
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 November 2018
Sec. Infectious Agents and Disease
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02895
 Zhihong Xu1
Zhihong Xu1 Aiping Zhou2
Aiping Zhou2 Jiawei Wu3
Jiawei Wu3 Aiwu Zhou3Jun Li4Shulin Zhang1
Aiwu Zhou3Jun Li4Shulin Zhang1 Wenjuan Wu2
Wenjuan Wu2 Petros C. Karakousis5
Petros C. Karakousis5 Yu-Feng Yao1,2*
Yu-Feng Yao1,2*Multidrug-resistant tuberculosis (TB), defined as TB resistant to the two first-line drugs, isoniazid and rifampin, is a serious challenge to global TB eradication efforts. Although mutations in rpoA or rpoC have been proposed to compensate for this fitness cost due to rpoB mutation in rifampicin-resistant Mycobacterium tuberculosis mutants, whether the compensatory effect exists and the underlying mechanisms of compensation remain unclear. Here, we used RNA sequencing to investigate the global transcriptional profiles of 6 rifampin-resistant clinical isolates with either single mutation in rpoB or dual mutations in rpoB/rpoC, as well as 3 rifampin-susceptible clinical isolates, trying to prove the potential compensatory effect of rpoC by transcriptomic alteration. In rifampin-free conditions, rpoC mutation was associated with M. tuberculosis upregulation of ribosomal protein-coding genes, dysregulation of growth-related essential genes and balancing the expression of arginine and glutamate synthesis-associated genes. Upon rifampin exposure of M. tuberculosis isolates, rpoC mutations were associated with the upregulation of the oxidative phosphorylation machinery, which was inhibited in the rpoB single mutants, as well as stabilization of the expression of rifampin-regulated essential genes and balancing the expression of genes involved in metabolism of sulfur-containing amino acids. Taken together, our data suggest that rpoC mutation may compensate for the fitness defect of rifampicin-resistant M. tuberculosis by altering gene expression in response to rifampin exposure.
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis and remains a major global health problem due to its high burden and death worldwide. Multidrug-resistant TB (MDR TB), defined as TB resistant to isoniazid and rifampin, is a continuing threat, with an estimated 600,000 new cases that were resistant to rifampin, the most effective first-line anti-TB drug (Vasilyeva et al., 2017).
Rifampin kills M. tuberculosis by binding to the β subunit of RNA polymerase, encoded by rpoB, where it exerts an early block of elongation of 2–3 nucleotide-long short RNA transcripts, thus inhibiting transcription (Campbell et al., 2001). Resistance to rifampin is primarily conferred by mutations in rpoB, which alter drug binding. Over 95% of rifampin-resistant M. tuberculosis clinical isolates harbor a mutation within the 81-bp region of rpoB known as the rifampin resistance-determining region (RRDR) (Ramaswamy and Musser, 1998), among which the most commonly observed mutations are S450L and H445Y, leading to high-level rifampin resistance (Mokrousov, 2004; Sandgren et al., 2009).
Given the essentiality of RNA polymerase for bacterial transcriptional processes, it stands to reason that mutations in rpoB could have profound effects on M. tuberculosis physiology or pathogenesis. Previous studies have reported that even in the absence of drug pressure, rpoB mutations confer a “fitness cost” in laboratory-derived strains of M. tuberculosis, including reduced growth rate and virulence compared to their drug-susceptible counterparts (Lloyd, 1987; Mariam et al., 2004; Gagneux et al., 2006; Rifat et al., 2017). However, Gagneux et al. (2006) reported previously that the same resistance mutations in rpoB are not necessarily associated with a fitness cost in M. tuberculosis clinical isolates, perhaps due to the presence of compensatory mutations, as have been reported in rifampin-resistant Escherichia coli (Reynolds, 2000). Specifically, mutations in the rpoC gene, encoding the β’ subunit of RNA polymerase, were associated with altered transcript elongation rate and gene expression, leading to metabolic changes and increased growth rate in rifampin-resistant E. coli (Conrad et al., 2010). Up to one third of rifampin-resistant M. tuberculosis rpoB mutant strains were found to harbor mutations in rpoC or rpoA, encoding the RNA polymerase α subunit (Comas et al., 2011). Song et al. (2014) reported that M. tuberculosis RpoC F452L and V483G alleles restored the transcription efficiency of RNA polymerase bearing RpoB S450L mutation. Although these findings suggest potential compensatory effects of rpoA or rpoC mutations for fitness costs associated with rpoB mutations in M. tuberculosis, the compensatory effect needs to be validated and the underlying molecular mechanisms remain to be elucidated.
In this study, we used next-generation sequencing to characterize the genomes and transcriptomes (with and without rifampin exposure) of 6 rifampin-resistant clinical isolates of M. tuberculosis, including 3 isolates with a single mutation in the rpoB and 3 isolates harboring rpoB and rpoC dual mutations, as well as 3 rifampin-susceptible isolates as reference. A list of differentially expressed genes (DEGs) was generated by comparing rpoB single mutant or rpoBC mutant isolates vs. drug-susceptible isolates before or after rifampin exposure. Dysregulated genes were further filtered by removing those displaying identical transcriptional regulation between the rpoB and rpoBC groups, as well as genes with missense mutations in open reading frames. The resulting gene candidate list was categorized using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Ogata et al., 1999) to identify metabolic pathways potentially critical for restoration of M. tuberculosis fitness in the context of rpoB mutation.
Mycobacterium tuberculosis isolates were collected from Suzhou Fifth People’s Hospital and Wuxi Fifth People’s Hospital between September 2013 and February 2014. Isolates were grown in Middlebrook 7H9 broth (Martin et al., 1989) supplemented with 10% Oleic acid-Albumin-Dextrose-Catalase, 0.5% glycerol, 0.05% Tween-80 at 37°C without shaking. Rifampin was added at 80 μg/mL.
Drug susceptibility testing of all the strains was performed as recommended by WHO/IUATLD (International union for tuberculosis and lung disease), as previously described (Ma et al., 2018), using the four first-line anti-TB drugs (ethambutol, isoniazid, rifampin, and streptomycin) and four second-line anti-TB drugs (amikacin, levofloxacin para-aminosalicylic acid, and prothionamide). The final concentrations of drugs in Lowenstein-Jensen media were as follows: isoniazid (ISN) 0.2 μg/mL, ethambutol (EMB) 2.0 μg/mL, rifampin (RIF) 40.0 μg/mL, streptomycin (STR) 4.0 μg/mL, amikacin (AMK) 100 μg/mL, levoflooxacin (LEV) 2.0 μg/mL, para-aminosalicylic acid (PA) 1.0 μg/mL, and prothionamide (PT) 25.0 μg/mL. The strain was considered to be resistant to the specific drug when the growth rate was >1% compared to the control group (without any drugs).
Genomic DNA was extracted from M. tuberculosis isolates as previously described (Song et al., 2014). Identification of the Beijing family was performed for all the strains collected in this study using the RD105 deletion-targeted multiplex PCR (DTM-PCR) method (Chen et al., 2007). The strains with no RD105 region amplification were classified as Beijing genotype, while the others containing RD105 region were classified as non-Beijing genotype.
Mycobacterium tuberculosis isolates were grown to mid-logarithmic phase, and DNA was isolated using Cetrimonium bromide as previously described (Song et al., 2014). Genome sequencing was performed by the Chinese National Human Genome Center in Shanghai using Illumina HiSeq 2500. Raw reads were aligned to the reference M. tuberculosis H37Rv genome (Camus et al., 2002) and assembled using Velvet version 1.2.03. Protein-coding genes were predicted using Glimmer version 3.02, while tRNA and rRNA were identified using tRNAscan-SE version 2.0 and RNAmmer version 1.2, respectively. Pairwise-alignment according to the reference M. tuberculosis H37Rv genome was performed using R package Biostrings version 2.46.0 to identify single nucleotide polymorphisms (SNPs) and insertion/deletions (INDELs). All genome sequence data could be accessible in NCBI BioProject database (Accession No. PRJNA497952, PRJNA497955, PRJNA497954, PRJNA497944, PRJNA497947, PRJNA497946, PRJNA497950, PRJNA497943, and PRJNA497939).
The atomic model of the M. tuberculosis RNA polymerase transcription initiation complex (TIC) was used for modeling (RCSB accession number: 6C04) (Boyaci et al., 2018). The mutations I491V, G594E, and A734V were then mapped onto the three-dimensional structure of the M. tuberculosis RNA polymerase and visualized using PyMOL version 2.1.
Mycobacterium tuberculosis isolates were grown to mid-logarithmic phase in the presence or absence of rifampin as indicated. Total RNA extraction was performed using the RiboPure-Bacteria Kit (Ambion) according to the manufacturer’s recommendations with modifications (Ren et al., 2016). RNA sequencing was performed on an Illumina HiSeq 2500 using the NEBNext Ultra RNA Library Prep Kit. Unsupervised hierarchical clustering and heatmap representation of genome-wide expression profiles of 9 clinical isolates by RNA-Seq were performed in R package gplots using Reads Per Kilobase per Million mapped reads (RPKM). Differential gene expression analysis was performed using R package DEGseq version 1.32.0 and processed using MA-plot-based method with random sampling model. RNA-Seq raw data were deposited in NCBI Sequence Read Archive (SRA) database (Accession No. SRR8149449, SRR8149447, SRR8149496, SRR8149483, SRR8149500, SRR8149502, SRR8149437, SRR8149444, SRR8149498, SRR8149478, and SRR8149497).
To ensure that the results of network construction were reliable, outlier samples were removed. An appropriate soft threshold power was selected in accordance with standard scale-free networks, with which adjacencies between all differential genes were calculated by a power function. Then, the adjacency was transformed into a topological overlap matrix (TOM), and the corresponding dissimilarity (1-TOM) was calculated. Module identification was accomplished with the dynamic tree cut method by hierarchically clustering genes using 1-TOM as the distance measure with a deepsplit value of 2 and a minimum size cutoff of 30 for the resulting dendrogram. Highly similar modules were identified by clustering and then merged together with a height cut-off of 0.42. To test the stability of each identified module, module preservation and quality statistics were computed with the module preservation function implemented in the R package WGCNA.
Statistical analysis of data was performed using GraphPad Prism version 6. Data were presented as mean values with SD. DEGs were identified with a Benjamini-Hochberg FDR < 0.05. Functional enrichment analysis of DEGs was performed according to KEGG and visualized using R package clusterProfiler version 3.6.0. Cutoff of P-value was set to 1 to obtain all the annotated information in the KEGG database. Results were visualized by barplot or dotplot. Statistical significant differences was determined by Student’s t-test or two-way ANOVA among different groups. P-values < 0.05 denoted statistical significance.
The study was approved by the ethical review boards of Suzhou Fifth People’s Hospital and Wuxi Fifth People’s Hospital.
Initially, 16 M. tuberculosis clinical isolates were evaluated for inclusion in this study. In order to gain primary data on these isolates and optimize group matching, antimicrobial phenotypic susceptibility testing, genotyping and rifampin resistance-associated gene sequencing were performed. As shown in Supplementary Table S1, susceptibility tests toward first-line and second-line anti-TB agents identified 9 rifampin-susceptible isolates and 7 rifampicin-resistant isolates. Genotyping (Supplementary Figure S1) revealed 11 Beijing family isolates and 5 non-Beijing family isolates. Sequence analyses of the rpoA, rpoB and rpoC genes for each isolate are listed in Supplementary Table S2. To optimize the paired design, isolates were classified into three groups, rifampin-resistant isolates with rpoB single mutation, rifampin-resistant isolates with rpoB/rpoC (rpoBC) double mutations and rifampin-susceptible isolates. To balance the sample size, three isolates were assigned to each group. The genotype of each isolate was also screened to ensure that both Beijing and non-Beijing genotype would be included in each group. Consequently, 6 rifampin-resistant isolates (3 rpoB single mutant and 3 rpoBC mutant isolates) and 3 rifampin-susceptible isolates were selected and subjected to genome sequencing. Characteristics, genome sequencing information and predicted ORFs of these 9 isolates are summarized in Table 1 and Supplementary Tables S3, S4, respectively.
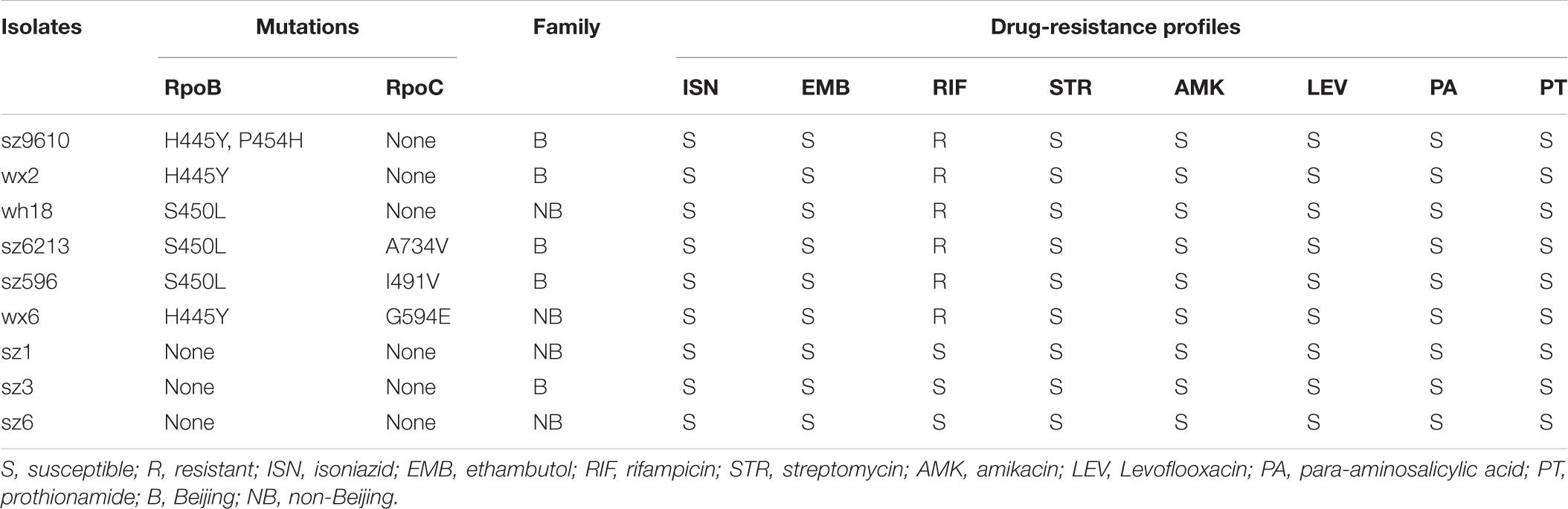
TABLE 1. Characteristics of clinical M. tuberculosis isolates used for this study.
Since the RNA polymerase subunits RpoA, RpoB, and RpoC interact with each other (Luo et al., 2005), we hypothesized that non-synonymous polymorphisms in rpoC occurring in rpoB mutant isolates were likely to be compensatory (Zhang et al., 1999). We then mapped the three amino acid substitutions of RpoC onto the three-dimensional structure of the M. tuberculosis RNA polymerase (Boyaci et al., 2018). As shown in Figure 1, I491V, also reported by Comas et al. (2011), is located at the interface between the α and β’ subunits; A734V is near the interface between the β and β’ subunits; G594E maps to a region between the two α helices, and the substitution of glycine with glutamate changes the charge of the side chain from neutral to negative. Interestingly, Stefan et al. recently reported similar findings that F452L and V483G, two mutants of RpoC near I491V, displayed a compensatory effect on RpoB S450L mutated RNA polymerase in vitro by recovering the defects of open-promoter complex stability and elongation rate, enhancing termination efficiency and RNA primer hydrolysis (Stefan et al., 2018). Their work demonstrates the possibility of and underlying mechanism of mutations in RpoC compensating for functional defects of RNA polymerase resulting from mutations in RpoB. Therefore, we speculate that the 3 mutations we identified also probably affect the interaction between the subunits of RNA polymerase, which requires confirmation in future studies.
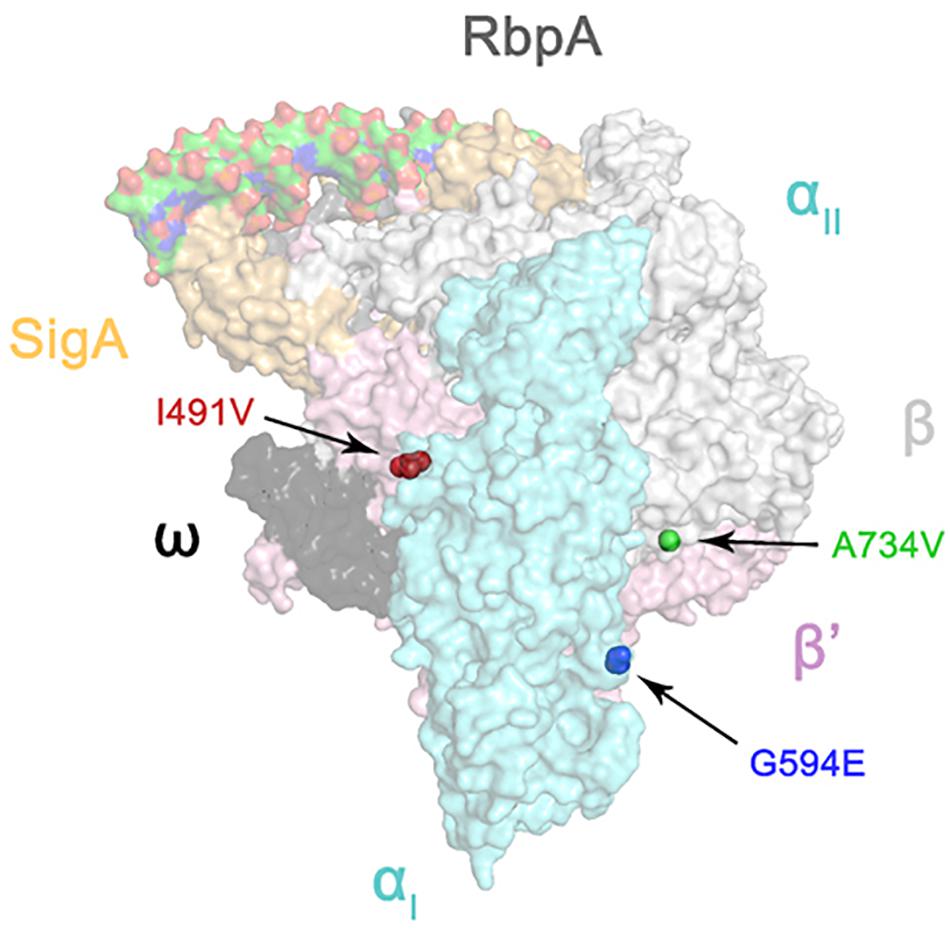
FIGURE 1. Three-dimensional representation of rpoC mutations on the atomic model of the M. tuberculosis RNA polymerase (represented by transparent molecular surfaces). The enzyme is depicted as α subunit (cyan), β subunit (white), β′ subunit (pink), ω subunit (black), SigA (yellow), and RbpA (gray). Compensatory mutations on β′ subunits are highlighted as red, blue, and green sphere pointed by arrow, respectively.
Since rpoC mutations alter the kinetic parameters of RNA polymerase in E. coli (Conrad et al., 2010), we hypothesized that rpoC mutations may similarly influence gene transcription in rifampin-resistant M. tuberculosis. To test this hypothesis, we performed RNA sequencing to determine the transcriptional profiles of the 9 clinical isolates in rifampin-free conditions.
We first performed unsupervised hierarchical clustering analysis to determine whether rpoC mutations affect the genome-wide expression profiles of rifampin-resistant isolates harboring rpoBC double mutations. The expression profiles of rifampin-resistant isolates harboring rpoB single mutation (sz9610, wx2, and wh18), rifampin-resistant isolates with rpoBC double mutations (sz6213, sz596, and wx6) and rifampin-susceptible isolates (sz1, sz3, and sz6) were subjected to clustering analysis and presented as heatmap using RPKM (Supplementary Figure S2). The expression profiles of 9 isolates were generally divided into two groups, rpoBC double mutants (sz596 and sz6213) and rifampin-susceptible isolates (sz1 and sz6) were clustered in one group, the other 5 isolates including rpoB single mutants (sz9610, wx2, and wh18), rpoBC double mutants (wx2) and rifampin-susceptible isolates (sz3) were clustered in the other group. These results suggest that rpoBC double mutants have more similar expression profiles to rifampin-susceptible isolates compared with rpoB single mutants.
Then differential gene expression analysis was performed for the following pairs: rpoB single mutant isolates vs. drug-susceptible isolates, and rpoBC isolates vs. drug-susceptible isolates. Supplementary Tables S5, S6 contains a summary of the dysregulated genes for each comparison.
To identify dysregulated genes associated with rpoC mutation, the genes were filtered using the following criteria: (1) Differential regulation in the comparison of rpoB single mutant isolates vs. drug-susceptible isolates and rpoBC isolates vs. drug-susceptible isolates; (2) Differential regulation with fold change ≥1.5 of at least one of 3 isolates in either the rpoB or rpoBC groups. A total of 474 genes that meet both criteria were subjected to further analyses.
M. tuberculosis has evolved significant genomic diversity, including multiple SNPs or INDELs, to adapt to the diverse environment encountered during natural infection of the human host (O’Neill et al., 2015). Therefore, to exclude the potential influence of genetic variation on transcription, comparative genomic analyses were performed to identify SNPs or INDELs in all 474 genes. These SNPs are listed in Supplementary Table S7. Genes with missense mutations were discarded. Finally, 360 DEGs between the rpoB and rpoBC groups were subjected to further study.
To explore the biological function of dysregulated genes associated with rpoC mutation, we performed KEGG enrichment analysis of the above 360 genes. 93 of these were enriched in 76 pathways (Supplementary Figure S3 and Figure 2A). The top 5 pathways represented include genes related to ribosome synthesis, arginine biosynthesis, vancomycin resistance, steroid degradation and sulfur metabolism.
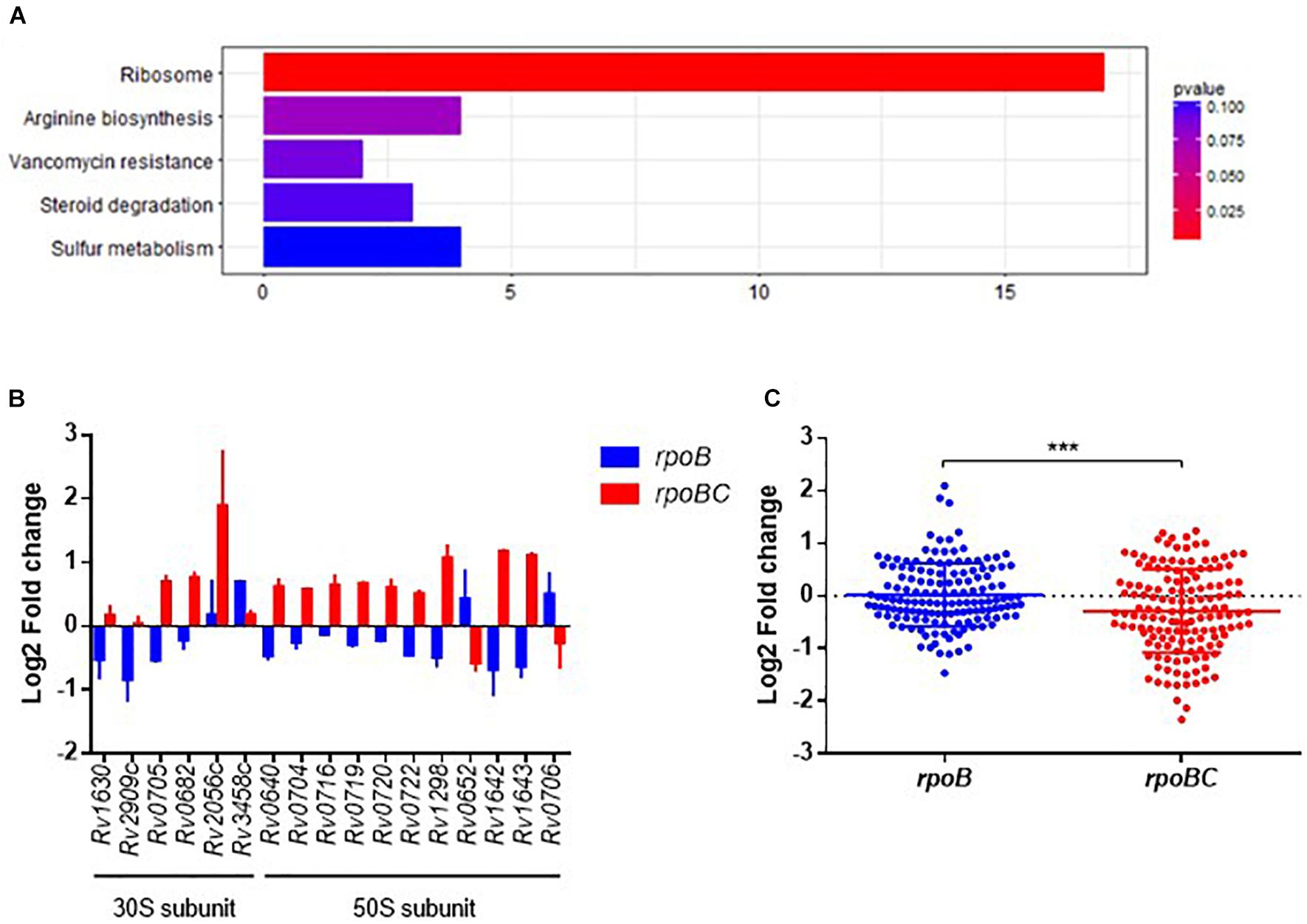
FIGURE 2. Global expression changes due to rpoC mutations in rifampicin-free conditions. (A) Top 5 KEGG enrichment of dysregulated genes. Genes that differentially regulated between comparisons of rpoB-mutated isolates vs. drug-susceptible isolates, rpoBC-mutated isolates vs. drug-susceptible isolates with fold change ≥1.5 of at least one of three isolates in either group remained. Genes with missense mutations were discarded. (B) Expression of dysregulated genes enriched in ribosome pathway. Fold change represents the average from two isolates in each group. (C) Fold change distribution of dysregulated essential genes due to rpoC mutations in rifampicin-free conditions. Dots represent the fold change of each gene from two isolates in each group. Student’s t-test, ∗∗∗P = 0.0002.
The most significantly enriched pathway was related to ribosomes (FDR = 0.0005, Figure 2A). As shown in Figure 2B, 17 genes (six 30S subunit-coding genes, eleven 50S subunit-coding genes) are assigned to this pathway. For example, relative to rifampin-susceptible isolates, Rv1630/rpsA and Rv2909c/rpsP were downregulated in the rpoB group and Rv0640/rplK, Rv0720/rplR, Rv0704/rplB, Rv0722/rpmD, Rv1298/rpmE, Rv0716/rplE, and Rv0719/rplF were upregulated in the rpoBC group. Rv1643/rplT and Rv1642/rpmI showed reduced expression in the rpoB group but increased expression in the rpoBC group compared to rifampin-susceptible isolates.
Sassetti et al. have identified 830 genes that are required for the in vitro growth and in vivo survival of M. tuberculosis (Sassetti and Rubin, 2003; Sassetti et al., 2003). To explore the potential role of rpoC mutation in mycobacterial growth, we investigated whether expression of these growth-required essential genes is affected by rpoC mutation. A total of 72 essential genes were differentially regulated in either the rpoB or rpoBC groups compared to rifampin-susceptible isolates (Supplementary Figure S4). Specifically, 20 genes were significantly upregulated and 9 genes were significantly downregulated in the rpoB group relative to rifampin-susceptible isolates, and 15 genes were significantly upregulated and 31 genes were significantly downregulated in the rpoBC group (Figure 2C).
To further screen for key dysregulated genes associated with rpoC mutation, a weighted gene co-expression network analysis (WGCNA) was applied to construct the gene co-expression networks among 360 genes differentially regulated in the 6 rifampin-resistant isolates. As shown in Figure 3A, we identified three co-expressed modules (110 genes in blue, 195 genes in royalblue and 55 genes in gray modules) within which genes displayed a similar expression pattern. Among these modules, dysregulation of genes within the royalblue module showed the closest relation to rpoB (R = 0.83, P = 0.04) and rpoBC (R = -0.83, P = 0.04) mutation isolates (Figure 3B). KEGG enrichment analysis was then performed on 195 genes in the royalblue module. The top 5 pathways represented include arginine biosynthesis, homologous recombination, fluorobenzoate degradation, toluene degradation, and ketone body synthesis and degradation (Figure 3C).
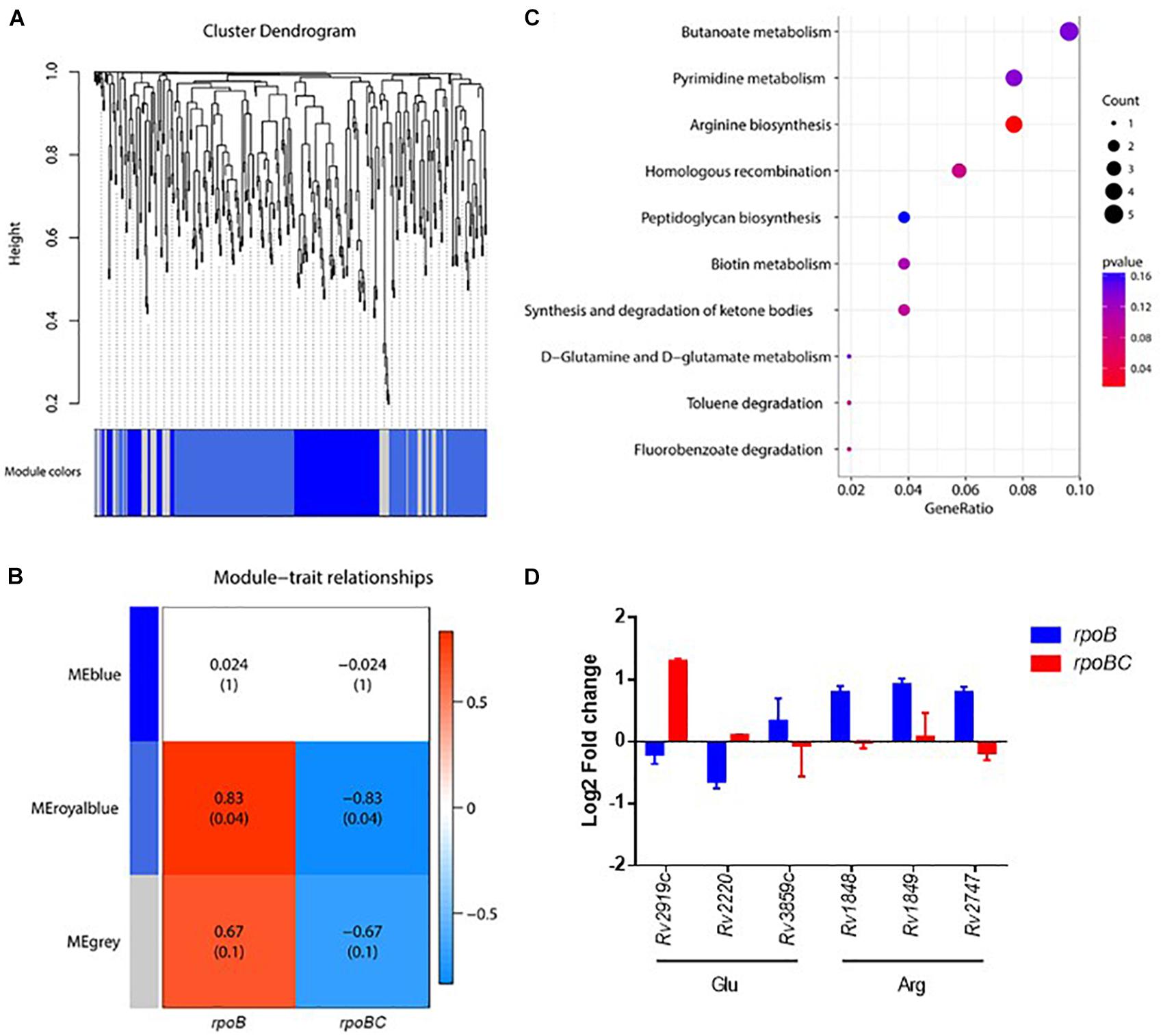
FIGURE 3. Co-expression networks of dysregulated genes due to rpoC mutations in rifampicin-free conditions. (A) Dendrogram of all differentially expressed genes clustered. Gene clustering tree (dendrogram) obtained by hierarchical clustering of adjacency-based dissimilarity. The colored row below the dendrogram indicates module membership identified by the dynamic tree cut method. Three kinds of color present three modules. (B) Correlation between each clustered module and mutation. Correlation index and P-value (parenthesized) are listed. Heatmap of the correlation between module eigengenes (MEs) and different group information (rpoB or rpoBC). (C) KEGG pathway enrichment analyses for genes in the royalblue module. The x-axis shows the ratio number of genes and the y-axis shows the KEGG pathway terms. The P-value of each term is colored according to the legend. (D) Expression of genes in glutamate and arginine synthesis. Fold change represents the average from two isolates in each group.
The arginine biosynthesis pathway was significantly enriched (P = 0.0114) (Figure 3D). Rv1848/ureA (encoding the urease γ subunit), Rv1849/ureB (encoding the urease β subunit), Rv2747/argA (encoding an N-acetylglutamate synthase) were upregulated in the rpoB group. Rv2220/glnA1 (encoding a glutamine synthetase) was downregulated in the rpoB group. These results suggest that rpoC mutation is responsible for stabilizing the expression of arginine synthesis-associated genes. Since arginine synthesis begins with the N-acetylation of L-glutamate in prokaryotes (Glansdorff and Xu, 2006), we therefore investigated whether genes involved in the synthesis of its precursor L-glutamate were also differentially regulated. As expected, Rv2919c/glnK (encoding a nitrogen assimilation regulatory protein, inhibiting ammonium uptake) was downregulated in the rpoB group but upregulated in the rpoBC group. Rv3859c/gltB (encoding the subunit of glutamine oxoglutarate aminotransferase for glutamate synthesis) was upregulated in the rpoB group (Figure 3D).
Since M. tuberculosis gene expression is altered following exposure to rifampin (Campbell et al., 2001), we next identified genes whose expression were affected by rifampin in the rpoB group but were reversed to homeostasis due to the presence of rpoC mutation. Differential gene expression analysis was performed pair-wise, i.e., pre-exposure vs. post-exposure of rpoB single mutant isolates and rpoBC mutant isolates, respectively. The numbers of dysregulated genes are summarized in Supplementary Tables S8, S9. To identify genes whose expression is dysregulated by rpoC mutation, the genes were further filtered using the same criteria as used for analysis in rifampin-free conditions. A total of 435 differentially regulated genes were identified. SNPs of these genes are listed in Supplementary Table S10.
KEGG enrichment analysis was then applied to obtain the functional annotation of these dysregulated genes. As a result, 120 of 435 candidates were enriched in 76 pathways (Figure 4A and Supplementary Figure S5). The top 5 pathways represented include oxidative phosphorylation, bacterial secretion systems, sulfur relay systems, protein export, and pathways involved in TB pathogenesis and host immune responses.
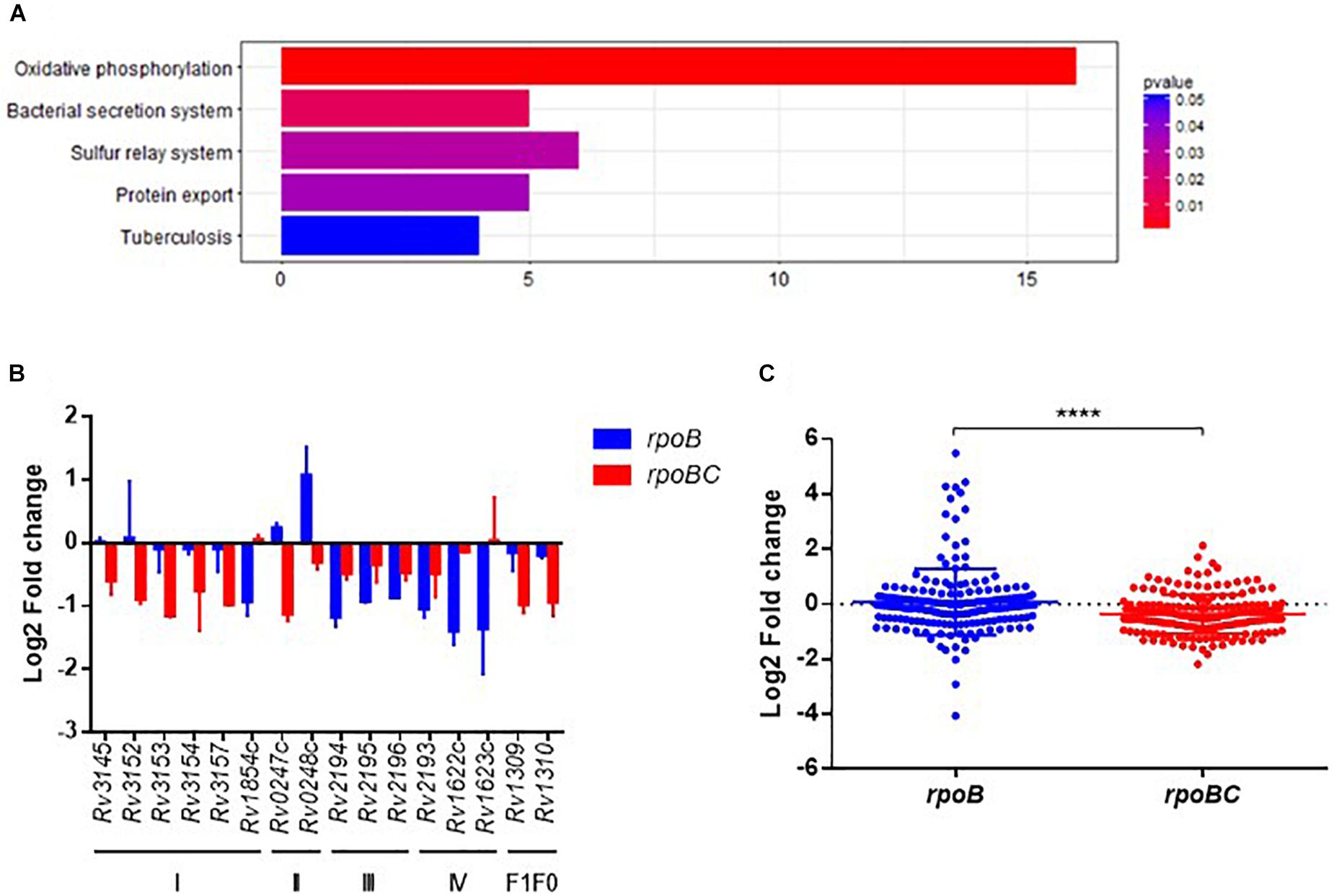
FIGURE 4. Global expression changes due to rpoC mutations after rifampicin stimulation. (A) Top 5 KEGG enrichment of dysregulated genes. Genes that differentially regulated between comparisons of pre-stimulation vs. post-stimulation of rpoB-mutated isolates and rpoBC-mutated isolates with fold change ≥1.5 of at least one of three isolates in either group remained. Genes with missense mutations were discarded. (B) Expression of dysregulated genes enriched in oxidative phosphorylation pathway. Fold change represents the average from two isolates in each group. (C) Fold change distribution of dysregulated essential genes due to rpoC mutations after rifampicin stimulation. Dots represent the fold change of each gene from two isolates in each group. ∗∗∗∗p < 0.01.
The Oxidative phosphorylation pathway was significantly enriched (FDR = 0.0016, Figure 4A). As demonstrated in Figure 4B, 16 genes are categorized to this pathway and can be sorted into several subsets of oxidative phosphorylation machinery. For example, in complex I, Rv3145/nuoA, Rv3152/nuoH, Rv3153/nuoI, Rv3154/nuoJ, Rv3157/nuoM [encoding a type I NADH dehydrogenase (Weinstein et al., 2005)] were downregulated in rpoBC group. Rv1854c/ndh [encoding a type II NADH dehydrogenase (Weinstein et al., 2005)] was downregulated in rpoB group.
Among the essential genes (Sassetti and Rubin, 2003; Sassetti et al., 2003), 91 genes were differentially expressed between the rpoB and rpoBC groups (Supplementary Figure S6). The global transcription profiles of the two groups are significantly different (Figure 4C). Thus, 14 genes were upregulated and 21 genes were downregulated in the rpoB group. 12 genes were upregulated and 47 genes were downregulated in the rpoBC group. However, the fold changes in expression of dysregulated genes in the rpoBC group are smaller than those in the rpoB group.
Key dysregulated genes associated with rpoC mutation were identified through gene construction of co-expression networks based on expression of 435 candidates. As shown in Figures 4, 5A co-expressed modules (107 genes in midnightblue, 163 genes in blue, 113 genes in royalblue and 52 genes in gray module) were identified. Dysregulated genes within the royalblue module showed the closest relation in rpoB (R = 0.8, P = 0.05) and rpoBC (R = -0.8, P = 0.05) mutant isolates (Figure 5B). The top 5 pathways emerging from KEGG enrichment analysis of the 113 genes in the royalblue module were oxidative phosphorylation, ABC transporters, sulfur relay system, cationic antimicrobial peptide resistance, and sulfur metabolism (Figure 5C).
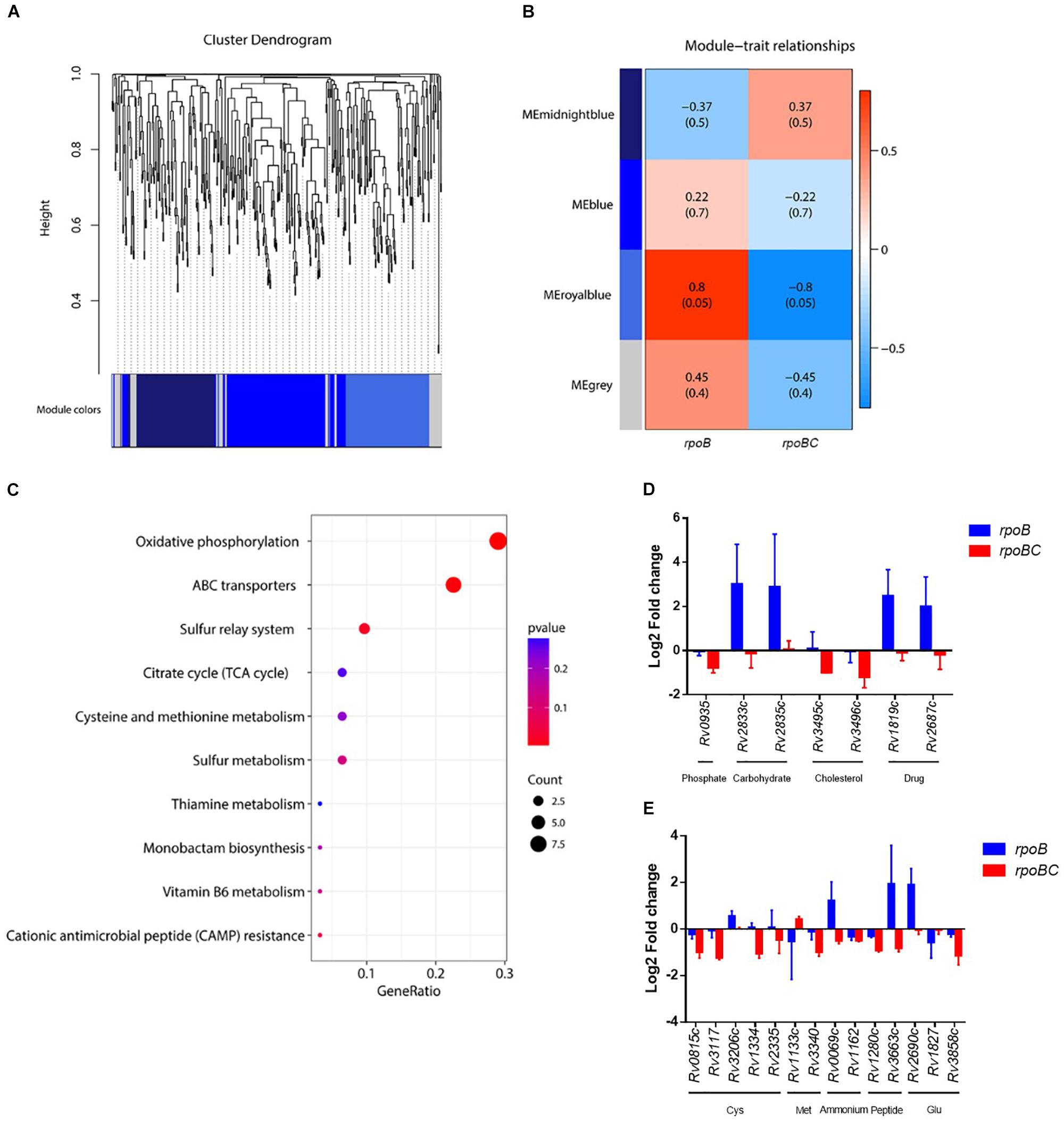
FIGURE 5. Co-expression networks of dysregulated genes due to rpoC mutations after rifampicin stimulation. (A) Dendrogram of all differentially expressed genes clustered. Gene clustering tree (dendrogram) obtained by hierarchical clustering of adjacency-based dissimilarity. The colored row below the dendrogram indicates module membership identified by the dynamic tree cut method. Four kinds of color present four modules. (B) Correlation between each clustered module and mutation. Correlation index and P-value (parenthesized) are listed. Heatmap of the correlation between module eigengenes (MEs) and different group information (rpoB or rpoBC). (C) KEGG pathway enrichment analyses for genes in the royalblue module. The x-axis shows the ratio number of genes and the y-axis shows the KEGG pathway terms. The P-value of each term is colored according to the legend. (D) Expression of genes coding ABC transporters. Fold change represents the average from two isolates in each group. (E) Expression of genes in cysteine and methionine metabolism. Fold change represents the average from two isolates in each group.
Several ABC transporters (Figure 5D) were found to be dysregulated in each mutant group. For example, Rv0935/pstC1 (encoding the subunit of an ABC transporter for phosphate uptake) was downregulated in the rpoBC group. Rv2833c/ugpB and Rv2835c/ugpA (encoding the subunits of an ABC transporter for carbohydrate uptake) were upregulated in the rpoB group. Rv3495c/lprN and Rv3496c/mce4D (encoding the subunits of an ABC transporter for cholesterol uptake) were downregulated in the rpoBC group. Rv1819c and Rv2687c (each encoding the subunit of a drug efflux pump) were upregulated in the rpoB group.
In the sulfur relay system pathway (Figure 5E), Rv0815c/cysA2 and Rv3117/cysA3 (each encoding a thiosulfate sulfurtransferase) were downregulated in the rpoBC group. Rv3206c/moeZ (encoding an adenylyltransferase) was upregulated in the rpoB group. Considering that proteins encoded by these genes are involved in sulfur assimilation, their reduced transcription levels in the rpoBC group compared to the rpoB group suggest that rpoC mutation might reduce sulfur assimilation following rifampin exposure.
Sulfur is mainly assimilated to synthesize cysteine and methionine in mycobacteria (Pinto et al., 2007). We therefore investigated whether genes in their synthesis were also dysregulated in the rpoB and rpoBC mutant isolates. As shown in Figure 5E, in the cysteine synthesis pathway, Rv1334/mec (encoding a Zn2+-dependent hydrolase) and Rv2335/cysE (encoding a serine acetyltransferase) were downregulated in rpoBC isolates, suggesting reduced cysteine synthesis in this group. In the methionine synthesis pathway, Rv1133c/metE (encoding a methionine synthase) was upregulated in the rpoBC isolates. On the other hand, Rv3340/metC (encoding a cystathionine β-lyase) was downregulated in the rpoBC isolates. Taken together, these results indicate an increase in methionine synthesis and consumption of homocysteine in rpoBC mutant isolates.
Mycobacterium tuberculosis is a highly aerobic bacterium that has adapted to inhabit a wide range of intracellular and extracellular environments (Cook et al., 2014). A fundamental feature of this adaptation is the utilization of variable sources for respiration and energy generation (Brodie and Gutnick, 1967). The inflow of electrons, the maintenance of proton motive force (PMF) and the synthesis of ATP, are essential for M. tuberculosis growth and survival (Sassetti et al., 2003; Tran and Cook, 2005; Rao et al., 2008). The transcription of the NDH-2 coding gene Rv1854c/ndh was significantly decreased in rpoB mutant isolates but remained stable in rpoBC mutant isolates following rifampin exposure. Considering the importance of NDH-2 activity for maintenance of the PMF, we propose that rpoC mutations might contribute to maintenance of PMF homeostasis by recovering the impaired expression of NDH-2. The inhibitor of succinate dehydrogenase, 3-nitropropionate, was able to dissipate membrane potential in M. smegmatis, and the transcription of the sdh1 operon was upregulated in response to such high PMF conditions (Pecsi et al., 2014). We also observed higher mRNA levels of the sdh1 operon, Rv0248c/sdhA and Rv0247c/sdhB, in the rpoB mutant isolates than in the rpoBC isolates, indicating that rpoB mutation might confer energy limitation in the absence of rpoC mutation.
Recent discoveries of small molecules targeting respiratory bc1 complex have triggered an interest in the cytochrome c pathway (Abrahams et al., 2012; Pethe et al., 2013; Arora et al., 2014; Rybniker et al., 2015). Our data show that the entire qcrCAB operon, together with another aa3-type cytochrome c oxidase coding gene, Rv2193/ctaE, were dramatically downregulated in rpoB single mutants, but less affected in rpoBC mutants following rifampin exposure. The impairment of the respiratory cytochrome c pathway suggests disrupted energy metabolism as a result of rpoB mutation, which might be rescued by compensatory rpoC mutations.
The gene encoding the alternative bd-type menaquinol oxidase displayed reduced expression in the rpoB single mutants. The role of this bd-type menaquinol oxidase (CydAB) is to protect M. tuberculosis from respiratory machinery inhibitors. Several studies in M. tuberculosis have demonstrated that inactivation of cydA made the bacilli hypersensitive to the ATP synthase inhibitor bedaquiline (Berney et al., 2014) and cytochrome c oxidoreductase inhibitors (Arora et al., 2014). Given the important role of CydAB for M. tuberculosis adaptation to stress conditions (Lu et al., 2015), the recovered expression of the cydAB operon resulting from rpoC mutations might compensate for the suppression in energy metabolism associated with rpoB mutation.
Amino acid metabolism in slow growing mycobacteria begins with the nitrogen assimilation pathways that incorporate ammonium or ammonia into glutamine and glutamate by glutamine synthetase and glutamine oxoglutarate aminotransferase (GOGAT) (Gillespie et al., 2011). Given that formation of glutamine from glutamate and ammonium is highly energy-consuming, as approximately 15% of the cell’s ATP supply is required (Reitzer, 2003), it is strictly regulated transcriptionally and/or post-translationally according to the cellular α-ketoglutarate/glutamine ratio (Gouzy et al., 2014). In the absence of rifampin exposure, Rv2919c/glnK, was slightly downregulated in the rpoB mutants, but dramatically upregulated in the rpoBC group. This suggests the constant demand for ammonium uptake observed in the rpoB group might decrease and provide a better intracellular nitrogen status in the rpoBC group. Rv2220/glnA1 was downregulated while Rv3859c/gltB was upregulated in the rpoB group, which might consequently promote the conversion of glutamine to glutamate. Depletion of glutamine and imbalance of the glutamine/glutamate ratio might occur in rpoB single mutant isolates, thus affecting bacterial growth and viability, since prior evidence suggests that supplementation of culture medium with glutamine is more favorable than glutamate for the growth of Mycobacterium avium (McCarthy, 1978). A M. tuberculosis mutant deficient in glnA1 became a glutamine auxotroph, requiring a relatively high level of exogenous L-glutamine for robust growth in vitro and exhibiting attenuated virulence in vivo (Tullius et al., 2003). Particularly, glutamine synthetase from pathogenic mycobacterial species, such as M. tuberculosis and M. bovis, was reported to be exported (Harth et al., 1994) and implicated in the synthesis of poly-L-glutamine-glutamate for maintaining the integrity of the mycobacterial cell wall (Harth et al., 2000). Inhibition of poly-L-glutamine-glutamate mRNA (Harth et al., 2000) or enzyme activity (Harth and Horwitz, 1999) reduced the growth of M. tuberculosis in vitro. Thus, we propose that downregulation of glnA1 contributes to the growth defect observed in rpoB single mutant isolates, and this effect is reversed by compensatory rpoC mutations.
Although the expression of Rv3858c/gltD remained stable in rpoB single mutants following rifampin exposure, that of Rv1827/garA, the activator of GOGAT, was significantly downregulated. These data are consistent with decreased activity of GOGAT, perhaps leading to the reduction of glutamate synthesis. Since GarA also functions as an inhibitor of glutamate dehydrogenase and 2-ketoglutarate dehydrogenase, its decreased expression likely results in increased glutamate degradation and, in turn, the entry of α-ketoglutarate into the TCA cycle. Along with the suppression in energy metabolism we observed in rpoB single mutant isolates, this could compensate for a potential energy shortage. Moreover, the diminished synthesis and enhanced degradation of glutamate would continuously reduce intracellular glutamate concentrations. However, in the rpoBC mutant isolates, the situation was reversed. Rv3858c/gltD was downregulated while Rv1827/garA was upregulated, maintaining the homeostasis of glutamate synthesis and degradation. In addition to the ammonium assimilation pathway, another two genes are involved in ammonium production. Rv0069c/sdaA encodes a serine deaminase to release ammonium from serine (Gouzy et al., 2014). Rv1162/narH is located in the narGHJI operon encoding a nitrate reductase, which reduces nitrates to nitrites (Khan and Sarkar, 2012). The dysregulation of these genes in the two groups suggests their disparate requirements for ammonium. Rv0069c/sdaA was upregulated and Rv1162/narH remained unchanged in the rpoB single mutant isolates, while expression of Rv0069c/sdaA was unchanged and Rv1162/narH was downregulated in rpoBC mutant isolates. These results indicate a higher demand for available ammonium in the rpoB single mutant isolates than in the rpoBC mutant isolates. Finally, several genes encoding transporters associated with peptide or amino acid uptake, including Rv1280c/oppA, Rv2690c, and Rv3663c/dppD (Cole et al., 1998; Braibant et al., 2000; Green et al., 2000; Flores-Valdez et al., 2009), were found to be upregulated in the rpoB group but downregulated in the rpoBC group, suggesting potential defective de novo synthesis of amino acids caused by rpoB mutation, and a requirement for exogenous supplementation of amino acids.
Our work has several limitations. First, we were able to study only 3 isolates in each mutant group, which may explain the relatively high variability of transcriptional profiles within groups. Second, due to the high genetic heterogeneity and incomplete genomic sequencing of the 9 M. tuberculosis clinical isolates studied, genome-scale SNPs or INDELs may not be fully identified, complicating a thorough analysis of the potential influence of genetic variation on transcription.
In summary, we studied the compensatory effect of rpoC mutations on rpoB mutant M. tuberculosis using RNA sequencing, and found that rpoC mutations were associated with the transcriptional regulation of genes encoding ribosomal proteins, proteins essential for M. tuberculosis growth, components of the oxidative phosphorylation machinery and amino acid synthesis pathways. But our work still has several limitations. Firstly, the M. tuberculosis isolate number is inadequate and needs to be enlarged in further studies, as only 3 isolates were studied in each group, which may explain the high variation of transcriptional profiles even among isolates within the same group. Secondly, due to the high genetic heterogeneity and incomplete genomic sequencing of the 9 M. tuberculosis clinical isolates, genome-scale SNPs or INDELs will not be fully identified, which makes it difficult to thoroughly exclude the potential influence of genetic variation on transcription. Also, our findings need to be validated using isogenic M. tuberculosis strains in further studies due to the non-isogenicity of clinical strains. Therefore, future genetic and biochemical studies will focus on further characterizing the contribution of each of these pathways in restoring rpoB mutation-associated fitness costs in M. tuberculosis laboratory strains.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81361120383, 81772140, and 81830068), the State Key Development Programs for Basic Research of China (973 Program No. 2015CB554203), Key Research and Development Project of China (No. 2016YFA0500600), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, a Municipal Human Resources Development Program for Outstanding Leaders in Medical Disciplines in Shanghai (2017BR032), Key Discipline Construction Project of Pudong Health Bureau of Shanghai (No. PWZxk201709), and grants AI106613 and HL106786 (NIH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02895/full#supplementary-material
Abrahams, K. A., Cox, J. A., Spivey, V. L., Loman, N. J., Pallen, M. J., Constantinidou, C., et al. (2012). Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One 7:e52951. doi: 10.1371/journal.pone.0052951
Arora, K., Ochoa-Montano, B., Tsang, P. S., Blundell, T. L., Dawes, S. S., Mizrahi, V., et al. (2014). Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6962–6965. doi: 10.1128/AAC.03486-14
Berney, M., Hartman, T. E., and Jacobs, W. R. (2014). A Mycobacterium tuberculosis cytochrome bd oxidase mutant is hypersensitive to bedaquiline. mBio 5:e1214. doi: 10.1128/mBio.01275-14
Boyaci, H., Chen, J., Lilic, M., Palka, M., Mooney, R. A., Landick, R., et al. (2018). Fidaxomicin jams Mycobacterium tuberculosis RNA polymerase motions needed for initiation via RbpA contacts. eLife 7:e34823. doi: 10.7554/eLife.34823
Braibant, M., Gilot, P., and Content, J. (2000). The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 24, 449–467. doi: 10.1111/j.1574-6976.2000.tb00550.x
Brodie, A. F., and Gutnick, D. L. (1967). Electron transport and oxidative phosphorylation in microbial systems. Methods Microbiol. 6, 383–393.
Campbell, E. A., Korzheva, N., Mustaev, A., Murakami, K., Nair, S., Goldfarb, A., et al. (2001). Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 104, 901–912. doi: 10.1016/S0092-8674(01)00286-0
Camus, J. C., Pryor, M. J., Medigue, C., and Cole, S. T. (2002). Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148(Pt 10), 2967–2973. doi: 10.1099/00221287-148-10-2967
Chen, J., Tsolaki, A. G., Shen, X., Jiang, X., Mei, J., and Gao, Q. (2007). Deletion-targeted multiplex PCR (DTM-PCR) for identification of Beijing/W genotypes of Mycobacterium tuberculosis. Tuberculosis 87, 446–449. doi: 10.1016/j.tube.2007.05.014
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Comas, I., Borrell, S., Roetzer, A., Rose, G., Malla, B., Kato-Maeda, M., et al. (2011). Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110. doi: 10.1038/ng.1038
Conrad, T. M., Frazier, M., Joyce, A. R., Cho, B. K., Knight, E. M., Lewis, N. E., et al. (2010). RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U.S.A. 107, 20500–20505. doi: 10.1073/pnas.0911253107
Cook, G. M., Hards, K., Vilcheze, C., Hartman, T., and Berney, M. (2014). Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2, 1–30. doi: 10.1128/microbiolspec.MGM2-0015-2013
Flores-Valdez, M. A., Morris, R. P., Laval, F., Daffe, M., and Schoolnik, G. K. (2009). Mycobacterium tuberculosis modulates its cell surface via an oligopeptide permease (Opp) transport system. FASEB J. 23, 4091–4104. doi: 10.1096/fj.09-132407
Gagneux, S., Long, C. D., Small, P. M., Van, T., Schoolnik, G. K., and Bohannan, B. J. (2006). The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312, 1944–1946. doi: 10.1126/science.1124410
Gillespie, J. J., Wattam, A. R., Cammer, S. A., Gabbard, J. L., Shukla, M. P., Dalay, O., et al. (2011). PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 79, 4286–4298. doi: 10.1128/IAI.00207-11
Glansdorff, N., and Xu, Y. (2006). “Microbial arginine biosynthesis: pathway, regulation and industrial production,” in Amino Acid Biosynthesis ~ Pathways, Regulation and Metabolic Engineering. Microbiology Monographs, Vol. 5, ed. V. F. Wendisch (Berlin: Springer), 219–257. doi: 10.1007/7171_2006_061
Gouzy, A., Poquet, Y., and Neyrolles, O. (2014). Nitrogen metabolism in Mycobacterium tuberculosis physiology and virulence. Nat. Rev. Microbiol. 12, 729–737. doi: 10.1038/nrmicro3349
Green, R. M., Seth, A., and Connell, N. D. (2000). A peptide permease mutant of Mycobacterium bovis BCG resistant to the toxic peptides glutathione and S-nitrosoglutathione. Infect Immun. 68, 429–436. doi: 10.1128/IAI.68.2.429-436.2000
Harth, G., Clemens, D. L., and Horwitz, M. A. (1994). Glutamine synthetase of Mycobacterium tuberculosis: extracellular release and characterization of its enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 91, 9342–9346. doi: 10.1073/pnas.91.20.9342
Harth, G., and Horwitz, M. A. (1999). An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J. Exp. Med. 189, 1425–1436. doi: 10.1084/jem.189.9.1425
Harth, G., Zamecnik, P. C., Tang, J. Y., Tabatadze, D., and Horwitz, M. A. (2000). Treatment of Mycobacterium tuberculosis with antisense oligonucleotides to glutamine synthetase mRNA inhibits glutamine synthetase activity, formation of the poly-L-glutamate/glutamine cell wall structure, and bacterial replication. Proc. Natl. Acad. Sci. U.S.A. 97, 418–423. doi: 10.1073/pnas.97.1.418
Khan, A., and Sarkar, D. (2012). Nitrate reduction pathways in mycobacteria and their implications during latency. Microbiology 158(Pt 2), 301–307. doi: 10.1099/mic.0.054759-0
Lloyd, D. G. (1987). Parallels between sexual strategies and other allocation strategies. Experientia Suppl. 55, 263–281. doi: 10.1007/978-3-0348-6273-8_12
Lu, P., Heineke, M. H., Koul, A., Andries, K., Cook, G. M., Lill, H., et al. (2015). The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 5:10333. doi: 10.1038/srep10333
Luo, M., Fadeev, E. A., and Groves, J. T. (2005). Mycobactin-mediated iron acquisition within macrophages. Nat. Chem. Biol. 1:149. doi: 10.1038/nchembio717
Ma, A. J., Wang, S. F., Fan, J. L., Zhao, B., He, G. X., and Zhao, Y. L. (2018). Genetic diversity and drug susceptibility of Mycobacterium tuberculosis isolates in a remote mountain area of China. Biomed. Environ. Sci. 31, 351–362. doi: 10.3967/bes2018.046
Mariam, D. H., Mengistu, Y., Hoffner, S. E., and Andersson, D. I. (2004). Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 48, 1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004
Martin, T., Cheke, D., and Natyshak, I. (1989). Broth culture: the modern ’guinea-pig’ for isolation of mycobacteria. Tubercle 70, 53–56. doi: 10.1016/0041-3879(89)90065-2
McCarthy, C. (1978). Ammonium ion requirement for the cell cycle of Mycobacterium avium. Infect Immun. 19, 304–311.
Mokrousov, I. (2004). Multiple rpoB mutants of Mycobacterium tuberculosis and second-order selection. Emerg. Infect Dis. 10, 1337–1338.
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
O’Neill, M. B., Mortimer, T. D., and Pepperell, C. S. (2015). Diversity of Mycobacterium tuberculosis across evolutionary scales. PLoS Pathog. 11:e1005257. doi: 10.1371/journal.ppat.1005257
Pecsi, I., Hards, K., Ekanayaka, N., Berney, M., Hartman, T., Jacobs, W. R., et al. (2014). Essentiality of succinate dehydrogenase in Mycobacterium smegmatis and its role in the generation of the membrane potential under hypoxia. mBio 5:e01093-14. doi: 10.1128/mBio.01093-14
Pethe, K., Bifani, P., Jang, J., Kang, S., Park, S., Ahn, S., et al. (2013). Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19, 1157–1160. doi: 10.1038/nm.3262
Pinto, R., Harrison, J. S., Hsu, T., Jacobs, W. R. Jr., and Leyh, T. S. (2007). Sulfite reduction in mycobacteria. J. Bacteriol. 189, 6714–6722. doi: 10.1128/JB.00487-07
Ramaswamy, S., and Musser, J. M. (1998). Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 79, 3–29. doi: 10.1054/tuld.1998.0002
Rao, S. P., Alonso, S., Rand, L., Dick, T., and Pethe, K. (2008). The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 105, 11945–11950. doi: 10.1073/pnas.0711697105
Reitzer, L. (2003). Nitrogen assimilation and global regulation in Escherichia coli. Ann. Rev. Microbiol. 57, 155–176. doi: 10.1146/annurev.micro.57.030502.090820
Ren, J., Sang, Y., Tan, Y., Tao, J., Ni, J., Liu, S., et al. (2016). Acetylation of lysine 201 inhibits the DNA-binding ability of PhoP to regulate Salmonella virulence. PLoS Pathog. 12:e1005458. doi: 10.1371/journal.ppat.1005458
Reynolds, M. G. (2000). Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156, 1471–1481.
Rifat, D., Campodonico, V. L., Tao, J., Miller, J. A., Alp, A., Yao, Y., et al. (2017). In vitro and in vivo fitness costs associated with Mycobacterium tuberculosis RpoB mutation H526D. Fut. Microbiol. 12, 753–765. doi: 10.2217/fmb-2017-0022
Rybniker, J., Vocat, A., Sala, C., Busso, P., Pojer, F., Benjak, A., et al. (2015). Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 6:7659. doi: 10.1038/ncomms8659
Sandgren, A., Strong, M., Muthukrishnan, P., Weiner, B. K., Church, G. M., and Murray, M. B. (2009). Tuberculosis drug resistance mutation database. PLoS Med. 6:e2. doi: 10.1371/journal.pmed.1000002
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Sassetti, C. M., and Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994. doi: 10.1073/pnas.2134250100
Song, T., Park, Y., Shamputa, I. C., Seo, S., Lee, S. Y., Jeon, H. S., et al. (2014). Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the beta’ subunit of RNA polymerase. Mol. Microbiol. 91, 1106–1119. doi: 10.1111/mmi.12520
Stefan, M. A., Ugur, F. S., and Garcia, G. A. (2018). Source of the fitness defect in rifamycin-resistant mycobacterium tuberculosis rna polymerase and the mechanism of compensation by mutations in the beta’ subunit. Antimicrob. Agents Chemother. 62:e00164-18. doi: 10.1128/AAC.00164-18
Tran, S. L., and Cook, G. M. (2005). The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 187, 5023–5028. doi: 10.1128/JB.187.14.5023-5028.2005
Tullius, M. V., Harth, G., and Horwitz, M. A. (2003). Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect Immun. 71, 3927–3936. doi: 10.1128/IAI.71.7.3927-3936.2003
Vasilyeva, I. À, Belilovsky, E. M., Borisov, S. E., and Sterlikov, S. À (2017). Who global tuberculosis reports: compilation and interpretation. Tuberc. Lung Dis. 95, 7–16. doi: 10.21292/2075-1230-2017-95-5-7-16
Weinstein, E. A., Yano, T., Li, L. S., Avarbock, D., Avarbock, A., Helm, D., et al. (2005). Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. U.S.A. 102, 4548–4553. doi: 10.1073/pnas.0500469102
Keywords: Mycobacterium tuberculosis, rifampicin resistance, fitness cost, rpoC mutation, RNA sequence
Citation: Xu Z, Zhou A, Wu J, Zhou A, Li J, Zhang S, Wu W, Karakousis PC and Yao Y-F (2018) Transcriptional Approach for Decoding the Mechanism of rpoC Compensatory Mutations for the Fitness Cost in Rifampicin-Resistant Mycobacterium tuberculosis. Front. Microbiol. 9:2895. doi: 10.3389/fmicb.2018.02895
Received: 16 June 2018; Accepted: 12 November 2018;
Published: 30 November 2018.
Edited by:
Yuji Morita, Meiji Pharmaceutical University, JapanReviewed by:
Sergei Borukhov, Rowan University, United StatesCopyright © 2018 Xu, Zhou, Wu, Zhou, Li, Zhang, Wu, Karakousis and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Feng Yao, eWZ5YW9Ac2p0dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.