 Gabriel R. de Abreu Cabral
Gabriel R. de Abreu Cabral Zi T. Wang1†
Zi T. Wang1† Renato A. DaMatta
Renato A. DaMatta
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 20 August 2018
Sec. Infectious Agents and Disease
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01936
Toxoplasma gondii, the causative agent of toxoplasmosis, is a widespread intracellular parasite able to infect virtually any nucleated cell. T. gondii infection of activated macrophages inhibits nitric oxide (NO) production; however, parasite effectors responsible for this block have not been defined. Macrophage populations are extremely heterogeneous, responding differently to stimuli and to parasite infection. Here we evaluated the inhibition of NO production caused by T. gondii infection of J774-A1 and RAW 264.7 macrophages and assessed the role of several known parasite virulence factors in this phenotype. Infection of activated macrophages from both macrophage lines reduced NO production, however, the mechanism of this decrease was different. Consistent with previous reports, infected J774-A1 macrophages had reduced iNOS expression and lower number of iNOS positive cells. In contrast, T. gondii infection of RAW 264.7 macrophages did not alter iNOS expression or the number of iNOS positive cells, and yet it led to lower levels of NO production. Deletion of a number of previously defined virulence factors including ROP kinases that disrupt innate immune factors, TgIST which blocks STAT1 activation, as well as the secretory trafficking proteins ASP5 and MYR1, did not alter the phenotype of decreased NO production. Taken together our findings indicate that T. gondii infection inhibits NO production of activated macrophages by different mechanisms that involve reduction of iNOS expression vs. iNOS impairment, and suggest that a novel parasite effector is involved in modulating this important host defense pathway.
Toxoplasmosis is a worldwide disease affecting about one-third of the human population (Tenter et al., 2000). Toxoplasma gondii, the causative agent of toxoplasmosis, is an obligate intracellular parasite that infects distinct host cells (Tenter et al., 2000). Macrophages are key players of the host immune system and are able to control T. gondii replication following activation by interferon gamma and a second signal provide by LPS or TNF-α (Adams et al., 1990; Sibley et al., 1991). One of the main components of antimicrobial activities of activated macrophages is the production of NO through the induction of iNOS (MacMicking et al., 1997). Generation of NO has been implicated in control of chronic toxoplasmosis (Chao et al., 1993; Khan et al., 1997; Scharton-Kersten et al., 1997; Roberts et al., 2000). However, T. gondii coevolved with its host and evasion mechanisms have emerged to thwart many of the effectors of activated macrophages. Included among these virulence factors are proteins released from rhoptries that block innate immunity (Hunter and Sibley, 2012) and dense granules that alter host cell transcription (Hakimi et al., 2017).
During host cell invasion, T. gondii secretes contents from specialized secretory organelles including rhoptries and dense granules that have a central role in parasitophorous vacuole formation and host immunity subversion (Carruthers and Sibley, 1997; Bougdour et al., 2013; Braun et al., 2013; Etheridge et al., 2014; Olias et al., 2016). For example, the ROP5-ROP17-ROP18 complex, which is secreted from rhoptries, blocks the assembly and function of vacuolar-targeted IRGs (Saeij et al., 2006; Taylor et al., 2006; Behnke et al., 2011; Reese et al., 2011; Etheridge et al., 2014). Recruitment of IRGs to the vacuole normally results in its destruction and death of the parasite (Zhao et al., 2009; Khaminets et al., 2010), but ROPs of virulent strains of the parasite are able to phosphorylate key IRG proteins, inhibiting their activity and assembly, protecting T. gondii (Fentress et al., 2010; Steinfeldt et al., 2010). Proteins from GRA are another important class of virulence factors secreted by T. gondii during and after host cell invasion that perform major roles in parasite survival and replication (Mercier et al., 2002). For example, GRA16 down-modulates host p53 expression changing the cell cycle (Bougdour et al., 2013), while GRA24 causes host p38α activation, leading to a strong proinflammatory response (Braun et al., 2013). Recently, another important T. gondii virulence factor known as inhibitor of STAT1-dependent transcription (IST) has been described (Gay et al., 2016; Olias et al., 2016). IST translocates to the host nucleus where it recruits a repressive complex of STAT1 promoters, blocking the IFN-γ dependent transcription, avoiding host cell activation (Olias et al., 2016).
Classically activated macrophages produce NO that control T. gondii replication (Adams et al., 1990; Bohne et al., 1994; Khan et al., 1997). NO is an important microbicidal molecule that is produced by iNOS (Stuehr et al., 1991; Xie et al., 1994; Lowenstein and Padalko, 2004). It is well known that T. gondii evades the cytotoxic effects of NO by inhibiting NO production of activated mice peritoneal macrophages (Dobbin et al., 2002; Seabra et al., 2002, 2004; Luder et al., 2003). Furthermore, in activated J774-A1 macrophages, infection causes iNOS degradation by the proteasome (Padrao Jda et al., 2014). Although TGF-β 1 is involved in the inhibition of NO production in infected activated macrophages (Seabra et al., 2004), the parasite effector responsible for iNOS degradation and NO inhibition still remains elusive. In addition, most of these studies have been done in vitro with a single cell type and without comparison of other macrophage cell lines.
Macrophages are an extremely heterogeneous cell population with many subpopulations that behave differently (Geissmann et al., 2010). Thus, T. gondii has to deal with many distinct macrophage subpopulations during host infection. To better understand how T. gondii copes with NO production of distinct activated macrophages lines, production of this microbicidal molecule and expression of iNOS were compared in two macrophage cell lines after infection. In addition, several previously described virulence factors were also analyzed as possible effectors responsible for NO inhibition of infected activated macrophages. This study reveals intrinsic differences between both macrophage cell lines in activation patterns and mechanisms by which T. gondii infection disrupted NO production. Furthermore, previously identified virulence effectors that were tested here did not alter the NO inhibition phenotype detected in both macrophages cell lines, indicating that a novel effector is responsible for the inhibition of this important host microbicidal molecule.
This study was carried out in accordance with the NIH standard biosecurity and institutional safety procedures of Washington University School of Medicine.
Knockout parasites used in this work were previously generated as reported: RHΔku80 (Fox et al., 2009); Δrop5 (Behnke et al., 2011); Δrop17, Δrop17/18, Δrop18 (Etheridge et al., 2014); ΔTgIST (Olias et al., 2016); (Δasp5) (Curt-Varesano et al., 2016); (Δmyr1) (Franco et al., 2016). Wild type T. gondii tachyzoites, deficient in Ku80 (RHΔku80) and knockout parasites, all of the RH strain, were maintained by serial passage in Human Foreskin Fibroblast (HFF) monolayers. Parasites were released from HFF using a cell scraper (TPP, Switzerland). The cell suspension was passed three times in a 10 ml syringe (BD, United States) with a 22-gauge needle (CML Supply, United States) and it was filtered on a 3 μm Whatman Nuclepore membrane (GE Healthcare Life Sciences, United States).
Human Foreskin Fibroblast cells were obtained from the Boothroyd laboratory at Stanford University. HFF cells and the mouse macrophage cell lines RAW 264.7 (ATCC TIB-71, United States, from BALB/c mice ascites after Abelson murine leukemia virus inoculation) and J774-A1 (ATCC TIB-67, United States, from BALB/c mice ascites after reticulum cell sarcoma inoculation) were cultivated in Dulbecco’s modified Eagle’s medium (DMEM - Invitrogen, United States) supplemented with 10% HyClone Fetal Bovine Serum (FBS - GE Healthcare Life Sciences, United States), 10 mM glutamine (Thermo Fisher Scientific, United States) and 10 μg/ml gentamicin (Gibco, United States) in an incubator (Thermo Fisher Scientific, United States) at 37°C in 5% CO2 atmosphere. Cultures were negative for Mycoplasma spp. contamination with the e-Myco plus PCR detection kit (Boca Scientific, United States).
The knockout plasmid construct for ROP16 was created using the three-fragment Gateway™ recombination system (Invitrogen, United States), joining two constructs homologous to the 5′ and 3′ flanks of ROP16 with a central HXGPRT expression cassette, as described previously (Etheridge et al., 2014). This plasmid was transfected into RHΔhxgprtΔku80 parasites, and resistant pools were expanded under treatment with mycophenolic acid (15 μg/ml) acid and xanthine (50 μg/ml). After PCR (Applied Biosystems, United States) confirmation of construct integration in the pool, parasites were subcloned by limiting dilution in 96-well plates (TPP, Switzerland) containing confluent monolayers of HFF cells. Clones were identified by visual confirmation of single plaques, screened by PCR to confirm replacement of the coding region of ROP16 with the HXGPRT cassette, expanded by growth on HFF monolayers, and cryo-preserved.
The RAW 264.7 and J774-A1 cells were seeded at the density of 5 × 104 cells per well in 96-well plates, activated with 200 U/ml of recombinant mouse IFN-γ (R&D Systems, United States) and 0.2 μg/ml of LPS from Escherichia coli 0111:B4 (Sigma-Aldrich, United States) for 24 h prior of the T. gondii infection. After 24 h of activation, cells were washed twice with sterile PBS, DMEM supplemented with 3% FBS was added, cells were infected with a 5 T. gondii per macrophage (5:1) ratio and incubated at 37°C for 2 h. The T. gondii-macrophage ratio used was determined by previous experiments examining NO production and number of adhered macrophages after 24 h infection. After infection, cells were washed twice in sterile PBS, and DMEM supplemented with 10% FBS with IFN-γ and LPS or these activators and L-arginine (Sigma-Aldrich, United States) at different concentration, was added. Supernatants were collected at 2, 4, 6, and 24 h after infection for the nitrite assay.
The NO produced by macrophages was assayed by the Griess reagent (Green et al., 1982). Briefly, 50 μl of cell supernatant from each well were collected and distributed in 96-well plates and 50 μl of Griess reagent (1:1, 0.1% N-(1-Naphthyl) ethylenediamine dihydrochloride (Sigma-Aldrich, United States) in distilled water and 1% sulfanilamide (Sigma-Aldrich, United States) in 5% phosphoric acid (Sigma-Aldrich, United States) were added. Plates were incubated at room temperature and nitrite was read in a plate reader (BioTek, United States) at 540 nm. The nitrite value was calculated from a calibrated standard curve using sodium nitrite ranging from 0 to 100 μM.
Infection of parental and knockout parasites into RAW 264.7 and J774-A1 macrophages was evaluated after 2 h of challenge in cells seeded in 96-well plates, activated and infected as described. After infection, cells were fixed and permeabilized for 30 min in PBS containing 4% formaldehyde (Polysciences, Inc., United States), 0.05% Triton X-100 (Fisher BioReagents, United States), washed with PBS and blocked for 30 min with 5% FBS and Normal Goat Serum (Sigma-Aldrich, United States) in PBS (FBS-NGS). Cells were incubated for 1 h with anti-RH (SAG1) rabbit antibody diluted 1:2000 in FBS-NGS, washed twice and incubated for 1 h with anti-rabbit HRP conjugated antibody (Life Technology, United States) diluted 1:4000 in FBS-NGS. Cells were washed three times, incubated for 15 min with 100 μl of TMB substrate (BD OptEIA, Thermo Fisher Scientific, United States), reaction stopped with 100 μl of 1M H2SO4 and the absorbance was read in a plate reader (BioTek, United States) at 450 nm. Non-infected macrophages were used as negative control.
RAW 264.7 and J774-A1 macrophages were seeded at the density of 5 × 105 cells per well over coverslips (Fisherbrand, United States) in 24-well plates (TPP, Switzerland), activated and infected as described. Cells were fixed for 30 min with PBS containing 4% formaldehyde, permeabilized for 15 min in PBS containing 0.1% Triton X-100, incubated for 30 min with PBS containing 100 mM of NH4Cl (Sigma-Aldrich, United States), and washed three times with PBS containing 1.5% Bovine Serum Albumin (PBS-BSA, Sigma-Aldrich, United States). Cells were incubated for 1 h with anti-iNOS mouse monoclonal antibody (sc-7271, Santa Cruz Biotechnology, United States) diluted 1:100 and anti-RH (SAG1) rabbit antibody diluted 1:2000 in PBS-BSA, washed once in PBS and twice in PBS-BSA and incubated with goat anti-mouse IgG monoclonal antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, United States) diluted 1:200 and goat anti-rabbit IgG monoclonal antibody conjugated to Alexa Fluor 594 (Thermo Fisher Scientific, United States) diluted 1:2000 in PBS-BSA. Coverslips containing cells were mounted with the Prolong Gold antifade reagent with DAPI (Life Technologies, United States) and visualized with a Zeiss Axioskop 2 MOT Plus epifluorescence microscope with a 63× Plan Apochromat lens (numerical aperture of 1.40; Carl Zeiss, Inc., Germany), equipped with an AxioCam MRm camera (Carl Zeiss, Inc., Germany). Images were acquired using Axiovision v4.6 (Carl Zeiss, Inc., Germany) and processed with Adobe Photoshop 6.0 (Adobe Systems Inc., United States).
To quantify the percentage of iNOS positive and iNOS negative cells in infected or non-infected cells, RAW 264.7 and J774-A1 macrophages were seeded in black 96-well cellstar microplates with clear bottom and condensation rings (BioTek, United States), activated and infected as described. Cells were fixed with PBS containing 4% formaldehyde for 15 min, washed three times with PBS and incubated for 15 min with FBS-NGS containing 5 μg/ml of Wheat Germ Agglutinin conjugated to Alexa Fluor 633 (Thermo Fisher Scientific, United States) for total cell staining. Cells were washed twice in PBS and incubated for 1 h with anti-iNOS mouse monoclonal antibody (Santa Cruz Biotechnology, United States) diluted 1:100 in FBS-NGS and anti-RH (SAG1) rabbit antibody diluted 1:10000 in 5% FBS-NGS. Cells were washed twice with PBS and incubated with goat anti-mouse IgG conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, United States) diluted 1:200 and goat anti-rabbit IgG conjugated to Alexa Fluor 594 (Thermo Fisher Scientific, United States) diluted 1:2000 in FBS-NGS. Cell analysis was performed with a Cytation 3 (BioTek, United States) multimode plate imager equipped with Gen5 software and 20× objective.
RAW 264.7 and J774-A1 macrophages were seeded at the density of 2 × 106 cells per well in 6-well plate (TPP, Switzerland), activated and infected as described. Cells were washed once in sterile PBS and lysed with 50 μl of lysing buffer containing 50 mM of Tris–HCL (Sigma-Aldrich, United States), 150 mM NaCl (Sigma-Aldrich, United States), 1% Triton X-100 (Sigma-Aldrich, United States) and protease inhibitor cocktail (Sigma-Aldrich, United States). Protein samples were frozen in liquid nitrogen three times, centrifuged (Eppendorf, Germany) at 5,000 g, for 3 min and protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, United States). Samples were diluted 4:1 in 5× Laemmli buffer containing 10 mM of dithiothreitol (Sigma-Aldrich, United States), boiled for 5 min, resolved in 8% polyacrylamide gels (Bio-Rad Laboratories, Inc., United States) by SDS–PAGE, and transferred to nitrocellulose membranes Amersham Protran 0.45 NC (GE Healthcare Life Sciences, United States) for 1 h. Membranes were blocked overnight at 4°C with 5% fat-free milk in PBS 0.1% Tween 20 (Sigma-Aldrich, United States), probed for 1 h with anti-iNOS mouse monoclonal antibody (Santa Cruz Biotechnology, United States) dilute 1:1000 and rabbit anti-β-actin (Cell Signaling Technology, United States) diluted 1:1000 in 5% fat-free milk in PBS 0.1% Tween 20. Membranes were washed with PBS 0.1% Tween 20 and labeled for 1 h with IR dye-conjugated secondary antibodies (LI-COR Biosciences, United States) against mouse and rabbit dilute 1:15000 and visualized on a LiCor Odyssey Imaging System (LI-COR Biosciences, United States). Western blots were quantified using the software ImageJ and intensity values were normalized to β-actin at each time interval.
Statistical analyses were conducted with Prism 7 (GraphPad Software Inc., United States), and differences in the means were assessed by one-way or two-way ANOVA with Tukey’s multiple comparison post-test, or unpaired Student’s t-test. P ≤ 0.05 was the cutoff considered minimum for significance.
To evaluate the ability of the parasite to inhibit NO production in different macrophage cell lines, activated J774-A1 or RAW 264.7 cells were infected with T. gondii and nitrite production was evaluated in culture supernatant at different time points. Differences in the timing of inhibition of NO production were observed between the two cell lines. In J774-A1 macrophages, inhibition of NO production occurred by 2 h post-infection (Figure 1A) and was sustained up to 24 h (Figure 1B). In contrast, inhibition of NO production in RAW 264.7 macrophages occurred only after 6 h post-infection (Figure 1C) and was sustained up to 24 h (Figure 1D).
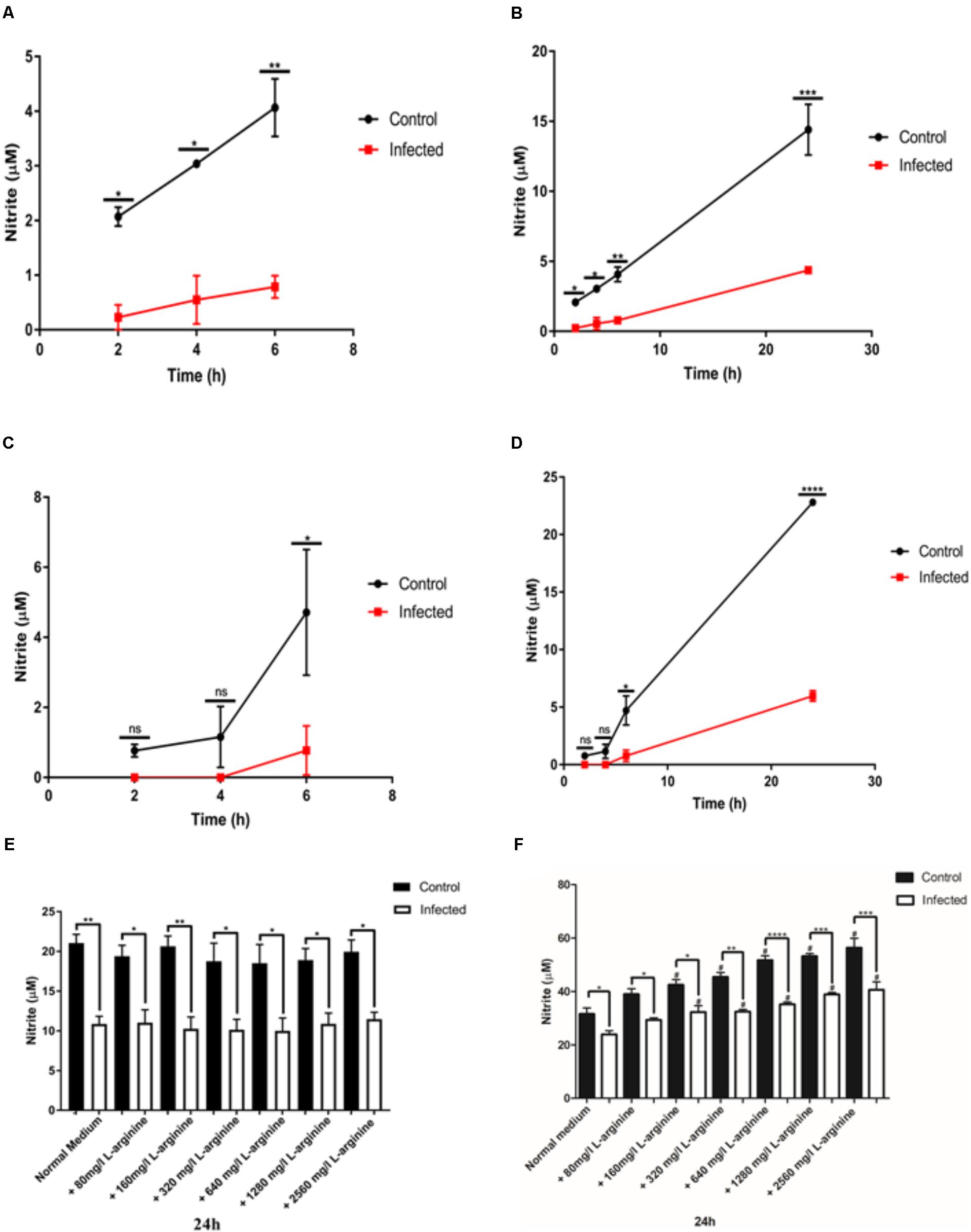
FIGURE 1. Nitric oxide (NO) production (nitrite in μM) in activated J774-A1 and RAW 264.7 cells macrophages after T. gondii infection. (A) NO production of non-infected (Control) or T. gondii (RH) infected J774-A1cells at 6 h and (B) 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). (C) NO production of non-infected (Control) or T. gondii (RH) infected RAW 264.7 cells at 6 h and (D) 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). ns (not significant), ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, two-way ANOVA with Tukey post-test. (E) NO production of non-infected (Control) or T. gondii (RH) infected J774-A1 (F) or RAW 264.7 cells for 24 h with normal medium or supplemented with different levels of L-arginine. Mean ± SEM (n = 3 experiments, each with 6 replicates). ∗P ≤ 0.05 and ∗∗P ≤ 0.01, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, ∗∗∗∗P ≤ 0.0001 one-way ANOVA with Tukey post-test, #P ≤ 0.05 comparing the “Control” or “Infected” bar with the respective “Normal medium” bar.
During host cell invasion T. gondii secretes important virulence factors including ROP16, which activates STAT 3 and STAT 6 in macrophages resulting in upregulation of arginase 1 (ARG1) (Butcher et al., 2011). Induction of ARG1 competes with iNOS for the substrate L-arginine (Butcher et al., 2011), hence this may be a mechanism of avoiding NO production. To determine if inhibition of NO production in activated macrophages was caused by substrate depletion, we supplemented the culture medium with increasing concentrations of L-arginine and evaluated NO production after 24 h of infection. Treatment of activated J774-A1 macrophages with increasing concentrations of L-arginine did not enhance NO production in control or infected cells, nor reverted the inhibition of NO production phenotype caused by T. gondii infection (Figure 1E). In contrast, incubation of activated RAW 264.7 macrophages with increasing concentrations of L-arginine enhanced NO production in control and infected cells, and yet it did not reverse the inhibition of NO production in infected cells (Figure 1F).
To examine whether the inhibition of NO production in activated J774-A1 macrophage was caused by protein degradation, iNOS expression was evaluated by different methods. iNOS showed variable levels of expression in activated non-infected J774-A1 macrophages based on IFA staining (Figure 2A). Following infection with T. gondii there were more iNOS negative cells at 24 h post-infection (Figure 2A). This finding was confirmed by analyzing the proportion of activated cells that were positive for iNOS in non-infected cells (Control) and T. gondii challenged cells (Non-infected or Infected) as depicted in Figure 2B. Although there was a tendency to decrease the number of iNOS positive cells in infected macrophages at 2 h, this effect was significant at 24 h post-infection (Figure 2B). Western blot (Figure 2C and Supplementary Figure 1) and densitometry analysis (Figure 2D) also confirmed inhibition of iNOS expression in cells infected for 24 h. However, reduction in iNOS expression was not observed at earlier time points after infection (Figure 2D).
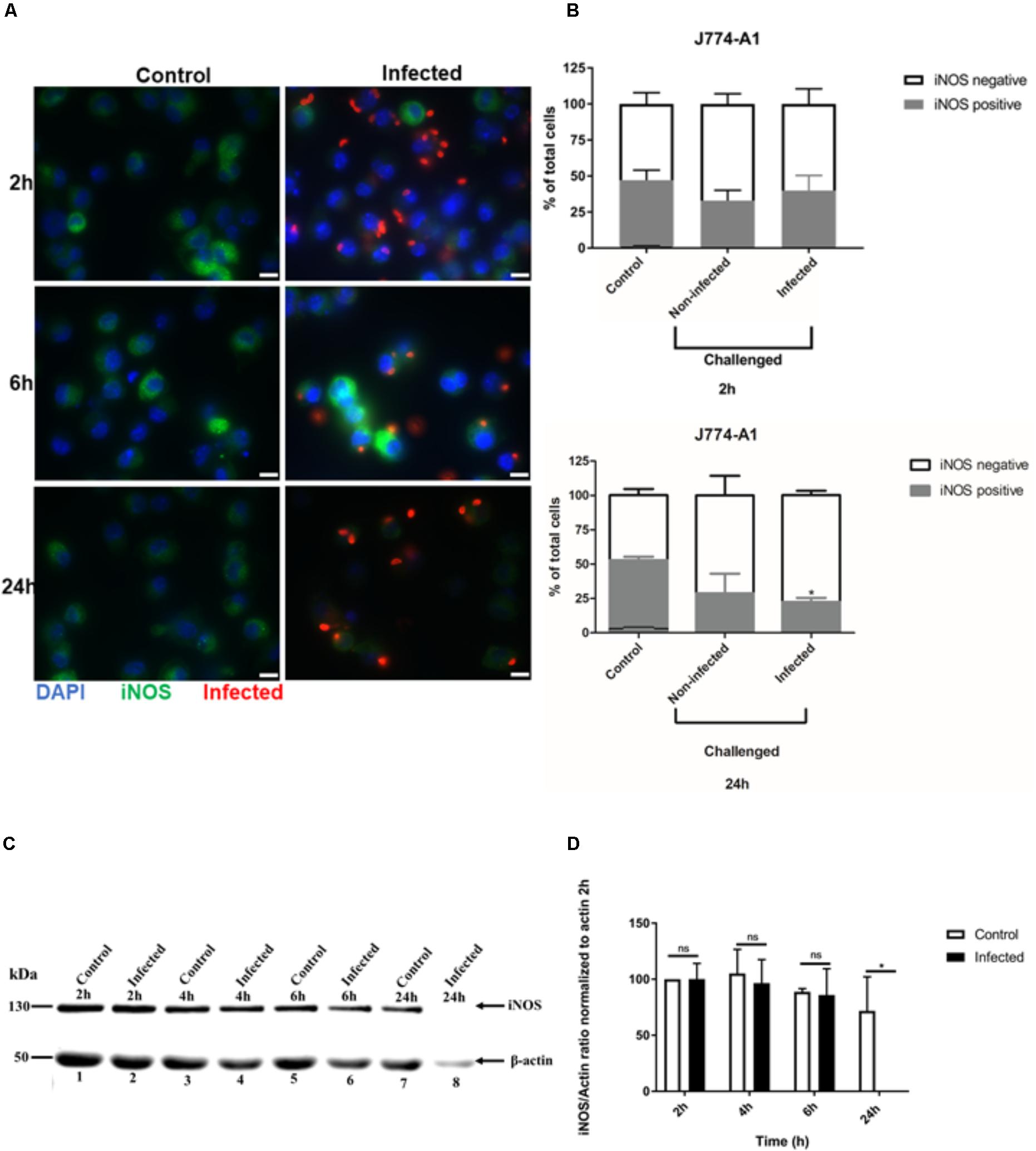
FIGURE 2. Immunofluorescence detection of iNOS in activated J774-A1 macrophages infected with T. gondii. (A) Detection of iNOS (green) in non-infected (Control) and in T. gondii (red) infected cells (DAPI - blue) at 2, 6, and 24 h post-infection. Scale bar = 10 μm. (B) Analysis of the proportion of iNOS positive or negative macrophages in non-infected (Control) and T. gondii infected cells at 2 and 24 h post-infection. Mean ± SEM (n = 4 experiments, each with 8 replicates). (C) Western blot detection of iNOS expression in non-infected (Control) and T. gondii infected (Infected) cells. β-actin was used as loading control. (D) Densitometry of western blots normalized to β-actin at 2 h post-infection. Mean ± SD (n = 3 experiments, each with 1 replicate). ∗P ≤ 0.05, two-way ANOVA with Tukey post-test, n.s (not significant).
We also analyzed iNOS expression after T. gondii infection of RAW 264.7 macrophages using similar IFA and Western blot analyses. The signal intensity of iNOS expression in RAW 264.7 cells was much higher, with all uninfected cells being uniformly positive (Figure 3A and Supplementary Figure 2). No difference in iNOS expression by IFA was observed between non-infected and infected RAW 264.7 macrophages at 24 h post-infection (Figure 3A). This finding was confirmed by analyzing the proportion of cells that were positive for iNOS. In non-infected and T. gondii challenged RAW 264.7 macrophage populations, most of the cells remained iNOS positive up to 24 h post-infection (Figure 3B). Similarly, no difference in iNOS expression between control and infected cells was observed by Western blot (Figure 3C) and densitometry analysis (Figure 3D) at different time points post-infection.
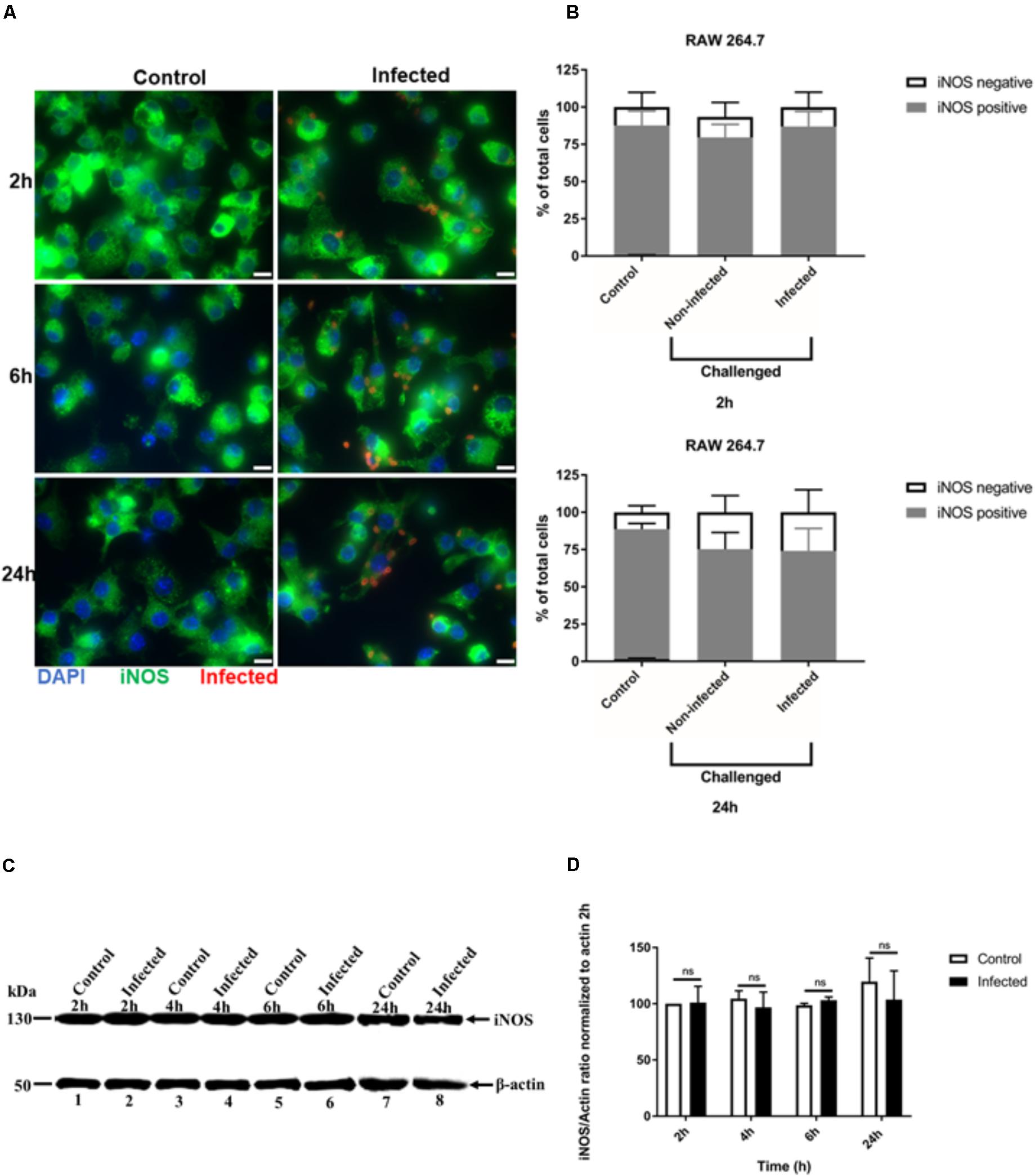
FIGURE 3. Immunofluorescence detection of iNOS in activated RAW 264.7 macrophages infected with T. gondii. (A) Detection of iNOS (green) in non-infected (Control) and in T. gondii (red) infected cells (DAPI - blue) at 2, 6, and 24 h post-infection. Scale bar - 10 μm. (B) Analysis of the proportion of iNOS positive or negative macrophages in non-infected (Control) and T. gondii infected cells at 2 and 24 h post-infection. Mean ± SEM (n = 4 experiments, each with 8 replicates). (C) Western blot detection of iNOS expression in non-infected (Control) and T. gondii - infected (Infected) cells at different time intervals post-infection. β-actin was used as loading control. (D) Densitometry of western bolts normalized to β-actin at 2 h post-infection. Mean ± SD (n = 3 experiments, each with 1 replicate), n.s (not significant).
A number of previous virulence factors have been identified in T. gondii including a complex of ROP kinases consisting of ROP5, ROP17, and ROP18 that participates in defense of the parasitophorous vacuole by thwarting IRGs (Hunter and Sibley, 2012). To determine whether the inhibition of NO production in infected activated J774-A1 and RAW 264.7 macrophages was dependent on the ROP kinases, we examined the inhibition of NO production in activated macrophages infected with a series of mutants. Inhibition of NO production was similar in both macrophage cell lines at 24 h infection when comparing the parent RH line to a series of ROP deletion mutants (Figures 4A,C). We also examined the ability of a Δrop16 mutant to alter this phenotype, since this kinase has previously been shown to activate STAT3/STAT6 and hence activate ARG1 (Butcher et al., 2011). The rop16 mutant showed a similar capacity to block NO production in activated J774-A1 and RAW 264.7 cells (Figures 4A,C). The various knockout parasites presented no deficiency in entry in activated J774-A1 or RAW 264.7 macrophages (Supplementary Figure 3).
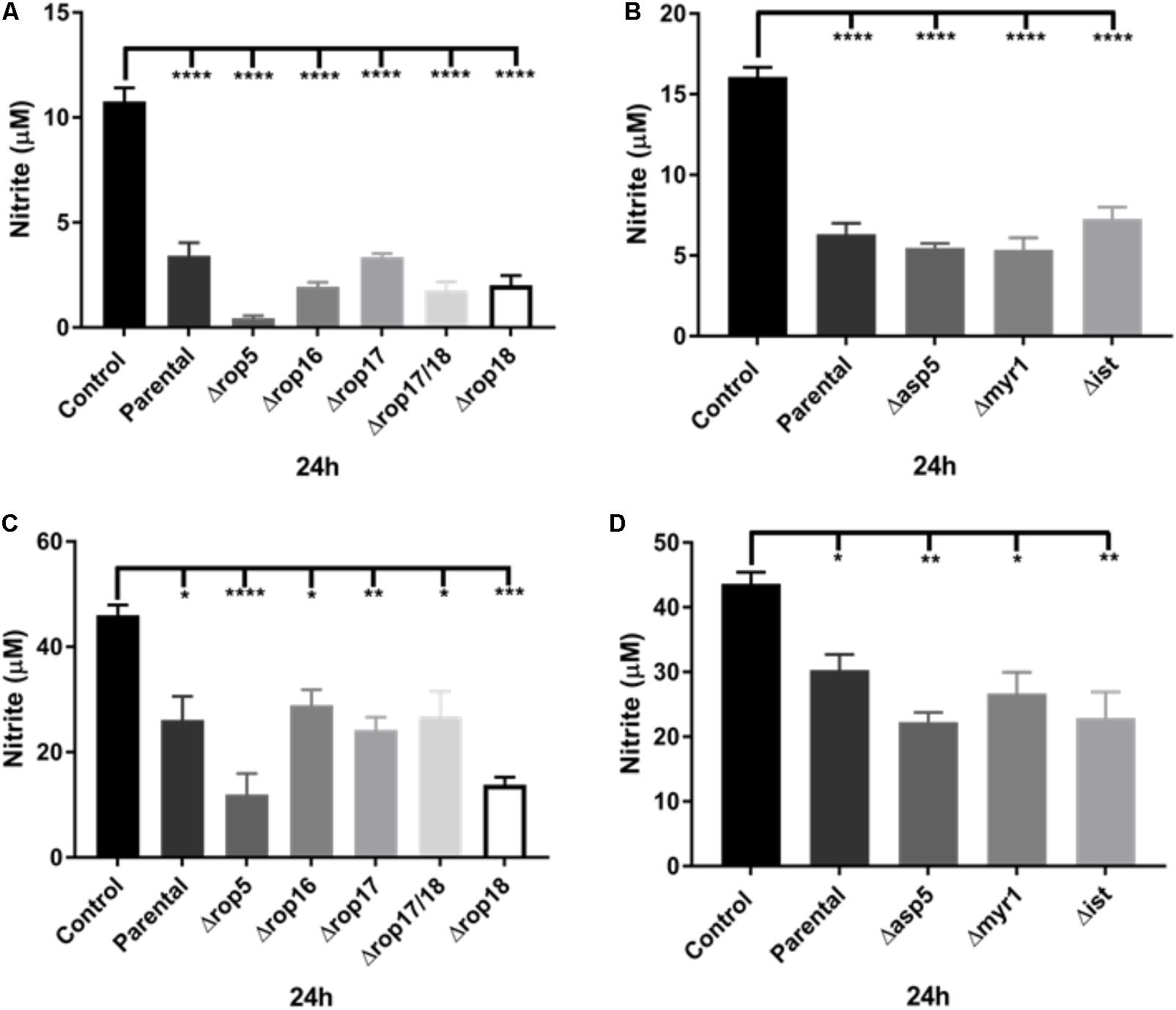
FIGURE 4. Analysis of ROP and ASP5, MYR1, and IST deletion mutants on the ability of T. gondii to inhibit NO production (nitrite in μM) of activated macrophages. (A) NO production of non-infected (Control) or activated J774-A1 cells infected with parental (RHΔku80 strain) or various ROP deletion strains of T. gondii at 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). ∗∗∗∗P ≤ 0.0001, one-way ANOVA with Tukey post-test. (B) NO production of non-infected (Control) or activated J774-A1 cells infected with parental (RHΔku80 strain) or Δasp5, Δmyr1, or Δist mutant strains of T. gondii at 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). ∗∗∗∗P ≤ 0.0001, one-way ANOVA with Tukey post-test. (C) NO production of non-infected (Control) or activated RAW 264.7 cells infected with parental (RHΔku80 strain) or various ROP deletion strains of T. gondii at 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, ∗∗∗∗P ≤ 0.0001, one-way ANOVA with Tukey post-test. (D) NO production of non-infected (Control) or activated RAW 264.7 cells infected with parental (RHΔku80 strain) or Δasp5, Δmyr1, and Δist mutant strains of T. gondii at 24 h post-infection. Mean ± SEM (n = 3 experiments, each with 12 replicates). ∗P ≤ 0.05, ∗∗P ≤ 0.01, one-way ANOVA with Tukey post-test.
We also tested T. gondii mutants in the modulator IST that inhibits STAT1 transcription (Hakimi et al., 2017). These mutants had no effect on the NO inhibition phenotype (Figures 4B,D). Recently, the major role of the Golgi-associated protein, ASP5, and the parasitophorous vacuole (PV) associated protein, MYR1, in the cleavage and export of some dense granule effector proteins across the vacuole membrane into the host cell has been demonstrated (Curt-Varesano et al., 2016; Franco et al., 2016). Therefore we examined the ability of mutants in these effectors to block the production of NO in activated macrophages. After infection, all knockout parasites were able to inhibit NO production of activated J774-A1 (Figure 4B) and RAW 264.7 macrophages (Figure 4D) similar to the parental parasite. The various knockout parasites presented no deficiency in entry in activated J774-A1 or RAW 264.7 macrophages (Supplementary Figure 3).
Toxoplasma gondii has many evasion mechanisms including the capacity to inhibit NO production of infected activated macrophages (Dobbin et al., 2002; Seabra et al., 2002, 2004; Luder et al., 2003; Padrao Jda et al., 2014). NO production inhibition, iNOS expression and the role of some T. gondii effectors were studied in parallel using two macrophage cell lines. Infection of both cell lines caused inhibition of NO production. However, only in J774-A1 macrophages was NO inhibition detected at early stages of infection (i.e., 2 h post-infection), while inhibition was seen starting at 6 h post-infection in both lines. Addition of extra L-arginine substrate to both macrophage lines did not change NO production inhibition, indicating that this result is not due to substrate limitation. Interestingly, reduction of iNOS expression after infection was only detected in J774-A1 cells, with RAW 264.7 presenting the same levels as non-infected cells. Finally, knockout parasites in known effectors were able to inhibit NO production similar to the parental strain. Our findings suggest that the inhibition of NO production of activated macrophages infected by T. gondii is a general phenomenon, but iNOS suppression varies depending on the macrophage cell line. In addition, it is likely that a novel parasite effector is responsible for this evasion mechanism.
Toxoplasma gondii infection causes ARG1 expression that competes with iNOS for L-arginine (El Kasmi et al., 2008; Butcher et al., 2011). However, extra L-arginine did not reverse NO production inhibition in either infected cell lines, indicating that the reduction in NO production is not due to substrate limitation. Following addition of L-arginine, J774-A1 produced the same amount of NO while RAW 264.7 macrophages responded to this addition by producing more NO. This finding is consistent with the differences in iNOS expression, and provides a further distinction in phenotypes between these two lines (Heming et al., 2001; Lindmark et al., 2004; El Aamri et al., 2015).
Previous report showed that NO production inhibition of T. gondii infected activated mice peritoneal macrophages was related to iNOS degradation (Seabra et al., 2002, 2004), which involves the proteasome in J774-A1 macrophages (Padrao Jda et al., 2014). Down modulation of iNOS expression was verified in infected J774-A1 but this was not observed in RAW 264.7 macrophages. These results suggest that the strategy adopted by T. gondii to inhibit NO production may be specific to the host cell, due to the intrinsic characteristics and origin of each macrophage cell line. The J774-A1 line was originally established from reticulum cell sarcoma (Hirst et al., 1971) while the RAW 264.7 line from a tumor induced by Abelson murine leukemia virus (Ralph and Nakoinz, 1977) both in BALB/c mice. Furthermore, exposure of both cell lines to Streptococcus iniae induces a higher respiratory burst response in RAW 264.7 than in J774-A1 macrophage (El Aamri et al., 2015). Moreover, RAW 264.7 produces 30-fold higher TNF-α mRNA than J774-A1 after LPS stimulation (Heming et al., 2001). These differences help to explain the higher expression of iNOS in RAW 264.7 compared to J774-A1 macrophages. In addition, a gene expression profile study (Lindmark et al., 2004) shows that J774-A1 is closer to peritoneal mice macrophages than RAW 264.7, despite the fact that both cell lines were derived from transformed cells obtained from ascites (Ralph and Nakoinz, 1977). Our findings reveal that T. gondii infection can down-regulate NO production in these different macrophage cell lines, albeit by different mechanisms.
During host cell infection, T. gondii secrets the content of rhoptries and dense granules ensuring the establishment of infection and hijacking host cell-autonomous immunity (Sibley, 2011). ROP and GRA proteins form complexes that protect T. gondii PV by avoiding recruitment of IRGs (Hunter and Sibley, 2012). GRA proteins are also exported across the PV altering important host functions (Hakimi et al., 2017). TgIST is a GRA effector that represses STAT1 transcription blocking gene expression induced by IFN-γ (Gay et al., 2016; Olias et al., 2016). In addition, the export and traffic of GRAs across the PV and into the host cell are dependent of ASP5 (Coffey et al., 2015; Curt-Varesano et al., 2016) and MYR1 (Franco et al., 2016). However, these virulence factors have not previously been evaluated for their ability to modulate other important host microbicidal systems such as NO production. Thus, knockout parasites were used to investigate whether some rhoptry proteins (ROP5, ROP16, ROP17, ROP18). The parasite ROP16 kinase has been implicated in modulating NO production in microglial cells and astrocytes (Butcher et al., 2011). However, the knockout in ROP16 did not affect the down modulation of NO production in either of the macrophage cells lines studied here. Additionally, the ROP5-ROP17-ROP18 complex, which has been implicated in blocking IRG-mediated clearance, did not affect the down modulation of NO production in T. gondii – infected cells. We also explored the roles of the GRA TgIST, or components of the PV membrane translocation system (ASP5, MYR1) for involvement in NO production control in infected macrophages. These GRA knockout parasites were able to inhibit NO production similar to the parental strain in J774-A1 and RAW 264.7 macrophages. These results indicate that the parasite effector that down modulates NO production is independent of TgIST, including other modulators that depend on the trafficking pathway based on ASP5 and MYR1.
Overall, our findings reveal that the strategy adopted by T. gondii to inhibit NO production in activated macrophages is independent of previously characterized virulence factors, such as the ROP5-ROP17-ROP18 complex, ROP16, GRA effectors, and the ASP5 and MYR1 export pathway. Thus, a possibly new parasite effector is involved in NO production inhibition in these cells. The mechanism causing NO production inhibition of macrophages infected by T. gondii varies depending on the host cell background: it involves reduction of iNOS expression in J774-A1 and iNOS impairment in RAW 264.7. This may be relevant to in vivo infections where T. gondii infects and needs to cope with distinct macrophage populations.
GC performed the experiments. ZW produced the ROP16 knockout. GC, LS, and RD wrote the manuscript. LS and RD designed the experiments and revised the manuscript critically. All authors read and approved the final manuscript.
GC was supported by a fellowship from the Brazilian Federal Agency, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (88881.132450/2016-01) and partially by grants from the NIH (AI118426) obtained by LS; RD is supported by a CNPq fellowship (307014/2015-0).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Jennifer Barks for support in cell culture and in-cell-ELISA experiments, Sumit Kumar for assistance in IFA and statistical analysis, Kevin Brown for total cell population analysis, Lisa Drewry for western blot assistance, and all of the LS’ lab members for helpful advice. We would like to thank Andrèa Carvalho César for proof reading the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01936/full#supplementary-material
ASP5, aspartyl protease 5; GRA, dense granule proteins; iNOS, inducible nitric oxide synthase; IRGs, immunity related GTPases; MYR1, myc regulation 1; NO, nitric oxide; ROP, rhoptry proteins; TgIST, Toxoplasma gondii inhibitor of STAT1-dependent transcription.
Adams, L. B., Hibbs, J. B. Jr., Taintor, R. R., and Krahenbuhl, J. L. (1990). Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from L-arginine. J. Immunol. 144, 2725–2729.
Behnke, M. S., Khan, A., Wootton, J. C., Dubey, J. P., Tang, K., and Sibley, L. D. (2011). Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. U.S.A. 108, 9631–9636. doi: 10.1073/pnas.1015338108
Bohne, W., Heesemann, J., and Gross, U. (1994). Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect. Immun. 62, 1761–1767.
Bougdour, A., Durandau, E., Brenier-Pinchart, M. P., Ortet, P., Barakat, M., Kieffer, S., et al. (2013). Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13, 489–500. doi: 10.1016/j.chom.2013.03.002
Braun, L., Brenier-Pinchart, M. P., Yogavel, M., Curt-Varesano, A., Curt-Bertini, R. L., Hussain, T., et al. (2013). A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J. Exp. Med. 210, 2071–2086. doi: 10.1084/jem.20130103
Butcher, B. A., Fox, B. A., Rommereim, L. M., Kim, S. G., Maurer, K. J., Yarovinsky, F., et al. (2011). Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 7:e1002236. doi: 10.1371/journal.ppat.1002236
Carruthers, V. B., and Sibley, L. D. (1997). Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 73, 114–123.
Chao, C. C., Anderson, W. R., Hu, S., Gekker, G., Martella, A., and Peterson, P. K. (1993). Activated microglia inhibit multiplication of Toxoplasma gondii via a nitric oxide mechanism. Clin. Immunol. Immunopathol. 67, 178–183. doi: 10.1006/clin.1993.1062
Coffey, M. J., Sleebs, B. E., Uboldi, A. D., Garnham, A., Franco, M., Marino, N. D., et al. (2015). An aspartyl protease defines a novel pathway for export of Toxoplasma proteins into the host cell. eLife 4:e10809. doi: 10.7554/eLife.10809
Curt-Varesano, A., Braun, L., Ranquet, C., Hakimi, M. A., and Bougdour, A. (2016). The aspartyl protease TgASP5 mediates the export of the Toxoplasma GRA16 and GRA24 effectors into host cells. Cell. Microbiol. 18, 151–167. doi: 10.1111/cmi.12498
Dobbin, C. A., Smith, N. C., and Johnson, A. M. (2002). Heat shock protein 70 is a potential virulence factor in murine Toxoplasma infection via immunomodulation of host NF-kappa B and nitric oxide. J. Immunol. 169, 958–965. doi: 10.4049/jimmunol.169.2.958
El Aamri, F., Remuzgo-Martinez, S., Acosta, F., Real, F., Ramos-Vivas, J., Icardo, J. M., et al. (2015). Interactions of Streptococcus iniae with phagocytic cell line. Microbes Infect. 17, 258–265. doi: 10.1016/j.micinf.2014.06.006
El Kasmi, K. C., Qualls, J. E., Pesce, J. T., Smith, A. M., Thompson, R. W., Henao-Tamayo, M., et al. (2008). Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9, 1399–1406. doi: 10.1038/ni.1671
Etheridge, R. D., Alaganan, A., Tang, K., Lou, H. J., Turk, B. E., and Sibley, L. D. (2014). The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 15, 537–550. doi: 10.1016/j.chom.2014.04.002
Fentress, S. J., Behnke, M. S., Dunay, I. R., Mashayekhi, M., Rommereim, L. M., Fox, B. A., et al. (2010). Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495. doi: 10.1016/j.chom.2010.11.005
Fox, B. A., Ristuccia, J. G., Gigley, J. P., and Bzik, D. J. (2009). Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 8, 520–529. doi: 10.1128/ec.00357-08
Franco, M., Panas, M. W., Marino, N. D., Lee, M. C., Buchholz, K. R., Kelly, F. D., et al. (2016). A novel secreted protein, MYR1, is central to toxoplasma’s manipulation of host cells. mBio 7:e02231-15. doi: 10.1128/mBio.02231-15
Gay, G., Braun, L., Brenier-Pinchart, M.-P., Vollaire, J., Josserand, V., Bertini, R.-L., et al. (2016). Toxoplasma gondii TgIST co-opts host chromatin repressors dampening STAT1-dependent gene regulation and IFN-γ–mediated host defenses. J. Exp. Med. 213, 1779–1798. doi: 10.1084/jem.20160340
Geissmann, F., Gordon, S., Hume, D. A., Mowat, A. M., and Randolph, G. J. (2010). Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 10, 453–460. doi: 10.1038/nri2784
Green, L. C., Wagner, D. A., Glogowski, J., Skipper, P. L., Wishnok, J. S., and Tannenbaum, S. R. (1982). Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126, 131–138. doi: 10.1016/0003-2697(82)90118-X
Hakimi, M. A., Olias, P., and Sibley, L. D. (2017). Toxoplasma effectors targeting host signaling and transcription. Clin. Microbiol. Rev. 30, 615–645. doi: 10.1128/cmr.00005-17
Heming, T. A., Tuazon, D. M., Dave, S. K., Chopra, A. K., Peterson, J. W., and Bidani, A. (2001). Post-transcriptional effects of extracellular pH on tumour necrosis factor-alpha production in RAW 246.7 and J774 A.1 cells. Clin. Sci. 100, 259–266. doi: 10.1042/cs1000259
Hirst, J. W., Jones, G. G., and Cohn, M. (1971). Characterization of a BALB/c myeloma library. J. Immunol. 107, 926–927.
Hunter, C. A., and Sibley, L. D. (2012). Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778. doi: 10.1038/nrmicro2858
Khaminets, A., Hunn, J. P., Konen-Waisman, S., Zhao, Y. O., Preukschat, D., Coers, J., et al. (2010). Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell. Microbiol. 12, 939–961. doi: 10.1111/j.1462-5822.2010.01443.x
Khan, I. A., Schwartzman, J. D., Matsuura, T., and Kasper, L. H. (1997). A dichotomous role for nitric oxide during acure Toxoplasma gondii infection in mice. Proc. Natl. Acad. Sci. U.S.A. 94, 13955–13960. doi: 10.1073/pnas.94.25.13955
Lindmark, H., Rosengren, B., Hurt-Camejo, E., and Bruder, C. E. (2004). Gene expression profiling shows that macrophages derived from mouse embryonic stem cells is an improved in vitro model for studies of vascular disease. Exp. Cell Res. 300, 335–344. doi: 10.1016/j.yexcr.2004.06.025
Lowenstein, C. J., and Padalko, E. (2004). iNOS (NOS2) at a glance. J. Cell Sci. 117(Pt 14), 2865–2867. doi: 10.1242/jcs.01166
Luder, C. G., Algner, M., Lang, C., Bleicher, N., and Gross, U. (2003). Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int. J. Parasitol. 33, 833–844. doi: 10.1016/S0020-7519(03)00092-4
MacMicking, J., Xie, Q. W., and Nathan, C. (1997). Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350. doi: 10.1146/annurev.immunol.15.1.323
Mercier, C., Dubremetz, J. F., Rauscher, B., Lecordier, L., Sibley, L. D., and Cesbron-Delauw, M. F. (2002). Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 13, 2397–2409. doi: 10.1091/mbc.E02-01-0021
Olias, P., Etheridge, R. D., Zhang, Y., Holtzman, M. J., and Sibley, L. D. (2016). Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-gamma-dependent gene expression. Cell Host Microbe 20, 72–82. doi: 10.1016/j.chom.2016.06.006
Padrao Jda, C., Cabral, G. R., da Silva Mde, F., Seabra, S. H., and DaMatta, R. A. (2014). Toxoplasma gondii infection of activated J774-A1 macrophages causes inducible nitric oxide synthase degradation by the proteasome pathway. Parasitol. Int. 63, 659–663. doi: 10.1016/j.parint.2014.05.003
Ralph, P., and Nakoinz, I. (1977). Antibody-dependent killing of erythrocyte and tumor targets by macrophage-related cell lines: enhancement by PPD and LPS. J. Immunol. 119, 950–954.
Reese, M. L., Zeiner, G. M., Saeij, J. P., Boothroyd, J. C., and Boyle, J. P. (2011). Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. U.S.A. 108, 9625–9630. doi: 10.1073/pnas.1015980108
Roberts, F., Roberts, C. W., Ferguson, D. J., and McLeod, R. (2000). Inhibition of nitric oxide production exacerbates chronic ocular toxoplasmosis. Parasite Immunol. 22, 1–5. doi: 10.1046/j.1365-3024.2000.00259.x
Saeij, J. P., Boyle, J. P., Coller, S., Taylor, S., Sibley, L. D., Brooke-Powell, E. T., et al. (2006). Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314, 1780–1783. doi: 10.1126/science.1133690
Scharton-Kersten, T. M., Yap, G., Magram, J., and Sher, A. (1997). Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185, 1261–1273. doi: 10.1084/jem.185.7.1261
Seabra, S. H., de Souza, W., and DaMatta, R. A. (2002). Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp. Parasitol. 100, 62–70. doi: 10.1006/expr.2001.4675
Seabra, S. H., de Souza, W., and DaMatta, R. A. (2004). Toxoplasma gondii exposes phosphatidylserine inducing a TGF-beta1 autocrine effect orchestrating macrophage evasion. Biochem. Biophys. Res. Commun. 324, 744–752. doi: 10.1016/j.bbrc.2004.09.114
Sibley, L. D. (2011). Invasion and intracellular survival by protozoan parasites. Immunol. Rev. 240, 72–91. doi: 10.1111/j.1600-065X.2010.00990.x
Sibley, L. D., Adams, L. B., Fukutomi, Y., and Krahenbuhl, J. L. (1991). Tumor necrosis factor-alpha triggers antitoxoplasmal activity of IFN-gamma primed macrophages. J. Immunol. 147, 2340–2345.
Steinfeldt, T., Konen-Waisman, S., Tong, L., Pawlowski, N., Lamkemeyer, T., Sibley, L. D., et al. (2010). Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 8:e1000576. doi: 10.1371/journal.pbio.1000576
Stuehr, D. J., Cho, H. J., Kwon, N. S., Weise, M. F., and Nathan, C. F. (1991). Purification and characterization of the cytokine-induced macrophage nitric oxide synthase: an FAD- and FMN-containing flavoprotein. Proc. Natl. Acad. Sci. U.S.A. 88, 7773–7777. doi: 10.1073/pnas.88.17.7773
Taylor, S., Barragan, A., Su, C., Fux, B., Fentress, S. J., Tang, K., et al. (2006). A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780. doi: 10.1126/science.1133643
Tenter, A. M., Heckeroth, A. R., and Weiss, L. M. (2000). Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258. doi: 10.1016/S0020-7519(00)00124-7
Xie, Q. W., Cho, H., Kashiwabara, Y., Baum, M., Weidner, J. R., Elliston, K., et al. (1994). Carboxyl terminus of inducible nitric oxide synthase. Contribution to NADPH binding and enzymatic activity. J. Biol. Chem. 269, 28500–28505.
Keywords: Toxoplasma gondii, virulence factors, macrophages, inducible nitric oxide synthase, nitric oxide
Citation: Cabral GRA, Wang ZT, Sibley LD and DaMatta RA (2018) Inhibition of Nitric Oxide Production in Activated Macrophages Caused by Toxoplasma gondii Infection Occurs by Distinct Mechanisms in Different Mouse Macrophage Cell Lines. Front. Microbiol. 9:1936. doi: 10.3389/fmicb.2018.01936
Received: 08 June 2018; Accepted: 31 July 2018;
Published: 20 August 2018.
Edited by:
Celio Geraldo Freire-de-Lima, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Shuai Wang, Xinxiang Medical University, ChinaCopyright © 2018 Cabral, Wang, Sibley and DaMatta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: L. D. Sibley, c2libGV5QHd1c3RsLmVkdQ== Renato A. DaMatta, cmVuYXRvQHVlbmYuYnI=
†Present address: Zi T. Wang, Customer Education and Training, MilliporeSigma, St. Louis, MO, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.