 Dimitry Y. Sorokin
Dimitry Y. Sorokin Maria S. Muntyan
Maria S. Muntyan Stepan V. Toshchakov
Stepan V. Toshchakov Aleksei Korzhenkov
Aleksei Korzhenkov Ilya V. Kublanov
Ilya V. Kublanov
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 November 2018
Sec. Extreme Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02672
This article is part of the Research Topic Living with Salt: Genetics and Ecology of Halophiles View all 17 articles
Stable development of a heterotrophic bacterial satellite with a peculiar cell morphology has been observed in several enrichment cultures of haloalkaliphilic benthic filamentous cyanobacteria from a hypersaline soda lake in Kulunda Steppe (Altai, Russia). The organism was isolated in pure culture (strain Omega) using sonicated cyanobacterial cells as substrate and it was identified as a deep phylogenetic lineage within the recently proposed phylum Balneolaeota. It is an obligately aerobic heterotroph utilizing proteins and peptides for growth. The cell morphology significantly varied from semicircles to long filaments depending on the growth conditions. The cultures are red-orange colored due to a presence of carotenoids. The isolate is an obligate alkaliphile with a pH range for growth from 8.5 to 10.5 (optimum at 9.5–10) and moderately salt-tolerant with a range from 0.3 to 3 M total Na+ (optimum at 1 M). The genome analysis of strain Omega demonstrated a presence of gene, encoding a proteorhodopsin forming a separate branch in the sodium-translocating proteorhodopsin family. Experiments with washed cells of Omega confirmed light-dependent sodium export. A possible physiological role of the sodium proteorhodopsin in strain Omega is discussed. Phylogenomic analysis demostrated that strain Omega forms an deep, independent branch of a new genus and family level within a recently established phylum Balneolaeota.
Soda lakes are a specific variety of salt lakes which brines are dominated by sodium carbonates resulting in molar alkalinity in solution and a stable pH around 10. Despite double extreme conditions of high salt-high pH, soda lakes, in general, are highly productive (Melack, 1981, 1988; Kompantseva et al., 2009) harboring diverse haloalkaliphilic communities dominated by prokaryotes responsible for the cycling of important biogenic elements, such as carbon, nitrogen and sulfur (last reviewed: Antony et al., 2013; Sorokin et al., 2014, 2015; Sorokin, 2017). Thus, during the last 30 years, the soda lake prokaryotes have been a subject of intense fundamental as well as biotechnological research, as a limits-of-life case and a source of salt-tolerant and alkali-stable exo-enzymes, respectively (last reviewed: Zhao et al., 2014; Amoozegar et al., 2017).
Haloalkaliphilic oxygenic cyanobacteria are the dominant primary producers in soda lakes. The evidences on their diversity are not abundant, however. Moderately saline tropical soda lakes in Africa are dominated by Spirulina-Cyanospira-Arthrospira clade (Krienitz et al., 2013; Krienitz and Schagerl, 2016), while in Siberian moderate and hypersaline soda lakes, at severe continental climate, two benthic filamentous forms belonging to the genera Nodosilinea and Geitlerinema have been consistently detected (Samylina et al., 2014). In contrast to algae, the cell wall of cyanobacterial cells lacks cellulose and their biomass is more rich in proteins, especially in filamentous diazotrophic species (Vargas et al., 1998). However, very little is still known about the identity of protein-utilizing microbial communities in soda lakes participating in cyanobacterial biomass mineralization. Recent studies identified two novel genera of aerobic extremely salt-tolerant alkaliphilic bacteria specialized on using various proteins as growth substrates, a gammaproteobacterium Natronospira proteinivora and Natronotalea proteinilytica, a representative of the phylum Rhodothermaeota (Sorokin et al., 2017a,b). Recently, a haloalkaliphilic anaerobic proteolytic clostridium, described as Proteivorax tanatarense gen. nov., sp. nov., was shown to utilize the cell proteins produced by a soda lake benthic cyanobacterium Geitlerinema sp. (Kevbrin et al., 2013). In addition, for hypersaline conditions, three natronoarchaeal species have been shown to produce haloalkali-stable proteases at salt-saturated conditions and high pH by members of the genera Natronococcus, Natrialba, and Natronolimnobius (Studdert et al., 2001; Selim et al., 2014; Derntl et al., 2015).
In this work, we describe phenotypic and genomic properties of a moderately salt-tolerant alkaliphilic aerobic protein-utilizing bacterium which developed in a stable co-culture with soda lake benthic filamentous cyanobacteria. The isolate belongs to a novel deep phylogenetic lineage within the recently suggested phylum Balneolaeota, a former part of the phylum Bacteroidetes (Hahnke et al., 2016; Munoz et al., 2016), forming a new genus and species candidate taxon “Ca. Cyclonatronum proteinivorum.”
An enrichment culture of filamentous benthic cyanobacteria was obtained from a biofilm developed in July 2011 on the surface of littoral sediments of the soda lake Bitter-3 (south Kulunda Steppe, Altai region, Russia; N51°40′/E79°53′). The brines total salinity was 90 g l-1 soluble carbonate alkalinity – 1.0 M and the pH 10.3. The enrichment culture was grown in 2 L Erlenmeyer flask containing 1 L medium mounted on a magnetic stirrer at 25°C and at ambient (day/night) light regiment. The basal mineral medium contained 1.3 M total Na+ at pH 10 with the following composition (g l-1): Na2CO3 – 57; NaHCO3 – 9; NaCl – 9, K2HPO4 – 1, KNO3 – 0.6, MgSO4 × H2O – 0.25, acidic trace metal solution (Pfennig and Lippert, 1966) – 1 ml. The fed-batch culture was maintained for 2 years aimed to produce biomass of the soda lake cyanobacteria. In several months the culture acquired a stable domination of two types of benthic filamentous haloalkaliphilic cyanobacteria with large and small cells identified as Geitlerinema sp. and Nodosilinea sp., respectively (Samylina et al., 2014).
Strain Omega was consistently developing as a heterotrophic satellite in several parallel cultures of haloalkaliphilic filamentous cyanobacteria enriched from a soda lake in S-W Siberia (Supplementary Figure S1). For isolation, a solid medium was prepared from the filter-sterilized cyanobacterial mineral medium, supplemented with sonicated and filter-sterilized cell-free extract of cyanobacteria after mixing 1:1 with 4% sterile agarose at 50°C. The inoculum was prepared from a stationary phase cyanobacterial culture after removing the cyanobacterial aggregates first by settling and the remaining suspended filaments – by a low-speed centrifugation. The inoculated plates were incubated up to 1 month in closed plastic bags at 25°C. A pure culture was isolated from a single colony after several rounds of restriking onto the solid medium. Further experiments with pure culture were performed in liquid salt media.
For standard cultivation and phenotypic characterization of strain Omega, a sodium carbonate-based medium buffered at pH 10 and containing 1 M total Na+ and casein peptone as substrate were used. For the salinity range (from 0.1 to 3 M total Na+, pH 10), the culture was pregrown at 1 M total Na+. An additional attempt to measure an absolute maximum of the salt tolerance was made afterward using a culture grown at maximum salinity in the first round. For the pH profiling, a range of pH from 6.5 to 11 with an increment of 0.5 unit was created using the following buffer systems containing 1 M total Na+: 0.08 M HEPES/0.05 M K-phosphate for pH from 6.5 to 8 and sodium bicarbonate-carbonate buffer system for pH 8-11. Growth (OD600) and the actual pH were monitored until the maximum OD values were reached. The temperature profile was measured at pH 10 and a total Na+ 1 M from 20 to 50°C with an increment of 5°C. Anaerobic growth either by fermentation or respiration with casein peptone carbon and energy source was tested in 10 ml cultures placed into 23 ml serum bottles closed with butyl rubber and made anoxic by 5 cycles of evacuation-flushing with sterile argon gas.
Biomass growth dynamics was followed by measuring optical density at 600 nm. Phase contrast microphotographs were produced with a Zeiss Axioplan Imaging 2 microscope (Göttingen, Germany). Pigments were extracted from wet cell biomass using 7:3 mixture of MeOH-aceton and 30 min vortexing. Absorption spectra were recorded on the UV-Visible diode-array HP 8453 spectrophotometer (Hewlett Packard, Amsterdam, Netherlands). The protease activity was tested qualitatively by diffusion-to-agar technique. For this, the culture supernatant was first passed through 0.22 μm syringe filter to remove residual cells and then 20 times concentrated using 20 ml Centricon tubes (Millipore) with 30 and 10 kDa membrane. The cell pellet was sonicated and the unbroken cells removed by 5 min centrifugation in 2 ml Eppendorf tube at 14,000 rpm, resulting in the cell-free extract fraction. 30 μl aliquats of each fraction were applied to wells cut into 1% casein agarose supplemented with sodium carbonate buffer containing 0.6 M total Na+ at pH 10. The plate was incubated for 72 h at 30°C and the hydrolysis zones were visualized by flooding with 10% (w/v) TCA solution.
The cells were grown aerobically in medium containing 1 M total Na+ (0.8 M Na carbonates/0.2 M NaCl), pH 10 at 30°C and harvested by centrifugation. The cell pellet was washed three times in a salt solution containing 0.25 M Na2SO4 and 0.55 M K2SO4, and finally resuspended in the same solution at concentration of × 20 times the original culture. In the experiments with “minus sodium” the washing salt solution contained 0.8 M K2SO4. All the experiments were performed in a completely darkened thermostated room at 20°C. To measure the light-induced proteorhodopsin responses, 400 μl of the washed cell suspension was placed into a 500 μl-transparent plastic vial mounted on a magnetic stirrer and covered with a thermally insulating plate. This photocell was illuminated by a “cold” light using a custom-made illuminator OVS-1M equipped with two fiber-optic cables and halogen incandescent lamp KGM (9 V × 70 W) with the light flux in each fiber-optic cable of 17 W (Lytkarino Optical Glass Factory of optical glass and materials, Moscow region, Russia). The light-induced pH changes in the bacterial suspensions were continuously monitored with a glass InLab pH-microelectrode (Ø 3 mm, Mettler Toledo) connected to PHM250 ion analyzer (Radiometer Analytical SAS, France). The light-dependent alkalinization of the medium was taken as an indication of the Na+ extrusion (Inoue et al., 2013).
The strain Omega genome was sequenced using both paired-end and mate-paired DNA libraries. Paired-end libraries were prepared using the Ultra-DNA library preparation kit (New England Biolabs, United States) according to manufacturer’s instructions to obtain mean library size of 500 bp. Mate-paired libraries were prepared with NexteraTM Mate Pair Library Prep Kit (Illumina Inc., United States). Finally, one paired-end and three mate-paired libraries were sequenced with MiSeqTM Personal Sequencing System (Illumina Inc., United States) resulting in 2 × 250 bp reads massive. After sequencing, all reads were subjected to stringent quality filtering with CLC Genomics Workbench 8.5 (Qiagen, Germany). After filtering, overlapping paired-end library reads were merged with SeqPrep tool1 resulting in 549,472 single merged reads and 178,732 read pairs with mean insert size of 602 bp. Mate paired reads were treated with NextClip tool (Leggett et al., 2014), resulting in 712,544 read pairs with mean insert size of 2233 bp. Reads were assembled with both ALLPATHS-LG (Butler et al., 2008) and SPADES 3.7.0 (Nurk et al., 2013) assemblers and refined manually using CLC Genomics Workbench 8.5 software (Qiagen, Germany), resulting in five high-quality contigs. Orientation of contigs and final filling of sequence gaps were made by PCR with outward-oriented primers to contigs termini and subsequent Sanger sequencing of resulting amplicons. Final average genome coverage of a single chromosome was 95.7 ×.
Gene prediction and primary annotation were performed with IMG/M ER System (Chen et al., 2017). Refining of the automated annotations and some particular predictions were done according to the protocol of Toshchakov et al. (2015). The annotated genome sequence of strain Omega has been deposited in the GenBank database under accession number CP027806. Corresponding Bioproject and Biosample IDs are PRJNA392178 and SAMN07284862, respectively.
16S- and 23S-rRNA gene sequences of representatives of Bacteroidetes and its two recently proposed sister phyla Rhodothermaeota and Balneolaeota, as well as of Chlorobi and Ignavibacteriae, were downloaded from the Silva (SSU_126 & LSU_126) databases. Together with the 16S- and 23S-rRNA gene sequences of strain Omega, they were concatenated and aligned in MAFFT v. 7 (G-INS-i algorithm, Katoh and Standley, 2013). JModel Test was used to test the best evolutionary model, which appeared to be the General Time Reversible (GTR) model (G + I, 4 categories) (Nei and Kumar, 2000). The tree with the model and 1000 bootstrap resamplings was inferred in MEGA 6.0 (Tamura et al., 2013).
Sequences of 27 ribosomal proteins universally conserved in 146 bacterial species (Supplementary Table S1) representing almost all currently known phyla with completely or almost completely sequenced genomes were aligned using MAFFT v. 7 (E-INS-i algorithm, Katoh and Standley, 2013). Alignments were concatenated and the phylogenetic tree was inferred using MEGA 6.0 (Tamura et al., 2013) with LG evolutionary model and gamma-distributed site rates.
For phylogene analysis of rhodopsin proteins, sequences of all characterized rhodopsins were searched using BLAST with a CYPRO_0974 as a query and Uniprot and SwissProt databases (16.05.2018) as the references. Altogether, 45 sequences were aligned in MAFFT v. 7 (L-INS-i algorithm, Katoh and Standley, 2013). The tree was constructed in MEGA 6.0 (Tamura et al., 2013) with LG evolutionary model and gamma-distributed site rates.
Subtilases phylogeny was performed for all S8 endopeptidases family proteins, found in in silico translated Omega proteome as well as from the SwissProt database (30.03.2018). Only proteins with >250 aa from SwissProt were chosen including 327 of the total 341 S8 peptidases. After clustering using 70% sequence identity threshold in CD-HIT (Huang et al., 2010), 161 Swissprot sequences remained and together with Omega proteins 170 S8 peptidase members were aligned in MAFFT v. 7 (L-INS-i algorithm, Katoh and Standley, 2013). The tree was constructed in MEGA 6.0 (Tamura et al., 2013) with LG evolutionary model and gamma-distributed site rates.
The S51 family peptidases phylogeny was performed using two sequences of strain Omega (CYPRO_0608 and CYPRO_2145), five characterized cyanophycinases and peptidases E (both belong to S51 family) from SwissProt, twenty two sequences from Pfam (seed sequences of PF03575 (Peptidase_S51) domain) and 100 first best BLAST hits of CYPRO_0608 and CYPRO_2145 (50 each) from Uniprot. After deleting duplicates, 129 sequences were aligned in MAFFT v. 7 (L-INS-i algorithm, Katoh and Standley, 2013). The tree was constructed in MEGA 6.0 (Tamura et al., 2013) with LG evolutionary model and gamma-distributed site rates.
Replicate enrichment cultures of filamentous benthic cyanobacteria from a soda lake in Kulunda Steppe (Altai, Russia) were developing in two phases. In the first phase, the cyanobacteria formed massive aggregated biomass layer floating on the top of incubation flask with no growth in the main medium body. In the second phase, the medium started to become turbid with pink coloration, while the cyanobacterial layer diminished and started to decompose (Supplementary Figure S1). We assumed that this pattern indicated a usual type of succession from a primary producer to heterotrophic degraders of the phototrophic biomass. Microscopic examination of the pink layer showed a domination of a bacterium with peculiar cell morphology in the form of semicircles with only a few other rod-shaped bacteria (Figure 1a). Since preliminary clone library (results are not shown) indicated that the dominant component belongs to an uncultured deep lineage bacteroidetes, attempts to isolate it in pure culture has been initiated using a solid medium containing sonicated cell extract from the cyanobacterial biomass as substrate. After a long incubation (3 weeks) small reddish colonies appeared which, after several rounds of resrtiking resulted in a pure culture, designated strain Omega, with a cell morphology resembling a dominant heterotrophic satellite (Figure 1b). However, the cell morphology varied depending on substrate and salinity, from semicircles to long “whip-like” cells (Figures 1c–f). The biomass was colored pink-orange due to a presence of pigments extractable with a mixture of methanol-aceton (Supplementary Figure S2), most probably belonging to carotenoids.
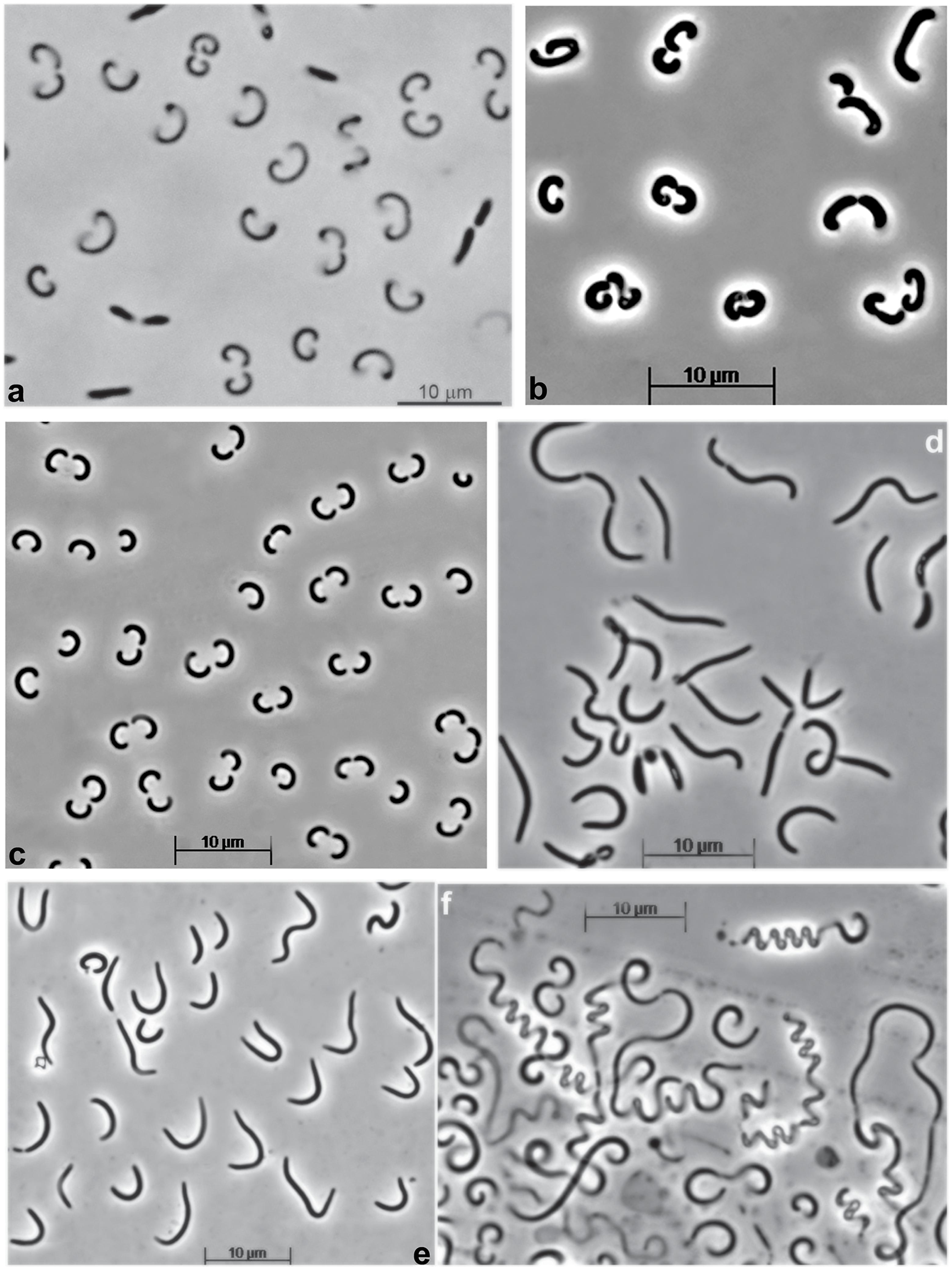
FIGURE 1. Cell morphology of strain Omega (phase contrast microphotographs) grow at pH 10 and 30°C in (a) a mix culture developing in supernatant of cyanobacterial enrichment from a soda lake, and (b-f) in pure culture. (b) Cells from colonies developing on solid medium with cyanobacterial cell extract as substrate; (c,d), cells grown with albumin in liquid culture at 2 M Na+ and 1 M Na+, respectively; (e,f), cells grown in liquid culture at 1 M Na+ with casein and caseinopepton, respectively.
Further work with the isolate demonstrated that it belongs to a functional group of obligate aerobic protein degraders. Best growth was observed with peptone from casein, but also other peptones, such as meat and soy, and yeast extract can serve as growth substrates. Among the proteins, the isolate was able to grow with heat-sterilized casein, bovine serum albumin and gelatin. The cells grown with casein showed a presence of cell-bound protease active at salt concentration from 0.2 to 1 M Na+ and with a pH optimum of 9–10 (Supplementary Figure S3). Sugars (glucose, fructose, galactose, mannose, lactose, xylose, sucrose, trehalose, cellobiose, maltose) and sugar polymers (such as starch, amorphous cellulose, chitin, pectin, alginate) did not support growth. Fermentative growth with peptone from casein and glucose was also not observed, as well as with added electron acceptors, such as nitrate, nitrite, sulfur, thiosulfate and fumarate. Strain Omega is a moderately salt-tolerant obligate alkaliphile with a pH range for growth from 8.5 to 10.5 (optimum at 9.5–10) and salt range at pH 10 from 0.3 to 2 M Na+ (Figure 2). In contrast to most of the moderately salt-tolerant bacterial isolates from soda lakes, strain Omega required cloride ion both for optimal growth and storage of the grown culture.
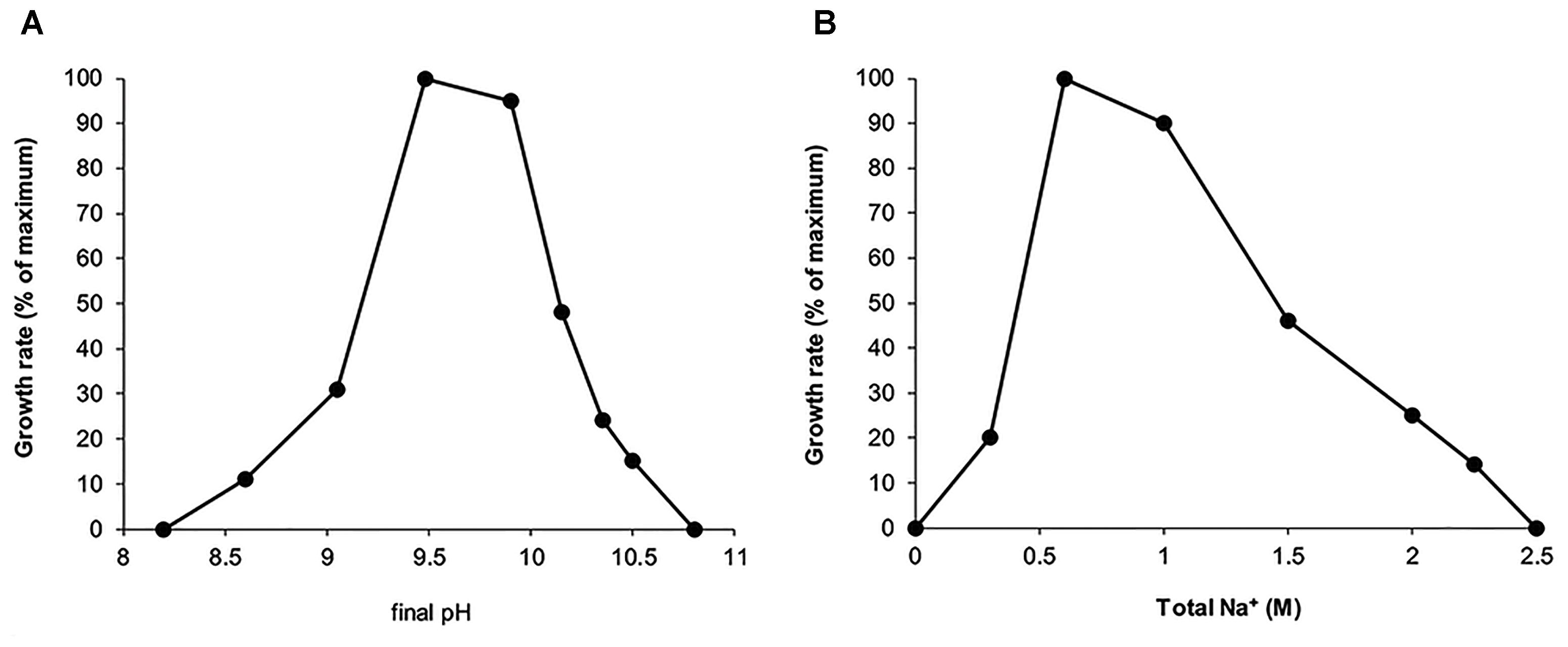
FIGURE 2. Influence of pH at 1 M total Na+ (A) and influence of total Na+ in the form of carbonates at pH 10 (B) on growth of strain Omega with caseinopepton. The actual pH values are presented on the X axis. The results are mean from a duplicate with the maximum deviation of less than 20%.
Assembled complete genome of strain Omega consists of 4294247 bp and does not contain any extrachromosomal elements (Supplementary Figure S4). Genome harbors forty three tRNA genes and three ribosomal operons with identical structure: 16S – tRNA-Ile – tRNA-Ala – 23S – 5S. Average G + C content is 51.39%. Genome contains 3284 protein-coding genes, 2349 (70.35%) of which were assigned with function. Two concatenated trees, based on 16S + 23S rRNA genes (Supplementary Figure S5) and on 27 conservative ribosomal proteins (Figure 3) placed strain Omega within the recently proposed phylum Balneolaeota (Hahnke et al., 2016) as a separate deep lineage. Distribution of Best BLAST Hits of in silico translated Omega proteome revealed strong domination of the proteins mostly related to the members of Balneolaeota (Figure 4; Supplementary Figure S6). Surprisingly, the number of hits, affiliated with the nearest phylum Rhodothermaeota was relatively low in comparison with Bacteroidetes sensu stricto and Proteobacteria. Yet, this is most probably due to a quantitative bias from the number of analyzed genomes of each phyla. 16S rRNA genes identity, calculated during pairwise alignment (BLAST) with validly published representatives of Balneolaeota was less than 88% with Balneola vulgaris strain 13IX (Urios et al., 2006) as the closest relative (87.7%), indicating that the soda lake isolate is forming a deep phylogenetic lineage at the level of a novel family. Furthermore, culture-independent surveys demonstrated a presence of two groups of clones related to strain Omega found previously in a low salt alkaline Lonar lake in India (clone SA_226, sequence identity 97%) (Antony et al., 2014) and a hypersaline soda lake Mono in California (clone ML602J-34, sequence identity 92%) (Humayoun et al., 2003).
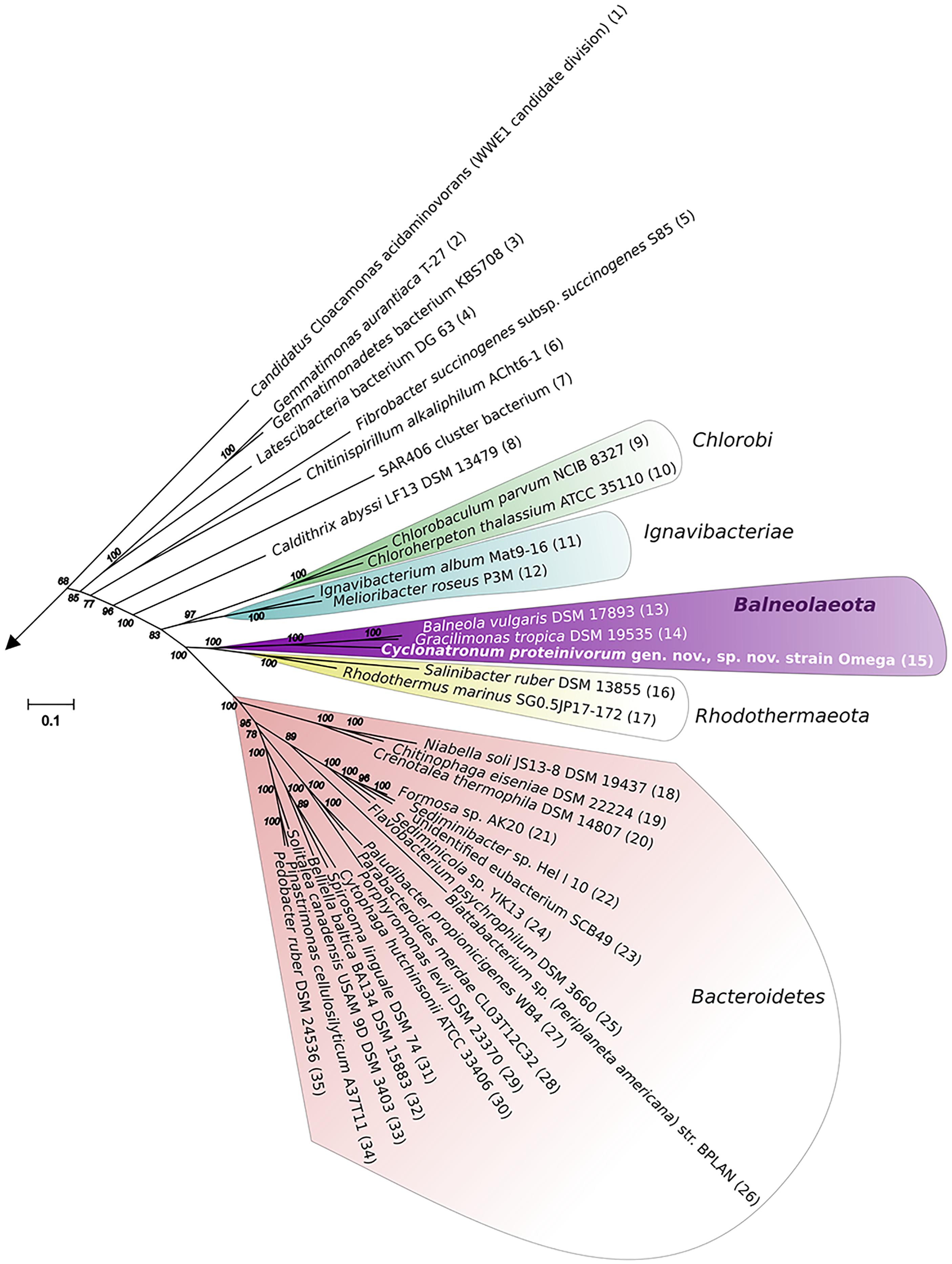
FIGURE 3. 27 concatenated conservative proteins-based phylogenetic analysis. The tree was constructed using Maximum-Likelihood method. The tree with the highest log likelihood is shown. The bootstrap values (100 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 136 amino acid sequences and 4318 positions. All positions with less than 95% site coverage were eliminated. The unrooted tree was generated in MEGA6 (Tamura et al., 2013). Only FCB superphylum representatives are shown. The sequences were obtained from IMG and have the following accession numbers: (1), 642560256; (2), 643773439; (3), 2578408359; (4), 2656359422; (5), 646369571; (6), 2638713609; (7), 265102961; (8), 2562178319; (9), 642718814; (10), 642716968; (11), 2514222721; (12), 2517204605; (13), 2515844915; (14), 2515866577; (15), 2695201269; (16), 637844027; (17), 2505982187; (18), 2506845178; (19), 2635114424; (20), 2601542687; (21), 2532033211; (22), 2561440247; (23), 641144478; (24), 2623579954; (25), 2623246297; (26), 646377621; (27), 649775995; (28), 2531043488; (29), 2515619891; (30), 638073153; (31), 646498001; (32), 2509560651; (33), 2509579588; (34), 2671580424; (35), 2677155390.
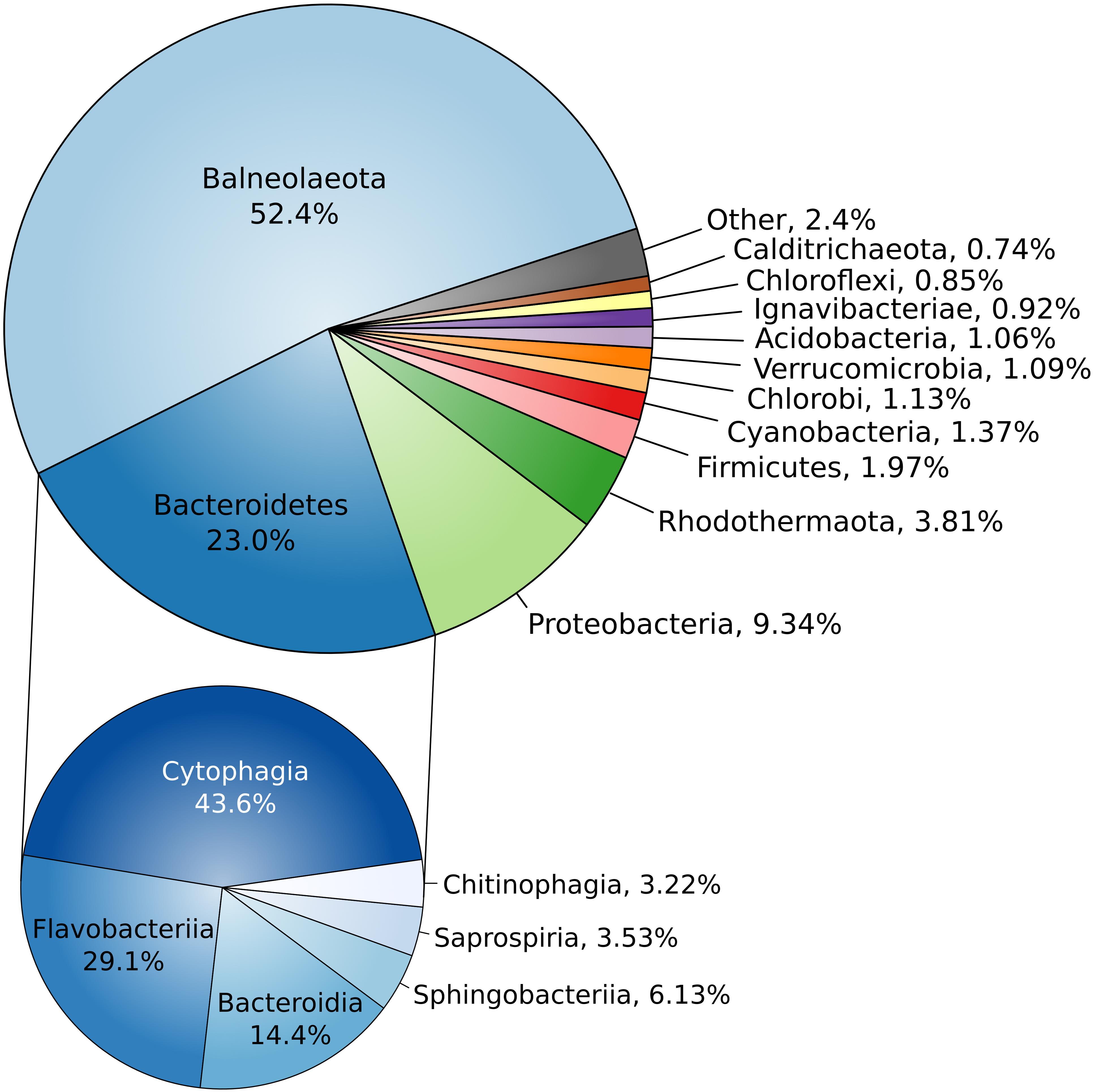
FIGURE 4. Blast-hit distribution of proteins. Taxonomic distribution of in silico translated proteome of strain Omega best BLAST hits. All hits are classified by phyla (top) and Bacteroidetes hits are classified by classes (bottom).
Results of the analysis of COG distribution performed by IMG-ER (Chen et al., 2017) supported phenotypic observations on the proteolytic lifestyle of strain Omega. High number of genes encoding proteins, participating in amino acid transport and metabolism (172 proteins, 8.56% of total proteome), as opposed to a lesser proportion of proteins, involved in carbohydrate transport and metabolism – 4.96%, has been observed in the genome which is significantly less than in Rhodothermus marinus, known for its active carbohydrate utilization (Supplementary Table S2). A comparatively high number of proteins involved in defense mechanisms (4.11%, the highest percentage among complete genomes of Balneolaeota and Rhodothermaeota) seems reasonable for an organism closely associated with cyanobacteria. High abundance and expression levels of proteins participating in defense mechanisms has recently been reported for cyanolytic bacterial heterotrophs (Osman et al., 2017).
It should be also noted, that among analyzed representatives of Balneolaeota and Rhodothermaeota, the genome of strain Omega encodes the highest number of proteins with only general function prediction (10.47%) and not assigned to any cluster of orthologous groups (44.95%), which reflects its deep-lineage phylogenetic position. The COG-based hierarchical clustering shows the Omega proteome is only distantly related to the genome-sequenced representatives of Balneolaeota and Rhodothermaeota. In contrast, hierarchical clustering by functional categories revealed that strain Omega located close to a Balneolaeota representative Gracilimonas tropica, which has also been isolated from cyanobacterial biomass (Supplementary Figure S6). This is an indication that despite their distinct phylogenetic positions both microorganisms independently adapted to their cyanobacteria-associated lifestyle. A recent metagenomic reconstructions from the Kulunda Steppe hypersaline soda lake brines also showed a prominent presence of a Balneola/Gracilimonas-related lineage there (T5-Br10_g13) (Vavourakis et al., 2016).
In prokaryotes, lateral gene transfer is the most widespread and universal mechanism of rapid adaptation to environmental changes (Madsen et al., 2012). Such adaptations might be particularly important for highly dynamic environments of hypersaline soda lakes in Central Asia, subjected to seasonal algal blooms, periods of extreme evaporation and dilution and a large annual temperature fluctuations. Omega genome was inspected for LGT events with Seqword Island Sniffer program, searching for atypical tetranucleotide usage (Bezuidt et al., 2009) and Island Viewer 3 package (Dhillon et al., 2015), implementing IslandPath-DIMOB (Waack et al., 2006), and SIGI-HMM, algorithms (Hsiao et al., 2003). 22 putative genomic islands (GI) of 377 kb total length were predicted (Supplementary Figure S5 and Supplementary Table S3). Most of genes of predicted GIs encoded proteins with unknown functions (55%), mobile element proteins (16%), proteins of defense systems (14%), which is typical for horizontally transferred genes. However, closer inspection of The Best BLAST hits of the majority of genes encoding these proteins showed that most of them have the best blast hits in closely related classes: Balneolia (24%), Flavobacteria (13%), or Cytophagia (11%), representatives of which could also be found in similar environments. Omega genome harbors 52 presumably functional or inactivated transposase-containing mobile elements, occupying totally over 67 Kbp. Analysis of transposase diversity with ISSaga showed that they belong to 12 different IS families. Two families, IS1380 and IS66 were not present either in Rhodothermaeota or Balneolaeota genomes, making them unique for the novel lineage within these Bacteroidetes-related phyla. While IS1380 elements contained only partial transposases, all four IS66 were complete, consisting of three ORFs which produce three functional proteins by translation coupling mechanism (Han et al., 2001). Interestingly, IS66 elements are mostly widespread in Proteobacteria, while only several IS66 lineages were detected in Bacteroidetes/Clorobi hosts (Gourbeyre et al., 2010).
The proteorhodopsin gene CYPRO_0974 identified in Omega genome has the NDQ motif unique for the Na+-translocating proteorhodopsin (NaR) recently discovered in a few members of salt-tolerant Bacteroidetes (Inoue et al., 2013; Kato et al., 2015; Pinhassi et al., 2016). It formed an independent lineage within the cluster of NaR with confirmed function (Figure 5A). Washed cells of strain Omega exhibited a light-induced alkalinization of the Na+-containing medium (Figure 5B), similar to the marine Krokinobacter eikastus (Inoue et al., 2013). The effects could be explained by functioning of a NaR or a halorhodopsin (HR). However, HR does not pump SO42- and since in this experiment we only used sodium/potassium sulfate in the incubation buffer, the HR possibility is eliminated. Moreover, when the sodium sulfate was replaced by potassium sulfate, the light-induced alkalinization in the Omega cell suspension ceased (Figure 5B). This differentiated the Omega proteorhodopsin from the xenorhodopsin behavior (Inoue et al., 2016; Shevchenko et al., 2017). Introduction of sodium ions in the cell suspension reactivated the Omega Na+-proteorhodopsin. Thus, the in vivo experiments demonstrated the expression of an active light-dependent primary sodium pump in strain Omega, which is only a second (to a low salt-tolerant Indibacter alkaliphilus) example of a haloalkaliphile from soda lakes with confirmed functional NaR. Taking into account that the pumping activity of NaR in the native cell suspensions of Omega is close to that of K. eikastus (Inoue et al., 2013), the Omega NaR can be rather assigned to a true primary ion pump than to a sensor protein. Thus, we can assume that it has an important role as an energy transducer. Such a pump might be profitable in a bacterial species associated with benthic cyanobacteria living at oxygen-limiting conditions on the surface of sulfidic sediments. At these conditions (O2 limitation + high alkalinity + unlimited sodium), Omega-like heterotrophic satellites should be at an advantage utilizing an O2-independent way of solar energy transduction by NaR, as it would allow such organisms to build up an additional sodium-based transmembrane difference of electrochemical potential (Banciu and Muntyan, 2015).
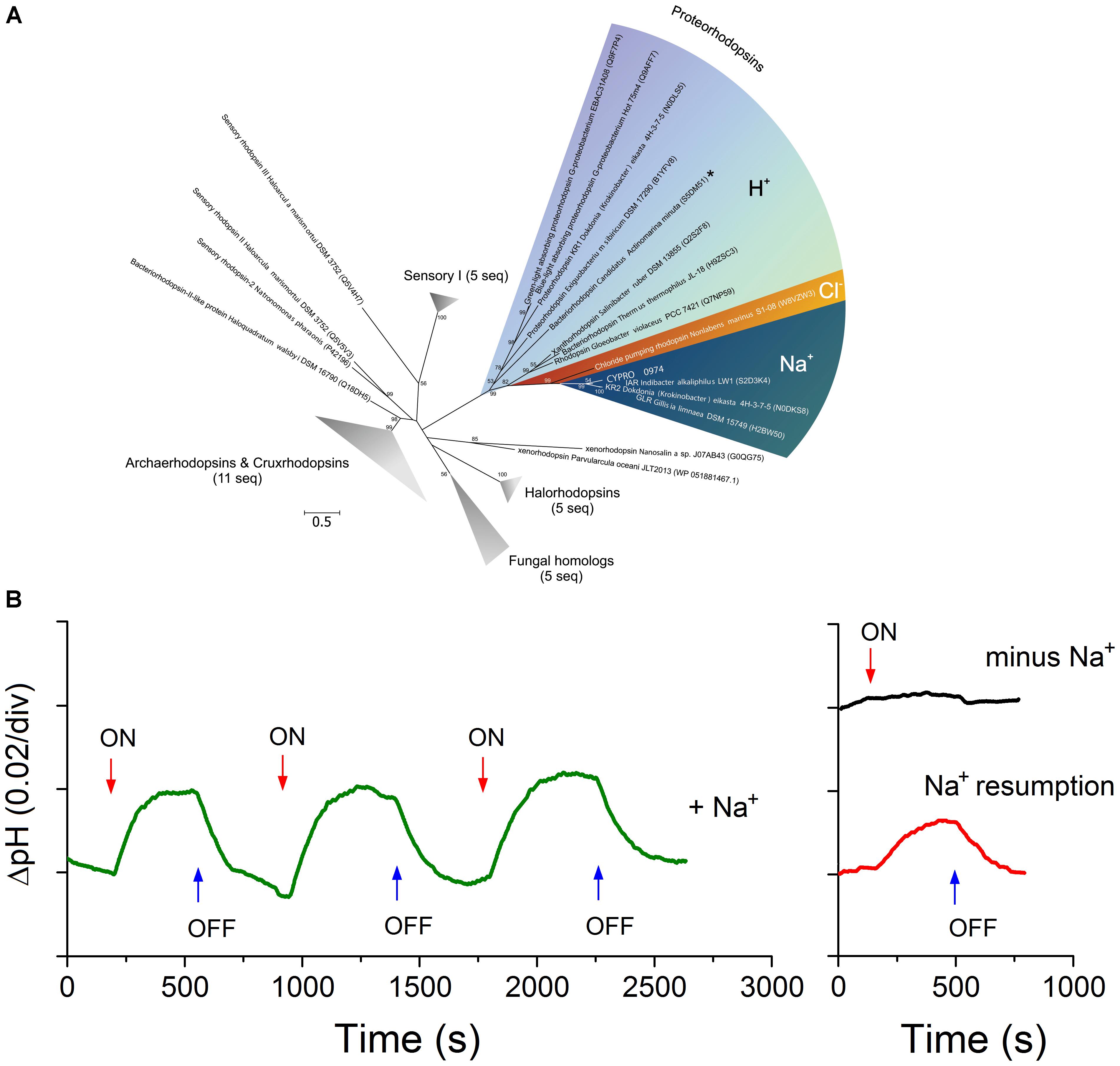
FIGURE 5. A presence of Na+-translocating light-driven primary ion pump in strain Omega. (A) Phylogenetic analysis of a rhodopsin protein encoded by the Omega genome. The tree was constructed using Maximum-Likelihood method. The tree with the highest log likelihood is shown. The bootstrap values (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 45 amino acid sequences and 206 positions. All positions with less than 95% site coverage were eliminated. The unrooted tree was generated in MEGA6 (Tamura et al., 2013). CYPRO_0974 is in bold. (B) Prove of functionality of the light-dependent primary sodium pump using washed cells of strain Omega. Light-induced alkalinization was registered by a pH microelectrode. The measuring medium (left graph) contained 0.25 M Na2SO4 and 0.55 M K2SO4. The “sodium free” medium (right graph) contained 0.8 M K2SO4. The illumination switches are indicated by arrows.
Another putative mechanism of Na+ export is based on the action of a multi-subunit Na+/H+ antiporter Mrp (Swartz et al., 2005), encoded in the Omega genome by Cypro_0615-0621. No genes for Rnf Na+/H+ exporter complex were found.
The genome of strain Omega encodes a classical pathway for synthesis of the typical organic osmolyte glycine betaine including oxidation of choline to betaine aldehyde and its further oxidation to betaine (Roberts, 2005). First step might be performed by either ferredoxin-dependent choline monooxygenase CYPRO_1993 or choline dehydrogenase CYPRO_1995. Second step, might be performed by one of five aldehyde dehydrogenases predicted in the Omega genome (CYPRO_1155, CYPRO_1282, CYPRO_1289, CYPRO_2064 and CYPRO_2065) among which CYPRO_2065 is being the most reliable candidate according to search of HMM profile of curated SwissProt betaine aldehyde dehydrogenase sequences as a query. The genes for sucrose and trehalose biosynthesis, that are often found in halophilic prokaryotes (as secondary) and eukaryotes (as primary, together with sugar alcohols) osmoprotectants (Oren, 2013; Gostinčar et al., 2011) have also been identified in the Omega genome. On the other hand, the genes, encoding proteins, related to ectoine biosynthesis, another osmolyte commonly found in halophilic and halotolerant bacteria, were not identified in the Omega genome.
The genome analysis revealed a presence of all genes encoding glycolysis/gluconeogenesis and TCA cycle enzymes. While no growth on sugars was detected, glycolysis/gluconeogenesis most probably takes a course on the direction of synthesis of glucose-1-phosphate, which is being a substrate for intracellular oligo- and polysaccharides biosynthesis. All genes, encoding proteins for glycogen biosynthesis were found. Surprisingly, relatively high number of CAZymes-coding genes (Lombard et al., 2014) were also found. Among them the most numerous families were glycosidases GH13, GH74, GH23, GH3, GH17, carbohydrate esterases CE1 and CE10 and polysaccharide lyases PL22. While this is quite common in marine Bacteroidetes associated with algae (Kappelmann et al., 2018), in the proteolytic strain Omega a potential for polysaccharide hydrolysis seems to be unnecessary. It is possible, however, that some of these (e.g., amylase-related proteins of the family GH13) are involved in intracellular oligo- and polysaccharide turnover as well as extracellular polysaccharide synthesis. Finally, one cannot exclude the probability of hydrolysis of unknown oligo- and polysaccharides (possibly of complex nature, e.g., components of cyanobacterial cell walls or extracellular matrix), that have not been tested during growth experiments.
According to the IMG ER pipelines, predicting amino acids auxotrophy/eutrophy, strain Omega is an auxotroph to the number of amino acids (arginine, histidine, isoleucine, leucine, lysine, phenylalanine, proline, serine, tyrosine, and valine) and apparently dependent on the external source, in accordance with its key physiology. Its genome contains 134 genes encoding proteins (Supplementary Figure S5 and Supplementary Table S4) of known MEROPS (Rawlings et al., 2014) families. Most of them are peptidases, yet the minor part represents peptidase homologs, lacking peptidase activities. Among the MEROPS enzymes, 51 were predicted to be extracellular using SinalP and SecretomeP prediction servers. Of these, the most represented families were S9 (18 total, 11 secreted proteins), S8 (all 9 are secreted proteins), S12 (5 total, 3 secreted proteins) and M28 (6 total, 4 secreted proteins). While currently known S8 and M28 peptidases are mainly represented by exo- (amino- or carboxy-) peptidases, the majority of S9 family of peptidases (subtilases) act as endopeptidases, being involved in hydrolysis of extracellular peptides and proteins. Phylogenetic analysis (Supplementary Figure S7) showed that Omega subtilases formed three distinct clusters, all of which are distant from biochemically characterized relatives. All four Omega S12 family proteins (CYPRO_0566, CYPRO_2116, CYPRO_2188, CYPRO_3057) have beta-lactamases and related proteins among the nearest characterized homologs, such as, for example, MlrB microcystine defensive system of Sphingomonas sp. (Bourne et al., 2001), suggesting their involvement in defense mechanisms, apparently beneficial for a satellite of cyanobacteria, that are known to synthesize various toxins. Yet, the mechanisms of this defense are vague, for example, the only currently known mechanism of aerobic degradation of cyanobacterial microcystine (Li et al., 2017) relies on four mlrA-D proteins, while Omega genome encodes only distant homologs of MlrB and no detectable homologs of other Mlr proteins. Furthermore, two peptidases of S51 family were found (CYPRO_0608 and CYPRO_2145) to be encoded in the Omega genome. The family includes cyanophycinases – exopeptidases, involved in cyanophycin (multi-L-arginyl-poly-L-aspartic acid, a cyanobacterial storage compound) degradation. Phylogenetic analysis (Supplementary Figure S8) showed that the Omega S51 peptidases fell into two distinct clusters. Both clusters have cyanobacterial representatives close to their nodes indicating independent HGT from different cyanobacteria to opportunistic bacteria, co-habiting with them or vice versa. Within the clusters, both Omega cyanophycinases have the nearest homologs from representatives of the FCB superphylum (Kublanov et al., 2017). Similarly to their close homologs from Salinivirga cyanobacteriivorans – a cyanobacteria-feeding Bacteroidetes member (Ben Hania et al., 2017), two Omega proteins were structurally different: CYPRO_2145 is being a monomeric protein, while CYPRO_0608 has an additional Por secretion system domain. However, the genomic context of CYPRO_0608 and CYPRO_2145 differs from their S. cyanobacteriivorans counterparts implying another mechanism of action.
A BLASTp of the bacteriocin sequences from BACTIBASE (Hammami et al., 2010) against the in silico translated Omega proteome resulted in 20 proteins, homologous to halocin, carocin, zoocin, colicin, linocin, helveticin, aerocin, dysgalacticin and pyocin. Manual verification revealed that at least half of them indeed are being bacteriocins and the most numerous (5 coding sequences) were zoocins – M23 peptidases, responsible for peptidoglycan cross-bridges degradation (Gargis et al., 2009). However, a search for secondary metabolite biosynthesis genes in the Omega genome using AntiSMASH software (Weber et al., 2015) did not reveal clusters for bacteriocin biosynthesis implicating putative novel biosynthetic pathways.
Overall, strain Omega represents a first example of an aerobic haloalkaliphilic protein-utilizing bacterium associated with a filamentous cyanobacterial bloom in hypersaline soda lakes. Its most prominent features include a peculiar cell morphology; the ability to utilize various proteins as carbon and energy source; a possession of an active light-dependent sodium-translocation primary ion pump representing a new lineage in the proteorodopsin family and deep-lineage phylogeny on the level of a new species-genus-family within the recently proposed phylum Balneolaeota. The genome analysis mostly confirmed the observed phenotypic properties, including proteolytic aerobic heterotrophic and haloalkaliphilic lifestyle. On the other hand, some of the functional genomic content (a presence of genes encoding polysaccharide-degrading GH family enzymes) was not validated by the in vivo tests. Furthermore, based on the overall results of phenotypic and phylogenomic analysis, strain Omega is suggested to form a novel candidate genus and species “Candidatus Cyclonatronum proteinivorum” in the phylum Balneolaeota.
DS isolated the culture and made its phenotypic characterization. MM tested cells for the activity of sodium rhodopsin. ST and AK sequenced and annotated the genome. IK, ST, and MM made phylogenetic analysis. All authors participated in writing the manuscript.
This work was supported by the Russian Science Foundation (RSF Grant 16-14-00121) to DS, IK, and ST. The work of MM was supported in parts by the RSF grant 14-50-00029 (genomic analysis) and by the RFBR grant 17-04-02173 (proteorhodopsin studies).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02672/full#supplementary-material
Amoozegar, M. A., Siroosi, M., Atashgahi, S., Smidt, H., and Ventosa, A. (2017). Systematics of haloarchaea and biotechnological potential of their hydrolytic enzymes. Microbiology 163, 623–645. doi: 10.1099/mic.0.000463
Antony, C. P., Kumaresan, D., Hunger, S., Drake, H. L., Murrell, J. C., and Shouche, Y. S. (2013). Microbiology of Lonar lake and other soda lakes. ISME J. 7, 468–476. doi: 10.1038/ismej.2012.137
Antony, C. P., Shimpi, G. G., Cockell, C. S., Patole, M. S., and Shouche, Y. S. (2014). Molecular characterization of prokaryotic communities associated with Lonar crater basalts. Geomicrobiol. J. 31, 519–528. doi: 10.1080/01490451.2013.849314
Banciu, H. L., and Muntyan, M. S. (2015). Adaptive strategies in the double-extremophilic prokaryotes inhabiting soda lakes. Curr. Opin. Microbiol. 25, 73–79. doi: 10.1016/j.mib.2015.05.003
Ben Hania, W., Joseph, M., Bunk, B., Spröer, C., Klenk, H. P., Fardeau, M. L., et al. (2017). Characterization of the first cultured representative of a Bacteroidetes clade specialized on the scavenging of cyanobacteria. Environ. Microbiol. 19, 1134–1148. doi: 10.1111/1462-2920.13639
Bezuidt, O., Lima-Mendez, G., and Reva, O. N. (2009). SeqWord Gene Island Sniffer: a program to study the lateral genetic exchange among Bacteria. World Acad. Sci. Eng. Technol. 58, 410–415.
Bourne, D. G., Riddles, P., Jones, G. J., Smith, W., and Blakeley, R. L. (2001). Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 16, 523–534. doi: 10.1002/tox.10013
Butler, J., MacCallum, I., Kleber, M., Shlyakhter, I. A., Belmonte, M. K., Lander, E. S., et al. (2008). ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820. doi: 10.1101/gr.7337908
Chen, I.-M. A., Markowitz, V. M., Chu, K., Palaniappan, K., Szeto, E., Pillay, M., et al. (2017). IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 45, D507–D516. doi: 10.1093/nar/gkw929
Derntl, C., Selb, R., Klein, R., Alte, B., and Witte, A. (2015). Genomic manipulations in alkaliphilic haloarchaea demonstrated by a gene disruption in Natrialba magadii. FEMS Microbiol. Lett. 362:fnv179. doi: 10.1093/femsle/fnv179
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. doi: 10.1093/nar/gkv401
Gargis, A. S., O’Rourke, A. L. D., Sloan, G. L., and Simmonds, R. S. (2009). Prevalence and acquisition of the genes for zoocin A and zoocin A resistance in Streptococcus equi subsp. zooepidemicus. J. Mol. Evol. 68, 498–505. doi: 10.1007/s00239-009-9221-x
Gostinčar, C., Lenassi, M., Gunde-Cimerman, N., and Plemenitaš, A. (2011). Fungal adaptation to extremely high salt concentrations. Adv. Appl. Microbiol. 77, 72–96. doi: 10.1016/B978-0-12-387044-5.00003-0
Gourbeyre, E., Siguier, P., and Chandler, M. (2010). Route 66: investigations into the organisation and distribution of the IS66 family of prokaryotic insertion sequences. Res. Microbiol. 161, 136–143. doi: 10.1016/j.resmic.2009.11.005
Hahnke, R. L., Meier-Kolthoff, J. P., García-López, M., Mukherjee, S., Huntemann, M., Ivanova, N. N., et al. (2016). Genome-based taxonomic classification of Bacteroidetes. Front. Microbiol. 7:2003. doi: 10.3389/fmicb.2016.02003
Hammami, R., Zouhir, A., Le Lay, C., Ben Hamida, J., and Fliss, I. (2010). BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 10:22. doi: 10.1186/1471-2180-10-22
Han, C. G., Shiga, Y., Tobe, T., Sasakawa, C., and Ohtsubo, E. (2001). Structural and functional characterization of IS679 and IS66-family elements. J. Bacteriol. 183, 4296–4304. doi: 10.1128/JB.183.14.4296-4304.2001
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. L. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Humayoun, S. B., Bano, N., and Hollibaugh, J. T. (2003). Depth distribution of microbial diversity in Mono lake, a meromictic soda lake in California. Appl. Environ. Microbiol. 69, 1030–1042. doi: 10.1128/AEM.69.2.1030-1042.2003
Inoue, K., Ito, S., Kato, Y., Nomura, Y., Shibata, M., Uchihashi, T., et al. (2016). A natural light-driven inward proton pump. Nat. Commun. 7:13415. doi: 10.1038/ncomms13415
Inoue, K., Ono, H., Abe-Yoshizumi, R., Yoshizawa, S., Ito, H., Kogure, K., et al. (2013). A light-driven sodium ion pump in marine bacteria. Nat. Commun. 4:1678. doi: 10.1038/ncomms2689
Kappelmann, L., Krüger, K., Hehemann, J.-H., Harder, J., Markert, S., Unfried, F., et al. (2018). Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 12. doi: 10.1038/s41396-018-0242-6
Kato, H. E., Inoue, K., Abe-Yoshizumi, R., Kato, Y., Ono, H., and Konno, M. (2015). Structural basis for Na + transport mechanism by a light-driven Na + pump. Nature 521, 48–53. doi: 10.1038/nature14322
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kevbrin, V., Boltyanskaya, Y., Zhilina, T., Kolganova, T., Lavrentjeva, E., and Kuznetsov, B. (2013). Proteinivorax tanatarense gen. nov., sp. nov., an anaerobic, haloalkaliphilic, proteolytic bacterium isolated from a decaying algal bloom, and proposal of Proteinivoraceae fam. nov. Extremophiles 17, 747–756. doi: 10.1007/s00792-013-0557-1
Kompantseva, E. I., Komova, A. V., Rusanov, I. I., Pimenov, N. V., and Sorokin, D. Y. (2009). Primary production of organic matter and phototrophic communities in the soda lakes of the Kulunda steppe (Altai, Russia). Microbiology 78, 643–649. doi: 10.1134/S002626170905018X
Krienitz, L., Dadheech, P. K., and Kotut, K. (2013). Mass developments of the cyanobacteria Anabaenopsis and Cyanospira (Nostocales) in the soda lakes of Kenya: ecological and systematic implications. Hydrobiologia 703, 79–93. doi: 10.1007/s10750-012-1346-z
Krienitz, L., and Schagerl, M. (2016). “Tiny and tough: microphytes of East African soda lakes,” in Soda lakes of East Africa, ed. M. Schagerl (Basel: Springer International Publishing), 149–177. doi: 10.1007/978-3-319-28622-8_6
Kublanov, I. V., Sigalova, O. M., Gavrilov, S. N., Lebedinsky, A. V., Rinke, C., Kovaleva, O., et al. (2017). Genomic analysis of Caldithrix abyssi, the thermophilic anaerobic bacterium of the novel bacterial phylum Calditrichaeota. Front. Microbiol. 8:195. doi: 10.3389/fmicb.2017.00195
Leggett, R. M., Clavijo, B. J., Clissold, L., Clark, M. D., and Caccamo, M. (2014). NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568. doi: 10.1093/bioinformatics/btt702
Li, J., Li, R., and Li, J. (2017). Current research scenario for microcystins biodegradation - A review on fundamental knowledge, application prospects and challenges. Sci. Total Environ. 595, 615–632. doi: 10.1016/j.scitotenv.2017.03.285
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Madsen, J. S., Burmølle, M., Hansen, L. H., and Sørensen, S. J. (2012). The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol. 65, 183–195. doi: 10.1111/j.1574-695X.2012.00960.x
Melack, J. M. (1981). Photosynthetic activity of phytoplankton in tropical African soda lakes. Hydrobiologia 81, 71–85. doi: 10.1007/BF00048707
Melack, J. M. (1988). Primary producer dynamics associated with evaporative concentration in a shallow, equatorial soda lake (Lake Elmenteita, Kenya). Hydrobiologia 158, 1–14. doi: 10.1007/BF00026264
Munoz, R., Rosselló-Móra, R., and Amann, R. (2016). Revised phylogeny of Bacteroidetes and proposal of sixteen new taxa and two new combinations including Rhodothermaeota phyl. nov. Syst. Appl. Microbiol. 39, 281–296. doi: 10.1016/j.syapm.2016.04.004
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Oren, A. (2013). “Life at high salt concentrations,” in The Prokaryotes—Prokaryotic Communities and Ecophysiology, 4th Edn, eds E. Rosenberg, E. F. DeLong, F. Thompson, S. Lory, and E. Stackebrandt (New York, NY: Springer).
Osman, O. A., Beier, S., Grabherr, M., and Bertilsson, S. (2017). Interactions of freshwater cyanobacteria with bacterial antagonists. Appl. Environ. Microbiol. 83:e02634-16. doi: 10.1128/AEM.02634-16
Pfennig, N., and Lippert, K. D. (1966). Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch. Mikrobiol. 55, 245–256. doi: 10.1007/BF00410246
Pinhassi, J., DeLong, E. F., Béjà, O., González, J. M., and Pedrós-Alióf, C. (2016). Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80, 929–954. doi: 10.1128/MMBR.00003-16
Rawlings, N. D., Waller, M., Barrett, A. J., and Bateman, A. (2014). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, D503–D509. doi: 10.1093/nar/gkt953
Roberts, M. F. (2005). Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Syst. 1:5. doi: 10.1186/1746-1448-1-5
Samylina, O. S., Sapozhnikov, F. V., Gainova, O. Y., Ryabova, A. V., Nikitin, M. A., and Sorokin, D. Y. (2014). Algo-bacterial phototrophic communities of soda lakes in Kulunda Steppe (Altai, Russia). Microbiology 83, 849–860. doi: 10.1134/S0026261714060162
Selim, S., Hagagy, N., Aziz, M. A., El-Meleigy, E. S., and Pessione, E. (2014). Thermostable alkaline halophilic-protease production by Natronolimnobius innermongolicus WN18. Nat. Prod. Res. 28, 1476–1479. doi: 10.1080/14786419.2014.907288
Shevchenko, V., Mager, T., Kovalev, K., Polovinkin, V., Alekseev, A., Juettner, J., et al. (2017). Inward H + pump xenorhodopsin: Mechanism and alternative optogenetic approach. Sci. Adv. 3:e1603187. doi: 10.1126/sciadv.1603187
Sorokin, D. Y. (2017). “Anaerobic haloalkaliphiles,” in Encyclopedia of Life Science, (Chichester: John Wiley & Sons, Ltd.), doi: 10.1002/9780470015902.a0027654
Sorokin, D. Y., Banciu, H. A., and Muyzer, G. (2015). Functional microbiology of soda lakes. Curr. Opin. Microbiol. 25, 88–96. doi: 10.1016/j.mib.2015.05.004
Sorokin, D. Y., Berben, T., Melton, E. D., Overmars, L., Vavourakis, C., and Muyzer, G. (2014). Microbial diversity and biogeochemical cycling in soda lakes. Extremophiles 18, 791–809. doi: 10.1007/s00792-014-0670-9
Sorokin, D. Y., Khijniak, T. V., Galinski, E. A., and Kublanov, I. V. (2017a). Natronotalea proteinilytica gen. nov., sp. nov, and Longimonas haloalkaliphilia sp. nov., extremely salt-tolerant alkaliphilic members of the phylum Rhodothermaeota isolated from hypersaline soda lakes. Int. J. Syst. Evol. Microbiol. 67, 4161–4167. doi: 10.1099/ijsem.0.002272
Sorokin, D. Y., Kublanov, I. V., and Khijniak, T. V. (2017b). Natronospira proteinivora gen. nov., sp. nov., an extremely salt tolerant alkaliphilic protein-utilizing gammaproteobacterium from hypersaline soda lakes. Int. J. Syst. Evol. Microbiol. 67, 2604–2608. doi: 10.1099/ijsem.0.001983
Studdert, C. A., Seitz, M. K. H., Gil, M. I. P., Sanchez, J. J., and de Castro, R. E. (2001). Purification and biochemical characterization of the haloalkaliphilic archaeon Natronococcus occultus extracellular serine protease. J. Basic Microbiol. 41, 375–383. doi: 10.1002/1521-4028(200112)41:6<375::AID-JOBM375>3.0.CO;2-0
Swartz, T. H., Ikewada, S., Ishikawa, O., Ito, M., and Krulwich, T. A. (2005). The Mrp system: A giant among monovalent cation/proton antiporters? Extremophiles 9, 345–354. doi: 10.1007/s00792-005-0451-6
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. doi: 10.1093/molbev/mst197
Toshchakov, S. V., Kublanov, I. V., Messina, E., Yakimov, M. M., and Golyshin, P. N. (2015). “Genomic analysis of pure cultures and communities,” in Hydrocarbon and Lipid Microbiology Protocols: Cultivation, eds T. J. McGenity, K. N. Timmis, and N. B. Fernández (Dordrecht: Springer), doi: 10.1007/8623_2015_126
Urios, L., Agogue, H., Lesongeur, F., Stackebrandt, E., Lebaron, P. (2006). Balneola vulgaris gen. nov., sp. nov., a member of the phylum Bacteroidetes from the north-western Mediterranean Sea. Int. J. Syst. Evol. Microbiol. 56, 1883–1887. doi: 10.1099/ijs.0.64285-0
Vargas, M. A., Rodríguez, H., Moreno, J., Olivares, H., Del Campo, J. A., and Rivas, J. (1998). Biochemical composition and fatty acid content of filamentous nitrogen-fixing cyanobacteria. J. Phycol. 34, 812–817. doi: 10.1046/j.1529-8817.1998.340812.x
Vavourakis, C. D., Ghai, R., Rodriguez-Valera, F., Sorokin, D. Y., Tringe, S. G., Hugenholtz, P., et al. (2016). Metagenomic insights into the microbial community structure and function of hypersaline soda lake brines. Front. Microbiol. 7:211. doi: 10.3389/fmicb.2016.00211
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., and Merkl, R. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 7:142. doi: 10.1186/1471-2105-7-142
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). AntiSMASH 3.0-A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Keywords: soda lakes, haloalkaliphilic, proteolytic, Balneolaeota, Na+-proteorhodopsin
Citation: Sorokin DY, Muntyan MS, Toshchakov SV, Korzhenkov A and Kublanov IV (2018) Phenotypic and Genomic Properties of a Novel Deep-Lineage Haloalkaliphilic Member of the Phylum Balneolaeota From Soda Lakes Possessing Na+-Translocating Proteorhodopsin. Front. Microbiol. 9:2672. doi: 10.3389/fmicb.2018.02672
Received: 19 July 2018; Accepted: 19 October 2018;
Published: 13 November 2018.
Edited by:
André Antunes, Edge Hill University, United KingdomReviewed by:
Mohammad Ali Amoozegar, University of Tehran, IranCopyright © 2018 Sorokin, Muntyan, Toshchakov, Korzhenkov and Kublanov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitry Y. Sorokin, c29yb2NAaW5taS5ydQ==; ZC5zb3Jva2luQHR1ZGVsZnQubmw=
†Present address: Aleksei Korzhenkov, National Research Centre, Kurchatov Institute, Moscow, Russia
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.