 Sean Storey
Sean Storey Mardiana Mohd Ashaari
Mardiana Mohd Ashaari Nicholas Clipson1,2
Nicholas Clipson1,2 Evelyn Doyle
Evelyn Doyle Alexandre B. de Menezes
Alexandre B. de Menezes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 21 November 2018
Sec. Microbiotechnology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02815
This article is part of the Research TopicUsing Genomics, Metagenomics and Other "Omics" to Assess Valuable Microbial Ecosystem Services and Novel Biotechnological ApplicationsView all 53 articles
Bioremediation offers a sustainable approach for removal of polycyclic aromatic hydrocarbons (PAHs) from the environment; however, information regarding the microbial communities involved remains limited. In this study, microbial community dynamics and the abundance of the key gene (PAH-RHDα) encoding a ring hydroxylating dioxygenase involved in PAH degradation were examined during degradation of phenanthrene in a podzolic soil from the site of a former timber treatment facility. The 10,000-fold greater abundance of this gene associated with Gram-positive bacteria found in phenanthrene-amended soil compared to unamended soil indicated the likely role of Gram-positive bacteria in PAH degradation. In contrast, the abundance of the Gram-negative PAHs-RHDα gene was very low throughout the experiment. While phenanthrene induced increases in the abundance of a small number of OTUs from the Actinomycetales and Sphingomonadale, most of the remainder of the community remained stable. A single unclassified OTU from the Micrococcaceae family increased ~20-fold in relative abundance, reaching 32% of the total sequences in amended microcosms on day 7 of the experiment. The relative abundance of this same OTU increased 4.5-fold in unamended soils, and a similar pattern was observed for the second most abundant PAH-responsive OTU, classified into the Sphingomonas genus. Furthermore, the relative abundance of both of these OTUs decreased substantially between days 7 and 17 in the phenanthrene-amended and control microcosms. This suggests that their opportunistic phenotype, in addition to likely PAH-degrading ability, was determinant in the vigorous growth of dominant PAH-responsive OTUs following phenanthrene amendment. This study provides new information on the temporal response of soil microbial communities to the presence and degradation of a significant environmental pollutant, and as such has the potential to inform the design of PAH bioremediation protocols.
Soil pollution is a global problem; there are about 342,000 contaminated sites in Europe alone, with this number set to increase to over 500,000 by 2025 (Anon, 2012). Polycyclic aromatic hydrocarbons (PAHs) are a class of toxic chemicals, composed of at least two benzene rings arranged in various conformations (Wickliffe et al., 2014). PAHs are released to the environment upon incomplete combustion of fossil fuels and are ubiquitous in nature, with soil as their major sink (Collins et al., 2013). Due to their carcinogenic and mutagenic potential, 16 of these compounds are listed as priority pollutants by the United States Environmental Protection Agency (Wang et al., 2010).
Bacterial communities are important components of soil environments and are essential in the provision of many ecosystem services (Brussaard, 2012), including degradation of toxic chemicals such as PAHs (Fernández-Luqueño et al., 2011). However, the hydrophobicity of PAHs renders them poorly available to microorganisms, leading to their persistence in the environment (Posada-Baquero and Ortega-Calvo, 2011). A wide range of bacteria and fungi capable of degrading PAHs have been isolated from contaminated sites (for reviews see Cerniglia, 1992, 1997; Doyle et al., 2008; Ghosal et al., 2016), with Pseudomonas, Sphingomonas, and Mycobacterium spp. amongst the most frequently isolated PAH degrading bacteria (Bastiaens et al., 2000; Johnsen and Karlson, 2005). Culture independent analyses have revealed that representatives of Proteobacteria, Actinobacteria, and Firmicutes are the bacteria most likely to increase during biodegradation of PAHs and petroleum hydrocarbons (Fuentes et al., 2014).
Although bioremediation approaches have been successfully applied to PAH-contaminated soils both in the laboratory (Sayara et al., 2011) and in the field (Lors et al., 2012), the process remains poorly understood, mainly in terms of the structure and function of microbial assemblages involved (Yang et al., 2015). PAH biodegradation studies have been analyzed using different molecular techniques (Muckian et al., 2009; Lors et al., 2010; Sun et al., 2012; Festa et al., 2016; Kuppusamy et al., 2016). In general, individual molecular microbial studies of PAH degradation targeted complex bioremediation conditions. For example, Jiao et al. (2017) showed that a variety of pollutants had consistent, broadly defined effects on soil microbial taxa, mostly repressing or inducing specific microbial populations. Zhao et al. (2016) showed that Mycobacterium contribute ring-hydroxylating dioxygenases involved in the initial steps of fluoranthene breakdown, while a more diverse group of bacteria contributed to the metabolism of downstream fluoranthene degradation products. Kuppusamy et al. (2016) demonstrated that metal-resistant, PAH-degrading Alphaproteobacteria can persist for longer in soils contaminated with heavy metals and PAHs compared to Gram-positive bacteria such as the Actinobacteria. Certain amendments can increase bioremediation efficiency, for example Wang et al. (2016) showed that surfactants increased PAH removal from soil and boosted the abundance of the Pseudomonas, Bacillus and Sphingomonas. The inoculation of PAH-degrading consortia into polluted soil can also enhance bioremediation and boost the abundance of genes associated with formation of PAH degradation products that can be further metabolized through the tricarboxylic acid cycle (Zafra et al., 2016). Festa et al. (2016) showed that inoculation of PAH-degrading Sphingobium increases the degradation of phenanthrene in soil but had no effect in the degradation of PAHs in a chronically contaminated soil. Other studies found less clear relationships between soil amendments, soil microbial communities and PAH degradation. For example, Thomas and Cébron (2016) found that phenanthrene amendment in the presence of plants did not lead to greater removal of phenanthrene from bulk soil, despite increases in the abundance of PAH-ring hydroxylating genes. Similarly, Delgado-Balbuena et al. (2016) did not find strong relationships between soil microbial community structure and anthracene removal from contaminated soil when applying different remediation strategies. Although these studies offer valuable insights into the microbial molecular ecology of PAH degradation in soils, they vary substantially in their objectives, target habitat, PAH type, and experimental design. Currently, studies examining the effects of a single PAH in the native soil microbiome without the confounding effects of multiple treatments and contaminants are limited.
PAH contamination causes physiological stress on the soil microbial communities and exposure to these compounds leads to the activation of detoxification and stress resistance (de Menezes et al., 2012). Therefore, PAH exposure not only triggers the growth of PAH-degrading microorganisms, but it may also affect the ecological stability of the soil microbiome, in turn potentially affecting a range of soil processes (Griffiths and Philipot, 2013). Microbial ecosystem stability is affected by species richness, evenness, and composition, however greater biodiversity levels are not necessarily a sign of a more stable ecosystem (Griffiths and Philipot, 2013; Shade, 2017). While the intermediate disturbance hypothesis was postulated to explain the frequent observation in macro-ecology of maximal biodiversity levels at intermediate levels of disturbance, the applicability of this hypothesis to microbial ecosystems is less certain (Gibbons et al., 2016). In microbial communities, disturbance is thought to cause changes in community composition, leading to a succession of species in which opportunistic, fast growing bacteria able to take advantage of transient conditions thrive initially, followed by slower growing, resource efficient microorganisms (Sigler and Zeyer, 2004). PAH contamination in soil therefore represents an ideal opportunity to study the effects of a disturbance on microbial community composition and succession.
Bacterial diversity in soil amended with the 3-ring PAH phenanthrene was compared with that in unamended soil during the course of phenanthrene degradation using 16S rRNA gene amplicon sequencing. The soil used in this experiment had previous history of exposure to PAH pollution due to the activities of a since-closed timber treatment facility, however at the time of sampling PAH levels in this soil were similar to uncontaminated soils. Although a previous study by de Menezes et al. (2012) examined gene expression in a phenanthrene-contaminated soil, only one time-point was examined and the effect of the PAH on soil microbial community structure was not assessed. We set up two sets of microcosms in triplicate, of which one set was amended with phenanthrene and the other set used as the unamended control. These microcosms were sampled at four time points during a 17-day incubation during which time phenanthrene levels were measured and bacterial diversity analyzed by high-throughput sequencing of the 16S rRNA gene. We also assessed the potential for PAH degradation in these soils by quantifying the abundance of the gene encoding the α subunit of a PAH ring hydroxylating dioxygenase (PAH-RHDα), a key gene involved in PAH degradation. Compared to the complex experimental design of most microbiome PAH bioremediation studies, the simple experimental design used in this study allows the investigation of phenanthrene as the single treatment, without the confounding effects of multiple treatments. We observed that dominant phenanthrene-responsive bacteria showed opportunistic traits, increasing substantially in abundance in the amended samples but also to a lesser extent in the control samples, likely as a result of their capacity to adapt and grow in ecologically disturbed environments.
Soil was collected from the site of a former timber treatment facility in Monard, Co. Tipperary, Ireland (52°30' N, 8°13' W). This site is elevated about 100 m above sea level and contains a mixture of gray and gray-brown podzolic soil with underlying limestone glacial till and limestone. Soil chemical analysis was carried out by ALcontrol Geochem, Dublin, Ireland, while soil particle size was determined following the method of Kettler et al. (2001). The soil had a moisture content of 18.45%, a pH of 6.58 and a total organic carbon content of 2% (w/v). The concentration of total background PAHs was 4.7 mg kg−1 (standard deviation 4.6%) as determined by GC-FID (Sawulski et al., 2014). The soil is a clay fine loamy drift with limestone soil, composed of 51.95% sand, 18.13% silt, and 29.91% and it is classified as luvisol.
A total of 24 microcosms were set up, 12 for each treatment (phenanthrene amendment and control) and 3 for each time point (de Menezes et al., 2012). Prior to microcosm set up, the soil was sieved to < 2 mm to remove plant matter and debris and amended with phenanthrene to a final concentration of 725 mg kg−1. Fifty grams of unamended or amended soil was then placed in black polyvinylchloride containers in triplicate and incubated at 22°C for the duration of the experiment (17 days). Soil moisture content was maintained at a constant level by gravimetric addition of sterile deionized water every 2–3 days. Pots were destructively sampled on days 0, 2, 7, and 17.
Phenanthrene was extracted from soil by adding acetone and hexane to soil samples and using mechanical agitation (Dean and Xiong, 2000). Phenanthrene concentration was determined by Gas Chromatography-Flame Ionization Detection as described by Storey et al. (2014).
DNA was extracted from triplicates of each treatment (3 microcosm pots were destructively harvested for each treatment at each time point) on days 0, 2, 7, and 17 of the experiment as outlined in de Menezes et al. (2012) using a modification of the phenol-chloroform method of Griffiths et al. (2000).
Amplicon sequencing was carried out using the procedure detailed by Martínez et al. (2009). Briefly, the V1-V3 regions of the 16S rRNA gene were amplified using 8F-518R primers (Lane et al., 1985; Muyzer et al., 1993) which contained specific adapter sequences and unique barcodes for each sample. GENETOOLS software (Syngene, Cambridge, UK) was used for quality control of PCR reactions. Equal amounts of amplicons from each PCR reaction were pooled, run on a gel to ensure purity and DNA concentrations then measured using the Quant-iT™ PicoGreen™ dsDNA Assay Kit (Thermo Fisher, Scientific, MS, USA) and a Qubit fluorimeter (Thermo Fisher, Scientific, MS, USA). Pyrosequencing was performed using the Roche-454 Titanium platform at the University of Nebraska-Lincoln Core for Applied Genomics and Ecology. Sequence files associated with each sample were deposited in the NCBI GenBank database under the BioProject accession number PRJNA284664.
Sequences were processed using mothur v.1.31.0. with default parameters for 454-Titanium sequence processing (Schloss et al., 2009). Briefly, after removing sequence noise, sequences smaller than 200 bp, with 1 or more nucleotide ambiguities or > 8 bp long homopolymers were removed from the dataset. Sequences were aligned against the Silva reference alignment and those sequences classified as plastid, mitochondrial, archaeal, eukaryotic or unknown at the kingdom level were discarded. Chimeras were detected using the UCHIME tool built within mothur (Edgar et al., 2011) and also removed from the dataset. After quality processing the number of sequences per sample ranged from 2,572 to 11,096 sequences. Operational taxonomic units (OTUs) were generated by calculating pairwise distances and clustering sequences with a distance cutoff of 0.03. OTUs present as a single sequence read in the dataset were removed. The OTUs were classified by obtaining the consensus taxonomy in mothur using the RDP reference files and a consensus confidence threshold cutoff of 0.8 (Schloss, 2009).
Relative abundances were determined in mothur as the abundance of each OTU to the total number of sequences in a sample. For beta-diversity analyses, the number of sequences across samples was subsampled using the sub.sample command in mothur to the number of sequences present in the sample with smallest number of sequences. The microbial community composition of one sample from day two of the control microcosms was substantially different from all other samples in the dataset and was no more similar to its two biological replicates than to samples from the other treatments. We therefore chose to remove this sample from the analysis. In order to identify individual OTUs that were enriched in PAH-amended microcosms, we used DESeq2, which has the advantage of not requiring rarefaction or subsampling of sequence data, a procedure that leads to loss of valid sequence information (McMurdie and Holmes, 2013, 2014; Love et al., 2014). DESeq2 tests were performed using the Wald test, automatic filtering of low abundance OTUs, and an alpha of 0.05 and multiple testing adjustment of p-values. Bacterial alpha diversity (Shannon index) was also calculated in Phyloseq, and paired t-tests using Bonferroni adjusted p-values were carried out to determine the significance of differences in Shannon index between treatments.
To generate standard curves of the RHDα-GP and the PAH-RHDα-GN genes, both genes were amplified from soil using the primer sets described in Cébron et al. (2008). End-point PCRs were performed initially in 25-μl volumes containing 12.5 μl 2X PCR master mix (Promega), 2.5 pmoles each primer, 0.2 μl ultrapure BSA (50 mg ml−1, Ambion), and 10 ng DNA template. The thermocycling conditions were as follows: 95°C for 5 min (one cycle); 95°C for 30 s, 57 (GP) or 54 °C (GN) for 30 s, 72°C for 30 s (30 cycles); 72°C for 7 min. PCR products were visualized on a 1.2% (w/v) agarose gel. The resulting products were cloned using the pGEM-T Easy Vector system (Promega), and clones containing both plasmid and insert were isolated and purified using the PureYield™ Plasmid Miniprep system (Promega). Plasmid DNA was quantified using a Nanodrop™ ND-1000 Spectrophotometer (Thermo Scientific). The copy number of the PAH-RHDα-GP and the PAH-RHDα-GN gene per volume was calculated using recombinant plasmid sizes of 3307 and 3321 bp, respectively, and a mass of 1.096 × 10–21 g bp−1. Serial dilutions were used to construct standard curves.
qPCR was then carried out using the Applied Biosystems Viia 7 qPCR machine (Life Technologies, Dublin, Ireland) in MicroAmp®optical 96-well reaction plates containing 6.25 μl of 2 × KAPA SYBR® FAST qPCR master mix, 0.25 μl ROX™ low passive reference dye (Anachem, Bedfordshire, UK), 0.2 pmoles each primer and 2 ng DNA. Nuclease-free water (Sigma-Aldrich, Arklow, Ireland) was added to give a final volume of 12.5 μl. No-template controls contained 1 μl nuclease-free water (Sigma-Aldrich, Arklow, Ireland) in place of DNA template. Amplification was carried out using a modification of the method of Cébron et al. (2008) as follows: 95°C for 1 min, then 40 cycles of 95°C for 30 s, annealing temperature for 30 s, then elongation at 72°C for 30 s. SYBR® Green signal intensity was then measured during a 10 s primer dissociation step at 80°C. A melting curve was included at the end of each reaction using a temperature increment of 0.05°C s−1 from 51–95°C. Standard curves were included in each experiment. All analyses were performed using ABI VIIA 7 software version 1.2.2 (Life Technologies, Carlsbad, USA), Microsoft Excel 2010 and SAS version 9.1 (SAS, Cary, USA).
Multivariate statistical analysis was carried out in PRIMER version 6.1.9 with PERMANOVA add-on version 1.0.1 (Primer-E Ltd., Plymouth, UK). Statistical analysis of the relative abundance of OTUs was assessed at genus level using PRIMER. Permutational multivariate analysis of variance (PERMANOVA) was performed using a Bray Curtis dissimilarity matrix in PRIMER using 9,999 unrestricted permutation of raw data. Analysis of variance (ANOVA) was carried out using SAS version 9.1. Principal coordinates analysis (PCoA) was performed using the capscale command in the vegan package in R (Oksanen et al., 2018), using square-root transformed genus-aggregated sequence abundances and a Bray-Curtis dissimilarity matrix, with overlaid arrows representing correlations > 0.4 between genera abundance profiles and PCoA ordination axes. Pearson correlation between selected OTUs and total abundance of the PAH-RHDα gene was calculated in R using the cor.test function.
The response of the indigenous soil bacterial community during phenanthrene degradation was examined using 454 amplicon sequencing. PERMANOVA analysis (Table 1) indicated that bacterial community structure changed both over time and in the presence of phenanthrene. Pairwise comparisons on bacterial community structures in phenanthrene amended soil revealed that this PAH had no significant effect on bacterial community structures on days 0 and 2, but bacterial community structures on day 7 of phenanthrene amended soil were significantly different from those on day 17. In addition, bacterial community structure in phenanthrene amended soil on days 7 and 17 were significantly different to those in the unamended control soil (P < 0.001; Supplementary Table 1). When temporal changes were examined, bacterial community structure was found to change significantly between each sampling day in the unamended control (P < 0.001), but no changes were observed in the phenanthrene-amended soil between days 0 and 2 (P = 0.09).

Table 1. PERMANOVA results for amplicon sequencing analysis of bacterial communities at different taxa levels in soil amended or unamended with phenanthrene on days 0, 2, 7, and 17.
Bacterial diversity was similar in both unamended and phenanthrene-amended soils on day 0, with soils dominated by two phyla the Actinobacteria and the Proteobacteria (Figure 1). Although a small increase in the relative abundance of Actinobacteria and concurrent decrease in the relative abundance of the Proteobacteria was observed between days 0 and 2 in the unamended soil, these changes were not statistically significant. In the phenanthrene amended soil, degradation proceeded rapidly following a brief lag period of approximately 5 days. 252 mg kg−1 (about 35%) of phenanthrene added remained after 7 days, with almost complete degradation observed by day 17 (de Menezes et al., 2012). In this soil, the relative abundance of the Actinobacteria was highest on day 7, representing 50% of the total amount of 16S rRNA gene sequences present. The relative abundance of this phylum was significantly (P < 0.05) lower in the unamended soil at the same time point. The relative abundance of sequences belonging to the Proteobacteria decreased significantly (P < 0.05) between days 2 and 7. By the end of the experiment (day 17), the relative abundance of the Actinobacteria in the phenanthrene-amended soil had returned to the levels observed on day 2, whereas, the relative abundance of the Proteobacteria returned to the levels observed initially (day 0).
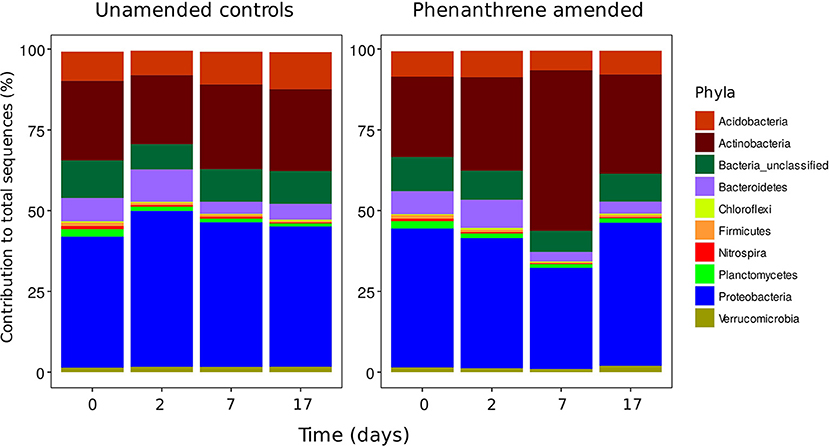
Figure 1. Phyla contributing >1% to the total number of sequences in unamended or phenanthrene-amended microcosm soils on days 0, 2, 7, and 17.
ANOVA analysis showed that on day 7 unclassified Micrococcaceae and Microbacteriaceae were significantly (P < 0.05) relatively more abundant in phenanthrene-amended soils compared to the unamended control soil (Table 2). The unclassified Micrococcaceae and Microbacteriaceae increased, respectively from 9 to 32% and from 1.4 to 3% of the total 16S rRNA gene sequences in the amended soils. The relative abundance of many genera, including a range of unclassified Bacteria belonging to the Alphaproteobacteria, Burkholderiales, and Bradyrhizobium (all members of the Proteobacteria), declined significantly (P < 0.05) in the presence of phenanthrene. Interestingly, many genera were not significantly affected by the presence of phenanthrene at this time point.
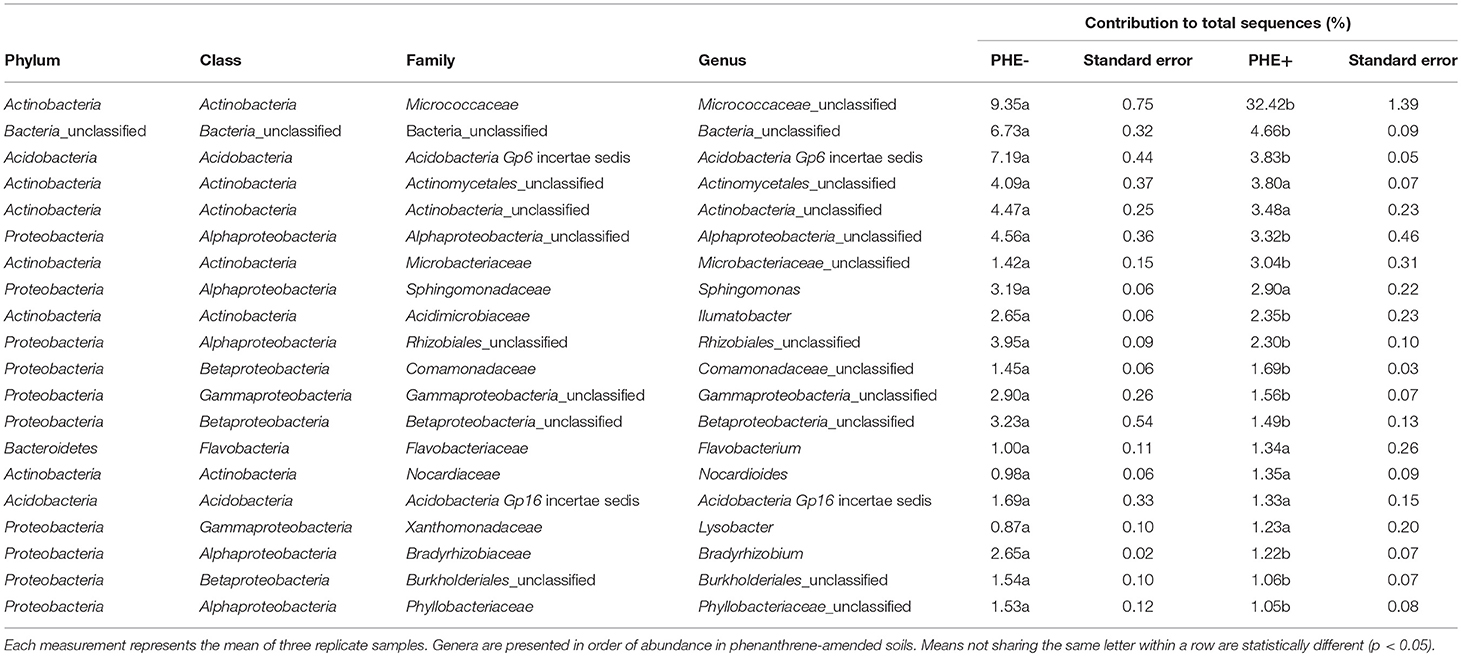
Table 2. Genera contributing >1 % to the total number of 16S rRNA genes sequences in phenanthrene-amended (PHE+) and unamended (PHE-) microcosm soils on day 7.
Principal coordinates analysis (PCoA) was used to correlate changes in community structure at genus level with the presence or absence of phenanthrene (Figure 2). The genus Flavobacterium and unclassified Actinobacteria were correlated with day 0 samples for both control and phenanthrene-amended samples. Two days after the start of the experiment, control and phenanthrene-amended samples were not forming clearly distinct clusters, however the unclassified Microbacteriaceae correlated more strongly with day 2 phenanthrene-amended samples. On day 7, control and phenanthrene-amended samples can be seen to be well separated, with the unclassified Micrococcaceae clearly correlating with the phenanthrene-amended samples, while day 7 and 17 control samples correlated primarily with the unclassified Myxococcales, the Acidobacteria Gp6 and unclassified Gammaproteobacteria. On day 17, phenanthrene-amended samples cluster more closely with the day 7 and 17 control samples.
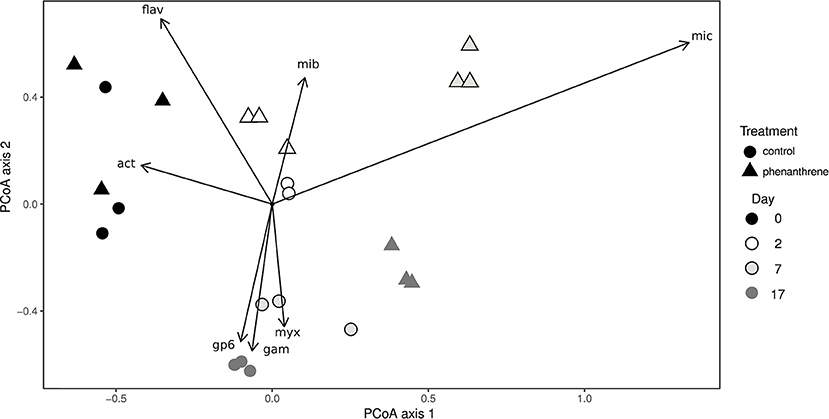
Figure 2. Principal coordinates analysis (PCoA) of bacterial diversity at genus level in soil amended (triangles) or unamended (circles) with phenanthrene on days 0, 2, 7, and 17. mic, Micrococcaceae_unclassified; mib; Microbacteriaceae_unclassified; act, Actinobacteria_unclassified; Gp6, Acidobacteria Gp6 incertae sedis; fla, Flavobacterium; gam, Gammaproteobacteria_unclassified; myx, Myxococcales_unclassified. Arrows represent correlations > 0.4 between genera and PCoA axes.
Although ANOVA showed which bacterial genera were more abundant at each treatment, it provided limited insight into changes in individual OTUs. DESeq2 analysis, which is a more appropriate statistical test for microbiome data than ANOVA, was therefore carried out to compare the differential abundance of individual OTUs in phenanthrene-amended to unamended soils at the same time points. No significantly differentially abundant OTUs were detected on either day 0 or day 2. However, on day 7, several OTUs classified to the Actinomycetales order, including Mycobacterium and unclassified Micrococcaceae were significantly more abundant in phenanthrene-amended soil than in its unamended counterpart (Table 2). One Mycobacterium OTU (OTU 82) was almost 200-fold more abundant on day 17 in phenanthrene-amended compared to unamended soil. OTUs classified to several other taxa, including Sphingomonas, unclassified Microbacteraceae and Pseudoxanthomonas were also significantly more abundant in the phenanthrene-amended soil compared to the unamended control soil. Interestingly, few OTUs responded negatively to phenanthrene, one unclassified OTU of the Burkholderiales and an unclassified Actinomycetales OTU were significantly less abundant in the phenanthrene-amended soil on day 17 of the experiment (Table 2).
When analyzing the relative abundance of those OTUs that were found to be significantly more abundant in the amended microcosms at any time point (phenanthrene-responsive OTUs), one single OTU classified to the Micrococcaceae family (OTU 1) increased in relative abundance from approximately 2.5% (day 0) to 32% (day 7) in the phenanthrene-amended microcosms, while in the unamended microcosms OTU 1 abundance also increased on between days 0 and 7 but to a lesser extent (10% of total sequences) (Figure 3). The second most relatively abundant phenanthrene-responsive OTU (OTU 2, Sphingomonas sp.) increased from ~2–5% of the total sequences in the amended microcosms toward the end of the experiment, while in the unamended microcosms OTU 2 increased in abundance earlier in the experiment at day 2 to approximately 5% followed by a decline to 2.5 % of total sequences (Figure 3). It is noteworthy that both dominant phenanthrene-responsive OTUs increased in relative abundance in the unamended controls from day 0 and 2. The relative abundance of two other phenanthrene-responsive actinobacterial OTUs (OTU 40, unclassified Actinomycetales, and OTU 82, Mycobacterium sp.) increased in relative abundance from days 7 to 17 in the amended microcosms however in the controls their abundance decreased in this period (Figure 4).
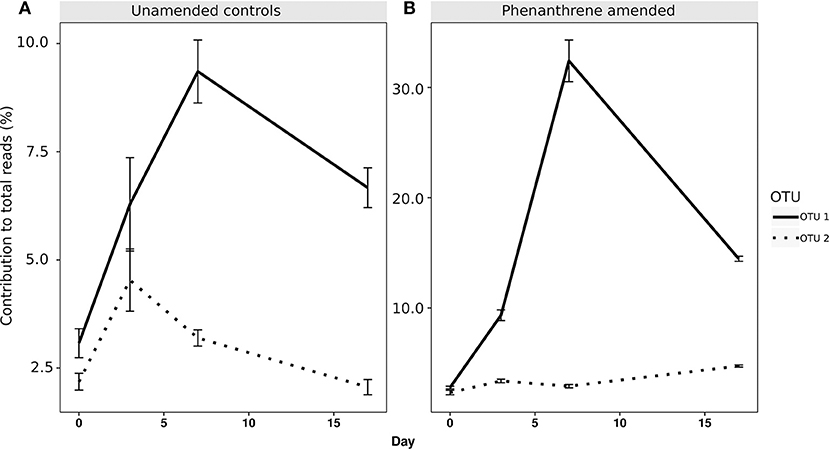
Figure 3. Percentage relative abundance of the two most abundant PAH-responsive OTUs in (A) unamended and (B) phenanthrene-amended microcosm soils on days 0, 2, 7, and 17. OTU 1, unclassified Micrococcaceae; OTU 2, Sphingomonas sp.
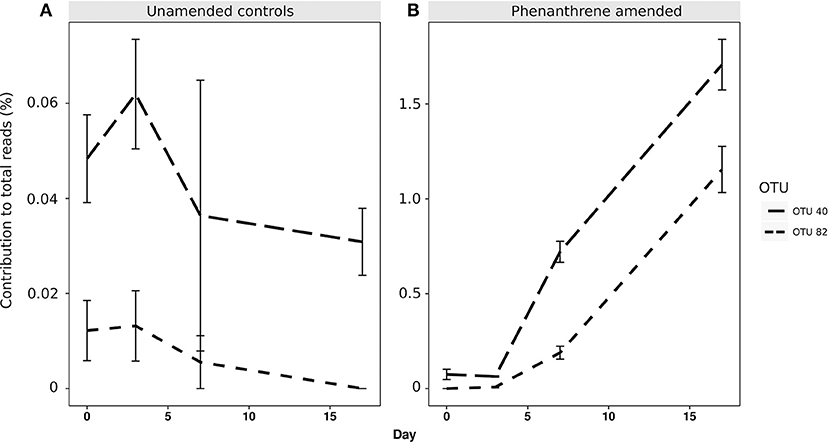
Figure 4. Percentage relative abundance of PAH-responsive OTUs classified to Actinomycetales (OTU 40) and Mycobacterium sp. (OTU 82) in (A) unamended and (B) phenanthrene-amended microcosm soils on days 0, 2, 7, and 17.
The Shannon index of the bacterial community did not change over the course of the experiment in the unamended control microcosms. However, it was significantly lower in the phenanthrene-amended microcosms on day 7 compared to days 0 (adjusted p-value < 0.001), 2 (adjusted p-value < 0.001), and 17 (adjusted p-value < 0.05; Figure 5).
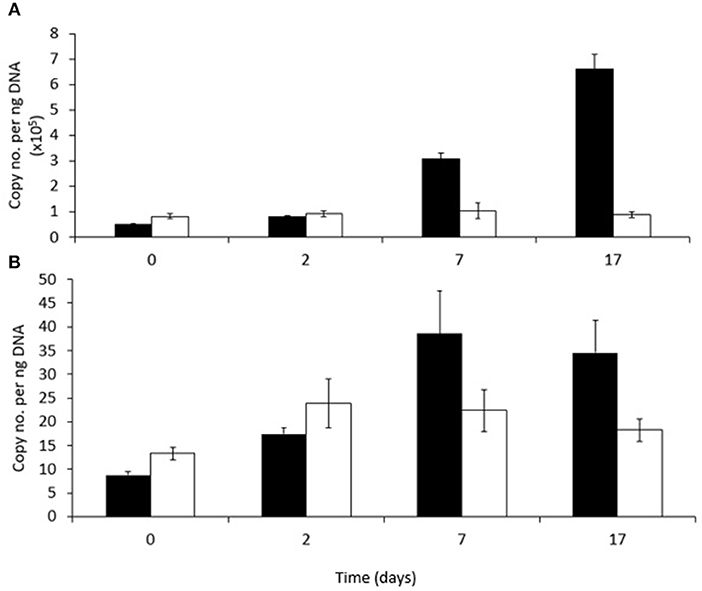
Figure 5. Abundance of (A) RHDα-GP and (B) RHDα-GN genes in phenanthrene-amended (black bars) or unamended (white bars) in microcosm soils on days 0, 2, 7, and 17. Each bar represents the mean of three replicates.
The abundance of a gene (PAH-RHDα) encoding the alpha subunit of a ring hydroxylating dioxygenase involved in the first step of PAH degradation by either Gram-positive (RHDα-GP) or Gram-negative (RHDα-GN) bacteria in both amended and unamended soils is shown in Figure 6, respectively. The abundance of this gene associated with Gram-positive bacteria (RHDα-GP) was significantly higher (between 103 and 104 times higher) in both unamended and phenanthrene-amended soils than the gene associated with Gram-negative bacteria (RHDα-GN) at all-time points. The relative abundance of RHDα-GP changed over time and in response to the presence of phenanthrene (P < 0.001). Abundance increased significantly between days 2 and 7, and again between days 7 and 17 in phenanthrene-amended samples. Abundance of RHDα-GP did not change significantly in unamended soil remaining at about 100,000 copies ng−1 soil throughout. RHDα-GN abundance did not change significantly either over time, or in response to phenanthrene amendment.
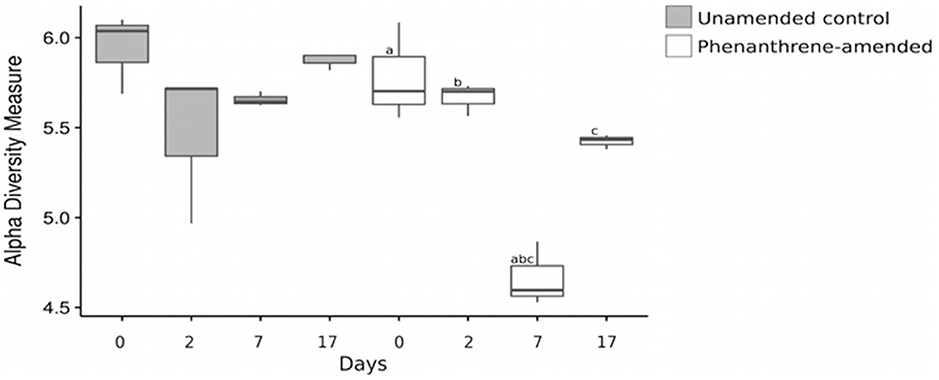
Figure 6. Shannon index of the bacterial community at all-time points. Shared letters represent significant adjusted p-values determined by pairwise t-tests. The lines extend to the largest value observed.
Analysis of the Pearson's correlation between RHDα-GP and the abundance of OTUs 40, 82, 1, and 2 showed that OTUs 40 and 82 were more strongly correlated with this gene (Pearson correlation coefficients of 0.99, p = 1.57−12 and 0.94, p = 2.94−06 for OTUs 40 and 82, respectively), compared to OTUs 1 and 2 (Pearson correlation coefficients of 0.38, p = 0.22 and 0.80, p value = 0.001 for OTUs 1 and 2, respectively).
PAH degradation has been shown to depend on the number of aromatic rings present, with the lower molecular weight PAHs (four or less aromatic rings) more susceptible to degradation than high molecular weight PAHs (Cerniglia, 1992). The rapid rate of degradation of the 3-ring PAH, phenanthrene, observed in this study was therefore not surprising, with similar results reported elsewhere. Sawulski et al. (2014) reported that 95% of phenanthrene (200 mg kg−1) was removed from a soil microcosm within two days. On day 2 of the experiment, the absence of a more pronounced phenanthrene toxic effect on the bacterial community, as indicated by the relatively small change in the relative abundances of the main bacterial phyla present, may be a result of previous exposure of these soils to creosote at the former timber treatment facility. Prior exposure to recalcitrant pollutants such as PAHs has been shown to impact soil microbial community structures (Johnsen and Karlson, 2005; Bargiela et al., 2015). Similarly, the fast rates of phenanthrene degradation observed on day 7 of the experiment are most likely a result of the prior exposure of this soil to PAHs. The constant leakage of PAHs into the soil from timber treatment may have stimulated any PAH degrading bacteria present or left a “seed” population of PAH-degrading bacteria capable of fast response to new pulses of PAH contamination. The greater phenanthrene degradation rate on day 7 coincided with a clear shift in bacterial community composition, indicating that phenanthrene degraders may have adapted and metabolized this compound. Actinobacteria and Proteobacteria are frequently observed as dominant phyla in soil (Janssen, 2006; Delgado-Baquerizo et al., 2016; Fierer, 2017) and representatives of both phyla have previously been associated with PAH degradation in soil (Mukherjee et al., 2014). In this study, both the bacterial diversity data and the abundance of the Gram-positive PAH-RHDα genes suggest that the Actinobacteria were the main contributors to phenanthrene degradation. This is supported by the previous study by de Menezes et al. (2012), which reported a significant increase in transcripts associated with dioxygenases from Actinobacteria but few from the Proteobacteria in the same soil on day 7.
The higher relative abundance of Actinobacteria in PAH amended soil appears to have been driven largely by an increase in the abundance of a single unclassified OTU from the family Micrococcaceae (OTU 1). Members of the Micrococcaceae such as Arthrobacter phenanthrneivorans (Kallimanis et al., 2009) and Arthrobacter oxydans (Thion et al., 2013) have been associated with PAH degradation, both in pure culture and in the environment (Aryal and Kyriakides-Liakopoulou, 2013). Although considerably less relatively abundant than the Micrococcaceae, unclassified Microbacteriaceae were also significantly more abundant in phenanthrene-amended soil, and this group has also been associated with PAH degradation in soil (Jacques et al., 2008; Jie et al., 2011). Although, based on relative abundance, the Proteobacteria appeared to play a smaller role in PAH-degradation in this study compared to the Actinobacteria, six OTUs classified to this phylum were significantly more abundant on days 7 and 17 in the contaminated soils compared to the controls, and this included the second most abundant phenanthrene-responsive OTU, which was classified to the Sphingomonas genus, often associated to PAH contamination (Leys et al., 2004). Both OTU 1 and OTU 2 were already amongst the most abundant in the test soils at the start of the experiment, which might have given them competitive advantage against other potential PAH-degrading taxa (O'Malley et al., 2010). Their original dominance in these soils may be connected to the previous history of PAH contamination of the source soil due to timber treatment, which is supported by the presence of PAH-RHDα genes in the unamended soil (Cerniglia, 1992; Johnsen and Karlson, 2005).
The involvement of OTU1 in phenanthrene degradation was suggested by the results of DeSeq2 analysis which demonstrated that the relative abundance of this OTU increased substantially at the same time as the PAH was being removed from soil. However, the relative abundance of OTU1 does not directly mirror the total abundance of the Gram-positive PAH-RHDα gene (Figures 3, 5). This discrepancy may be due to changes in the total abundance of the remainder of the bacterial community in these soils, or due to this OTU expressing a PAH-RHDα gene which is not amplified by the qPCR primers used. A further possibility is that this OTU was not directly involved in the phenanthrene ring hydrolysis, but that it utilized its degradation products generated by other members of the microbial community (Festa et al., 2013). The relative abundance of two other actinobacterial OTUs (OTU 40, unclassified Actinomycetales and OTU 82, Mycobacterium sp.) showed stronger correlations with the total abundance of the PAH-RHDα gene than OTUs 1 and 2, indicating that OTUs 40 and 82 may therefore have been the primary taxa involved in the initial stages of phenanthrene degradation. Actinobacteria have been reported to be the primary taxa involved in PAH degradation (Uyttebroek et al., 2006; Cébron et al., 2008; Marcos et al., 2009) and a metatranscriptomic analysis of one time point during PAH degradation in soil reported that transcripts of PAH hydroxylase genes present were primarily from the Actinobacteria (de Menezes et al., 2012). The increase in the abundance of some Gram-negative OTUs of taxa associated with PAH degradation (e.g., Sphingomonas) without a concurrent increase in the abundance of the Gram-negative PAH-RHDα gene could also be explained by cross feeding between ring-hydroxylating bacteria and other bacteria capable of utilizing phenanthrene breakdown products (McDonald et al., 2005).
The considerable decrease in the relative abundance of OTU 1 and the concurrent rise in the relative abundance of the Proteobacteria between days 7 and 17 in the phenanthrene-amended microcosms indicate that the community was returning to its original composition. This conclusion is further supported by the closer proximity of phenanthrene-amended day 17 samples to day 0 and 2 sample data points from phenanthrene-amended and control samples in the PCoA plot. The return of the bacterial community to its original composition demonstrates the resilience of the bacteria to the impact of phenanthrene, which is likely due to their previous exposure to PAHs in the original site from where the soil was sourced, as discussed above. Bacterial communities have been reported to recover from exposure to pollutants such as PAHs, but the length of time this recovery may take will depend on a range of factors including concentration and type of pollutant (Bordenave et al., 2007; Rodriguez et al., 2015). It may be that the growth of OTUs 1, 40, and 82 was key to the resilience of the overall bacterial community to phenanthrene on account of their possible role in removing this pollutant from the soil. With most of phenanthrene exhausted from the soil, OTU 1 may have lost its competitive advantage, and other microorganisms were then able to grow and return to their original relative abundances.
Interestingly, the same unclassified Micrococcaceae and Sphingomonas OTUs that responded positively to phenanthrene amendment also increased in relative abundance to a lesser extent in the control microcosms, following similar abundance temporal patterns as in the amended microcosms. The increase in abundance of the two dominant phenanthrene-responsive OTUs in both the PAH amended and control microcosms suggests that the ability to withstand PAH toxicity and use these compounds as growth substrates may be related to their general ecological opportunism. Although widely used to generate useful information, microcosm-based studies have several limitations (Carpenter, 1996). Harvesting of soil and set-up of microcosms can introduce oxygen and affect soil structure, which are known to affect microbial communities (Fierer et al., 2003). Opportunistic bacteria are often the first to colonize a habitat following a disturbance and show adaptations such as fast growth on limiting substrates and high tolerance to environmental stress (Sigler et al., 2002; Sigler and Zeyer, 2004; Fierer et al., 2010; Lozupone et al., 2012). It would appear that disturbance-driven ecological succession may have occurred in the phenanthrene-amended and to a lesser extent in the control soil microbiome in this experiment (Sigler and Zeyer, 2004), however the presence of phenanthrene enhanced the dominance of these opportunistic strains. The presence of an opportunistic phenotype is supported by results from the metatranscriptomic study undertaken on the same soils. Transcripts classified as heat shock proteins from Actinobacteria and Alphaproteobacteria were observed to increase when phenanthrene was added to the soil (de Menezes et al., 2012) and similar genes were identified as a genetic signature of gut early-colonizing opportunistic bacteria by Lozupone et al. (2012).
The growth and decline of the dominant PAH-responsive bacteria in the current study decreased community evenness, in agreement with the study of Thomas and Cébron (2016) who observed a decrease in bacterial community evenness two days following phenanthrene soil amendment. However, relationships between microbial evenness (Shannon diversity) and disturbance vary depending on the frequency, intensity and type of disturbance (Gibbons et al., 2016). These relationships were not tested specifically in the current experiment and warrant further investigation.
The presence of phenanthrene led to significant shifts in the structure of soil bacterial communities, and this change was dominated by an increase in the relative abundance of a small number of opportunistic taxa. The prior exposure to pollution of the soil used in this experiment may have played a role in conditioning the soil community for fast responses to pulses of PAH contamination through the growth of opportunistic, fast-growing PAH-degrading bacteria mainly from the Actinobacteria phylum. The fact that 8 out of the13 PAH-responsive bacterial OTUs in this experiment were not classified to genus level (Table 3) highlights the need to better characterize uncultured soil microorganisms which have beneficial roles in bioremediation. Finally, the recognition that the soil bacterial community's response to phenanthrene contamination is at least in some cases restricted to a small number of bacterial taxa may simplify the ecological modeling and design of PAH-remediation strategies.
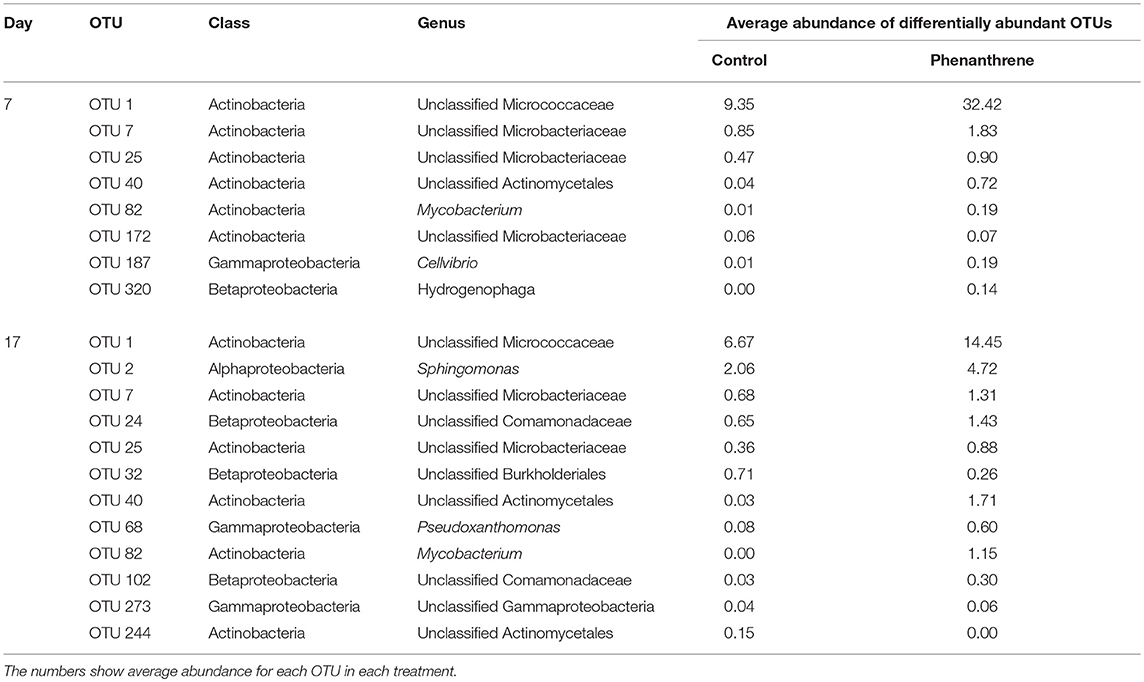
Table 3. Results from differential abundance analysis using DESeq2 (alpha = 0.05) showing the number of OTUs that are significant more abundant in either phenanthrene-amended or unamended samples at days 7 and 17 of the experiment.
SS is a postdoctoral fellow and contributed to qPCR analysis, amplicon data analysis and preparation of the manuscript. MA contributed to the qPCR analysis. ED and NC are the PIs and were responsible for experimental design. ED also contributed to the writing of the paper. AdM contributed to experimental design, set up the microcosm experiment, carried out the amplicon sequencing and the majority of the data analysis and contributed to preparation of the manuscript.
This work was funded under the Irish Environmental Protection Agency STRIVE programme 2007–2013 (grant no. 2008-PhD-WRM-1), the Ireland-Wales Programme 2007-2013 (GIFT project), and the SLAB scholarship programme of the Malaysian Ministry of Higher Education and the International Islamic University, Malaysia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02815/full#supplementary-material
Anon (2012). Soil Contamination: A Severe Risk for the Environment and Human Health. Available online at: http://globalsoilweek.org/wpcontent/uploads/2014/11/GSW_factsheet_Contamination_en.pdf
Aryal, M., and Kyriakides-Liakopoulou, M. (2013). Biodegradation and kinetics of phenanthrene and pyrene in the presence of non-ionic surfactants by Arthrobacter strain Sphe3. Water Air Soil Pollut. 224, 1426–1436. doi: 10.1007/s11270-012-1426-8
Bargiela, R., Mapelli, F., Rojo, D., Chouaia, B., Tornés, J., Borin, S., et al. (2015). Bacterial population and biodegradation potential in chronically crude oilcontaminated marine sediments are strongly linked to temperature. Sci. Rep. 5:11651. doi: 10.1038/srep11651
Bastiaens, L., Springael, D., Wattiau, P., Harms, H., deWachter, R., Verachtert, H., et al. (2000). Isolation of adherent polycyclic aromatic hydrocarbon (PAH)-degrading bacteria using PAH-sorbing carriers. Appl. Environ. Microbiol. 66, 1834–1843. doi: 10.1128/AEM.66.5.1834-1843.2000
Bordenave, S., Goñi-Urriza, M. S., Caumette, P., and Duran, R. (2007). Effects of heavy fuel oil on the bacterial community structure of a pristine microbial mat. Appl. Environ. Microbiol. 73, 6089–6097. doi: 10.1128/AEM.01352-07
Brussaard, L. (2012). “Ecosystem services provided by the soil biota,” in Soil Ecology and Ecosystem Services, eds R. D. Bardgett, J. E. Herrick, J. Six, T. Heffin Jones, D.R. Strong, and W. H. van der Putten (Oxford, UK: Oxford University Press), 45–58.
Carpenter, S. R. (1996). Microcosm experiments have limited relevance for community and ecosystem ecology. Ecology 77, 677–680. doi: 10.2307/2265490
Cébron, A., Norini, M. P., Beguiristain, T., and Leyval, C. (2008). Real-Time PCR quantification of PAH-ring hydroxylating dioxygenase genes from Gram-positive and Gram-negative bacteria in soil and sediment samples. J. Microbiol. Methods 73, 148–159. doi: 10.1016/j.mimet.2008.01.009
Cerniglia, C. E. (1992). Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation 3, 351–368. doi: 10.1007/BF00129093
Cerniglia, C. E. (1997). Fungal metabolism of polycyclic aromatic hydrocarbons: past, present and future applications in bioremediation. J. Ind. Microbiol. Biotechnol. 19, 324–333. doi: 10.1038/sj.jim.2900459
Collins, C. D., Mosquera-Vazquez, M., Gomez-Eyles, J., Mayer, P., Gouliarmou, V., and Blum, F. (2013). Is there sufficient ‘sink' in current bioaccessibility determinations of organic pollutants in soils? Environ. Pollut. 181, 128–132. doi: 10.1016/j.envpol.2013.05.053
de Menezes, A., Clipson, N., and Doyle, E. M. (2012). Comparative metatranscriptomics reveals widespread community responses during phenanthrene degradation in soil. Environ. Microbiol. 14, 2577–2588. doi: 10.1111/j.1462-2920.2012.02781.x
Dean, J. R., and Xiong, G. H. (2000). Extraction of organic pollutants from environmental matrices: selection of extraction technique. Trac. Trends Anal. Chem. 19, 553–564. doi: 10.1016/S0165-9936(00)00038-8
Delgado-Balbuena, L., Bello-Lopez, J. M., Navarro-Noya, Y. E., Rodríguez-Valentín, A., Luna-Guido, M. L., and Dendooven, L. (2016). Changes in the bacterial community structure of remediated anthracene-contaminated soils. PLoS ONE 11:e0160991. doi: 10.1371/journal.pone.0160991
Delgado-Baquerizo, M., Maestre, F. T., Reich, P. B., Jeffries, T. C., Gaitan, J. J., Encinar, D., et al. (2016). Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 7:10541. doi: 10.1038/ncomms10541
Doyle, E., Muckian, L., Hickey, A. M., and Clipson, N. (2008). Microbial PAH degradation. Adv. Appl. Microbiol. 65, 27–66. doi: 10.1016/S0065-2164(08)00602-3
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fernández-Luqueño, F., Valenzuela-Encinas, C., Marsch, R., Martínez-Suárez, C., Vázquez-Núñez, E., and Dendooven, L. (2011). Microbial communities to mitigate contamination of PAHs in soil—possibilities and challenges: a review. Environ. Sci. Pollut. Res. 18, 12–30. doi: 10.1007/s11356-010-0371-6
Festa, S., Coppotelli, B. M., and Morelli, I. S. (2013). Bacterial diversity and functional interactions between bacterial strains from a phenanthrene-degrading consortium obtained from a chronically contaminated-soil. Int. Biodeterior. Biodegrad. 85, 42–51. doi: 10.1016/j.ibiod.2013.06.006
Festa, S., Macchi, M., Cortes, F., Morelli, I. S., and Coppotelli, B. M. (2016). Monitoring the impact of bioaugmentation with a PAH-degrading strain on different soil microbiomes using pyrosequencing. FEMS Microbiol. Lett. 92:125. doi: 10.1093/femsec/fiw125
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Micro. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., Nemergut, D., Knight, R., and Craine, J. M. (2010). Changes through time: integrating microorganisms into the study of succession. Res. Microbiol. 161, 635–642. doi: 10.1016/j.resmic.2010.06.002
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 35, 167–176. doi: 10.1016/S0038-0717(02)00251-1
Fuentes, S., Mendez, V., Aguila, P., and Seeger, R. M. (2014). Bioremediation of petroleum hydrocarbons: catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 98, 4781–4794. doi: 10.1007/s00253-014-5684-9
Ghosal, D., Ghosh, S., Dutta, T. K., and Ahn, Y. (2016). Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front. Microbiol. 7:1369. doi: 10.3389/fmicb.2016.01369
Gibbons, S. M., Scholz, M., Hutchison, A. L., Dinner, A. R., Gilbert, J. A., and Coleman, M. L. (2016). Disturbance regimes predictably alter diversity in an ecologically complex bacterial system. Mbio 7:e01372. doi: 10.1128/mBio.01372-16
Griffiths, B. S., and Philipot, L. (2013). Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol. Rev. 37, 112–129. doi: 10.1111/j.1574-6976.2012.00343.x
Griffiths, R. I., Whiteley, A. S., O'Donnell, G., and Bailey, M. J. (2000). Rapid method for coextraction of DNA and RNA from natural environments for analsis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66, 5488–5491. doi: 10.1128/AEM.66.12.5488-5491.2000
Jacques, R. J. S., Okeke, B. C., Bento, F. M., Teixeira, A. S., Peralba, M. C. R., and Camargo, F. A. O. (2008). Microbial consortium bioaugmentation of a PAH-contaminated soil. Biores. Technol. 99, 2637–2643. doi: 10.1016/j.biortech.2007.04.047
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006
Jiao, S., Zhang, Z., Yang, F., Lin, Y., Chen, W., and Wei, G. (2017). Temporal dynamics of microbial communities in microcosms in response to pollutants. Mol. Ecol. 26, 923–936. doi: 10.1111/mec.13978
Jie, H. L., Yun, Y. C., Yun, T., Hui, L. G., and Ling, Z. T. (2011). Using population dynamics analysis by DGGE to design the bacterial consortium isolated from mangrove sediments for biodegradation of PAHs. Int. Biodeterior. Biodegradation. 65, 269–275. doi: 10.1016/j.ibiod.2010.11.010
Johnsen, A. R., and Karlson, U. (2005). PAH degradation capacity of soil microbial communities – does it depend on PAH exposure? Microb. Ecol. 50, 488–495. doi: 10.1007/s00248-005-0022-5
Kallimanis, A., Kavakiotis, K., Perisynakis, A., Spröer, C., Pukall, R., Drainas, C., et al. (2009). Arthrobacter phenanthrenivorans sp. nov., to accomodate the phenanthere-degrading bacterium Arthrobacter sp. strain Sphe3. Int. J. Syst. Evol. Microbiol. 24, 569–581. doi: 10.1099/ijs.0.000984-0
Kettler, T., Doran, J., and Gilbert, T. (2001). Simplified method for soil particle-size determination to accompany soil-quality analyses. Soil Sci. Soc. Am. J. 65, 849–852. doi: 10.2136/sssaj2001.653849x
Kuppusamy, S., Thavamani, P., Megharaj, M., Venkateswarlu, K., Lee, Y. B., and Naidu, R. (2016). Pyrosequencing analysis of bacterial diversity in soils contaminated long-term with PAHs and heavy metals: implications to bioremediation. J. Hazard. Mater. 317, 169–179. doi: 10.1016/j.jhazmat.2016.05.066
Lane, D. J., Pace, B., Olsen, G. J., Stahl, D. A., Sogin, M. L., and Pace, N. R. (1985). Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. U.S.A. 82, 6955–6959. doi: 10.1073/pnas.82.20.6955
Leys, N., Ryngaert, A., Bastiaens, L., Verstraete, W., Top, E. M., and Springael, D. (2004). Occurrence and phylogenetic diversity of Sphingomonas strains in soils contaminated with polycyclic aromatic hydrocarbons. Appl Environ. Microbiol. 70, 1944–1955. doi: 10.1128/AEM.70.4.1944-1955.2004
Lors, C., Damidot, D., Ponge, J. F., and Périé, F. (2012). Comparison of a bioremediation process of PAHs in a PAH-contaminated soil at field and laboratory scales. Environ. Pollut. 165, 11–17. doi: 10.1016/j.envpol.2012.02.004
Lors, C., Ryngaert, A., Périé, F., Diels, L., and Damidot, D. (2010). Evolution of bacterial community during bioremediation of PAHs in a coal tar contaminated soil. Chemosphere 81, 1263–1271. doi: 10.1016/j.chemosphere.2010.09.021
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold-change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–571. doi: 10.1186/s13059-014-0550-8
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansen, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Marcos, M. S., Lozada, M., and Dionisi, H. M. (2009). Aromatic hydrocarbon degradation genes from chronically polluted subantarctic marine sediments. Lett. Appl. Microbiol. 49, 602–608. doi: 10.1111/j.1472-765X.2009.02711.x
Martínez, I., Wallace, G., Zhang, C. M., Legge, R., Benson, A. K., Carr, T. P., et al. (2009). Diet induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl. Environ. Microbiol. 75, 4175–4184. doi: 10.1128/AEM.00380-09
McDonald, I. R., Radajewski, S., and Murrell, J. C. (2005). Stable isotope probing of nucleic acids in methanotrophs and methylotrophs: a review. Org. Geochem. 36, 779–787. doi: 10.1016/j.orggeochem.2005.01.005
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: An R package for reroducible interactive ananlysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
McMurdie, P. J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10:e1003531. doi: 10.1371/journal.pcbi.1003531
Muckian, L. M., Grant, R. J., Clipson, N. J., and Doyle, E. M. (2009). Bacterial community dynamics during bioremediation of phenanthrene-and fluoranthene-amended soil. Int. Biodeterior. Biodegr. 63, 52–56. doi: 10.1016/j.ibiod.2008.04.005
Mukherjee, S., Juottenen, H., Siivonen, P., Quesada Lloret, C., Tuomi, P., Pulkkinen, P., et al. (2014). Spatial patterns of microbial diversity and activity in an aged creosote-contaminated site. ISME J. 8, 2131–2142. doi: 10.1038/ismej.2014.151
Muyzer, G., Dewaal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S ribosomal RNA. Appl. Environ. Microbiol. 59, 695–700.
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, P. R., et al. (2018). vegan: Community Ecology Package version 2.4-2. Available online at: https://cran.r-project.org/web/packages/vegan/index.html.
O'Malley, L., Korniss, G., and Mungara, S. S. P. (2010). Spatial competition and the dynamics of rarity in a temporally varying environment. Evol. Ecol. Res. 12, 279–305. Available online at: http://www.evolutionary-ecology.com/issues/v12/n03/ccar2546.pdf
Posada-Baquero, R., and Ortega-Calvo, J. J. (2011). Recalcitrance of polycyclic aromatic hydrocarbons in soil contributes to background pollution. Environ. Pollut. 159, 3692–3699. doi: 10.1016/j.envpol.2011.07.012
Rodriguez, R. L., Overholt, W. A., Hagan, C., Huettel, M., Kostka, J. E., and Konstantinidis, K. T. (2015). Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9, 1928–1940. doi: 10.1038/ismej.2015.5
Sawulski, P., Clipson, N., and Doyle, E. (2014). Effects of polycyclic aromatic hydrocarbons on microbial community structure and PAH ring hydroxylating dioxygenase gene abundance in soil. Biodegradation 25, 835–847. doi: 10.1007/s10532-014-9703-4
Sayara, T., Borràs, E., Caminal, G., Sarr,à, M., and Sánchez, A. (2011). Bioremediation of PAH-contaminated soil through composting: Influence of bioaugmentation and biostimulation on contaminant biodegradation. Int. Biodeterior. Biodegr. 65, 859–865. doi: 10.1016/j.ibiod.2011.05.006
Schloss, P. D. (2009). A high-throughput DNA sequence aligner for microbial ecology studies. PLoS ONE 4:e8230. doi: 10.1371/journal.pone.0008230
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shade, A. (2017). Diversity is the question, not the answer. ISME J. 11, 1–6. doi: 10.1038/ismej.2016.118
Sigler, W. V., Crivii, S., and Zeyer, J. (2002). Bacterial succession in glacial forefield soils characterized by community structure, activity and opportunistic growth dynamics. Microb. Ecol. 44, 306–316. doi: 10.1007/s00248-002-2025-9
Sigler, W. V., and Zeyer, J. (2004). Colony-forming analysis of bacterial community succession in deglaciated soils indicates pioneer stress-tolerant opportunists. Microb. Ecol. 48, 316–323. doi: 10.1007/s00248-003-0189-6
Storey, S., Ashaari, M. M., McCabe, G., Harty, M., Dempsey, R., Doyle, O., et al. (2014). Microbial community structure during fluoranthene degradation in the presence of plants. J. Appl. Microbiol. 117, 74 – 84. doi: 10.1111/jam.12518
Sun, F. L., Wang, Y. S., Sun, C. C., Peng, Y. L., and Deng, C. (2012). Effects of three different PAHs on nitrogen-fixing bacterial diversity in mangrove sediment. Ecotoxicology 21, 1651–1660. doi: 10.1007/s10646-012-0946-8
Thion, C., Cébron, A., Beguiristain, T., and Leyval, C. (2013). Inoculation of PAH-degrading microbial interactions and PAH dissipation. Biodegradation 24, 569–581. doi: 10.1007/s10532-013-9628-3
Thomas, F., and Cébron, A. (2016). Short-term rhizosphere effect on available carbon sources, phenanthrene degradation, and active microbiome in an aged-contaminated industrial soil. Front. Microbiol. 7:92. doi: 10.3389/fmicb.2016.00092
Uyttebroek, M., Breugelmans, P., Janssen, M., Wattiau, P., Joffe, B., Karlson, U., et al. (2006). Distribution of the Mycobacterium community and polycyclic aromatic hydrocarbons (PAHs) among different size. Environ. Microbiol. 8, 836–847. doi: 10.1111/j.1462-2920.2005.00970.x
Wang, L. W., Li, F., Zhan, Y., and Zhu, L. (2016). Shifts in microbial community structure during in situ surfactant-enhanced bioremediation of polycyclic aromatic hydrocarbon-contaminated soil. Environ. Sci. Pollut. Res. 23, 14451–14461. doi: 10.1007/s11356-016-6630-4
Wang, W., Massey Simonich, S., Xue, M., Zhao, J., Zhang, N., Wang, R., et al. (2010). Concentrations, sources and spatial distribution of polycyclic aromatic hydrocarbons in soils from Beijing, Tianjin and surrounding areas, North China. Environ. Pollut.158, 1245–1251. doi: 10.1016/j.envpol.2010.01.021
Wickliffe, J., Overton, E., Frickel, S., Howard, J., Wilson, M., Simon, B., et al. (2014). Evaluation of polycyclic aromatic hydrocarbons using analytical methods, toxicology, and risk assessment research: seafood safety after a petroleum spill as an example. Environ. Health Perspect. 122, 6–9. doi: 10.1289/ehp.1306724
Yang, Y., Wang, J., Liao, J., Xie, S., and Huang, Y. (2015). Abundance and diversity of soil petroleum hydrocarbon-degrading microbial communities in oil exploring areas. Appl. Microbiol. Biotechnol. 99, 1935–1946. doi: 10.1007/s00253-014-6074-z
Zafra, G., Taylor, T. D., Absalón, A. E., and Cortés-Espinosa, D. V. (2016). Comparative metagenomic analysis of PAH degradation in soil by a mixed microbial consortium. J. Hazard. Mater. 318, 702–710. doi: 10.1016/j.jhazmat.2016.07.060
Keywords: polycyclic aromatic hydrocarbons, microbiome, bioremediation, soil, phenanthrene
Citation: Storey S, Ashaari MM, Clipson N, Doyle E and de Menezes AB (2018) Opportunistic Bacteria Dominate the Soil Microbiome Response to Phenanthrene in a Microcosm-Based Study. Front. Microbiol. 9:2815. doi: 10.3389/fmicb.2018.02815
Received: 13 December 2017; Accepted: 02 November 2018;
Published: 21 November 2018.
Edited by:
Diana Elizabeth Marco, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Wenli Chen, Huazhong Agricultural University, ChinaCopyright © 2018 Storey, Ashaari, Clipson, Doyle and de Menezes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre B. de Menezes, YWxleGFuZHJlLmRlbWVuZXplc0BudWlnYWx3YXkuaWU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.