
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 16 July 2018
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01595
 Hsin-Yu Li1†
Hsin-Yu Li1† Cheng-Yen Kao1†‡
Cheng-Yen Kao1†‡ Wei-Hung Lin2
Wei-Hung Lin2 Po-Xing Zheng3
Po-Xing Zheng3 Jing-Jou Yan4Ming-Cheng Wang2
Jing-Jou Yan4Ming-Cheng Wang2 Ching-Hao Teng5
Ching-Hao Teng5 Chin-Chung Tseng2
Chin-Chung Tseng2 Jiunn-Jong Wu1*
Jiunn-Jong Wu1*Prokaryotic CRISPR-Cas systems limit the acquisition of genetic elements and provide immunity against invasive bacteriophage. The characteristics of CRISPR-Cas systems in clinical Klebsiella pneumoniae isolates are still unknown. Here, 97 K. pneumoniae genomes retrieved from the Integrated Microbial Genomes & Microbiomes genome database and 176 clinical isolates obtained from patients with bloodstream (BSI, n = 87) or urinary tract infections (UTI, n = 89) in Taiwan, were used for analysis. Forty out of ninety-seven genomes (41.2%) had CRISPR-Cas systems identified by the combination of CRISPRFinder and cas1 gene sequence alignment. The phylogenetic trees revealed that CRISPR-Cas systems in K. pneumoniae were divided into two types (type I-E, 23; subtype I-E∗, 17) based on the sequences of Cas1 and Cas3 proteins and their location in the chromosome. The distribution of type I-E and I-E∗ CRISPR-Cas systems was associated with the multilocus sequence typing and the pulsed-field gel electrophoresis results. Importantly, no CRISPR-Cas system was identified in published genomes of clonal complex 258 isolates (ST11 and ST258), which comprise the largest multi-drug resistant K. pneumoniae clonal group worldwide. PCR with cas-specific primers showed that 30.7% (54/176) of the clinical isolates had a CRISPR-Cas system. Among clinical isolates, more type I-E CRISPR-Cas systems were found in UTI isolates (BSI, 5.7%; UTI, 11.2%), and subtype I-E∗ CRISPR-Cas systems were dominant in BSI isolates (BSI, 28.7%; UTI, 15.7%) (p = 0.042). Isolates which had subtype I-E∗ CRISPR-Cas system were more susceptible to ampicillin-sulbactam (p = 0.009), cefazolin (p = 0.016), cefuroxime (p = 0.039), and gentamicin (p = 0.012), compared to the CRISPR-negative isolates. The strains containing subtype I-E∗ CRISPR-Cas systems had decreased numbers of plasmids, prophage regions, and acquired antibiotic resistance genes in their published genomes. Here, we first revealed subtype I-E∗ CRISPR-Cas system in K. pneumoniae potentially interfering with the acquisition of phages and plasmids harboring antibiotic resistance determinants, and thus maintained these isolates susceptible to antibiotics.
During bacterial evolution, the ability of bacteria to adapt to various hosts and environments has been favored by the acquisition of DNA elements through horizontal gene transfer (HGT) (Frost et al., 2005; Ilyas et al., 2017). An adaptive immune system, clustered regularly interspaced short palindromic repeats and their associated Cas proteins (CRISPR-Cas), which has been identified in many species of bacteria, allows bacteria to limit the acquisition of external genetic elements, and thus balances the need to acquire beneficial characteristics through HGT with the need to defend against infection by bacteriophage (Barrangou, 2015; Jiang et al., 2016; Hoyland-Kroghsbo et al., 2017). Moreover, the CRISPR-Cas systems have been shown to play a critical role in the exchange of genetic elements, biofilm formation, colonization, and virulence regulation in multiple pathogenic bacteria (Shah and Garrett, 2011; Louwen et al., 2013, 2014; Almendros et al., 2014).
The CRISPR elements are composed of short direct repeat sequences (DR) separated by unique sequences (spacers) that range in size from 26 to 72 bp and originate from mobile genetic elements, such as phages, transposons, or plasmids (Barrangou, 2015; Koonin et al., 2017). Based on the cas gene content, cas operon architecture, Cas protein sequences, and processes that underlie the aforementioned steps, CRISPR-Cas systems are divided into two main classes, which encompass 6 major types and 33 different subtypes (Makarova and Koonin, 2015; Makarova et al., 2015; Koonin et al., 2017). The number of cas genes varies from 4 to over 20, and the Cas proteins contain a variety of enzymatic domains with nuclease, helicase, or polymerase activity (Makarova et al., 2011; Koonin et al., 2017). Therefore, Cas proteins with enzymatic activities are required for the acquisition of spacers as well as for marking the invader elements (Nunez et al., 2014).
Klebsiella pneumoniae is a Gram-negative bacteria belonging to the family Enterobacteriaceae, closely related to the pathogens Salmonella enterica and Escherichia coli. K. pneumoniae has been the most frequent pathogen causing hospital-acquired infections and has high rates of antibiotic resistance (Gomez-Simmonds and Uhlemann, 2017). The multi-drug resistance (MDR) and hypervirulent phenotype of K. pneumoniae are commonly associated with the presence of high molecular weight plasmids (Mathers et al., 2015; Lee et al., 2017). CRISPR-Cas systems are shown to be associated with antibiotic susceptibility in Streptococcus pyogenes and E. coli (Zheng et al., 2014; Aydin et al., 2017). The CRISPRI-F system is found potentially interfering with the acquisition of antibiotic-resistant plasmids, and thus CRISPRI-F system is dominant in antibiotic-susceptible E. coli (Aydin et al., 2017). However, the association of CRISPR-Cas systems and antibiotic resistance in K. pneumoniae is still unclear.
The prevalence of CRISPR-Cas systems among Klebsiella genomes was very different using CRISPERFinder analysis (Ostria-Hernandez et al., 2015; Shen et al., 2017). Ostria-Hernandez et al. (2015) reported that CRISPR-Cas systems were identified in 6 out of the 52 (11.5%) K. pneumoniae genome sequences by using CRISPRFinder. In contrast, a total of 37 (54.4%) CRISPR-Cas systems were found by CRISPERFinder among the 68 Klebsiella genomes retrieved from the NCBI genome database (Shen et al., 2017). Moreover, three different CRISPR-Cas systems (type I-E, type I-F, and subtype I-E∗) were found in the published Klebsiella genomes (Shen et al., 2017). Type I-E and subtype I-E∗ CRISPR-Cas systems were found in K. pneumoniae and K. oxytoca, and type I-F CRISPR-Cas system was identified in only K. oxytoca (Shen et al., 2017). In this study, we characterized the CRISPR-Cas systems in K. pneumoniae published genomes and clinical isolates obtained from patients with bloodstream infection (BSI) or urinary tract infection (UTI) in Taiwan and found that isolates having subtype I-E∗ CRISPR-Cas system are more susceptible to certain antibiotics.
A total of 97 complete genome sequences of K. pneumoniae (including chromosome and plasmids sequences), were retrieved from the Integrated Microbial Genomes & Microbiomes (IMG/M) genome database1. CRISPRFinder was used to identify the presence of CRISPR-Cas systems and spacers among the genomes2. Given that Cas1 is considered a genetic and universal marker for the CRISPR-Cas systems, BLASTn of cas1 and cas3 genes was used to validate the results of CRISPRFinder3. Sequence types (STs) were determined based on a comparative analysis of seven housekeeping genes (rpoB, gapA, mdh, pgi, phoE, infB, and tonB) against the K. pneumoniae PubMLST database4. The software MEGA X was used to perform a Maximum Likelihood-based phylogenetic tree (Jukes–Cantor model) based on aligned nucleotide sequences of concatenated multilocus sequence typing (MLST) genes (12,096 bp) of CRISPR-Cas positive K. pneumoniae genomes (Kumar et al., 2018).
PHAge Search Tool (PHAST) was used to identify, annotate, and graphically display prophage sequences within K. pneumoniae genomes5 (Zhou et al., 2011). Acquired antibiotic resistance genes carried on plasmids, including aminoglycoside, β-lactam, fluoroquinolones, MLS (macrolides, lincosamides, and streptogramines), rifampicin, phenicols, sulfonamide, tetracycline, and trimethoprim resistance genes, were identified by ResFinder 3.0 using BLAST for identification of acquired antibiotic resistance genes in plasmid sequences of the 97 genomes6 (Zankari et al., 2012).
Eighty-seven and eighty-nine non-consecutive, non-duplicate clinical isolates were obtained from patients with BSI and UTI respectively, at the National Cheng Kung University Hospital, Taiwan. These isolates were randomly collected and excluded the outbreak strains over a period of 6 years, between 2011 and 2016. K. pneumoniae isolates were identified by colony morphology, Gram stain, biochemical tests, and the Vitek 2 system (bioMérieux, Marcy-l’Etoile, France) according to the manufacturer’s recommendations. K. pneumoniae was grown on Luria-Bertani (LB) agar or broth. The isolates were stored at -80°C in LB broth containing 20% glycerol (v/v) until used.
Primers used in this study are listed in Supplementary Table S1. DNA was extracted with the boiling method. In brief, 5 mL bacterial culture was grown for 16 h, and was centrifuged (3,500 rpm, 5 min). The pellet was resuspended in 1.0 mL of TE buffer (EDTA 1.0 mM; Tris-HCl 10 mM, pH = 8.0), boiled for 15 min, and was centrifuged (8,000 rpm, 5 min). The supernatant containing genomic DNA was collected and stored at 4°C until used. The PCR reactions were carried out in a total volume of 25 μL containing 1x PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 1 unit of GoTaqDNA polymerases (Promega, Madison, WI, United States), 10 pmol of each primer and 1 μL DNA template. The PCR amplifications were performed using an ABI 2720 Thermal Cycler (Applied Biosystems, Foster City, CA, United States). The PCR conditions forcas1 and cas3 genes were: 95°C for 5 min, followed by 30 repeated cycles of 30 s at 95°C, 30 s of annealing at 60°C and 1 min of extension at 72°C, followed by 7 min as final extension at 72°C.
Amplification of seven housekeeping genes, rpoB, gapA, mdh, pgi, phoE, infB, and tonB, was accomplished using PCR. Primer sets for gene amplification and sequencing were described previously (Diancourt et al., 2005), and the annealing temperatures of each primer were described at the K. pneumoniae PubMLST database. However, primers for pgi amplification showed poor specificity in this study, therefore, primers pgi-1 (GGCCGTTAGTAGAGCTGTCG) and pgi-2 (GAAGAACGTGAATCCGGAAA) were designed and used for 1,040 bp pgi fragment amplification and sequencing (annealing temperature: 60°C). STs were determined by comparing the nucleotide sequences against the K. pneumoniae PubMLST database.
Pulsed-field gel electrophoresis (PFGE) of XbaI-digested genomic DNA was carried out with a CHEF Mapper XA apparatus (Bio-Rad Laboratories, Inc., Hercules, CA, United States) using the following parameters: separation on a 1% agarose gel (Seakem Gold agarose; FMC Bio Products) in 0.5× Tris-Borate-EDTA for 19 h at 14°C with pulsed times ranging from 5 to 35 s at 6 V/cm. The gels were stained with ethidium bromide and photographed with UV transillumination. PFGE profiles were analyzed and compared using the GelCompar II software, version 2.0 (Unimed Healthcare, Inc., Houston, TX, United States).
Antibiotic susceptibilities were determined by the disk diffusion method on Mueller-Hinton agar according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (Clinical and Laboratory Standards Institute [CLSI], 2009). All clinical isolates were tested for the susceptibilities of nine antibiotics, including amikacin (30 μg/mL), ampicillin-sulbactam (10/10 μg/mL), cefotaxime (30 μg/mL), cefazolin (30 μg/mL), cefuroxime (30 μg/mL), ertapenem (10 μg/mL), gentamicin (10 μg/mL), levofloxacin (5 μg/mL), and sulfamethoxazole-trimethoprim (23.75/1.25 μg/mL) (BD BioSciences, Cockeysville, MD, United States). E. coli ATCC 25922 was used as quality control strain. Antibiotic susceptibility was interpreted according to the CLSI guidelines (Clinical and Laboratory Standards Institute [CLSI], 2016).
The Student’s t-test and paired t-tests were applied as appropriate for the parametric differences. Chi-square tests were used to test the association of a set of counts or frequencies. All tests with a p-value < 0.05 were taken as significant.
Forty-three out of the ninety-seven genomes (44.3%) had confirmed CRISPR loci determined by CRISPERFinder. However, no cas1 gene was found in strains YH43, KPNIH29, DHQP1002001, and Kpn555. Therefore, these four strains were defined as CRISPR-Cas system negative in the following analysis. In contrast, strain SB3432, which had a questionable CRISPR-Cas system as determined by CRISPERFinder, was defined as CRISPR-Cas system positive strain with the Cas proteins present in the genome. Therefore, a total of 40 (41.2%) CRISPR-Cas system positive genomes were found in this study (Table 1).
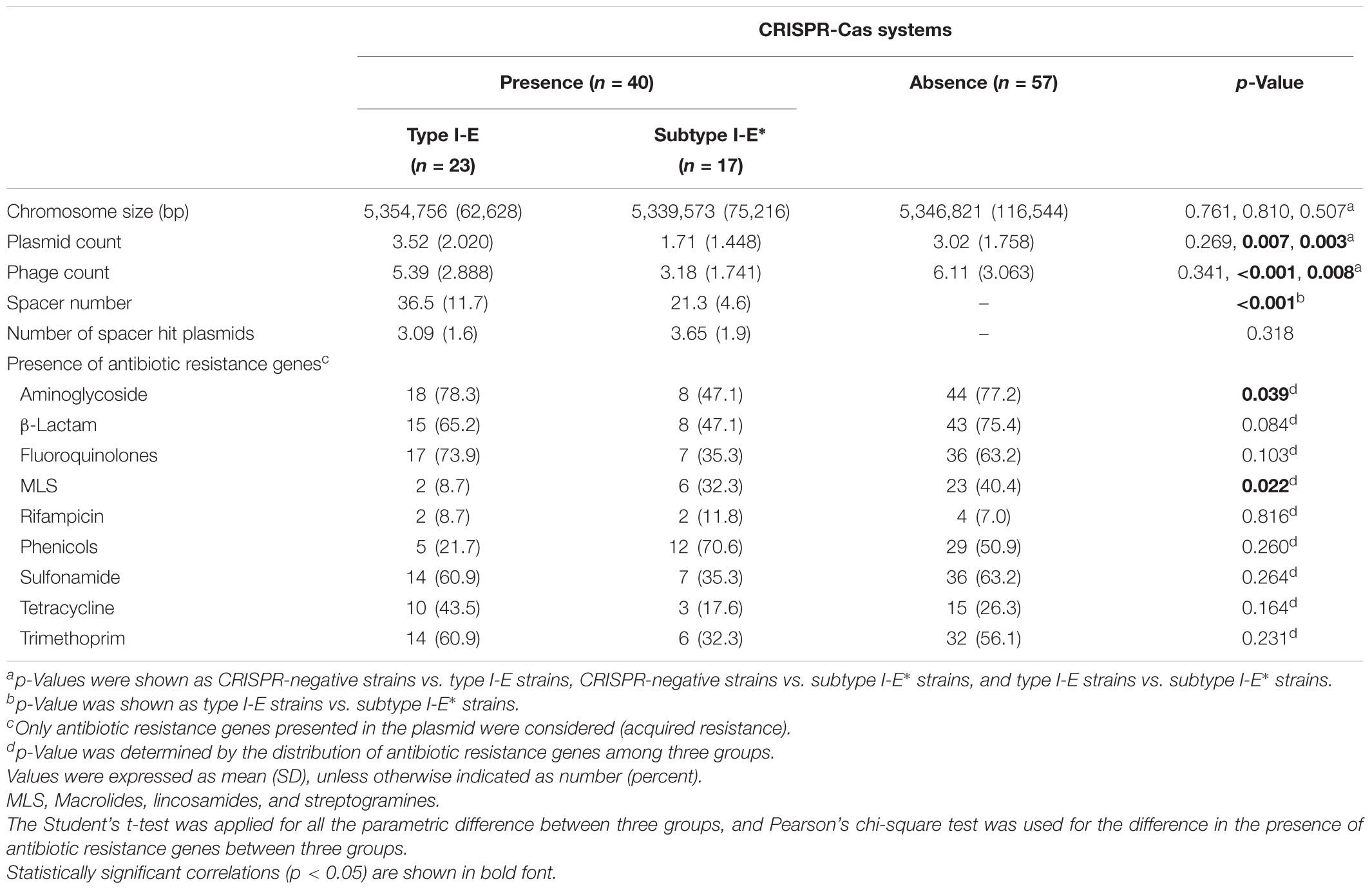
TABLE 1. The association of CRISPR-Cas systems with chromosome size, plasmid count, phage count, spacer numbers, number of spacer hit plasmids, and the presence of antibiotic resistance genes among 97 K. pneumoniae genomes.
The prevalence of CRISPR-Cas systems among Klebsiella strains was very diverse using CRISPERFinder analysis (Ostria-Hernandez et al., 2015; Shen et al., 2017). CRISPERFinder with the Vmatch program to browse the maximal repeats to get possible CRISPR localization, following by direct consensus DR selection according to candidate occurrences and a score computation. After DR and spacer size check, the tandem repeats are eliminated using ClustalW for aligning spacers (Grissa et al., 2007). The presence of cas genes was not verified by CRISPERFinder. Previous report indicated that Cas1-Cas2 complex formation played a critical role in mediating spacer acquisition during CRISPR-Cas adaptive immunity (Nunez et al., 2014). Therefore, we suggested that CRISPERFinder is useful for screening for CRISPR-Cas system, however, the presence of Cas genes/proteins should be validated.
The phylogenetic trees showed two types of Cas1 or Cas3 proteins (Supplementary Figure S1). Twenty-three strains (23/40, 57.5%) had Cas1 and Cas3 alleles (type A) corresponding to CRISPR-Cas type I-E, while the remaining strains (17/40, 42.5%) had Cas1 and Cas3 alleles (type B) consistent with CRISPR-Cas I-E∗ (Table 1 and Supplementary Figure S1). Moreover, the identity of protein sequences between type A Cas1 and type B Cas1 was 35%, and the identity of protein sequences between type A Cas3 and type B Cas3 was 33%. Therefore, the CRISPR type-specific primers for cas1 and cas3 genes were designed for determining the distribution of CRISPR-Cas systems in clinical K. pneumoniae isolates (Supplementary Table S1).
Shen et al. (2017) reported that type I-E and subtype I-E∗ CRISPR-Cas systems were detected in K. pneumoniae with different locations in the chromosome. Type I-E CRISPR-Cas system was located in the iap-cysH region, and subtype I-E∗ CRISPR-Cas system was located in the ABC transport system-glyoxalase region (Shen et al., 2017). Here, we revealed that type I-E and subtype I-E∗ CRISPR-Cas systems in K. pneumoniae could be typed based on the amino sequences of Cas1 and Cas3. However, the function/activity of Cas proteins in different CRISPR-Cas type systems in K. pneumoniae remains to be determined.
Type I-E CRISPR-Cas was found in ST34, 45, 66, 67, 147, 273, 383, 392, and 941, while subtype I-E∗ CRISPR-Cas was present in ST14, 15, 23, 28, 111, 374, 493, and 505 (Supplementary Table S2). The distribution of CRISPR-Cas systems was strongly associated with certain STs (Supplementary Table S2, p < 0.001). To better understand any MLST relationship with the CRISPR-Cas types across the K. pneumoniae, phylogenetic tree of 40 published K. pneumoniae genomes based on aligned nucleotide sequences of concatenated MLST sequences is shown in Figure 1. Thirty-seven isolates were assigned to four MLST clusters (Figure 1). Nearly all cluster 1 and cluster 2 isolates contained type I-E∗ CRISPR-Cas system (except cluster 2 isolates, TGH8 and TGH10). All cluster 3 (n = 6) and cluster 4 (n = 13) isolates contained type I-E CRISPR-Cas system (Figure 1). Three unclustered isolates, KP5-1, J1, and CAV1016, contained type I-E, type I-E∗, and type I-E CRISPR-Cas system, respectively (Figure 1). Moreover, all ST14 (n = 5), 15 (n = 4), and 23 (n = 4) isolates contained type I-E∗ CRISPR-Cas system. In contrast, all ST147 (n = 11), 383 (n = 2), 392 (n = 2), and 941 (n = 2) isolates contained type I-E CRISPR-Cas system. These results showed that there was a significant MLST association with the distribution of the type I-E and I-E∗ CRISPR-Cas systems across K. pneumoniae (Figure 1).
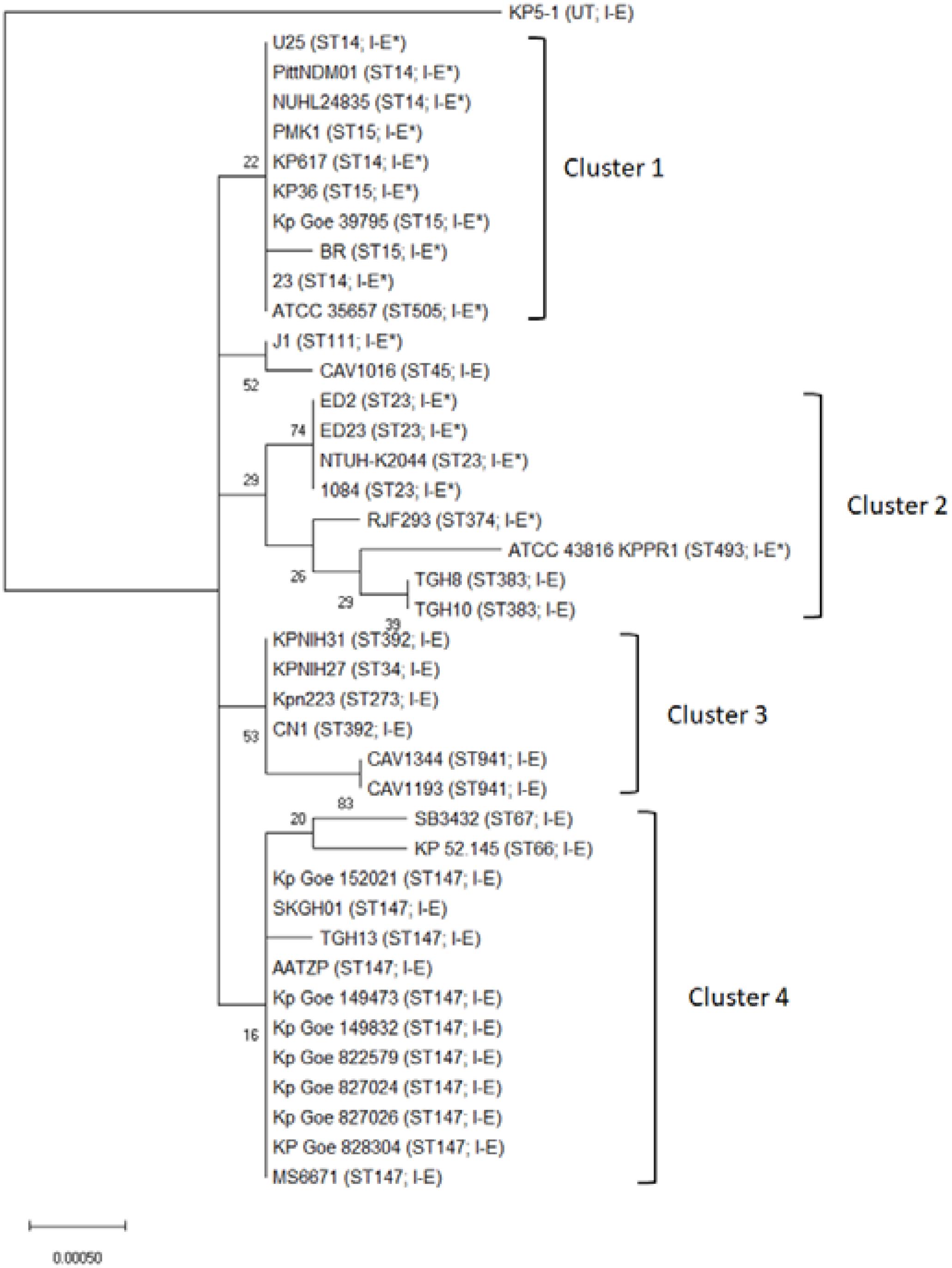
FIGURE 1. Phylogenetic tree of the concatenated MLST sequences of 40 Klebsiella pneumoniae isolates. The phylogenetic tree was based on the aligned nucleotide sequences of concatenated MLST sequences of 40 K. pneumoniae isolates by using the Maximum Likelihood method. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Sequence types and CRISPR-Cas subtypes appear in brackets. UT, untypable.
Overall, 30.7% of clinical K. pneumoniae isolates had CRISPR-Cas systems in 176 K. pneumoniae strains isolated from patients with BSI (n = 87) or UTI (n = 89) in Taiwan. There were no significant differences in the frequency of presence of CRISPR-Cas systems in K. pneumoniae between BSI (34.5%, 30/87) or UTI (27.0%, 24/89) strains (p > 0.05). However, more type I-E CRISPR-Cas systems were found in UTI isolates and more subtype I-E∗ CRISPR-Cas systems were found in BSI isolates (p = 0.042). The type I-E CRISPR-Cas systems had 5 (5.7%) and 10 (11.2%) isolates in BSI and UTI isolates, respectively. The subtype I-E∗ CRISPR-Cas systems had 25 (28.7%) and 14 (15.7%) in BSI and UTI isolates, respectively.
We further determined whether the PFGE typing was associated with the presence of CRISPR-Cas subtypes (Figure 2). The PFGE patterns of 54 isolates showed high diversity, and only 25 isolates (46.3%) were assigned to six clusters based on >80% pattern similarity (Figure 2). All clusters 1 (n = 2), 2 (n = 12), 3 (n = 3), 5 (n = 4), and 6 (n = 2) isolates contained type I-E∗ CRISPR-Cas system. Two cluster 4 isolates contained type I-E CRISPR-Cas system. The PFGE results suggest that the distribution of CRISPR-Cas subtypes was associated with PFGE typing (Figure 2).

FIGURE 2. Pulsed-field gel electrophoresis (PFGE) patterns, CRISPR-Cas systems, and STs among 54 clinical K. pneumoniae isolates. The first letter of the name of isolates is based on the origins of isolate. U, urinary tract infection; B, bloodstream infection; UT, untypable.
We next performed MLST analysis to understand the association of STs with the distribution of CRISPR-Cas types among clinical isolates (Figure 2). The results showed that 52 isolates studied comprised 24 different MLST types (isolates B2213 and U1741 were untypable). The most prevalent sequence type was ST23 (11 BSI isolates and 4 UTI isolates), followed by ST15 (3 BSI isolates and 6 UTI isolates). Type I-E CRISPR-Cas was found in ST147, 592, 867, and 873, while subtype I-E∗ CRISPR-Cas was present in ST1, 11, 13, 15, 23, 35, 76, 607, 1544, and 2668 (Figure 2). The results were consistent with the distribution of CRISPR-Cas systems among published genomes which revealed that ST34, ST45, and ST147 strains contained Type I-E CRISPR-Cas and ST15 and ST23 strains contained subtype I-E∗ CRISPR-Cas (Figure 1). Interestingly, no CRISPR-Cas system was identified in eight ST11 published genomes but isolate B2206 (ST11) contained subtype I-E∗ CRISPR-Cas (Figure 2 and Supplementary Table S2). Karah et al. (2015) reported that a vertical transmission of the CRISPR-Cas subtype I-Fb in a global collection of 76 Acinetobacter baumannii isolates with occasional events of horizontal transfer have increased the diversification and facilitated further dissemination of subtype I-Fb. Taken together, these results raised the possibility of horizontal transfer of CRISPR-Cas systems among K. pneumoniae isolates. Moreover, CRISPR-based genotyping approach has been expanded to the analysis of populations and dissemination routes of particular isolates, with the ability to truly assess genetic diversity even within relatively clonal set of bacteria (Karah et al., 2015; Barrangou and Dudley, 2016). Therefore, it is interesting to determine the efficacy of CRISPR typing in epidemiological surveillance and outbreak investigation of K. pneumoniae in the future.
Our results revealed that PFGE-clusters 1, 4, 5, and 6, were composed of single sequence type (Figure 2). Cluster 2 was composed of ST23 and ST2944 isolates, and cluster 3 was composed of ST1 and ST23 isolates (Figure 2). However, 1 ST1544 (U1145), 2 ST23 (B2012 and U911), 1 ST592 (U1411), and 3 ST15 isolates (B2006, U693, and U698), were not included in six PFGE-clusters. These results suggest that MLST had greater discriminatory ability than PFGE to detect the presence of CRISPR-Cas types.
Isolates having the subtype I-E∗ CRISPR-Cas system were more susceptible to ampicillin-sulbactam (76.9%), cefazolin (79.5%), cefuroxime (79.5%), cefotaxime (82.1%), gentamicin (87.2%), and sulfamethoxazole-trimethoprim (76.9%), compared to CRISPR-negative and type I-E CRISPR-Cas isolates (Table 2). The antibiotic susceptibilities to ampicillin-sulbactam (p = 0.033) and gentamicin (p = 0.032) were statistically different between three CRISPR-Cas groups (Table 2). Moreover, isolates having the subtype I-E∗ CRISPR-Cas system were more susceptible to ampicillin-sulbactam (p = 0.009), cefazolin (p = 0.016), cefuroxime (p = 0.039), and gentamicin (p = 0.012), compared to CRISPR-negative isolates (Table 2). In contrast, isolates having the type I-E CRISPR-Cas system showed no significant difference of nine antibiotic susceptibilities compared to CRISPR-negative isolates.
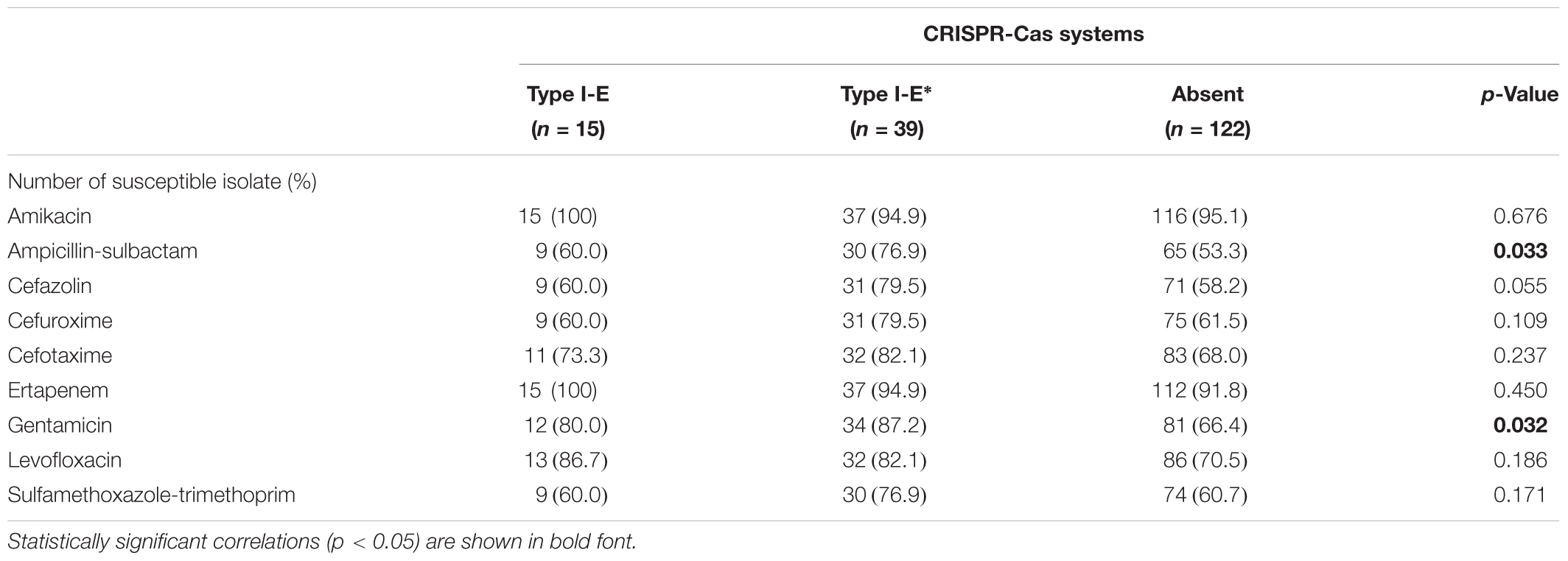
TABLE 2. The association of nine antibiotic susceptibilities with CRISPR-Cas systems in clinical K. pneumoniae isolates.
Our data showed that strains having subtype I-E∗ CRISPR-Cas systems were more susceptible to antibiotics (Table 2). The results raised the possibility that the subtype I-E∗ CRISPR-Cas system efficiently limits the acquisition of acquired antibiotic resistance genes and external DNA fragments. Therefore, the association of chromosome size, numbers of plasmid and phage with the presence of CRISPR-Cas systems in published K. pneumoniae genomes were analyzed. The results showed that the average chromosome size between three groups were similar (p > 0.05) (Table 1). Importantly, strains that had subtype I-E∗ CRISPR-Cas systems had significantly decreased numbers of plasmid and phage counts compared with CRISPR type I-E strains or CRISPR-negative strains (Table 1). However, the numbers of spacer hit plasmids in subtype I-E∗ CRISPR-strains were similar to type I-E CRISPR strains (p = 0.318) (Table 1 and Supplementary Table S3). We next analyzed the spacer sequences of subtype I-E∗ CRISPR-Cas systems in 17 published genomes. The results showed that the spacers of subtype I-E∗ CRISPR-Cas systems targeted sequences match genes encoded phage-related proteins (including phage integrase, tail, portal, and holin family proteins), conjugal transfer proteins, and IS3 family transposase. These results suggested that subtype I-E∗ CRISPR-Cas system efficiently limited the acquisition of external DNA fragments and phage infections.
We next examined the distribution of acquired antibiotic resistance genes among 97 published genomes (Table 1). The results showed a low frequency of aminoglycoside and MLS resistance genes among CRISPR subtype I-E∗ and type I-E strains, respectively (Table 1). Moreover, strains containing CRISPR subtype I-E∗ had lower numbers of β-lactam (p = 0.026) and fluoroquinolone resistance genes (p = 0.037) compared with CRISPR-negative and type I-E strains, respectively (Table 1). However, no subtype I-E∗ CRISPR-Cas-targeting spacer sequences complementary to any known antibiotic resistance genes were identified. A small number of spacer target sequences did match known and previously studied antibiotic resistance-encoding plasmids. Among these spacers, spacer sequences CCGGCATCCGTCAGCTCGACGGCCAGCTGCAG (and its complementary sequence) and CCGCCGTTTAATCGCGGTGATGATATCCGGCA (and its complementary sequence) were found in 13 (76.5%) and 10 (58.8%) subtype I-E∗ CRISPR-Cas systems, respectively (Supplementary Table S3). Moreover, nucleotide BLAST results showed that these two spacers matched short regions within conjugatable IncFIB-, IncFII, IncN-, and IncQ1-type plasmids carrying several antibiotic resistance genes.
ST11 and ST258, belonged to clonal complex 258 (CC258), are well-established to comprise the largest MDR K. pneumoniae clonal group worldwide (Pitout et al., 2015; Lee et al., 2016). ST258 is the predominant clone associated with the spread of KPC-producing K. pneumoniae observed in European countries and the United States (Bowers et al., 2015), and ST11 is the prevalent KPC-producing clone in China and Taiwan (Tseng et al., 2015; Dong et al., 2018). Here, we revealed no CRISPR-Cas system was identified in ST11 (n = 8) and ST258 (n = 16) published genomes (Supplementary Table S2). Moreover, isolate B2206 (ST11) contained subtype I-E∗ CRISPR-Cas showed susceptibility to amikacin, ertapenem, gentamicin, and levofloxacin. Taken together, these results suggest the direct association between absence of CRISPR-Cas system and MDR-phenotype of CC258 K. pneumoniae isolates.
CRISPR-Cas systems are associated with antibiotic susceptibility in S. pyogenes, Pseudomonas aeruginosa and E. coli (Zheng et al., 2014; van Belkum et al., 2015; Aydin et al., 2017). Aydin et al. (2017) reported that five Type I-F spacers matched conserved regions within IncFII-, IncFIB-, and IncI1-type plasmids which are associated with the spread of antibiotic resistance genes. Taken together, these results suggest that CRISPR-Cas systems, especially subtype I-E∗, potentially interfere with the acquisition of antibiotic resistance plasmids, maintaining susceptibility in K. pneumoniae isolates. However, the sequences of spacers among clinical K. pneumoniae isolates having subtype I-E∗ CRISPR systems remain to be determined to clarify their roles in limiting external DNA acquisition.
In summary, we first characterized the CRISPR-Cas systems in clinical K. pneumoniae isolates. The results showed that the distribution of CRISPR-Cas subtypes was directly associated with PFGE typing. Importantly, isolates that had subtype I-E∗ CRISPR-Cas system were more susceptible to certain antibiotics, compared to CRISPR-negative isolates, and the results were consistent with our observation from published genomes.
H-YL and C-YK designed the study and performed analyses. W-HL, P-XZ, J-JY, M-CW, C-HT, C-CT, and J-JW participated, coordinated, and supervised the study. H-YL, C-YK, and J-JW wrote the manuscript. All authors approved the final manuscript.
This study was supported by grant MOST 104-2320-B-010-046-MY3 from the Ministry of Science and Technology, Taiwan and by grant NCKUH-10509002 from the National Cheng Kung University Hospital, Tainan, Taiwan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Robert Jonas for helpful comments on this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01595/full#supplementary-material
Almendros, C., Mojica, F. J., Diez-Villasenor, C., Guzman, N. M., and Garcia-Martinez, J. (2014). CRISPR-Cas functional module exchange in Escherichia coli. mBio 5, e00767-13. doi: 10.1128/mBio.00767-13
Aydin, S., Personne, Y., Newire, E., Laverick, R., Russell, O., Roberts, A. P., et al. (2017). Presence of Type I-F CRISPR/Cas systems is associated with antimicrobial susceptibility in Escherichia coli. J. Antimicrob. Chemother. 72, 2213–2218. doi: 10.1093/jac/dkx137
Barrangou, R. (2015). Diversity of CRISPR-Cas immune systems and molecular machines. Genome Biol. 16:247. doi: 10.1186/s13059-015-0816-9
Barrangou, R., and Dudley, E. G. (2016). CRISPR-based typing and next-generation tracking technologies. Annu. Rev. Food Sci. Technol. 7, 395–411. doi: 10.1146/annurev-food-022814-015729
Bowers, J. R., Kitchel, B., Driebe, E. M., MacCannell, D. R., Roe, C., Lemmer, D., et al. (2015). Genomic analysis of the emergence and rapid global dissemination of the clonal group 258 Klebsiella pneumoniae pandemic. PLoS One 10:e0133727. doi: 10.1371/journal.pone.0133727
Clinical and Laboratory Standards Institute [CLSI] (2009). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard M07-A8, 8th Edn. Wayne, PA: Clinical Laboratory Standard Institute.
Clinical and Laboratory Standards Institute [CLSI] (2016). Performance Standards for Antimicrobial Susceptibility Testing, Twenty-Third Informational Supplement, M100-S26. Wayne, PA: Clinical Laboratory Standard Institute.
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A., and Brisse, S. (2005). Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J. Clin. Microbiol. 43, 4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005
Dong, N., Zhang, R., Liu, L., Li, R., Lin, D., Chan, E.W., et al. (2018). Genome analysis of clinical multilocus sequence Type 11 Klebsiella pneumoniae from China. Microb. Genom. doi: 10.1099/mgen.0.000149 [Epub ahead of print].
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Gomez-Simmonds, A., and Uhlemann, A. C. (2017). Clinical implications of genomic adaptation and evolution of carbapenem-resistant Klebsiella pneumoniae. J. Infect. Dis. 215(Suppl. 1), S18–S27. doi: 10.1093/infdis/jiw378
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hoyland-Kroghsbo, N. M., Paczkowski, J., Mukherjee, S., Broniewski, J., Westra, E., Bondy-Denomy, J., et al. (2017). Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. U.S.A. 114, 131–135. doi: 10.1073/pnas.1617415113
Ilyas, B., Tsai, C. N., and Coombes, B. K. (2017). Evolution of Salmonella-host cell interactions through a dynamic bacterial genome. Front. Cell. Infect. Microbiol. 7:428. doi: 10.3389/fcimb.2017.00428
Jiang, W., Samai, P., and Marraffini, L. A. (2016). Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR-Cas immunity. Cell 164, 710–721. doi: 10.1016/j.cell.2015.12.053
Karah, N., Samuelsen, O., Zarrilli, R., Sahl, J. W., Wai, S. N., and Uhlin, B. E. (2015). CRISPR-cas subtype I-Fb in Acinetobacter baumannii: evolution and utilization for strain subtyping. PLoS One 10:e0118205. doi: 10.1371/journal.pone.0118205
Koonin, E. V., Makarova, K. S., and Zhang, F. (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. doi: 10.1016/j.mib.2017.05.008
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee, C. R., Lee, J. H., Park, K. S., Jeon, J. H., Kim, Y. B., Cha, C. J., et al. (2017). Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front. Cell. Infect. Microbiol. 7:483. doi: 10.3389/fcimb.2017.00483
Lee, C. R., Lee, J. H., Park, K. S., Kim, Y. B., Jeong, B. C., and Lee, S. H. (2016). Global dissemination of carbapenemase-producing Klebsiella pneumoniae: epidemiology, genetic context, treatment options, and detection methods. Front. Microbiol. 7:895. doi: 10.3389/fmicb.2016.00895
Louwen, R., Horst-Kreft, D., de Boer, A. G., van der Graaf, L., de Knegt, G., Hamersma, M., et al. (2013). A novel link between Campylobacter jejuni bacteriophage defence, virulence and Guillain-Barre Syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 32, 207–226. doi: 10.1007/s10096-012-1733-4
Louwen, R., Staals, R. H., Endtz, H. P., van Baarlen, P., and van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88. doi: 10.1128/MMBR.00039-13
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Makarova, K. S., and Koonin, E. V. (2015). Annotation and classification of CRISPR-Cas systems. Methods Mol. Biol. 1311, 47–75. doi: 10.1007/978-1-4939-2687-9_;4
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Mathers, A. J., Peirano, G., and Pitout, J. D. (2015). The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clin. Microbiol. Rev. 28, 565–591. doi: 10.1128/CMR.00116-14
Nunez, J. K., Kranzusch, P. J., Noeske, J., Wright, A. V., Davies, C. W., and Doudna, J. A. (2014). Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 21, 528–534. doi: 10.1038/nsmb.2820
Ostria-Hernandez, M. L., Sanchez-Vallejo, C. J., Ibarra, J. A., and Castro-Escarpulli, G. (2015). Survey of clustered regularly interspaced short palindromic repeats and their associated Cas proteins (CRISPR/Cas) systems in multiple sequenced strains of Klebsiella pneumoniae. BMC Res. Notes 8:332. doi: 10.1186/s13104-015-1285-7
Pitout, J. D., Nordmann, P., and Poirel, L. (2015). Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob. Agents Chemother. 59, 5873–5884. doi: 10.1128/AAC.01019-15
Shah, S. A., and Garrett, R. A. (2011). CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res. Microbiol. 162, 27–38. doi: 10.1016/j.resmic.2010.09.001
Shen, J., Lv, L., Wang, X., Xiu, Z., and Chen, G. (2017). Comparative analysis of CRISPR-Cas systems in Klebsiella genomes. J. Basic Microbiol. 57, 325–336. doi: 10.1002/jobm.201600589
Tseng, I. L., Liu, Y. M., Wang, S. J., Yeh, H. Y., Hsieh, C. L., Lu, H. L., et al. (2015). Emergence of carbapenemase producing Klebsiella pneumoniae and spread of KPC-2 and KPC-17 in Taiwan: a nationwide study from 2011 to 2013. PLoS One 10:e0138471. doi: 10.1371/journal.pone.0138471
van Belkum, A., Soriaga, L. B., LaFave, M. C., Akella, S., Veyrieras, J. B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio 6:e01796-15. doi: 10.1128/mBio.01796-15
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zheng, P. X., Chiang-Ni, C., Wang, S. Y., Tsai, P. J., Kuo, C. F., Chuang, W. J., et al. (2014). Arrangement and number of clustered regularly interspaced short palindromic repeat spacers are associated with erythromycin susceptibility in emm12, emm75 and emm92 of group a streptococcus. Clin. Microbiol. Infect. 20, 516–523. doi: 10.1111/1469-0691.12379
Keywords: CRISPR-Cas systems, Klebsiella pneumoniae, urinary tract infection, bacteremia, antibiotic susceptibility
Citation: Li H-Y, Kao C-Y, Lin W-H, Zheng P-X, Yan J-J, Wang M-C, Teng C-H, Tseng C-C and Wu J-J (2018) Characterization of CRISPR-Cas Systems in Clinical Klebsiella pneumoniae Isolates Uncovers Its Potential Association With Antibiotic Susceptibility. Front. Microbiol. 9:1595. doi: 10.3389/fmicb.2018.01595
Received: 26 February 2018; Accepted: 26 June 2018;
Published: 16 July 2018.
Edited by:
Miklos Fuzi, Semmelweis University, HungaryReviewed by:
Vartul Sangal, Northumbria University, United KingdomCopyright © 2018 Li, Kao, Lin, Zheng, Yan, Wang, Teng, Tseng and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiunn-Jong Wu, amp3dTEwMTlAeW0uZWR1LnR3
†These authors have contributed equally to this work.
‡Present address: Cheng-Yen Kao, Department of Medical Microbiology and Immunology, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.