
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 20 November 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02811
 Alex Ranieri Jerônimo Lima1*
Alex Ranieri Jerônimo Lima1* Andrei Santos Siqueira1
Andrei Santos Siqueira1 Janaina Mota de Vasconcelos1James Siqueira Pereira2Juliana Simão Nina de Azevedo2Pablo Henrique Gonçalves Moraes1Délia Cristina Figueira Aguiar1Clayton Pereira Silva de Lima3João Lídio Silva Gonçalves Vianez-Júnior3Márcio Roberto Teixeira Nunes3Luciana Pereira Xavier4
Janaina Mota de Vasconcelos1James Siqueira Pereira2Juliana Simão Nina de Azevedo2Pablo Henrique Gonçalves Moraes1Délia Cristina Figueira Aguiar1Clayton Pereira Silva de Lima3João Lídio Silva Gonçalves Vianez-Júnior3Márcio Roberto Teixeira Nunes3Luciana Pereira Xavier4 Leonardo Teixeira Dall’Agnol5Evonnildo Costa Goncalves1
Leonardo Teixeira Dall’Agnol5Evonnildo Costa Goncalves1Currently only four genome sequences for Limnothrix spp. are publicly available, and information on the genetic properties of cyanobacteria belonging to this genus is limited. In this study, we report the draft genome of Limnothrix sp. CACIAM 69d, isolated from the reservoir of a hydroelectric dam located in the Amazon ecosystem, from where cyanobacterial genomic data are still scarce. Comparative genomic analysis of Limnothrix revealed the presence of key enzymes in the cyanobacterial central carbon metabolism and how it is well equipped for environmental sulfur and nitrogen acquisition. Additionally, this work covered the analysis of Limnothrix CRISPR-Cas systems, pathways related to biosynthesis of secondary metabolites and assembly of extracellular polymeric substances and their exportation. A trans-AT PKS gene cluster was identified in two strains, possibly related to the novel toxin Limnothrixin biosynthesis. Overall, the draft genome of Limnothrix sp. CACIAM 69d adds new data to the small Limnothrix genome library and contributes to a growing representativeness of cyanobacterial genomes from the Amazon region. The comparative genomic analysis of Limnothrix made it possible to highlight unique genes for each strain and understand the overall features of their metabolism.
The Cyanobacteria phylum (domain Bacteria) is one of the oldest and most morphologically diverse on the planet. The emergence of oxygen in the atmosphere, around 2.22–2.45 billion years ago, is attributed to the ability of cyanobacteria to perform photosynthesis (Schirrmeister et al., 2011). Given their metabolic diversity, cyanobacteria have a ubiquitous distribution, inhabiting various environments including freshwater, oceans, and soils. They also can be found in association with other organisms and in extreme environments, such as deserts or Antarctica (Vis, 2005; Komárek et al., 2012; Rigonato et al., 2012). Taxonomic studies, based on morphological criteria, have organized cyanobacteria into five subsections: (I) Chroococcales, (II) Pleurocapsales, (III) Oscillatoriales, (IV) Nostocales, and (V) Stigonematales (Rippka et al., 1979).
Cyanobacteria have been widely studied and exploited because of their biotechnological potential. Some cyanobacterial compounds have exhibited antibacterial, immunosuppressant, anticancer, anti-HIV and anti-fungal activities (Vijayakumar and Menakha, 2015). This ability to produce several bioactive compounds is mainly due to two biosynthetic enzyme complexes: NRPS (non-ribosomal peptide synthetases) and PKS (polyketide synthases). The NRPS complex can incorporate either proteinogenic or non-proteinogenic amino acids into the final peptide structure. Polyketides are biosynthesized by the PKS complex from acyl-CoA. Both enzyme complexes are similar in their biochemical logic, composing enzymes containing several modules, with each module performing a biochemical reaction in a sequential order. Thus, the order of modules determines the peptide or polyketide chain sequence (Micallef et al., 2015). One known exception is the trans-AT PKS system, which has no AT (acyltransferase) domain arranged in a modular fashion. Instead, the synthesized molecule receives an acyl building block through a free-standing AT (Helfrich and Piel, 2016). Additionally, the great diversity of genes encoding these enzymes can be combined to produce different metabolites (Nunnery et al., 2010).
Given their environmental and biotechnological importance, the number of available cyanobacterial genome sequences in GenBank has increased recently, from 340 reported genomes in 2015 (Land et al., 2015) to 838 as of April 2018 (GenBank prokaryotes.txt file). However, this number represents only 0.61% of the total bacterial genomes available. Despite recent attempts to increase the amount of available genomes in the five cyanobacteria subsections (Shih et al., 2013; Zhu et al., 2017), public genomic databases are lacking in geographical diversity, since many sequenced cyanobacteria are of European origin (Alvarenga et al., 2017).
Although the Brazilian Amazon is known to harbor vast biodiversity, cyanobacterial genomic data are scarce for this biome (Rigonato et al., 2017). Previous metagenomic analysis conducted in Tucuruí Hydroelectric Reservoir (3°49′55″S, 49°38′50″W), which has an area of 1,783 km2 of flooded primary Amazonian forest (Fearnside, 2001) (Figure 1), revealed a high diversity of cyanobacteria (Baraúna et al., 2013; Das Graças et al., 2015). The reservoir has little human interference in its water quality (Curtarelli et al., 2016), and rainfall is the main source of nutrients when it is full, during the wet period from January to April; from March to December (the dry period) the nutrients are released from the lake bottom (Pettersson and Pozdnyakov, 2013). Due to rainfall changes, the pH and some nutrients such as nitrite, nitrate and phosphate may vary according to the seasons, affecting the average cyanobacterial abundance, which is higher in the wet period. Although the reservoir presents quite different physicochemical characteristics depending on sampling point, can be classified, in general, as mesotrophic with few oligotrophic sites (Brandão et al., 2017).
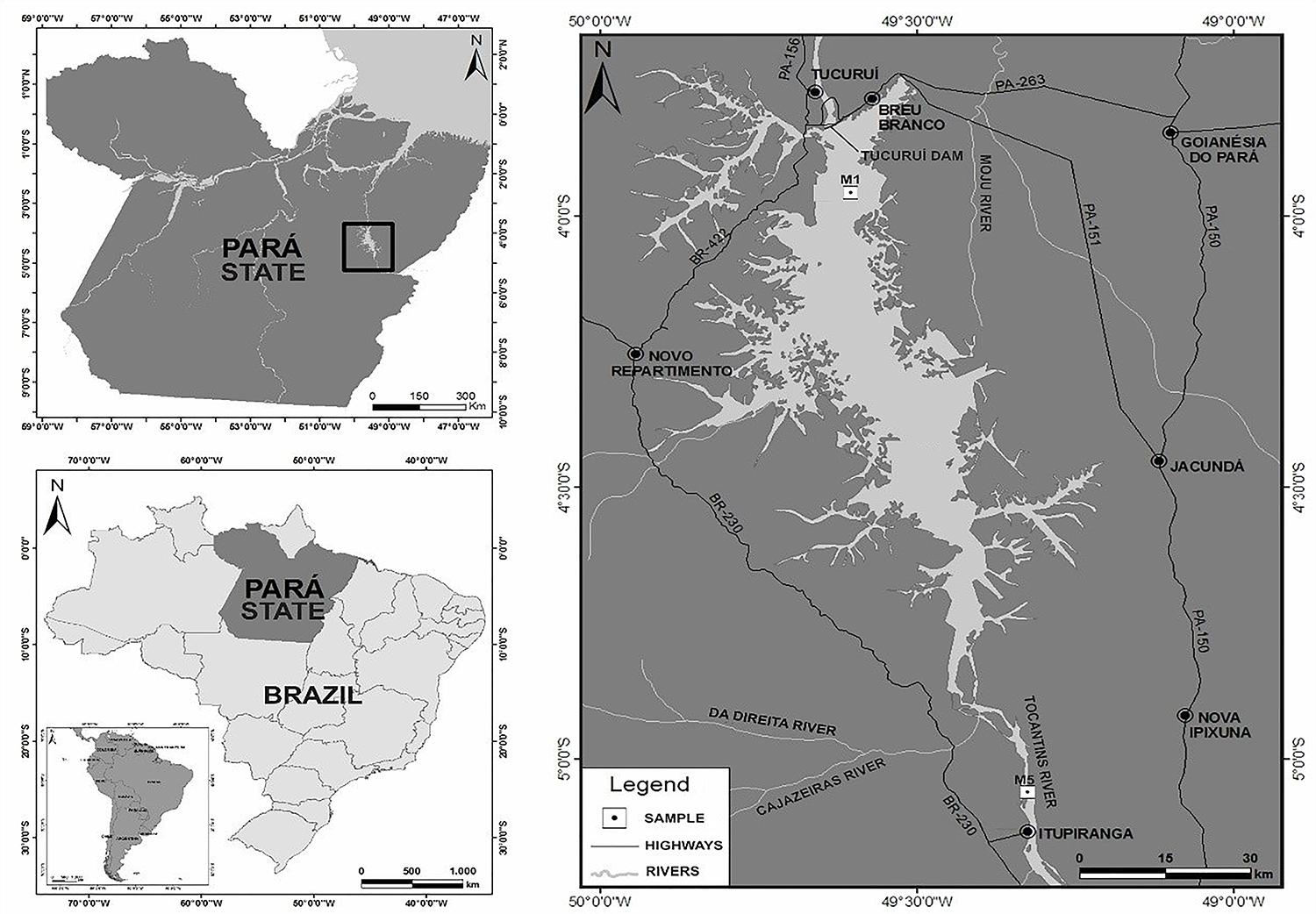
FIGURE 1. Map of Tucuruí Hydroelectric Dam, showing the M1 sampling point.
The high diversity of cyanobacteria, along with the distinct environmental characteristics of Tucuruí Hydroelectric Reservoir, led our group to isolate these microorganisms from that area, depositing them in the CACIAM collection (in Portuguese: Coleção Amazônica de Cianobactérias e Microalgas or Amazonian Collection of Cyanobacteria and Microalgae). Some representatives of the CACIAM collection have already been sequenced and published (Lima et al., 2014; Castro et al., 2016), including the genome of the newly described Alkalinema genus (Lima et al., 2017). Until the present study there were only three Limnothrix genomes available in public databases: Limnothrix sp. P13C2 (Tan et al., 2016), Limnothrix sp. PR1529 (GenBank: NZ_LIRO00000000) and Limnothrix rosea IAM M-220 NIES-208 (Zhu et al., 2017). In this work, we present the genome of Limnothrix sp. CACIAM 69d, isolated from Tucuruí Hydroelectric Reservoir (Figure 1).
The genus Limnothrix includes filamentous cyanobacteria, with no cellular differentiation or branching in their trichomes, presenting narrow cylindrical cells with polar and/or central aerotopes and very slight or non-existent motility (Zhu et al., 2012). This genus belongs to the cyanobacteria subsection III and has been classified within the order Oscillatoriales, being morphologically similar and phylogenetically closely related to Pseudanabaena spp. (Castenholz et al., 2015b). Some studies have been conducted with representatives of the genus Limnothrix in order to verify exopolysaccharides (EPS) (Khattar et al., 2010) and lipid production (Economou et al., 2015; Aboim et al., 2016; Hong et al., 2016; Oliveira et al., 2018), suggesting that this cyanobacteria could be used as a source for compounds of biotechnological interest. Besides that, Limnothrix (strain AC0243) is able to produce a toxin which causes severe toxicity to developing vertebrates with injuries particularly noted in the brain, notochord and pancreas, showing strong similarities to amino acid beta-N-methylamino-L-alanine (BMAA) intoxication (Daniels et al., 2014). Furthermore, another study revealed that the toxin produced by Limnothrix (strain AC0243) is capable of damaging the liver, lungs and gastrointestinal tract of mice. However, the authors were unable to isolate and identify this toxin (Humpage et al., 2012).
All these studies have been published in the absence of available genomes for Limnothrix, which may have limited the search for specific metabolites or their genes. Since the genomes are now available, it is possible to improve knowledge about this genus through genomic analysis. This approach is useful to gain insight into the diversity of cyanobacterial metabolism (Beck et al., 2012) and make it possible to identify several biosynthetic gene clusters which produce structurally diverse metabolites, such as alkaloids, terpenes, fatty acids, non-ribosomal peptides, UV-absorbing compounds, polyketides and ribosomal peptides (Micallef et al., 2015). Considering all those aspects, this work aims to perform a comparative analysis on the four available Limnothrix genomes, describing their metabolic characteristics and genome mining findings, emphasizing the strain CACIAM 69d.
Limnothrix genomes analyzed where obtained from GenBank, using the accession numbers: MBRF00000000 (Limnothrix sp. P13C2), NZ_LIRO00000000 (Limnothrix sp. PR1529) and NZ_MRBY00000000 (Limnothrix rosea IAM M-220 NIES-208).
The genome sequence of Limnothrix sp. CACIAM 69d has been deposited at DDBJ/ENA/GenBank under the accession MKGP00000000. The described in this paper is version MKGP00000000.2.
The isolate Limnothrix sp. strain CACIAM 69d was obtained from a water sample collected in December, 2010 from the M1 point of Tucuruí Hydroelectric Reservoir (3°49′55″S, 49°38′50″W), Pará, Brazil (Figure 1). Using a sterile plastic bottle, five liters of water were collected 50 cm below the lake surface. The water sample was sequentially filtered by negative pressure in four steps: prefiltration with qualitative filter paper grade 5 (Whatman, United Kingdom) and three consecutive filtrations using nitrocellulose membranes with pore diameters of 0.8 μm, 0.45 μm (Millipore, Brazil) and 0.2 μm (Whatman, United Kingdom); next, the 0.2 μm membrane containing the retained microorganisms was inoculated in BG-11 medium (Allen, 1968). After biomass had increased, the sample was inoculated in a cultivation medium containing cycloheximide (18.5 mg/mL) and then submitted to a five-step serial dilution process with factor 10. The sample was maintained in Erlenmeyer flasks at 29°C with a 13 h light/11 h dark regime. Monthly, an inoculum of Limnothrix sp. strain CACIAM 69d was transferred to another Erlenmeyer flask containing BG-11 medium.
The total DNA was extracted after 1 month of cultivation using a modified phenol–chloroform based protocol (Lin et al., 2010). One sequencing run was performed on a GS FLX 454 platform using a non-paired library, using standard protocols in 2011. The raw reads obtained were quality-filtered with a minimum Phred score of 20. Assembly was carried out using two tools: (i) Newbler v2.9 (Margulies et al., 2005), parameterized with minimum overlap of 40 bp, minimum overlap identity of 90%, heterozygote mode and extended low-depth overlap options; (ii) SPAdes 3.12.0 (Bankevich et al., 2012), using error correction and automatic k-mer estimation size. The assembly results were compared using QUAST (Gurevich et al., 2013).
Contigs from both assemblers were submitted to MaxBin 2.2 (Wu et al., 2015), MyCC (Lin and Liao, 2016), and MetaBAT 2 (Kang et al., 2015) for binning. CheckM (Parks et al., 2015) was used to obtain taxonomic classification of bins generated for each binning tool. Individually, for each assembly tool, bins identified as cyanobacteria were compared using checkM and QUAST to determine the best binning tool. Next, the best results of the best binning tool of each assembly were compared to choose the final representation of the assembly and binning process.
The cyanobacterial bin contamination assessment was performed by CheckM, identifying possible duplicated gene markers. BLASTp (Altschul et al., 1990) was used to identify which of the duplicated markers was a contamination. Next, genomic regions upstream and downstream of the contaminated marker gene were checked for contamination, using a preliminary annotation from RAST (Aziz et al., 2008) and BLASTp. Contaminated sequences were removed from the cyanobacterial bin and then a new contamination assess was performed by CheckM.
For Limnothrix sp. CACIAM 69d genome, the annotation was performed by NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). BLAST searches in BSRD database (Li et al., 2013) were executed to identify sRNA.
For all genomes, CRISPR arrays were identified by CRISPRDetect (Biswas et al., 2016), using default parameters. All unquestionable arrays considered by CRISPRDetect where selected for target prediction with CRISPRTarget (Biswas et al., 2013), comparing the spacer sequences against GenBank-Phage and RefSeq-Plasmid databases using default parameters. Whole genome BLAST search was performed against the COG database (Tatusov et al., 2003), using RPSBLAST with e-value of 1E-02. The genome mining process was carried out by a complete antiSMASH 4.0 (Blin et al., 2017) analysis, including whole-genome Pfam (Finn et al., 2014) searches. KS domain analysis was performed by NaPDoS (Ziemert et al., 2012), using the amino acid sequences of CDS presented in Table 4. Additional domain searches were performed by DELTA-BLAST against the NCBI’s Conserved Domain Database (CDD) (Marchler-Bauer et al., 2015). Metabolic pathway analysis was achieved by results obtained from a KEGG automatic annotation server KAAS (Moriya et al., 2007) and Reconstruct Pathway tool of KEGG Mapper. Additional searches where performed by BLAST using the amino acid sequences of Limnothrix sp. KNUA012 for AAR/ADO genes (GenBank accessions: KU341740 and KU341741) queries against translated CDS databases of each Limnothrix genome. For toxins, the following sequences where used as query (GenBank accession numbers): FJ477836.2 and JF803645.1 (anatoxin), KJ139742.1 (cylindrospermopsin), AY588942.1 (lyngbyatoxin), AY212249.1 and AF183408.1 (microcystin), AY210783.2 (nodularin), DQ787200.1 (saxitoxin). Pan-genome analysis was performed using the Bacterial Pan Genome Analysis Tool (BPGA) (Chaudhari et al., 2016), with default parameters.
To generate the Bayesian tree, 16S rRNA sequences were obtained from NCBI and Limnothrix genomes. GenBank accession numbers for each sequence are shown in Figure 2. The sequences were aligned with an online version of MAFFT (Katoh et al., 2017). The nucleotide substitution model used was SYM+G, determined by PAUP 4.0b10 (Swofford, 2002) and MrModeltest 2 (Nylander, 2004). The tree was constructed using MrBayes 3.2.6 (Ronquist et al., 2012), running with 107 generations, sampling every 100th iteration. TRACER 1.6 (Rambaut and Drummond, 2013) was used to check the performance of tree construction.
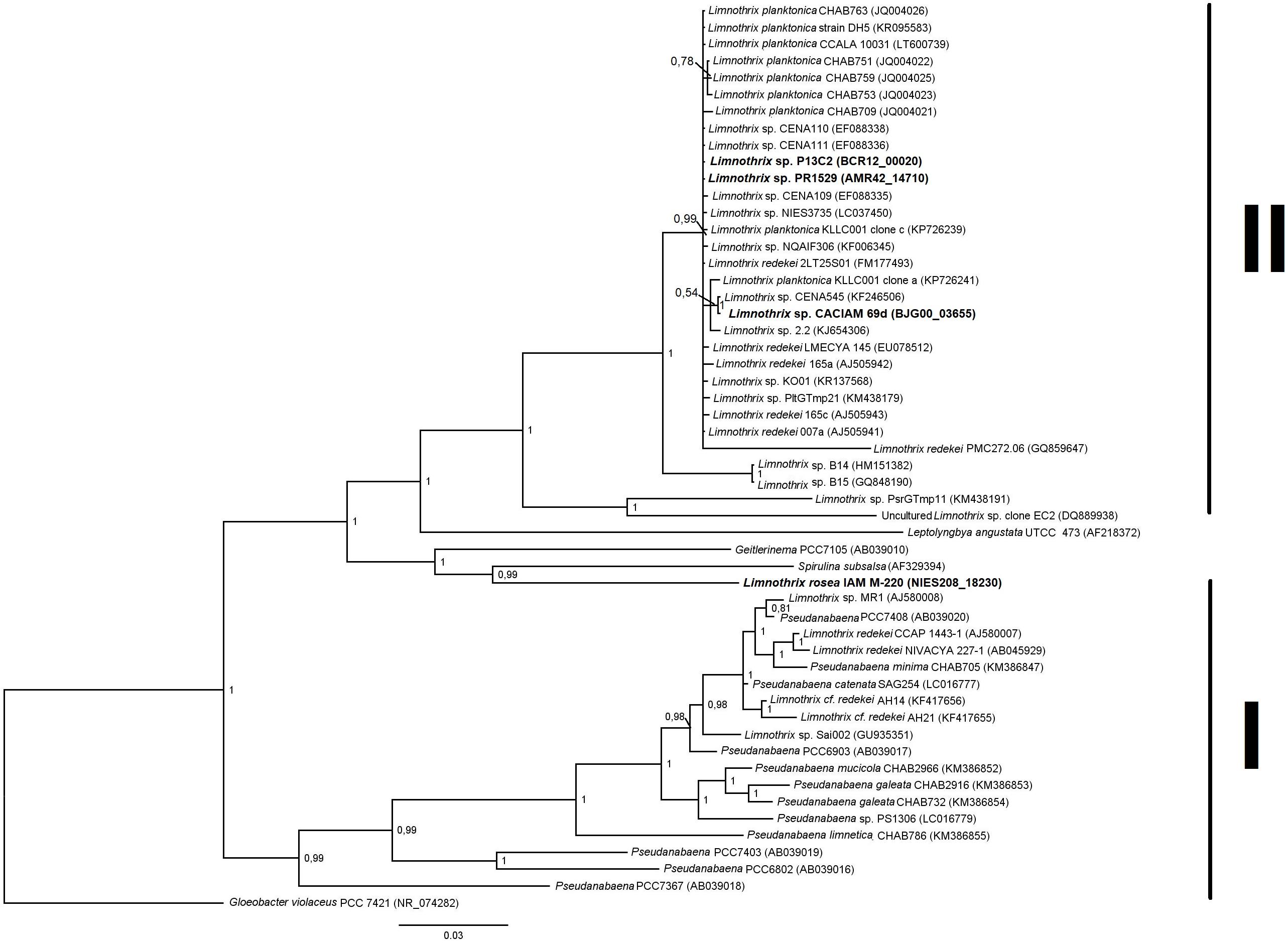
FIGURE 2. Bayesian phylogenetic tree based on 16S rRNA sequences from Limnothrix strains and other Oscillatorian genera. Limnothrix strains with sequenced genomes are shown in bold. Gloeobacter violaceus PCC 7421 was used as outgroup. GenBank accession numbers are in parentheses.
A phylogenetic tree of closely related genomes was constructed using the four Limnothrix genomes by the Species Tree App present in KBase. Briefly, this tool uses 49 highly conserved COG families to perform an alignment. The closest neighbors are extracted and concatenated to generate the tree using FastTree2, which is an approximation of the maximum likelihood method (Arkin et al., 2018).
Results of CACIAM 69d assembly are shown in Supplementary Table 1. SPAdes assemblies were performed using 21, 33, 55, 77, 99, and 127 k-mer sizes due to the auto parameter. SPAdes k-127 assembly produced more contigs, a higher number of bases and the longest contig. Newbler 2.9 presented a larger N50 value, but also presented some mismatches (630 N’s) in the assembled contigs. Since the goal was to assemble the cyanobacterial genome, contigs from both Newbler 2.9 and SPAdes k-127 were selected to perform the binning process.
All three binning tools were able to recover a single cyanobacterial bin using contigs from both assemblers as input, showing the same percentage of genome completeness (99.64%) and contamination level (0.34%) (Bin01 in Table 1). Other bins are predominantly proteobacteria, presenting coverage values lower than 8x, completeness levels ranging from 6.8 to 99.73%, and contamination estimation from 0.11 to 45.53%.
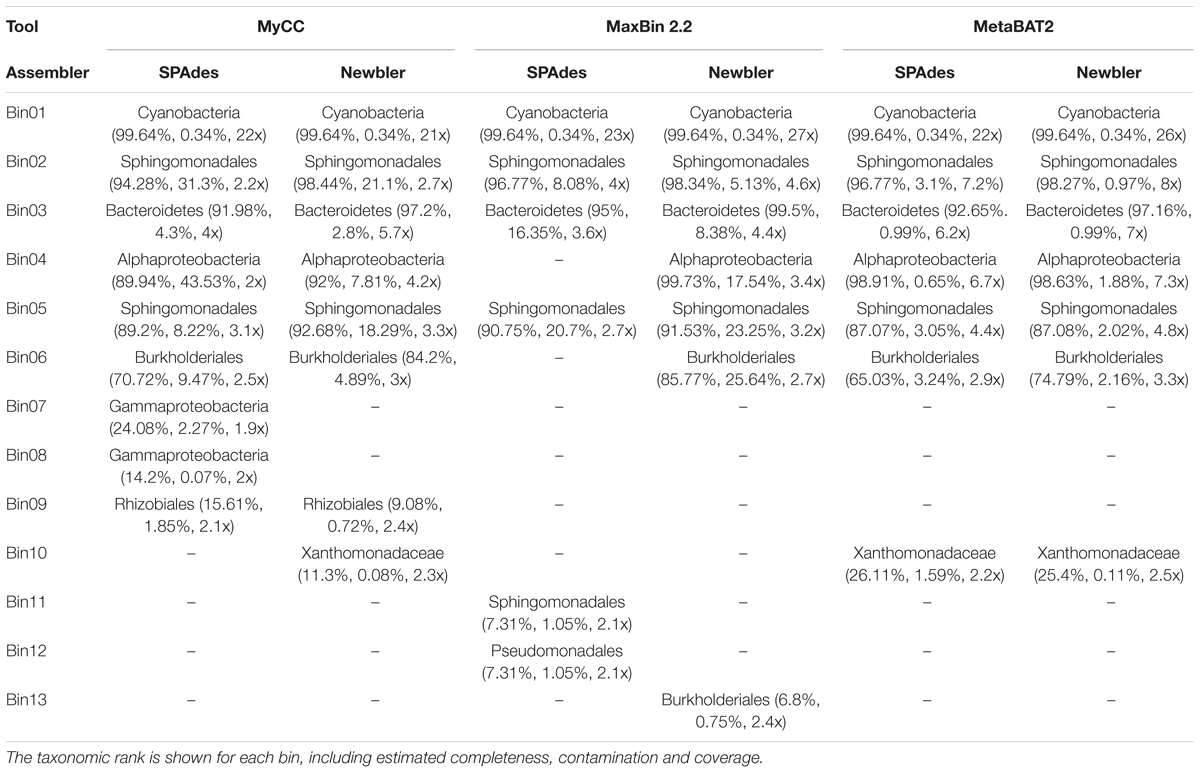
TABLE 1. CheckM results for each assembler and binning tool.
After the comparative analyzes of the binning tools, MyCC revealed the best set of contigs for the cyanobacterial bins for both Newbler and SPAdes. This means that MyCC recovered a higher number of contigs and bases, besides containing sequences from other binning tools. Binning tool comparisons for Newbler contigs are available in Supplementary File 1; while the binning tool comparisons for SPAdes contigs are present in Supplementary File 2.
Metrics of the cyanobacterial bins recovered by MyCC from both Newbler and SPAdes contigs are shown in Supplementary Table 2. The bin from Newbler has more contigs and a higher number of bases, while the bin presented by SPAdes has the longest contig and a larger N50 value. An attempt to join both assemblies using Geneious 9.1.8 de novo Assemble tool (Kearse, 2012) revealed that 82 contigs from Newbler are contained in SPAdes contigs with more than 98% of similarity; 15 contigs from SPAdes are contained in Newbler contigs; and only 15 contigs overlapped, although with low similarity or adding only a few bases. In this way, the contigs assembled by SPAdes and binned by MyCC were chosen for further analysis.
Before the final version of the genome was obtained, checkM pointed out two duplicated marker genes suggesting a possible contamination. The marker PF09285.6 present at one end of contig MKGP02000001 was identified as a contamination and therefore removed, along with downstream and upstream regions not belonging to cyanobacteria. The other marker, TIGR01394, was found in the final portion of the second longest contig. After removing the contaminated region, the contig was split in MKGP02000002 and MKGP02000003. A new checkM analysis revealed no contamination present in the genome.
The draft genome of Limnothrix sp. strain CACIAM 69d has a total of 4.5 Mb distributed among 100 contigs, which range from 594 to 251,784 bp, with GC content of 55.23%, N50 of 86,917 bp and mean coverage of 22×. The NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016) identified 3,789 CDS, 42 tRNA, 2 rRNA, and 4 ncRNA. Additionally, an Yfr1 sRNA homolog with 64 bp was identified by BLAST searches.
The 16S rRNA gene present in Limnothrix sp. CACIAM 69d was identical to the one found in strain CENA545 and had 99.8% of identity with the 16S rRNA of Limnothrix planctonica KLLC001 clone a. Both Limnothrix sp. P13C2 and Limnothrix sp. PR1529 showed identical 16S rRNA sequences. These Limnothrix strains were grouped in the larger cluster II in a Bayesian tree (Figure 2). This cluster was formed mainly by strains from Asia, Europe, Africa, and America (Zhu et al., 2012).
Regarding Limnothrix rosea IAM M-220, it clustered together with Spirulina subsalsa, outside of the two formed clusters (Figure 2). A BLASTn search of Limnothrix rosea IAM M-220 16S rRNA sequence against the NCBI non-redundant nucleotide database revealed a 99% similarity with Leptolyngbya sp. PCC 7376, and also 98% of similarity with some Synechococcus strains, such as PCC 7117, PCC 8807 and PCC 73109. In this sense, Limnothrix rosea IAM M-220 was shown to be more similar to Leptolyngbya sp. PCC 7376 than other Limnothrix strains (Figure 3). The Species Tree reflects the same results obtained from BLASTn using the Limnothrix rosea IAM M-220 16S rRNA sequence.
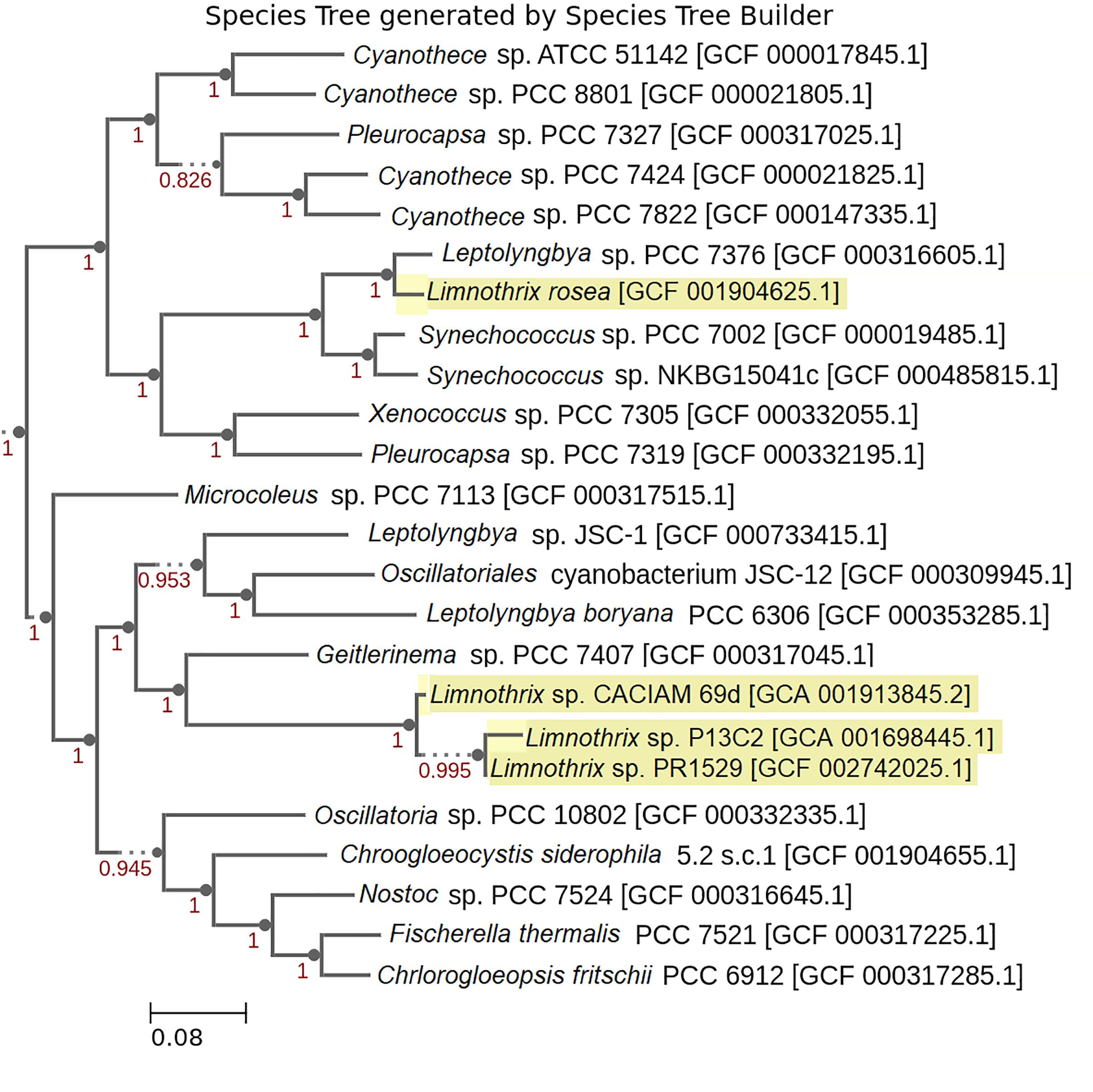
FIGURE 3. Phylogenetic Species Tree based on genomic sequences. Limnothrix strains are highlighted with a yellow background. The numbers presented are confidence values used by FastTree 2 (Price et al., 2010) to estimate maximum likelihood. GenBank and RefSeq assembly accessions are provided within the brackets.
The four Limnothrix genomes were scanned for Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) revealing the presence of four to seven arrays with multiple spacers: seven arrays, containing a total of 24 spacers, were identified in Limnothrix sp. CACIAM 69d; five arrays and 57 total spacers are present in Limnothrix sp. PR1529; four arrays were identified in Limnothrix sp. P13C2 (66 total spacers) and Limnothrix rosea IAM M-220 (316 spacers). Alignments of the spacers and their potential targets can be found in Supplementary Figures 1–4.
The CRISPR-Cas systems identified in Limnothrix genomes were classified according to the classification criteria most often adopted (Makarova et al., 2015) and are shown in Figure 4. With exception of Limnothrix rosea IAM M-220, only two CRISPR-Cas systems were identified in all Limnothrix, one of them classified as type I-A. Besides that, both Limnothrix sp. P13C2 and Limnothrix sp. PR1529 showed identical results in CRISPR-Cas system classification and gene order.
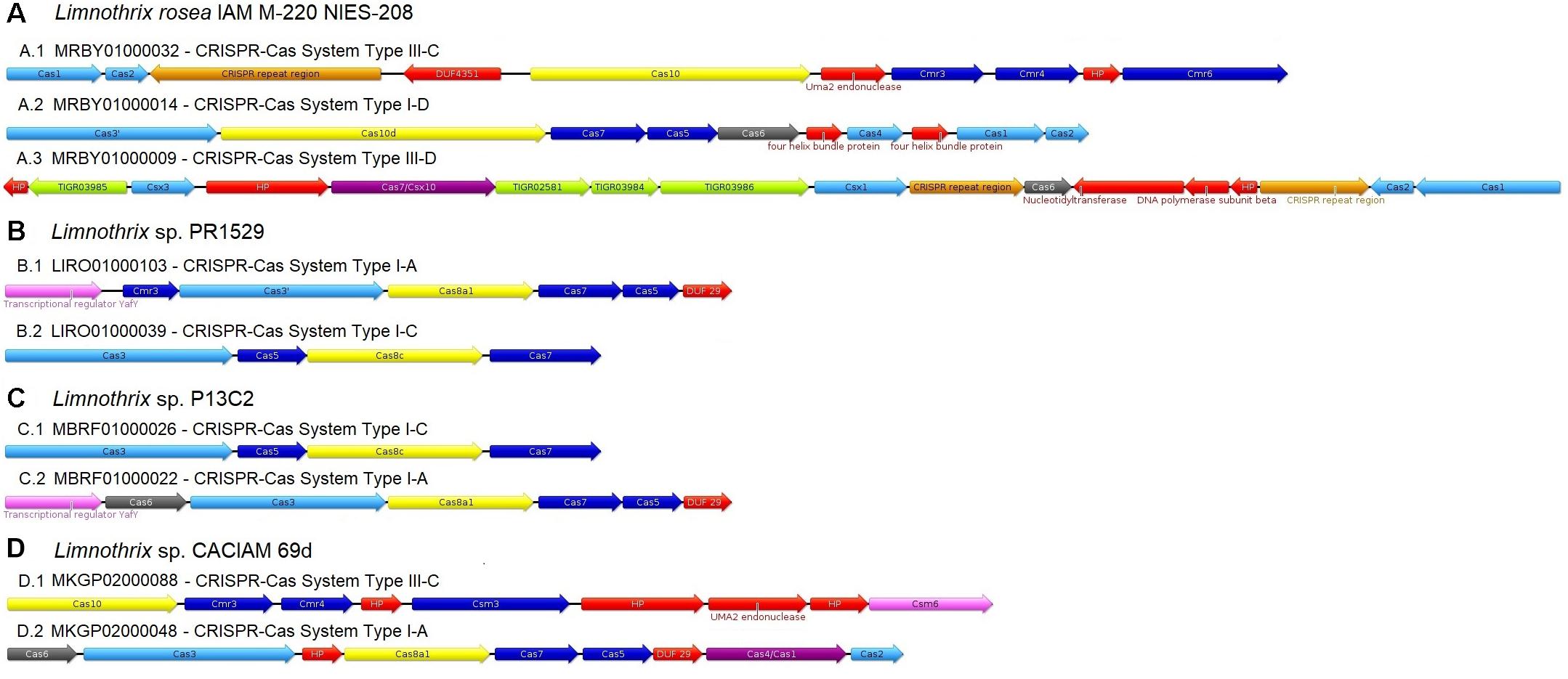
FIGURE 4. Classification of CRISPR-Cas systems found in all four analyzed Limnothrix genome (A–D). The colors indicate the following categories: repeat-associated mysterious protein (RAMP) family RNases involved in crRNA process (gray), CRISPR-Cas protein with no major category (light blue), large CRISPR-associated complex for antiviral defense (Cascade) subunits (yellow), RAMP Cascade subunits (dark blue), fused Cas protein (purple), transcriptional regulator (light pink), protein containing a CRISPR-associated domain (green), CRISPR repeat region (orange) and domain of unknown function (DUF) or hypothetical protein (HP) or other (red).
Clusters of Orthologous Group (COG) analyses have been performed on cyanobacterial genomes to obtain their functional profiling (Prabha et al., 2016), study cyanobacteria metabolism via comparative genomics (Leao et al., 2017) and to obtain the composition features of their core genomes (Simm et al., 2015). In this way, to gain further understanding of the general Limnothrix metabolism, a COG comparison was performed between all four Limnothrix genomes and Synechocystis sp. PCC 6803, which is a cyanobacterial model for several studies (Figure 5). Relevant differences were observed; strains CACIAM 69d, P13C2 and Limnothrix rosea IAM M-220 presented more genes related to cell wall/membrane/envelope biogenesis (M) than PCC 6803 and PR1529. Moreover, the number of hits in categories R (general function prediction only) and S (function unknown) are higher in Limnothrix in comparison to Synechocystis sp. PCC 6803. Remarkably, the strains CACIAM 69d and P13C2 had a number of hits greater than 500 for category S.
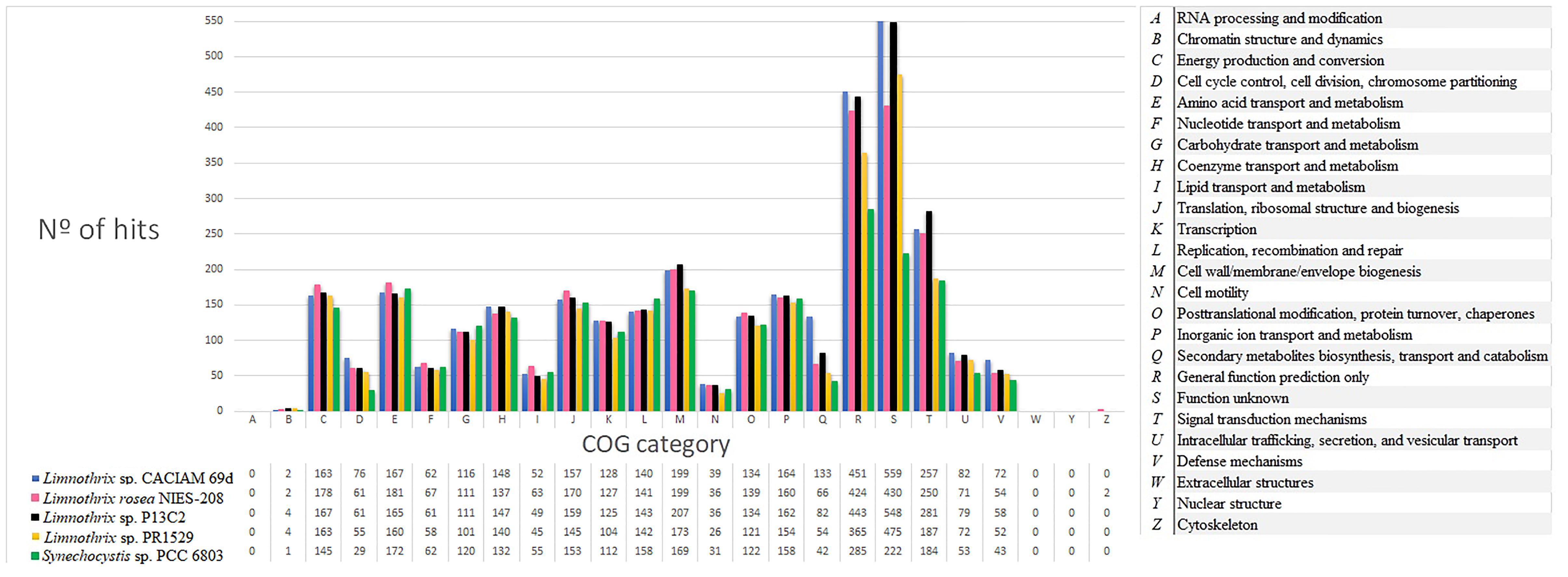
FIGURE 5. Number of hits per COG category for all four Limnothrix genomes and Synechocystis sp. PCC 6803.
In a pan-genome analysis, the core genome is the set of genes shared by all strains, being responsible for major phenotypic traits and basic metabolism of the analyzed group (Chaudhari et al., 2016). In this way, the four analyzed Limnothrix strains share approximately 30% of the total coding CDS content (Table 2), most of them related to energy metabolism (Figure 6). There are as well many accessory genes, i.e., dispensable genes that occur in two or more strains (Chaudhari et al., 2016), present in CACIAM 69d, P13C2 and PR1529. Due to the close phylogenetic relation of these strains (Figures 2, 3), we hypothesized that many of these accessory genes are shared among them, since Limnothrix rosea IAM M-220 presented a high number (64.8%) of unique genes (Table 2). A new pan-genome analysis excluding Limnothrix rosea IAM M-220 revealed a different core genome size, with approximately 74% of total CDS content (Table 3). Thus, many accessory genes present in strains CACIAM 69d, P13C2 and PR1529 showed in Table 2 (the first analysis) were grouped in the core genome in the second analysis (Table 3), since the number of unique genes did not vary much in both investigations. Additional pan-genome analysis of Limnothrix rosea IAM M-220 (Supplementary Tables 3–6) revealed that it shares 77% of its genes with Leptolyngbya sp. PCC 7376; the latter shares 64.5% with this Limnothrix. The differences between Limnothrix rosea IAM M-220 and the three other Limnothrix strains became clearer when some individual aspects of the genomes were analyzed, as further discussed below.

TABLE 2. Number of protein sequences for representatives of core, accessory and unique orthologous clusters present in all four Limnothrix genomes.
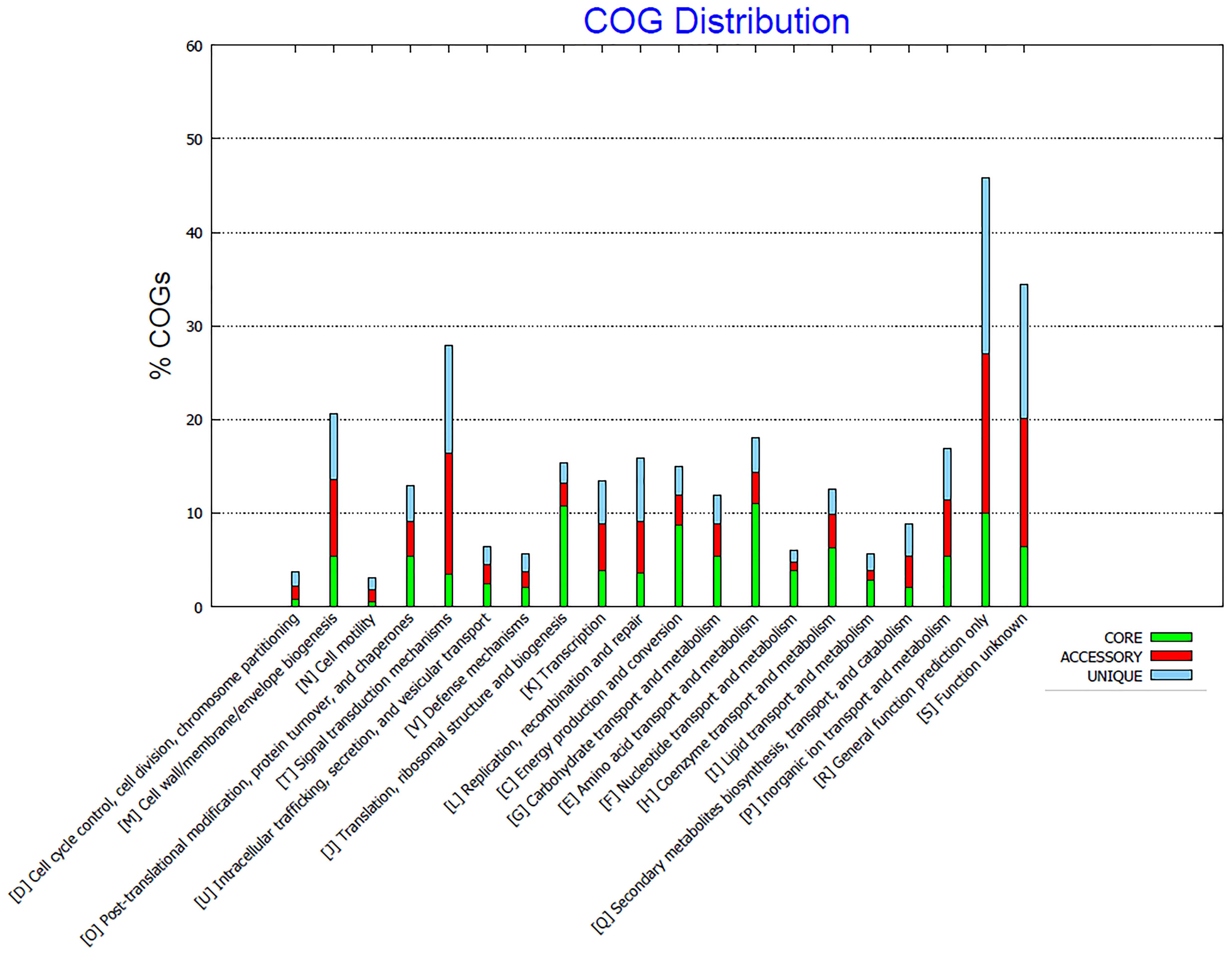
FIGURE 6. COG distribution of core, accessory and unique genes present in all four analyzed Limnothrix genomes generated by Bacterial Pan Genome Analysis Tool (BPGA).

TABLE 3. Number of protein sequences for representatives of core, accessory and unique orthologous clusters present in Limnothrix genomes, excluding Limnothrix rosea IAM M-220.
Comparing the COG distribution of the genetic content present in Limnothrix genomes, it was possible to identify conserved categories associated with the core genome, such as energy production and conversion (C), carbohydrate (G), amino acid (E), nucleotide (F), coenzyme (H), and lipid (I) transport and metabolisms (Figure 6). On the other hand, some categories, such as M, S and R have a high percentage of accessory and unique genes. The exclusion of Limnothrix rosea IAM M-220 from this analysis revealed a decrease of unique and accessory COGs in category C. Furthermore, no unique COGs were identified in category I (Supplementary Figure 5).
Despite the central carbon metabolism, the complete pathways of glycolysis, Calvin-Benson cycle and pentose phosphate were identified and all core enzymes of the cyanobacterial pyruvate metabolism (Beck et al., 2012) were present in Limnothrix genomes analyzed here. The tricarboxylic acid (TCA) cycle is completed through conversion of 2-oxoglutarate to succinic semialdehyde, by 2-oxoglutarate decarboxylase, and then to succinate by a succinic semialdehyde dehydrogenase (Figure 7). The cyanobacterial glyoxylate shunt is not present in Limnothrix and the γ-aminobutyrate (GABA) shunt is incomplete due to the absence of glutamate decarboxylase (GadA). Limnothrix rosea IAM M-220 is the only genome that has malate dehydrogenase (EC 1.1.1.37). The malic enzyme (EC 1.1.1.38) compensates for the lack for malate dehydrogenase in strains CACIAM 69d, PR1529 and P13C2, converting malate to pyruvate; it is also present in Limnothrix rosea IAM M-220, acting as an alternative pathway for obtaining pyruvate.
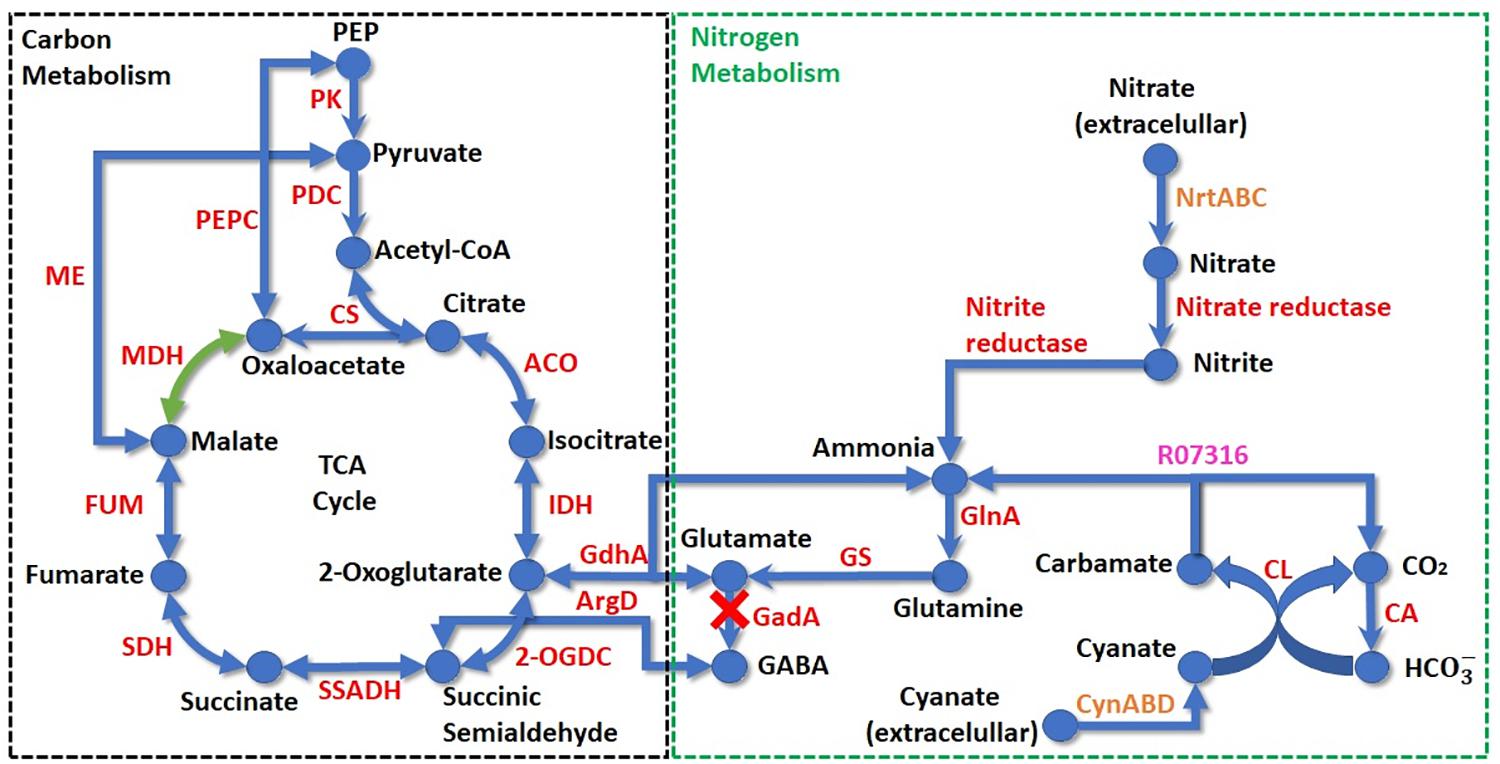
FIGURE 7. Schematic showing TCA cycle and nitrogen acquisition pathways in Limnothrix. The dotted boxes delimit carbon (black) and nitrogen (green) metabolisms. Compounds are represented by circles and their respective names indicated in black. Arrows indicate reactions; enzyme reactions are shown in red, transport reactions in orange and a spontaneous KEGG reaction is shown in purple. The green reaction occurs exclusively in Limnothrix rosea IAM M-220 NIES-208. ACO, aconitase; ArgD, γ-aminobutyric acid aminotransferase; CA, carbonic anhydrase; CL, cyanate lyase; CS, citrate synthase; FUM, fumarase; GadA, glutamate decarboxylase; GdhA, glutamate dehydrogenase; GlnA, glutamine synthase; GS, glutamine synthase; IDH, isocitrate dehydrogenase; MDH, malate dehydrogenase; ME, malic enzyme; 2-OGDC, 2-oxoglutarate decarboxylase; PDC, pyruvate dehydrogenase complex; PEP, phosphoenolpyruvate; PEPC, phosphoenolpyruvate carboxylase; PK, pyruvate kinase; SDH, succinate dehydrogenase; SSADH, succinic semialdehyde dehydrogenase.
The MEP (methylerythritol-phosphate) pathway, which uses glyceraldehyde 3-phosphate and pyruvate to synthesize terpenoids (Pattanaik and Lindberg, 2015), is present in all four Limnothrix. Additionally, the CACIAM 69d, P13C2 and PR1529 strains are capable of producing hydrocarbons from fatty acids using the AAR/ADO (acyl-ACP reductase/aldehyde-deformylating oxygenase) pathway, while Limnothrix rosea IAM M-220 appears to achieve the same using the OLS (α-olefin synthase) pathway (Zhu et al., 2018), which involves the participation of PKS modules (Coates et al., 2014).
In the carbon fixation pathway of photosynthetic organisms, a difference in erythrose-4P synthesis was observed between all Limnothrix and Synechocystis sp. PCC 6803: the former has glycoaldehydetransferase (EC 2.2.1.1), which uses D-fructose 6P and glyceraldehyde-3P, producing xylulose-5P, while the latter has fructose-phosphate phosphoketolase (EC 4.1.2.22), which acts on D-fructose 6P and phosphate. Regarding the storage metabolism of Limnothrix, genes involved in synthesis and mobilization of glycogen and cyanophycin were found in the four genomes.
Analysis of the nitrogen metabolism revealed that Limnothrix can use nitrate and cyanate as nitrogen sources (Figure 7), and all genomes lack the genes for nitrogenases. Nitrogen and carbon metabolisms are connected through ammonia by glutamate dehydrogenase, which produces 2-oxoglutarate for the TCA cycle.
Further analysis revealed that the strains CACIAM 69d, PR1529 and P13C2 can use organosulfonated compounds to acquire sulfur under limiting conditions due to presence of the ssu operon in their genomes (Eichhorn et al., 2000; Ellis, 2011). Besides that, those strains can also incorporate sulfur in their metabolism by using thiosulfate as substrate for thiosulfate sulfurtransferase (EC 2.8.1.1).
Cyanobacteria harbors genes for three main pathways related to assembly and export of extracellular polymeric substances: Wzy-, ABC transporter- and Synthase dependent (Pereira et al., 2015). A search for protein domains present in those mechanisms revealed similar results for all Limnothrix (Supplementary Table 7). In this way, there is a functional Wzy pathway, along with some identified components of ABC transporter (KpsD, KpsE, KpsM, KpsT, and KpsU) and Synthase dependent (Alg8/BcsA, AlgG, AlgI and ExoD) pathways, with multiple results for Alg8/BcsA.
The antiSMASH tool (Blin et al., 2017) detected a piricyclamide gene cluster in Limnothrix sp. CACIAM 69d genome, which was previously described only in the genus Microcystis (Leikoski et al., 2012). Manual curation pointed to the location of one precursor gene (pirE3) in contig MKGP02000097 (Figure 8A). Additional searches performed by HMMER 3 (Eddy, 2011) using TIGR04446 (prenylated cyclic peptide, anacyclamide/piricyclamide family) against all CACIAM 69d CDS returned no results other than the previously identified pirE3. In comparison to Microcystis aeruginosa PCC7005 (Figure 8B), Limnothrix sp. CACIAM 69d presents: (i) no hypothetical proteins; (ii) a transposase IS200 like (PF01797) inserted between pirF and a precursor gene pirE3; and (iii) only one precursor gene pirE.
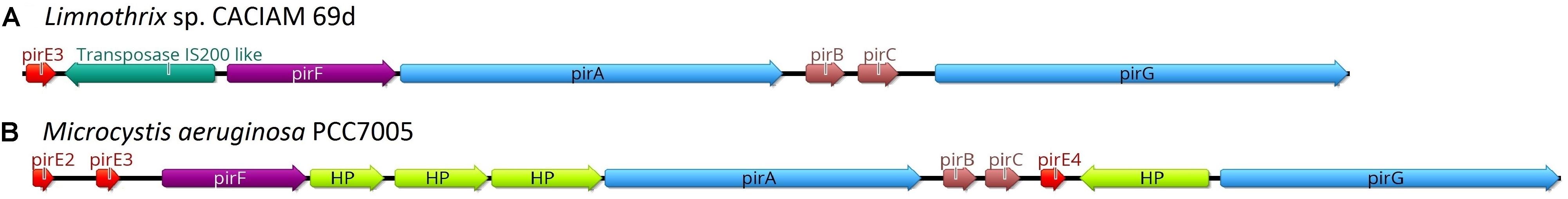
FIGURE 8. Piricyclamide gene cluster schematics. (A) Cluster found in Limnothrix sp. CACIAM 69d contig MKGP02000097 and (B) cluster previously described in Microcystis aeruginosa PCC7005. The precursor genes are shown in red, proteases are blue, prenyltransferase is purple, genes with no prediction function are brown, hypothetical proteins (HP) are light green, a transposase is dark green, and R is for repeat region.
A trans-AT PKS cluster was detected in contig MKGP02000067 of Limnothrix sp. CACIAM 69d and contig MBRF01000028 of Limnothrix sp. P13C2, both showing similar module compositions (Figure 9). A NaPDoS (Ziemert et al., 2012) analysis for its KS domain containing proteins returned hits for domains present in the genes responsible for the biosynthesis of virginiamycin, leinamycin and kirromycin (Table 4). Despite showing higher identity for virginiamycin and kirromycin, the proteins clustered together with genes related to biosynthesis of leinamycin, LnmI and LnmJ (Figure 10). However, they displayed a long phylogenetic distance from the related genes.
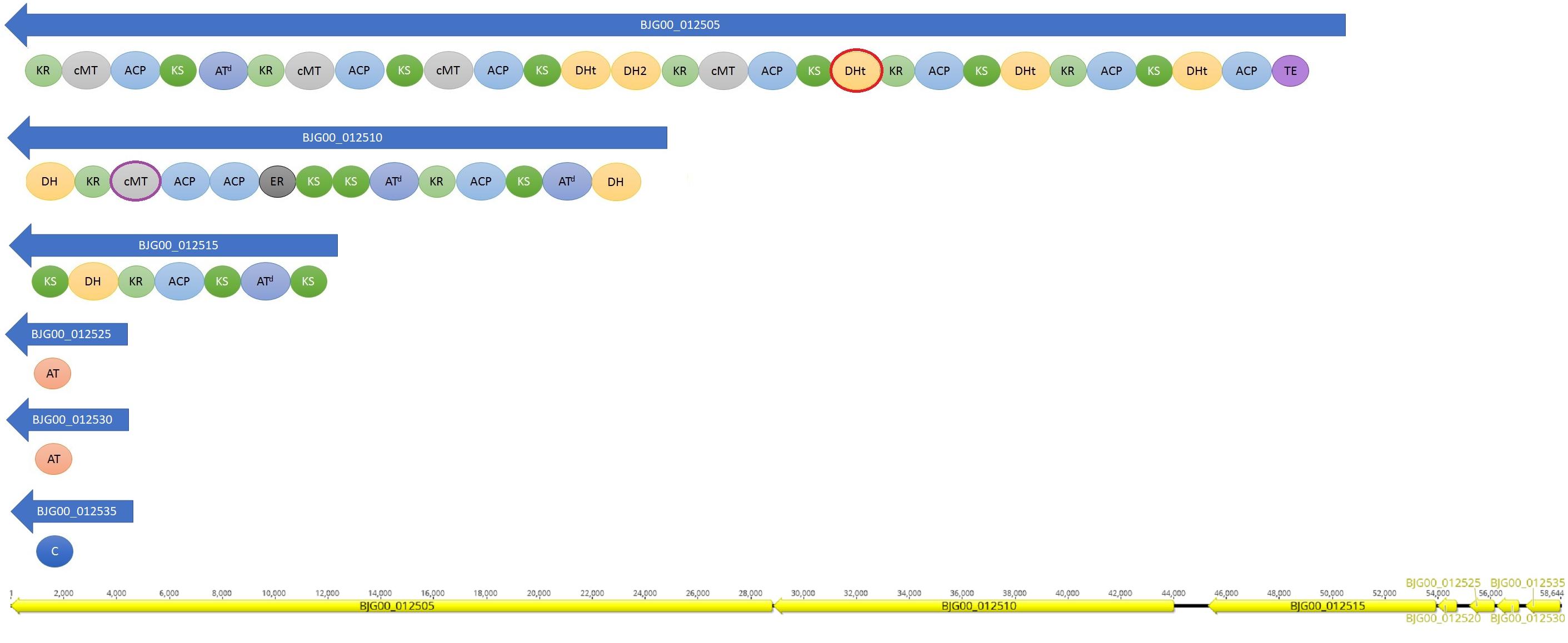
FIGURE 9. Domains present in trans-AT PKS cluster found in Limnothrix sp. CACIAM 69d and Limnothrix sp. P13C2. The results are presented according to antiSMASH 4.0 output. The red circle indicates a domain present only in P13C2 strain and the purple circle a domain found only in CACIAM 69d strain. The order of the CDS for CACIAM 69d and P13C2 is shown at the bottom. ACP, acyl carrier protein; AT, acyltransferase domain; C, condensation domain; DH, dehydratase domain; ER, enoyl reductase domain; KR, keto reductase domain; KS, keto synthase domain; MT, methyl transferase domain; TE, thioesterase domain.
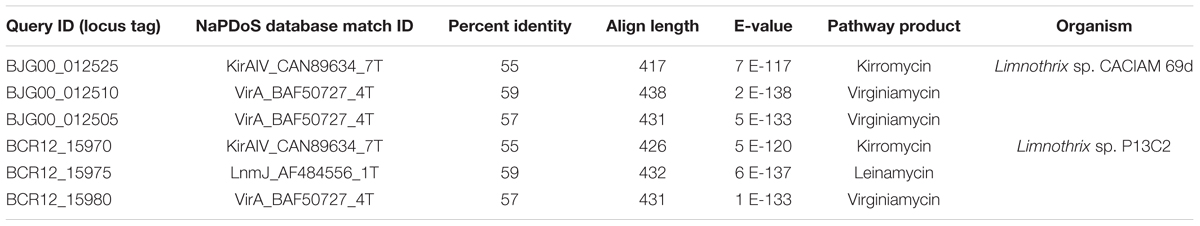
TABLE 4. NaPDoS results for KS domain containing proteins present in trans-AT PKS clusters found in Limnothrix sp. CACIAM 69d and Limnothrix sp. P13C2.
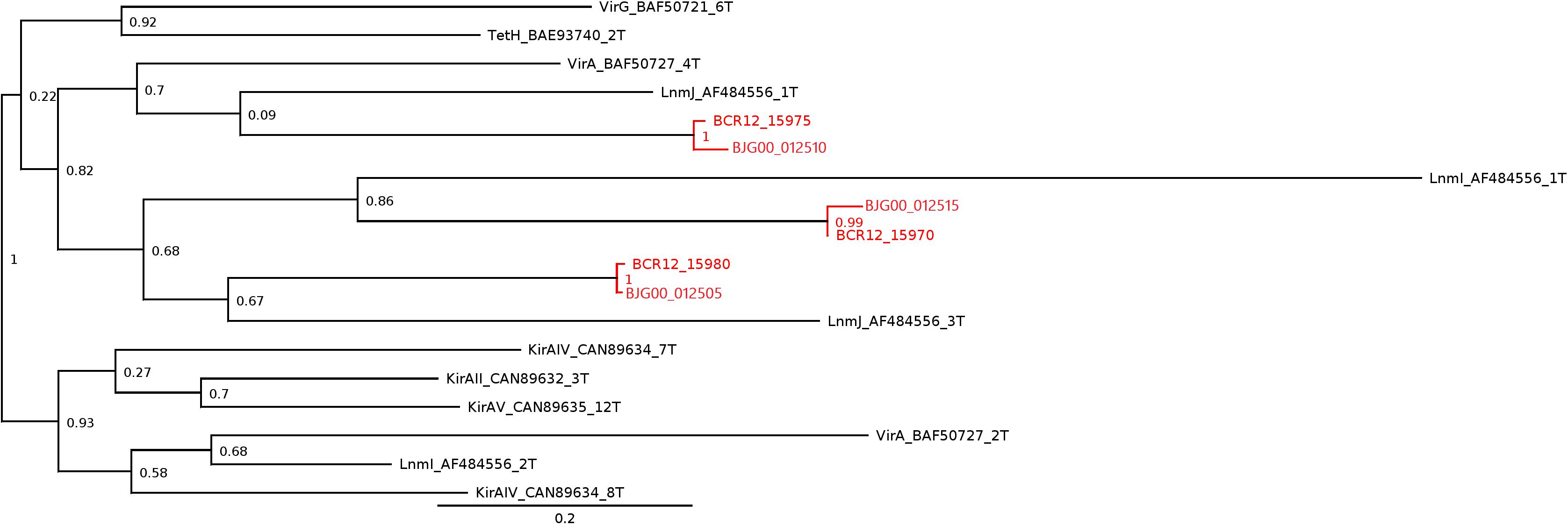
FIGURE 10. Subtree generated by NaPDoS for Limnothrix sp. CACIAM 69d and Limnothrix sp. P13C2 trans-AT PKS cluster analysis. Limnothrix domain sequences are in red and named based on Table 4; the initials BCR12 and BJG00 are, respectively, from Limnothrix sp. P13C2 and Limnothrix sp. CACIAM 69d. Other domain sequences are named according to the NaPDoS database: Vir, virginiamycin; Tet, tetronomycin; Lnm, leinamycin; Kir, kirromycin. The numbers presented are confidence values generated in the NaPDoS tree reconstruction process, which uses the FastTree (Guindon and Gascuel, 2003) to estimate maximum likelihood. The complete tree is available in Supplementary File 3.
Additional searches with BLAST in Limnothrix genomes returned no results for anatoxin, cylindrospermopsin, lyngbyatoxin, microcystin, nodularin and saxitoxin gene clusters.
Genome assembly using different algorithms followed by a comparison binning process was capable of reconstructing a nearly complete and representative uncontaminated genome of Limnothrix sp. CACIAM 69d, which is phylogenetically close to Limnothrix sp. CENA545 from a Brazilian Pantanal saline-alkaline lake (Andreote et al., 2014) (Figure 2). Thus, it was possible to analyze the actual gene content in order to reveal important aspects of this cyanobacterial metabolism, both central and secondary. Several cyanobacterial genomes display a high number of repeats due to insertion sequences (Vigil-Stenman et al., 2015), which can be a problem because assembly algorithms cannot characterize repeat elements that span longer than read length (Alkan et al., 2010). Nevertheless, the assembly of some regions could have been affected by repetition sequences, which may have prevented the formation of longer contigs. A potential solution for this problem is to increase read length using new technologies such as PacBio or Oxford Nanopore MinION (Koren and Phillippy, 2015).
Regarding Limnothrix rosea IAM M-220, its position within both phylogenetic trees (Figures 2, 3) raised the suspicions as to whether this strain really belongs to the Limnothrix genus. This strain was originally named Oscillatoria rosea NIES-208 when the genome was published by Zhu et al. (2017). However, the GenBank record (MRBY00000000) provided by that work points to Limnothrix rosea IAM M-220. Furthermore, the NCBI Taxonomy Browser lists Oscillatoria rosea NIES-208 as a synonym for Limnothrix rosea IAM M-220 (Taxonomy ID: 454133). A comparison of Limnothrix rosea IAM M-220 and three other available Oscillatoria genomes (Supplementary Table 5) revealed that they share only 961 genes, which is only 19.5% of the former total coding CDS. An additional comparison of Limnothrix rosea IAM M-220 and other two Spirulina genomes also resulted in a low number of shared genes: 1,313, representing 37% of the former total coding CDS (Supplementary Table 6). This last value, however, is higher than the one presented in Table 2, which considers the three other Limnothrix genomes. Based on the Species Tree generated by KBase (Figure 3), we have assumed that perhaps Limnothrix rosea IAM M-220 was close to the genus Leptolyngbya, which is also from the Cyanobacteria Subsection III (Castenholz et al., 2015a), since it shares 77% of its genes with Leptolyngbya sp. PCC 7376 (Supplementary Table 3). Nevertheless, this hypothesis was discarded when another pan-genome analysis was performed adding other genomes of Leptolyngbya returned none core genes (data not shown). Limnothrix rosea IAM M-220 has 1,002 genes shared with some strains of Leptolyngbya (PCC 7376, PCC 6406 and O 77) in a pan-genome analysis (Supplementary Table 4). Given these data, we were not able to determinate if Limnothrix rosea IAM M-220 belongs to the genus Leptolyngbya and concluded that this strain is not a Limnothrix. We even raise suspicion regarding the Leptolyngbya sp. PCC 7376 taxonomic classification. In this sense, only a polyphasic approach could be able to correctly classify these cyanobacteria taxonomically (Komárek et al., 2014).
Many bacteria and most archaea have an adaptive immune system known as CRISPR-Cas. The CRISPR locus contain a sequence array, comprising short direct repeats separated by short variable DNA sequences, which are called spacers. Aided by Cas protein, the spacers specifically target invading virus or plasmid sequences (i.e., the protospacers) and cleave their nucleic acids. Thus, they are responsible for the sequence memory of this immune system (Makarova et al., 2015). Regarding the CRISPR search in Limnothrix genomes, many spacer sequences were found, but only a few of them had potential matches (protospacers) when compared to GenBank-phage and plasmid RefSeq databases (Supplementary Figures 1–4). For strains CACIAM 69d and PR1529, about 12.5% and 9% of spacers, respectively, had potential targets; about 11% and 4% spacers from, respectively, strain P13C2 and Limnothrix rosea IAM M-220. Most of the protospacers sequences found belong to plasmids and are probably unrelated to cyanobacteria. Cyanophage protospacers were only found in Limnothrix rosea IAM M-220, and yet they are related to Synechococcus phages (Supplementary Figure 4). Cyanophages are important agents in cyanobacterial population control and evolution, since they can transfer genes among cyanobacteria and also act as an environmental gene storage (Shestakova and Karbysheva, 2015). The fact that most of the spacers present in Limnothrix genomes have no known targets indicates the lack of representation of these cyanophages in public databases. This could be due to scarce genomic data available from these Limnothrix environments.
Cai et al. (2013) addressed the distribution of CRISPR-Cas systems in cyanobacteria and found that, in general, subsection III has a median of about 100 spacer sequences and tends to have a large number of them, with a maximum around 600 sequences. Considering the results found in Limnothrix (subsection III), only Limnothrix rosea IAM M-220 presented a high number of spacer sequences, which could be related to the presence of Cas1 and Cas2 in its CRISPR-Cas systems (Figure 4A). These two proteins are responsible for forming an adaptation module complex required for the insertion of spacers into CRISPR arrays (Makarova et al., 2015). Thus, when comparing the number of spacers in Limnothrix rosea IAM M-220 genome with the other three Limnothrix genomes, which CRISPR-Cas systems lack Cas1 and Cas2 (Figures 4B–D), it is expected that the former will have more spacers due to Cas1 and Cas2 function. However, it is possible that these three Limnothrix genomes have another way to acquire new protospacers via a mechanism yet to be discovered. Since the incorporation of new spacers and the loss of existing spacer sequences is related to a highly dynamic response to the environment (Kuno et al., 2012), another explanation is that there is not enough pressure in their environment to acquire new sequences; or there is pressure for them to lose their previously acquired spacers.
Regarding the central carbon metabolism, all Limnothrix genomes and Synechocystis sp. PCC 6803 investigated have the key enzymes, highlighting that the former (except for Limnothrix rosea IAM M-220) are better equipped for environmental sulfur acquisition in limiting conditions. Inorganic sulfate is the second most abundant ion present in aquatic environments, acting as a main source of sulfur for microorganisms (Loka Bharathi, 2008). However, under sulfur-limiting conditions, bacteria are capable of using organosulfonates as sulfur sources (Ellis, 2011). Due to the ssu operon, Limnothrix sp. CACIAM 69d is able to acquire sulfur from organic substrates, such as C-2 to C-10 alkanesulfonates (Eichhorn et al., 1999), anaerobically released from the bottom of the lake (Pereira et al., 1994; Fonseca et al., 2012), thus adapting to the seasonal changes affecting sulfate levels on Tucuruí Hydroelectric Reservoir. Limnothrix sp. P13C2 may face similar sulfate limitations in Pandan Reservoir (1°18′51.8″ N, 103°44′29.2″ E), Singapore, as a spatiotemporal analysis of waterways flowing from this reservoir showed different levels of sulfate along the water course (Saxena et al., 2015).
Limnothrix genomes harbor transporters for nitrate, ammonium and cyanate, indicating that they can use these compounds as nitrogen sources. Due to rainfall changes, the concentration of nitrate is affected in Tucuruí Hydroelectric Reservoir (Brandão et al., 2017), reaching low levels at the water surface in dry periods (Baraúna et al., 2013). This suggests that ammonium may be used as the primary nitrogen source during low rain season. Nonetheless, the presence of cyanate transporter, allied with a higher density of rotifers (zooplankton) in the dry period (Francineli et al., 2015), could indicate that cyanate has an important role in Limnothrix nitrogen metabolism. It is known that zooplankton can excrete nutrients to support primary production in lakes (Vanni, 2002). In this way, the excreted urea can undergo spontaneous dissociation to produce cyanate (Kamennaya et al., 2008), which can be acquired by Limnothrix and then converted to ammonium (Figure 7). Additionally, urea can also be supplied by the decomposing organic matter at the bottom of the lake. Even if there is not enough nitrogen available, Limnothrix is able to overcome this using the stored cyanophycin, which is a polymer composed of aspartate and arginine, as nitrogen and carbon source (Beck et al., 2012).
Cyanobacteria have an universal ability to produce hydrocarbons (Han et al., 1968, 1969; Winters et al., 1969), which can be achieved via AAR/ADO and OLS pathways. Naturally, these pathways do not occur in the same organism, with the former presenting the largest taxonomical distribution (Coates et al., 2014). Since the AAR/ADO pathway was found only in strains CACIAM 69d, P13C2 and PR1529, we suppose that Limnothrix rosea IAM M-220 possesses the OLS pathway. A closer look at contig MRBY01000033 from the latter revealed a beta-ketoacyl synthase (NIES208_12590) that has the same domains when compared to OLS PKS modules that were presented by Coates et al. (2014). A recent study also reported the presence of an OLS pathway in Limnothrix rosea IAM M-220 using bioinformatic analyses (Zhu et al., 2018).
The production of many EPS by Limnothrix redekei PUPCCC 16 has been reported, and most of them are released into the cultivation medium, making the isolation process easy (Khattar et al., 2010). Given the high number of genes present in the COG category cell wall/membrane/envelope biogenesis (Figure 5) and multiple hits for Alg8/BcsA (Supplementary Table 7), we supposed that Limnothrix can produce a broad variety of extracellular polymeric substances that could be EPS. In a previous study (Pereira et al., 2015), AlgG was detected in a very restricted number of cyanobacterial strains and the absence of AlgI was observed in Synechocystis sp. PCC 6803. These enzymes are involved in the synthesis of alginate (Franklin et al., 2011), an EPS that confers a mucoid phenotype (Evans and Linker, 1973). However, the other enzymes required for alginate biosynthesis were not found in Limnothrix genomes (Supplementary Table 7). This investigation of these differently sequenced and assembled genomes suggests that the full mechanism of alginate biosynthesis may be not truly present in Limnothrix. Furthermore, it is speculated that EPS production in cyanobacteria may not follow existing patterns (Pereira et al., 2015). Therefore, more Limnothrix genomes are needed to solve this question.
Only recently, have some studies begun investigating the toxicity of Limnothrix (Bernard et al., 2011; Humpage et al., 2012; Daniels et al., 2014). These works have not detected the presence of previously described cyanobacterial toxins (microcystins, saxitoxins, and cylindrospermopsins). BLAST searches in Limnothrix genomes returned no results for the aforementioned toxins and were also negative for anatoxin and lyngbyatoxin. A novel toxin called Limnothrixin, which has been associated to damage in liver, lungs and gastrointestinal tract of mice (Humpage et al., 2012) as well as toxic activity against Bufo marinus larvae (Daniels et al., 2014), has been proposed. However, since there is no resolved structure for this toxin, the identification of genes responsible for its biosynthesis is difficult.
Cyanobacterial toxins have different chemical structures (Bláha et al., 2009) (peptides, alkaloids, lipidic compounds and lipopolysaccharides), which can be produced by a variety of mechanisms and pathways (Dittmann et al., 2013). The trans-AT PKS gene cluster detected in both Limnothrix sp. CACIAM 69d and Limnothrix sp. P13C2 may be a potential candidate for Limnothrixin biosynthesis, since it appears to be conserved among the genomes (Figure 9), presenting a long phylogenetic distance (Figure 10) and less than 80% identity with other known compounds present in the NaPDoS database; moreover, the trans-AT PKS systems are capable of producing many structurally complex, bioactive natural compounds (Helfrich and Piel, 2016).
Cyanobactins are small cyclic peptides ribosomally produced by many cyanobacteria using solely proteinogenic amino acids (Sivonen et al., 2010). Piricyclamide is a cyanobactin previously identified in strains of Microcystis (Leikoski et al., 2012), but no toxicity assays have been performed on it so far. The cluster identified in Limnothrix sp. CACIAM 69d (Figure 8A) has one precursor gene, and although it was not possible to find other precursor genes, they could be located in an unsequenced and/or unassembled region of the genome. Since cyanobactins have a broad toxic activity (Sivonen et al., 2010), it is possible that they may contribute, along with lipopolysaccharides, to Limnothrix toxicity.
Although only four genomes are currently available for the genus Limnothrix, it was possible to analyze their metabolic diversity, which is a key feature present in Cyanobacteria phylum. The draft genome of Limnothrix sp. CACIAM 69d adds a new member to the small Limnothrix genome library and contributes to a growing representativeness of cyanobacterial genomes from Amazon region. The comparative genomic analysis of Limnothrix made it possible to highlight unique genes for each strain and to understand the overall features of their metabolism. Furthermore, it was important to show inconsistencies in cyanobacteria taxonomical classification of Limnothrix.
AL, AS, JA, DA, PM, LD, and EG designed the project and the analysis procedures. AS collected, cultivated, and extracted the DNA from the sample. CdL performed the sequencing protocol. AL and JP executed the bioinformatics tools and pipelines. AL, AS, JV, PM, LX, LD, and EG analyzed the final data and wrote the manuscript. DA, JV-J, MN, and EG contributed with materials, reagents, and equipment. All the authors read and approved the final version of the manuscript.
This work was funded by Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (grants 556851/2009-9 and 554321/2010-6) and Fundação Amazônia Paraense (FAPESPA) (grant ICAAF 006/2012). This work was financially supported through grants from CAPES (Coordination for the Improvement of Higher Education Personnel, Brazil), PROPESP/UFPA (Pró-Reitoria de Pesquisa e Pós-Graduação/Universidade Federal do Pará, Brazil), FAPEMA (Maranhão State Research Foundation, Brazil), and Instituto Evandro Chagas (IEC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AL and handling Editor declared their shared affiliation.
We are grateful to Centrais Elétricas do Norte do Brasil S/A (Eletronorte) for providing the logistics during collection of strains. We are grateful to Fábio Daniel Florêncio da Silva from Laboratório de Tecnologia Biomolecular for elaborate Figure 1 and allowing the use of it in this work. We are also grateful to the reviewers for their collaboration in the process of reviewing and analyzing this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02811/full#supplementary-material
Aboim, J. B., Oliveira, D., Ferreira, J. E., Siqueira, A. S., Dall’Agnol, L. T., Rocha Filho, G. N., et al. (2016). Determination of biodiesel properties based on a fatty acid profile of eight Amazon cyanobacterial strains grown in two different culture media. RSC Adv. 6, 109751–109758. doi: 10.1039/c6ra23268j
Alkan, C., Sajjadian, S., and Eichler, E. E. (2010). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi: 10.1038/nmeth.1527
Allen, M. M. (1968). Simple conditions for growth of unicellular blue-green algae on plates. J. Phycol. 4, 1–4. doi: 10.1111/j.1529-8817.1968.tb04667.x
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alvarenga, D. O., Fiore, M. F., and Varani, A. M. (2017). A metagenomic approach to cyanobacterial genomics. Front. Microbiol. 8:809. doi: 10.3389/fmicb.2017.00809
Andreote, A. P., Vaz, M. G., Genuário, D. B., Barbiero, L., Rezende-Filho, A. T., and Fiore, M. F. (2014). Nonheterocytous cyanobacteria from Brazilian saline-alkaline lakes. J. Phycol. 50, 675–684. doi: 10.1111/jpy.12192
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: the United States department of energy systems biology knowledgebase. Nat. Biotechnol. 36, 566–569. doi: 10.1038/nbt.4163
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baraúna, R. A., Graças, D. A., Miranda, P. R., Ghilardi, R., Barbosa, M. S., Schneider, M. P. C., et al. (2013). Prokaryotic diversity of the Tucuruí hydropower plant reservoir in the Brazilian Amazon. Aquat. Sci. Technol. 1, 181–199. doi: 10.5296/ast.v1i1.2981
Beck, C., Knoop, H., Axmann, I. M., and Steuer, R. (2012). The diversity of cyanobacterial metabolism: genome analysis of multiple phototrophic microorganisms. BMC Genomics 13:56. doi: 10.1186/1471-2164-13-56
Bernard, C., Froscio, S., Campbell, R., Monis, P., Humpage, A., and Fabbro, L. (2011). Novel toxic effects associated with a tropical Limnothrix/Geitlerinema-like cyanobacterium. Environ. Toxicol. 26, 260–270. doi: 10.1002/tox.20552
Biswas, A., Gagnon, J. N., Brouns, S. J. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget. RNA Biol. 10, 817–827. doi: 10.4161/rna.24046
Biswas, A., Staals, R. H. J., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0
Bláha, L., Babica, P., and Maršálek, B. (2009). Toxins produced in cyanobacterial water blooms - toxicity and risks. Interdiscip. Toxicol. 2, 36–41. doi: 10.2478/v10102-009-0006-2
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). AntiSMASH 4.0 - improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Brandão, I. D. S., Mannaerts, C. M., Verhoef, W., Saraiva, A. C. F., Paiva, R. S., and da Silva, E. V. (2017). Using synergy between water limnology and satellite imagery to identify algal blooms extent in a Brazilian Amazonian reservoir. Sustain 9:2194. doi: 10.3390/su9122194
Cai, F., Axen, S. D., and Kerfeld, C. A. (2013). Evidence for the widespread distribution of CRISPR-Cas system in the Phylum Cyanobacteria. RNA Biol. 10, 687–693. doi: 10.4161/rna.24571
Castenholz, R. W., Rippka, R., Herdman, M., and Wilmotte, A. (2015a). “Form-leptolyngbya,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds G. Garrity, D. R. Boone, and R. W. Castenholz (Hoboken, NJ: Wiley), doi: 10.1002/9781118960608.gbm00437
Castenholz, R. W., Rippka, R., Herdman, M., Wilmotte, A., Castenholz, R. W., Rippka, R., et al. (2015b). “Form-Limnothrix,” in Bergey’s Manual of Systematics of Archaea and Bacteria, ed. W. B. Whitman (Chichester: John Wiley & Sons, Ltd), 1–2. doi: 10.1002/9781118960608.gbm00438
Castro, W. O., Lima, A. R. J., Moraes, P. H. G., Siqueira, A. S., Aguiar, D. C. F., Baraúna, A. R. F., et al. (2016). Draft genome sequence of Microcystis aeruginosa CACIAM 03, a cyanobacterium isolated from an Amazonian freshwater environment. Genome Announc. 4:e01299-16. doi: 10.1128/genomeA.01299-16
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Coates, R. C., Podell, S., Korobeynikov, A., Lapidus, A., Pevzner, P., Sherman, D. H., et al. (2014). Characterization of cyanobacterial hydrocarbon composition and distribution of biosynthetic pathways. PLoS One 9:e85140. doi: 10.1371/journal.pone.0085140
Curtarelli, M. P., Ogashawara, I., de Araújo, C. A. S., Lorenzzetti, J. A., Leão, J. A. D., Alcântara, E., et al. (2016). Carbon dioxide emissions from Tucuruí reservoir (Amazon biome): new findings based on three-dimensional ecological model simulations. Sci. Total Environ. 551–552, 676–694. doi: 10.1016/j.scitotenv.2016.02.001
Daniels, O., Fabbro, L., and Makiela, S. (2014). The effects of the toxic cyanobacterium Limnothrix (Strain AC0243) on Bufo marinus larvae. Toxins 6, 1021–1035. doi: 10.3390/toxins6031021
Das Graças, D. A., Ramos, R. T. J., Sá, P. G., Baraúna, R. A., Ghilardi, R., Schneider, M. P. C., et al. (2015). Semiconductor sequencing reveals the diversity of bacterial communities in an Amazonian reservoir. Aquat. Sci. Technol. 3, 18–32. doi: 10.5296/ast.v3i1.6468
Dittmann, E., Fewer, D. P., and Neilan, B. A. (2013). Cyanobacterial toxins: biosynthetic routes and evolutionary roots. FEMS Microbiol. Rev. 37, 23–43. doi: 10.1111/j.1574-6976.2012.12000.x
Economou, C. N., Marinakis, N., Moustaka-Gouni, M., Kehayias, G., Aggelis, G., and Vayenas, D. V. (2015). Lipid production by the filamentous cyanobacterium Limnothrix sp. growing in synthetic wastewater in suspended- and attached-growth photobioreactor systems. Ann. Microbiol. 65, 1941–1948. doi: 10.1007/s13213-014-1032-7
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Eichhorn, E., Van Der Ploeg, J. R., and Leisinger, T. (1999). Characterization of a two-component alkanesulfonate monooxygenase from Escherichia coli. J. Biol. Chem. 274, 26639–26646. doi: 10.1074/jbc.274.38.26639
Eichhorn, E., van der Ploeg, J. R., and Leisinger, T. (2000). Deletion analysis of the Escherichia coli taurine and alkanesulfonate transport systems. J. Bacteriol. 182, 2687–2695. doi: 10.1128/JB.182.10.2687-2695.2000
Ellis, H. R. (2011). Mechanism for sulfur acquisition by the alkanesulfonate monooxygenase system. Bioorg. Chem. 39, 178–184. doi: 10.1016/j.bioorg.2011.08.001
Evans, L. R., and Linker, A. (1973). Production and characterization of the slime polysaccharide of Pseudomonas aeruginosa. J. Bacteriol. 116, 915–924.
Fearnside, P. M. (2001). Environmental impacts of Brazil’s Tucuruí Dam: unlearned lessons for hydroelectric development in Amazonia. Environ. Manage. 27, 377–396. doi: 10.1007/s002670010156
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fonseca, R., Patinha, C., Barriga, F., and Morais, M. (2012). Role of the sediments of two tropical dam reservoirs in the flux of metallic elements to the water column. Water Sci. Technol. 66, 254–266. doi: 10.2166/wst.2012.169
Francineli, M., Sena, B. A., and Eduardo, J. (2015). Composição e variabilidade da comunidade de rotifera em um reservatório tropical. Bol. Inst. Pesca 41, 493–506.
Franklin, M. J., Nivens, D. E., Weadge, J. T., and Lynne Howell, P. (2011). Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2:167. doi: 10.3389/fmicb.2011.00167
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Han, J., Chan, H. W., and Calvin, M. (1969). Biosynthesis of alkanes in Nostoc muscorum. J. Am. Chem. Soc. 91, 5156–5159. doi: 10.1021/ja01046a037
Han, J., McCarthy, E. D., Van Hoeven, W., Calvin, M., and Bradley, W. H. (1968). Organic geochemical studies, II. A preliminary report on the distribution of aliphatic hydrocarbons in algae, in bacteria, and in a recent lake sediment. Proc. Natl. Acad. Sci. U.S.A. 59, 29–33. doi: 10.1073/pnas.59.1.29
Helfrich, E. J. N., and Piel, J. (2016). Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 33, 231–316. doi: 10.1039/C5NP00125K
Hong, J. W., Jo, S., Kim, O. H., Jeong, M. R., Kim, H., Park, K. M., et al. (2016). Characterization of a Korean domestic cyanobacterium Limnothrix sp. KNUA012 for biofuel feedstock. J. Life Sci. 26, 460–467. doi: 10.5352/JLS.2016.26.4.460
Humpage, A., Falconer, I., Bernard, C., Froscio, S., and Fabbro, L. (2012). Toxicity of the cyanobacterium Limnothrix AC0243 to male Balb/c mice. Water Res. 46, 1576–1583. doi: 10.1016/j.watres.2011.11.019
Kamennaya, N. A., Chernihovsky, M., and Post, A. F. (2008). The cyanate utilization capacity of marine unicellular Cyanobacteria. Limnol. Oceanogr. 53, 2485–2494. doi: 10.4319/lo.2008.53.6.2485
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. doi: 10.1093/bib/bbx108 [Epub ahead of print].
Kearse, M. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Khattar, J. I. S., Singh, D. P., Jindal, N., Kaur, N., Singh, Y., Rahi, P., et al. (2010). Isolation and characterization of exopolysaccharides produced by the cyanobacterium Limnothrix redekei PUPCCC 116. Appl. Biochem. Biotechnol. 162, 1327–1338. doi: 10.1007/s12010-010-8922-3
Komárek, J., Kaštovský, J., Mareš, J., and Johansen, J. R. (2014). Taxonomic classification of cyanoprokaryotes (cyanobacterial genera) 2014, using a polyphasic approach. Preslia 86, 295–335.
Komárek, J., Nedbalová, L., and Hauer, T. (2012). Phylogenetic position and taxonomy of three heterocytous cyanobacteria dominating the littoral of deglaciated lakes, James Ross Island, Antarctica. Polar Biol. 35, 759–774. doi: 10.1007/s00300-011-1123-x
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Kuno, S., Yoshida, T., Kaneko, T., and Sako, Y. (2012). Intricate interactions between the bloom-forming cyanobacterium Microcystis aeruginosa and foreign genetic elements, revealed by diversified clustered regularly interspaced short palindromic repeat(CRISPR) signatures. Appl. Environ. Microbiol. 78, 5353–5360. doi: 10.1128/AEM.00626-12
Land, M., Hauser, L., Jun, S.-R., Nookaew, I., Leuze, M. R., Ahn, T.-H., et al. (2015). Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 15, 141–161. doi: 10.1007/s10142-015-0433-4
Leao, T., Castelão, G., Korobeynikov, A., Monroe, E. A., Podell, S., Glukhov, E., et al. (2017). Comparative genomics uncovers the prolific and distinctive metabolic potential of the cyanobacterial genus Moorea. Proc. Natl. Acad. Sci. U.S.A. 114, 3198–3203. doi: 10.1073/pnas.1618556114
Leikoski, N., Fewer, D. P., Jokela, J., Alakoski, P., Wahlsten, M., and Sivonen, K. (2012). Analysis of an inactive Cyanobactin biosynthetic gene cluster leads to discovery of new natural products from strains of the genus Microcystis. PLoS One 7:e43002. doi: 10.1371/journal.pone.0043002
Li, L., Huang, D., Cheung, M. K., Nong, W., Huang, Q., and Kwan, H. S. (2013). BSRD: a repository for bacterial small regulatory RNA. Nucleic Acids Res. 41, D233–D238. doi: 10.1093/nar/gks1264
Lima, A. R. J., Castro, W. O., Moraes, P. H. G., Siqueira, A. S., Aguiar, D. C. F., de Lima, C. P. S., et al. (2017). Draft genome sequence of Alkalinema sp. Strain CACIAM 70d, a Cyanobacterium isolated from an Amazonian freshwater environment. Genome Announc. 5:e00635-17. doi: 10.1128/genomeA.00635-17
Lima, A. R. J., Siqueira, A. S., Dos Santos, B. G. S., da Silva, F. D. F., Lima, C. P., Cardoso, J. F., et al. (2014). Draft genome sequence of the Brazilian Cyanobium sp. Strain CACIAM 14. Genome Announc. 2:e00669-14. doi: 10.1128/genomeA.00669-14
Lin, H.-H., and Liao, Y.-C. (2016). Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 6:24175. doi: 10.1038/srep24175
Lin, S., Wu, Z., Yu, G., Zhu, M., Yu, B., and Li, R. (2010). Genetic diversity and molecular phylogeny of Planktothrix (Oscillatoriales, cyanobacteria) strains from China. Harmful Algae 9, 87–97. doi: 10.1016/j.hal.2009.08.004
Loka Bharathi, P. A. (2008). “Sulfur cycle,” in Encyclopedia of Ecology, eds S. E. Jorgensen and B. Fath (Oxford: Elsevier), 3424–3431. doi: 10.1016/B978-008045405-4.00761-8
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Micallef, M. L., D’Agostino, P. M., Al-Sinawi, B., Neilan, B. A., and Moffitt, M. C. (2015). Exploring cyanobacterial genomes for natural product biosynthesis pathways. Mar. Genomics 21, 1–12. doi: 10.1016/j.margen.2014.11.009
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Nunnery, J. K., Mevers, E., and Gerwick, W. H. (2010). Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotechnol. 21, 787–793. doi: 10.1016/j.copbio.2010.09.019
Nylander, J. A. A. (2004). MrModeltest v2. Program distributed by the author. Evol. Biol. Cent. Uppsala Univ. 2, 1–2.
Oliveira, D. T. D., Turbay Vasconcelos, C., Feitosa, A. M. T., Aboim, J. B., Oliveira, A. D. N., Xavier, L. P., et al. (2018). Lipid profile analysis of three new Amazonian cyanobacteria as potential sources of biodiesel. Fuel 234, 785–788. doi: 10.1016/j.fuel.2018.07.080
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pattanaik, B., and Lindberg, P. (2015). Terpenoids and their biosynthesis in cyanobacteria. Life 5, 269–293. doi: 10.3390/life5010269
Pereira, A., Tassin, B., and Jørgensen, S. E. (1994). A model for decomposition of the drown vegetation in an Amazonian reservoir. Ecol. Modell. 75–76, 447–458. doi: 10.1016/0304-3800(94)90039-6
Pereira, S. B., Mota, R., Vieira, C. P., Vieira, J., and Tamagnini, P. (2015). Phylum-wide analysis of genes/proteins related to the last steps of assembly and export of extracellular polymeric substances (EPS) in cyanobacteria. Sci. Rep. 5:14835. doi: 10.1038/srep14835
Pettersson, L. H., and Pozdnyakov, D. (2013). Monitoring of Harmful Algal Blooms. Heidelberg: Springer-Verlag GmbH, doi: 10.1007/978-3-540-68209-7
Prabha, R., Singh, D. P., Somvanshi, P., and Rai, A. (2016). Functional profiling of cyanobacterial genomes and its role in ecological adaptations. Genomics Data 9, 89–94. doi: 10.1016/j.gdata.2016.06.005
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rambaut, A., and Drummond, A. J. (2013). Tracer v1.6. Available at: http://tree.bio.ed.ac.uk/software/tracer
Rigonato, J., Alvarenga, D. O., Andreote, F. D., Dias, A. C. F., Melo, I. S., Kent, A., et al. (2012). Cyanobacterial diversity in the phyllosphere of a mangrove forest. FEMS Microbiol. Ecol. 80, 312–322. doi: 10.1111/j.1574-6941.2012.01299.x
Rigonato, J., Alvarenga, D. O., and Fiore, M. F. (2017). “Tropical cyanobacteria and their biotechnological applications,” in Diversity and Benefits of Microorganisms from the Tropics, eds J. L. de Azevedo and M. C. Quecine (Cham: Springer International Publishing), 139–167. doi: 10.1007/978-3-319-55804-2_7
Rippka, R., Deruelles, J., Waterbury, J. B., Herdman, M., and Stanier, R. Y. (1979). Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111, 1–61. doi: 10.1099/00221287-111-1-1
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Saxena, G., Marzinelli, E. M., Naing, N. N., He, Z., Liang, Y., Tom, L., et al. (2015). Ecogenomics reveals metals and land-use pressures on microbial communities in the waterways of a megacity. Environ. Sci. Technol. 49, 1462–1471. doi: 10.1021/es504531s
Schirrmeister, B. E., Antonelli, A., and Bagheri, H. C. (2011). The origin of multicellularity in cyanobacteria. BMC Evol. Biol. 11:45. doi: 10.1186/1471-2148-11-45
Shestakova, S. V., and Karbysheva, E. A. (2015). The role of viruses in the evolution of Cyanobacteria. Biol. Bull. Rev. 5, 527–537. doi: 10.1016/S0306-9877(76)80009-6
Shih, P. M., Wu, D., Latifi, A., Axen, S. D., Fewer, D. P., Talla, E., et al. (2013). Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 1053–1058. doi: 10.1073/pnas.1217107110
Simm, S., Keller, M., Selymesi, M., and Schleiff, E. (2015). The composition of the global and feature specific cyanobacterial core-genomes. Front. Microbiol. 6:219. doi: 10.3389/fmicb.2015.00219
Sivonen, K., Leikoski, N., Fewer, D. P., and Jokela, J. (2010). Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 86, 1213–1225. doi: 10.1007/s00253-010-2482-x
Swofford, D. L. (2002). PAUP∗. Phylogenetic Analysis using Parsimony (∗and other Methods). Version 4. Sunderland, MA: Sinauer Associates, doi: 10.1159/000170955
Tan, B. F., Te, S. H., Gin, K. Y.-H., and Thompson, J. R. (2016). Draft genome sequence of a tropical freshwater cyanobacterium, Limnothrix sp. Strain P13C2. Genome Announc. 4:e01117-16. doi: 10.1128/genomeA.01117-16.Copyright
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vanni, M. J. (2002). Nutrient cycling by animals in freshwater ecosystems. Annu. Rev. Ecol. Syst. 33, 341–370. doi: 10.1146/annurev.ecolsys.33.010802.150519
Vigil-Stenman, T., Larsson, J., Nylander, J. A. A., and Bergman, B. (2015). Local hopping mobile DNA implicated in pseudogene formation and reductive evolution in an obligate cyanobacteria-plant symbiosis. BMC Genomics 16:193. doi: 10.1186/s12864-015-1386-7
Vijayakumar, S., and Menakha, M. (2015). Pharmaceutical applications of cyanobacteria-A review. J. Acute Med. 5, 15–23. doi: 10.1016/j.jacme.2015.02.004
Vis, M. L. (2005). Freshwater algae of North America: ecology and classification. J. Torrey Bot. Soc. 132, 533–534. doi: 10.3159/1095-5674(2005)132[533b:FAONAE]2.0.CO;2
Winters, K., Parker, P. L., and Van Baalen, C. (1969). Hydrocarbons of blue-green algae: geochemical signfficance. Science 163, 467–468. doi: 10.1126/science.163.3866.467
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Zhu, M., Yu, G., Li, X., Tan, W., and Li, R. (2012). Taxonomic and phylogenetic evaluation of Limnothrix strains (Oscillatoriales, Cyanobacteria) by adding Limnothrix planktonica strains isolated from central China. Hydrobiologia 698, 367–374. doi: 10.1007/s10750-012-1127-8
Zhu, T., Hou, S., Lu, X., and Hess, W. R. (2017). Draft genome sequences of nine cyanobacterial strains from diverse habitats. Genome Announc. 5:e01676-16. doi: 10.1128/genomeA.01676-16
Zhu, T., Scalvenzi, T., Sassoon, N., Lu, X., and Gugger, M. (2018). Terminal olefin profiles and phylogenetic analyses of olefin synthases in diversified cyanobacterial species. Appl. Environ. Microbiol. 84:e00425-18. doi: 10.1128/AEM.00425-18
Keywords: Amazonian, cyanobacteria, Limnothrix, comparative genomics, genome mining
Citation: Lima ARJ, Siqueira AS, Vasconcelos JM, Pereira JS, Azevedo JSN, Moraes PHG, Aguiar DCF, de Lima CPS, Vianez-Júnior JLSG, Nunes MRT, Xavier LP, Dall’Agnol LT and Goncalves EC (2018) Insights Into Limnothrix sp. Metabolism Based on Comparative Genomics. Front. Microbiol. 9:2811. doi: 10.3389/fmicb.2018.02811
Received: 04 May 2018; Accepted: 01 November 2018;
Published: 20 November 2018.
Edited by:
Johann Peter Gogarten, University of Connecticut, United StatesReviewed by:
Artemis S. Louyakis, University of Connecticut, United StatesCopyright © 2018 Lima, Siqueira, Vasconcelos, Pereira, Azevedo, Moraes, Aguiar, de Lima, Vianez-Júnior, Nunes, Xavier, Dall’Agnol and Goncalves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex Ranieri Jerônimo Lima, YWxleC5saW1hQGljYi51ZnBhLmJy; YWxleHJhbmllcmlAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.