 Sabina Horvat
Sabina Horvat Maja Rupnik
Maja Rupnik- 1Department of Microbiology, Faculty of Medicine, University of Maribor, Maribor, Slovenia
- 2Centre for Medical Microbiology, National Laboratory for Health, Environment and Food, Maribor, Slovenia
Disturbance in gut microbiota is crucial for the development of Clostridioides difficile infection (CDI). Different mechanisms through which gut microbiota influences C. difficile colonization are known. However, C. difficile could also affect gut microbiota balance as previously demonstrated by cultivation of fecal microbiota in C. difficile conditioned medium. In current study, the interactions of C. difficile cells with gut microbiota were addressed. Three different strains (ribotypes 027, 014/020, and 010) were co-cultivated with two types of fecal microbiota (healthy and dysbiotic) using in vitro batch model. While all strains showed higher sporulation frequency in the presence of dysbiotic fecal microbiota, the growth was strain dependent. C. difficile either proliferated to comparable levels in the presence of dysbiotic and healthy fecal microbiota or grew better in co-culture with dysbiotic microbiota. In co-cultures with any C. difficile strain fecal microbiota showed decreased richness and diversity. Dysbiotic fecal microbiota was more affected after co-culture with C. difficile than healthy microbiota. Altogether, 62 OTUs were significantly changed in co-cultures of dysbiotic microbiota/C. difficile and 45 OTUs in co-cultures of healthy microbiota/C. difficile. However, the majority of significantly changed OTUs in both types of microbiota belonged to the phylum Firmicutes with Lachnospiraceae and Ruminococcaceae origin.
Introduction
Clostridium difficile, recently reclassified as Clostridioides difficile (Lawson et al., 2016), is an ubiquitous, anaerobic, spore-forming, Gram-positive bacterium that is currently the leading cause of nosocomial and community associated diarrhea worldwide (Martin et al., 2016). C. difficile infection (CDI) outcome can range from asymptomatic colonization to diarrhea and more severe, life-threatening pseudomembranous colitis (Rupnik et al., 2009; Leffler and Lamont, 2015). CDI is a toxin mediated disease. Two toxins, A and B, are well established as virulence factors, while the presence of the third toxin, binary toxin CDT, is less clear but seems to contribute to disease severity (Gerding et al., 2014).
Success of enteric pathogen to cause a disease depends mainly on two phases, i.e., the initial colonization phase of pathogen in the gut, followed by pathogen-promoting physiological changes in the gut important for its maintenance (Ferreyra et al., 2014). The colonization of C. difficile is strongly associated with disturbance in gut microbiota (i.e., dysbiosis) and several studies explored the mechanisms through which gut microbiota influences C. difficile (Britton and Young, 2012; Pérez-Cobas et al., 2015). While for some enteric pathogens (e.g., Salmonella) mechanisms involved in the pathogen-promoting physiological changes in the gut have already been described (Stecher et al., 2007), little is known about possible impact of C. difficile on gut microbiota. In our previous work, we have shown that C. difficile conditioned medium has specific effect on fecal microbiota and that nutrient competition is the most likely mechanism (Horvat et al., 2017). Additionally, changed bacterial groups detected in in vitro model using C. difficile conditioned medium were comparable to those described in CDI patients.
In this study, we have investigated the interactions between C. difficile vegetative cells and fecal bacterial communities in a simple in vitro model. Several lines of evidence supports the need for dysbiotic gut microbiota for successful C. difficile colonization, such as low carriage rates in healthy population, need of antibiotic pretreatment of animals in animal models of CDI and association of dysbiotic gut microbiota with CDI in humans. However, recent increase in community cases without previous antibiotic exposure (Gupta and Khanna, 2014) suggests that C. difficile can colonize gut also in the presence of non-dysbiotic microbiota. In line with this observations are in vivo and in vitro studies demonstrating capability of C. difficile to colonize the gut even in the presence of complex microbiota (Robinson et al., 2014) and showing that in the absence of some protective species (i.e., C. scindens) colonization with C. difficile even without antibiotic treatment can result in a significant compositional perturbation of gut microbiota in animal models (Studer et al., 2016). We have therefore compared two types of fecal samples, one representing healthy microbiota and the other representing dysbiotic microbiota. Healthy microbiota was obtained from healthy donors. As a proxy for dysbiotic microbiota fecal samples sent to routine diagnostic laboratory for testing for C. difficile were used. Such samples were shown before to clearly group separately from healthy volunteer samples (Skraban et al., 2013; Horvat et al., 2017). The interactions of both fecal microbiota types were tested with three different C. difficile strains as representatives of globally widespread PCR ribotypes (027, 014/020, and 010) and three different toxin production types (A+B+CDT+, A+B+CDT-, A-B-CDT-). The strains were selected with the objective to cover large diversity within C. difficile species. The aim was not to compare ribotype or toxinotype association with changes in interactions with gut microbiota as only a single representative was used.
Materials and Methods
All described cultivation procedures were performed anaerobically at 37°C in an anaerobic workstation (10% CO2, 10% H2, and 80% N2) (Don Whitley Scientific).
C. difficile Strain Selection and Preparation of Vegetative Cells
Three different C. difficile strains, belonging to PCR ribotypes 010, 014/020, and 027, were selected from our C. difficile strain collection (strain designations ZZV15-6684, ZZV11-3236, ZZV09-2033). Selected strains originated from humans and were isolated between the years 2009 and 2015. Strains were plated onto Columbia agar with 5% horse blood (BioMerieux) and incubated for 48 h. Single colony of each strain was transferred into 5 ml of pre-reduced Wilkins Chalgren Anaerobe Broth (WCAB) (Oxoid) and incubated for 17 h. Overnight cultures of each strain were once again transferred to freshly prepared and pre-reduced WCAB growth medium (1:500, v:v) and incubated for 17 h to obtain suspensions of C. difficile vegetative cells with up to 2 × 108 colony forming units (CFUs) per ml, respectively.
Fecal Samples Collection and Preparation of Fecal Inocula
C. difficile negative fecal samples were randomly selected from (i) non-diarrheal samples sent for C. difficile testing (male: n = 5, female: n = 5, all aged under 65 years) (dysbiotic microbiota) and (ii) samples of healthy individuals (male: n = 3, female: n = 2, all aged under 65 years) (healthy microbiota). Healthy individuals had not taken antibiotics for at least 3 months prior to donation. Healthy individuals have given informed consent and agreed to use the samples for the experiments. The use for laboratory samples without informed consent was approved by National Medical Ethic Committee.
After donation samples were stored at 4°C for maximally 24 h. Aliquots of 0.2 g of each specimen were prepared and frozen at -80°C until further processing. Based on our previous comparisons of individual and pooled samples and published studies (Aguirre et al., 2014) we used pooled samples for experiments with fecal microbiota. For pooled slurry preparation each specimen was thawed and diluted in pre-reduced WCAB to obtain 10% fecal slurry. Equal amounts of fecal slurries from each group (i.e., dysbiotic and healthy) were pooled, added to pre-reduced WCAB growth medium (1:100, v:v) and incubated overnight to prepare fecal inocula for in vitro cultivation.
In Vitro Model Set Up of C. difficile and Fecal Microbiota Co-cultivation
Overnight culture of fecal inoculum (50 μl) was combined with suspensions of C. difficile vegetative cells (50 μl) in pre-reduced WCAB growth medium (4.9 ml) in a 6-well plate (Sarstedt). Initial concentration of C. difficile in co-cultures was approximately 2 × 106 CFU/ml. Cultures were gently mixed and incubated for 3 days. Monocultures of fecal microbiota or C. difficile grown in freshly prepared and pre-reduced WCAB growth medium were used as controls. Each described growth condition was done in triplicate. Samples (5 ml) were taken after 72 h incubation period and screened for total viable cell count and spore count of C. difficile (described below). After centrifugation at 10,000 rpm for 10 min, pellets were used for total bacterial DNA extraction. Additionally, fecal pooled sample before any cultivation (i.e., fecal input sample) was included as a control for 16S rDNA sequencing to test for changes of microbiota due to in vitro cultivation.
C. difficile Growth and Sporulation
For C. difficile total viable cell count serial dilutions were prepared in pre-reduced 0.9% sterile saline and plated onto pre-reduced chromID C. difficile agar plates (BioMerieux). C. difficile spores were defined as CFUs on chromID C. difficile agar plates after treatment with 100% ethanol for 30 min. The frequency of sporulation was determined as the percentage of ethanol resistant cell count relative to total cell count. One-way ANOVA was used in the statistical analysis with Bonferroni correction to determine statistically significantly different samples.
Isolation of the Total Bacterial DNA and 16S rDNA Amplicon Sequencing of Microbiota
The extraction of bacterial DNA was performed with the QIAamp Fast Stool DNA Mini Kit (Qiagen) after the mechanical disruption (speed 7,000 for 70 s) with the SeptiFast Lys Kit (Roche) on MagNA Lyser (Roche). The DNA concentration was verified using Quant-iT PicoGreen dsDNA Kit (Thermo Fisher Scientific). Bacterial community composition was determined by paired-end sequencing on an Illumina MiSeq platform, targeting the V3–V4 hypervariable region of the 16S rRNA gene. Library preparation was carried out using the 341F (5′-CCTACGGGNGGCWGCAG-3′) – 805R (5′-GACTACHVGGGTATCTAATCC-3′) set of primers. On the same run a template-free sample was included as a negative sequencing control.
Sequencing data was deposited at metagenomic analysis server MG-RAST (Meyer et al., 2008) and is accessible at https://www.mg-rast.org/linkin.cgi?project=mgp85501.
Analysis of Sequence Data
MiSeq output data was analyzed with statistical tools included in the mothur software (v.1.36.1) (Schloss et al., 2009), according to the MiSeq standard operating procedure (SOP) for Illumina paired end reads (Kozich et al., 2013). Sequences with any ambiguous base or more than eight homopolymers were removed from downstream analysis. The remaining sequences were aligned against the Silva reference alignment (Quast et al., 2013). Chimeras were searched with the UCHIME algorithm (Edgar et al., 2011) and removed. Classification of sequences was performed using the RDP training set (v.9) with an 80% confidence threshold. Sequences restricted to bacterial origin only were clustered into operational taxonomic units (OTUs) at a 3% dissimilarity level. To adjust for the influence of the number of OTUs in a sample on diversity and other statistical tests, any OTUs with relative abundance lower than 0.001% were removed from further analyses. Chao1 and Shannon indices were chosen to characterize alpha samples diversity. For beta diversity estimation weighted UniFrac measure was used (Lozupone et al., 2007). Samples hierarchical clustering was obtained with MEGA software (v.7.0.26) (Kumar et al., 2016) using Bray–Curtis distances as input. OTUs that differed between treatments were selected with respect to linear discriminant analysis (LDA) effect size (LEfSe) method (Segata et al., 2011).
Results
C. difficile Can Grow in the Presence of Dysbiotic and Healthy Fecal Microbiota in Vitro
Two types of fecal microbiota were tested. As explained previously one pooled sample was collected from healthy individuals while the other originated from samples subjected to C. difficile testing and were having negative result. Microbiota in such samples was previously and in this study shown to differ from microbiota in healthy samples (see below). Because the differences include characteristics associated with dysbalanced microbiota such as lower diversity and higher proportion of Proteobacteria, we are referring to this sample as to dysbiotic.
Each tested C. difficile strain showed specific pattern of growth in the presence of healthy or dysbiotic microbiota. Strains of ribotypes 010 and 027 grew best in the presence of dysbiotic microbiota, while ribotype 014/020 strain growth was similar in both microbiota types (Figure 1A).
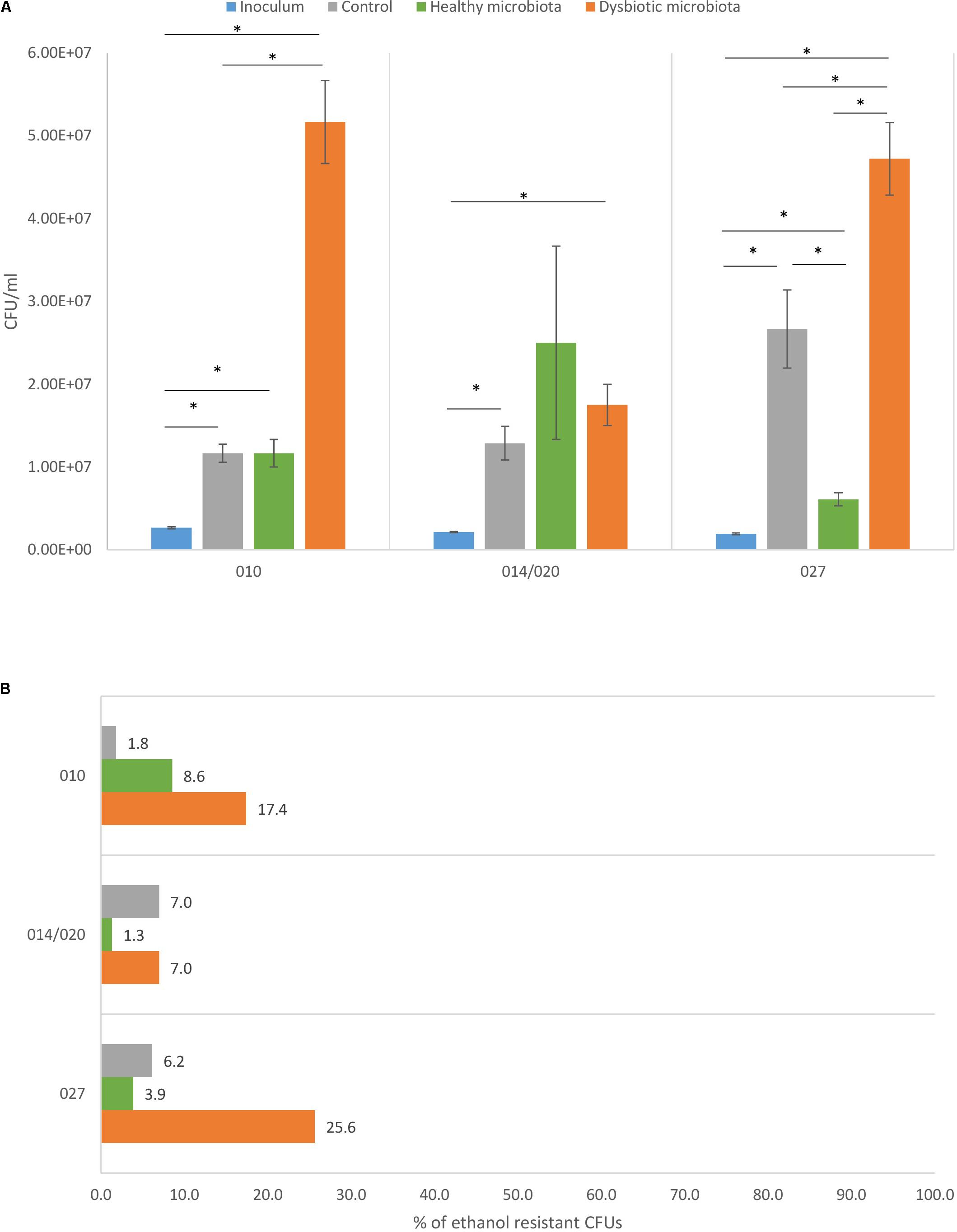
FIGURE 1. (A) Total viable cell count with corresponding standard deviation for C. difficile ribotypes 010, 014/020, and 027 strains in the inoculum (blue), in the control (gray), in co-culture with healthy fecal microbiota (green) and in co-culture with dysbiotic fecal microbiota (orange). ∗Statistically significant difference. (B) Percentage of C. difficile spores detected as ethanol resistant CFUs in proportion to total CFUs for ribotypes 010, 014/020, and 027 strains in the control (gray), in combination with healthy microbiota (green) and in combination with dysbiotic microbiota (orange).
In both microbiota types an increase in C. difficile total cell count was observed compared to original inoculum, indicating that all three strains were able to proliferate in the presence of disturbed and undisturbed microbiota. Difference between growth in control (growth medium only) and in combination with microbiota (any type) was significant only for PCR ribotype 027 strain (Figure 1A).
C. difficile Sporulation Is Better in the Presence of Dysbiotic Fecal Microbiota
The C. difficile sporulation frequency in co-cultures with fecal microbiota varied between 1.3 and 25.6% (Figure 1B). All three strains formed higher percentage of spores in co-culture with dysbiotic microbiota (7.0–25.6%), while in the co-culture with healthy microbiota spore percentage was lower (1.3–8.6%). Statistically significantly higher amount of spores in dysbiotic microbiota was observed only for ribotype 010 and 027 strains (one-way ANOVA: p < 0.05). Strains of ribotypes 014/020 and 027 displayed similar pattern of sporulation as for both the percentage of spores was the lowest in combination with healthy microbiota. For ribotype 010 strain the lowest sporulation was observed in control samples without fecal microbiota (Figure 1B).
Bacterial Richness and Diversity of Fecal Microbiota Decrease in the Presence of C. difficile
Sequencing yielded a total of 1,480,790 sequences of bacterial origin, with an average depth of 50,385 sequences per sample. Sequences were classified into 365 OTUs with relative abundance higher than 0.001%.
Input sample of healthy microbiota initially contained lower levels of Proteobacteria and higher proportions of Actinobacteria and Firmicutes than input sample of dysbiotic microbiota (Figure 2). After 3 days of in vitro incubation the control samples of both types of microbiota displayed similar composition at the phylum level (Figure 2). Nevertheless, statistical analysis revealed statistically higher bacterial richness and diversity in samples of healthy microbiota than in samples of dysbiotic microbiota (p < 0.05; Supplementary Table S1).
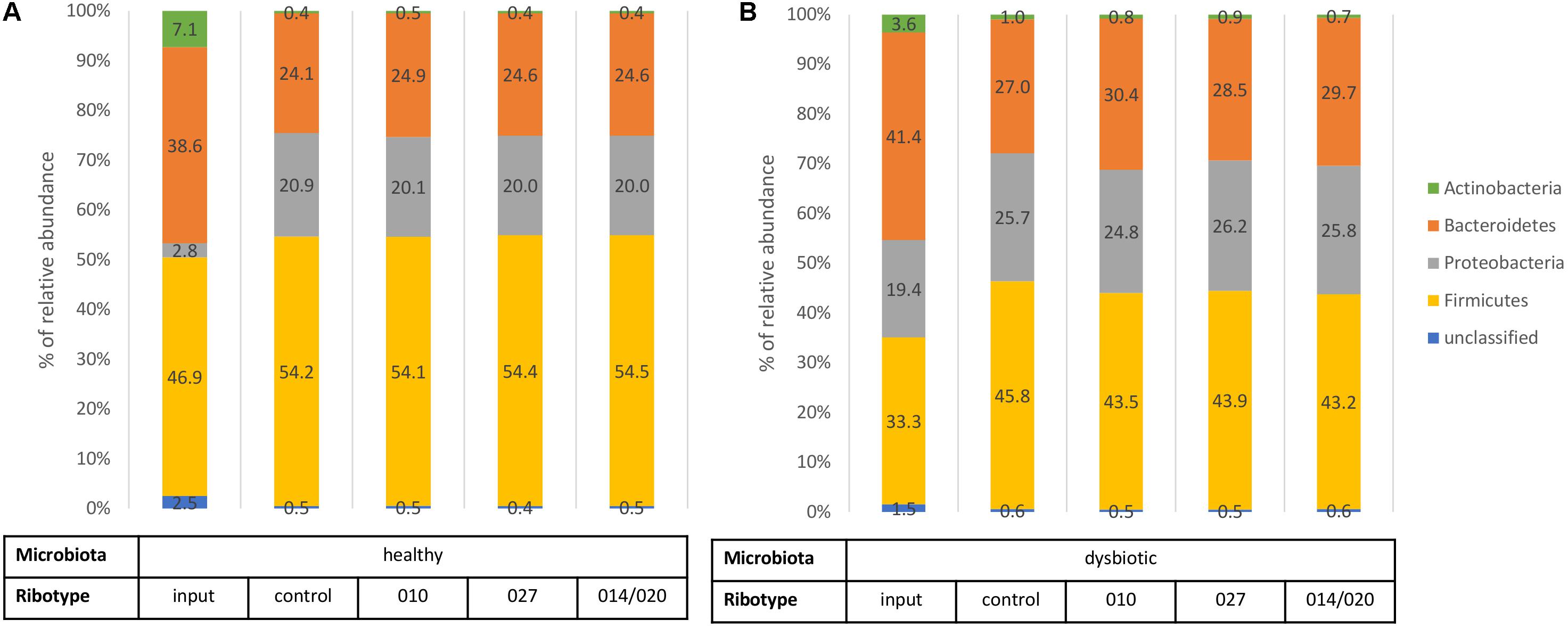
FIGURE 2. Bacterial phylum level assignments of average relative abundances for fecal input samples (before any cultivation), control samples (fecal microbiota only), and samples of fecal microbiota in combination with C. difficile ribotypes 010, 027, and 014/020 strains. Chart (A) represents samples of healthy fecal microbiota and chart (B) represents samples of dysbiotic fecal microbiota.
Also in comparison of control samples and co-cultures microbiota/C. difficile no statistically significant differences (p > 0.05) were detected at the phylum level (Figure 2), but significant changes were observed in bacterial richness and diversity (Figure 3) and on lower taxonomic levels (Table 1).
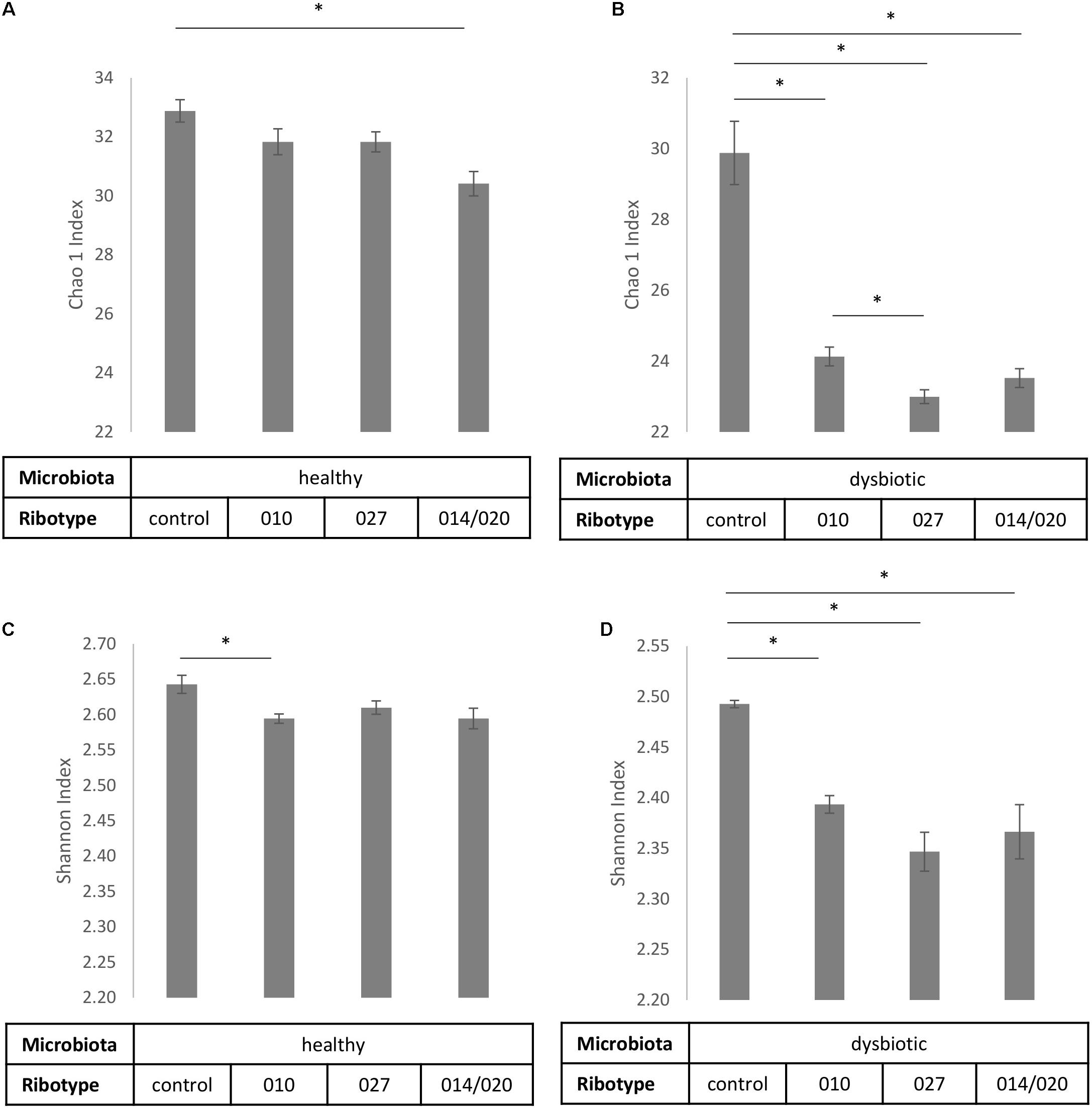
FIGURE 3. Alpha diversity estimates Chao1 (A,B) and Shannon index (C,D) with corresponding standard deviations for control samples of fecal microbiota and samples of fecal microbiota in combination with C. difficile ribotypes 010, 027, and 014/020 strains. Charts (A,C) represent samples of healthy fecal microbiota and charts (B,D) represent samples of dysbiotic fecal microbiota. ∗Statistically significant difference.
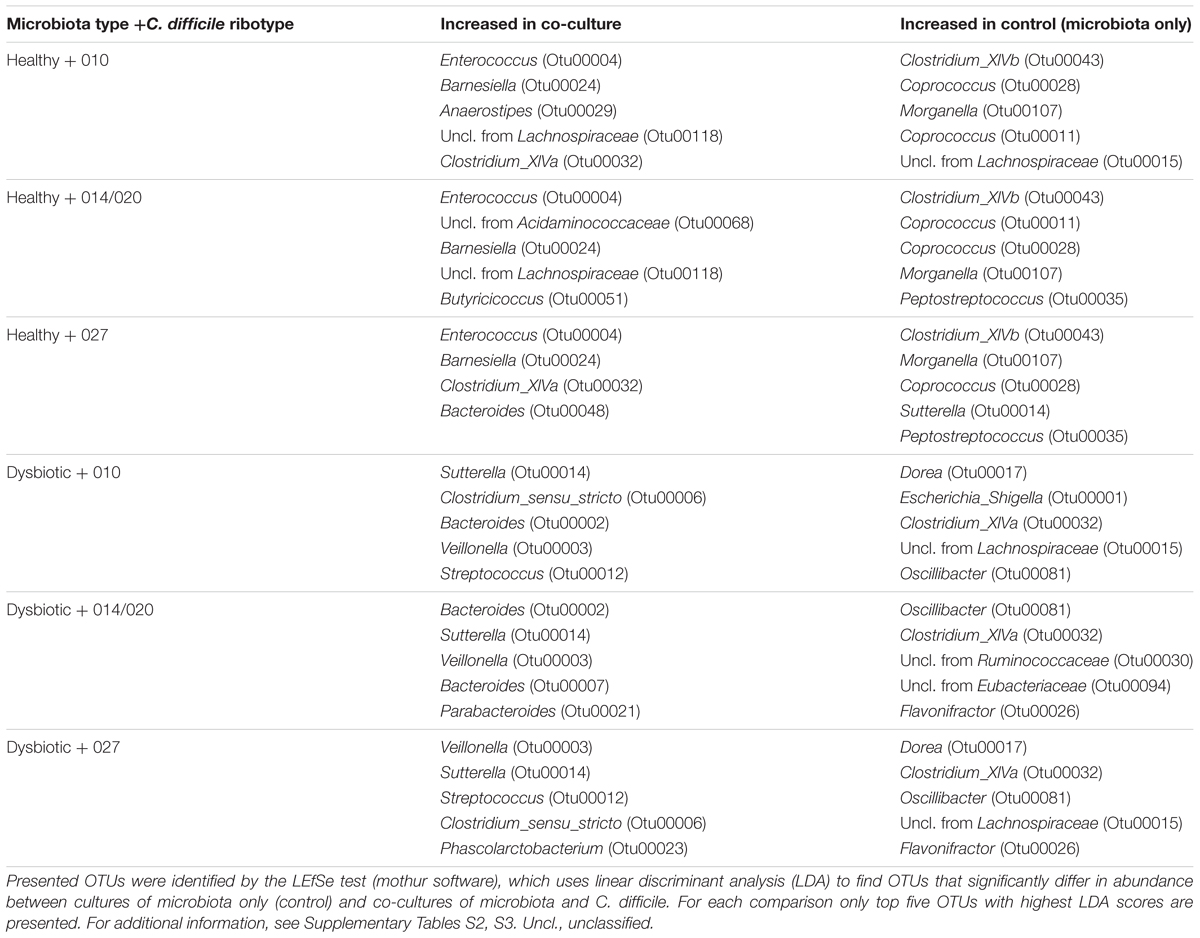
TABLE 1. Comparison of significantly increased OTUs in control samples (healthy or dysbiotic microbiota only) and in samples of co-cultures (healthy microbiota/C. difficile or dysbiotic microbiota/C. difficile).
To estimate the microbial richness we used the Chao1 index. According to control samples with healthy microbiota alone significantly lower bacterial richness was observed only if healthy microbiota was co-cultured with ribotype 014/020 strain (Figure 3A), whereas bacterial richness of dysbiotic microbiota was lowered in the presence of all three strains (Figure 3B). In addition, significant differences in bacterial richness were observed between co-culture of dysbiotic microbiota with ribotype 027 strain and co-culture of dysbiotic microbiota with ribotype 010 strain (Figure 3B).
Shannon index, predicting the microbial diversity, was significantly higher in control samples than in co-cultures microbiota/C. difficile (Figures 3C,D). For dysbiotic microbiota that trend was noticeable for all three strains, whereas bacterial diversity of healthy microbiota significantly decreased only in co-culture with ribotype 010 strain.
Dysbiotic Microbiota Is More Affected by C. difficile Than Healthy Microbiota
Bacterial community patterns of control and co-culture samples congregated in two major groups by hierarchical clustering; i.e., samples of dysbiotic microbiota (Figure 4, segment A) and samples of healthy microbiota (Figure 4, segment B). The deepest branching in both groups clearly divided control samples of microbiota only from co-cultures microbiota/C. difficile (Figure 4). According to weighted UniFrac test statistically significant differences in OTU composition were observed between control samples (dysbiotic or healthy microbiota only; Figure 4, segments a and b) and samples of microbiota (dysbiotic or healthy) in co-culture with all three C. difficile ribotypes (Figure 4, segments a1–3 and b1–3).
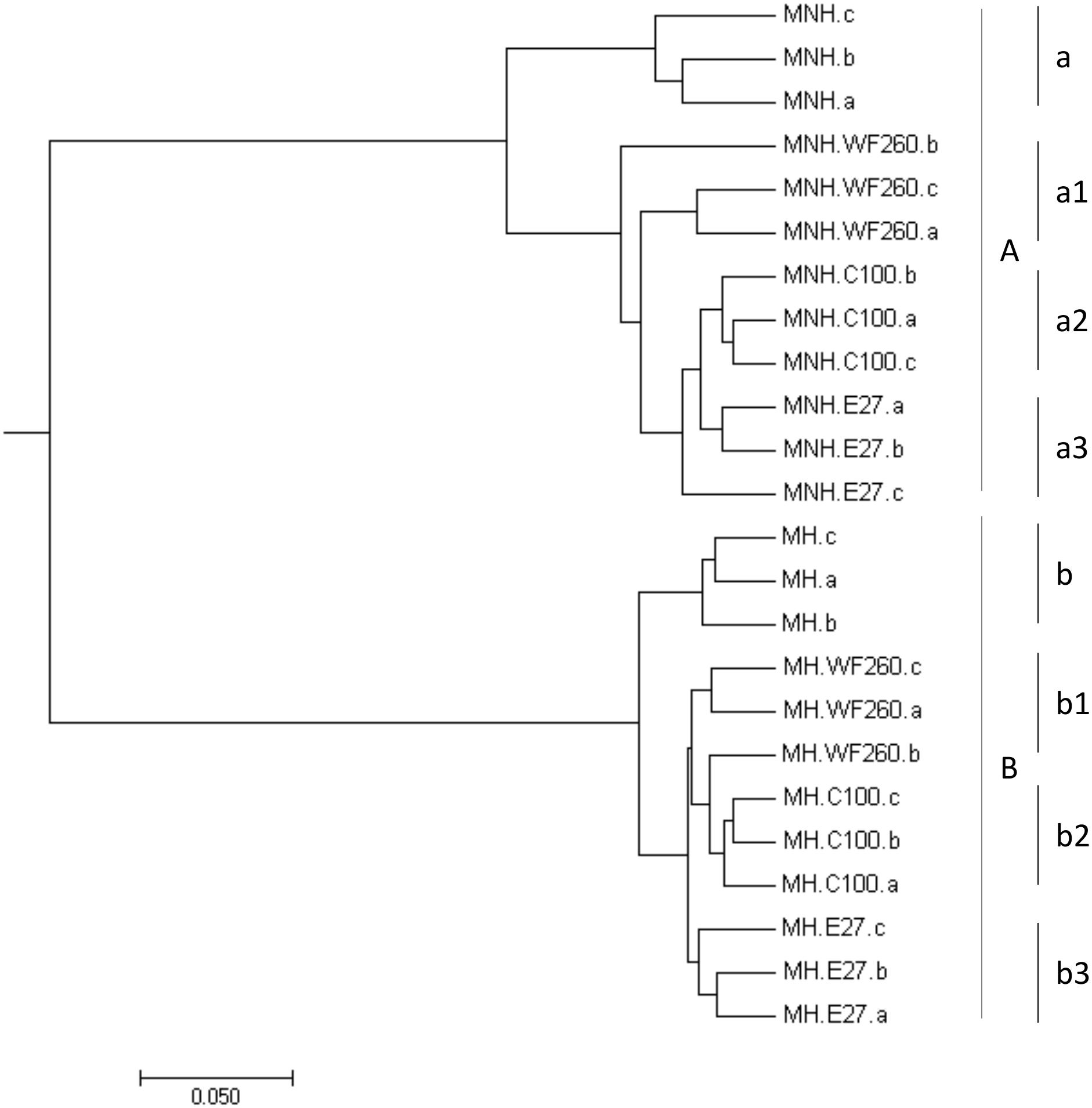
FIGURE 4. Hierarchical clustering of samples of dysbiotic fecal microbiota (MNH; segment A) and samples of healthy fecal microbiota (MH; segment B) without and with the addition of C. difficile ribotype 010 strain (C100), ribotype 027 strain (E27), and ribotype 014/020 strain (WF260), obtained by MEGA software (v.7.0.26). Statistically significant differences were observed between control samples (dysbiotic or healthy fecal microbiota only; segments a and b) and samples of fecal microbiota (dysbiotic or healthy) with added C. difficile ribotypes 010 (segments a2 and b2), 014/020 (segments a1 and b1), and 027 (segments a3 and b3) strains.
To identify significantly changed OTUs we have used the LEfSe test. Altogether, 62 OTUs were affected in co-cultures of dysbiotic microbiota and 45 OTUs in co-cultures of healthy microbiota (Supplementary Tables S2, S3). The smallest number of OTUs was affected in co-culture of healthy microbiota and C. difficile ribotype 027 strain (Supplementary Table S3). In general, the majority of significantly changed OTUs in both types of microbiota belonged to the phylum Firmicutes, with Lachnospiraceae and Ruminococcaceae origin. OTUs from both families were mostly associated with control samples of microbiota only (Table 1 and Supplementary Tables S2, S3). For Clostridium_XIVa, member of Lachnospiraceae family, an increase was observed in control samples of dysbiotic microbiota only as well as in co-cultures of healthy microbiota and C. difficile (Table 1 and Supplementary Tables S2, S3). Members of Bacteroidetes (particularly Bacteroides), Firmicutes (e.g., Veillonella) and Proteobacteria (i.e., Sutterella) were mostly associated with co-cultures of dysbiotic microbiota/C. difficile, whereas in co-cultures of healthy microbiota/C. difficile taxa from Bacteroidetes (i.e., Barnesiella) and Firmicutes (e.g., Enterococcus) were enriched (Table 1).
Discussion
Widespread use of antibiotics in hospitalized as well as in non-hospitalized patients leads to the disturbance of normal gut microbiota and allows C. difficile to expand in the gut. However, CDI might occur also in the individuals with no previous exposure to antibiotics or other apparent reason for microbiota disruption (Bauer et al., 2008; Wilcox et al., 2008).
To partially address the issue of C. difficile proliferation in dysbiotic and non-dysbiotic gut microenvironment, we explored C. difficile growth in the presence of two different types of microbiota. As previously C. difficile conditioned medium was shown to cause changes in gut microbiota in vitro (Horvat et al., 2017), we have studied if comparable changes are observed also with C. difficile vegetative cells.
All tested C. difficile strains successfully proliferated in our in vitro model with both types of fecal microbiota. That is in contrast to results obtained with some other in vitro gut models, where the inability of C. difficile to proliferate in the absence of antibiotic disturbance of microbiota was demonstrated (Borriello and Barclay, 1986; Wilson and Freter, 1986; Drummond et al., 2003; Freeman et al., 2003). Yet, the capability of C. difficile to colonize gut microenvironments even in the presence of complex microbial communities was demonstrated for ribotype 027 strains (Robinson et al., 2014). Therefore, it seems likely that ability of C. difficile to compete with complex non-dysbiotic microbial communities is strain dependent.
With the exception of ribotype 014/020 strain, C. difficile proliferated to a significantly greater extent in the presence of dysbiotic microbiota than in the presence of healthy microbiota (Figure 1A). In addition, greater proportion of OTUs was affected in co-cultures dysbiotic microbiota/C. difficile than in co-cultures healthy microbiota/C. difficile (Supplementary Tables S2, S3). Interestingly, the presence of ribotype 027 in healthy microbiota affected only small proportion of OTUs (Supplementary Table S3). That is in contrast with several previous studies on symptomatic humans or animals where ribotype 027 correlated with more disturbed microbiota (Lawley et al., 2012; Rea et al., 2012; Skraban et al., 2013).
Considering sporulation, higher proportion of C. difficile spores was observed in co-cultures with dysbiotic microbiota. That trend was apparent for all three strains (Figure 1B). This indicates, that conditions in dysbiotic microbiota support formation of greater amount of spores, which are important for further C. difficile transmission and spread. Additionally, an increased spore production appears to be relevant in severe CDI cases (Carlson et al., 2013).
The explanation of mechanism underlying the observed increased sporulation could be only speculative, as sporulation signals for C. difficile are not known (Zhu et al., 2018) in contrast to well understood germination signals (Theriot and Young, 2014). Nutrient starvation, quorum sensing and other environmental stimuli are most likely involved in sporulation signaling through several regulatory networks (Zhu et al., 2018). All of these three potential factors could be also involved in changes of microbiota dependent sporulation frequency.
Presence of C. difficile in co-cultures significantly decreased microbial richness and diversity in both types of fecal microbiota, but dysbiotic microbiota seemed to be more responsive and healthy microbiota more robust (Figure 3). Diversity was decreased in the presence of all three tested strains. The association of C. difficile colonization and/or infection with a decrease in gut microbiota diversity is well documented, but was so far only viewed as a predisposing condition (reviewed in Vincent and Manges, 2015) and not also as a consequence of C. difficile presence as suggested here and elsewhere (Horvat et al., 2017).
In general, potentially beneficial autochthonous bacteria, particularly butyrate-producing bacteria (Lachnospiraceae and Ruminococcaceae) were present in larger proportions in microbiota cultivated without C. difficile. These bacteria are usually found in healthy subjects and are thought to provide colonization resistance against CDI (Ross et al., 2016).
The presence of C. difficile in any type of microbiota resulted in increased proportions of opportunistic pathogens (i.e., Sutterella in co-culture with dysbiotic microbiota and Enterococcus in co-culture with healthy microbiota). Previous studies already reported about an increased proportion of Sutterella in C. difficile positive subjects (Perez-Cobas et al., 2014; Milani et al., 2016). Also, the presence of Sutterella was linked to several other human diseases, where gut microbiota seems to play a crucial role in their development (e.g., metabolic syndrome and autism) (Wang et al., 2013; Lim et al., 2017). An association between C. difficile colonization/infection and enterococci or vancomycin-resistant enterococci (VRE) is also well documented (Ozaki et al., 2004; Choi et al., 2011; Fujitani et al., 2011; Stripling et al., 2015; McKinley et al., 2016). Moreover, our previous study demonstrated undisturbed growth of representative strain of Enterococcus in co-culture with C. difficile on agar plates (Horvat et al., 2017). Veillonella was another genus that increased in the presence of C. difficile in dysbiotic microbiota. That is in agreement with two other studies (Antharam et al., 2016; Khanna et al., 2016) and in contrast to our previous study, where reduced proportions were found in C. difficile conditioned medium (Horvat et al., 2017). Decreased levels of Veillonella were also found in oncological patients colonized with C. difficile (Zwielehner et al., 2011).
Our results further support the observation that C. difficile could have an impact on gut microbiota and that the impact is strain dependent. Dysbiotic microbiota is in vitro more susceptible for disturbances caused by co-culture with C. difficile and is also associated with better sporulation frequency of C. difficile. Better competitive ability, inhibition of certain bacterial groups associated with colonization resistance and better sporulation in the presence of dysbiotic microbiota possibly contribute to endemic spread of some strains.
Author Contributions
SH performed the experiments and data analysis. MR contributed to the data analysis. Both authors designed the study and contributed to the text preparation and reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding P3-0387 and Ph.D. program ‘Mladi raziskovalci’).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01633/full#supplementary-material
References
Aguirre, M., Ramiro-Garcia, J., Koenen, M. E., and Venema, K. (2014). To pool or not to pool? impact of the use of individual and pooled fecal samples for in vitro fermentation studies. J. Microbiol. Methods 107, 1–7. doi: 10.1016/j.mimet.2014.08.022
Antharam, V., McEwen, D., Garrett, T., Dossey, A., Li, E., Kozlov, A., et al. (2016). An integrated metabolomic and microbiome analysis identified specific gut microbiota associated with fecal cholesterol and coprostanol in Clostridium difficile infection. PLoS One 11:e0148824. doi: 10.1371/journal.pone.0148824
Bauer, M. P., Goorhuis, A., Koster, T., Numan-Ruberg, S. C., Hagen, E. C., Debast, S. B., et al. (2008). Community-onset Clostridium difficile-associated diarrhoea not associated with antibiotic usage–two case reports with review of the changing epidemiology of Clostridium difficile-associated diarrhoea. Neth. J. Med. 66, 207–211.
Borriello, S. P., and Barclay, F. E. (1986). An in-vitro model of colonisation resistance to Clostridium difficile infection. J. Med. Microbiol. 21, 299–309. doi: 10.1099/00222615-21-4-299
Britton, R. A., and Young, V. B. (2012). Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 20, 313–319. doi: 10.1016/j.tim.2012.04.001
Carlson, P. E., Walk, S. T., Bourgis, A. E., Liu, M. W., Kopliku, F., Lo, E., et al. (2013). The relationship between phenotype, ribotype, and clinical disease in human Clostridium difficile isolates. Anaerobe 24, 109–116. doi: 10.1016/j.anaerobe.2013.04.003
Choi, H., Kim, K., Lee, S., and Lee, S. (2011). Risk factors for recurrence of Clostridium difficile infection: effect of vancomycin-resistant Enterococci colonization. J. Korean Med. Sci. 26, 859–864. doi: 10.3346/jkms.2011.26.7.859
Drummond, L. J., Smith, D. G., and Poxton, I. R. (2003). Effects of sub-MIC concentrations of antibiotics on growth of and toxin production by Clostridium difficile. J. Med. Microbiol. 52(Pt 12), 1033–1038. doi: 10.1099/jmm.0.05387-0
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Ferreyra, J. A., Ng, K. M., and Sonnenburg, J. L. (2014). The enteric two-step: nutritional strategies of bacterial pathogens within the gut. Cell. Microbiol. 16, 993–1003. doi: 10.1111/cmi.12300
Freeman, J., O’Neill, F. J., and Wilcox, M. H. (2003). Effects of cefotaxime and desacetylcefotaxime upon Clostridium difficile proliferation and toxin production in a triple-stage chemostat model of the human gut. J. Antimicrob. Chemother. 52, 96–102. doi: 10.1093/jac/dkg267
Fujitani, S., George, W., Morgan, M., Nichols, S., and Murthy, A. (2011). Implications for vancomycin-resistant Enterococcus colonization associated with Clostridium difficile infections. Am. J. Infect. Control 39, 188–193. doi: 10.1016/j.ajic.2010.10.024
Gerding, D. N., Johnson, S., Rupnik, M., and Aktories, K. (2014). Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5, 15–27. doi: 10.4161/gmic.26854
Gupta, A., and Khanna, S. (2014). Community-acquired Clostridium difficile infection: an increasing public health threat. Infect. Drug Resist. 7, 63–72. doi: 10.2147/IDR.S46780
Horvat, S., Mahnic, A., Breskvar, M., Dzeroski, S., and Rupnik, M. (2017). Evaluating the effect of Clostridium difficile conditioned medium on fecal microbiota community structure. Sci. Rep. 7:16448. doi: 10.1038/s41598-017-154341
Khanna, S., Montassier, E., Schmidt, B., Patel, R., Knights, D., Pardi, D., et al. (2016). Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Aliment. Pharmacol. Ther. 44, 715–727. doi: 10.1111/apt.13750
Kozich, J., Westcott, S., Baxter, N., Highlander, S., and Schloss, P. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lawley, T. D., Clare, S., Walker, A. W., Stares, M. D., Connor, T. R., Raisen, C., et al. (2012). Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995. doi: 10.1371/journal.ppat.1002995
Lawson, P., Citron, D., Tyrrell, K., and Finegold, S. (2016). Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) prevot 1938. Anaerobe 40, 95–99. doi: 10.1016/j.anaerobe.2016.06.008
Leffler, D., and Lamont, J. (2015). Clostridium difficile infection. New Eng. J. Med. 372, 1539–1548. doi: 10.1056/NEJMra1403772
Lim, M. Y., You, H. J., Yoon, H. S., Kwon, B., Lee, J. Y., Lee, S., et al. (2017). The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 66, 1031–1038. doi: 10.1136/gutjnl-2015-311326
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Martin, J., Monaghan, T., and Wilcox, M. (2016). Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 13, 206–216. doi: 10.1038/nrgastro.2016.25
McKinley, L., Becerra, B., Moriarty, H., Short, T. H., Hagle, M., Reymann, A., et al. (2016). Vancomycin-resistant Enterococcus co-colonization rates with methicillin-resistant Staphylococcus aureus and Clostridium difficile in critically ill veterans. Am. J. Infect. Control 44, 1047–1049. doi: 10.1016/j.ajic.2016.02.005
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 19:386. doi: 10.1186/1471-2105-9-386
Milani, C., Ticinesi, A., Gerritsen, J., Nouvenne, A., Lugli, G., Mancabelli, L., et al. (2016). Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci. Rep. 6:25945. doi: 10.1038/srep25945
Ozaki, E., Kato, H., Kita, H., Karasawa, T., Maegawa, T., Koino, Y., et al. (2004). Clostridium difficile colonization in healthy adults: transient colonization and correlation with Enterococcal colonization. J. Med. Microbiol. 53, 167–172. doi: 10.1099/jmm.0.05376-0
Perez-Cobas, A., Artacho, A., Ott, S., Moya, A., Gosalbes, M., and Latorre, A. (2014). Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Front. Microbiol. 5:335. doi: 10.3389/fmicb.2014.00335
Pérez-Cobas, A., Moya A., Gosalbes, M., and Latorre, A. (2015). Colonization resistance of the gut microbiota against Clostridium difficile. Antibiotics 4, 337–357. doi: 10.3390/antibiotics4030337
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rea, M. C., O’Sullivan, O., Shanahan, F., O’Toole, P. W., Stanton, C., Ross, R. P., et al. (2012). Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J. Clin. Microbiol. 50, 867–875. doi: 10.1128/JCM.05176-11
Robinson, C., Auchtung, J., Collins, J., and Britton, R. (2014). Epidemic Clostridium difficile strains demonstrate increased competitive fitness compared to nonepidemic isolates. Infect. Immun. 82, 2815–2825. doi: 10.1128/IAI.01524-14
Ross, C. L., Spinler, J. K., and Savidge, T. C. (2016). Structural and functional changes within the gut microbiota and susceptibility to Clostridium difficile infection. Anaerobe 41, 37–43. doi: 10.1016/j.anaerobe.2016.05.006
Rupnik, M., Wilcox, M., and Gerding, D. (2009). Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536. doi: 10.1038/nrmicro2164
Schloss, P., Westcott, S., Ryabin, T., Hall, J., Hartmann, M., Hollister, E., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Skraban, J., Dzeroski, S., Zenko, B., Mongus, D., Gangl, S., and Rupnik, M. (2013). Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One 8:e58005. doi: 10.1371/journal.pone.0058005
Stecher, B., Robbiani, R., Walker, A. W., Westendorf, A. M., Barthel, M., Kremer, M., et al. (2007). Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5:e244. doi: 10.1371/journal.pbio.0050244
Stripling, J., Kumar, R., Baddley, J., Nellore, A., Dixon, P., Howard, D., et al. (2015). Loss of vancomycin-resistant Enterococcus fecal dominance in an organ transplant patient with Clostridium difficile colitis after fecal microbiota transplant. Open Forum Infect. Dis. 2:ofv078. doi: 10.1093/ofid/ofv078
Studer, N., Desharnais, L., Beutler, M., Brugiroux, S., Terrazos, M. A., Menin, L., et al. (2016). Functional intestinal bile acid 7α-Dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model. Front. Cell. Infect. Microbiol. 6:191. doi: 10.3389/fcimb.2016.00191
Theriot, C. M., and Young, V. B. (2014). Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 5, 86–95. doi: 10.4161/gmic.27131
Vincent, C., and Manges, A. (2015). Antimicrobial use, human gut microbiota and Clostridium difficile colonization and infection. Antibiotics 4, 230–253. doi: 10.3390/antibiotics4030230
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T., and Conlon, M. A. (2013). Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 4:42. doi: 10.1186/2040-2392-4-42
Wilcox, M. H., Mooney, L., Bendall, R., Settle, C. D., and Fawley, W. N. (2008). A case-control study of community-associated Clostridium difficile infection. J. Antimicrob. Chemother. 62, 388–396. doi: 10.1093/jac/dkn163
Wilson, K. H., and Freter, R. (1986). Interaction of Clostridium difficile and Escherichia coli with microfloras in continuous-flow cultures and gnotobiotic mice. Infect. Immun. 54, 354–358
Zhu, D., Sorg, J. A., and Sun, X. (2018). Clostridioides difficile biology: sporulation, germination, and corresponding therapies for C. difficile Infection. Front. Cell. Infect. Microbiol. 8:29. doi: 10.3389/fcimb.2018.00029
Keywords: Clostridioides (Clostridium) difficile, gut microbiota, sporulation, colonization, pathogenesis, CDI
Citation: Horvat S and Rupnik M (2018) Interactions Between Clostridioides difficile and Fecal Microbiota in in Vitro Batch Model: Growth, Sporulation, and Microbiota Changes. Front. Microbiol. 9:1633. doi: 10.3389/fmicb.2018.01633
Received: 23 February 2018; Accepted: 29 June 2018;
Published: 24 July 2018.
Edited by:
Meina Neumann-Schaal, Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), GermanyReviewed by:
Jozsef Soki, University of Szeged, HungaryTill Strowig, Helmholtz-Gemeinschaft Deutscher Forschungszentren (HZ), Germany
Copyright © 2018 Horvat and Rupnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maja Rupnik, bWFqYS5ydXBuaWtAbmx6b2guc2k=