
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 31 October 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02596
 Cyril Barbezange1,2,3,4*†
Cyril Barbezange1,2,3,4*† Louis Jones2,3,4,5
Louis Jones2,3,4,5 Hervé Blanc1,3Ofer Isakov6Gershon Celniker6Vincent Enouf2,3,4
Hervé Blanc1,3Ofer Isakov6Gershon Celniker6Vincent Enouf2,3,4 Noam Shomron6Marco Vignuzzi1,3‡
Noam Shomron6Marco Vignuzzi1,3‡ Sylvie van der Werf2,3,4*†
Sylvie van der Werf2,3,4*†After a pandemic wave in 2009 following their introduction in the human population, the H1N1pdm09 viruses replaced the previously circulating, pre-pandemic H1N1 virus and, along with H3N2 viruses, are now responsible for the seasonal influenza type A epidemics. So far, the evolutionary potential of influenza viruses has been mainly documented by consensus sequencing data. However, like other RNA viruses, influenza A viruses exist as a population of diverse, albeit related, viruses, or quasispecies. Interest in this quasispecies nature has increased with the development of next generation sequencing (NGS) technologies that allow a more in-depth study of the genetic variability. NGS deep sequencing methodologies were applied to determine the whole genome genetic heterogeneity of the three categories of influenza A viruses that circulated in humans between 2007 and 2012 in France, directly from clinical respiratory specimens. Mutation frequencies and single nucleotide polymorphisms were used for comparisons to address the level of natural intrinsic heterogeneity of influenza A viruses. Clear differences in single nucleotide polymorphism profiles between seasons for a given subtype also revealed the constant genetic drift that human influenza A virus quasispecies undergo.
The development of deep sequencing technology, also known as next generation sequencing (NGS), offers a powerful tool to study the intrinsic heterogeneity of nucleic acids. For RNA viruses, it represents a major improvement in the study of their quasispecies nature over cloning and clone sequencing. The notion of quasispecies refers to the fact that RNA viruses exist as heterogeneous populations of closely related genetic variants (Domingo, 2006; Lauring and Andino, 2010), because their polymerase lacks fidelity and introduces point mutations during replication (Steinhauer et al., 1989a,b). Although the number of studies on RNA virus quasispecies evaluated by deep sequencing technology has dramatically increased in the past few years (Quinones-Mateu et al., 2014), there are still a limited number of such studies for influenza viruses.
Influenza A viruses belong to the Orthomyxoviridae viral family. One particularity of influenza A viruses is the segmented nature of their genome, which is composed of eight single-stranded RNA molecules of negative polarity. Each genomic segment encodes one major protein: the three polymerase sub-units PB2, PB1, and PA; the nucleoprotein (NP) associated with each of the RNA segments; the two envelope glycoproteins hemagglutinin (HA) and neuraminidase (NA); the matrix protein M1; and the multifunctional non-structural protein NS1. In addition, several segments encode one or more supplementary proteins through mechanisms of alternative splicing (PB2, MP/M, NS segments), alternative translation initiation (PB1, PA segments), or ribosomal frame-shift (PA segment), among which the ion channel protein M2 encoded by the M segment and the nuclear export protein NEP/NS2 encoded by the NS segment are best characterized (Wise et al., 2011; Jagger et al., 2012; Muramoto et al., 2013; Shaw and Palese, 2013; Yamayoshi et al., 2015). The two glycoproteins in the viral envelope, HA and NA, are used to define subtypes among influenza A viruses (Munster et al., 2007; Tong et al., 2013). Continuous genetic variation of influenza viruses due to the low fidelity of the viral polymerase may translate into antigenic variations through mutations in the HA and/or NA, a phenomenon also known as antigenic drift. In addition, introductions into the human population of new influenza A viruses, or antigenic shift, can occur through cross-species transmission from the animal reservoir (mainly poultry and pigs), and occasionally lead to devastating pandemics. After a pandemic wave, the newly introduced viruses gradually become seasonal (Tscherne and Garcia-Sastre, 2011). They are then responsible for yearly epidemics, mostly causing a relatively mild disease of the upper respiratory tract, that nonetheless represent a major burden for public health worldwide, with significant morbidity and mortality (Molinari et al., 2007; Ortega-Sanchez et al., 2012). Since 1977, two influenza A virus subtypes, H1N1 and H3N2, co-circulate in the human population. While undergoing genetic variation, H1N1 viruses with the H275Y mutation in NA that confers resistance to antiviral oseltamivir treatment emerged in late 2007 in the human population and circulated independently of the use of antiviral treatment to eventually become dominant over the sensitive H1N1 viruses by late 2008 (Meijer et al., 2009; Yang et al., 2011). These H1N1 viruses were fully replaced by the H1N1pdm09 viruses that originated from pigs and resulted in the 2009 pandemics (Neumann et al., 2009). The H1N1pdm09 viruses became established in the human population and currently co-circulate with H3N2 viruses.
Deep sequencing technology for influenza virus has mainly been used to determine the consensus sequence more rapidly than conventional methods and for a large number of samples simultaneously (Hoper et al., 2011; Ren et al., 2013; Rutvisuttinunt et al., 2013; Lin et al., 2014; Monne et al., 2014; Lee et al., 2016; Zhao et al., 2016), and to study the intrinsic heterogeneity of the virus genome on cell-adapted viruses (Watson et al., 2013; Thyagarajan and Bloom, 2014) or in clinical samples but focusing primarily on the HA and NA genes (Chen et al., 2010; Ghedin et al., 2011; Fordyce et al., 2013; Téllez-Sosa et al., 2013; Pizzorno et al., 2014; Dinis et al., 2016; Poon et al., 2016; Pichon et al., 2017). Deep sequencing was also applied to monitor the emergence of antiviral resistance in patients treated with oseltamivir (Ghedin et al., 2012; Rogers et al., 2015) or to understand transmission pathways and investigate local outbreaks (Ghedin et al., 2012; Poon et al., 2016; Meinel et al., 2018). Here, by evaluating the virus quasispecies diversity directly in infected human respiratory specimens, we demonstrated differences in the intrinsic genetic diversity between subtypes and showed that the composition of the quasispecies evolves season after season.
The samples (Supplementary Table 1) used in this study were collected during five consecutive seasons of influenza surveillance by the Northern France National Influenza Center (from 2007–2008 to 2011–2012). Respiratory specimens were selected from the collection of samples that tested positive for influenza A virus and for which the virus subtype was determined. For each season, more samples were selected from the dominant subtype that was circulating: pre-pandemic H1N1 (sH1N1) in 2007–2008, H3N2 (sH3N2) in 2008–2009 and 2011–2012, and pandemic H1N1pdm09 (pH1N1) in 2009–2010 and 2010–2011. For sH1N1 positive samples, selection was also based on the presence or absence of the H275Y mutation in NA, which confers resistance to oseltamivir. In addition, the selection included samples from mild cases collected through the GROG community surveillance network, and from severe cases defined as requiring external respiratory assistance and collected through the RENAL network of hospital laboratories. The median patient age was 22, 41, and 29 years old for sH1N1, H3N2 and pH1N1, respectively (95% CI of median: 5–38, 30–50, and 16–36 years old, respectively), with the patient age ranging from a minimum of 2, 0.8, and 3 years-old to a maximum of 81, 91, and 83 years old, respectively. The interval from onset of symptoms to sample collection was similar for the three subtypes, with a median interval of 1, 1.5, and 1 day for sH1N1, H3N2, and pH1N1, respectively (95% CI of median: 1–2 days for the three subtypes). The maximum sampling time from symptoms onset was 9, 5, and 6 days for sH1N1, H3N2, and pH1N1, respectively. PCRs were performed on cDNA synthesized from RNA directly extracted from the clinical specimen in order to avoid artifacts due to virus amplification in cell culture or in eggs. The strategy implemented to cover as much as possible of the coding regions was based on 12 specific PCRs, the four larger segments being amplified by two overlapping PCRs. Moreover, high-fidelity enzymes for both cDNA synthesis and PCR were used to reduce the incorporation of errors during viral genome amplification.
For each sample, reads obtained by Illumina sequencing were checked for quality (Supplementary Table 1) and cleaned by three successive steps using FastQC and fqcleaner Galaxy tools (Mareuil et al., 2017): first, reads were cleaned of Illumina adaptors and based on Phred scores (retained bases had a score above 30); then reads were cleaned of PCR primer sequences and non-PCR target contaminants; finally the remaining reads were cleaned of non-influenza virus contaminants, using the sequence of other viruses manipulated in the laboratory. For all samples, this last cleaning showed that very few contaminations occurred during sample preparation. For good runs, between 60 and 70% of the reads were retained after the three steps of cleaning. One run for pH1N1 was characterized by an excess of Illumina adaptor contaminants, but the remaining cleaning steps led to proportions of retained reads similar to what was observed for good runs, even if the overall retain proportion was of course lower. The reads obtained after the three steps of cleaning were deposited in the European Nucleotide Archive database for NGS sequences1 (Accession No. ERP012790). The consensus sequence was extracted for each segment of each sample and was deposited in the Global Initiative on Sharing All Influenza Data (GISAID) database (accession numbers in Supplementary Table 1).
To analyze the intrinsic genetic diversity, two pipelines were used to process the cleaned reads and identify positions with significant variants, using each sample’s own consensus sequence as reference. Both pipelines were developed by independent groups and use different algorithms to identify significant variants. The ViVAN pipeline is an all-inclusive pipeline (its own mapper is included) and was recently developed specifically for virus NGS data (Isakov et al., 2015). LoFreq was also specifically designed for virus NGS data and is already recognized within the scientific community (Wilm et al., 2012); it was used in conjunction with Bowtie mapper but we will refer to “LoFreq pipeline” for the whole process of mapping and extracting variant positions.
We first compared the “depth of sequencing” obtained by both pipelines. Mapping of the cleaned reads on the reference sequence allowed to extract the number of times each position was covered, i.e., the depth of sequencing. For both pipelines, it was found to be homogeneous along each PCR product. The mean depth of sequencing was calculated for each viral genomic segment sequence of each sample and ranged from around 200 to around 105 reads per position, with the majority of the mean depth values being above 103 reads per position. Both pipelines gave similar results for a given sample, except above 104 reads per position where the LoFreq pipeline was more powerful to map cleaned reads (Figure 1A and Supplementary Table 1). Consequently, the median of the mean depth values was higher with LoFreq (in reads per position: for sH1N1, median 11989, 95% CI of median 9894–14065, min 269, max 89267; for sH3N2, median 11567, 95% CI of median 10237–12293, min 273, max 94094; for pH1N1, median 3790, 95% CI of median 2816–4781, min 175, max 110570) than with ViVAN (in reads per position: for sH1N1, median 6228, 95% CI of median 5796–6471, min 268, max 7747; for sH3N2, median 6398, 95% CI of median 6034–6703, min 271, max 7633; for pH1N1, median 3698, 95% CI of median 2785–4343, min 173, max 7751).
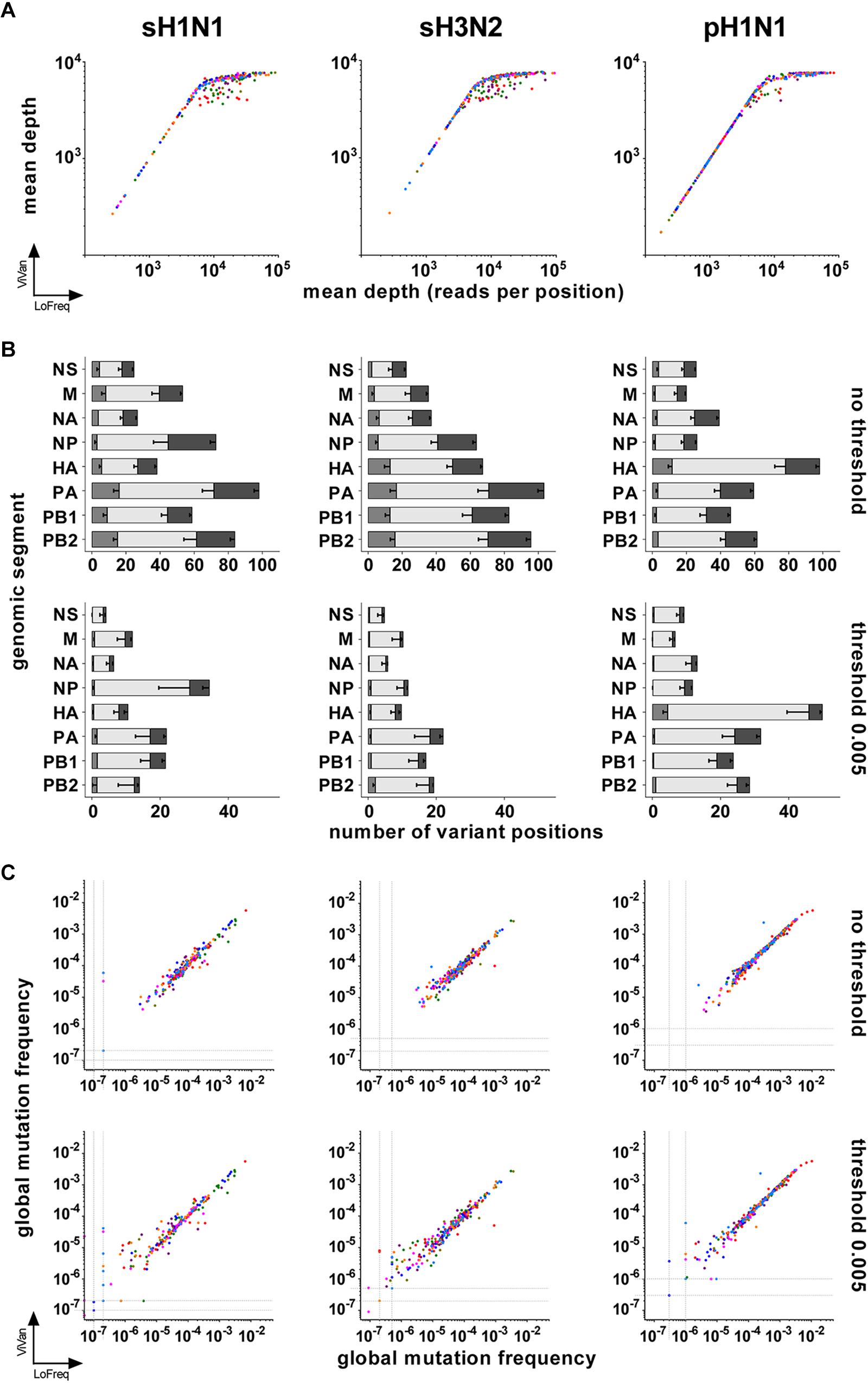
FIGURE 1. Comparison of LoFreq and ViVAN. (A) Correlation of the mean depths of sequencing (in number of reads per position) obtained by both pipelines. X-axis: LoFreq; Y-axis: ViVAN. Each dot represents one segment of one sample: polymerase sub-unit PB2: olive; polymerase sub-unit PB1: green; polymerase sub-unit PA: purple; hemagglutinin HA: red; nucleoprotein NP: dark blue; neuraminidase NA: pink; matrix and ion channel proteins M: orange; non-structural protein and nuclear export protein NS: light blue. (B) Mean number of variant positions identified by the pipelines, without or with setting a mutation frequency threshold. For each bar, from left to right: gray, specific to LoFreq; light gray, common to LoFreq and ViVAN; dark gray, specific to ViVAN. Error bars represent the Standard Error to the Mean. (C) Correlation of the global mutation frequencies obtained with both pipelines with or without setting a mutation frequency threshold. X-axis: LoFreq; Y-axis: ViVAN. Each dot represents one segment of one sample: PB2: olive; PB1: green; PA: purple; HA: red; NP: dark blue; NA: pink; M: orange; NS: light blue.
Both pipelines include tests that take into account the depth of sequencing, the complementarity of read orientation, the error rate of the Illumina method and the nature of the nucleotides in the sequence surrounding the position of interest, in order to identify positions with significant variants, even if those are present at a very low frequency (for a given position, number of times a base is different from the reference sequence – here the consensus sequence of the given sample – divided by depth of sequencing). It is commonly accepted that very low frequency variants might be unreliable. We compared the numbers of significant variants obtained by both pipelines without a threshold or after setting a mutation frequency threshold at 0.005 for retaining significant variants (meaning that only positions with a significant variant representing more than 0.5% of the reads would be counted). This threshold represents five times the error rate recognized for the Illumina method using standard, non-high-fidelity enzymes to prepare PCR products from RNA (Nakamura et al., 2011). While an important number of variant positions were specific of each pipeline when no threshold was set, Figure 1B shows that a 0.005 frequency threshold reduced the number of pipeline-specific variant positions (Supplementary Tables 1, 2). This was particularly true for the LoFreq pipeline, for which only a few pipeline-specific variants remained for the three subtypes.
We further estimated the viral intrinsic genetic diversity by calculating, for each variant caller pipeline, the “global mutation frequency” for each segment of each sample, using the data generated by the ViVAN and LoFreq pipelines when no threshold or a 0.005 threshold was used (Supplementary Table 1). Basically, the number of variant bases corresponding to all identified or retained variant positions was divided by the total number of sequenced bases (i.e., the sum of the depth of sequencing) covering a given item (segment-sample). When there was no variant identified in a segment (especially when the 0.005 threshold was applied), we allocated an arbitrary value for global mutation frequency slightly below the lowest calculated value among the samples of the same group (one genomic segment of one subtype), in order to avoid null global mutation frequencies. A good correlation between the two pipelines was found when the global mutation frequencies were compared (Figure 1C and Table 1). No correlation was found between possible biases (mean depth, age of patient, virus load) and the global mutation frequency (Supplementary Figure 1 and Supplementary Table 3).
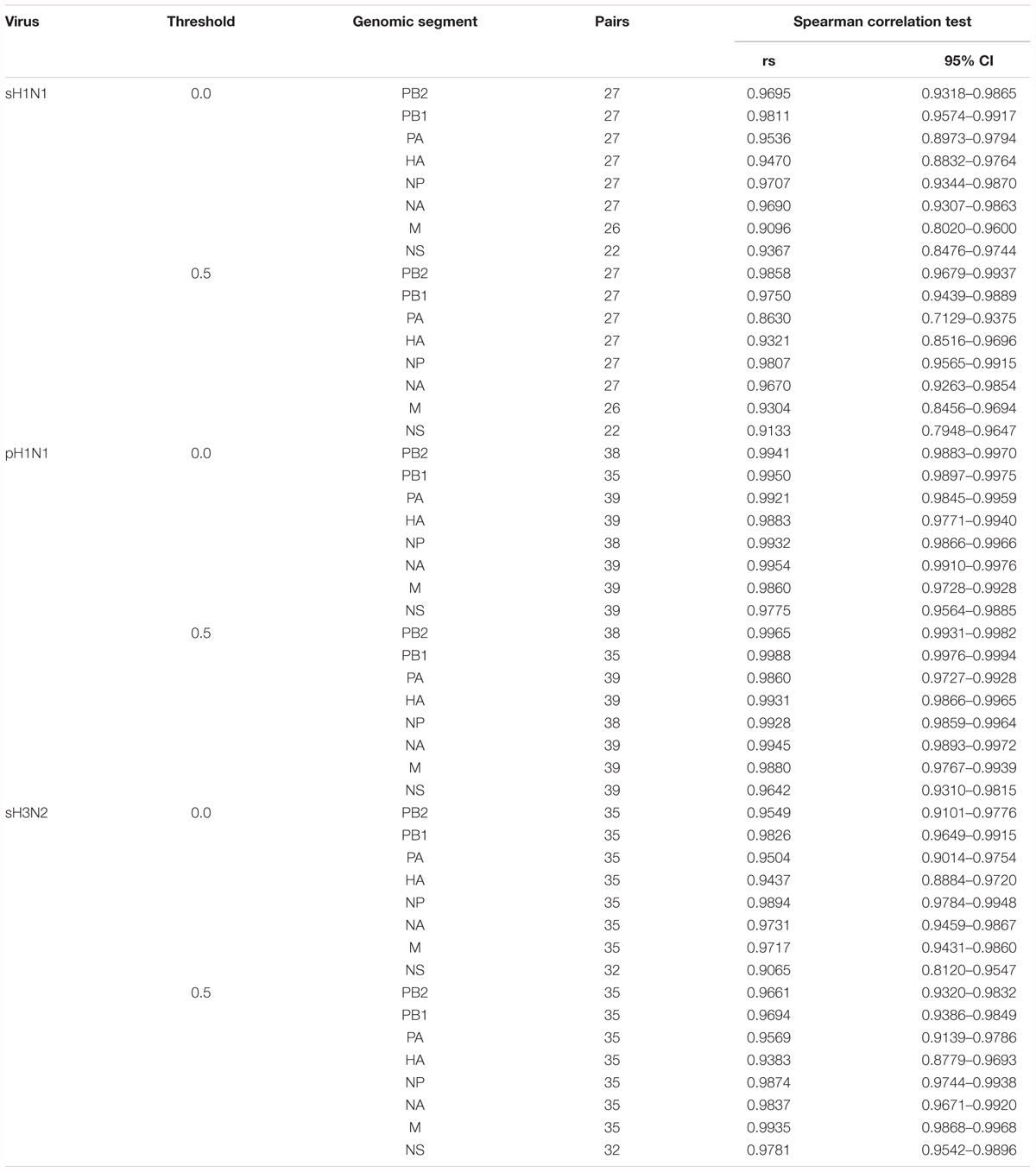
TABLE 1. Correlation between global mutation frequencies obtained with LoFreq and ViVAN, evaluated by Spearman non-parametric test.
Global mutation frequency data generated with the LoFreq pipeline at a 0.005 threshold were used to analyze the differences between different groups. Mann–Whitney–Wilcoxon non-parametric test was used for all pairwise comparisons. Three subtypes were considered: sH1N1, sH3N2, and pH1N1. Within sH1N1, subgroups were defined, based on the mutation conferring resistance to oseltamivir (H275Y in the neuraminidase) and on the season. Within sH3N2 and pH1N1, subgroups were defined, based on the severity status (mild and severe cases) and on the season. The few samples of the non-dominant subtype during a season were not included and only the samples belonging to the specified groups within a season were used for comparison. The global mutation frequencies calculated for each segment of each sample of each group are shown in Figure 2.
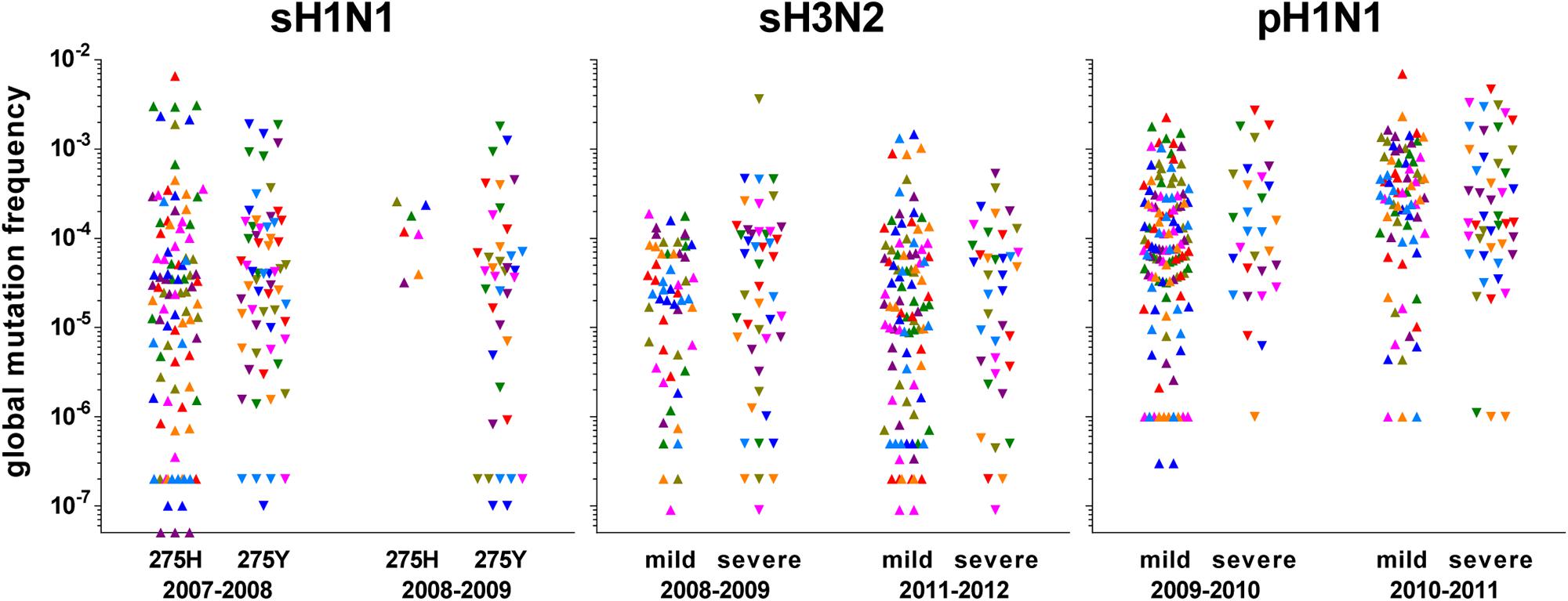
FIGURE 2. Representation of the global mutation frequencies according to season and group of interest. Values were calculated from the data obtained with LoFreq at a 0.005 threshold. Each symbol represents one segment of one sample: polymerase sub-unit PB2: olive; polymerase sub-unit PB1: green; polymerase sub-unit PA: purple; hemagglutinin HA: red; nucleoprotein NP: dark blue; neuraminidase NA: pink; matrix and ion channel proteins M: orange; non-structural protein and nuclear export protein NS: light blue. For pre-pandemic sH1N1 viruses, groups were based on the oseltamivir-resistance mutation H275Y (275H: sensitive; 275Y: resistant) in the neuraminidase; for both H3N2 and pandemic pH1N1 viruses, groups were based on the severity according to the network of sampling (mild through community physicians, severe through hospitals).
Whatever the genomic segment, no significant difference in the global mutation frequency distribution between any subgroups was observed for sH1N1 samples; they were pooled to form a unique group “sH1N1.” The median global mutation frequency for sH1N1 ranged from 6.8 mutations per 106 bases for the NS segment to 1.4 mutations per 104 bases for the PB1 segment (Table 2). Similarly for sH3N2, whatever the genomic segment, no significant difference was observed in the global mutation frequency distribution between the mild and severe cases or between the two studied seasons; all samples were pooled to form the “sH3N2” group. The median global mutation frequency for sH3N2 ranged from one mutation per 105 bases for the NA segment to 3.2 mutations per 105 bases for the NP segment (Table 2). For pH1N1, whatever the genomic segment, no significant difference was observed between mild and severe cases in each studied season. When comparing seasons, a significant difference was observed in the global mutation frequency distribution for genomic segment PA encoding the PA polymerase sub-unit (Mann–Whitney–Wilcoxon U = 61; N2009-2010 = 20; N2010-2011 = 16; actual difference = 2.6 × 10-4; 95% CI of difference = 6.4 × 10-5 to 8.9 × 10-4) and for segment MP encoding the M1 matrix and M2 ion channel proteins (Mann–Whitney–Wilcoxon U = 95; N2009-2010 = 20; N2010-2011 = 16; actual difference = 1.9 × 10-4; 95% CI of difference = 0 to 3.9 × 10-4). The samples were pooled to form the “pH1N1” group, except for PA and MP, for which each season was kept separated for the subtype comparison. The pH1N1 median global mutation frequency ranged from 6.1 mutations per 105 bases for the MP segment of season 2009–2010 to 4.2 mutations per 104 bases for the PB2 segment encoding the PB2 polymerase sub-unit (Table 2).
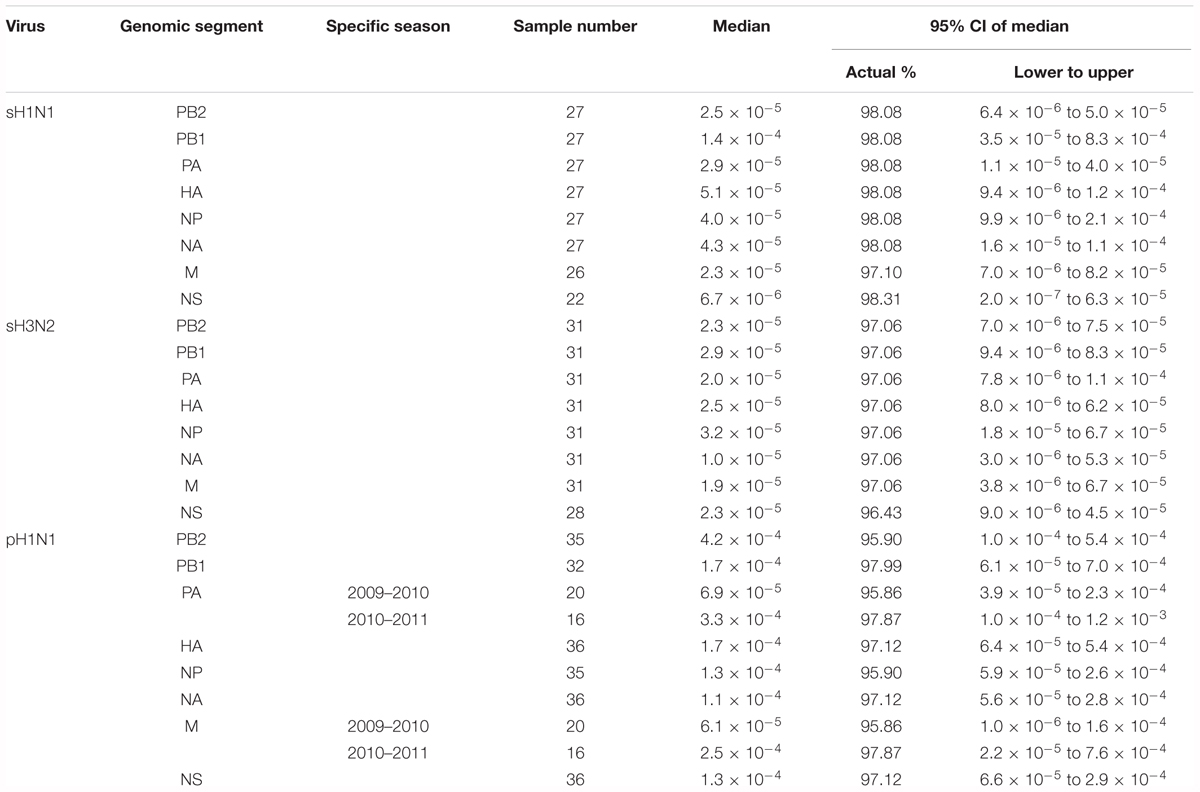
TABLE 2. Median global mutation frequency per segment for each virus.
No significant difference in the global mutation frequency distribution was observed between sH1N1 and sH3N2, except for the PB1 polymerase sub-unit genomic segment. On the contrary, pH1N1 was found significantly different from both sH1N1 and sH3N2 for most genomic segments (Figure 3 and Supplementary Table 4). The pH1N1 subtype clearly differed from both sH1N1 and sH3N2 subtypes, and was found to be more heterogeneous with higher global mutation frequencies in most genomic segments.
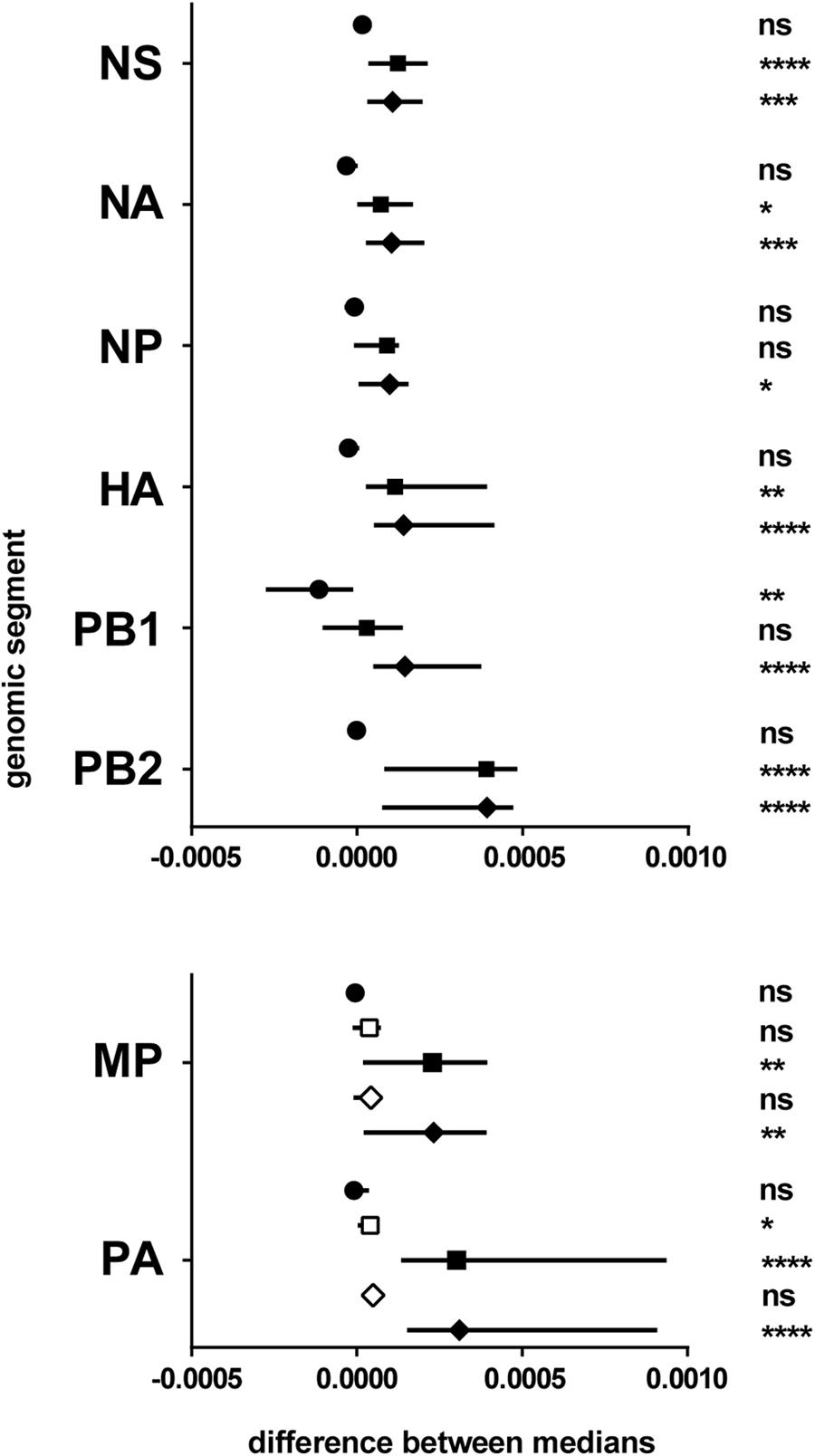
FIGURE 3. Differences between subtypes. Results of the pairwise comparisons by Mann–Whitney–Wilcoxon test of the global mutation frequency distributions (Supplementary Table 4). The genomic segments considered are mentioned on the left. X-axis: difference between the medians (symbol) and 95% CI of the difference (line). Symbols: circle, sH1N1 vs. sH3N2; square, sH1N1 vs. pH1N1 (for MP and PA, open square for pH1N1 season 2009–2010, plain square for pH1N1 season 2010–2011); diamond, sH3N2 vs. pH1N1 (for MP and PA, open square for pH1N1 season 2009–2010, plain square for pH1N1 season 2010–2011). ns, non-significant; ∗0.05 ≥p-value > 0.01; ∗∗0.01 ≥p-value > 0.001; ∗∗∗0.001 ≥p-value > 0.0001; ∗∗∗∗p-value ≤ 0.0001.
To further analyze the quasispecies structure for the different viruses, we examined the distribution of the positions with a significant variant along each of the genomic segments. A viral single nucleotide polymorphism (vSNP) was defined as a significant variant position that was shared by more than 15% of the samples within a subtype, within a given season. Differences in the vSNP patterns were observed between the two studied seasons for both sH3N2 and pH1N1 subtypes (Figure 4). Subtype sH3N2 was characterized by very few vSNPs in the HA and NA segments, and vSNPs in the other segments that corresponded to variants that were mainly shared by less than 40% of the samples (with the exceptions of position 1731 in the PB1, 1849 in the PA, and 761 in the NS segments). Subtype pH1N1 was characterized by many vSNPs in the HA and NA segments. During season 2009–2010, most vSNPs were variants that were shared by less than 50% of the samples (Figure 4B, green). During season 2010–2011, several vSNPs in PB2, PA, HA, NP, NA, M, and NS segments were variants shared by 50–80% of the samples, whereas most vSNPs in the PB1 segment were shared by less than 50% of the samples (Figure 4B, orange).
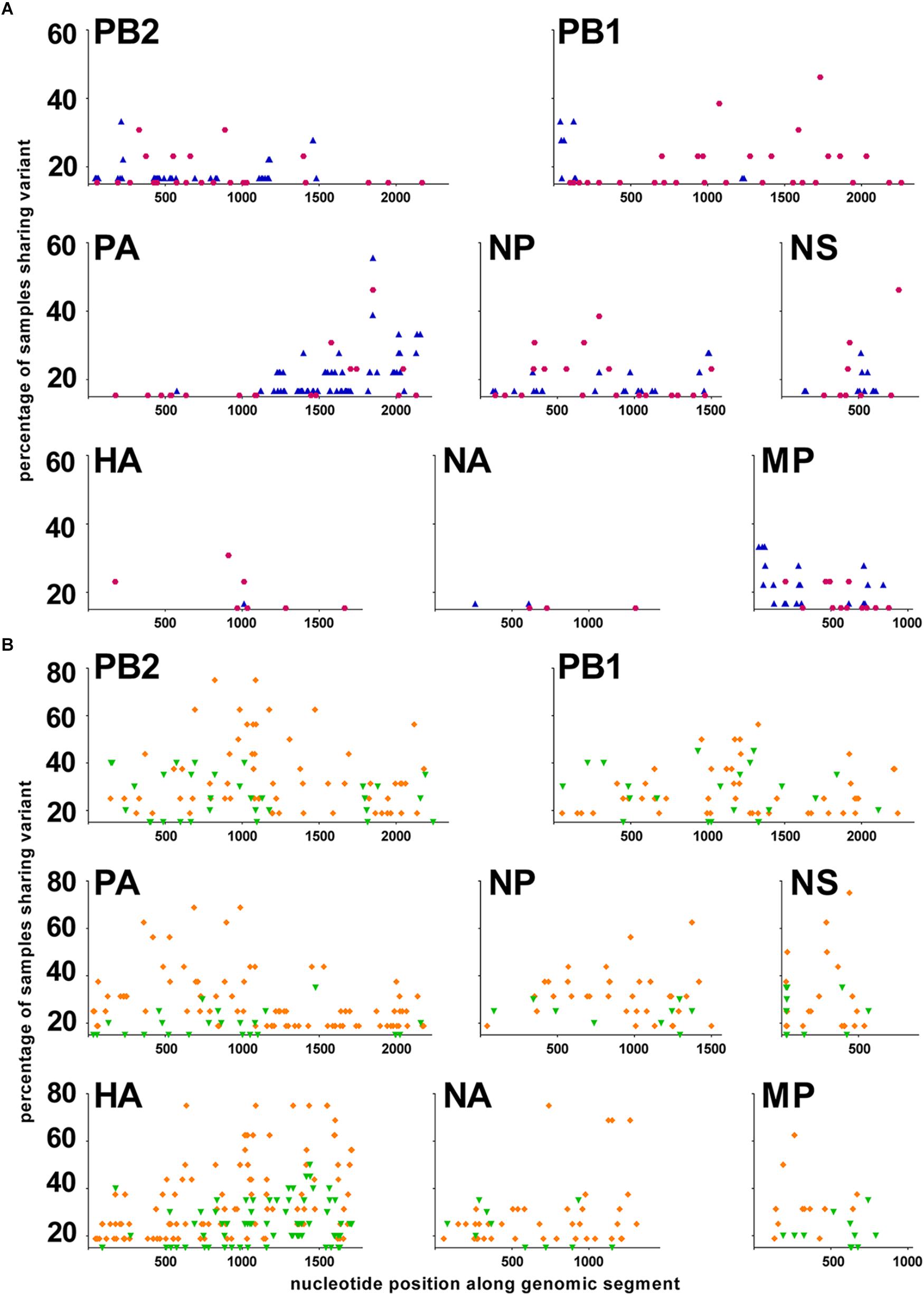
FIGURE 4. Distribution of vSNPs along the genome. A viral Single Nucleotide Polymorphism (vSNP) is a position with a significant variant that has been identified in at least 15% of the samples in a group. X-axis: nucleotide position along each genomic segment; Y-axis: percentage of samples sharing a significant variant. (A) sH3N2 samples. In purple: season 2008–2009; in blue: season 2011–2012. (B) pH1N1 samples. In green: season 2009–2010; in orange: season 2010–2011.
The intrinsic heterogeneity of nearly the entire viral genome of influenza A viruses was evaluated directly in respiratory specimens collected from a large number of infected patients over five consecutive seasons in a defined geographic area (Northern half of France). Depth of sequencing was not only high, with a coverage largely above 1000 for most PCR products, but it was also found to be extremely homogeneous along the PCR products, which represented an improvement compared to some published data (Kuroda et al., 2010; Kampmann et al., 2011; Lin et al., 2014; Welkers et al., 2015). Differences in the library generation protocols probably explain those results and highlight the fact that many parameters are still not totally controlled when deep sequencing technology is applied (Beerenwinkel et al., 2012). The use of high fidelity enzymes for both the reverse transcription and the PCR steps allowed to limit the introduction of errors during the viral genome amplification, with an estimated overall error reduction by 150 times compared to classical enzymes according to the manufacturers’ data. The introduction of errors during the preparation of libraries for deep sequencing runs is a well-known limitation of NGS that needs to be taken into account (Willerth et al., 2010; Acevedo and Andino, 2014). An 0.001 mutation frequency has been used by others as a threshold to define viral subpopulations based on Illumina technology results (Nakamura et al., 2011). We decided to use, for specific comparisons, only positions with a mutation frequency above 0.005 and that have been identified by two independent pipelines that were specifically designed for the identification of virus quasispecies, making our results extremely reliable and comparable to previous studies (Welkers et al., 2015). However, the physiological relevance of low mutation frequencies is still unknown and it is thus difficult to determine the impact of any subpopulations described by deep sequencing.
The global mutation frequency that we calculated for the influenza virus samples appeared slightly lower than the published data on RNA virus polymerase error-rates (Parvin et al., 1986; Drake and Holland, 1999; Sanjuan et al., 2010), suggesting that some bottleneck events might occur between replication of the genome in the cells of the respiratory tract and excretion of the virus as collected in respiratory specimens (Xue et al., 2018). The fact that no bias was observed demonstrated that the level of heterogeneity detected by deep sequencing was not correlated to the virus load in the sample or to the age of the patient. This is an important point, as one could have hypothesized that the virus variability could be higher in infected children or that a higher mutation frequency could artificially be the consequence of more virus genomes being present in the sample. Deep sequencing approaches based on clinical samples instead of cell culture-amplified virus to evaluate the viral quasispecies have long focused on HIV and hepatitis viruses (Quinones-Mateu et al., 2014). Most studies concerned the appearance of resistance and the evolution of specific related mutations under antiviral treatments (Simen et al., 2009; Nasu et al., 2011; Fisher et al., 2012; Nishijima et al., 2012; Svarovskaia et al., 2012; Li et al., 2013; Rodriguez et al., 2013). A few studies were in experimental in vivo conditions, using animal models (Moncla et al., 2013; Sutton et al., 2014), and some focused on the intrinsic heterogeneity of the virus as it is excreted, following, for example, HIV1/HCV quasispecies in the semen of naturally (co-)infected males (Paranjpe et al., 2002; Briat et al., 2005). The importance of studying the quasispecies composition of RNA viruses was recently highlighted when deep sequencing analysis allowed to follow the virus evolutionary trajectories upon the emergence of a Chikungunya virus variant responsible for an epidemic in the Indian Ocean in 2006 (Stapleford et al., 2014).
The description of vSNPs common to several samples within a subtype has been previously reported for influenza viruses mainly at positions involved in resistance to antivirals (Téllez-Sosa et al., 2013; Pizzorno et al., 2014). In the present study, vSNPs in the whole genome could be identified because a relatively large number of samples were analyzed for each subtype and for two seasons for sH3N2 and pH1N1. The physiological importance of the vSNPs we found in the different genomic segments is totally unknown and will require further investigation with the help of reverse genetics systems. Most of the positions with vSNPs concerned the third nucleotide of codons, and the variant mutations were consequently mainly synonymous (Supplementary Table 5), meaning that the phenotype of the variant subpopulations was probably not different from that of the main subpopulation. If intrinsic genetic variability does not confer variability in phenotypes, i.e., potentially immediate fitness advantages (Debbink et al., 2017; McCrone et al., 2017), it must have an importance in terms of evolution and adaptability. A heterogeneous population, even with variants at very low frequency, could facilitate or speed up evolution. Comparison of two seasons for a given subtype strikingly highlighted the evolution dynamics and quasispecies plasticity of influenza A viruses. Whereas no difference was observed in the global mutation frequency distribution between the two seasons for both sH3N2 and pH1N1, the vSNPs dispersal along the genome clearly showed differences between the two seasons.
Interestingly, contrary to sH3N2 for which only a few positions were vSNPs in the HA and NA gene sequences, pH1N1 was characterized by the presence of many vSNPs in the glycoprotein gene sequences. Together with the higher level of global heterogeneity compared to sH1N1 and sH3N2, and despite the relative stability at the consensus sequence level since its introduction in human in 2009 (Klein et al., 2014), it demonstrated that pH1N1, a virus recently introduced in the human population, is still adapting to the human host, exploring different areas of the sequence space. This plasticity of the quasispecies was nonetheless also observed for sH3N2 viruses, which have been circulating in the human population since the late 1960s, since vSNP differences were identified between seasons. This phenomenon was nonetheless less pronounced than for pH1N1, but it illustrated the permanent, underlying genetic drift occurring in human influenza viruses.
Two intriguing observations will require further investigations. First, for both pH1N1 and sH3N2, non-synonymous vSNPs were identified in viral components involved in intracellular steps of the virus replication, such as the polymerase sub-units, the nucleoprotein, the matrix M1 protein, and the non-structural NS1 protein. We hypothesize that these vSNPs might provide some flexibility in different processes, either directly by modulating the activity of a given protein, or indirectly by affecting the interaction with cellular factors. The second point concerns the main determinants of influenza virus antigenic evolution: the envelope glycoproteins HA and NA. It is unclear why so few vSNPs were identified in the HA and NA for sH3N2. We wonder whether a similar situation would be observed for sH3N2 samples collected during a season when sH3N2 does not dominate and whether there could be a relationship between the level of intrinsic heterogeneity and the alternate dominance observed between subtypes. We also noticed that several variants that were identified as minority mutations in the samples used in this study were later detected as dominant in other parts of the world and used to define some genetic clades/groups. Thus, for pH1N1, mutations A134T/S183P in the HA and Q313R/V394I in the NA, that we identified as minority mutations during the 2010–2011 season, were used the following season by WHO Collaborating Centers (Barr et al., 2014) to define group 3 sequences (represented by A/Hong-Kong/3934/2011). Similarly, mutations S143G/A197T in the HA and N44S in the NA were used to define group 7 sequences (represented by A/St-Petersburg/100/2011).
Studying the intrinsic heterogeneity of influenza A viruses by deep sequencing directly from respiratory specimens clearly gave interesting insights into the virus evolution dynamics by adding a new dimension to analyses performed on consensus sequences. It would now be interesting to process samples from more epidemic seasons to better describe the phenomenon of evolutionary plasticity. Studies by other groups have not been able to address this question due to the limited number of seasons (Dinis et al., 2016; Poon et al., 2016) or samples (McCrone et al., 2017) that were under consideration or available. Thus, analyzing future seasons of pH1N1 dominance would show if the virus would maintain its high level of heterogeneity and continue to explore the sequence space, or if some equilibrium might soon be reached with a level of heterogeneity similar to that observed for sH3N2. Given the large dispersion observed in the global mutation frequency distribution within a group, it nonetheless seems necessary to process a large set of samples in order to identify statistically significant differences; this will be more achievable as the NGS technologies evolve and the cost decreases.
Samples were selected among influenza A virus-positive respiratory specimens available in the collection of the National Influenza Centre (NIC) for Northern France. For yearly epidemiological surveillance of influenza viruses circulating during seasonal epidemics, nasopharyngeal swabs were regularly collected by physicians of the GROG network from patients with ARI (acute respiratory illness) and sent to the NIC for virus detection and characterization. As part of its routine surveillance activities, the NIC also received respiratory specimens (nasopharyngeal swabs or nasopharyngeal aspirates) from the hospital laboratories belonging to the RENAL network, which mainly sent samples from hospitalized patients. In this study, for H3N2 (sH3N2) and pandemic 2009 H1N1 (pH1N1), mild cases were defined as patients from the community sampled by the GROG network, while severe cases were patients with severe acute respiratory distress requiring external respiratory assistance and were sampled by the RENAL network. For pre-pandemic H1N1 (sH1N1), sensitivity and resistance to Oseltamivir were based on the amino acid nature at position 275 in the neuraminidase NA protein (H and Y, respectively). All specimens were declared to the Ministère de l’Enseignement Supérieur et de la Recherche (French Research and Higher Education Ministry) as a collection of samples that may be used for research activities including viral quasispecies genetic characterization (Number DC-2010-1197, Collection Number 4). Access to personal data was limited to the patient age.
Viral RNA was extracted from 140 μL of respiratory specimens with Qiagen QIAamp Viral RNA kit (Cat# 52904). Ten microliters of purified RNA were then reverse transcribed with Agilent AccuScript High Fidelity 1st strand cDNA Synthesis kit (Cat# 200820) using Uni1 (5′-AGCRAAAGCAGG-3′) primer. The viral genome was then amplified by PCR using Thermo Scientific Phusion High-Fidelity DNA Polymerase (Cat# F-530). All reactions were performed according to the manufacturers’ instructions. Twelve PCRs were designed to cover as much as possible of the coding regions of the eight genomic segments (primer sequences and specific annealing temperature and elongation time conditions are available in Supplementary Table 6).
After purification with Macherey-Nagel Nucleospin Gel and PCR Clean-up kit (Cat# 740609), the PCR products were quantified with Invitrogen Quant-iT Picogreen dsDNA Assay Kit (Cat# P7589). For each sample, two pools were prepared with the 12 PCRs covering the genome, fragmented (New England BioLabs NEBNext dsDNA Fragmentase, Cat# M0348), multiplexed, sequenced with Illumina cBot and GAIIX technology (Illumina TruSeq SR Cluster Kit v2-cBot-GA, Cat# GD-300-2001; Illumina TruSeq SBS Kit v5-GA, Cat# FC-105-5001), and analyzed with established deep sequencing data analysis tools and in-house scripts. Briefly, clipping was performed using the Galaxy tools (Mareuil et al., 2017), removing common adapter and other contaminants and trimming low quality bases (Phred < 30). Clipped reads were aligned to sequences of sH1N1 A/New Caledonia/20/1999 (GISAID Accession Nos. EPI_ISL_649) or sH3N2 A/Brisbane/10/2007 (GISAID Accession Nos. EPI_ISL_25019) or pH1N1 A/California/7/2009 (GISAID Accession Nos. EPI_ISL_159427) as reference, to extract the consensus viral sequence for each sample. For the ViVAN pipeline (Isakov et al., 2015), clipped reads of a given sample were aligned to the consensus sequence of that sample using Burrows Wheeler Aligner BWA v0.5.9 (Li and Durbin, 2009), then alignments were processed using SAMTools (Li et al., 2009) to obtain a pileup of the called bases at each position and statistically significant variants were identified above the background noise due to sequencing error, in every sufficiently covered site (>100×). In parallel, the clipped reads were aligned to the consensus sequence using Bowtie Aligner (Langmead et al., 2009) and significant variants were identified with LoFreq (Wilm et al., 2012).
Correlations were evaluated using Spearman non-parametric tests (Kornbrot, 2014). Differences in global mutation frequency distributions between two groups (pairwise comparisons) were compared by the two-tailed Mann–Whitney–Wilcoxon test, a non-parametric test based on rank-sums (Moses, 2014).
The cleaned reads (as FASTQ files) were deposited in the European Nucleotide Archive database for NGS sequences https://www.ebi.ac.uk/ena; accession number ERP012790). In addition, the original reads (prior to cleaning) will be made available by the authors, without undue reservation, to any qualified researcher.
CB, SW, and MV conceptualized the study. CB and HB performed the template preparations and NGS sequencing. LJ, OI, GC, and NS designed the softwares and performed the bio-informatics analyses with the Variant Callers. VE gathered the resources. CB contributed to formal analysis and visualization of the data, and wrote the original draft of the manuscript. CB, SW, and MV wrote, reviewed, and edited the manuscript. SW, MV, and NS acquired the funding.
The research leading to these results has received funding from the Israeli Ministry of Science, Technology and Space, and the French Ministries of Foreign Affairs and of National Education, Higher Education and Research, through the Israeli-French High Council for Scientific and Technological Research (program “Complexity in Biology: molecular aspects of infectious diseases”) and from the European Union Seventh Framework Programme [FP7/2007–2013] (Grant Agreement n°278433-PREDEMICS: Preparedness, Prediction, and Prevention of Emerging Zoonotic Viruses with Pandemic Potential using Multidisciplinary Approaches).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the National Influenza Centre for Northern France, particularly Sylvie Behillil for extracting a selection list of positive samples, and Vanessa Roca, Mathilde Benassaya, Marie Lazzerini, and Sébastien Le Gal for retrieving the samples from the collection. We would also like to thank Professor Nir Ben-Tal from Georges S. Wise Faculty of Life Sciences, Tel Aviv University, Israel, for helpful discussion and critical reading. The results of initial analyses of the data were presented as posters in 2013 at the 8th “Options for the Control of Influenza” meeting organized by the International Society for Influenza and Other Respiratory Viruses (ISIRV) in Cape Town, South Africa, and at the 5th European Congress of Virology organized by the European Society for Virology (ESV) in Lyon, France, and in 2014 at the 5th Influenza Conference organized by the European Scientific Working group on Influenza (ESWI) in Riga, Latvia, and at the 4th International Influenza Meeting organized by FluResearchNet in Muenster, Germany.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02596/full#supplementary-material
Acevedo, A., and Andino, R. (2014). Library preparation for highly accurate population sequencing of RNA viruses. Nat. Protoc. 9, 1760–1769. doi: 10.1038/nprot.2014.118
Barr, I. G., Russell, C., Besselaar, T. G., Cox, N. J., Daniels, R. S., Donis, R., et al. (2014). WHO recommendations for the viruses used in the 2013-2014 Northern Hemisphere influenza vaccine: epidemiology, antigenic and genetic characteristics of influenza A(H1N1)pdm09, A(H3N2) and B influenza viruses collected from October 2012 to January 2013. Vaccine 32, 4713–4725. doi: 10.1016/j.vaccine.2014.02.014
Beerenwinkel, N., Gunthard, H. F., Roth, V., and Metzner, K. J. (2012). Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 3:329. doi: 10.3389/fmicb.2012.00329
Briat, A., Dulioust, E., Galimand, J., Fontaine, H., Chaix, M. L., Letur-Konirsch, H., et al. (2005). Hepatitis C virus in the semen of men coinfected with HIV-1: prevalence and origin. AIDS 19, 1827–1835. doi: 10.1097/01.aids.0000189847.98569.2d
Chen, H., Wen, X., To, K. K., Wang, P., Tse, H., Chan, J. F., et al. (2010). Quasispecies of the D225G substitution in the hemagglutinin of pandemic influenza A(H1N1) 2009 virus from patients with severe disease in Hong Kong, China. J. Infect. Dis. 201, 1517–1521. doi: 10.1086/652661
Debbink, K., McCrone, J. T., Petrie, J. G., Truscon, R., Johnson, E., Mantlo, E. K., et al. (2017). Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog. 13:e1006194. doi: 10.1371/journal.ppat.1006194
Dinis, J. M., Florek, N. W., Fatola, O. O., Moncla, L. H., Mutschler, J. P., Charlier, O. K., et al. (2016). Deep sequencing reveals potential antigenic variants at low frequencies in influenza A virus-infected humans. J. Virol. 90, 3355–3365. doi: 10.1128/JVI.03248-15
Domingo, E. (2006). Quasispecies: concept and implication for virology. Curr. Top. Microbiol. Immunol. 299. doi: 10.1007/b137531
Drake, J. W., and Holland, J. J. (1999). Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 96, 13910–13913. doi: 10.1073/Pnas.96.24.13910
Fisher, R., van Zyl, G. U., Travers, S. A., Kosakovsky Pond, S. L., Engelbrech, S., Murrell, B., et al. (2012). Deep sequencing reveals minor protease resistance mutations in patients failing a protease inhibitor regimen. J. Virol. 86, 6231–6237. doi: 10.1128/jvi.06541-11
Fordyce, S. L., Bragstad, K., Pedersen, S. S., Jensen, T. G., Gahrn-Hansen, B., Daniels, R., et al. (2013). Genetic diversity among pandemic 2009 influenza viruses isolated from a transmission chain. Virol. J. 10:116. doi: 10.1186/1743-422x-10-116
Ghedin, E., Holmes, E. C., DePasse, J. V., Pinilla, L. T., Fitch, A., Hamelin, M.-E., et al. (2012). Presence of oseltamivir-resistant pandemic A/H1N1 minor variants before drug therapy with subsequent selection and transmission. J. Infect. Dis. 206, 1504–1511. doi: 10.1093/infdis/jis571
Ghedin, E., Laplante, J., DePasse, J., Wentworth, D. E., Santos, R. P., Lepow, M. L., et al. (2011). Deep sequencing reveals mixed infection with 2009 pandemic Influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J. Infect. Dis. 203, 168–174. doi: 10.1093/infdis/jiq040
Hoper, D., Hoffmann, B., and Beer, M. (2011). A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS One 6:e19075. doi: 10.1371/journal.pone.0019075
Isakov, O., Borderia, A. V., Golan, D., Hamenahem, A., Celniker, G., Yoffe, L., et al. (2015). Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics 31, 2141–2150. doi: 10.1093/bioinformatics/btv101
Jagger, B. W., Wise, H. M., Kash, J. C., Walters, K. A., Wills, N. M., Xiao, Y. L., et al. (2012). An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science (80-) 337, 199–204. doi: 10.1126/science.1222213
Kampmann, M. L., Fordyce, S. L., Avila-Arcos, M. C., Rasmussen, M., Willerslev, E., Nielsen, L. P., et al. (2011). A simple method for the parallel deep sequencing of full influenza A genomes. J. Virol. Methods 178, 243–248. doi: 10.1016/j.jviromet.2011.09.001
Klein, E. Y., Serohijos, A. W., Choi, J. M., Shakhnovich, E. I., and Pekosz, A. (2014). Influenza A H1N1 pandemic strain evolution–divergence and the potential for antigenic drift variants. PLoS One 9:e93632. doi: 10.1371/journal.pone.0093632
Kornbrot, D. (2014). Spearman’s Rho: Wiley StatsRef Stat. Ref. Online. Hoboken, NJ: John Wiley & Sons.
Kuroda, M., Katano, H., Nakajima, N., Tobiume, M., Ainai, A., Sekizuka, T., et al. (2010). Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS One 5:e10256. doi: 10.1371/journal.pone.0010256
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lauring, A. S., and Andino, R. (2010). Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 6:e1001005. doi: 10.1371/journal.ppat.1001005
Lee, H. K., Lee, C. K., Tang, J. W.-T., Loh, T. P., and Koay, E. S.-C. (2016). Contamination-controlled high-throughput whole genome sequencing for influenza A viruses using the MiSeq sequencer. Sci. Rep. 6:33318. doi: 10.1038/srep33318
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J. Z., Paredes, R., Ribaudo, H. J., Kozal, M. J., Svarovskaia, E. S., Johnson, J. A., et al. (2013). Impact of minority nonnucleoside reverse transcriptase inhibitor resistance mutations on resistance genotype after virologic failure. J. Infect. Dis. 207, 893–897. doi: 10.1093/infdis/jis925
Lin, Z., Farooqui, A., Li, G., Wong, G. K., Mason, A. L., Banner, D., et al. (2014). Next-generation sequencing and bioinformatic approaches to detect and analyze influenza virus in ferrets. J. Infect Dev. Ctries 8, 498–509. doi: 10.3855/jidc.3749
Mareuil, F., Doppelt-Azeroual, O., and Ménager, H. (2017). “A public Galaxy platform at Pasteur used as an execution engine for web services” in Poster at the F1000Research, 6:1030. doi: 10.7490/F1000RESEARCH.1114334.1
McCrone, J. T., Woods, R. J., Martin, E. T., Malosh, R. E., Monto, A. S., and Lauring, A. S. (2017). Stochastic processes dominate the within and between host evolution of influenza virus. Elite 7:e35962. doi: 10.1101/176362
Meijer, A., Lackenby, A., Hungnes, O., Lina, B., van der Werf, S., Schweiger, B., et al. (2009). Oseltamivir-resistant influenza Virus A (H1N1), Europe, 2007–08 Season. Emerg. Infect. Dis. 15, 552–560. doi: 10.3201/eid1504.081280
Meinel, D. M., Heinzinger, S., Eberle, U., Ackermann, N., Schönberger, K., and Sing, A. (2018). Whole genome sequencing identifies influenza A H3N2 transmission and offers superior resolution to classical typing methods. Infection 46, 69–76. doi: 10.1007/s15010-017-1091-3
Molinari, N. A., Ortega-Sanchez, I. R., Messonnier, M. L., Thompson, W. W., Wortley, P. M., Weintraub, E., et al. (2007). The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25, 5086–5096. doi: 10.1016/j.vaccine.2007.03.046
Moncla, L. H., Ross, T. M., Dinis, J. M., Weinfurter, J. T., Mortimer, T. D., Schultz-Darken, N., et al. (2013). A novel nonhuman primate model for influenza transmission. PLoS One 8:e78750. doi: 10.1371/journal.pone.0078750
Monne, I., Fusaro, A., Nelson, M. I., Bonfanti, L., Mulatti, P., Hughes, J., et al. (2014). Emergence of a highly pathogenic avian influenza virus from a low-pathogenic progenitor. J. Virol. 88, 4375–4388. doi: 10.1128/JVI.03181-13
Moses, L. E. (2014). Wilcoxon-Mann-Whitney Test: Definition and Example: Wiley StatsRef Stat. Ref. Online. Hoboken, NJ: John Wiley & Sons. doi: 10.1002/9781118445112.stat06898
Munster, V. J., Baas, C., Lexmond, P., Waldenstrom, J., Wallensten, A., Fransson, T., et al. (2007). Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 3:e61. doi: 10.1371/journal.ppat.0030061
Muramoto, Y., Noda, T., Kawakami, E., Akkina, R., and Kawaoka, Y. (2013). Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 87, 2455–2462. doi: 10.1128/JVI.02656-12
Nakamura, K., Oshima, T., Morimoto, T., Ikeda, S., Yoshikawa, H., Shiwa, Y., et al. (2011). Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 39:e90. doi: 10.1093/nar/gkr344
Nasu, A., Marusawa, H., Ueda, Y., Nishijima, N., Takahashi, K., Osaki, Y., et al. (2011). Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLoS One 6:e24907. doi: 10.1371/journal.pone.0024907
Neumann, G., Noda, T., and Kawaoka, Y. (2009). Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939. doi: 10.1038/nature08157
Nishijima, N., Marusawa, H., Ueda, Y., Takahashi, K., Nasu, A., Osaki, Y., et al. (2012). Dynamics of hepatitis B virus quasispecies in association with nucleos(t)ide analogue treatment determined by ultra-deep sequencing. PLoS One 7:e35052. doi: 10.1371/journal.pone.0035052
Ortega-Sanchez, I. R., Molinari, N. A., Fairbrother, G., Szilagyi, P. G., Edwards, K. M., Griffin, M. R., et al. (2012). Indirect, out-of-pocket and medical costs from influenza-related illness in young children. Vaccine 30, 4175–4181. doi: 10.1016/j.vaccine.2012.04.057
Paranjpe, S., Craigo, J., Patterson, B., Ding, M., Barroso, P., Harrison, L., et al. (2002). Subcompartmentalization of HIV-1 quasispecies between seminal cells and seminal plasma indicates their origin in distinct genital tissues. AIDS Res. Hum. Retrovir. 18, 1271–1280. doi: 10.1089/088922202320886316
Parvin, J. D., Moscona, A., Pan, W. T., Leider, J. M., and Palese, P. (1986). Measurement of the mutation rate of animal viruses: influenza A virus and poliovirus type 1. J. Virol. 59, 377–383.
Pichon, M., Gaymard, A., Josset, L., Valette, M., Millat, G., Lina, B., et al. (2017). Characterization of oseltamivir-resistant influenza virus populations in immunosuppressed patients using digital-droplet PCR: comparison with qPCR and next generation sequencing analysis. Antiviral Res. 145, 160–167. doi: 10.1016/J.ANTIVIRAL.2017.07.021
Pizzorno, A., Abed, Y., Plante, P. L., Carbonneau, J., Baz, M., Hamelin, M. E., et al. (2014). Evolution of oseltamivir resistance mutations in Influenza A(H1N1) and A(H3N2) viruses during selection in experimentally infected mice. Antimicrob. Agents Chemother. 58, 6398–6405. doi: 10.1128/aac.02956-14
Poon, L. L. M., Song, T., Rosenfeld, R., Lin, X., Rogers, M. B., Zhou, B., et al. (2016). Quantifying influenza virus diversity and transmission in humans. Nat. Genet. 48, 195–200. doi: 10.1038/ng.3479
Quinones-Mateu, M. E., Avila, S., Reyes-Teran, G., and Martinez, M. A. (2014). Deep sequencing: becoming a critical tool in clinical virology. J. Clin. Virol. 61, 9–19. doi: 10.1016/j.jcv.2014.06.013
Ren, X., Yang, F., Hu, Y., Zhang, T., Liu, L., Dong, J., et al. (2013). Full genome of influenza A (H7N9) virus derived by direct sequencing without culture. Emerg. Infect. Dis. 19, 1881–1884. doi: 10.3201/eid1911.130664
Rodriguez, C., Chevaliez, S., Bensadoun, P., and Pawlotsky, J. M. (2013). Characterization of the dynamics of hepatitis B virus resistance to adefovir by ultra-deep pyrosequencing. Hepatology 58, 890–901. doi: 10.1002/hep.26383
Rogers, M. B., Song, T., Sebra, R., Greenbaum, B. D., Hamelin, M. E., Fitch, A., et al. (2015). Intrahost dynamics of antiviral resistance in influenza a virus reflect complex patterns of segment linkage, reassortment, and natural selection. MBio 6:e02464-14. doi: 10.1128/mBio.02464-14
Rutvisuttinunt, W., Chinnawirotpisan, P., Simasathien, S., Shrestha, S. K., Yoon, I.-K., Klungthong, C., et al. (2013). Simultaneous and complete genome sequencing of influenza A and B with high coverage by Illumina MiSeq platform. J. Virol. Methods 193, 394–404. doi: 10.1016/J.JVIROMET.2013.07.001
Sanjuan, R., Nebot, M. R., Chirico, N., Mansky, L. M., and Belshaw, R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. doi: 10.1128/JVI.00694-10
Shaw, M. L., and Palese, P. (2013). “Orthomyxoviridae,” in Fields Virology, 6th Edn, eds D. M. Knipe and P. M. Howley (Philadelphia: Lippincott Williams & Wilkins), 1151–1185.
Simen, B. B., Simons, J. F., Hullsiek, K. H., Novak, R. M., Macarthur, R. D., Baxter, J. D., et al. (2009). Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 199, 693–701. doi: 10.1086/596736
Stapleford, K. A., Coffey, L. L., Lay, S., Borderia, A. V, Duong, V., Isakov, O., et al. (2014). Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microbe 15, 706–716. doi: 10.1016/j.chom.2014.05.008
Steinhauer, D. A., de la Torre, J. C., and Holland, J. J. (1989a). High nucleotide substitution error frequencies in clonal pools of vesicular stomatitis virus. J. Virol. 63, 2063–2071.
Steinhauer, D. A., de la Torre, J. C., Meier, E., and Holland, J. J. (1989b). Extreme heterogeneity in populations of vesicular stomatitis virus. J. Virol. 63, 2072–2080.
Sutton, T. C., Finch, C., Shao, H., Angel, M., Chen, H., Capua, I., et al. (2014). Airborne transmission of highly pathogenic H7N1 influenza virus in ferrets. J. Virol. 88, 6623–6635. doi: 10.1128/JVI.02765-13
Svarovskaia, E. S., Martin, R., McHutchison, J. G., Miller, M. D., and Mo, H. (2012). Abundant drug-resistant NS3 mutants detected by deep sequencing in hepatitis C virus-infected patients undergoing NS3 protease inhibitor monotherapy. J. Clin. Microbiol. 50, 3267–3274. doi: 10.1128/jcm.00838-12
Téllez-Sosa, J., Rodríguez, M. H., Gómez-Barreto, R. E., Valdovinos-Torres, H., Hidalgo, A. C., Cruz-Hervert, P., et al. (2013). Using high-throughput sequencing to leverage surveillance of genetic diversity and oseltamivir resistance: a pilot study during the 2009 Influenza A(H1N1) pandemic. PLoS One 8:e67010. doi: 10.1371/journal.pone.0067010
Thyagarajan, B., and Bloom, J. D. (2014). The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. Elife 3:e03300. doi: 10.7554/eLife.03300
Tong, S., Zhu, X., Li, Y., Shi, M., Zhang, J., Bourgeois, M., et al. (2013). New world bats harbor diverse influenza A viruses. PLoS Pathog. 9:e1003657. doi: 10.1371/journal.ppat.1003657
Tscherne, D. M., and Garcia-Sastre, A. (2011). Virulence determinants of pandemic influenza viruses. J. Clin. Invest. 121, 6–13. doi: 10.1172/jci44947
Watson, S. J., Welkers, M. R., Depledge, D. P., Coulter, E., Breuer, J. M., de Jong, M. D., et al. (2013). Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R Soc. L B Biol. Sci. 368:20120205. doi: 10.1098/rstb.2012.0205
Welkers, M. R. A., Jonges, M., Jeeninga, R. E., Koopmans, M. P. G., and de Jong, M. D. (2015). Improved detection of artifactual viral minority variants in high-throughput sequencing data. Front. Microbiol. 5:804. doi: 10.3389/fmicb.2014.00804
Willerth, S. M., Pedro, H. A., Pachter, L., Humeau, L. M., Arkin, A. P., and Schaffer, D. V. (2010). Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS One 5:e13564. doi: 10.1371/journal.pone.0013564
Wilm, A., Aw, P. P. K., Bertrand, D., Yeo, G. H. T., Ong, S. H., Wong, C. H., et al. (2012). LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201. doi: 10.1093/nar/gks918
Wise, H. M., Barbezange, C., Jagger, B. W., Dalton, R. M., Gog, J. R., Curran, M. D., et al. (2011). Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res. 39, 7775–7790. doi: 10.1093/nar/gkr487
Xue, K. S., Moncla, L. H., Bedford, T., and Bloom, J. D. (2018). Within-host evolution of human influenza virus. Trends Microbiol. 26, 781–793. doi: 10.1016/J.TIM.2018.02.007
Yamayoshi, S., Watanabe, M., Goto, H., and Kawaoka, Y. (2015). Identification of A novel viral protein expressed from the PB2 segment of influenza A virus. J. Virol. 90, 444–456. doi: 10.1128/jvi.02175-15
Yang, J. R., Lin, Y. C., Huang, Y. P., Su, C. H., Lo, J., Ho, Y. L., et al. (2011). Reassortment and mutations associated with emergence and spread of oseltamivir-resistant seasonal influenza A/H1N1 viruses in 2005-2009. PLoS One 6:e18177. doi: 10.1371/journal.pone.0018177
Zhao, J., Liu, J., Vemula, S. V., Lin, C., Tan, J., Ragupathy, V., et al. (2016). Sensitive detection and simultaneous discrimination of influenza A and B viruses in nasopharyngeal swabs in a single assay using next-generation sequencing-based diagnostics. PLoS One 11:e0163175. doi: 10.1371/journal.pone.0163175
Keywords: influenza virus, quasispecies, NGS, genetic drift, influenza season
Citation: Barbezange C, Jones L, Blanc H, Isakov O, Celniker G, Enouf V, Shomron N, Vignuzzi M and van der Werf S (2018) Seasonal Genetic Drift of Human Influenza A Virus Quasispecies Revealed by Deep Sequencing. Front. Microbiol. 9:2596. doi: 10.3389/fmicb.2018.02596
Received: 06 August 2018; Accepted: 11 October 2018;
Published: 31 October 2018.
Edited by:
Andrew S. Lang, Memorial University of Newfoundland, CanadaReviewed by:
Thomas Paul Fabrizio, St. Jude Children’s Research Hospital, United StatesCopyright © 2018 Barbezange, Jones, Blanc, Isakov, Celniker, Enouf, Shomron, Vignuzzi and van der Werf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cyril Barbezange, Y3lyaWwuYmFyYmV6YW5nZUBzY2llbnNhbm8uYmU= Sylvie van der Werf, c3lsdmllLnZhbi1kZXItd2VyZkBwYXN0ZXVyLmZy
†Present address: Cyril Barbezange, Viral Diseases, Sciensano, Brussels, Belgium
‡Co-last authors
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.