
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 August 2018
Sec. Food Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01786
 Anja Brickwedde†
Anja Brickwedde† Nick Brouwers†
Nick Brouwers† Marcel van den Broek
Marcel van den Broek Joan S. Gallego Murillo
Joan S. Gallego Murillo Julie L. Fraiture
Julie L. Fraiture Jack T. Pronk
Jack T. Pronk Jean-Marc G. Daran*
Jean-Marc G. Daran*Saccharomyces pastorianus lager brewing yeasts are domesticated hybrids of Saccharomyces cerevisiae and cold-tolerant Saccharomyces eubayanus. To understand the contribution of both parental genomes to maltose metabolism in brewing wort, this study focuses on maltose transport in the S. eubayanus type strain CBS 12357T/FM1318. To obtain complete sequences of the MAL loci of this strain, a near-complete genome assembly was generated using the Oxford Nanopore Technology MinION sequencing platform. Except for CHRXII, all sixteen chromosomes were assembled as single contigs. Four loci harboring putative maltose transporter genes (SeMALT1-4), located in subtelomeric regions of CHRII, CHRV, CHRXIII, and CHRXVI, were completely resolved. The near-identical loci on CHRV and CHRXVI strongly resembled canonical S. cerevisiae MAL loci, while those on CHRII and CHRXIII showed different structures suggestive of gene loss. Overexpression of SeMALT1-4 in a maltose-transport-deficient S. cerevisiae strain restored growth on maltose, but not on maltotriose, indicating maltose-specific transport functionality of all four transporters. Simultaneous CRISPR-Cas9-assisted deletion of only SeMALT2 and SeMALT4, which shared 99.7% sequence identity, eliminated growth of S. eubayanus CBS 12357T on maltose. Transcriptome analysis of S. eubayanus CBS 12357T established that SeMALT1 and SeMALT3, are poorly expressed in maltose-grown cultures, while SeMALT2 and SeMALT4 were expressed at much higher levels than SeMALT1 and SeMALT3, indicating that only SeMALT2/4 are responsible for maltose consumption in CBS 12357T. These results represent a first genomic and physiological characterization of maltose transport in S. eubayanus CBS 12357T and provides a valuable resource for further industrial exploitation of this yeast.
Saccharomyces eubayanus was first isolated from Nothofagus trees and stromata of Cyttaria harioti in North-Western Patagonia (Libkind et al., 2011). Strains of S. eubayanus have subsequently been also isolated from locations in North America (Peris et al., 2014), Asia (Bing et al., 2014), and Oceania (Gayevskiy and Goddard, 2016). Initial physiological characterization of the Patagonian S. eubayanus strain CBS 12357T revealed that it grows faster than S. cerevisiae at temperatures below 10°C (Hebly et al., 2015), shows poor flocculation (Krogerus et al., 2015), and consumes maltose but not maltotriose (Hebly et al., 2015; Gibson et al., 2017).
Isolation and characterization of S. eubayanus provided a strong impetus for research on S. pastorianus lager brewing yeasts. The hybrid nature of lager yeast genomes was already shown by Southern hybridization (Tamai et al., 1998; Yamagishi et al., 1999); RFLP genotyping, Sanger sequencing (Casaregola et al., 2001; Rainieri et al., 2006), and comparative proteomics (Joubert et al., 2000; Caesar et al., 2007) However, release of the first S. eubayanus genome sequence (Libkind et al., 2011) unequivocally established that this cold-tolerant Saccharomyces species contributed the non-cerevisiae part of S. pastorianus genomes (Nakao et al., 2009; Hewitt et al., 2014; Walther et al., 2014; van den Broek et al., 2015) Access to this genome sequence and its updates (Baker et al., 2015; Hebly et al., 2015) proved invaluable for resolving the complex structure of aneuploid S. pastorianus genomes. Moreover, access to S. eubayanus strains stimulated vigorous research into de novo generation of hybrids between S. cerevisiae and S. eubayanus in the laboratory (Steensels et al., 2014; Hebly et al., 2015; Krogerus et al., 2015, 2017a; Magalhães et al., 2017) This approach has the potential to increase our understanding of the domestication process of lager brewing strains and, moreover, to strongly increase the genetic and phenotypic variety of lager yeast strains available to the brewing industry. De novo constructed S. cerevisiae × S. eubayanus hybrids have been demonstrated to combine advantageous brewing-related properties of both parents (cryo-tolerance, maltotriose utilization, and strong flocculation) and even exhibited best parent heterosis also referred to as hybrid vigor (Steensels et al., 2014; Hebly et al., 2015; Krogerus et al., 2017a,b; Peris et al., 2017). However, generation of new hybrids is, by itself, not sufficient to understand the genetic basis for the exceptional performance of S. pastorianus under brewing conditions.
Lager brewing strains of S. pastorianus have, over several centuries, been selected for rapid, near-complete fermentation of all-malt brewer's wort fermentable sugars, which typically comprise 60% maltose, 25% maltotriose, and 15% glucose, with trace amounts of fructose (Zastrow et al., 2001). Lager brewing therefore critically depends on the capacity of S. pastorianus strains to efficiently take up and ferment the wort α-glucosides maltose and maltotriose. The required maltose fermentation characteristics of S. pastorianus strains are conferred by genes originating from each of the parents and from a set that likely arose during its domestication history (e.g., MTT1) (Chow et al., 1989; Dietvorst et al., 2005; Salema-Oom et al., 2005; Vidgren et al., 2005; Alves et al., 2008; Vidgren and Londesborough, 2012; Cousseau et al., 2013; Magalhães et al., 2016).
In S. cerevisiae, maltose metabolism and the responsible MAL genes are well characterized in term of sequence, genetics, regulation and biochemistry. S. cerevisiae MAL loci harbor the three key genes essential for maltose utilization, encoding a transcriptional activator (MALx3), a maltose permease (MALx1) and a maltase (MAx2) (Charron et al., 1989). Numbers and identities of MAL loci are highly strain dependent, with up to five MAL loci (MAL1, 2, 3, 4, and 6) occurring in haploid S. cerevisiae genomes. MAL loci are typically located in subtelomeric regions, with the structurally identical MAL1, 2, 3, 4, and 6 located near telomeres of CHRVII, III, II, XI, and VIII, respectively (Cohen et al., 1985; Dubin et al., 1985; Charron et al., 1989; Chow et al., 1989; Michels et al., 1992; Han et al., 1995; Day et al., 2002b).
Maltose is transported across the S. cerevisiae plasma membrane by maltose-proton symport, mediated by Malx1 transporters (Serrano, 1977; van Leeuwen et al., 1992) and, to a lesser extent, by facilitated diffusion (Day et al., 2002a). All MALx1 genes are highly similar, with the exception of MAL11 and its allele AGT1, whose DNA sequence shows only 57% identity to the other four MALx1 transporter genes (Han et al., 1995; Vidgren et al., 2009). This sequence difference is accompanied by a difference in substrate range, with Agt1 also being able to transport other α-glucosides, such as trehalose (Plourde-Owobi et al., 1999), sucrose (Basso et al., 2011; Marques et al., 2017) and, importantly for brewing applications, maltotriose (Alves et al., 2008; Vidgren et al., 2009). The S. cerevisiae genome harbors two additional maltose permease genes, MPH2 and MPH3, which are located subtelomerically on CHRIV and X, respectively. Although the transport mechanisms of Mph2 and Mph3 have not been experimentally established, both carriers were shown to transport of a range of substrates including glucose, maltose, maltotriose, α-methylglucoside, and turanose (Day et al., 2002a).
In contrast to the wealth of information on S. cerevisiae, knowledge on maltose transport in S. eubayanus is limited. The type strain S. eubayanus CBS 12357T grows on maltose, but not on maltotriose (Hebly et al., 2015). Annotation of its genome sequence revealed four open reading frames sharing similarity with S. cerevisiae MAL31 (SeMALT1; SeMALT2, SeMALT3, and SeMALT4) (Baker et al., 2015). The hybrid S. pastorianus genome harbors two additional maltose transporter gene variants that were not found in either of the reference parental genomes. The first of these, MTT1/MTY1, shares 90 and 54% DNA sequence identity with S. cerevisiae MAL31 and MAL11, respectively (Dietvorst et al., 2005; Salema-Oom et al., 2005). The second S. pastorianus-specific maltose-transporter gene, SeAGT1, shared significant identity with S. cerevisiae AGT1 (85% ScAGT1; Vidgren and Londesborough, 2012). SeAGT1 was unexpectedly found to be located on the S. eubayanus-derived CHRVIII-XV, suggesting a S. eubayanus origin, despite the absence of similar genes in currently available S. eubayanus genome sequences (Nakao et al., 2009; van den Broek et al., 2015) However, genome assembly of an Asian S. eubayanus strain (CDFM21L.1; Bing et al., 2014) revealed short (<200 bp) sequences reminiscent of a putative SeAGT1 gene (Hebly et al., 2015). Both MTT1/MTY1 and SeAGT1 were shown to confer low- temperature dependent transport of both maltose and maltotriose (Vidgren et al., 2014). To illustrate the complexity of α-glucoside transport in S. pastorianus, the model strain Weihenstephan 34/70 contains all S. cerevisiae MAL loci except for MAL2, a single MPH gene (MPH2), as well as all four S. eubayanus genes (MALT1 to 4) (Nakao et al., 2009; van den Broek et al., 2015) and the two S. pastorianus-specific genes MTT1 and SeAGT1 (Magalhães et al., 2016). In this S. pastorianus background, the S. cerevisiae allele of AGT1 (MAL11) carries a nonsense mutation (Vidgren et al., 2009).
Hitherto, no study has systematically investigated the functionality of the individual α-glucoside transporters in S. pastorianus. In addition to the complexity of maltose metabolism in S. pastorianus strains, genetic analysis is complicated by the limited genetic accessibility of industrial lager brewing yeasts (Bolat et al., 2013). However, availability of the S. eubayanus type strain and of its genome sequence offers an alternative approach to fill existing knowledge gaps on transport of wort sugars. The aim of this study was therefore to investigate the contribution of individual putative maltose-transporter (SeMALT) genes in S. eubayanus CBS 12357T. To this end, a new near-complete genome sequence of the strain CBS 12357T was assembled using Oxford Nanopore Technology's MinION long-read sequencing platform. Subsequently, CRISPR-Cas9 gene editing was used to systematically delete the MALT genes in S. eubayanus. In a complementary approach, all four S. eubayanus MALT open reading frames were cloned and constitutively expressed alongside the S. cerevisiae MAL12 maltase gene in a S. cerevisiae strain lacking all maltose utilization genes (MALx1, MALx2, and MALx3), MPH1/2, SUC2, and IMA1-5 genes; (Marques et al., 2018). Subsequently, growth of the genetically modified yeast strains was analyzed on different carbon sources. Finally, RNA sequencing was performed on glucose- and maltose-grown cultures to study differential expression of the S. eubayanus MALT genes.
S. eubayanus strain CBS 12357T (alias FM1318, Libkind et al., 2011) was obtained from the Westerdijk Fungal Biodiversity Institute (Utrecht, the Netherlands, http://www.westerdijkinstitute.nl/). The S. cerevisiae strain IMZ616 (Marques et al., 2018) was derived from the CEN.PK lineage (Entian and Kötter, 2007; Salazar et al., 2017). All strains used in this study are listed in Table 1. Stock cultures of S. eubayanus and S. cerevisiae strains were grown in YPD (10 g L−1 yeast extract, 20 g L−1 peptone, and 20 g L−1 glucose) until late exponential phase, complemented with sterile glycerol to a final concentration of 30% (v/v) and stored at −80°C as 1.5 ml aliquots until further use.
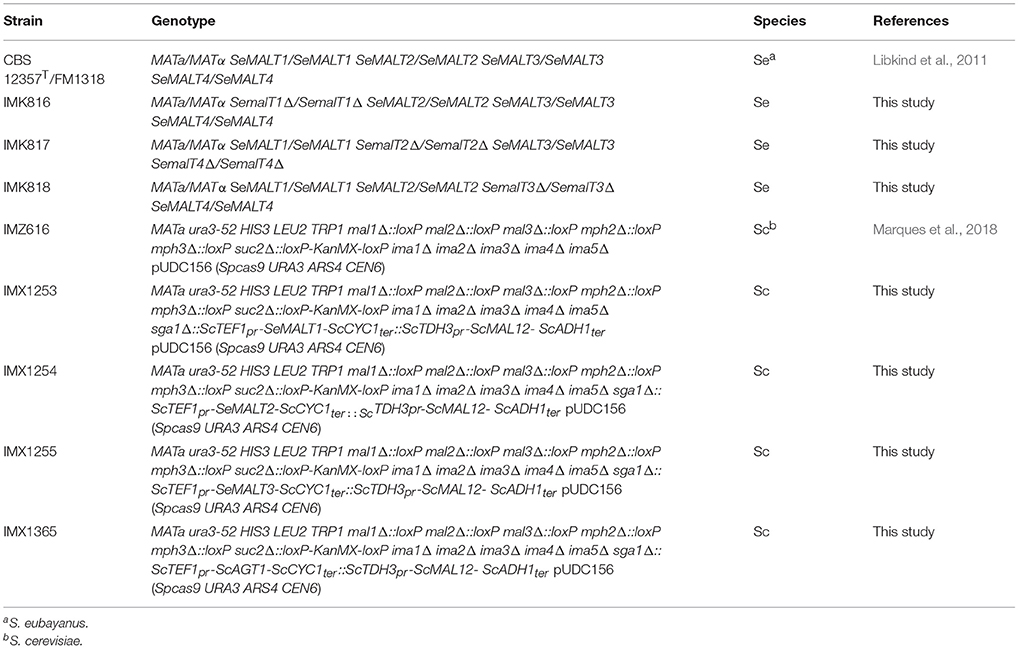
Table 1. Strains used in this study.
S. eubayanus batch cultures were grown on synthetic medium (SM) containing 3.0 g L−1 KH2PO4, 5.0 g L−1 (NH4)2SO4, 0.5 g L−1 MgSO4, 7 H2O, 1 mL L−1 trace element solution, and 1 mL L−1 vitamin solution (Verduyn et al., 1992). The pH was set to 6 with 2 M KOH prior to autoclaving at 120°C for 20 min. Vitamin solutions (Verduyn et al., 1992) were sterilized by filtration and added to the sterile medium. Concentrated sugar solutions were autoclaved at 110°C for 20 min and added to the sterile flasks to give a final concentration of 20 g L−1 carbon source [glucose (SMG), maltose (SMM) or maltotriose (SMMt)]. S. cerevisiae batch cultures were grown on SM supplemented with 150 mg L−1 uracil to compensate for loss of plasmid pUDC156 that carried the cas9 endonuclease gene, and supplemented with 20 g L−1 carbon source [glucose (SMuG), maltose (SMuM) or maltotriose (SMuMt)]. All batch cultures were grown in 500 mL shake flasks with a working volume of 100 mL. The cultures were inoculated at an initial OD660nm of 0.1 and incubated under an air atmosphere and shaken at 200 rpm and at 20°C in a New Brunswick™ Innova44 incubator (Eppendorf Nederland B.V, Nijmegen, The Netherlands).
Selection of the S. eubayanus strains transformed with plasmids pUDP062 (gRNASeMALT1), pUDP063 (gRNASeMALT2), and pUDP064 (gRNASeMALT3) was carried out on a modified SMG medium, in which (NH4)2SO4 was replaced by 5 g.L−1 K2SO4 and 10 mM acetamide (SMAceG) (Solis-Escalante et al., 2013). SM- based solid medium contained 2% Bacto Agar (BD, Franklin Lakes, NJ). Selection of S. cerevisiae integration strains was carried out on SMAceG. For plasmid propagation, E. coli XL1-Blue-derived strains (Agilent Technologies, Santa Clara, CA) were grown in Lysogeny Broth medium (LB, 10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl) supplied with 100 mg L−1 ampicillin.
Guide-RNA (gRNA) sequences for deletion of SeMALT1, SeMALT2/T4 and SeMALT3 were designed following the guiding principles recommended in Gorter de Vries et al. (2017). The DNA sequences encoding these gRNAs were synthesized at GeneArt (Thermo Fisher Scientific, Waltham, MA) and were delivered in pUD631, pUD632, and pUD633 respectively (Table 2). The gRNA spacer sequences (SeMALT1 5′ATTCCAAACGACAATAAAGA3′, SeMALT2/T4 5′-TACAGGAGAATGGGAGATTT-3′ and SeMALT3 5′- GTTTTCAAAGCTTGCAGAAG-3′) and the structural gRNA sequence were flanked at their 5′ ends by the Hammerhead ribozyme (HH) and at their 3′ ends by the Hepatitis Delta Virus ribozyme (HDV) (Gao and Zhao, 2014). The HH-gRNA-HDV fragment was flanked on both ends with a BsaI site for further cloning (Gorter de Vries et al., 2017; Juergens et al., 2018). In the next step, the gRNAs were transferred into the pUDP004 plasmid (Gorter de Vries et al., 2017), which enables combined expression of the gRNA cassette and of Spcas9D147YP411T (Bao et al., 2015). The plasmid pUDP062, expressing gRNASeMALT1 was constructed in a one-pot reaction by digesting pUDP004 and pUD631 using BsaI and ligating with T4 ligase. Similarly, pUDP063, expressing gRNASeMAT2/T4 and Spcas9D147YP411T was assembled from pUDP004 and pUD632. The plasmid pUDP064 expressing gRNASeMALT3 and Spcas9D147YP411T was assembled from pUDP004 and pUD633. Correct assembly of pUDP062-064 was verified by restriction analysis with SspI and PdmI.

Table 2. Plasmids used in this study.
The coding regions of SeMALT1, SeMALT2 and SeMALT3 were amplified from CBS 12357T genomic DNA with Phusion High-Fidelity DNA polymerase (ThermoFisher Scientific), according to the supplier's instructions with primers pairs 10491/10492, 10632/10633, and 10671/10672 (Table S5), respectively. The coding sequence of ScAGT1 was amplified from CEN.PK113-7D genomic DNA with Phusion High-Fidelity DNA polymerase (ThermoFisher Scientific), according to the supplier's instructions with primers pairs 9940/9941. Each primer carried a 40 bp extension complementary to the plasmid backbone of p426-TEF-amdS (Solis-Escalante et al., 2013; Marques et al., 2018), which was PCR amplified using Phusion High-Fidelity DNA polymerase (ThermoFisher Scientific) and primers 7812 and 5921 (Table S5). p426-TEF-amdS is an expression plasmid that harbors the promoter of the translational elongation factor EF-1 alpha (TEF1) of S. cerevisiae. Each SeMALT fragment was assembled with the p426-TEF-amdS backbone fragment using NEBuilder HiFi DNA Assembly (New England Biolabs, Ipswich, MA), resulting in plasmids pUD479 (SeMALT1), pUD480 (SeMALT2/T4), pUD481 (SeMALT3), and pUD445 (ScAGT1) (Table 2).
S. eubayanus IMK816 (SemalT1Δ) was constructed by transforming CBS 12357T by electroporation (Gorter de Vries et al., 2017) with 200 ng of pUDP062 and 1 μg of 120 bp repair fragment obtained by mixing an equimolar amount of primers 11850 and 11851 (Table S5) (Mans et al., 2015) (Figure 1). As control, the same transformation was performed without including the repair DNA fragment. Transformants were selected on SMAceG plates. Strain IMK817 (SemalT2Δ SemalT4Δ) and IMK818 (SemalT3Δ) were constructed in the same way. The SeMALT2/T4 deletion was constructed by co-transforming pUDP063 and a repair DNA fragment formed by primers 11328 and 11329, while the SeMALT3 deletion involved pUDP064 and a repair DNA formed by primers 11330 and 11331 (Table S5). Deletion of SeMALT1, SeMALT2/T4, and SeMALT3 was verified by diagnostic PCR, using primers pairs 11671/11672, 11673/11674, and 11675/11676 (Table S5), respectively (Figure 1C). Prior to storing at −80°C, transformants were successively streaked on SMAceG and YPD plates. The genotype was verified after each plating round with the primers pairs mentioned above.
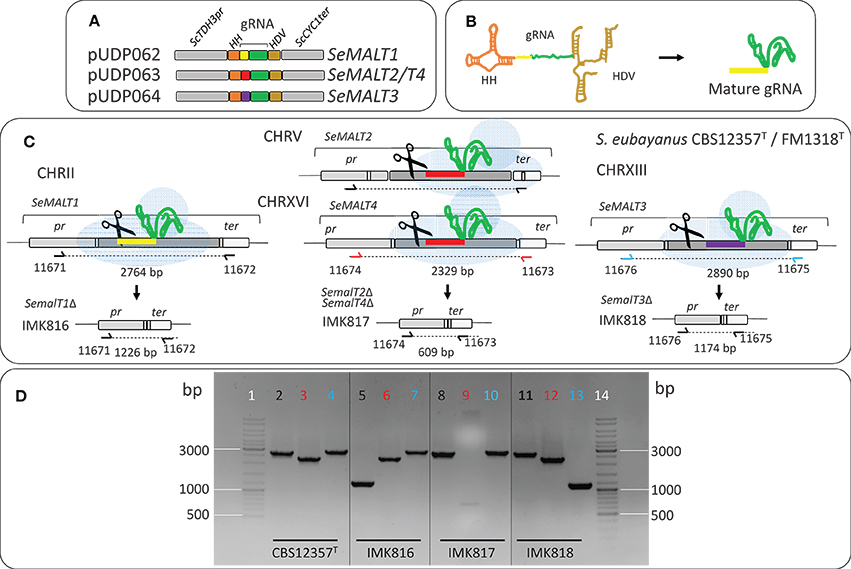
Figure 1. Deletion of SeMALT genes using CRISPR-Cas9-assisted genome editing in S. eubayanus CBS 12357T. (A) Representation of the gRNA expression cassette in pUDP062, pUDP063, and pUDP064. gRNAs targeting either SeMALT1, SeMALT2/T4 or SeMALT3 were flanked by a 5′ hammerhead ribozyme (HH, orange) and a 3′ hepatitis-δ virus ribozyme (HD, bronze). These constructs were expressed from the RNA polymerase II ScTDH3 promoter and the ScCYC1 terminator. (B) Upon ribozyme self-cleavage, a mature gRNA comprising the SeMALT guiding spacer (yellow) and the constant structural gRNA fragment (green) is released. (C) Schematic representation of SeMALT gene editing upon transformation of pUDP062 or pUDP063 or pUDP064 into S. eubayanus CBS 12357T. Primers used for verification of transformants from transformation are indicated together with the size of the expected PCR products. (D) Validation of transformants derived from transformations of S. eubayanus CBS 12357T with either pUDP062, pUDP063 or pUDP064 in presence of the corresponding 120 bp repair DNA fragments. Lanes 1 and 14 GeneRuler DNA Ladder Mix (Thermo Fischer Scientific). Lanes 2, 5, 8, and 11 fragments amplified with primers 11671 and 11672 black label). Lanes 3, 6, 9, 12 fragments amplified with primers 11674 and 11673 (red label). Lanes 4, 7, 10, and 13 fragments amplified with primers 11676 and 11675 (blue label) from genomic DNA from CBS 12357T (Lanes 2, 3, and 4), from IMK816 (SemalT1Δ) (Lanes 5, 6, and 7), from IMK817 (SemalT2Δ/ SemalT4Δ) (Lanes 8, 9, and 10), and from IMK818 (SemalT3Δ) (Lanes 11, 12, and 13).
S. cerevisiae IMZ616 [mal1Δ mal2Δ mal3Δ mph2Δ mph3Δ suc2Δ ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ pUDC156 (Spcas9 URA3 ARS4 CEN6)], which cannot grow on α-glucosides (Marques et al., 2018) was used as a host to test the functionality of individual S. eubayanus (putative) maltose transporter genes. S. cerevisiae IMX1253 was constructed by integrating the S. cerevisiae maltase gene ScMAL12 and the SeMALT1 transporter gene at the ScSGA1 locus of strain IMZ616 (Figure 2). The ScSGA1 gene encodes an intracellular sporulation-specific glucoamylase (Yamashita and Fukui, 1985) that is not expressed during vegetative growth (Knijnenburg et al., 2009). This integration site was shown suitable for expression of single or multiple genes as previously demonstrated in Mans et al. (2015), Kuijpers et al. (2016), Verhoeven et al. (2017), and Bracher et al. (2018) The fragment containing ScMAL12 was PCR amplified using Phusion High-Fidelity DNA polymerase (Thermo FisherScientific) from pUDE044 (Basso et al., 2011) with primers 9596 and 9355, which included a 5′ extension homologous to the upstream region of the S. cerevisiae SGA1 locus and an extension homologous to the co-transformed transporter fragment, respectively. The DNA fragment carrying the S. eubayanus SeMALT1 maltose symporter was PCR amplified from pUD479 using primers 9036 and 9039, which included a 5′ extension homologous to the co-transformed transporter fragment and an extension homologous to the downstream region of the S. cerevisiae SGA1 locus, respectively. To facilitate integration in strain IMZ616, the two PCR fragments were co-transformed with plasmid pUDR119, which expressed a gRNA targeting ScSGA1 (spacer sequence: 5′-ATTGACCACTGGAATTCTTC-3′) (van Rossum et al., 2016) (Figure 2A). The plasmid and repair fragments were transformed using the LiAc protocol (Gietz and Schiestl, 2007) and transformed cells were plated on SMAceG. Correct integration was verified by diagnostic PCR with primers pairs 4226/5043 and 942/4224 (Figure 2, Table S5). Strains S. cerevisiae IMX1254, IMX1255, and IMX1365 were constructed following the same principle, but instead of using pUD479 to generate the transporter fragment, pUD480 pUD481 and pUD445 were used to PCR amplify SeMALT2/T4, SeMALT3, and ScAGT1 respectively. Correct integration was verified by diagnostic PCR with primers pairs 4226/5043 and 942/4224 (Figure 2, Table S5). All PCR-amplified gene sequences were Sanger sequenced (Baseclear, Leiden, The Netherlands).
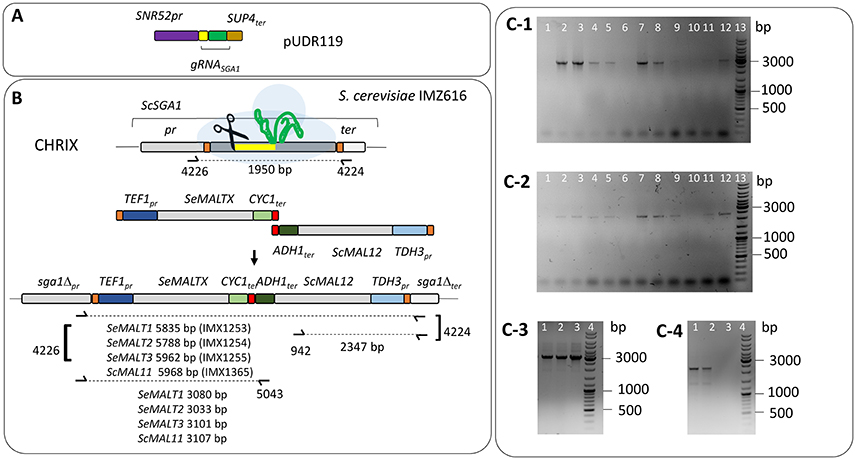
Figure 2. Integration of S. eubayanus CBS 12357T maltose transporter genes at the ScSGA1 locus of S. cerevisiae IMZ616 (mal1Δ mal2Δ mal3Δ mph2Δ mph3Δ::suc2Δ ima1Δ ima2Δ ima3Δ ima4Δ ima5Δ Spcas9) (Marques et al., 2018). (A) Integration at the ScSGA1 locus by Cas9-assisted genome editing. The Cas9-targeting gRNA was expressed from pUDR119 (van Rossum et al., 2016). (B) Schematic representation of the integration of SeMALT expression cassettes at the ScSGA1 locus. Upon cleavage, the Cas9-induced double strand break was repaired by the two co-transformed fragments harboring a transporter gene expression cassette and the S. cerevisiae maltase gene MAL12, respectively. Primers used for verification of transformants from transformation are indicated together with the size of the expected PCR products. Integration of SeMALT1, SeMALT2, SeMALT3 or ScMAL11 resulted in S. cerevisiae strains IMX1253, IMX1254, IMX1255, and IMX1365 respectively. (C) Validation of the S. cerevisiae IMX1253, IMX1254, IMX1255, and IMX1365. (C-1) Lane 13 GeneRuler DNA Ladder Mix (Thermo Fischer Scientific). Lanes 1 to 12 fragments amplified with primers 4226 and 5043. Lanes 1, 2, 3, and 4 fragments amplified from clones transformed with SeMALT1. The strain corresponding to lane 3 was renamed IMX1253. Lanes 5, 6, 7, and 8 fragments amplified from clones transformed with SeMALT2. The strain corresponding to lane 7 was renamed IMX1254. Lanes 9, 10, 11, and 12 fragments amplified from clones transformed with SeMALT3. The strain corresponding to lane 12 was renamed IMX1255. (C-2) Lane 13 GeneRuler DNA Ladder Mix (Thermo Fischer Scientific). Lanes 1 to 12 fragments amplified with primers 942 and 4224. Lanes 1, 2, 3, and 4 fragments amplified from clones transformed with SeMALT1. Lane 3 corresponds to IMX1253. Lanes 5, 6, 7, and 8 fragments amplified from clones transformed with SeMALT2. Lane 7 corresponds to IMX1254. Lanes 9, 10, 11, and 12 fragments amplified from clones transformed with SeMALT3. Lane 12 corresponds to IMX1255. (C-3) Lanes 1 to 3 fragments amplified with primers 4226 and 5043. Lanes 1, 2, 3, and 4 fragments amplified from clones transformed with ScMAL11. The strain corresponding to lane 3 was renamed IMX1365. Lane 4 GeneRuler DNA Ladder Mix (Thermo Fischer Scientific). (C-4) Lanes 1 to 4 fragments amplified with primers 942 and 4224. Lanes 1, 2, and 3 fragments amplified from clones transformed with ScMAL11. Lane 3 corresponds to IMX1365. Lane 4 GeneRuler DNA Ladder Mix (Thermo Fischer Scientific).
Genomic DNA from S. eubayanus CBS 12357T was isolated as previously described in van den Broek et al. (2015). Paired-end sequencing (2-fold 150 bp) was performed on a 350 bp PCR-free insert library using Illumina HiSeq2500 (San Diego, CA) by Novogene (HK) Company Ltd (Hong Kong, China) with a sample size of 3.2 Gbase. Sequence data are available at NCBI under Bioproject accession number PRJNA450912.
For Nanopore sequencing, a 1D sequencing library (SQK-LSK108) was prepared according to the manufacturer's recommendation and loaded onto an FLO-MIN106 (R9.4) flow cell, connected to a MinION Mk1B unit (Oxford Nanopore Technology, Oxford, United Kingdom). MinKNOW software (version 1.5.12; Oxford Nanopore Technology) was used for quality control of active pores and for sequencing. Raw files generated by MinKNOW were base called using Albacore (version 1.1.0; Oxford Nanopore Technology). Reads, in fastq format, with a minimum length of 1,000 bp were extracted, yielding 3.26 Gb of sequence with an average read length of 8.07 kb. Sequencing data are available at NCBI under Bioproject accession number PRJNA450912.
De novo assembly of the Oxford Nanopore MinION dataset was performed using Canu (v1.4, setting: genomesize = 12 m; Koren et al., 2017). Assembly correctness was assessed using Pilon (Walker et al., 2014) and further correction “polishing” of sequencing/assembly errors was performed by aligning Illumina reads with BWA (Li and Durbin, 2010) using correction of only SNPs and short indels (–fix bases parameter). Genome assembly gene annotation was performed with the MAKER2 annotation pipeline (version 2.31.9) (Holt and Yandell, 2011) using SNAP (version 2013–11-29) (Korf, 2004) and Augustus (version 3.2.3) (Stanke et al., 2006) as ab initio gene predictors. S. cerevisiae S288C EST and protein sequences were obtained from SGD (Saccharomyces Genome Database, http://www.yeastgenome.org/) and were aligned using BLASTX on the obtained polished sequence assembly (BLAST version 2.2.28+) (Camacho et al., 2009). Predicted translated protein sequences of the final gene model were aligned to the S. cerevisiae S288C protein Swiss-Prot database using BLASTP (http://www.uniprot.org/). Custom-made Perl scripts were used to map systematic names to the annotated gene names. Error rates in the nanopore-sequencing data were estimated from the q score (Phred scaled) per read, as calculated by the base caller Albacore (version 1.1.0) (Oxford Nanopore Technology). Average q score was used to calculate the error P = 10q/10.
S. eubayanus CBS 12357T was grown in either SMG or SMM until mid-exponential phase (OD660nm of 12.5). Culture samples corresponding to ca. Two Hundred and Forty Milligram of biomass wet weight were directly quenched in liquid nitrogen. The resulting frozen pellet was gently thawed on ice and spun down at 4700 × g for 5 min at 0°C. Pellets were then resuspended in 1.2 mL of ice-cold AE buffer (50 mM sodium acetate and 10 mM EDTA, pH 5.0), followed by addition of 1.2 mL of acid phenol/chloroform/isoamyl alcohol mix and 0.12 mL 10% sodium dodecyl sulfate. The resulting mix was vortexed for 30 s and incubated for 5 min at 65°C. After homogenizing for 30 s by vortexing, 800 μL aliquots were distributed in RNase-free screw-cap tubes (Tai et al., 2005). After centrifugation (15 min at 10,000 × g), the aqueous phase was transferred to a new tube containing 0.4 mL of acid phenol/chloroform. The mix was vortexed for 30 s, centrifuged (15 min at 10,000 × g) and the aqueous phase was again transferred to a new tube. RNA was then ethanol precipitated and re-suspended in RNAse-free water. Prior to cDNA synthesis, purity, concentration, and integrity of the RNA in the samples was assessed with the Nanodrop (Thermo Fisher Scientific), Qubit (Thermo Fisher Scientific), and Tapestation 220 with RNA Screen Tape (Agilent Technologies), respectively, according the manufacturers' recommendations. cDNA libraries were prepared using the TruSeq RNA V2 kit (Illumina) and sequenced on HISeq 2500 (Illumina) at Novogene (HK) Company Ltd (Hong Kong, China).
Libraries with 300 bp insert size were paired end sequenced (150 bp). Duplicate biological samples were processed, generating an average sequence quantity of 23.7 M reads per sample. Reads were aligned to the Oxford Nanopore CBS 12357T reference assembly using a two-pass STAR (Dobin and Gingeras, 2016) procedure. In the first pass, splice junctions were assembled and used to inform the second round of alignments. Introns between 15 and 4,000 bp were allowed, and soft clipping was disabled to prevent low-quality reads from being spuriously aligned. Ambiguously mapped reads were removed from the dataset. Expression level for each transcript were quantified using htseq-count (Anders et al., 2015) in union mode. Fragments per kilo-base of feature (gene) per million reads mapped (FPKM) values were calculated by “Applying the rpkm method” from the edgeR package (Robinson et al., 2010; McCarthy et al., 2012) Differential expression analysis was performed using DESeq (Anders et al., 2013). Transcript data can be retrieve at the Genome Omnibus Database (GEO: https://www.ncbi.nlm.nih.gov/geo/) under accession number: GSE117246.
Optical densities of yeast cultures were measured with a Libra S11 spectrophotometer (Biochrom, Cambridge, UK) at a wavelength of 660 nm. Biomass dry weight was measured by filtering 10-mL culture samples over pre-weighed nitrocellulose filters with a pore size of 0.45 μm. Filters were washed with 10 mL water, dried in a microwave oven (20 min at 350 W) and reweighed. Each measurement was performed in duplicate. For glucose, maltose, maltotriose, and ethanol analysis, culture samples were centrifuged 5 min at 10,000 g and supernatants were analyzed by high-performance liquid chromatography (HPLC) analysis on an Agilent 1260 HPLC equipped with a Bio-Rad HPX 87 H column (Bio-Rad, Hercules, CA). Elution was performed at 65°C with 5 mM H2SO4 at a flow rate of 0.8 mL min−1. Detection was by means of an Agilent refractive-index detector and an Agilent 1260 VWD detector.
Cultures were analyzed on a BD FACSAria™ II SORP Cell Sorter (BD Biosciences, Franklin Lakes, NJ) equipped with 355, 445, 488, 561, and 640 nm lasers and a 70 μm nozzle, and operated with filtered FACSFlow™ (BD Biosciences). Correct cytometer performance was evaluated prior to each experiment by running a CST cycle with corresponding CS&T Beads (BD Biosciences). Drop delay for sorting was determined by running an Auto Drop Delay cycle with Accudrop Beads (BD Biosciences). Morphology of the cells was analyzed by plotting forward scatter (FSC) against side scatter (SSC). Ninety-Six single cells were sorted onto 96-well format Nunc omnitray (Thermo Scientific) plates containing YPD agar using a “single cell” sorting mask, corresponding to a yield mask of 0, a purity mask of 32 and a phase mask of 16. Viability was measured as the average percentage of sorted cells able to form a colony after 48 h incubation at 30°C on three triplicate plates.
Owing to advances in genome sequencing technology, the quality of genome sequence data of S. eubayanus CBS 12357T/FM1318 has gradually improved (Libkind et al., 2011; Baker et al., 2015; Hebly et al., 2015; Okuno et al., 2016) The currently available reference sequence is based on second generation sequencing technology (Illumina generated data), obtained from libraries with different insert sizes that were co-assembled into a 11.66 Mb genome, comprising 144 contigs forming 22 scaffolds. While representing an important resource for research on S. eubayanus and S. pastorianus, this most advanced draft genome sequence is incomplete (Baker et al., 2015). In particular multiple repeated regions, such as subtelomeric regions, are not yet fully resolved due to limitations of short-read sequencing technology. In total, approximately 122 kb of the scaffolded genome remain undefined.
To generate a near-complete, chromosome-level de novo assembly of S. eubayanus CBS 12357T, we used long-read sequencing with third-generation single-molecule technology (Oxford Nanopore Technology MinION platform). A single flow cell was used to generate 3.3 Gb of sequence reads, with an average read length of 8 kb and an estimated average error rate of 9.6%. These data represented a genome coverage of 135 fold of the estimated diploid genome size (24 Mb). An assembly exclusively based on the MinION reads was generated with the Canu program (Koren et al., 2017). This assembly yielded 19 contigs, which is 8-fold fewer than obtained in the short-read-only assembly of the latest CBS 12357T draft genome (Baker et al., 2015). In the MinION-based assembly, the mitochondrial genome and all chromosomes except for CHRXII were assembled as single contigs. CHRXII was manually reconstructed by joining three contigs, with a 1,000 N residues gaps introduced between the contigs. The sequence discontinuity was caused by the inability of the assembly software to handle the highly repetitive DNA organization of the rDNA locus (Figure 3). This approach yielded a nearly complete 11.9 Mb genome assembly.
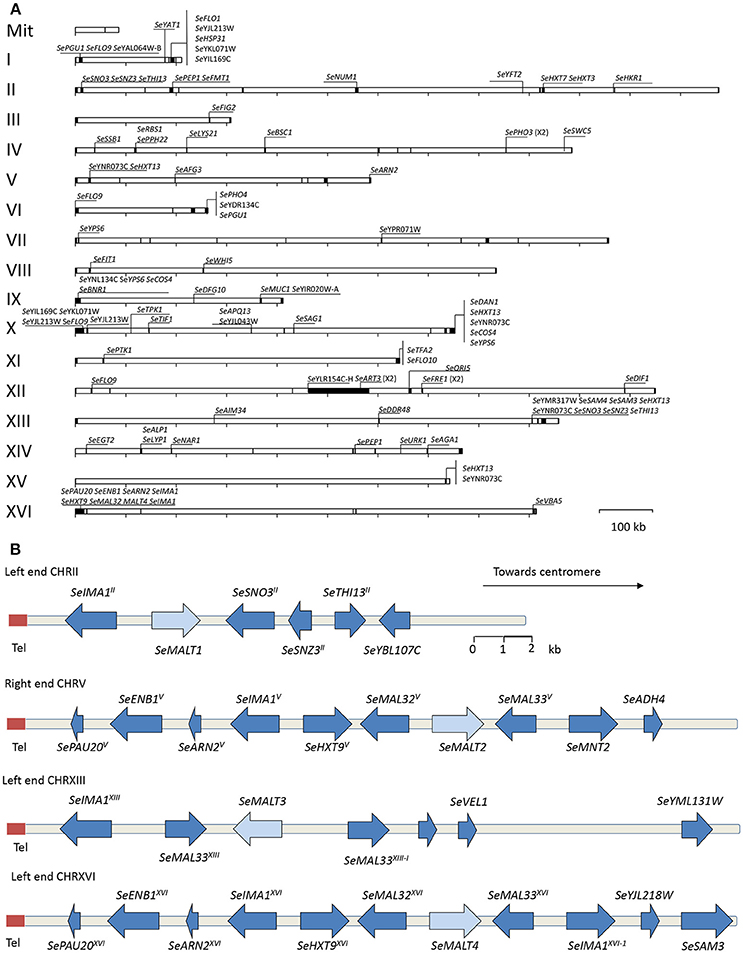
Figure 3. High-quality S. eubayanus CBS12357T genome assembly with 330 kb of unexplored sequence including four MAL loci. (A) Representation of the assembled S. eubayanus chromosomes, the black boxes denote newly added sequences. New annotated open reading frames and gene entries modified relative to the earlier draft genome (Baker et al., 2015) often leading to a redefinition of start and stop codons. (B) Organization of subtelomeric regions including maltose metabolism genes. Arrows denote the direction of transcription. The label “Tel” indicates the position of the telomere. The SeMALT genes are indicated in light blue. The gene and interval sizes are approximately to scale.
Prior to annotation, the assembly based on MinION sequencing data was “polished” with additional Illumina sequencing data using Pilon (Walker et al., 2014). In fine this polished genome assembly included 330 kb of sequence which were not assembled in the previous genome assembly. With the exception of a region on chromosome XII that corresponded to a partially reconstructed rDNA locus, additional sequences were mainly located in the subtelomeric regions (Figure 3A). A total of 5,444 ORFs were annotated, including 41 previously unassembled ORFs (Baker et al., 2015). Additionally, 60 ORFs were modified relative to the earlier draft genome, often leading to a redefinition of start and stop codons (Figure 3A, Table S6). For 65 of the 101 new genes, a paralog or an identical copy of the gene had already been assembled at a different location in the previous assembly. Gene ontology analysis using Fischer's Exact Test revealed an overrepresentation among the new genes of three GO categories related to sugar transport and cell wall (Table 3). This is not that surprising since, subtelomeres are acknowledged to be the major chromosomal regions involved functional evolution as they are sites for large rearrangement in term of structure, gene content and copy number (not only gain but also loss-of-function variants; Brown et al., 2010; Bergström et al., 2014).
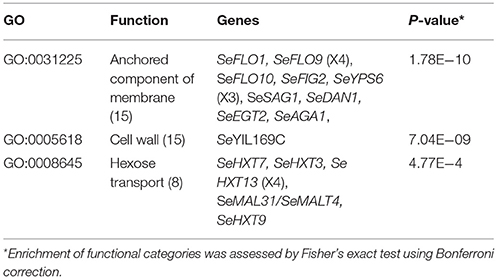
Table 3. Overrepresented GO functional categories among 101 newly identified genes in a MinION-sequencing based S. eubayanus CBS 12357 genome assembly annotation.
Four subtelomeric regions harbored complete sequences of putative maltose transporters. Two of these, which contained SeMALT1 and SeMALT3, showed structural features that differed from those of canonical S. cerevisiae MAL loci. The CHRII locus only contained a transporter gene (SeMALT1) while the CHRXIII “SeMAL locus” consisted of a transporter gene (SeMALT3) flanked by two non-identical genes that strongly resembled the S. cerevisiae regulator genes MAL33 and MAL63 (Figure 3B). In contrast, the S. eubayanus MAL loci on CHRV and CHRXVI showed the same organization as the well described S. cerevisiae MAL loci. Starting from their telomeric ends, they contained a maltase gene (SeMAL32), followed by the transporter gene (SeMALT2 on CHRV and SeMALT4 on CHRXVI), which shared a bi-directional promoter with the maltase gene, and a MAL regulator gene (Figure 3B, Figure S8). Similarity between the right-arm CHRV and left-arm CHRXVI subtelomeric regions extended beyond the SeMAL genes, with a sequence identity of 94% and shared gene synteny over a 20 kb region (Figure 3B). The fully assembled SeMALT4 gene shared 99.7% identity with SeMALT2, from which it differed by only five nucleotides. None of these five nucleotide variations affected the predicted amino acid sequence of the encoded transporters.
To explore the contribution of the four S. eubayanus MALT genes to maltose consumption, deletion strains were constructed. Because the high sequence similarity of SeMALT2 and SeMALT4 complicated individual deletion of these genes, three strains were constructed with either a single deletion of SeMALT1 or SeMALT3 or a double deletion of SeMALT2 and SeMALT4 (Figure 1). The option offered by CRISPR-Cas9 to simultaneously delete of multiple gene copies in a single transformation step (Mans et al., 2015) is especially helpful in diploid strains such as S. eubayanus CBS 12357T. To explore the use of this methodology in S. eubayanus, we used a broad-host-range yeast plasmid for co-expression of Spcas9 and a cassette encoding a ribozyme-flanked gRNA, which was successfully used in the Saccharomycotina yeasts S. pastorianus (Gorter de Vries et al., 2017), Kluyveromyces sp. and Ogataea sp. (Juergens et al., 2018). Cloning of specific gRNA cassettes targeting SeMALT1, SeMALT2/T4, and SeMALT3 in pUDP002 resulted in pUDP062, pUDP063, and pUDP064, respectively. These plasmids were then transformed into S. eubayanus CBS 12357T, either alone or in combination with a 120-bp double stranded repair DNA fragment for the targeted SeMALT gene (Figure 1). In the absence of a repair fragment, transformation with a gRNA-expressing construct was expected to be fatal if both gene copies were cut, unless both breaks were repaired by non-homologous end joining (NHEJ) of the induced double strand breaks. However, transformation of S. eubayanus CBS 12357T with pUDP062, pUDP063 or pUDP064 alone yielded 1100, 128, and 9 transformants, respectively. These numbers were not substantially different from those observed upon co-transformation of the corresponding repair fragments (3000, 114, and 13 colonies, respectively). Based on a set of 30 transformants, genome editing with the gRNASeMALT1 yielded the lowest frequency of transformants in which both gene copies were deleted (3%). The SeMALT3 gRNA performed better with a 7% frequency out of 13 transformants tested, while the gRNA targeting SeMALT2/T4 showed an efficiency of 40% of accurate deletion of both copies of the two genes out of a set of eight transformants.
The resulting S. eubayanus deletion strains IMK816 (SemalT1Δ), IMK817 (SemalT2Δ SemalT4Δ), and IMK818 (SemalT3Δ), as well as the wild-type strain CBS 12357T, were grown in SMG and SMM media. While specific growth rates of all four strains in SMG were the same (0.22 h−1 at 20°C, Table 4), strain IMK187 (SemalT2Δ SemalT4Δ) did not grow on maltose (Figure 4). Conversely, strains IMK816 and IMK818 exhibited the same specific growth rate on maltose as the reference strain (0.17 h−1 at 20°C, Table 4). These data suggested that only SeMALT2 and/or SeMALT4 only contributed to growth on maltose of wild- type S. eubayanus CBS 12357T.
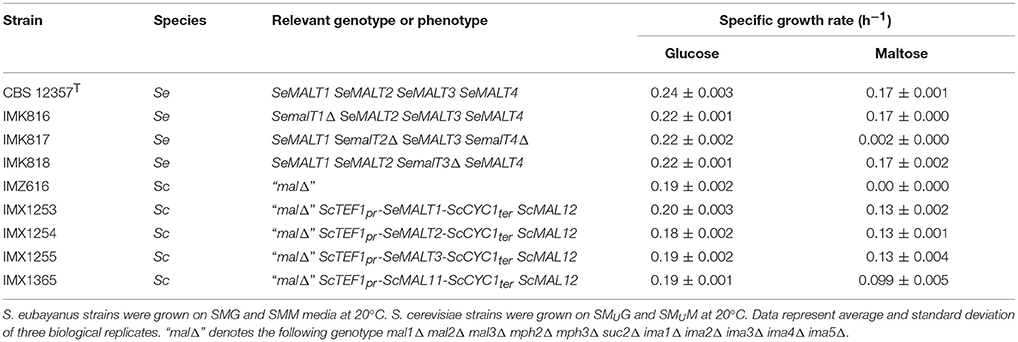
Table 4. Specific growth rates (h−1) of S. eubayanus CBS 12357T (Libkind et al., 2011), S. eubayanus (Se) maltose transporter deletion mutants, S. cerevisiae (Sc) strains overexpressing individual S. eubayanus maltose transporter genes and the maltose-consumption-deficient host strain S. cerevisiae IMZ616 (Marques et al., 2017).
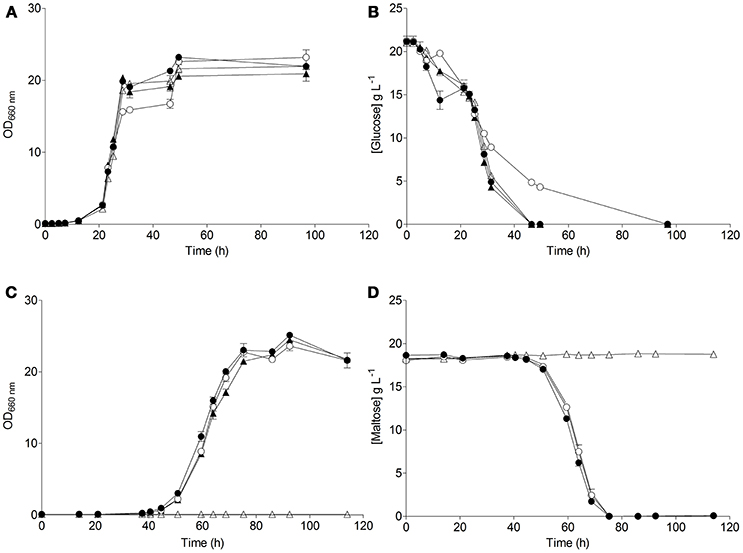
Figure 4. Characterization of the S. eubayanus strains ( ) CBS 12357T, (
) CBS 12357T, ( ) IMK816 (SemalT1Δ), (
) IMK816 (SemalT1Δ), ( ) IMK817 (SemalT2Δ SemalT4Δ), and (
) IMK817 (SemalT2Δ SemalT4Δ), and ( ) IMK818 (SemalT3Δ) during growth on glucose and maltose as sole carbon source. S. eubayanus strains were grown on SMG or SMM at 20°C. Growth on glucose (A) and on maltose (C) was monitored based on optical density measurement at 660 nm (OD660nm). Concentrations of glucose (B) and maltose (D) in culture supernatants were measured by HPLC. Data are presented as average and standard deviation of three biological replicates.
) IMK818 (SemalT3Δ) during growth on glucose and maltose as sole carbon source. S. eubayanus strains were grown on SMG or SMM at 20°C. Growth on glucose (A) and on maltose (C) was monitored based on optical density measurement at 660 nm (OD660nm). Concentrations of glucose (B) and maltose (D) in culture supernatants were measured by HPLC. Data are presented as average and standard deviation of three biological replicates.
For further functional analysis, SeMALT1, SeMALT2, and SeMALT3 were expressed in a maltose-transporter negative S. cerevisiae strain background. SeMALT4 was not included in this comparison, as it encodes a protein with the same amino acid sequence as SeMALT2. The maltose-negative S. cerevisiae strain IMZ616 originates from S. cerevisiae CEN.PK102-3A, which carries three MAL loci (MAL1, MAL2, and MAL3) (Basso et al., 2011). To eliminate growth on α-glucosides, these three MAL loci, MPH2, and MPH3 as well as the α-glucoside hydrolase-encoding genes SUC2 and IMA1-5 were deleted, yielding S. cerevisiae IMK291 (Marques et al., 2017). Introduction of cas9 into this strain yielded IMZ616 (Marques et al., 2018).
Restoration of growth on maltose of S. cerevisiae IMZ616 requires simultaneous expression of a maltose transporter and a maltase. Therefore, the three S. eubayanus transporter genes were cloned behind the constitutive ScTEF1 promoter and, together with an expression cassette for the ScMAL12 maltase gene, integrated at the SGA1 locus of S. cerevisiae IMZ616 (Figure 2). The resulting S. cerevisiae strains IMX1253, IMX1254, IMX1255, which expressed SeMALT1, SeMALT2, and SeMALT3, respectively, were grown on SMUM. As expected, the host strain IMZ616 did not show any growth on maltose, while the three SeMALT-expressing strains showed different growth profiles on this disaccharide. Strain IMX1255 (SeMALT3) resumed growth after a lag phase of ~10 h and consumed half of the maltose supplied (Figure 5). Strains IMX1253 (SeMALT1) and IMX1254 (SeMALT2) showed lag phases of 100 and 250 h, respectively. However, after these lag phases, maltose was consumed. Strain IMX1254 (SeMALT2) consumed 75% of the supplied maltose in 150 h. In the same conditions the control strain IMX1365 co expressing ScAGT1 and ScMAL12 showed a short lag phase of 10 h and that was immediately followed by exponential growth, IMX1365 reached stationary phase and full maltose consumption in less than 100 h a performance comparable to the IMX1255 (SeMALT3) (Figure 5).
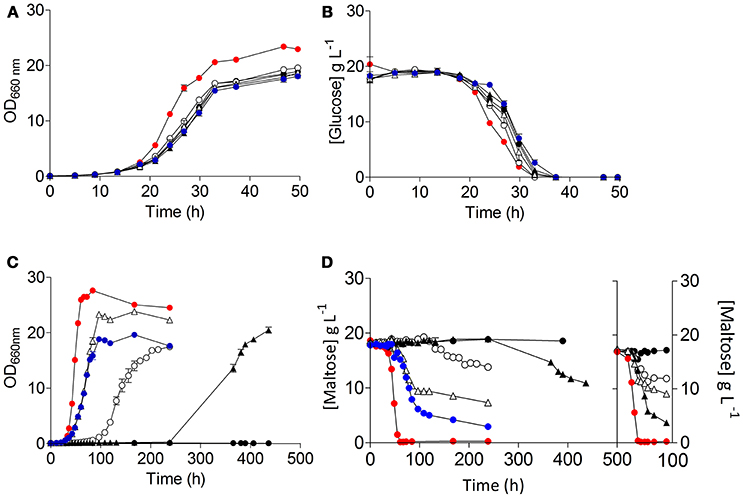
Figure 5. Characterization of the S. cerevisiae strains () IMZ616 (Marques et al., 2018), () IMX1253 (ScTEF1pr-SeMALT1-ScCYC1ter), () IMX1254 (ScTEF1pr-SeMALT2-ScCYC1ter), () IMX1255 (ScTEF1pr-SeMALT3-ScCYC1ter), ( ) IMX1365 (ScTEF1pr-ScMAL11-SeCYC1ter), and (
) IMX1365 (ScTEF1pr-ScMAL11-SeCYC1ter), and ( ) S. eubayanus CBS 12357T. The strains were grown in SMUG and SMUM at 20°C. Growth on glucose (A) and on maltose (C) was monitored based on optical density measurement at 660 nm (OD660nm). Concentrations of glucose (B) and maltose (D) in culture supernatants were measured by HPLC. (D) Shows the data of two consecutive batches. Data are presented as average and standard deviation of three biological replicates.
) S. eubayanus CBS 12357T. The strains were grown in SMUG and SMUM at 20°C. Growth on glucose (A) and on maltose (C) was monitored based on optical density measurement at 660 nm (OD660nm). Concentrations of glucose (B) and maltose (D) in culture supernatants were measured by HPLC. (D) Shows the data of two consecutive batches. Data are presented as average and standard deviation of three biological replicates.
These delayed growth phenotypes of the SeMALT-expressing strains resemble those observed during growth on lactose of S. cerevisiae strains expressing the β-galactosidase (LAC4) and the lactose permease (LAC12) genes of Kluyveromyces marxianus and Kluyveromyces lactis (Domingues et al., 1999; Guimarães et al., 2008) In those studies, lactose utilization first had to be improved by laboratory evolution which resulted in lowering copy number of the plasmid harboring the permease and hydrolase genes as well as a short internal deletion located in the bi-directional promoter driving expression of the two genes (Guimarães et al., 2008). The same way, an adaptation step have been included in the workflow for complementation of the hexose transporter null (hxt0) strain EBYWV4000 with human glucose transporter (GLUT) (Boles and Oreb, 2018) suggesting that swapping transporter or implementing new assimilatory pathway remains non-trivial.
When, at the end of a first round of batch cultivation on maltose, cells of the SeMALT-expressing strains were transferred to fresh maltose medium, they all showed instantaneous growth at a specific growth rate of 0.13 h−1 (Table 4). Under the same conditions, S. eubayanus CBS 12357T grew on maltose at a specific growth rate of 0.17 h−1 (Figure 5, Table 4). Even after transfer to fresh maltose medium, none of the heterologously expressed SeMALT transporters enabled full maltose consumption in these cultures. Similarly to the first cycle of batch cultivation on maltose, strain IMX1254 (MALT2) consumed 75% of the maltose supplied, while strains IMX1255 (SeMALT3) and IMX1253 (SeMALT1) consumed ca. 50 and 35 percentage even after prolonged incubation, none of the S. cerevisiae strains expressing an SeMALT gene nor S. eubayanus CBS 12357T showed growth on maltotriose, while the positive control IMX1365 (ScAGT1) showed growth at a rate of 0.08 ± 0.007 h−1 (Figure S9).
In contrast to the deletion study, which suggested that only SeMalt2 and/or SeMalt4 were able to transport maltose, heterologous expression in the Mal− S. cerevisiae strain IMZ616 showed that all four transporter genes encode transporters that, in combination with a maltase, allow growth on maltose.
Since maltose is imported by proton symporters energized by the plasma-membrane proton-motive force (Serrano et al., 1986; van den Broek et al., 1994), an unrestricted influx of maltose can lead to a fast influx of protons (Jansen et al., 2004). Unless the resulting rate of proton influx can be countered by the proton-pumping plasma-membrane ATPase (Pma1; Serrano et al., 1986), dissipation of the proton motive force and cytosolic acidification can cause maltose-induced cell death. Indeed, pronounced maltose-accelerated death has been observed S. cerevisiae evolved an increased maltose-transport capacity (Jansen et al., 2004). To test whether this phenomenon was responsible for the observed delayed growth of S. cerevisiae strains expressing S. eubayanus maltose transporters, the corresponding S. cerevisiae strains (IMX1243, IMX1254, and IMX1255) were first grown on SMG. Upon reaching late exponential phase, cells after washing were transferred to SM medium (without C-source) to give an OD660 of 1.0. The resulting cell suspension was then sampled before and 30, 120, and 270 min after addition of 20 g.L−1maltose. From each sample, 96 cells were sorted using gated Forward scatter signal and side scatter signal intensities on SMG medium and the viability was estimated based on the number of growing colonies. Neither the three SeMALT-expressing S. cerevisiae strains, nor S. eubayanus CBS 12357T or the Mal− S. cerevisiae IMZ616 showed a decreased viability over a period of 270 min exposure to maltose (Figure 6). This result indicated that delayed growth of the SeMALT-expressing S. cerevisiae strains was not due to maltose accelerated death.

Figure 6. Cell viability after exposure to maltose of glucose-pregrown cultures of S. eubayanus CBS 12357T ( ), S. cerevisiae IMZ616 (
), S. cerevisiae IMZ616 ( ), IMX1253 (SeMALT1) (
), IMX1253 (SeMALT1) ( ), IMX1254 (SeMALT2) (
), IMX1254 (SeMALT2) ( ) and IMX1255 (SeMALT3) (
) and IMX1255 (SeMALT3) ( ). Cells from glucose-grown batch cultures were resuspended in SM. Prior addition of 20 g L−1 of maltose, the initial viability was measured by sorting 96 cells per strain on SMUG plates. The SMUG cultures were sampled after 30, 120 and 270 min. The viability data are represented as averages ± mean deviations of three independent experiments for each strain.
). Cells from glucose-grown batch cultures were resuspended in SM. Prior addition of 20 g L−1 of maltose, the initial viability was measured by sorting 96 cells per strain on SMUG plates. The SMUG cultures were sampled after 30, 120 and 270 min. The viability data are represented as averages ± mean deviations of three independent experiments for each strain.
The inability of the double deletion mutant S. eubayanus IMK817 (SemalT2Δ-SemalT4Δ) to grow on maltose, despite the demonstration that SeMALT1 and SeMALT3 have the potential to encode functional maltose transporters, might indicate that SeMALT1 and SeMALT3 are not expressed in maltose-grown cultures. To investigate the impact of carbon sources on genome-wide transcript profiles and, more specifically, on transcriptional regulation of maltose metabolism genes, duplicate cultures of the S. eubayanus wild-type strain CBS 12357T were grown on SMG and SMM and sampled in mid-exponential phase (OD660nm = 12.5 ± 1.0). After mRNA isolation and processing, cDNA libraries were sequenced with Illumina sequencing technology. cDNA sequencing reads were mapped onto the newly annotated S. eubayanus CBS 12357T genome assembly and used to calculate FPKM (fragments per kilobase of feature (gene) per million reads mapped) expression values. FPKM results represent normalized expression values that take into account gene length and sequencing depth. Statistical analysis showed that 125 genes were differentially expressed in the glucose- and maltose-grown cultures with a fold difference > 4 (Table S7). All four S. eubayanus MALT transporters were significantly upregulated during growth on maltose and three (SeMALT2, SeMALT4, and SeMALT1) were found among the ten most upregulated genes (Table 5). SeMALT2 and SeMALT4 exhibited 262- and 244-fold higher transcript levels during growth on maltose than during growth on glucose. SeMALT1 and SeMALT3 represented a substantially lower fold-difference between maltose- and glucose-grown cultures (67- and 6.8- fold, respectively; Figure 7, Table 5). The most pronounced difference between SeMALT2/SeMALT4 and SeMALT1/SeMALT3 concerned their expression level. The FPKM value of SeMALT1 in maltose-grown cultures was 48-fold lower than that of SeMALT2 and SeMALT4 value (FPKMSeMALT1 = 30; FPKMSeMALT2 = 1,683, FPKMSeMALT4 = 1451). Similarly SeMALT3 exhibited a FPKM value of only 200. In the same analysis, SeACT1 and the glycolytic gene SeTDH3, genes commonly used as internal standard in transcript analysis exhibited substrate-independent FPKM values of 1600 and 6000, respectively. The maltase genes that were physically associated to SeMALT2 and SeMALT4 (Figure 3) were also strongly overexpressed in maltose-grown cultures, representing the highest upregulation fold difference in expression in glucose- and maltose-grown cultures of S. eubayanus (Table 5). This result confirms the functionality of the bidirectional promoters controlling the maltase and transporter genes in the S. eubayanus MAL loci harboring SeMALT2 and SeMALT4.
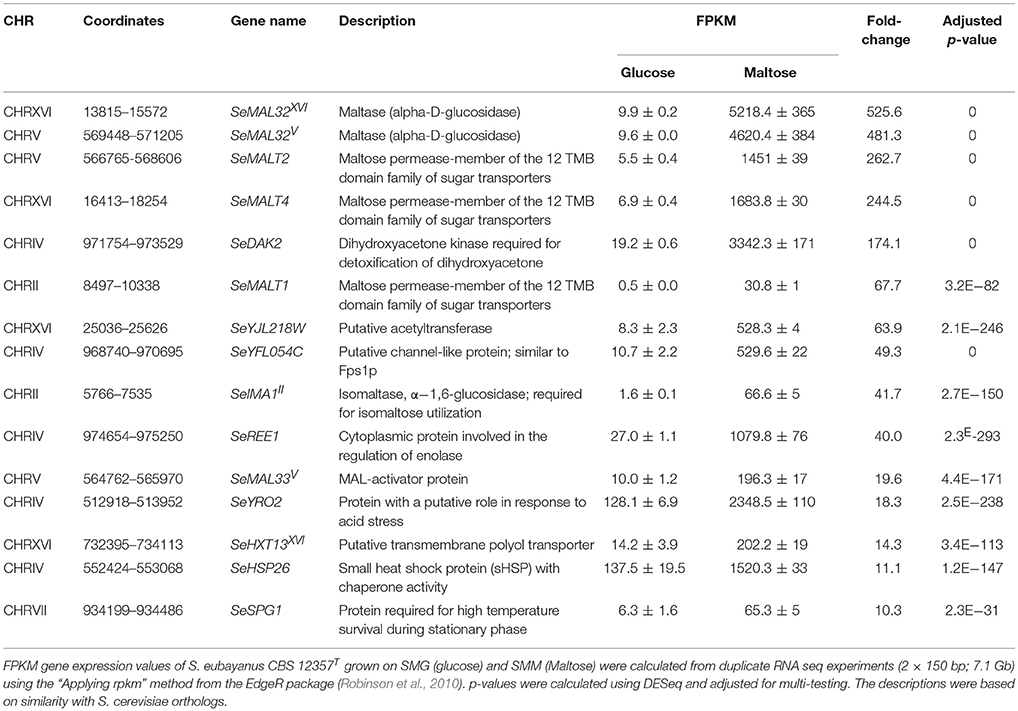
Table 5. Transcript level of the 15 most strongly upregulated genes in maltose-grown S. eubayanus CBS 12357T.
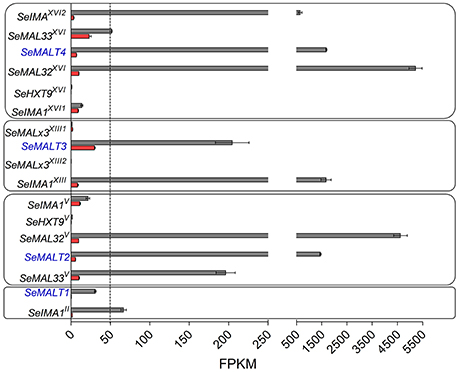
Figure 7. Expression of the maltose metabolism associated genes in S. eubayanus CBS 12357T. Transcript levels of maltose metabolism genes located at subtelomeric regions of CHRII, V, XII and XVI. FPKM gene expression values of S. eubayanus CBS 12357T grown on SMG [glucose, ()] and SMM [maltose, ()] at 20°C were calculated from duplicate RNA seq experiments (2 × 150 bp; 7.1 Gb) using the “Applying rpkm” method in the EdgeR package (Robinson et al., 2010). Data represent average and mean deviation of two biological replicates.
S. eubayanus is not only a key contributor to the hybrid genomes of current S. pastorianus lager brewing strain and a basis for developing new brewing yeast hybrids (Gibson et al., 2017) but is also directly used for brewing specialty lager beer (https://www.cnbc.com/2017/09/26/heineken-unveils-h41-a-beer-that-puts-yeast-into-focus.html) (Hittinger et al., 2018). This study provided an upgraded, near-complete genome sequence of the S. eubayanus type strain CBS 12357Tand the first characterization of its four maltose transporter genes.
Complete and accurate de novo assembly of eukaryotic genomes has long been a major challenge. While short-read, high-coverage technologies such as the popular Illumina platforms enable accurate sequencing and assembly of unique genomic sequences, they cannot resolve repetitive sequences. As previously demonstrated for S. cerevisiae CEN.PK113-7D (Nijkamp et al., 2012; Salazar et al., 2017; Jenjaroenpun et al., 2018), use of the Oxford Nanopore MinION platform generated long-reads that enabled a near-complete assembly of 15 of the 16 chromosomes of S. eubayanus CBS 12357T (Figure 3A). Correct assembly of subtelomeric regions, which are known for their high sequence redundancy, was especially important in the context of this study in view of the subtelomeric location of the four MAL loci in this strain (Figure 3B). Despite the massive sequencing coverage yielded by a single flow cell, which was invaluable for genome scaffold construction, the intrinsic higher error rate of MinION sequencing (Goodwin et al., 2015) required polishing with additional Illumina sequencing data. Although highly effective, as illustrated by the complete assembly of the four MAL loci and the use of resulting high-quality genome sequence information for transcriptome analysis, this approach could not correct all errors. In particular, INDELs causing omissions of single nucleotides in homopolymer regions were left in the final assembly, which is a known pitfall of the single molecule nanopore sequencing (Oxford Nanopore Technology MinIOn). Manual curation and, in particular, validation of relevant sequences by Sanger sequencing will be required to further refine the current genome sequence of S. eubayanus CBS 12357T.
Also the first application of CRISPR-Cas9-assisted genome editing in S. eubayanus reported in this study was greatly facilitated by the availability of an accurate genome sequence. Resolution of the sequences of the four SeMALT genes enabled optimization of the gRNA spacer selection, thereby minimizing the risk of undesirable off-target events. Although targeting efficiencies of the employed ribozyme-flanked-gRNA expression system (Gorter de Vries et al., 2017), which ranged from 3 to 40% were not ideal, Cas9-assisted gene deletion offered clear advantages over traditional methods that rely on a double crossover event that inserts a DNA fragment containing a selection marker in the recipient strain's genome (Baudin et al., 1993; Wilson et al., 2000). Cas9-assisted genome editing in S. eubayanus did not make use of a marker cassette and, most importantly, enabled simultaneous marker-free editing both alleles of the SeMALT1 or the SeMALT3 gene. In the case of SeMALT2 and SeMALT4, a single transformation was even sufficient to delete both alleles of two genes (DiCarlo et al., 2013; Mans et al., 2015). Achieving these objectives with conventional techniques would have been extremely time consuming as multiple rounds of transformation and marker recovery would be required. Depending on consumer acceptance and regulations in place (Hall, 2016; Waltz, 2016), marker-free, Cas9-assisted genome editing of S. eubayanus may be combined with the generation of new Saccharomyces hybrids to accelerate development to novel brewing strains.
Cas9-mediated gene disruption and transcriptome analysis in S. eubayanus CBS 12357 showed that, in maltose-grown cultures, SeMALT2 and SeMALT4 were predominantly responsible for maltose uptake. These transporters are located within two nearly identical (97% identity) SeMAL loci that strongly resemble canonical S. cerevisiae MAL loci (Charron et al., 1989; Chow et al., 1989; Vidgren et al., 2005). Although two other genes, SeMALT3 and SeMALT1, could restore growth upon their expression in the Mal− S. cerevisiae strain IMZ616, their low expression levels in S. eubayanus CBS 12357T were apparently not sufficient to support growth on maltose when SeMALT2 and SeMALT4 were both deleted (Figures 3, 7). This study did not provide new insights into the origin of MTT1/MTY1 (Dietvorst et al., 2005; Salema-Oom et al., 2005) and SeAGT1 (Nakao et al., 2009; Vidgren and Londesborough, 2012; Hebly et al., 2015) in industrial S. pastorianus strains as, consistent with earlier observations (Baker et al., 2015), no genes with strong sequence similarity to these maltose transporter genes were identified in the improved genome sequence of S. eubayanus CBS 12357T.
The low expression levels of SeMALT1 and SeMALT3 may be related to their genomic context, as they were located in atypical SeMAL loci on CHRII and CHRXIII, respectively (Figure 3B). Assuming that these two atypical SeMAL loci evolved from a complete MAL locus, loss of the maltase gene may have disrupted the bi-directional promoter that, in S. cerevisiae, controls expression of both the maltose and the maltose transporter genes (Levine et al., 1992). In S. cerevisiae, the maltose regulator Mal63 binds two regulatory sites (5′MGSN9MGS3′) located between positions −465 and −579 in the region separating the two divergent genes (Sirenko et al., 1995). While two of these elements were also found in the promoter regions of SeMALT2 and SeMALT4 (Figure S8), the SeMALT1 and SeMALT3 promoters each harbored only a single element (Figure S8). Alternatively, low expression of SeMALT1 and SeMALT3 in maltose-grown cultures may reflect a sub-functionalization that, during evolution, led to a different regulation and/or catalytic properties of the encoded transporters (Ohno, 1970; Hughes, 1994). Indeed, such a sub-functionalization has been experimentally reconstructed for yeast α-glucoside hydrolases (Voordeckers et al., 2012).
None of the four SeMALT genes identified in S. eubayanus CBS 12357T were found to encode a functional maltotriose transporter. Although, the S. eubayanus Patagonian lineage is unlikely to have contributed the S. eubayanus subgenome of S. pastorianus lager brewing strains (Nakao et al., 2009), this observation would be consistent with the notion that S. cerevisiae has contributed the vital ability to ferment this trisaccharide, which is abundantly present in wort as in contrast multiple S. cerevisiae ale strains mainly issued from the beer 1 and 2 groups have been shown to use this sugar (Gallone et al., 2016). However, CBS 12357 is a representative of sole the Patagonia B group, one of five groups defined based of the phylogenetic distribution of S. eubayanus strains isolated so far (Peris et al., 2016). Therefore, we cannot at all exclude the possibility that this brewing relevant phenotypic trait of lager yeast would originate from the S. eubayanus parent. The only elements so far that could tilt toward this hypothesis are very fragmented sequencing data of an isolate from the Holartic group, that suggested occurrence of S. eubayanus ortholog of the S. cerevisiae AGT1/MAL11 gene (Hebly et al., 2015). Therefore, the resource and methodology used in this study paved the way for further exploration of the diversity of S. eubayanus population and elucidation of S. eubayanus parental lineage of S. pastorianus.
J-MD, JP, AB, NB designed experiments. AB, NB, JG performed experiments. MB, NB, and J-MD performed bioinformatics work. J-MD, JP, AB, NB, JG analyzed result data. J-MD and JP wrote the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This project was funded by the Seventh Framework Programme of the European Union in the frame of the SP3 people support for training and career development of researchers (Marie Curie), Networks for Initial Training (PITN-GA-2013 ITN-2013-606795) YeastCell (https://yeastcell.eu/) and the BE-Basic R&D Program (http://www.be-basic.org/), which was granted an FES subsidy from the Dutch Ministry of Economic Affairs, Agriculture and Innovation (EL&I). We thank Alex Salazar, Niels Kuijpers (Heineken Supply Chain B.V.) and Jan-Maarten Geertman (Heineken Supply Chain B.V.) for their support and Arthur Gorter de Vries for critically reading the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01786/full#supplementary-material
Alves, S. L., Herberts, R. A., Hollatz, C., Trichez, D., Miletti, L. C., de Araujo, P. S., et al. (2008). Molecular analysis of maltotriose active transport and fermentation by Saccharomyces cerevisiae reveals a determinant role for the AGT1 permease. Appl. Environ. Microbiol. 74, 1494–1501. doi: 10.1128/AEM.02570-07
Anders, S., McCarthy, D. J., Chen, Y., Okoniewski, M., Smyth, G. K., Huber, W., et al. (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786. doi: 10.1038/nprot.2013.099
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Baker, E., Wang, B., Bellora, N., Peris, D., Hulfachor, A. B., Koshalek, J. A., et al. (2015). The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeasts. Mol. Biol. Evol. 32, 2818–2831. doi: 10.1093/molbev/msv168
Bao, Z., Xiao, H., Lang, J., Zhang, L., Xiong, X., Sun, N., et al. (2015). Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 4, 585–594. doi: 10.1021/sb500255k
Basso, T. O., de Kok, S., Dario, M., do Espirito-Santo, J. C., Müller, G., Schlölg, P. S., et al. (2011). Engineering topology and kinetics of sucrose metabolism in Saccharomyces cerevisiae for improved ethanol yield. Metab. Eng. 13, 694–703. doi: 10.1016/j.ymben.2011.09.005
Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F., and Cullin, C. (1993). A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 21, 3329–3330. doi: 10.1093/nar/21.14.3329
Bergström, A., Simpson, J. T., Salinas, F., Barré, B., Parts, L., Zia, A., et al. (2014). A high-definition view of functional genetic variation from natural yeast genomes. Mol. Biol. Evol. 31, 872–888. doi: 10.1093/molbev/msu037
Bing, J., Han, P. J., Liu, W. Q., Wang, Q. M., and Bai, F. Y. (2014). Evidence for a far East Asian origin of lager beer yeast. Curr. Biol. 24, R380–R381. doi: 10.1016/j.cub.2014.04.031
Bolat, I., Romagnoli, G., Zhu, F., Pronk, J. T., and Daran, J. M. (2013). Functional analysis and transcriptional regulation of two orthologs of ARO10, encoding broad-substrate-specificity 2-oxo-acid decarboxylases, in the brewing yeast Saccharomyces pastorianus CBS1483. FEMS Yeast Res. 13, 505–517. doi: 10.1111/1567-1364.12051
Boles, E., and Oreb, M. (2018). A growth-based screening system for hexose transporters in yeast. Methods Mol. Biol. 1713, 123–135. doi: 10.1007/978-1-4939-7507-5_10
Bracher, J. M., Verhoeven, M. D., Wisselink, H. W., Crimi, B., Nijland, J. G., Driessen, A. J. M., et al. (2018). The Penicillium chrysogenum transporter PcAraT enables high-affinity, glucose-insensitive l-arabinose transport in Saccharomyces cerevisiae. Biotechnol. Biofuels 11:63. doi: 10.1186/s13068-018-1047-6
Brown, C. A., Murray, A. W., and Verstrepen, K. J. (2010). Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr. Biol. 20, 895–903. doi: 10.1016/j.cub.2010.04.027
Caesar, R., Palmfeldt, J., Gustafsson, J. S., Pettersson, E., Hashemi, S. H., and Blomberg, A. (2007). Comparative proteomics of industrial lager yeast reveals differential expression of the cerevisiae and non-cerevisiae parts of their genomes. Proteomics 7, 4135–4147. doi: 10.1002/pmic.200601020
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Casaregola, S., Nguyen, H. V., Lapathitis, G., Kotyk, A., and Gaillardin, C. (2001). Analysis of the constitution of the beer yeast genome by PCR, sequencing and subtelomeric sequence hybridization. Int. J. Syst. Evol. Microbiol. 51, 1607–1618. doi: 10.1099/00207713-51-4-1607
Charron, M. J., Read, E., Haut, S. R., and Michels, C. A. (1989). Molecular evolution of the telomere-associated MAL loci of Saccharomyces. Genetics 122, 307–316.
Chow, T. H., Sollitti, P., and Marmur, J. (1989). Structure of the multigene family of MAL loci in Saccharomyces. Mol. Gen. Genet. 217, 60–69. doi: 10.1007/BF00330943
Cohen, J. D., Goldenthal, M. J., Chow, T., Buchferer, B., and Marmur, J. (1985). Organization of the MAL loci of Saccharomyces. Physical identification and functional characterization of three genes at the MAL6 locus. Mol. Gen. Genet. 200, 1–8. doi: 10.1007/BF00383304
Cousseau, F. E., Alves, S. L., Trichez, D., and Stambuk, B. U. (2013). Characterization of maltotriose transporters from the Saccharomyces eubayanus subgenome of the hybrid Saccharomyces pastorianus lager brewing yeast strain Weihenstephan 34/70. Lett. Appl. Microbiol. 56, 21–29. doi: 10.1111/lam.12011
Day, R. E., Higgins, V. J., Rogers, P. J., and Dawes, I. W. (2002a). Characterization of the putative maltose transporters encoded by YDL247w and YJR160c. Yeast 19, 1015–1027. doi: 10.1002/yea.894
Day, R. E., Rogers, P. J., Dawes, I. W., and Higgins, V. J. (2002b). Molecular analysis of maltotriose transport and utilization by Saccharomyces cerevisiae. Appl. Environ. Microbiol. 68, 5326–5335. doi: 10.1128/AEM.68.11.5326-5335.2002
DiCarlo, J. E., Norville, J. E., Mali, P., Rios, X., Aach, J., and Church, G. M. (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336–4343. doi: 10.1093/nar/gkt135
Dietvorst, J., Londesborough, J., and Steensma, H. Y. (2005). Maltotriose utilization in lager yeast strains: MTT1 encodes a maltotriose transporter. Yeast 22, 775–788. doi: 10.1002/yea.1279
Dobin, A., and Gingeras, T. R. (2016). Optimizing RNA-Seq Mapping with STAR. Methods Mol. Biol. 1415, 245–262. doi: 10.1007/978-1-4939-3572-7_13
Domingues, L., Teixeira, J. A., and Lima, N. (1999). Construction of a flocculent Saccharomyces cerevisiae fermenting lactose. Appl. Microbiol. Biotechnol. 51, 621–626. doi: 10.1007/s002530051441
Dubin, R. A., Needleman, R. B., Gossett, D., and Michels, C. A. (1985). Identification of the structural gene encoding maltase within the MAL6 locus of Saccharomyces carlsbergensis. J. Bacteriol. 164, 605–610.
Entian, K.-D., and Kötter, P. (2007). “25 yeast genetic strain and plasmid collections,” in Methods in Microbiology, Vol. 36, eds Stansfield, I., and Stark, M. J. R. (London, UK: Academic Press), 629–66.
Gallone, B., Steensels, J., Prahl, T., Soriaga, L., Saels, V., Herrera-Malaver, B., et al. (2016). Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 166, 1397–1410.e16. doi: 10.1016/j.cell.2016.08.020
Gao, Y., and Zhao, Y. (2014). Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Int. Plant Biol. 56, 343–349. doi: 10.1111/jipb.12152
Gayevskiy, V., and Goddard, M. R. (2016). Saccharomyces eubayanus and Saccharomyces arboricola reside in North Island native New Zealand forests. Environ. Microbiol. 18, 1137–1147. doi: 10.1111/1462-2920.13107
Gibson, B., Geertman, J. A., Hittinger, C. T., Krogerus, K., Libkind, D., Louis, E. J., et al. (2017). New yeasts-new brews: modern approaches to brewing yeast design and development. FEMS Yeast Res. 17:fox038. doi: 10.1093/femsyr/fox038
Gietz, R. D., and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34. doi: 10.1038/nprot.2007.13
Goodwin, S., Gurtowski, J., Ethe-Sayers, S., Deshpande, P., Schatz, M. C., and McCombie, W. R. (2015). Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 25, 1750–1756. doi: 10.1101/gr.191395.115
Gorter de Vries, A. R., de Groot, P. A., van den Broek, M., and Daran, J. G. (2017). CRISPR-Cas9 mediated gene deletions in lager yeast Saccharomyces pastorianus. Microb. Cell Fact. 16:222. doi: 10.1186/s12934-017-0835-1
Guimarães, P. M., François, J., Parrou, J. L., Teixeira, J. A., and Domingues, L. (2008). Adaptive evolution of a lactose-consuming Saccharomyces cerevisiae recombinant. Appl. Environ. Microbiol. 74, 1748–1756. doi: 10.1128/AEM.00186-08
Hall, S. S. (2016). Editing the mushroom. Sci. Am. 314, 56–63. doi: 10.1038/scientificamerican0316-56
Han, E. K., Cotty, F., Sottas, C., Jiang, H., and Michels, C. A. (1995). Characterization of AGT1 encoding a general alpha-glucoside transporter from Saccharomyces. Mol. Microbiol. 17, 1093–1107. doi: 10.1111/j.1365-2958.1995.mmi_17061093.x
Hebly, M., Brickwedde, A., Bolat, I., Driessen, M. R., de Hulster, E. A., van den Broek, M., et al. (2015). S. cerevisiae x S. eubayanus interspecific hybrid, the best of both worlds and beyond. FEMS Yeast Res 15:fov005. doi: 10.1093/femsyr/fov005
Hewitt, S. K., Donaldson, I. J., Lovell, S. C., and Delneri, D. (2014). Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS ONE 9:e92203. doi: 10.1371/journal.pone.0092203
Hittinger, C. T., Steele, J. L., and Ryder, D. S. (2018). Diverse yeasts for diverse fermented beverages and foods. Curr. Opin. Biotechnol. 49, 199–206. doi: 10.1016/j.copbio.2017.10.004
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491. doi: 10.1186/1471-2105-12-491
Hughes, A. L. (1994). The evolution of functionally novel proteins after gene duplication. Proc. Biol. Sci. 256, 119–124. doi: 10.1098/rspb.1994.0058
Jansen, M. L., Daran-Lapujade, P., de Winde, J. H., Piper, M. D., and Pronk, J. T. (2004). Prolonged maltose-limited cultivation of Saccharomyces cerevisiae selects for cells with improved maltose affinity and hypersensitivity. Appl. Environ. Microbiol. 70, 1956–1963. doi: 10.1128/AEM.70.4.1956-1963.2004
Jenjaroenpun, P., Wongsurawat, T., Pereira, R., Patumcharoenpol, P., Ussery, D. W., Nielsen, J., et al. (2018). Complete genomic and transcriptional landscape analysis using third-generation sequencing: a case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 46:e38. doi: 10.1093/nar/gky014
Joubert, R., Brignon, P., Lehmann, C., Monribot, C., Gendre, F., and Boucherie, H. (2000). Two-dimensional gel analysis of the proteome of lager brewing yeasts. Yeast 16, 511–222. doi: 10.1002/(SICI)1097-0061(200004)16:6<511::AID-YEA544>3.0.CO;2-I
Juergens, H., Varela, J. A., de Vries, A. R. G., Perli, T., Gast, V. J. M., Gyurchev, N. Y., et al. (2018). Genome editing in Kluyveromyces and Ogataea yeasts using a broad-host-range Cas9/gRNA co-expression plasmid. FEMS Yeast Res. 18:foy012. doi: 10.1093/femsyr/foy012
Knijnenburg, T. A., Daran, J. M., van den Broek, M. A., Daran-Lapujade, P. A., de Winde, J. H., Pronk, J. T., et al. (2009). Combinatorial effects of environmental parameters on transcriptional regulation in Saccharomyces cerevisiae: a quantitative analysis of a compendium of chemostat-based transcriptome data. BMC Genomics 10:53. doi: 10.1186/1471-2164-10-53
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Krogerus, K., Magalhães, F., Vidgren, V., and Gibson, B. (2015). New lager yeast strains generated by interspecific hybridization. J. Ind. Microbiol. Biotechnol. 42, 769–778. doi: 10.1007/s10295-015-1597-6
Krogerus, K., Magalhães, F., Vidgren, V., and Gibson, B. (2017a). Novel brewing yeast hybrids: creation and application. Appl. Microbiol. Biotechnol.101, 65–78. doi: 10.1007/s00253-016-8007-5
Krogerus, K., Seppänen-Laakso, T., Castillo, S., and Gibson, B. (2017b). Inheritance of brewing-relevant phenotypes in constructed Saccharomyces cerevisiae x Saccharomyces eubayanus hybrids. Microb. Cell Fact. 16:66. doi: 10.1186/s12934-017-0679-8
Kuijpers, N. G., Solis-Escalante, D., Luttik, M. A., Bisschops, M. M., Boonekamp, F. J., van den Broek, M., et al. (2016). Pathway swapping: toward modular engineering of essential cellular processes. Proc. Natl. Acad. Sci. U.S.A. 113, 15060–15065. doi: 10.1073/pnas.1606701113
Levine, J., Tanouye, L., and Michels, C. A. (1992). The UAS(MAL) is a bidirectional promotor element required for the expression of both the MAL61 and MAL62 genes of the Saccharomyces MAL6 locus. Curr. Genet. 22, 181–189. doi: 10.1007/BF00351724
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Libkind, D., Hittinger, C. T., Valério, E., Gonçalves, C., Dover, J., Johnston, M., et al. (2011). Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc. Natl. Acad. Sci. U.S.A. 108, 14539–14544. doi: 10.1073/pnas.1105430108
Magalhães, F., Krogerus, K., Vidgren, V., Sandell, M., and Gibson, B. (2017). Improved cider fermentation performance and quality with newly generated Saccharomyces cerevisiae x Saccharomyces eubayanus hybrids. J. Ind. Microbiol. Biotechnol. 44, 1203–1213. doi: 10.1007/s10295-017-1947-7
Magalhães, F., Vidgren, V., Ruohonen, L., and Gibson, B. (2016). Maltose and maltotriose utilisation by group I strains of the hybrid lager yeast Saccharomyces pastorianus. FEMS Yeast Res. 16:fow053. doi: 10.1093/femsyr/fow053
Mans, R., van Rossum, H. M., Wijsman, M., Backx, A., Kuijpers, N. G., van den Broek, M., et al. (2015). CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 15:fov004. doi: 10.1093/femsyr/fov004
Marques, W. L., Mans, R., Henderson, R. K., Marella, E. R., Horst, J. T., Hulster, E., et al. (2018). Combined engineering of disaccharide transport and phosphorolysis for enhanced ATP yield from sucrose fermentation in Saccharomyces cerevisiae. Metab. Eng. 45, 121–133. doi: 10.1016/j.ymben.2017.11.012
Marques, W. L., Mans, R., Marella, E. R., Cordeiro, R. L., van den Broek, M., Daran, J.-M., et al. (2017). Elimination of sucrose transport and hydrolysis in Saccharomyces cerevisiae, a platform strain for engineering sucrose metabolism. FEMS Yeast Res. 17:fox006. doi: 10.1093/femsyr/fox006
McCarthy, D. J., Chen, Y., and Smyth, G. K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. doi: 10.1093/nar/gks042
Michels, C. A., Read, E., Nat, K., and Charron, M. J. (1992). The telomere-associated MAL3 locus of Saccharomyces is a tandem array of repeated genes. Yeast 8, 655–665. doi: 10.1002/yea.320080809
Nakao, Y., Kanamori, T., Itoh, T., Kodama, Y., Rainieri, S., Nakamura, N., et al. (2009). Genome sequence of the lager brewing yeast, an interspecies hybrid. DNA Res. 16, 115–129. doi: 10.1093/dnares/dsp003
Nijkamp, J. F., van den Broek, M., Datema, E., de Kok, S., Bosman, L., Luttik, M. A., et al. (2012). De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb. Cell Fact. 11:36. doi: 10.1186/1475-2859-11-36
Okuno, M., Kajitani, R., Ryusui, R., Morimoto, H., Kodama, Y., and Itoh, T. (2016). Next-generation sequencing analysis of lager brewing yeast strains reveals the evolutionary history of interspecies hybridization. DNA Res. 23, 67–80. doi: 10.1093/dnares/dsv037
Peris, D., Langdon, Q. K., Moriarty, R. V., Sylvester, K., Bontrager, M., Charron, G., et al. (2016). Complex ancestries of lager-brewing hybrids were shaped by standing variation in the wild yeast Saccharomyces eubayanus. PLoS Genet. 12:e1006155. doi: 10.1371/journal.pgen.1006155
Peris, D., Moriarty, R. V., Alexander, W. G., Baker, E., Sylvester, K., Sardi, M., et al. (2017). Hybridization and adaptive evolution of diverse Saccharomyces species for cellulosic biofuel production. Biotechnol Biofuels 10:78.
Peris, D., Sylvester, K., Libkind, D., Gonçalves, P., Sampaio, J. P., Alexander, W. G., et al. (2014). Population structure and reticulate evolution of Saccharomyces eubayanus and its lager-brewing hybrids. Mol. Ecol. 23, 2031–2045. doi: 10.1111/mec.12702
Plourde-Owobi, L., Durner, S., Parrou, J. L., Wieczorke, R., Goma, G., and Francois, J. (1999). AGT1, encoding an alpha-glucoside transporter involved in uptake and intracellular accumulation of trehalose in Saccharomyces cerevisiae. J. Bacteriol. 181, 3830–3832.
Rainieri, S., Kodama, Y., Kaneko, Y., Mikata, K., Nakao, Y., and Ashikari, T. (2006). Pure and mixed genetic lines of Saccharomyces bayanus and Saccharomyces pastorianus and their contribution to the lager brewing strain genome. Appl. Environ. Microbiol. 72, 3968–3974. doi: 10.1128/AEM.02769-05
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Salazar, A. N., Gorter de Vries, A. R., van den Broek, M., Wijsman, M., de la Torre Cortés, P., Brickwedde, A., et al. (2017). Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D. FEMS Yeast Res. 17:fox074. doi: 10.1093/femsyr/fox074
Salema-Oom, M., Valadao Pinto, V., Goncalves, P., and Spencer-Martins, I. (2005). Maltotriose utilization by industrial Saccharomyces strains: characterization of a new member of the alpha-glucoside transporter family. Appl. Environ. Microbiol. 71, 5044–5049. doi: 10.1128/AEM.71.9.5044-5049.2005
Serrano, R., Kielland-Brandt, M. C., and Fink, G. R. (1986). Yeast plasma membrane ATPase is essential for growth and has homology with (Na+, K+), K+- and Ca2+-ATPases. Nature 319, 689–693. doi: 10.1038/319689a0
Serrano, R. (1977). Energy-requirements for maltose transport in yeast. Eur. J. Biochem. 80, 97–102. doi: 10.1111/j.1432-1033.1977.tb11861.x
Sirenko, O. I., Ni, B., and Needleman, R. B. (1995). Purification and binding properties of the Mal63p activator of Saccharomyces cerevisiae. Curr. Genet. 27, 509–516. doi: 10.1007/BF00314440
Solis-Escalante, D., Kuijpers, N. G., Bongaerts, N., Bolat, I., Bosman, L., Pronk, J. T., et al. (2013). amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res. 13, 126–139. doi: 10.1111/1567-1364.12024
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Steensels, J., Meersman, E., Snoek, T., Saels, V., and Verstrepen, K. J. (2014). Large-scale selection and breeding to generate industrial yeasts with superior aroma production. Appl. Environ. Microbiol. 80, 6965–6975. doi: 10.1128/AEM.02235-14
Tai, S. L., Boer, V. M., Daran-Lapujade, P., Walsh, M. C., de Winde, J. H., Daran, J. M., et al. (2005). Two-dimensional transcriptome analysis in chemostat cultures - combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J. Biol. Chem. 280, 437–447. doi: 10.1074/jbc.M410573200
Tamai, Y., Momma, T., Yoshimoto, H., and Kaneko, Y. (1998). Co-existence of two types of chromosome in the bottom fermenting yeast, Saccharomyces pastorianus. Yeast 14, 923–933. doi: 10.1002/(SICI)1097-0061(199807)14:10<923::AID-YEA298>3.0.CO;2-I
van den Broek, M., Bolat, I., Nijkamp, J. F., Ramos, E., Luttik, M. A. H., Koopman, F., et al. (2015). Chromosomal copy number variation in Saccharomyces pastorianus is evidence for extensive genome dynamics in industrial lager brewing strains. Appl. Environ. Microbiol. 81, 6253–6267. doi: 10.1128/AEM.01263-15
van den Broek, P. J., van Leeuwen, C. C., Weusthuis, R. A., Postma, E., van Dijken, J. P., Karssies, R. H., et al. (1994). Identification of the maltose transport protein of Saccharomyces cerevisiae. Biochem. Biophys. Res. Com. 200, 45–51. doi: 10.1006/bbrc.1994.1411
van Leeuwen, C. C. M., Weusthuis, R. A., Postma, E., van den Broek, P. J. A., and Van Dijken, J. P. (1992). Maltose proton cotransport in Saccharomyces cerevisiae- comparative-study with cells and plasma-membrane vesicles. Biochem. J. 284, 441–445. doi: 10.1042/bj2840441
van Rossum, H. M., Kozak, B. U., Niemeijer, M. S., Duine, H. J., Luttik, M. A., Boer, V. M., et al. (2016). Alternative reactions at the interface of glycolysis and citric acid cycle in Saccharomyces cerevisiae. FEMS Yeast Res. 16:fow017. doi: 10.1093/femsyr/fow017
Verduyn, C., Postma, E., Scheffers, W. A., and Vandijken, J. P. (1992). Effect of benzoic-acid on metabolic fluxes in yeasts - a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8, 501–517. doi: 10.1002/yea.320080703
Verhoeven, M. D., Lee, M., Kamoen, L., van den Broek, M., Janssen, D. B., Daran, J. G., et al. (2017). Mutations in PMR1 stimulate xylose isomerase activity and anaerobic growth on xylose of engineered Saccharomyces cerevisiae by influencing manganese homeostasis. Sci. Rep. 7:46155. doi: 10.1038/srep46155
Vidgren, V., Huuskonen, A., Virtanen, H., Ruohonen, L., and Londesborough, J. (2009). Improved fermentation performance of a lager yeast after repair of its AGT1 maltose and maltotriose transporter genes. Appl. Environ. Microbiol. 75, 2333–2345. doi: 10.1128/AEM.01558-08
Vidgren, V., and Londesborough, J. (2012). Characterization of the Saccharomyces bayanus-type AGT1 transporter of lager yeast. J. Inst. Brew. 118, 148–151. doi: 10.1002/jib.22
Vidgren, V., Ruohonen, L., and Londesborough, J. (2005). Characterization and functional analysis of the MAL and MPH loci for maltose utilization in some ale and lager yeast strains. Appl. Environ. Microbiol. 71, 7846–7857. doi: 10.1128/AEM.71.12.7846-7857.2005
Vidgren, V., Viljanen, K., Mattinen, L., Rautio, J., and Londesborough, J. (2014). Three Agt1 transporters from brewer's yeasts exhibit different temperature dependencies for maltose transport over the range of brewery temperatures (0–20 degrees C). FEMS Yeast Res. 14, 601–613. doi: 10.1111/1567-1364.12147
Voordeckers, K., Brown, C. A., Vanneste, K., van der Zande, E., Voet, A., Maere, S., et al. (2012). Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol. 10:e1001446.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Walther, A., Hesselbart, A., and Wendland, J. (2014). Genome sequence of Saccharomyces carlsbergensis, the world's first pure culture lager yeast. G3 4, 783–793. doi: 10.1534/g3.113.010090
Waltz, E. (2016). Gene-edited CRISPR mushroom escapes US regulation. Nature 532:293. doi: 10.1038/nature.2016.19754
Wilson, R. B., Davis, D., Enloe, B. M., and Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16, 65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508>3.0.CO;2-M
Yamagishi, H., Otsuta, Y., Funahashi, W., Ogata, T., and Sakai, K. (1999). Differentiation between brewing and non-brewing yeasts using a combination of PCR and RFLP. J. Appl. Microbiol. 86, 505–513. doi: 10.1046/j.1365-2672.1999.00691.x
Yamashita, I., and Fukui, S. (1985). Transcriptional control of the sporulation-specific glucoamylase gene in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 5, 3069–3073. doi: 10.1128/MCB.5.11.3069
Keywords: yeast, brewing, domestication, gene regulation, wort, transport, Saccharomyces eubayanus
Citation: Brickwedde A, Brouwers N, van den Broek M, Gallego Murillo JS, Fraiture JL, Pronk JT and Daran J-MG (2018) Structural, Physiological and Regulatory Analysis of Maltose Transporter Genes in Saccharomyces eubayanus CBS 12357T. Front. Microbiol. 9:1786. doi: 10.3389/fmicb.2018.01786
Received: 12 May 2018; Accepted: 17 July 2018;
Published: 10 August 2018.
Edited by:
Vittorio Capozzi, University of Foggia, ItalyReviewed by:
Diego Libkind, CONICET Instituto Andino Patagónico en Tecnologías Biológicas y Geoambientales (IPATEC), ArgentinaCopyright © 2018 Brickwedde, Brouwers, van den Broek, Gallego Murillo, Fraiture, Pronk and Daran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Marc G. Daran, ai5nLmRhcmFuQHR1ZGVsZnQubmw=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.