Commentary: Study the Features of 57 Confirmed CRISPR Loci in 38 Strains of Staphylococcus aureus
 Xihong Zhao
Xihong Zhao Zhixue Yu
Zhixue Yu Zhenbo Xu2*
Zhenbo Xu2*- 1Research Center for Environmental Ecology and Engineering, Key Laboratory for Green Chemical Process of Ministry of Education, Key Laboratory for Hubei Novel Reactor & Green Chemical Technology, School of Environmental Ecology and Biological Engineering, Wuhan Institute of Technology, Wuhan, China
- 2School of Food Science and Engineering, South China University of Technology, Guangzhou, China
Staphylococcus aureus is a foodborne pathogen that causes food contamination and food poisoning, which poses great harm to health, agriculture and other hosts. Clustered regularly interspaced short palindromic repeats (CRISPR) are a recently discovered bacterial immune system that resists foreign genes such as phage DNA. This system inhibits the transfer of specific movable genetic elements that match the CRISPR spacer sequences, thereby preventing the spread of drug-resistant genes between pathogens. In this study, 57 CRISPR loci were screened from 38 strains of S. aureus based on the CRISPR database, and bioinformatics tools were used to investigate the structural features and potential functions of S. aureus CRISPR loci. The results showed that most strains contained only one CRISPR locus, a few strains contained multiple loci with sparsely distributed sites. These loci mainly included highly conserved direct repeat sequences and highly variable spacer sequences, as well as polymorphic cas genes. In addition, the analysis of secondary structure of direct repeat RNA showed that all sites can form stable RNA secondary structure. The results of constructing phylogenetic tree based on spacer sequence showed that some strains contained a high degree of phylogenetic relationship, while the differences among other strains in evolutionary processes were quite obvious. Of the 57 CRISPR loci identified, only the cas gene was found near the 4 CRISPR loci.
Introduction
In the past, it was known that vertebrates have the ability to resist and eliminate foreign pathogens, and could form a highly effective secondary immune mechanism to prevent the re-invasion of pathogens. Until the advent of clustered regularly interspaced short palindromic repeats (CRISPR), researchers realized that prokaryotes also had an adaptive secondary immune system similar to animals (Morange, 2015). CRISPR was the product of the evolution of life in the history of bacterial invasion against viruses, the bacteria to remove the virus invasive alien genes, evolved this powerful immune defense system. The main fundamental principle of the CRISPR system: firstly, the characteristic gene (proto-spacer) was extracted from the invaded foreign DNA and embedded in the CRISPR locus. Secondly, when the exogenous phage invaded again, this prokaryotic immune system used characteristic genes (spacers) to rapidly target and recognized foreign DNA. Finally, with the participation of a Cas protein complex, the invading phage DNA sequence was targeted and interfered, and the recognized foreign DNA was excised to eliminate exogenous invasion. The prokaryotic immune system, both acquired and heritable, is widespread in about 47% of bacteria and 84% of archaebacterial, which truly documents the pathways of bacterial evolution and the history of confrontations with foreign invaders (such as phages) (Lillestøl et al., 2006; Wakefield et al., 2015; Yang et al., 2015). Staphylococcus aureus, as an important foodborne pathogen in humans, is widely found in nature, air, water, dust, and human and animal excrement (Zhao et al., 2016). Furthermore, S. aureus is the most common pathogen in human purulent infection, in addition to local purulent infection, but also can cause pneumonia, pseudomembranous colitis, pericarditis, and even systemic infection such as sepsis (Larkin et al., 2009; Lindsay, 2014; Pérez-Montarelo et al., 2017). The pathogenicity of S. aureus mainly depends on the toxins and invasive enzymes it produces (Zhao X. H. et al., 2017). One of the most important toxins is enterotoxin, a protein toxin that can cause acute gastroenteritis. The enterotoxin can tolerate boiling at 100°C for 30 min without being destroyed, and the food poisoning symptoms causes are vomiting and diarrhea (Zhang and Zhao, 2017; Zhang et al., 2017; Zhao Y. et al., 2017). At present, S. aureus can be divided into five groups and 26 types using phage typing (Dua et al., 1982), most of which have high environmental adaptability. It is tempting to speculate that CRISPR, as prokaryotic immune system, may be closely related to the high environmental adaptability of S. aureus (Schröder et al., 2013).
The typical genomic architecture of a CRISPR-Cas system is consists of a CRISPR locus, a series of cas genes, and a leader region. One of the major system components is the CRISPR locus, which is characterized by a series of tandem repeats separated with a unique spacer sequence (Horvath and Barrangou, 2010; Koonin and Makarova, 2013). The spacers of the CRISPR locus are highly specific, while the repeats are almost identical in same CRISPR locus (Guzina et al., 2017; Rossi et al., 2017). The direct repeat contains palindrome, which can form RNA secondary hairpin structure. Further study found that the hairpin structure was the recognition and binding site of Cas protein in the process of interference function (Wang et al., 2011). The spacer sequences were shown to be derived from previously encountered phages (Bolotin et al., 2005), and a small proportion came from the same bacterial genome, suggesting that there is horizontal gene transfer between homologous species in CRISPR system (Grissa et al., 2008; Rossi et al., 2017). In addition to the CRISPR locus, CRISPR-Cas system also includes a series of Cas proteins with multiple nuclease activities, as well as the leader region identified as having promoter function. In general, the CRISPR-Cas system can be divided into two categories based on the Cas protein. Class 1 systems perform functions through a multi-subunit Cas protein complex, whereas the Class 2 systems only require a single Cas protein (Cas9 or Cpfl) in the crRNA effector complex. Class 1 includes Type I, Type III, and Type IV systems, and Class 2 includes Type II and Type V systems (Quan and Ye, 2017). It illustrates the complexity of the CRISPR system from another perspective.
At present, traditional pathogen detection methods include PCR, bacterial identification media, latex agglutination test, and traditional bacterial typing methods such as serotyping, phage typing, and drug resistance profile typing (Sabat et al., 2013; Zhong and Zhao, 2017, 2018; Wei et al., 2018; Zhao et al., 2018). It has been difficult for them to meet the current requirements for accurate diagnosis, traceable typing, and epidemiological studies of pathogens. For instance, foodborne pathogens once induced into the viable but non-culturable state (VBNC) cannot be detected by routine bacterial culture assays and can easily lead to undetected pathogens caused by the VBNC status, posing a serious threat to food safety and human health (Zhao et al., 2014; Ding et al., 2017; Liu et al., 2017; Zhao X. et al., 2017). Therefore, exploring how to identify pathogens more accurately and rapidly at the genetic level becomes a new direction for CRISPR systems in the field of microbiology. Currently, researchers have proposed to apply the diversity and specificity of spacers in CRISPR to genotyping techniques and have been well-established in some bacteria. For instance, Shariat et al. (2013) have proposed a new typing method CRISPR-MVLST which was a combination of bacterial marker gene, CRISPR and multiple site sequence typing. This typing method not only showed high degree consistency in epidemiology, but also higher resolution than pulsed-field gel electrophoresis, which distinguished highly cloned strains during pathogen outbreaks. In fact, the focus of CRISPR research is very prominent, especially as a faster and simpler application of gene editing tools (Doudna and Charpentier, 2014; Zhang et al., 2014), which can be used in biotechnology and medicine, and has great potential in gene and cell therapy (Maeder and Gersbach, 2016). The third generation of gene editing CRISPR-Cas9 technology is used to study the genetic engineering of various organisms such as eukaryotes, bacteria and viruses (Penewit et al., 2018). As CRISPR technology continues to improve, replacing Cas9 with Cpf1 or xCas9 may provide more opportunities for these applications (Hu et al., 2018). Given the diversity of CRISPR-Cas systems in different prokaryotes (Koonin et al., 2017; Wexler and Tajkarimi, 2017), researchers have been working on these sites in various bacteria in recent years. At present, bioinformatics tools can help researchers expand their knowledge in different fields without the need for laborious laboratory experiments. Based on this method, there have been some studies on the CRISPR system in several bacteria (Hidalgo-Cantabrana et al., 2017; Negahdaripour et al., 2017; Tomida et al., 2017; Hao et al., 2018). Bioinformatics Analysis of CRISPR in foodborne pathogens is crucial for assessing the potential evolution of foodborne pathogens to predict the outbreak of food borne pathogens, which is of great importance to food safety. In order to investigate the presence of CRISPR in different strains and the range of possible immune defenses, the effects of different CRISPR/Cas systems on the pathogenicity of S. aureus were explored, and accurately and quickly detect foodborne pathogenic bacteria, prevent and eliminate S. aureus caused by a variety of foodborne infections. It is significant to decipher the CRISPR locus that has now been identified. Therefore, systematic research on CRISPR structures helps us to better explore other functions of the CRISPR system in addition to bacteriophage immunity.
In this study, 57 identified CRISPR loci from 38 species of S. aureus were selected for experimental subjects. By bioinformatics analysis of its structural characteristics and potential functional activity in S. aureus, based on the similarity of the phylogenetic relationships between spacer and direct repeat sequences, the 57 CRISPR loci were classified and verified that the spacer sequence was derived from a phage or exogenous plasmid. In addition, bioinformatics tools were used to predict RNA secondary structure formed by direct repeat sequences and their stability. Finally, the presence and distribution of the cas gene near the CRISPR locus were analyzed. The aim is to provide a new strategy for the control of foodborne pathogens S. aureus resistance studies, genotyping, traceability analysis, food safety and prevention.
Materials and Methods
Data Sources
The different S. aureus strain genomes were searched by the National Center for Biotechnology Information (NCBI) nucleotide database (http://www.ncbi.nlm.nih.gov/) with default parameters; then S. aureus CRISPR loci were searched by the CRISPR Finder server (E-value ≤ 0.001) (http://crispr.i2bc.paris-saclay.fr/Server/)(Last updated on May 9, 2017). The CRISPR loci contained in 38 stains of S. aureus were classified as suspected CRISPR loci and identified CRISPR loci. All the identified 57 CRISPR loci were selected for study.
Analysis Method
CRISPR loci were grouped according to the similarity of the common direct sequence repeats (CDRs) of the CRISPR locus. First, using the MEGA6.06 software (https://www.megasoftware.net/) for direct sequences multiple sequence alignment (MSA), similar repeat sequences were clustered into the same group, and then the RNA secondary structures and minimum free energy (MFE) of each direct repeat sequence were predicted by the RNA fold Web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). As for the algorithm for secondary structure folding and MFE, the output option was set to default. The prediction of these structures are based on the cyclic energy model and the dynamic program definition algorithm (Zuker and Stiegler, 1981).
In addition, since the CRISPR system may contain numerous spacers, the principal component analysis (PCA) was used to screen representative spacers from each CRISPR system, then MEGA6.06 software (https://www.megasoftware.net/) was used to import the selected spacer sequences to describe phylogenetic tree and identify the phylogenetic relationships among the stains, the genotyping and phylogenetic relationship of 38 strains of S. aureus were predicted, and the highly homologous sequence sources were searched by NCBI BLASTN search pattern (default parameters, nr database, mismatches ≤ 3). Finally, the cas gene near the CRISPR locus was searched in CRISPRs database (http://crispr.i2bc.paris-saclay.fr/crispr/). In addition, multiple sequence aligned proteins were searched by the BLASTP platform in NCBI (identity ≥ 90%, coverage > 90%). The above results are used to describe the type of CRISPR system and the distribution of cas genes in order to achieve bioinformatic analysis of the CRISPR locus identified in the selected strain.
Results
CRISPR Locus of Staphylococcus aureus in CRISPR Database
CRISPR database is a relational database implemented using mysql 4.1 (http://www.mysql.com/). Its implementation of related Web services is based on Perl 5.8.8 (http://www.perl.org/) and the Linux operating system (debian Sarge 3.1). Run on the Apache 2.0 web server (http://www.apache.org/). And use some modules to process the sequence. The database core application consists of two main programs: (a) CRISPR Finder for detecting CRISPRs and extracting them from the genome sequence. (b) Database tools for downloading prokaryotic genomes from the NCBI ftp site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria), save CRISPRs, and update them (Grissa et al., 2007a). Currently, CRISPR database analyzes the genomes of 232 archaea by CRISPR Finder and obtains 870 CRISPR loci, 202 of which are convincing. In addition, 8,690 CRISPR loci were obtained by analyzing the genomes of 6,786 bacterial species, of which 3,059 loci were identified. In the CRISPR database, CRISPR loci contained in 38 S. aureus were searched. Among them, 57 CRISPR loci have been confirmed and others were suspicious CRISPR loci (last updated: 9 May, 2017), 38 strains of S. aureus have been confirmed in 57 CRISPR loci to carry out bioinformatics analysis of the components. As shown in Table 1, 22 strains of S. aureus contained only one CRISPR locus, 14 strains of S. aureus contained 2 CRISPR loci, and the other 2 strains contained 3 and 4 CRISPR loci. This was in comparison to other CRISPR loci on the distribution of the number of species was rare, it came down to the fact that the study of the S. aureus CRISPR system was not extensive enough, there may be some CRISPR loci hidden in the large number of suspicious CRISPR sequences. The number of spacer sequences in each CRISPR loci was between 1 and 15, the length of spacer sequence was concentrated in 25–31 bp, the number of direct repeats was between 2 and 16, and the length of the sequence was concentrated in 23–26 bp.
TABLE 1
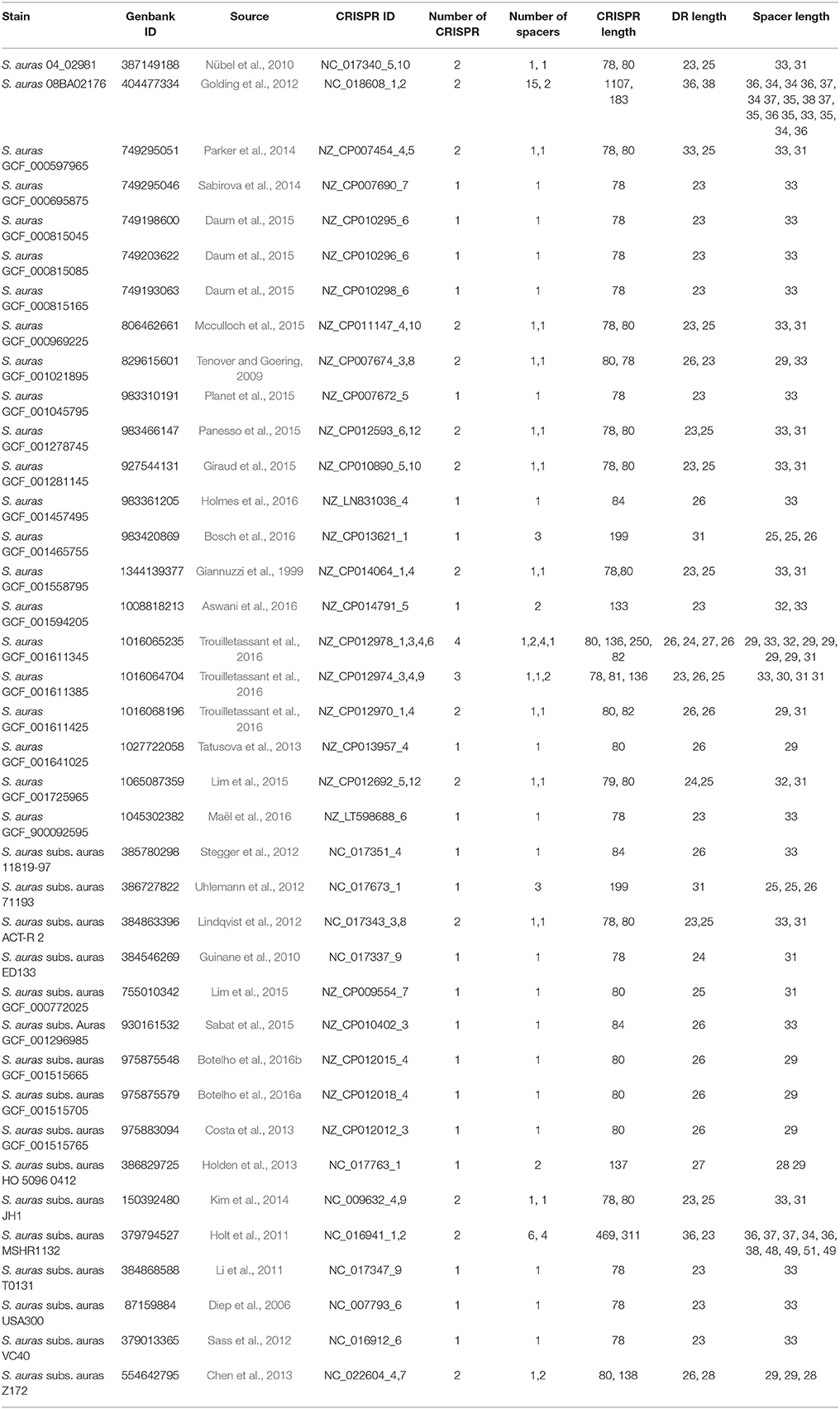
Table 1. Statistical table of 38 CRISPR loci of Staphylococcus aureus.
Repeat Sequences
Direct repeat sequences (DR) are always highly similar or identical at the same CRISPR locus, the consensus direct repeat sequence (CDR) of each CRISPR locus for multiple sequence alignment analysis was chosen. Based on the results of the comparison, 57 CRISPR loci in 38 strains of S. aureus were divided into 25 groups. As shown in Table 2, each group was composed of the same DR sequence. In order to facilitate multiple sequence comparison and clustering, one DR sequence for homology analysis was chosen in each group. In addition, the RNA secondary structure of each repeat of 25 sets of DR sequences was predicted and recorded its MFE by RNA fold web server (Grissa et al., 2007b). In all groups, the RNA secondary structure was bound at both ends, forming stems in the middle. As shown in Figure 1, group 2, group 14, group 16, and group 22 were predicted to have a repeat length of 4 bp for the RNA secondary structure, 3 bp for the 9th group, the stem length of the secondary structure of group 19 and group 24 was 6 bp, the length of the 6th and 12th groups was 7 bp, the length of the secondary structure formed by group 21 was 8 bp, and that of other groups was 5 bp. Due to the formation of difference algorithm and system structure uncertainty, in partial red graphic representation form different secondary structure types, the structure formation probability prediction system was the possibility of relatively large, While the greenish-green indicated that the RNA secondary structure formed by the repeated sequences was still predicted to have a low probability of formation after the system optimization algorithm (Mathews, 2004). In addition, the sequence stability and the degree of conservation of the DR can be predicted by RNA secondary structure diagram and MFE value. Due to possible systematic errors in the prediction of the green group with a lower probability of being predicted, the group with the largest MFE value was group 22, which meant that it was not only the most unstable in the green marker group but also the least stable in all RNA secondary structures. The 21th group had a minimum MFE value of −13.2 kcal/mol and the longest stem formed in all RNA secondary structures, which was in line with the secondary structure of RNA stability and the formation of the stem length showed a certain linear positive correlation theory. In the same cluster, in addition to stem length can affect the stability of the secondary structure; other factors include the length of the repeat sequence and the “GC” content. In general, repeats with higher “GC” content at the same length are more stable; the longer the repeats, the more stable the secondary structure may be. Overall, the DR sequence with lower MFE value was more stable than the DR sequence with high MFE value.
TABLE 2
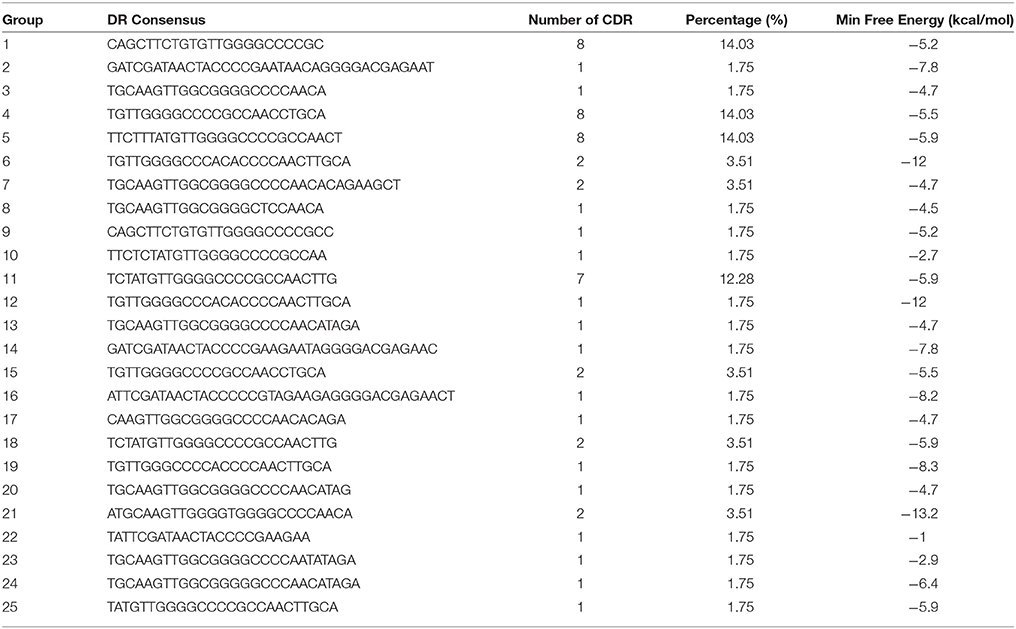
Table 2. DR Sequence and RNA secondary structure MFE value statistics.
FIGURE 1
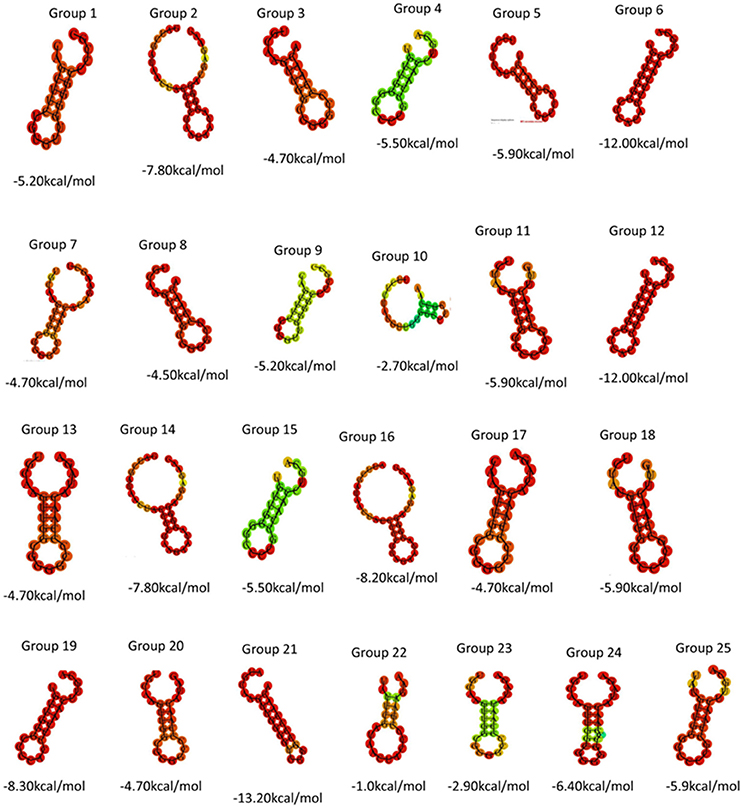
Figure 1. Using DR sequences to generate 25 sets of RNA secondary structure prediction and MFE values.
Spacers
Based on multi-sequence alignment of spacer sequences, a total of 92 spacers were found in 57 CRISPR loci in 38 stains of S. aureus and the spacer sequences present at the CRISPR locus appear to be highly homologous, contrary to the diversity necessary as a bacterial self-defense system. But from another perspective, they seemed to have undergone the same phage invasion or the process of gene transfer. After further analysis of all spacers sequences present in the CRISPR system by MSA, the CRISPR loci were classified into 22 groups, What's more, on the basis of the MSA analysis, it was found that there was no highly conserved nucleotide in the spacer sequences of different CRISPR loci. Finally, homologous matching of 92 spacer sequences from 57 CRISPR loci by NCBI blast showed that most of the spacers matched the corresponding exogenous elements. Strikingly, 11 spacers from 2 strains (08BA02176 and MSHR1132) appeared to be highly homologous to exogenous phage or plasmid (Table 3). Besides, PCA was used for each of the 57 CRISPR loci after optimization based on MSA data. In the process of dimensionality reduction of the experimental data, the spacer components of each CRISPR locus were projected onto the same analysis plane for each CRISPR locus, and in the principal component data on the overall contribution of the calculation results were consistent with the required precision. Through such a simplified analysis of the data, it avoided the systematic error of deducting the principle of deduction too much when constructing the phylogenetic tree due to the uneven length of the nucleotide sequence (Moore, 1981). The evolutionary relationship between strains was explored by using the difference between the access and deletion of the spacer sequences. The phylogenetic tree of 57 groups of spacer components was constructed by using mega6.06 software (Figure 2), which enabled the homology analysis of the selected 57 CRISPR loci.
TABLE 3

Table 3. Statistics of phage or plasmid highly homologous to spacers.
FIGURE 2
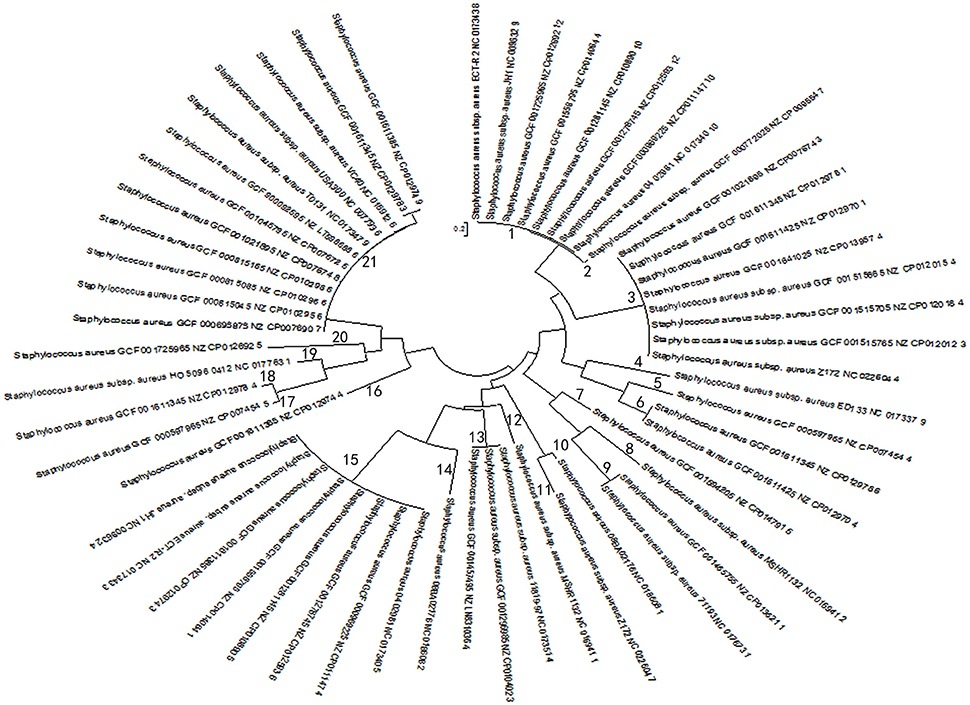
Figure 2. Biological evolution tree generated by principal component spacer sequences. Evolutionary tree results are grouped based on evolutionary relationships. The numbers from 1 to 21 represent 21 groups. Strains in the same group indicate higher evolutionary similarity, and the closer the distance, the higher the affinity is. The evolutionary distance scale is 0.2.
Cas Genes Near CRISPR Loci
For the CRISPR-Cas system, the cas1 and cas2 genes were essential elements of a normally active CRISPR system and were located near the CRISPR locus. Therefore, the presence of the cas1 and cas2 genes were searched in the range of 10,000 bp upstream and 10,000 bp downstream of all 57 CRISPR loci. The result found that the presence of two core cas genes only 3 strains of S. aureus were S. aureus 08BA02176 (CRISPR ID: NC_018608_1) and S. aureus GCF_001611345 (CRISPR ID: NC_CP012978_4) and S. aureus MSHR1132 (CRISPR ID: NC_016941_1, NC_016941_2), the other 35 strains did not exist in these two kinds of cas gene. Based on this result, cas genes contained in these three strains were described (Figure 3). The CRISPR of S. aureus 08BA02176 NC_018608_1 belongs to the subtype I-C, its related proteins near CRISPR were endonuclease Cas1, integrase Cas2, helicase Cas3, protein Cas4, protein Cas5 and protein Cas7. The CRISPR of S. aureus MSHR1132 NC_016941_1 belongs to the subtype III-A and the CRISPR-related proteins in the vicinity of NC_016941_1 were nuclease Cas9, endonuclease Cas1, integrase Cas2 and protein Csn2. CRISPR's associated proteins near CRISPR of MSHR1132 NC_016941_2 (subtype III-A) were Cas6, protein Cas10, protein Csm10, protein Csm4, endonuclease Cas1 and integrase Cas2. CRISPR-related proteins near CRISPR (GCF_001611345 NC_CP012978_4, subtype III-A) were endonuclease Cas1, integrase Cas2, protein Cas6, protein Cas10, protein Csm2, and protein Csm6. From the observation, it showed that similar proteins of the same CRISPR type can be found on CRISPR loci with 4 or more flanking proteins.
FIGURE 3
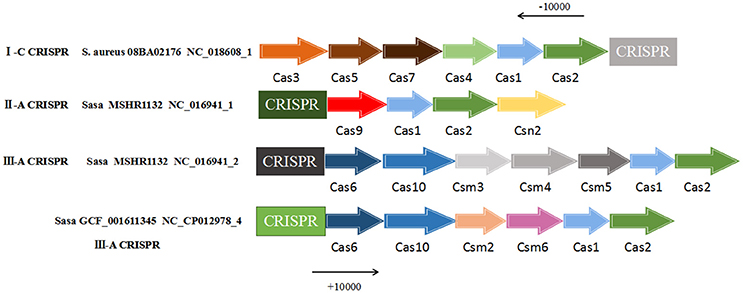
Figure 3. The cas genes in vicinity of CRISPR loci. Cas genes were searched from 10,000 bp upstream to 10,000 bp downstream the CRISPR sequence. “Sasa” represents Staphylococcus aureus subsp. aureus. “S. aureus” represents Staphylococcus aureus.
Discussion
Staphylococcus aureus is currently one of the major pathogenic microorganisms that cause infectious diseases in humans worldwide. In recent years, due to the widespread use of multiple antibiotics, various resistance genes (such as mec gene-encoding penicillin-binding protein) were horizontally transferred between Staphylococcus strains resulted in multi-drug resistance of S. aureus (Chuang et al., 2014; Lin et al., 2016; Miao et al., 2016). One study analyzed 370 CRISPR-Cas systems from 148 prokaryotic genomes and constructed a phylogenetic tree based on the cas1, cas2, cas3, and cas4 genes present in these systems. Surprisingly, the species that are supposed to be closely related to each other are on separate branches of the phylogenetic tree. Further analysis revealed 10 large plasmids with a CRISPR system, suggesting that the CRISPR-Cas system was a mobile genetic element and that many horizontal gene transfer events have occurred through binding transfer, allowing the system to spread among distantly related species (Borowski, 2017). In addition, Bikard et al. (2012) constructed a CRISPR locus targeting a drug resistance gene on the chromosome of the recipient bacteria and introduced the plasmid containing the CRISPR gene into the chromosome of the recipient bacteria, resulting in the death of the bacteria containing the drug resistance gene. Based on this, a mobile CRISPR element targeting a drug-resistant or virulence gene was designed to not only kill pathogens with the target genes but also prevent the spread of drug-resistant or virulence genes among bacteria. The development of this mobile CRISPR element as an antimicrobial agent will provide a new strategy for the control of foodborne pathogens.
Some studies have found that about 40% of the bacterial genome contains the CRISPR locus (Hakim et al., 2016). Most of the CRISPR loci of prokaryotes are located on their chromosomes, rarely on plasmids (Sorek et al., 2008). The main reason for this phenomenon is that the CRISPR system could provide the scavenging effect on the foreign bacteriophage and plasmid through the targeting interference mechanism of RNA, which endowed prokaryotes with strong adaptability in the face of different evolutionary environments (Koonin and Wolf, 2015). If CRISPR exists on the plasmid, it will be detrimental to the heritability of this immunity. In this study, there were 16 kinds of S. aureus in all 38 kinds of S. aureus contains 2 or more CRISPR loci, and they may present different CRISPR system type. It directly proved that a bacterial genome containing multiple CRISPR loci. There were great distinctions in the number and characteristics of the CRISPR loci contained among different species, which was also a necessary condition for bacterial genotyping (Horvath et al., 2009). In general, direct repeats of a single CRISPR locus are extremely conserved; however, there exist in some modified nucleotides in direct repeats within different CRISPR loci. When a new spacer is formed, it always remove the internal spacer by homologous recombination between direct repeats to limit the size of the CRISPR. The terminal repeats were frequently observed to be polymorphic, which was well-explained by the frequent loss of spacer/repeats containing the last of the terminal repeats (Vahidi and Honda, 1991; Horvath et al., 2008). The position of the repeat appears to be consistent with the location of nearby cas gene. In addition, variations within repetitive sequences often occur throughout the CRISPR locus (Horvath et al., 2008). In the 38 strains of S. aureus, repeats of most strains were highly conserved and were located in groups 1, 4, and 5. Therefore, it could be inferred that these direct repetitive mutations were less likely to occur in other groups, suggesting that the presence of cas around the CRISPR locus in these clusters is probably less.
Due to the presence of short palindromic sequences in direct repeats, the secondary structures of RNA that may be formed by these direct repeats during transcription were investigated. This structure could cooperate with the crRNA transcribed from the entire CRISPR sequence to form a bimodal structure, guiding the Cas protein to target the site. Kunin et al. (2007) indicated that the stem-loop structure of some repeats may contribute to recognition-mediated contact between a gap-targeted exogenous RNA or DNA and a Cas-encoded protein, suggested that the stability of RNA secondary structure may affect CRISPR function. In addition, the MFE values of RNA secondary structures formed by all 25 different direct repeats were predicted. By comparing their MFE, secondary structures of RNA with lower minimum free energies were more stable. The stability of secondary structure of RNA was found depends on the length of the repeat and by the number of bp as well as the content of “GC” in stem. By homologous comparison of the spacer sequences in the NCBI Database, the source of the known genes highly homologous thereto was queried. Boch et al. (2009) indicated that the spacer sequence was proved from the exogenous gene components, CRISPR got the spacer sequence from the new invasion of exogenous DNA by some means. A homology survey of all the spacer sequences of this experiment by NCBI blast showed that most of the spacers of S. aureus strains can be matched to the foreign phage or plasmid in NCBI. In particular, 11 spacers from 2 strains of S. aureus presented highly matched exogenous phage or plasmids (as shown in Table 3). This strongly proves that the spacer is derived from exogenous elements. To a certain extent, the higher the number of phages with higher similarity to the spacers, the higher the frequency of phages attacking to the bacterium, and the more important the survival of the CRISPR system for bacteria in the environment of phage invasion. In addition, the phylogenetic tree of the selected strains was constructed by using the spacer sequences to provide reference value for the genotyping of S. aureus. In fact, the CRISPR-based typing has been well-established in some bacteria, the most representative of which is the application of Salmonella typing (Liu et al., 2011; Fabre et al., 2012). Even recent studies have demonstrated that Helicobacter pylori can be typed using CRISPR-like sequences in combination with virulence genes. The CRISPR-virulence technique was constructed for the 20 Helicobacter pylori strains obtained and compared with the phenotype obtained by the random amplified polymorphic DNA technique. There is no difference between the discrimination of the CRISPR typhoid typing and the RAPD typing (Bangpanwimon et al., 2017). Thus, the CRISPR-based typing method has a broad application prospect in the investigation of foodborne pathogens. However, at the moment, the data of the research on the typing of foodborne pathogenic bacteria by this method is still not enough, the database is not perfect, and no uniform standard. Due to the increasingly serious S. aureus infection and the relatively few studies on the CRISPR of S. aureus, which means that extensive research on the CRISPR typing of S. aureus is particularly urgent. Through the establishment of a database to explore the occurrence of foodborne pathogens CRISPR, diversity and activity in the relevant pathogens, in order to solve the problem of lasting food safety (Nikki and Dudley, 2014).
In this paper, the 92 spacers were found in 38 species of S. aureus, of which only 9 species of S. aureus contained 2 or more spacers, and other strains containing only one spacer, the 92 spacers with an average length of about 32 bp, the shortest 25 bp, the longest 51 bp, and the diversity of spacer length and sequence will affect the bacterial activity in the CRISPR system. Horvath et al. (2008) studies showed that longer CRISPR sequences may be more active than short ones. Di et al. (2014) studies indicated that CRISPR loci containing more number of the spacers with length of 30 bp were more active than sites containing a small amount of 36 bp. Therefore, the CRISPR loci in the strains of our choice may be more active than the previously studied strains with shorter spacers. Additionally, the same nucleic acid sequence was observed in the spacers of different strains, which suggested that these strains may be attacked by several phages with higher relatives or due to horizontal gene transfer.
For the 57 CRISPR loci contained in 38 S. aureus strains, each of the 10,000 bp upstream and downstream of these CRISPR loci was queried for the presence of cas1 and cas2 genes. Three S. aureus strains were screened to meet the requirements. The three S. aureus strains contained cas1 and cas2 genes to systematically search for the distribution of other cas genes in the vicinity of the CRISPR sequence. Wherein in the CRISPR system of S. aureus 08BA02176, the cas gene have located in 10,000 bp downstream of the CRISPR sequence. However, in the two other strains of S. aureus (S. aureus GCF_001611345 and S. aureus subsp. aureus MSHR1132), the cas gene have located in upstream of the CRISPR sequence, it proved that CRISPR-Cas system could transfer between the same strains (Borowski, 2017). In summary, the other strains of S. aureus that did not detect the cas1 and cas2 genes were considered inactive by the CRISPR system they contained, because when these key cas genes in certain CRISPR loci inactivated or not present, bacterial drug resistance and its ability to integrate new spacer sequence will be lost.
At present, research on the CRISPR system of pathogenic bacteria of foodborne origin has not paid enough attention. In fact, in addition to the immune defense function, the CRISPR system has also been found to have the functions of regulating the virulence of bacteria and influencing the formation of biofilms of foodborne pathogens. Zegans et al. (2009) found that the CRISPR system was involved in the lysogenic infection of bacteriophage DMS3 in Pseudomonas aeruginosa PA14, resulting in the inhibition of P. aeruginosa biofilm formation and decreased the swarming motility. Palmer and Gilmore (2010) indicated that multidrug-resistant Enterococcus strains were usually missing the CRISPR system, and thus speculated that the CRISPR system may be involved in the interference of drug-resistance gene capture. Lovley and Muktak (2010) showed that the spacer1 in the CRISPR system is homologous to the histidine-tRNA synthetase of the bacterium in Pelobacter carbinolicus, which prompted P. carbinolicus to selectively delete the genes with more histidine codons during evolution. As a result, P. carbinolicus differentiated from other geobacteraceae to form new species. Babu et al. (2011) demonstrated that Cas1 (Ygb T) in the CRISPR I-E system of E. coli K12 can act on the ss DNA or branched DNA in the Holiday model, replication fork, 5′-flaps structure, and cleave them to affect the recombination and repair of the host genome. These studies revealed that the CRISPR system still has many unknown functions in regulating bacterial physiological activities. Most of the current research is based on bacterial genotyping, drug resistance, epidemiological studies, and traceability analysis. Therefore, in this study, S. aureus, the representative of foodborne pathogens, was selected as the research target, and the basic structure of the CRISPR system contained in the 38 strains of S. aureus was analyzed by means of bioinformatics. The results of bioinformatics analysis can provide a data reference for a broader range of CRISPR studies in S. aureus. At present, there are still many foodborne pathogens CRISPR system research is not involved, which need to be based on large data genotyping and foodborne pathogenic mechanism of pathogenesis is unfavorable, in this regard, put more effort to construct extensive research on CRISPR system of food borne pathogenic bacteria is imperative. With the rapid development of CRISPRs technology, from the CRISPR-Cas9 DNA-editing system to the discovery of the CRISPR-Cas13 RNA-editing system that is now replacing RNAi technology (Doudna and Charpentier, 2014; Abudayyeh et al., 2017), there is no doubt that CRISPR system have underestimated in the future various fields of research potential and application value. It is believed that as the structure of the CRISPR system is further revealed, which will change the research pattern in the life sciences.
Conclusion
Most strains of S. aureus contain only one CRISPR site, and a few strains contain multiple sites with sparse distribution. These loci mainly include highly conserved direct repeats and highly variable spacers, as well as polymorphic cas genes. In addition, all direct repeats can form stable RNA secondary structures, and spacer sequences have been shown to originate from exogenous phage or plasmids. Moreover, the specificity of spacer sequences can serve as a basis for accurate genotyping techniques. Three different CRISPR system subtypes were found in 38 S. aureus strains, including 4 active CRISPR-Cas systems. The analysis and comparison of the CRISPR/Cas system can help to understand the environmental adaptability of each S. aureus evolutionary lineage that represents different pathogenicity. Bioinformatics analysis provides data support for bacterial typing, traceability analysis, and exploration of CRISPR other than immune. The CRISPR study provides new ideas for preventing the spread of resistant genes between S. aureus and eliminating drug resistance genes.
Author Contributions
XZ designed this study and wrote the manuscripts. ZY finished the experiments and collected the data. XZ, ZY, and ZX revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (Grant No. 31501582) and Hubei Provincial Natural Science Foundation of China (2018CFB514), Science Research Fund of Wuhan Institute of Technology (17QD01 and 16QD04) and Open Project Program of Key Laboratory for Green Chemical Process of Ministry of Education in Wuhan Institute of Technology (2017007).
References
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., et al. (2017). RNA targeting with CRISPR–Cas13. Nature 550, 280–284. doi: 10.1038/nature24049
Aswani, V., Mau, B., and Shukla, S. K. (2016). Complete genome sequence of Staphylococcus aureus MCRF184, a necrotizing fasciitis-causing methicillin-sensitive sequence type 45 Staphylococcus strain. Genome Announc. 4, e00374-16.doi: 10.1128/genomeA.00374-16
Babu, M., Beloglazova, N., Flick, R., Graham, C., Skarina, T., Nocek, B., et al. (2011). A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 79, 484–502. doi: 10.1111/j.1365-2958.2010.07465.x
Bangpanwimon, K., Sottisuporn, J., Mittraparp-Arthorn, P., Ueaphatthanaphanich, W., Rattanasupar, A., Pourcel, C., et al. (2017). CRISPR-like sequences in Helicobacter pyloriand application in genotyping. Gut Pathog. 9:65. doi: 10.1186/s13099-017-0215-8
Bikard, D., Hatoum-Aslan, A., Mucida, D., and Marraffini, L. A. (2012). CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12, 177–186. doi: 10.1016/j.chom.2012.06.003
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi: 10.1126/science.1178811
Bolotin, A., Quinquis, B., Sorokin, A., and Ehrlich, S. D. (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. doi: 10.1099/mic.0.28048-0
Borowski, J. D. (2017). Analysis of CRISPR-Cas Systems Within the Bacterial Family Vibrionaceae Uncovered Significant Diversity and Evidence of Horizontal Transfer. University of Delaware.
Bosch, T., Witteveen, S., Haenen, A., Landman, F., and Schouls, L. M. (2016). Next-generation sequencing confirms presumed nosocomial transmission of livestock-associated methicillin-resistant Staphylococcus aureus in the Netherlands. Appl. Environ. Microbiol. 82:4081. doi: 10.1128/AEM.00773-16
Botelho, A. M. N., Costa, M. O. C., Beltrame, C. O., Ferreira, F. A., Côrtes, M. F., Bandeira, P. T., et al. (2016a). Complete genome sequence of an agr-dysfunctional variant of the ST239 lineage of the methicillin-resistant Staphylococcus aureus strain GV69 from Brazil. Standards Genomic Sci. 11:34. doi: 10.1186/s40793-016-0154-x
Botelho, A. M. N., Costa, M. O. C., Beltrame, C. O., Ferreira, F. A., Lima, N. C. B., Costa, B. S. S., et al. (2016b). Complete genome sequence of the MRSA isolate HC1335 from ST239 lineage displaying a truncated AgrC histidine kinase receptor. Genome Biol. Evol. 8, 3187–3192.doi: 10.1093/gbe/evw225
Chen, F.-J., Lauderdale, T.-L., Wang, L.-S., and Huang, I.-W. (2013). Complete genome sequence of Staphylococcus aureus Z172, a vancomycin-intermediate and daptomycin-nonsusceptible methicillin-resistant strain isolated in Taiwan. Genome Announc. 1, e01011-13.doi: 10.1128/genomeA.01011-13
Chuang, T. L., Chang, C. C., Chu-Su, Y., Wei, S. C., Zhao, X. H., Hsueh, P. R., et al. (2014). Disposable surface plasmon resonance aptasensor with membrane-based sample handling design for quantitative interferon-gamma detection. Lab Chip 14, 2968–2977. doi: 10.1039/C4LC00249K
Costa, M. O., Beltrame, C. O., Ferreira, F. A., Botelho, A. M., Lima, N. C., Souza, R. C., et al. (2013). Complete genome sequence of a variant of the methicillin-resistant Staphylococcus aureus ST239 lineage, strain BMB9393, displaying superior ability to accumulate ICA-independent biofilm. Genome Announc. 1:e00576-13. doi: 10.1128/genomeA.00576-13
Daum, L. T., Bumah, V. V., Masson-Meyers, D. S., Khubbar, M., Rodriguez, J. D., Fischer, G. W., et al. (2015). Whole-genome sequence for methicillin-resistant Staphylococcus aureus strain ATCC BAA-1680. Genome Announc. 3:e00011-15. doi: 10.1128/genomeA.00011-15
Di, H., Ye, L., Yan, H., Meng, H., Yamasak, S., and Shi, L. (2014). Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem. Biophys. Res. Commun. 454, 399–403. doi: 10.1016/j.bbrc.2014.10.018
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/S0140-6736(06)68231-7
Ding, T., Suo, Y. J., Xiang, Q. S., Zhao, X. H., Chen, S. G., Ye, X. Q., et al. (2017). Significance of viable but nonculturable Escherichia coli: induction, detection, and control. J. Microbiol. Biotechnol. 27, 417–428. doi: 10.4014/jmb.1609.09063
Doudna, J. A., and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Dua, M., Agarwal, D. S., and Natarajan, R. (1982). Phage typing of Staphylococcus aureus using phages other than those of basic set and new methods. Indian J. Med. Res. 75:348.
Fabre, L., Zhang, J., Guigon, G., Le, H. S., Guibert, V., Accoudemartin, M., et al. (2012). CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS ONE 7:e36995. doi: 10.1371/annotation/e79cea9a-6716-4519-9e96-31b17bf6a4fb
Giannuzzi, L., Contreras, E., and Zaritzky, N. (1999). Modeling the aerobic growth and decline of Staphylococcus aureus as affected by pH and potassium sorbate concentration. J. Food Protect. 62, 356–362. doi: 10.4315/0362-028X-62.4.356
Giraud, C., Hausmann, S. P., Lemeille, S., Prados, J., Redder, P., and Linder, P. (2015). The C-terminal region of the RNA helicase CshA is required for the interaction with the degradosome and turnover of bulk RNA in the opportunistic pathogen Staphylococcus aureus. RNA Biol. 12, 658–674. doi: 10.1080/15476286.2015.1035505
Golding, G. R., Bryden, L., Levett, P. N., Mcdonald, R. R., Wong, A., Graham, M. R., et al. (2012). whole-genome sequence of livestock-associated st398 methicillin-resistant Staphylococcus aureus Isolated from Humans in Canada. J. Bacteriol. 194, 6627–6628. doi: 10.1128/JB.01680-12
Grissa, I., Vergnaud, G., and Pourcel, C. (2007a). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172
Grissa, I., Vergnaud, G., and Pourcel, C. (2007b). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Grissa, I., Vergnaud, G., and Pourcel, C. (2008). CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 36(Suppl. 2), W145–W148. doi: 10.1093/nar/gkn228
Guinane, C. M., Ben Zakour, N. L., Tormo-Mas, M. A., Weinert, L. A., Lowder, B. V., Cartwright, R. A., et al. (2010). Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol. Evol. 2, 454–466. doi: 10.1093/gbe/evq031
Guzina, J., Rodić, A., Blagojević, B., and Ðorðević, M. (2017). Modeling and bioinformatics of bacterial immune systems: understanding regulation of CRISPR/Cas and restriction-modification systems. Biol. Serb. 39, 112–122. doi: 10.5281/zenodo.827157
Hakim, J. A., Koo, H., van Elsas, J. D., Trevors, J. T., and Bej, A. K. (2016). “CRISPR-Cas system: a new paradigm for bacterial stress response through genome rearrangement,” in Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria, ed F. J. de Bruijn (Hoboken, NJ: John Wiley & Sons, Inc.), 146–160.
Hao, M., Cui, Y., and Qu, X. (2018). Analysis of CRISPR-cas system in Streptococcus thermophilus and its application. Front. Microbiol. 9:257. doi: 10.3389/fmicb.2018.00257
Hidalgo-Cantabrana, C., Crawley, A. B., Sanchez, B., and Barrangou, R. (2017). Characterization and exploitation of CRISPR Loci in Bifidobacterium longum. Front. Microbiol. 8:1851. doi: 10.3389/fmicb.2017.01851
Holden, M. T., Hsu, L.-Y., Kurt, K., Weinert, L. A., Mather, A. E., Harris, S. R., et al. (2013). A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 23, 653–664.doi: 10.1101/gr.147710.112
Holmes, M. A., Harrison, E. M., Fisher, E. A., Graham, E. M., Julian, P., Geoffrey, F., et al. (2016). Genomic analysis of companion rabbit Staphylococcus aureus. PLoS ONE 11:e0151458. doi: 10.1371/journal.pone.0151458
Holt, D. C., Holden, M. T., Tong, S. Y., Castillo-Ramirez, S., Clarke, L., Quail, M. A., et al. (2011). A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol. Evol. 3, 881–895. doi: 10.1093/gbe/evr078
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Horvath, P., Coûté-Monvoisin, A. C., Romero, D. A., Boyaval, P., Fremaux, C., and Barrangou, R. (2009). Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. 131, 62–70. doi: 10.1016/j.ijfoodmicro.2008.05.030
Horvath, P., Romero, D. A., Coûté-Monvoisin, A. C., Richards, M., Deveau, H., Moineau, S., et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190, 1401–1412. doi: 10.1128/JB.01415-07
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63. doi: 10.1038/nature26155
Kim, B. S., Yi, H., Chun, J., and Cha, C. J. (2014). Genome sequence of type strain of Staphylococcus aureus subsp. aureus. Gut Pathog. 6:6. doi: 10.1186/1757-4749-6-6
Koonin, E. V., and Makarova, K. S. (2013). CRISPR-Cas: evolution of an RNA-based adaptive immunity system in prokaryotes. RNA Biol. 10, 679–686. doi: 10.4161/rna.24022
Koonin, E. V., and Wolf, Y. I. (2015). Evolution of the CRISPR-Cas adaptive immunity systems in prokaryotes: models and observations on virus-host coevolution. Mol. Biosyst. 11, 20–27. doi: 10.1039/C4MB00438H
Koonin, E. V., Makarova, K. S., and Zhang, F. (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. doi: 10.1016/j.mib.2017.05.008
Kunin, V., Sorek, R., and Hugenholtz, P. (2007). Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 8:R61. doi: 10.1186/gb-2007-8-4-r61
Larkin, E. A., Carman, R. J., Krakauer, T., and Stiles, B. G. (2009). Staphylococcus aureus: the toxic presence of a pathogen extraordinaire. Curr. Med. Chem. 16, 4003–4019. doi: 10.2174/092986709789352321
Li, Y., Cao, B., Zhang, Y., Zhou, J., Yang, B., and Wang, L. (2011). Complete genome sequence of Staphylococcus aureus T0131, an ST239-MRSA-SCCmec type III clone isolated in China. J. Bacteriol. 05135–05111. doi: 10.1128/JB.05135-11
Lillestøl, R. K., Redder, P., Garrett, R. A., and Brügger, K. (2006). A putative viral defence mechanism in archaeal cells. Archaea Int. Microbiol. J. 2, 59–72. doi: 10.1155/2006/542818
Lim, S., Lee, D. H., Kwak, W., Shin, H., Ku, H. J., Lee, J. E., et al. (2015). Comparative genomic analysis of Staphylococcus aureus FORC_001 and S. aureus MRSA252 reveals the characteristics of antibiotic resistance and virulence factors for human infection. J. Microbiol. Biotechnol. 25, 98–108. doi: 10.4014/jmb.1410.10005
Lin, S. Q., Li, L., Li, B., Zhao, X. H., Lin, C. W., Deng, Y., et al. (2016). Development and evaluation of quantitative detection of Nε-carboxymethyl-lysine in lococcus aureus biofilm by LC-MS method. Basic Clin. Pharmacol. Toxicol. 118, 33–33.
Lindqvist, M., Isaksson, B., Grub, C., Jonassen, T., and Hällgren, A. (2012). Detection and characterisation of SCCmec remnants in multiresistant methicillin-susceptible Staphylococcus aureus causing a clonal outbreak in a Swedish county. Eur. J. Clin. Microbiol. Infect. Dis. 31, 141–147. doi: 10.1007/s10096-011-1286-y
Lindsay, J. A. (2014). Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int. J. Med. Microbiol. 304, 103–109. doi: 10.1016/j.ijmm.2013.11.010
Liu, F., Barrangou, R., Gernersmidt, P., Ribot, E. M., Knabel, S. J., and Dudley, E. G. (2011). Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl. Environ. Microbiol. 77, 1946–1956. doi: 10.1128/AEM.02625-10
Liu, J., Zhou, R., Li, L., Peters, B. M., Li, B., Lin, C.- w., et al. (2017). Viable but non-culturable state and toxin gene expression of enterohemorrhagic Escherichia coli O157 under cryopreservation. Res. Microbiol. 168, 188–193. doi: 10.1016/j.resmic.2016.11.002
Lovley, D. R., and Muktak, A. (2010). Interference with histidyl-tRNA synthetase by a CRISPR spacer sequence as a factor in the evolution of Pelobacter carbinolicus. BMC Evol. Biol. 10:230. doi: 10.1186/1471-2148-10-230
Maeder, M. L., and Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Mol. Ther. 24, 430–446. doi: 10.1038/mt.2016.10
Maël, B., Sadeuh-Mba, S. A., Marie-Line, J., Richter, R., Patsy, P., Romain, V., et al. (2016). Whole genome sequencing of enterovirus species C isolates by high-throughput sequencing: development of generic primers. Front. Microbiol. 7:1294. doi: 10.3389/fmicb.2016.01294
Mathews, D. H. (2004). Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 10, 1178–1190. doi: 10.1261/rna.7650904
Mcculloch, J. A., Silveira, A. C., Lima, C. M. A., Pérezchaparro, P. J., Ferreira, M. S., Almeida, L. M., et al. (2015). Complete genome sequence of Staphylococcus aureus FCFHV36, a methicillin-resistant strain heterogeneously resistant to vancomycin. Genome Announc. 3:e00893-15. doi: 10.1128/genomeA.00893-15
Miao, J., Peters, B. M., Li, L., Li, B., Zhao, X. H., Xu, Z. B., et al. (2016). Evaluation of ERIC-PCR for fingerprinting merhicillin-resistant Staphylococcus aureus strains. Basic Clin. Pharmacol. Toxicol. 118, 33–33.
Moore, B. (1981). Principal component analysis in linear systems: controllability, observability, and model reduction. Autom. Control IEEE Trans. 26, 17–32. doi: 10.1109/TAC.1981.1102568
Morange, M. (2015). What history tells us XXXVII. CRISPR-Cas: the discovery of an immune system in prokaryotes. J. Biosci. 40, 221–223. doi: 10.1007/s12038-015-9532-6
Negahdaripour, M., Nezafat, N., Hajighahramani, N., Rahmatabadi, S. S., and Ghasemi, Y. (2017). Investigating CRISPR-Cas systems in Clostridium botulinum via bioinformatics tools. Infect. Genet. Evol. 54, 355–373. doi: 10.1016/j.meegid.2017.06.027
Nikki, S., and Dudley, E. G. (2014). CRISPRs: molecular signatures used for pathogen subtyping. Appl. Environ. Microbiol. 80, 430–439. doi: 10.1128/AEM.02790-13
Nübel, U., Dordel, J., Kurt, K., Strommenger, B., Westh, H., Shukla, S. K., et al. (2010). A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 6:e1000855.doi: 10.1371/journal.ppat.1000855
Palmer, K. L., and Gilmore, M. S. (2010). Multidrug-resistant enterococci lack CRISPR-cas. Mbio 1, 516–524. doi: 10.1128/mBio.00227-10
Panesso, D., Planet, P. J., Diaz, L., Hugonnet, J. E., Tran, T. T., Narechania, A., et al. (2015). Methicillin-susceptible, vancomycin-resistant Staphylococcus aureus, Brazil. Emerg. Infect. Dis. 21, 1844–1848. doi: 10.3201/eid2110.141914
Parker, D., Narechania, A., Sebra, R., Deikus, G., Larussa, S., Ryan, C., et al. (2014). Genome sequence of bacterial interference strain Staphylococcus aureus 502A. Genome Announc 2:e00284-14. doi: 10.1128/genomeA.00284-14
Penewit, K., Holmes, E. A., Mclean, K., Ren, M., Waalkes, A., and Salipante, S. J. (2018). Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. Mbio 9, e00067-18. doi: 10.1128/mBio.00067-18
Pérez-Montarelo, D., Viedma, E., Murcia, M., Muñoz-Gallego, I., Larrosa, N., Brañas, P., et al. (2017). Pathogenic characteristics of Staphylococcus aureus endovascular infection isolates from different clonal complexes. Front. Microbiol. 8:917. doi: 10.3389/fmicb.2017.00917
Planet, P. J., Diaz, L., Kolokotronis, S. O., Narechania, A., Reyes, J., Xing, G., et al. (2015). Parallel epidemics of community-associated methicillin-resistant Staphylococcus aureus USA300 infection in North and South America. J. Infect. Dis. 212, 1874–1882. doi: 10.1093/infdis/jiv320
Quan, Z., and Ye, Y. (2017). Not all predicted CRISPR–Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics 18:92. doi: 10.1186/s12859-017-1512-4
Rossi, C. C., Souzasilva, T., Araújoalves, A. V., and Giambiagidemarval, M. (2017). CRISPR-cas systems features and the gene-reservoir role of coagulase-negative Staphylococci. Front. Microbiol. 8:1545. doi: 10.3389/fmicb.2017.01545
Sabat, A. J., Budimir, A., Nashev, D., Sáleão, R., Jm, V. D., Laurent, F., et al. (2013). Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 18:20380. doi: 10.2807/ese.18.04.20380-en
Sabat, A. J., Pournaras, S., Akkerboom, V., Tsakris, A., Grundmann, H., and Friedrich, A. W. (2015). Whole-genome analysis of an oxacillin-susceptible CC80 mecA-positive Staphylococcus aureus clinical isolate: insights into the mechanisms of cryptic methicillin resistance. J. Antimicrob. Chemother. 70, 2956–2964. doi: 10.1093/jac/dkv210
Sabirova, J. S., Xavier, B. B., Hernalsteens, J. P., De Greve, H., Ieven, M., Goossens, H., et al. (2014). Complete genome sequences of two prolific biofilm-forming Staphylococcus aureus isolates belonging to USA300 and EMRSA-15 clonal lineages. Genome Announc. 2:e00610-14. doi: 10.1128/genomeA.00610-14
Sass, P., Berscheid, A., Jansen, A., Oedenkoven, M., Szekat, C., Strittmatter, A., et al. (2012). Genome sequence of Staphylococcus aureus VC40, a vancomycin-and daptomycin-resistant strain, to study the genetics of development of resistance to currently applied last-resort antibiotics. J. Bacteriol. 194, 2107–2108. doi: 10.1128/JB.06631-11
Schröder, W., Goerke, C., and Wolz, C. (2013). Opposing effects of aminocoumarins and fluoroquinolones on the SOS response and adaptability in Staphylococcus aureus. J. Antimicrob. Chemother 68, 529–538. doi: 10.1093/jac/dks456
Shariat, N., Kirchner, M. K., Sandt, C. H., Trees, E., Barrangou, R., and Dudley, E. G. (2013). Subtyping of Salmonella enterica serovar newport outbreak isolates by CRISPR-MVLST and determination of the relationship between CRISPR-MVLST and PFGE results. J. Clin. Microbiol. 51, 2328–2336. doi: 10.1128/JCM.00608-13
Sorek, R., Kunin, V., and Hugenholtz, P. (2008). CRISPR–a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 6, 181–186. doi: 10.1038/nrmicro1793
Stegger, M., Price, L. B., Larsen, A. R., Gillece, J. D., Waters, A. E., Skov, R., et al. (2012). Genome sequence of Staphylococcus aureus strain 11819-97, an ST80-IV European community-acquired methicillin-resistant isolate. J. Bacteriol. 194, 1625–1626. doi: 10.1128/JB.06653-11
Tatusova, T., Ciufo, S., Fedorov, B., O'Neill, K., and Tolstoy, I. (2013). RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42, D553–D559. doi: 10.1093/nar/gkv278
Tenover, F. C., and Goering, R. V. (2009). Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J. Antimicrob. Chemother. 64, 441–446. doi: 10.1093/jac/dkp241
Tomida, J., Morita, Y., Shibayama, K., Kikuchi, K., Sawa, T., Akaike, T., et al. (2017). Diversity and microevolution of CRISPR loci in Helicobacter cinaedi. PLoS ONE 12:e0186241. doi: 10.1371/journal.pone.0186241
Trouilletassant, S., Lelièvre, L., Martinssimões, P., Gonzaga, L., Tasse, J., Valour, F., et al. (2016). Adaptive processes of Staphylococcus aureus isolates during the progression from acute to chronic bone and joint infections in patients. Cell. Microbiol. 18, 1405–1414. doi: 10.1111/cmi.12582
Uhlemann, A.-C., Porcella, S. F., Trivedi, S., Sullivan, S. B., Hafer, C., Kennedy, A. D., et al. (2012). Identification of a highly transmissible animal-independent Staphylococcus aureus ST398 clone with distinct genomic and cell adhesion properties. MBio 3:e00027-12. doi: 10.1128/mBio.00027-12
Vahidi, H., and Honda, B. M. (1991). Repeats and subrepeats in the intergenic spacer of rDNA from the nematode Meloidogyne arenaria. Mol. Gen. Genet. 227, 334. doi: 10.1007/BF00259687
Wakefield, N., Rajan, R., and Sontheimer, E. J. (2015). Primary processing of CRISPR RNA by the endonuclease Cas6 in Staphylococcus epidermidis. FEBS Lett. 589, 3197–3204. doi: 10.1016/j.febslet.2015.09.005
Wang, R., Gan, P., Terns, M. P., Terns, R. M., and Li, H. (2011). Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure 19, 257–264. doi: 10.1016/j.str.2010.11.014
Wei, C. J., Zhong, J. L., Hu, T., and Zhao, X. H. (2018). Simultaneous detection of Escherichia coli O157:H7, Staphylococcus aureus and Salmonella by multiplex PCR in milk. 3 Biotech 8, 76. doi: 10.1007/s13205-018-1086-5
Wexler, H., and Tajkarimi, M. (2017). CRISPR-Cas systems in Bacteroides fragilis, an important pathobiont in the human gut microbiome. Front. Microbiol. 8:2234. doi: 10.3389/fmicb.2017.02234
Yang, S., Liu, J., Shao, F., Wang, P., Duan, G., and Yang, H. (2015). Analysis of the features of 45 identified CRISPR loci in 32 Staphylococcus aureus. Biochem. Biophys. Res. Commun. 464, 894–900. doi: 10.1016/j.bbrc.2015.07.062
Zegans, M. E., Wagner, J. C., Cady, K. C., Murphy, D. M., Hammond, J. H., and O'Toole, G. A. (2009). Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 191, 210–219. doi: 10.1128/JB.00797-08
Zhang, F., Wen, Y., and Guo, X. (2014). CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum. Mol. Genet. 23, R40–R46. doi: 10.1093/hmg/ddu125
Zhang, J. J., and Zhao, X. H. (2017). Changes in protein hydrolysates during processing of chinese traditional dry-cured Bacon (Laru) production. J. Food Biochem. 41:e12304. doi: 10.1111/jfbc.12304
Zhang, J. J., Xu, D. L., Zhao, X. H., Mo, H. Z., and Fang, Z. X. (2017). Effect of Zanthoxylum bungeanum maxim on the lipid oxidation and fatty acid composition of dry-cured fish during processing. J. Food Process. Preserv. 41:e12894. doi: 10.1111/jfpp.12894
Zhao, X., Lin, C. W., Wang, J., and Oh, D. H. (2014). Advances in rapid detection methods for foodborne pathogens. J. Microbiol. Biotechnol. 24, 297–312. doi: 10.4014/jmb.1310.10013
Zhao, X., Zhong, J., Wei, C., Lin, C. W., and Ding, T. (2017). Current perspectives on viable but non-culturable state in foodborne pathogens. Front. Microbiol. 8:580. doi: 10.3389/fmicb.2017.00580
Zhao, X. H., Li, M., and Xu, Z. B. (2018). Detection of foodborne pathogens by surface enhanced raman spectroscopy. Front. Microbiol. 9:1236. doi: 10.3389/fmicb.2018.01236
Zhao, X. H., Wei, C. J., Zhong, J. L., and Jin, S. W. (2016). Research advance in rapid detection of foodborne Staphylococcus aureus. Biotechnol. Biotechnol. Equipment 30, 827–833. doi: 10.1080/13102818.2016.1209433
Zhao, X. H., Zhao, F. H., Wang, J., and Zhong, N. J. (2017). Biofilm formation and control strategies of foodborne pathogens: food safety perspectives. RSC Adv. 7, 36670–36683. doi: 10.1039/C7RA02497E
Zhao, Y., Zhu, A., Tang, J., Tang, C., and Chen, J. (2017). Identification and measurement of staphylococcal enterotoxin M from Staphylococcus aureus isolate associated with staphylococcal food poisoning. Lett. Appl. Microbiol. 65, 27–34. doi: 10.1111/lam.12751
Zhong, J. L., and Zhao, X. H. (2017). Detection of viable but non-culturable Escherichia coli O157:H7 by PCR in combination with propidium monoazide. 3 Biotech 8, 28. doi: 10.1007/s13205-017-1052-7
Zhong, J. L., and Zhao, X. H. (2018). Isothermal amplification technologies for the detection of foodborne pathogens. Food Anal. Methods 11, 1543–1560. doi: 10.1007/s12161-018-1177-2
Keywords: CRISPR, direct repeat, Staphylococcus aureus, spacer, cas, food safety
Citation: Zhao X, Yu Z and Xu Z (2018) Study the Features of 57 Confirmed CRISPR Loci in 38 Strains of Staphylococcus aureus. Front. Microbiol. 9:1591. doi: 10.3389/fmicb.2018.01591
Received: 09 February 2018; Accepted: 26 June 2018;
Published: 26 July 2018.
Edited by:
Qingli Dong, University of Shanghai for Science and Technology, ChinaReviewed by:
Mehrdad Mark Tajkarimi, Brentwood Biomedical Research Institute, United StatesBeatrix Stessl, Veterinärmedizinische Universität Wien, Austria
Copyright © 2018 Zhao, Yu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xihong Zhao, eGh6aGFvMjAwNkBnbWFpbC5jb20=
Zhenbo Xu, emhlbmJvLnh1QGhvdG1haWwuY29t