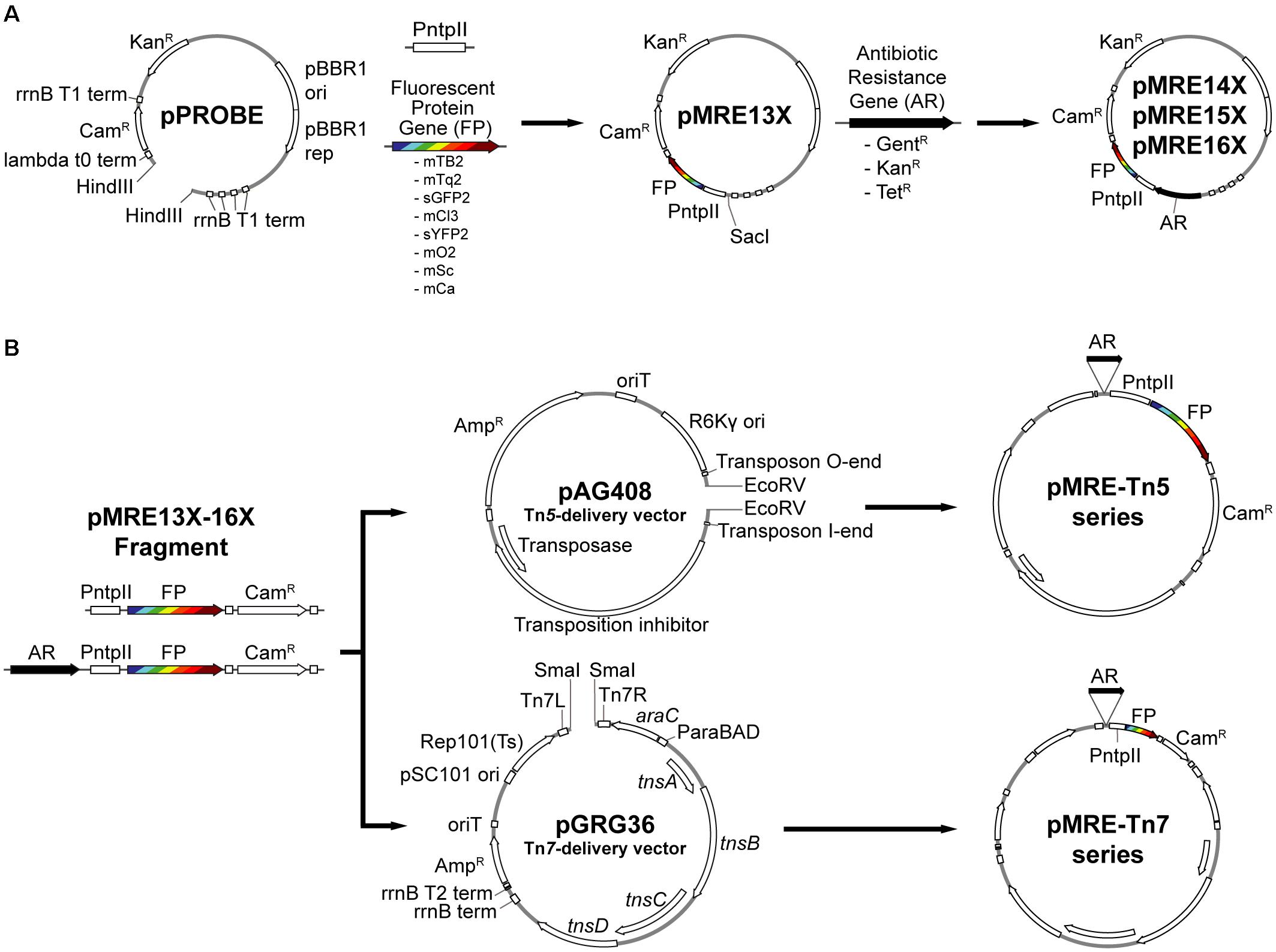
Figure 1. Overview of cloning procedures. (A) Construction of the pMRE plasmid series. To obtain the pMRE13X plasmid series, HindIII-digested pPROBE-derived pFru97 was isothermally assembled with a fragment containing the ntpII promoter (PntpII) and a fragment carrying one of eight fluorescent protein genes (FP) – mTagBFP2, mTurquoise2, sGFP2, mClover3, sYFP2, mOrange2, mScarlet-I, and mCardinal. The rest of the pMRE series were constructed through the assembly of SacI-digested pMRE13X and one of three additional antibiotic resistance gene (AR) – Gentamicin (GentR), Kanamycin (KanR), and Tetracycline (TetR). (B) Construction of the transposon delivery plasmid series pMRE-Tn5 and pMRE-Tn7. PCR products using the pMRE plasmid series as templates containing AR, FP and a Chloramphenicol resistance (CamR) were isothermally inserted into EcoRV digested pAG408 to yield the pMRE-Tn5 series. Similarly, PCR products containing AR, FP and a Chloramphenicol resistance (CamR) were blunt-end cloned into SmaI digested pGRG36 to yield the pMRE-Tn7 series.
Bacterial strains and their growth media are shown in Table 1. Growth media were prepared according to the manufacturer’s recommendations and supplemented with 1.5% agar (Agar No.1, Oxoid) where commercial broth media was used as a base: lysogeny broth agar (LBA, Lysogeny broth Miller, Merck), tryptic soy broth agar (TSA, Merck), nutrient agar (NA, Nutrient broth, Oxoid), or Reasoner’s 2a agar (R2A, HiMedia). E. coli carrying pMRE or pMRE-Tn5 plasmids were cultivated at 37°C, whilst pMRE-Tn7 plasmid-carrying cells were cultivated at 30°C to prevent plasmid loss. All other strains used in this study were cultivated at 30°C. For counterselection after conjugation, auxotroph E. coli S17-1 was grown on either MM2 agar medium (Zengerer et al., 2018) (4.0 g L-1 L-asparagine, 2.0 g L-1 K2HPO4, 0.2 g L-1 MgSO4 ⋅ 7H2O, 3.0 g L-1 NaCl, 10.0 g L-1 sorbitol, 15 g L-1 agar) or minimal agar medium (Harder et al., 1973) (1.62 g L-1 NH4Cl, 0.2 g L-1 MgSO4, 1.59 g L-1 K2HPO4, 1.8 g L-1 NaH2PO4 ⋅ 2H2O, 15 g L-1 agar, with the following trace elements: 15 mg L-1 Na2EDTA2 ⋅ H2O, 4.5 mg L-1 ZnSO4 ⋅ 7H2O, 3 mg L-1 CoCl2 ⋅ 6H2O, 0.6 mg L-1 MnCl2, 1 mg L-1 H3BO3, 3.0 mg L-1 CaCl2, 0.4 mg L-1 Na2MoO4 ⋅ 2H2O, 3 mg L-1 FeSO4 ⋅ 7H2O, and 0.3 mg L-1 CuSO4 ⋅ 5H2O) supplemented with 0.4% w/v succinate or glucose were used, depending on the recipient strain (Table 1). Where appropriate, media were supplemented with antibiotics in the following concentrations: 100 mg L-1 ampicillin, 50 mg L-1 kanamycin, 20 mg L-1 gentamicin, 15 mg L-1 chloramphenicol, or 15 mg L-1 tetracycline.
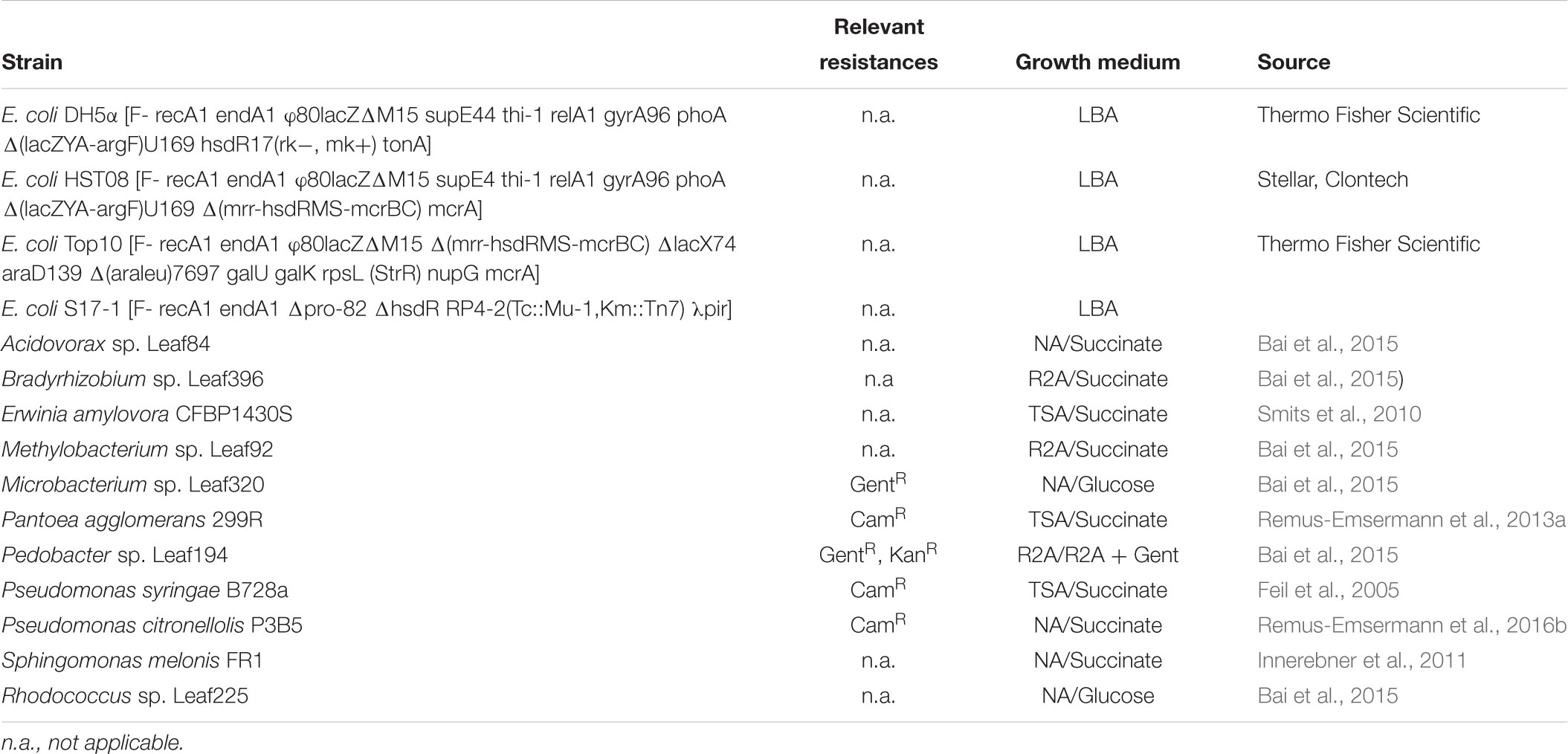
Table 1. Bacterial strains, their relevant naturally-occurring antibiotic resistances, standard growth media, and conditions for selection after conjugation experiments.
All plasmids used or constructed in this study are shown in Table 2. Generic plasmid maps and cloning procedures are presented in Figure 1. For plasmid construction, PCRs were performed using Phusion High-Fidelity DNA polymerase (Thermo Scientific) following the manufacturer’s recommendations. Annealing temperatures (Ta) were chosen based on the respective melting temperature (tm) of the primers (Table 3). Touchdown PCRs were performed to amplify PCR products with overlapping ends for isothermal assemblies. To that end, the initial Ta was set to 10°C above the lowest tm of the respective primers and the Ta was gradually reduced 1°C per cycle for a total of 10 cycles. After the 10th cycle, a two-step PCR with Ta set to 72°C was performed for 25 cycles.

Table 2. Plasmids used in this work.
Table 3. Primer used in this work.
Several plasmids that were used as source for fluorescent protein genes were acquired from Addgene, a non-profit plasmid repository1: pGRG36 was a gift from Nancy Craig (Addgene plasmid #16666). pTriEx-RhoA-wt_mScarlet-I_SGFP2 (Addgene plasmid #85071) and pLifeAct-mTurquoise2 (Addgene plasmid #36201) were gifts from Dorus Gadella. pNCS-mClover3 was a gift from Michael Lin (Addgene plasmid #74236). Plasmids mCardinal-pBAD (Addgene plasmid #54800) and mTagBFP2-pBAD (Addgene plasmid #54572) were gifts from Michael Davidson and mOrange2-pBAD was a gift from Michael Davidson and Roger Tsien (Addgene plasmid #54531). pMREtn5-Ptuf-sYFP2 was a gift from Raphael Ledermann and Hans-Martin Fischer. pFru97 was a gift from Johan Leveau (Tecon and Leveau, 2012). All plasmids that were used for PCR amplification were isolated using the DNA-spin plasmid DNA purification kit (INtRON biotechnology) following the manufacturer’s recommendations. The designated plasmid backbones of the herein constructed plasmids, pFru97, pGRG36, and pAG408, were isolated using the Zyppy plasmid midiprep kit (Zymo) following the manufacturer’s recommendations. Gel purification and PCR clean-up was performed using the Monarch DNA Gel Extraction Kit (NEB) or the DNA Clean & Concentrator-5 Kit (Zymo), respectively.
To construct the pMRE130 series, the promoter of the nptII gene (PnptII) was amplified from pRJPaph_gfp+ using primer FWD_PnptII, which contains overlapping sequences to HindIII-digested pFru97, and either primer REV_PnptII_1, REV_PnptII_2, or REV_PnptII_3, which contain overlapping sequences to the different fluorescent protein genes. The fluorescent protein genes were amplified using primer FWD_FP1, FWD_FP2, or FWD_FP3 with overlapping sequences to the PnptII fragment and primer REV_FP1, REV_FP2, REV_FP3, REV_FP4, REV_FP5, or REV_FP6 with overlapping sequences to HindIII-digested pFru97. The two resulting fragments were mixed with the HindIII-digested pFru97 backbone and isothermally assembled to yield plasmids pMRE130, pMRE131, pMRE132, pMRE133, pMRE134, pMRE135, pMRE136, and pMRE137. To construct the pMRE140 plasmid series, the gentamicin resistance gene (GentR) was amplified from pAG408 using primers FWD_Gent and REV_Gent with overlaps to SacI-digested plasmids of the pMRE13X series, yielding pMRE140, pMRE141, pMRE142, pMRE143, pMRE144, pMRE145, pMRE146, and pMRE147. To construct the pMRE150 plasmid series, the kanamycin resistance gene (KanR) was amplified from pAG408 using primers FWD_Kan and REV_Kan with overlapping sequences to SacI-digested plasmids of the pMRE13X series, yielding pMRE150, pMRE151, pMRE152, pMRE153, pMRE154, pMRE155, pMRE156, and pMRE157. To construct the pMRE16X plasmid series, the tetracycline resistance genes (TetR) were amplified from pTE105-mChe using primers FWD_Tet and REV_Tet with overlapping sequences to SacI-digested plasmids of the pMRE13X series, yielding pMRE160, pMRE161, pMRE162, pMRE163, pMRE164, pMRE165, pMRE166, and pMRE167.
Isothermal assembly was performed as described previously (Gibson et al., 2009; Benoit et al., 2016; Remus-Emsermann et al., 2016a). In short, plasmid backbones and inserts with overlapping sequences were mixed in a 1:3 molar ratio (20–100 ng backbone) to reach a total volume of 5 μL. Then, 15 μL of isothermal assembly mix were added and the reaction was incubated for 15 min at 50°C. Transformation of chemically competent E. coli strains was performed using standard procedures as recommended by the respective suppliers or as described in Sambrook et al. (1989) (see Table 1).
To construct the pMRE-Tn5 plasmid series, pMRE1XX plasmids were used as a template to amplify the desired DNA fragments, which include one of the eight fluorescent protein gene and one of the four antibiotic resistance marker combinations. Cassettes were amplified as two fragments using the primers FWD1_pMRE_pAG408 and RV1_pMRE_pAG408 or FWD2_pMRE_pAG408 and RV2_pMRE_pAG408. Primers FWD1_pMRE_pAG408 and RV2_pMRE_pAG408 contain overlapping sequences to EcoRV-digested pAG408. For isothermal assemblies, EcoRV-digested pAG408 was mixed with the insert fragments in a 1:3:3 ratio as described above and assembled plasmids were transformed into chemically competent E. coli S17-1.
To construct the pMRE-Tn7 family, fluorescent protein genes and antibiotic resistance genes were amplified from the pMRE1XX series using primers FWD_pMRE-Tn7 and REV_pMRE-Tn7. The amplicons were gel purified using the Monarch DNA Gel Extraction Kit (NEB) and phosphorylated using T4 polynucleotide kinase (Life Technologies). pGRG36 was SmaI digested and dephosphorylated using thermosensitive alkaline phosphatase according to the manufacturers’ recommendations (Fast AP, Life Technologies). Following dephosphorylation, plasmids were purified using the DNA Clean & ConcentratorTM-5 Kit (Zymo). Amplicons were then cloned into linearized and dephosphorylated pGRG36 using Quick-Stick T4 Ligase (Bioline). Ligations were purified using the DNA Clean & ConcentratorTM-5 Kit and transformed into chemically competent E. coli (Top10, One ShotTM MAX EfficiencyTM DH5TMα-T1R, or HST08).
All pMRE1XX series plasmids were verified by Sanger sequencing using primers FWD_seq and REV_seq at Macrogen (South Korea). pMRE-Tn5-1XX and pMRE-Tn7-1XX series plasmids were verified by PvuII restriction digests at 37°C for 1 h. To provide a convenient means of plasmid delivery, all plasmids were subsequently transformed into E. coli S17-1, which allows conjugations to recipient strains.
Recipients were grown on standard agar media for up to 4 days, depending on the growth rate of each environmental strain (see Table 1). Donor strains E. coli S17-1 were grown overnight on LBA supplemented with either Kan or Amp to maintain pMRE or pMRE-Tn5 and pMRE-Tn7 plasmids, respectively (see Table 1). Plasmids delivered into recipient strains include pMRE135, pMRE145, pMRE165, pMRE-Tn5-143, pMRE-Tn5-145, pMRE-Tn5-165, pMRE-Tn7-145, or pMRE-Tn7-165. Freshly grown bacteria were harvested using a loop and resuspended in 1× phosphate buffered saline (1×PBS; 8 g L-1 NaCl, 0.24 g L-1 KCl, 1.42 g L-1 Na2HPO4, 0.24 g L-1 KH2PO4) to reach an OD600 nm of 1. Each recipient strain was mixed with their respective donor strains in 1:1 ratio and the mix was then concentrated by centrifugation (4000 g, 5 min) to reach an estimated OD600 nm of 20. Bacterial mixes were drop spotted on NA and incubated for 18 h at 30°C. For conjugations using pMRE-Tn7, 0.1% w/v arabinose was added to the medium, as the Tn7 transposase genes are under the control of an arabinose-inducible promoter. After incubation, the cells were harvested and resuspended in 1 mL 1× PBS. The bacterial mix was spread onto appropriate minimal agar media containing a sole carbon source (either 0.4% w/v succinate or 0.4% w/v glucose, see Table 1) and appropriate antibiotics. Depending on the recipient strain, transconjugants appeared within 5–10 days. To further counterselect against E. coli, single colonies were restreaked at least twice onto fresh minimal media before growing them on complex media.
To provide a convenient tool to assess successful insertions of Tn5 and Tn7 transposons, a multiplex PCR was designed to determine the presence of plasmid backbones and fluorescent protein genes in one reaction. Primers FWD_Tn5/7_gt and RV_Tn5/7_gt were designed to amplify the fluorescent protein genes for both Tn5 and Tn7 delivery systems, while the combinations FWD_Tn5_gt and RV_Tn5_gt and FWD_Tn7_gt, RV_Tn7_gt, and Tn7_gt amplify a specific fragment of the backbone of Tn5 and Tn7 plasmids, respectively. For Tn5 insertions, the primer mix FWD_tn5/7_gt, RV_tn5/7_gt, FWD_tn5_gt, and RV_tn5_gt was used. For Tn7 insertions, primer mix FWD_tn5/7_gt and RV_tn5/7_gt and primer mix FWD_tn7_gt, RV_tn7_gt, and Tn7_gt were used. Reactions were performed using the KAPA2G Fast HotStart ReadyMix PCR kit (Kapa Biosystems) following the manufacturer’s instructions.
The absorption and emission spectra of the different proteins were measured in a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies). To that end, overnight cultures of E. coli strains expressing the different fluorescent proteins were harvested by centrifugation (4000 g, 3 min), washed in 1× PBS and resuspended to reach an OD600 nm of 1. Then, 1 mL of each culture was measured in a polystyrene cuvette (VWR), except for cultures expressing mTagBFP2 and mCardinal, which were measured in a far-UV quartz cuvette (Agilent Technologies) due to their spectral properties. Absorption and emission were measured in the spectrophotometer in 1 nm intervals. The absorption and emission spectra of the proteins used in this study are available as “protein collection” on the webpage fpbase.org (1Lambert, 2018). This online platform allows for a convenient comparison of existing microscopy hardware and fluorophore properties.
To determine brightness of E. coli DH5α carrying plasmids or transposon insertions, respective strains were grown in eight biological replicates in LB broth (300 rpm, 37°C) to reach an OD600 nm of 1. Fluorescence intensity of cultures were measured in a flat bottom 96-well plate (Costar) using a FLUOstar Omega plate reader (BMG Labtech) equipped with an excitation filter (band pass 580/10) and an emission filter (band pass 620/10). Fluorescence intensity was corrected by background subtraction.
Fluorescent protein-expressing E. coli were grown overnight in LB broth (210 rpm, 37°C). Cultures were harvested by centrifugation (4000 g, 3 min) and washed in 1× PBS. Bacterial cultures were resuspended in 1× PBS to an OD600 nm of 0.01, i.e., ∼107 bacteria mL-1, and were mounted on agarose slabs (0.8% w/v agarose in H2O). Fluorescence microscopy was performed on a Zeiss AxioImager.M1 fluorescent widefield microscope equipped with Zeiss filter sets 38HE, 43HE, 46HE, 47HE, and 49 (BP 470/40-FT 495-BP 525/50, BP 550/25-FT 570-BP 605/70, BP 500/25-FT 515-BP 535/30, BP 436/25-FT 455-BP 480/40, and G 365-FT 395-BP 445/50, respectively), an Axiocam 506 and the software Zeiss Zen 2.3. For confocal microscopy, a Leica SP5 confocal laser scanning microscope equipped with a 405 nm UV laser, an Argon laser line 458, 476, 514 nm, and a diode-pumped solid-state laser line 561 nm and software Leica LAS AF (version 2.6.3.8173) was used. Suspensions containing six or seven different fluorescent protein-tagged E. coli were detected through confocal microscopy using sequential scanning mode. For six color mixtures consisting of mTB2, mTq2, mCl3, mO2, mSc, and mCa-expressing E. coli, a first scan using laser line 561 nm was used for emission windows of 570–599, 600–630, and 650–750 nm. Then, a second scan using laser lines 405, 458, and 476 nm were used for emission windows 440–460, 480–500, and 520–540 nm. For seven color mixtures consisting of mTB2, mTq2, sGFP2, sYFP2, mO2, mSc, and mCa-expressing E. coli, a first scan using laser line 561 nm was used for emission windows of 570–599, 600–630, and 650–750 nm. A second scan using line 514 nm was used for the emission windows 540–560 nm. A third scan using lines 405, 458, 476 nm, and were used for the emission windows 440–460, 480–500, and 520–540 nm.
All image analysis and processing were carried out in ImageJ/FIJI (Schindelin et al., 2012). Signal bleed-through was corrected using a channel subtraction method. First, individual channels were thresholded (ImageJ/Fiji threshold using Otsu) to produce binary images. Binary template images were then dilated (ImageJ/Fiji command dilate) to accommodate spherical aberrations and subtracted from channels with signal bleed-through. All channels were background subtracted.
Single-cell fluorescence was analyzed as described previously (Remus-Emsermann et al., 2016a). In short, bacteria were mounted on an agar slab as described above and samples were analyzed using a Zeiss AxioImager.M1 at 100x magnification. Multichannel images were acquired using phase contrast and appropriate fluorescent filters (see above). Using ImageJ/Fiji, phase contrast channels were thresholded using standard settings and used as a mask to determine the mean fluorescence signal of individual particles in the respective fluorescent channels.
In this work, a total of 96 plasmids were constructed (Table 2). The plasmids constitute three sets, including (i) 32 broad-host range plasmids based on the pBBR1 origin of replication, (ii) 32 suicide plasmids based on the R6K origin of replication, which are maintained solely in hosts carrying the π factor, and a Tn5-based transposon system, and (iii) 32 narrow host-range plasmids harboring a pSC101 temperature-sensitive origin of replication and the Tn7-based transposon system. Each set consists of plasmids that carry one of eight different fluorescent protein genes and one of four different combinations of antibiotic resistances. We used a simple naming convention and separated the plasmid into the pMRE13X-series, which all confer chloramphenicol resistance, the pMRE14X-series, which all confer gentamicin and chloramphenicol resistance, the pMRE15X-series, which all confer kanamycin and chloramphenicol resistance, and the pMRE16X-series, which all confer tetracycline and chloramphenicol resistance. The pFru97-based pMRE series contains an additional kanamycin resistance. Furthermore, we named plasmids based on the contained fluorescent protein, i.e., pMRE1X0 denotes mTagBFP2, pMRE1X1 denotes mTurquoise2, pMRE1X2 denotes sGFP2, pMRE1X3 denotes sYFP2, pMRE1X4 denotes mOrange2, pMRE1X5 denotes mScarlet-I, pMRE1X6 denotes mCardinal, pMRE1X7 denotes mClover3. Transposons are indicated as pMRE-Tn5-1XX for Tn5 transposons and pMRE-Tn7-1XX for Tn7 transposons.
Fluorescent protein-expressing bacteria were found to exhibit the expected fluorescence absorption and emission spectra (Figure 2) (Kremers et al., 2007; Subach et al., 2011; Goedhart et al., 2012; Chu et al., 2014; Bindels et al., 2017), suggesting that no changes in their spectral properties were introduced by cloning procedures. However, it was noticeable that bacterial cells harboring the pMRE14X-series exhibited increased fluorescence intensities than in the rest of the plasmid series, likely due to readthrough of the gentamicin promoter which was cloned upstream the fluorescent protein gene. Therefore, the fluorescence intensity of the pMRE-series carrying the mScarlet-I gene in combination with all antibiotic resistance genes was determined in E. coli DH5α. Additionally, the emitted fluorescence was assessed in four independent E. coli DH5α::MRE-Tn5-145 Tn5 insertions mutants and four independent E. coli DH5α::MRE-Tn7-145 Tn7 insertion mutants. Plasmids pMRE135, pMRE155, and pMRE165 led to similar fluorescence in E. coli DH5α, however, plasmid pMRE145 resulted in significantly brighter fluorescence intensities (Figure 3A). After Tn5 and Tn7 transposition, E. coli DH5α exhibited similar fluorescence intensities, however, transposon-conferred fluorescence intensities were signifi-cantly lower than plasmid-conferred fluorescence intensities (Figure 3B).
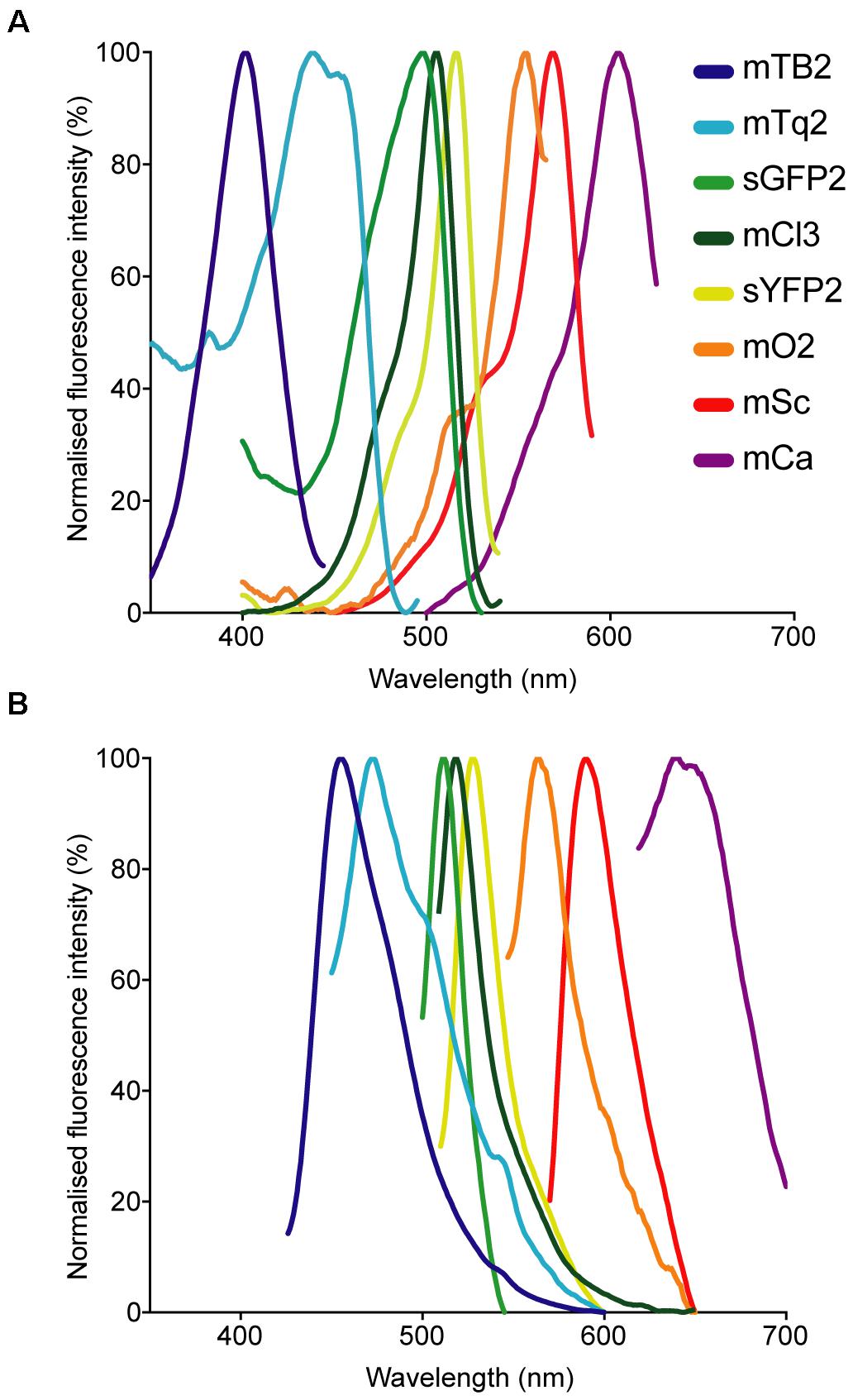
Figure 2. Normalized (A) absorption and (B) emission spectra of the fluorescent proteins used in this work.
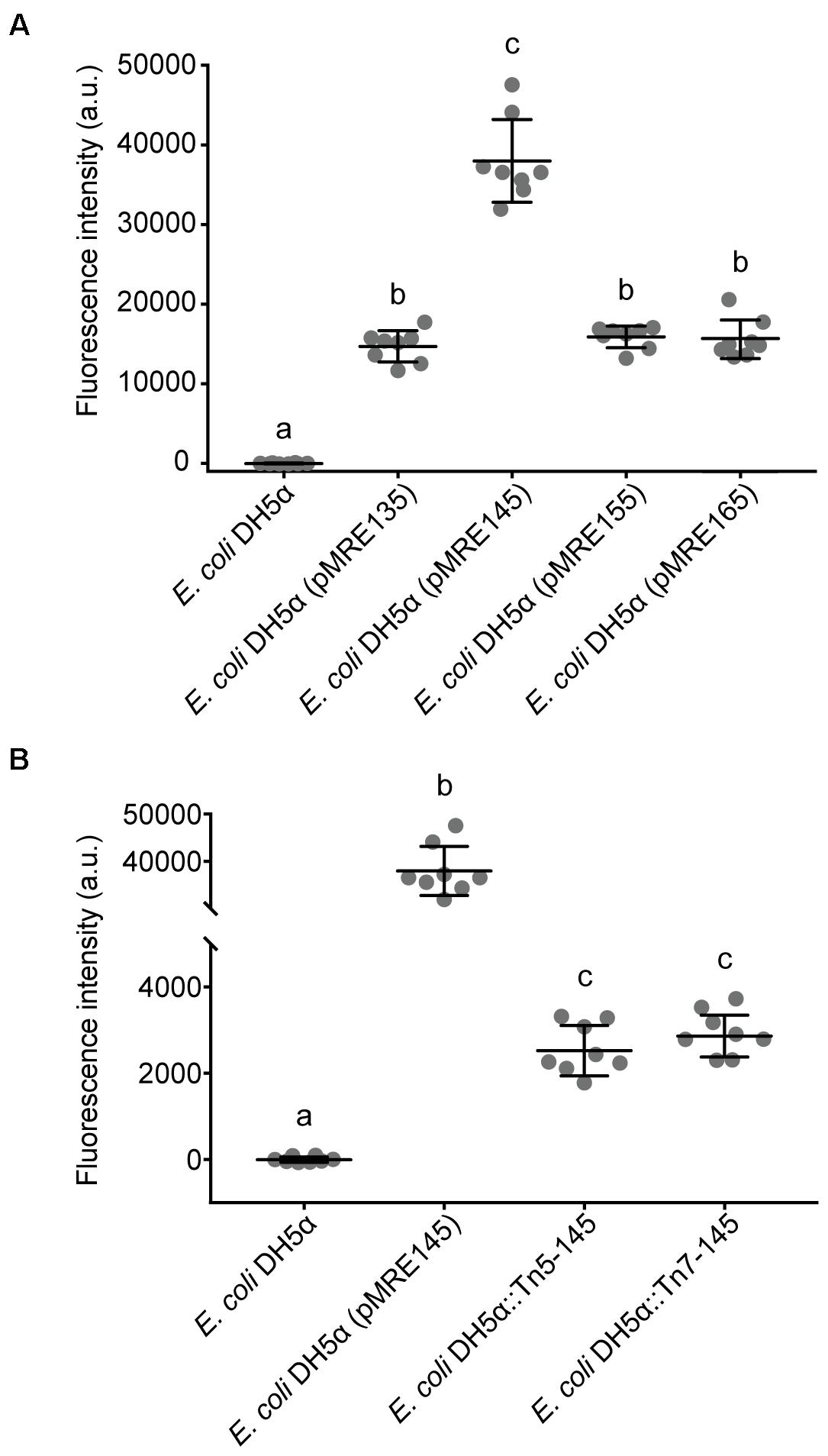
Figure 3. (A) Fluorescence intensity of Escherichia coli DH5α cultures expressing mScarlet-I from different pMRE plasmid series (pMRE135, pMRE145, pMRE155, pMRE165). (B) Fluorescence intensity of mScarlet-I in plasmid-borne E. coli DH5α cultures (pMRE145), Tn5- and Tn7-insertion mutants. E. coli DH5α was used as control. One-way ANOVA and Tukey’s post hoc test were used to infer statistical differences between groups. Letters indicate differences among means with a 99.99% confidence interval (p < 0.0001). A.u., arbitrary units.
Plasmid functionality and fluorescence protein expression was tested in a phylogenetically broad range of bacteria that were previously isolated from plant leaves (Table 4). The pMRE-series plasmids were successfully conjugated and maintained in Sphingomonas melonis FR1, Erwinia amylovora CFBP1430S, Pantoea agglomerans 299R, and Pseudomonas syringae B728a. Tn5 transposon plasmid were delivered by conjugation and integrated into the genome of Bradyrhizobium sp. Leaf396, Methylobacterium sp. Leaf92, S. melonis FR1, Sphingomonas phyllosphaerae FA2, E. amylovora CFBP1430S, P. agglomerans 299R, and Pseudomonas citronellolis P3B5. Tn7 transposon plasmids were delivered by conjugation and integrated into the genome of S. melonis FR1, E. amylovora CFBP1430S, and P. agglomerans 299R.
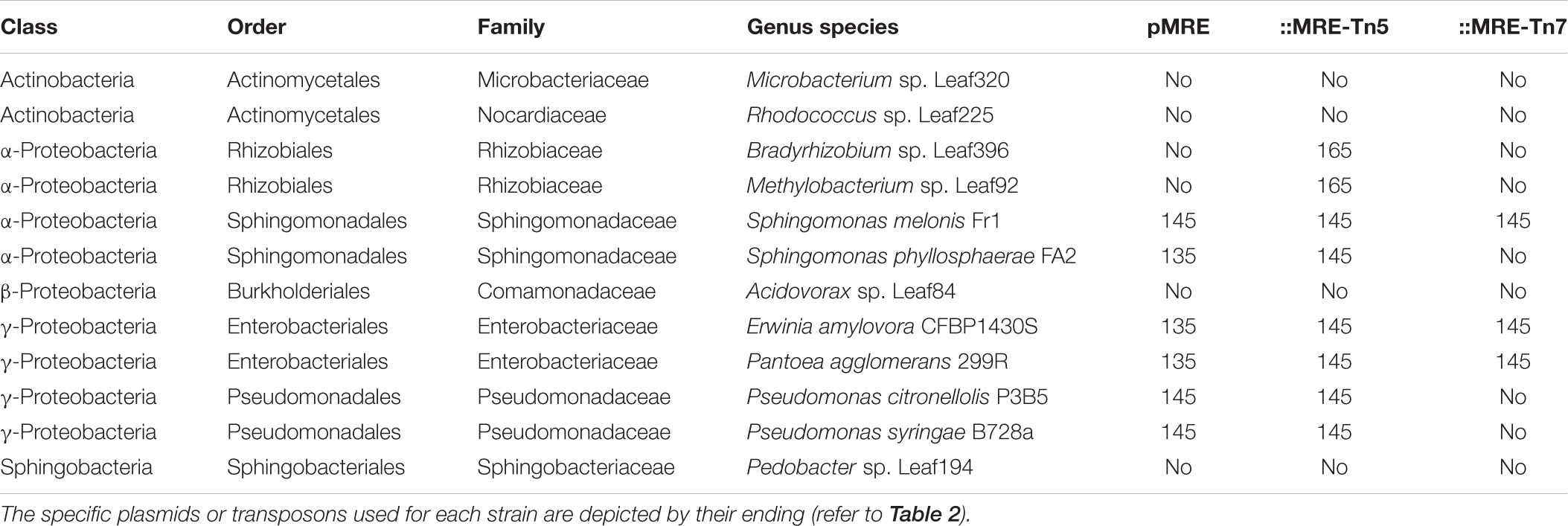
Table 4. Bacterial strains used as recipients for conjugation of pMRE, pMRE-Tn5, and pMRE-Tn7.
For a fast assessment of transposon-mediated insertions, multiplex PCRs were developed to amplify a region of the insert (i.e., fluorescent protein gene) and plasmid backbone simultaneously, i.e., a PCR on the donor plasmid resulted in two PCR products, while a genomic integration of the transposon resulted in only one PCR product. As expected, successfully integrated insertions into the genome of all tested strains were corroborated, as no plasmid backbone was detected whilst the fluorescent protein gene was correctly amplified (Supplementary Figure 1).
To test for stability and fitness cost of the plasmids and transposon insertions, populations of P. agglomerans 299R (Pa299R) wild type, Pa299R (pMRE), Pa299R::MRE-Tn5, and Pa299R::MRE-Tn7 were grown in rich media supplemented with or without chloramphenicol. The bacteria were then plated onto media containing antibiotics to detect subpopulations that lost their plasmid or transposon and differences in single cell fluorescence was assessed using fluorescence microscopy. Other than a reduction in the wild type Pa299R populations in presence of chloramphenicol, no statistical differences were found between the rest of the strains and growth conditions (Supplementary Figure 2A). In addition, fluorescence intensity of single cells was higher in Pa299R(pMRE135) compared to transposon insertion mutants in both growth conditions. Also, a decrease in fluorescence intensity in Pa299R(pMRE135) was observed when grown without chloramphenicol compared to the same strain grown with antibiotic pressure (Supplementary Figure 2B), indicating that plasmid loss occurred. Fluorescence did not decline in Tn5 or Tn7 transposon mutants growing without antibiotic pressure, indicating that no loss of transposon insertions occurred.
Using standard fluorescence microscopy equipment, it was possible to track up to seven bacterial strains in parallel. Employing widefield fluorescence microscopy and widely distributed fluorescence filter sets (i.e., standard blue, green, and red fluorescence emission filters) three bacterial strains could be unambiguously identified, allowing to monitor three strains equipped with mTB2, mCl3, and mSc (Figure 4A), respectively. Less common combination of filters (i.e., blue, cyan, yellow, and red fluorescence emission filters) allows monitoring of up to four strains expressing mTB2, mTq2, sYFP2, and mSc, respectively (Figure 4B).
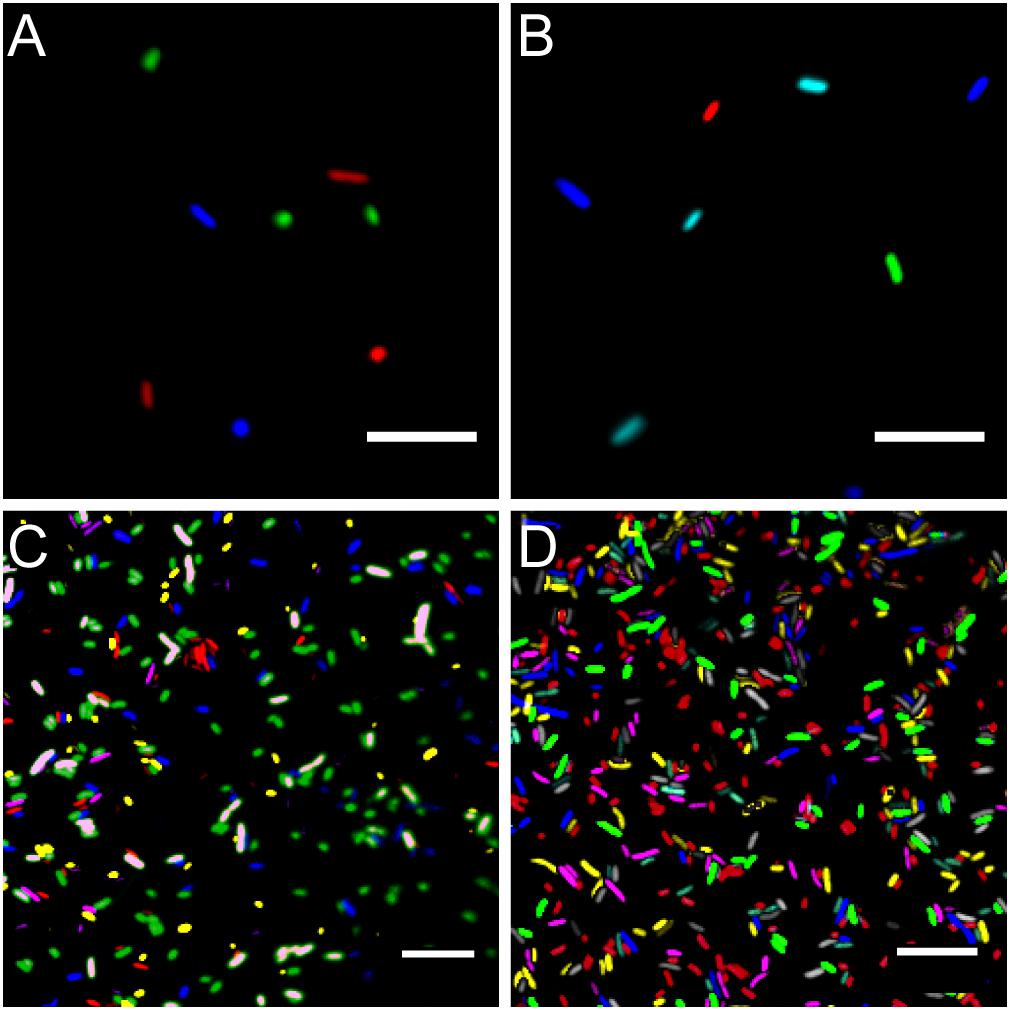
Figure 4. Microscopy images of fluorescent bacteria. (A) Widefield epifluorescence micrograph of E. coli expressing either mTB2 (blue), mCl3 (green), or mSc (red). (B) Widefield epifluorescence micrograph of E. coli expressing either mTB2 (blue), mTq2 (cyan), sYFP2 (green), or mSc (red). (C) Confocal microscopy of a mixed E. coli expressing either mTB2 (blue), mTq2 (magenta), mCl3 (yellow), mO2 (green), mSc (white), or mCa (red). (D) Confocal microscopy of a mixed E. coli expressing either mTB2 (cyan), mTq2 (magenta), sGFP2 (yellow), sYFP2 (gray), mO2 (red), mSc (green), or mCa (blue). In all cases, E. coli harboring pMRE14X plasmids series were used. Scale bar: 10 μm.
Using filter free confocal laser scanning microscopy, it was possible to detect six different fluorescently tagged cell populations without further image processing (Figure 4C). After bleedthrough correction (see section “Materials and Methods,” i.e., for sYFP2 and sGFP2, mO2 and mSc, and mSc and mCa) seven different fluorescently tagged cell populations could be distinguished (Figure 4D).
All bacteria that were successfully equipped with plasmids or transposons (Table 4) were grown on agar media plates and epifluorescence widefield microscopy was performed to determine fluorescence at the single-cell resolution. All bacterial strains exhibited fluorescence that could be determined at the single-cell resolution with a high signal-to-noise ratio (Figure 5). The range of fluorescence intensity of the environmental strains was comparable to E. coli DH5α and yielded signals in the same magnitude. In general, bacterial strains harboring pMRE-series plasmids showed higher variations in fluorescence intensity than Tn5 and Tn7 transposon mutants. Additionally, P. syringae B728a yielded an exceptionally bright signal compared to other bacterial strains (Figure 6).
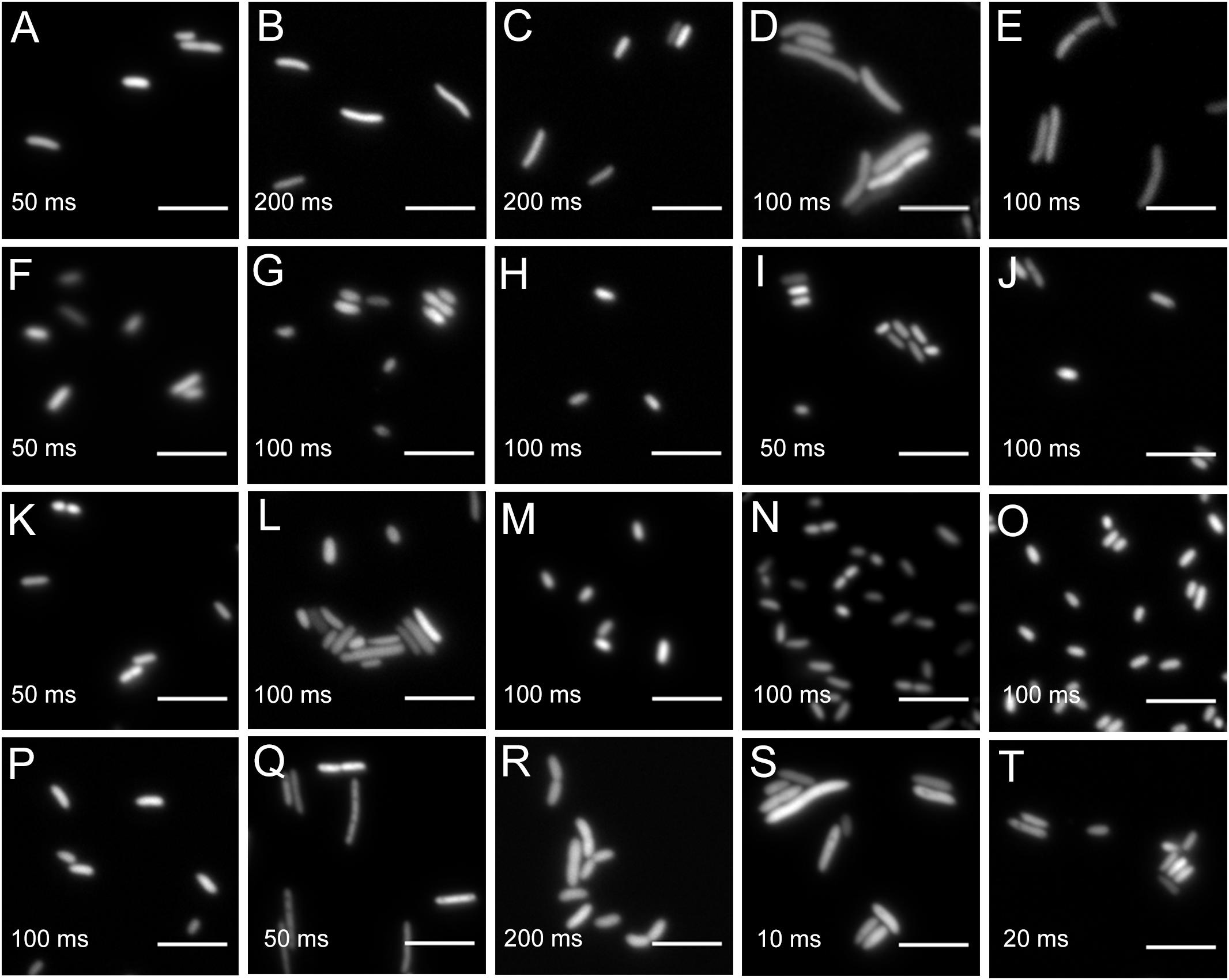
Figure 5. Widefield microscopy of environmental bacteria expressing fluorescent proteins. (A) E. coli DH5α (pMRE145). (B) E. coli DH5α::MRE-Tn5-145. (C) E. coli DH5α::MRE-Tn7-145. (D) Bradyrhizobium sp. Leaf396::MRE-Tn5-165. (E) Methylobacterium sp. Leaf92::MRE-Tn5-165. (F) Sphingomonas melonis FR1 (pMRE145). (G) S. melonis FR1::MRE-Tn5-145. (H) S. melonis FR1::MRE-Tn7-145. (I) Sphingomonas phyllosphaerae FA2 (pMRE135). (J) S. phyllosphaerae FA2::MRE-Tn5-145. (K) Erwinia amylovora CFBP1430S (pMRE135). (L) E. amylovora CFBP1430S::MRE-Tn5-145. (M) E. amylovora CFBP1430S::MRE-Tn7-145. (N) Pantoea agglomerans 299R (pMRE135). (O) P. agglomerans 299R::MRE-Tn5-145. (P) P. agglomerans 299R::MRE-Tn7-145. (Q) Pseudomonas citronellolis P3B5 (pMRE145). (R) P. citronellolis P3B5::MRE-Tn5-145. (S) Pseudomonas syringae pv. syringae B728a (pMRE145). (T) P. syringae B728a::MRE-Tn5-145. Exposure times used during image acquisition are depicted in the corresponding images. Scale bars represent 5 μm.
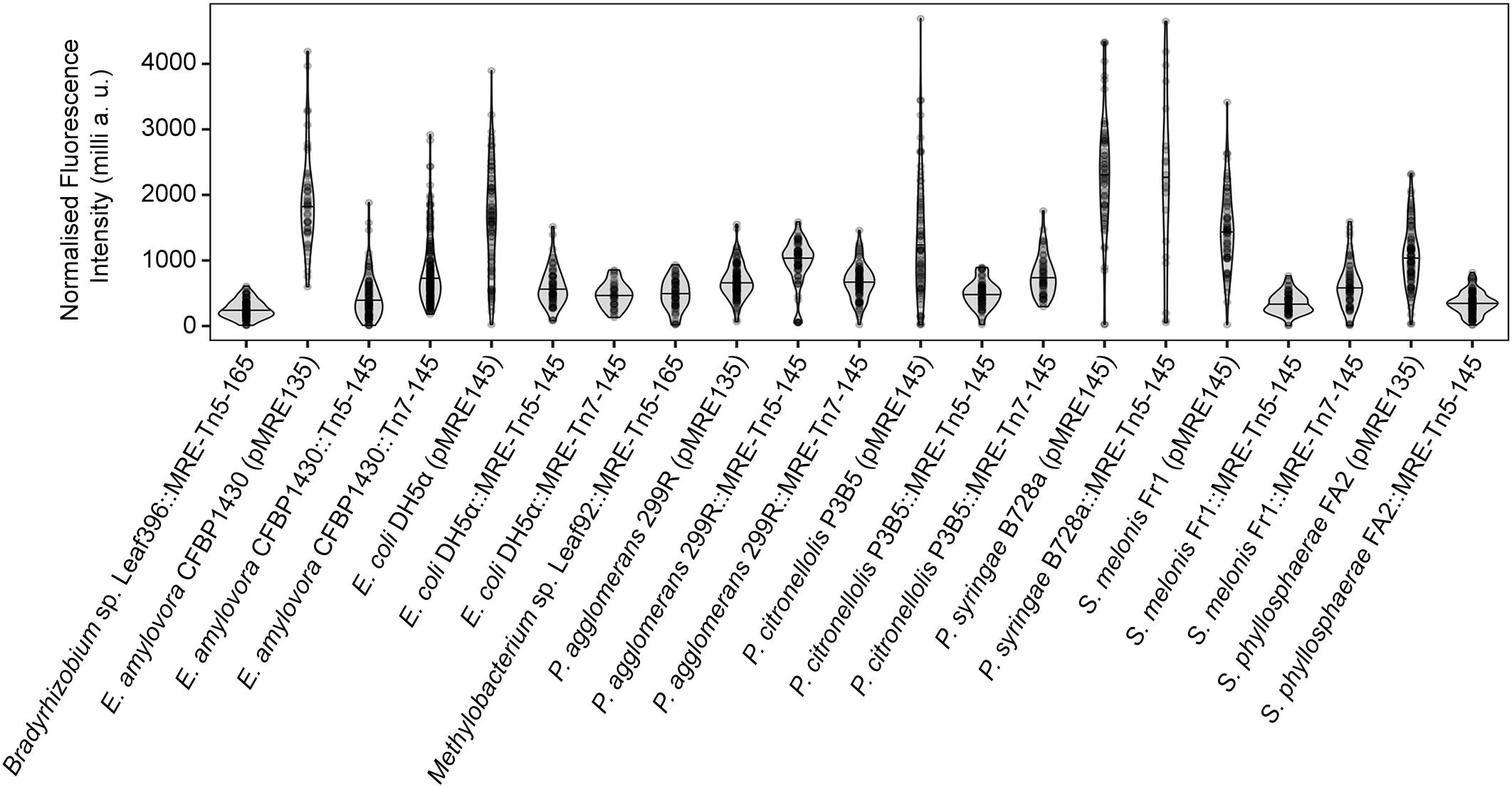
Figure 6. Single-cell fluorescence intensity of bacterial strains expressing fluorescent proteins. Data were normalized by exposure time and expressed as milli arbitrary units.
A set of plasmids was constructed conferring eight fluorescent phenotypes in a wide range of bacteria. The fluorescent proteins used in this work were selected to have minimal spectral overlap, which allows parallel detection of up to four fluorescent proteins on a standard widefield microscopy system and seven fluorescent proteins on filter free confocal microscopy systems. Additionally, the series of plasmids and transposons carry four different antibiotic resistance combinations which allows the selection of bacterial mutants while accommodating their requirements of naturally-occurring antibiotic resistance.
The strong, constitutive promoter of the nptII gene is active in a wide range of taxa and has been shown to be suitable to drive fluorescent protein expression from single chromosomal insertions (Ledermann et al., 2015). All bacterial strains that carried either a plasmid of the pMRE-series, Tn5 or Tn7-based transposons, exhibited visible fluorescence at a single-cell resolution. This shows the application of the here constructed toolbox and its potential use to generate fluorescently-tagged bacterial communities.
The plasmids were tested in a wide range of bacterial taxa that originated from aboveground surfaces of plants (Feil et al., 2005; Smits et al., 2010; Innerebner et al., 2011; Remus-Emsermann et al., 2013a, 2016b; Bai et al., 2015; Schmid et al., 2018). For efficient delivery of the plasmids, conjugation using E. coli S17-1 was performed. The presence of the tra operon in this strain allows for the mobilization of plasmids to recipient strains (Simon et al., 1983), bypassing the need to produce and optimize competent cells for every recipient strain used, which can be very time and labor intensive for strains that were recently isolated. However, optimization of conjugation conditions might be necessary to increase conjugation efficiencies.
pFru97-based pMRE plasmids could be conjugated into Pseudomonads and Enterobacteriaceae supporting previous findings (Table 4) (Miller et al., 2000; Nakai et al., 2001). In contrast to previous findings, the Microbacterium sp. Leaf320 tested in this study did not readily accept pMRE plasmids compared to other Microbacterium sp. tested previously (Lin et al., 2012). For short term studies, or providing that the pMRE plasmids are highly stable in the used bacterial strains, they could be advantageous to other systems, since fluorescence is generally higher than after chromosomal insertion (Figure 3B and Supplementary Figure 2).
The delivery of fluorescent protein genes into bacterial isolates was most successful using the Tn5 transposon compared to the other two delivery systems. Most members of the Proteobacteria, including members of the α- and γ-Proteobacteria, were integrating the Tn5 transposon into their genome, which is in line with previous observations (Andersen et al., 1998; Schada von Borzyskowski et al., 2015). The Tn5 transposome system, employing Tn5 transposase bound to transposon DNA flanked by Tn5 inverted repeats, has been shown to be suitable to construct random insertion mutations in Gram-positive bacteria (Fernandes et al., 2001; Vidal et al., 2009). Consequently, Tn5 transposon integration was tested in Gram-positive bacteria as well. Since the purpose of the constructs described here is not to generate mutant libraries but to fluorescently tag bacteria, Tn5 insertions at low frequencies would be sufficient to serve the purpose as a gene delivery tool. However, we failed to identify positive mutants in the two Gram-positive strains tested. Even though this strain selection is not exhaustive, the lack of evidence of plasmid-based Tn5 transposon delivery into Gram-positive strains underlines the slim chances of successfully using the here proposed system in Gram-positives. In the future, additional plasmids will be developed to allow fluorescent protein gene delivery into Gram-positive bacteria. The here constructed plasmids will serve as a foundation for this work. Transposon mediated delivery tools such as Himar will be employed to deliver fluorescent protein genes to widened bacterial host range (Nilsson et al., 2014).
The functionality of Tn7 transposons is well-documented in Proteobacteria. However, the here employed plasmid backbone has been predominantly used in E. coli and very close relatives such as Salmonella (McKenzie and Craig, 2006; Remus-Emsermann et al., 2016a). In E. coli, pMRE-Tn7 plasmids are conditional suicide plasmids that fail to replicate when cells are cultivated at temperatures above 32°C. In other bacteria, this plasmid is a suicide plasmid and is not replicating. pMRE-Tn7 plasmids carry the complete Tn7 transposase machinery in cis (McKenzie and Craig, 2006). This is a great advantage compared to classical Tn7 systems, which usually involve helper plasmids that provide the transposase machinery in trans (Lambertsen et al., 2004; Choi and Schweizer, 2006). As the transposon machinery has to be provided on a second plasmid, the probability for both plasmids to be electroporated or conjugated into the same target cell is expected to be lower than in the system established by McKenzie and Craig (2006). Without having compared the systems of Lambertsen et al. (2004) and Choi and Schweizer (2006), we expect that transposon delivery will be more successful using the here used delivery system. Even though the delivery system has previously been used in Pseudomonads (Klümper et al., 2014), to our knowledge, this is the first successful application of the system as a suicide plasmid in Enterobacteriaceae and S. melonis FR1 that do not replicate the plasmid backbone.
To determine the successful genomic integration of the Tn5 or Tn7 transposons, we provide a convenient PCR-based tool that allows for screening integration events. However, mapping of transposon insertion sites will be important if the aim of a study is functional studies, especially if the Tn5 transposon system, which occur randomly in the bacterial genome and may disrupt essential genes of the studied species, is used. Due to this property, Tn5 transposon systems have previously not only been used to deliver DNA fragments into bacterial species, but also to create mutant pools and transposon libraries for genome functional analyses (Winterberg et al., 2005; Gallagher et al., 2011). Thus, an arbitrary PCR as described by Das et al. (2005) should be performed. It is furthermore advisable to assess if there are any fitness differences between Tn5 insertion mutants and wild type strains. In the case of Tn7 transposition, insertions occur in specific attachment sites (attTn7), which have been mapped at the 3′ end of the glmS gene and has been shown to pose no fitness cost to the bacterial host (Enne et al., 2005; McKenzie and Craig, 2006).
Using standard epifluorescence widefield microscopy, we were able to distinguish four differentially labeled bacterial strains (Figure 4). Using less common fluorescent filters and/or spectral linear unmixing approaches (Zimmermann, 2005), it should be possible to unambiguously identify five different fluorescently tagged populations. Next to E. coli DH5α, environmental strains also expressed fluorescent proteins to a similar degree that enabled observation of bacteria at the single-cell resolution (Figure 5 and Supplementary Figure 2).
Recently, many techniques have been developed to investigate fluorescently-tagged bacteria in situ, which for instance allowed the detection of 15 differentially tagged bacterial strains from dental plaques (Valm et al., 2012). In combination with other experimental advances such as removing autofluorescent background from environmental samples (Remus-Emsermann et al., 2014; Peredo and Simmons, 2017) and spatially explicit analysis of bacteria in situ (Daims et al., 2006; Schmidt et al., 2018), the here constructed molecular tools set the foundation to further advance in situ investigation of environmental bacteria.
We present a plasmid toolbox suitable to fluorescently label a broad range of bacteria for research in microbiology and microbial ecology that will be provided to the scientific community. The use of these plasmids will enable convenient tagging of many environmental bacteria and will thereby facilitate several disciplines of microbiology, such as single-cell microbiology, synthetic community, biofilm, host–microbe and microbe–microbe interactions. The ability to tag bacteria with unambiguous fluorescent colors in combination with antibiotic resistance markers to track bacterial population development at the single-cell resolution as well as on the whole population scale will be invaluable for many studies.
All plasmids constructed in this work have been made available in the convenient conjugation strain E. coli S17-1 that allows conjugation of the mobilisable plasmids into other bacteria at the non-profit plasmid repository Addgene2.
Plasmids and plasmid maps are available at the repository Addgene (http://www.addgene.org).
RS constructed the pMRE-Tn5 plasmid series, performed most other experiments, analyzed the data, and wrote the manuscript. HJ constructed the pMRE plasmid series. MB constructed the pMRE-Tn7 plasmid series. SO performed genotyping PCRs. EB performed several mating experiments with γ-proteobacteria and contributed to other experiments. DM-L constructed pMRE134 and pMRE137. RD contributed materials and facilities. DR contributed to experiments, supervision, discussion, and writing. MR-E conceived and supervised the study, wrote the manuscript, contributed to some experiments, and analyzed data. All authors have read and approved the manuscript.
This work was funded by a seed grant of the Biomolecular Interaction Centre of the University of Canterbury to RD and MR-E and the Royal Society of New Zealand Marsden Fast Start grant (UOC1704) to MR-E. RS was supported by a NZIDRS doctoral scholarship. MB was supported by a University of Canterbury Doctoral Scholarship and a Biomolecular Interaction Centre doctoral scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Raphael Ledermann and Hans-Martin Fischer (ETH Zurich) are acknowledged for contributing to the construction of pMRE-Tn5-sYFP.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03052/full#supplementary-material
Andersen, J. B., Sternberg, C., Poulsen, L. K., Bjorn, S. P., Givskov, M., and Molin, S. (1998). New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 64,break 2240–2246.
Andersson, D. I., and Hughes, D. (2011). Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 35, 901–911. doi: 10.1111/j.1574-6976.2011.00289.x
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Bajar, B. T., Wang, E. S., Lam, A. J., Kim, B. B., Jacobs, C. L., Howe, E. S., et al. (2016). Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 16:20889. doi: 10.1038/srep20889
Barbier, M., and Heath Damron, F. (2016). Rainbow vectors for broad-range bacterial fluorescence labeling. PLoS One 11:e0146827. doi: 10.1371/journal.pone.0146827
Benoit, R. M., Ostermeier, C., Geiser, M., Li, J. S. Z., Widmer, H., and Auer, M. (2016). Seamless insert-plasmid assembly at high efficiency and low cost. PLoS One 11:e0153158. doi: 10.1371/journal.pone.0153158
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56. doi: 10.1038/nmeth.4074
Bloemberg, G. V., Wijfjes, A. H., Lamers, G. E., Stuurman, N., and Lugtenberg, B. J. (2000). Simultaneous imaging of Pseudomonas fluorescens WCS365 populations expressing three different autofluorescent proteins in the rhizosphere: new perspectives for studying microbial communities. Mol. Plant Microbe Interact. 13, 1170–1176. doi: 10.1094/MPMI.2000.13.11.1170
Carroll, D. (2011). Genome engineering with zinc-finger nucleases. Genetics 188, 773–782. doi: 10.1534/genetics.111.131433
Choi, K.-H., and Schweizer, H. P. (2006). mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1, 153–161. doi: 10.1038/nprot.2006.24
Christen, B., Abeliuk, E., Collier, J. M., Kalogeraki, V. S., Passarelli, B., Coller, J. A., et al. (2011). The essential genome of a bacterium. Mol. Syst. Biol. 7:528. doi: 10.1038/msb.2011.58
Chu, J., Haynes, R. D., Corbel, S. Y., Li, P., González-González, E., Burg, J. S., et al. (2014). Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat. Methods 11, 572–578. doi: 10.1038/nmeth.2888
Daims, H., Lücker, S., and Wagner, M. (2006). daime, a novel image analysis program for microbial ecology and biofilm research. Environ. Microbiol. 8, 200–213. doi: 10.1111/j.1462-2920.2005.00880.x
Das, S., Noe, J. C., Paik, S., and Kitten, T. (2005). An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 63, 89–94. doi: 10.1016/j.mimet.2005.02.011
de Lorenzo, V., Herrero, M., Jakubzik, U., and Timmis, K. N. (1990). Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 172, 6568–6572. doi: 10.1128/jb.172.11.6568-6572.1990
Diard, M., Garcia, V., Maier, L., Remus-Emsermann, M. N. P., Regoes, R. R., Ackermann, M., et al. (2013). Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 494, 353–356. doi: 10.1038/nature11913
Enne, V. I., Delsol, A. A., Davis, G. R., Hayward, S. L., Roe, J. M., and Bennett, P. M. (2005). Assessment of the fitness impacts on Escherichia coli of acquisition of antibiotic resistance genes encoded by different types of genetic element. J. Antimicrob. Chemother. 56, 544–551. doi: 10.1093/jac/dki255
Feil, H., Feil, W. S., Chain, P., Larimer, F., DiBartolo, G., Copeland, A., et al. (2005). Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. U.S.A. 102, 11064–11069. doi: 10.1073/pnas.0504930102
Fernandes, P. J., Powell, J. A., and Archer, J. A. (2001). Construction of Rhodococcus random mutagenesis libraries using Tn5 transposition complexes. Microbiology 147, 2529–2536. doi: 10.1099/00221287-147-9-2529
Gallagher, L. A., Shendure, J., and Manoil, C. (2011). Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio 2, 315–310. doi: 10.1128/mBio.00315-10
Gibson, D. G., Young, L., Chuang, R.-Y., Venter, J. C., Hutchison, C. A. III, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Goedhart, J., von Stetten, D., Noirclerc-Savoye, M., Lelimousin, M., Joosen, L., Hink, M. A., et al. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 3:751. doi: 10.1038/ncomms1738
Harder, W., Attwood, M. M., and Quayle, J. R. (1973). Methanol assimilation by Hyphomicrobium sp. Microbiology 78, 155–163.
Innerebner, G., Knief, C., and Vorholt, J. A. (2011). Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 77, 3202–3210. doi: 10.1128/AEM.00133-11
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Klümper, U., Droumpali, A., Dechesne, A., and Smets, B. F. (2014). Novel assay to measure the plasmid mobilizing potential of mixed microbial communities. Front. Microbiol. 5:730. doi: 10.3389/fmicb.2014.00730
Kremers, G.-J., Goedhart, J., van den Heuvel, D. J., Gerritsen, H. C., and Gadella, T. W. Jr. (2007). Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 46, 3775–3783. doi: 10.1021/bi0622874
Kroupitski, Y., Golberg, D., Belausov, E., Pinto, R., Swartzberg, D., Granot, D., et al. (2009). Internalization of Salmonella enterica in leaves is induced by light and involves chemotaxis and penetration through open stomata. Appl. Environ. Microbiol. 75, 6076–6086. doi: 10.1128/AEM.01084-09
Lagendijk, E. L., Validov, S., Lamers, G. E. M., de Weert, S., and Bloemberg, G. V. (2010). Genetic tools for tagging Gram-negative bacteria with mCherry for visualization in vitro and in natural habitats, biofilm and pathogenicity studies. FEMS Microbiol. Lett. 305, 81–90. doi: 10.1111/j.1574-6968.2010.01916.x
Lambertsen, L., Sternberg, C., and Molin, S. (2004). Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ. Microbiol. 6, 726–732. doi: 10.1111/j.1462-2920.2004.00605.x
Lau, B. T. C., Malkus, P., and Paulsson, J. (2013). New quantitative methods for measuring plasmid loss rates reveal unexpected stability. Plasmid 70, 353–361. doi: 10.1016/j.plasmid.2013.07.007
Ledermann, R., Bartsch, I., Remus-Emsermann, M. N., Vorholt, J. A., and Fischer, H.-M. (2015). Stable fluorescent and enzymatic tagging of Bradyrhizobium diazoefficiens to analyze host-plant infection and colonization. Mol. Plant Microbe Interact. 28, 959–967. doi: 10.1094/MPMI-03-15-0054-TA
Lin, L., Guo, W., Xing, Y., Zhang, X., Li, Z., Hu, C., et al. (2012). The actinobacterium Microbacterium sp. 16SH accepts pBBR1-based pPROBE vectors, forms biofilms, invades roots, and fixes N2 associated with micropropagated sugarcane plants. Appl. Microbiol. Biotechnol. 93, 1185–1195. doi: 10.1007/s00253-011-3618-3
Liu, H., Bouillaut, L., Sonenshein, A. L., and Melville, S. B. (2013). Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J. Bacteriol. 195, 629–636. doi: 10.1128/JB.01288-12
McKenzie, G. J., and Craig, N. L. (2006). Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 6:39. doi: 10.1186/1471-2180-6-39
Miller, W. G., Leveau, J. H., and Lindow, S. E. (2000). Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant Microbe Interact. 13, 1243–1250. doi: 10.1094/MPMI.2000.13.11.1243
Million-Weaver, S., Alexander, D. L., Allen, J. M., and Camps, M. (2012). Quantifying plasmid copy number to investigate plasmid dosage effects associated with directed protein evolution. Methods Mol. Biol. 834, 33–48. doi: 10.1007/978-1-61779-483-4_3
Monier, J.-M., and Lindow, S. E. (2005). Spatial organization of dual-species bacterial aggregates on leaf surfaces. Appl. Environ. Microbiol. 71, 5484–5493. doi: 10.1128/AEM.71.9.5484-5493.2005
Nakai, J., Ohkura, M., and Imoto, K. (2001). A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 19, 137–141. doi: 10.1038/84397
Nilsson, M., Christiansen, N., Høiby, N., Twetman, S., Givskov, M., and Tolker-Nielsen, T. (2014). A mariner transposon vector adapted for mutagenesis in oral streptococci. Microbiologyopen 3, 333–340. doi: 10.1002/mbo3.171
Parks, A. R., and Peters, J. E. (2007). Transposon Tn7 is widespread in diverse bacteria and forms genomic islands. J. Bacteriol. 189, 2170–2173. doi: 10.1128/JB.01536-06
Peredo, E. L., and Simmons, S. L. (2017). Leaf-FISH: microscale imaging of bacterial taxa on phyllosphere. Front. Microbiol. 8:2669. doi: 10.3389/fmicb.2017.02669
Peters, J. (2015). “Tn7”, in Mobile DNA III, eds N. Craig, M. Chandler, M. Gellert, A. Lambowitz, P. Rice, S. Sandmeyer (Washington, DC: ASM Press), 647–667. doi: 10.1128/microbiolspec.MDNA3-0010-2014
Ramirez-Mata, A., Pacheco, M. R., Moreno, S. J., Xiqui-Vazquez, M. L., and Baca, B. E. (2018). Versatile use of Azospirillum brasilense strains tagged with egfp and mCherry genes for the visualization of biofilms associated with wheat roots. Microbiol. Res. 215, 155–163. doi: 10.1016/j.micres.2018.07.007
Remus-Emsermann, M. N. P., Gisler, P., and Drissner, D. (2016a). MiniTn7-transposon delivery vectors for inducible or constitutive fluorescent protein expression in Enterobacteriaceae. FEMS Microbiol. Lett. 363:fnw178. doi: 10.1093/femsle/fnw178
Remus-Emsermann, M. N. P., Schmid, M., Gekenidis, M.-T., Pelludat, C., Frey, J. E., Ahrens, C. H., et al. (2016b). Complete genome sequence of Pseudomonas citronellolis P3B5, a candidate for microbial phyllo-remediation of hydrocarbon-contaminated sites. Stand. Genomic Sci. 11:75. doi: 10.1186/s40793-016-0190-6
Remus-Emsermann, M. N. P., Kim, E. B., Marco, M. L., Tecon, R., and Leveau, J. H. J. (2013a). Draft genome sequence of the phyllosphere model bacterium Pantoea agglomerans 299R. Genome Announc. 1:e00036-13. doi: 10.1128/genomeA.00036-13
Remus-Emsermann, M. N. P., Kowalchuk, G. A., and Leveau, J. H. J. (2013b). Single-cell versus population-level reproductive success of bacterial immigrants to pre-colonized leaf surfaces. Environ. Microbiol. Rep. 5, 387–392. doi: 10.1111/1758-2229.12040
Remus-Emsermann, M. N. P., Lücker, S., Müller, D. B., Potthoff, E., Daims, H., and Vorholt, J. A. (2014). Spatial distribution analyses of natural phyllosphere-colonizing bacteria on Arabidopsis thaliana revealed by fluorescence in situ hybridization. Environ. Microbiol. 16, 2329–2340. doi: 10.1111/1462-2920.12482
Remus-Emsermann, M. N. P., and Schlechter, R. O. (2018). Phyllosphere microbiology: at the interface between microbial individuals and the plant host. New Phytol. 218, 1327–1333. doi: 10.1111/nph.15054
Reznikoff, W. S. (2008). Transposon Tn5. Annu. Rev. Genet. 42, 269–286. doi: 10.1146/annurev.genet.42.110807.091656
Rodriguez, M. D., Paul, Z., Wood, C. E., Rice, K. C., and Triplett, E. W. (2017). Construction of stable fluorescent reporter plasmids for use in Staphylococcus aureus. Front. Microbiol. 8:2491. doi: 10.3389/fmicb.2017.02491
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold spring harbor laboratory press.
Schada von Borzyskowski, L., Remus-Emsermann, M., Weishaupt, R., Vorholt, J. A., and Erb, T. J. (2015). A set of versatile brick vectors and promoters for the assembly, expression, and integration of synthetic operons in Methylobacterium extorquens AM1 and other alphaproteobacteria. ACS Synth. Biol. 4, 430–443. doi: 10.1021/sb500221v
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schmid, M., Frei, D., Patrignani, A., Schlapbach, R., Frey, J. E., Remus-Emsermann, M. N. P., et al. (2018). Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Res. 46, 8953–8965. doi: 10.1093/nar/gky726
Schmidt, H., Nunan, N., Höck, A., Eickhorst, T., Kaiser, C., Woebken, D., et al. (2018). Recognizing patterns: spatial analysis of observed microbial colonization on root surfaces. Front. Environ. Sci. Eng. 6:61. doi: 10.3389/fenvs.2018.00061
Simon, R., Priefer, U., and Pühler, A. (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology 1:784. doi: 10.1038/nbt1183-784
Smith, M. A., and Bidochka, M. J. (1998). Bacterial fitness and plasmid loss: the importance of culture conditions and plasmid size. Can. J. Microbiol. 44, 351–355. doi: 10.1139/w98-020
Smits, T. H. M., Rezzonico, F., Kamber, T., Blom, J., Goesmann, A., Frey, J. E., et al. (2010). Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol. Plant Microbe Interact. 23, 384–393. doi: 10.1094/MPMI-23-4-0384
Stewart, P. S., and Costerton, J. W. (2001). Antibiotic resistance of bacteria in biofilms. Lancet 358, 135–138. doi: 10.1016/S0140-6736(01)05321-1
Suarez, A., Güttler, A., Strätz, M., Staendner, L. H., Timmis, K. N., and Guzmán, C. A. (1997). Green fluorescent protein-based reporter systems for genetic analysis of bacteria including monocopy applications. Gene 196, 69–74. doi: 10.1016/S0378-1119(97)00197-2
Subach, O. M., Cranfill, P. J., Davidson, M. W., and Verkhusha, V. V. (2011). An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 6:e28674. doi: 10.1371/journal.pone.0028674
Summers, D. K. (1991). The kinetics of plasmid loss. Trends Biotechnol. 9, 273–278. doi: 10.1016/0167-7799(91)90089-Z
Tecon, R., and Leveau, J. H. J. (2012). The mechanics of bacterial cluster formation on plant leaf surfaces as revealed by bioreporter technology. Environ. Microbiol. 14, 1325–1332. doi: 10.1111/j.1462-2920.2012.02715.x
Tolker-Nielsen, T., and Molin, S. (2000). Spatial organization of microbial biofilm communities. Microb. Ecol. 40, 75–84.
Valm, A. M., Mark Welch, J. L., and Borisy, G. G. (2012). CLASI-FISH: principles of combinatorial labeling and spectral imaging. Syst. Appl. Microbiol. 35, 496–502. doi: 10.1016/j.syapm.2012.03.004
Vidal, J. E., Chen, J., Li, J., and McClane, B. A. (2009). Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS One 4:e6232. doi: 10.1371/journal.pone.0006232
Whitaker, W. R., Shepherd, E. S., and Sonnenburg, J. L. (2017). Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell 169, 538.e12–546.e12. doi: 10.1016/j.cell.2017.03.041
Winterberg, K. M., Luecke, J., Bruegl, A. S., and Reznikoff, W. S. (2005). Phenotypic screening of Escherichia coli K-12 Tn5 insertion libraries, using whole-genome oligonucleotide microarrays. Appl. Environ. Microbiol. 71, 451–459. doi: 10.1128/AEM.71.1.451-459.2005
Zengerer, V., Schmid, M., Bieri, M., Müller, D. C., Remus-Emsermann, M. N. P., Ahrens, C. H., et al. (2018). Pseudomonas orientalis F9: a potent antagonist against phytopathogens with phytotoxic effect in the apple flower. Front. Microbiol. 9:145. doi: 10.3389/fmicb.2018.00145
Keywords: fluorophore, fluorescent labeling, tagging, Tn5, Tn7, transposon
Citation: Schlechter RO, Jun H, Bernach M, Oso S, Boyd E, Muñoz-Lintz DA, Dobson RCJ, Remus DM and Remus-Emsermann MNP (2018) Chromatic Bacteria – A Broad Host-Range Plasmid and Chromosomal Insertion Toolbox for Fluorescent Protein Expression in Bacteria. Front. Microbiol. 9:3052. doi: 10.3389/fmicb.2018.03052
Received: 14 September 2018; Accepted: 27 November 2018;
Published: 12 December 2018.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, ChinaReviewed by:
Yongtao Zhu, Minnesota State University, Mankato, United StatesCopyright © 2018 Schlechter, Jun, Bernach, Oso, Boyd, Muñoz-Lintz, Dobson, Remus and Remus-Emsermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitja N. P. Remus-Emsermann, bWl0amEucmVtdXMtZW1zZXJtYW5uQGNhbnRlcmJ1cnkuYWMubno=; bWl0amEucmVtdXMuZW1zZXJtYW5uQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.