 Wei Li1†
Wei Li1† Amira Yazidi2,3†Amitkumar N. Pandya4†Pooja Hegde4Weiwei Tong1Vinicius Calado Nogueira de Moura1E. Jeffrey North4*
Amira Yazidi2,3†Amitkumar N. Pandya4†Pooja Hegde4Weiwei Tong1Vinicius Calado Nogueira de Moura1E. Jeffrey North4* Jurgen Sygusch2,3*
Jurgen Sygusch2,3* Mary Jackson1*
Mary Jackson1*- 1Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO, United States
- 2Biochimie et Médecine Moléculaire, Université de Montréal, Montréal, QC, Canada
- 3Groupe d'Étude des Protéines Membranaires, Université de Montréal, Montréal, QC, Canada
- 4Department of Pharmacy Sciences, School of Pharmacy and Health Professions, Creighton University, Omaha, NE, United States
Nontuberculous mycobacterial (NTM) pulmonary infections are emerging as a global health problem and pose a threat to susceptible individuals with structural or functional lung conditions such as cystic fibrosis, chronic obstructive pulmonary disease and bronchiectasis. Mycobacterium avium complex (MAC) and Mycobacterium abscessus complex (MABSC) species account for 70–95% of the pulmonary NTM infections worldwide. Treatment options for these pathogens are limited, involve lengthy multidrug regimens of 12–18 months with parenteral and oral drugs, and their outcome is often suboptimal. Development of new drugs and improved regimens to treat NTM infections are thus greatly needed. In the last 2 years, the screening of compound libraries against M. abscessus in culture has led to the discovery of a number of different chemotypes that target MmpL3, an essential inner membrane transporter involved in the export of the building blocks of the outer membrane of all mycobacteria known as the mycolic acids. This perspective reflects on the therapeutic potential of MmpL3 in Mycobacterium tuberculosis and NTM and the possible reasons underlying the outstanding promiscuity of this target. It further analyzes the physiological and structural factors that may account for the apparent looser structure-activity relationship of some of these compound series against M. tuberculosis compared to NTM.
Introduction
The prevalence of pulmonary nontuberculous mycobacterial (NTM) infections caused by Mycobacterium avium complex (MAC) and Mycobacterium abscessus complex (MABSC) species is increasing worldwide and poses a particular threat to susceptible individuals with structural or functional lung conditions such as cystic fibrosis (CF), chronic obstructive pulmonary disease, and bronchiectasis (Park and Olivier, 2015; Parkins and Floto, 2015; Bryant et al., 2016; Floto et al., 2016; Martiniano et al., 2016). Treatment options for NTM pulmonary infections involve lengthy (12–18 months) combination regimens with antibiotics that lack bactericidal activity and are associated with significant toxicity. For pulmonary MAC, the recommended treatment includes a macrolide, rifamycin, and ethambutol to which intravenous amikacin may be added. Treatment of pulmonary MABSC typically consists of an oral macrolide in conjunction with intravenous or inhaled amikacin, and one or more of the following drugs: intravenous cefoxitin, imipenem, or tigecycline, in addition to oral antibiotics (minocycline, clofazimine, moxifloxacin, linezolid; Floto et al., 2016). The impermeability of the cell envelopes of NTM to drugs and the high number of efflux systems and antibiotic inactivation mechanisms with which NTM are typically endowed confer upon these microorganisms high intrinsic protection against antibiotics (Brown-Elliott et al., 2012). There is clearly an urgent need for more active and better-tolerated drugs to improve therapeutic outcome (Jarand et al., 2011; Maurer et al., 2014; Park and Olivier, 2015; Martiniano et al., 2016).
In last 3 years, the phenotypic screening of compound libraries against NTM has yielded a number of hits with activity against MABSC, MAC, or both complexes. Interestingly, several of these compounds appear to kill NTM through the inhibition of MmpL3, an essential mycolic acid transporter present in all mycobacteria whose therapeutic potential in the treatment of M. tuberculosis infections was highlighted in a number of recent studies (Sacksteder et al., 2012; Kondreddi et al., 2013; Lun et al., 2013; Rao et al., 2013; Remuinan et al., 2013; Yokokawa et al., 2013; Li et al., 2016, 2017; Poce et al., 2016, 2018; Stec et al., 2016; Degiacomi et al., 2017). The availability of cidal inhibitors against this new target, some of which have already demonstrated activity in in vivo models of MABSC infection (Dupont et al., 2016; De Groote et al., in revision; Pandya et al., in revision), provides much-needed novel opportunities for the treatment of pulmonary NTM infections.
This perspective reflects on the therapeutic potential and promiscuity of MmpL3 in NTM, and discusses recent findings from our laboratories toward understanding the basis for the better activity and looser structure-activity relationship of MmpL3 inhibitors against M. tuberculosis compared to NTM.
The Phenotypic Screening of Compound Libraries Against NTM Identifies Inhibitors of MmpL3
In the last 3 years, the screening of compound libraries, including libraries of TB actives, against MABSC and MAC, has yielded a number of potent hits that appear to target the mycolic acid transporter MmpL3. These include indole-2-carboxamides (ICs) (Franz et al., 2017; Kozikowski et al., 2017; Low et al., 2017), benzothiazole amides (De Groote et al., in revision), and a piperidinol derivative (PIPD1) (Dupont et al., 2016; Low et al., 2017). Earlier work on analogs of the M. tuberculosis MmpL3 inhibitor BM212 had further highlighted the activity of pyrole derivatives against a variety of NTM including M. avium, M. gordonae, M. smegmatis, and M. marinum (Biava et al., 1999, 2007; Biava, 2002). A subset of these hits and their MIC against M. tuberculosis, M. avium and MABSC isolates (including M. abscessus subsp. abscessus, M. abscessus subsp. Massiliense, and M. abscessus subsp. bolletti) is presented in Table 1 along with that of other chemotypes reported to inhibit MmpL3 activity in M. tuberculosis (i.e., the 1,2-ethylene diamine SQ109, the tetrahydropyrazolopyrimide carboxamide THPP1, and the adamantyl urea AU1235) (Grzegorzewicz et al., 2012; La Rosa et al., 2012; Tahlan et al., 2012; Remuinan et al., 2013).
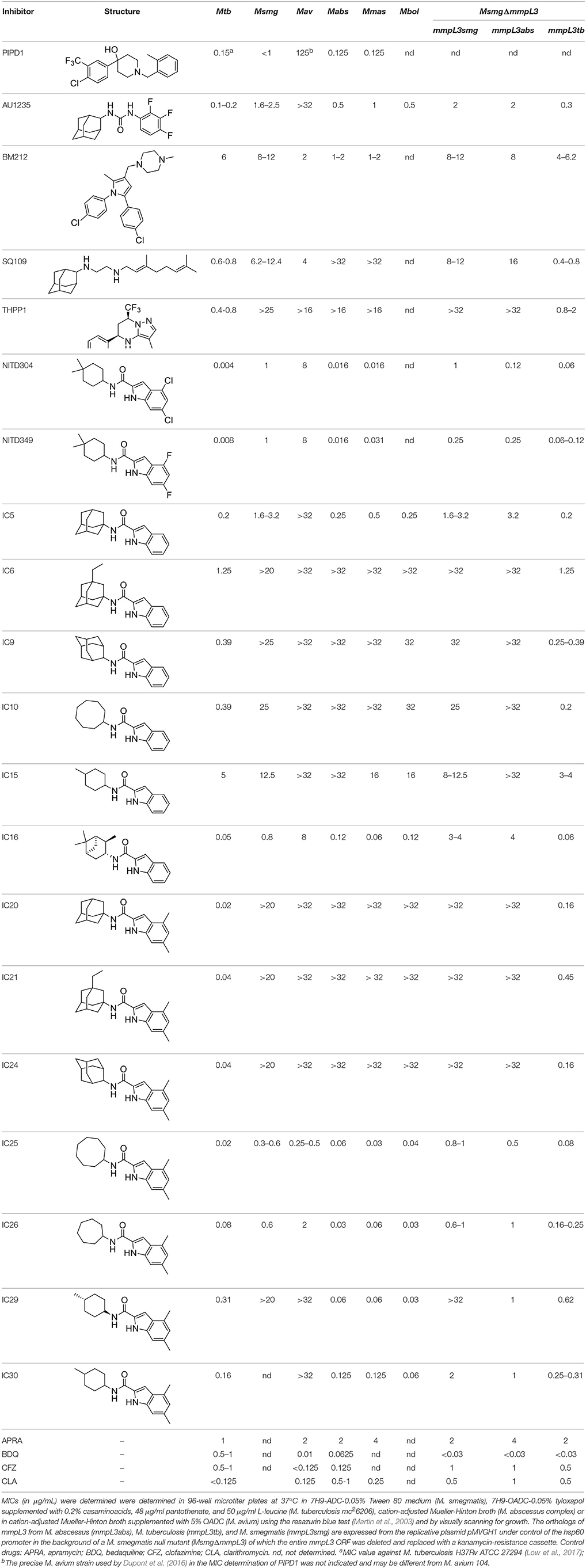
Table 1. MICs of MmpL3 inhibitors against M. tuberculosis [Mtb], M. abscessus complex species (M. abscessus subsp. abscessus ATCC 19977 [Mabs]; M. abscessus subsp. massiliense CIP 108297 [Mmas]; M. abscessus subsp. bolletii ATCC 14472 [Mbol]), M. avium 104 [Mav], and M. smegmatis recombinant strains expressing different mmpL3 orthologs.
ICs have previously been identified as a novel chemical scaffold showing promise in the treatment of tuberculosis (Kondreddi et al., 2013; Lun et al., 2013; Rao et al., 2013; Stec et al., 2016). Based on their high anti-MABSC potency, bactericidal activity on extracellularly- and intracellularly-grown bacilli and promising safe pharmacological profile (Franz et al., 2017; Kozikowski et al., 2017; Pandya et al., in revision), two lead molecules were advanced for efficacy studies in a mouse model of MABSC infection. Oral administration of the lead compounds showed a statistically significant reduction in bacterial load in the lungs, spleen and liver of MABSC-infected mice compared to an untreated control group, with one of the two compounds (compound # IC25; see Table 1) showing similar efficacy to amikacin (Pandya et al., in revision). The intrapulmonary delivery of a lead benzothiazole amide compound also demonstrated in vivo efficacy in a mouse model of chronic MABSC lung infection (De Groote et al., in revision), whereas the piperidinol-based compound PIPD1 was reported to restrict bacterial growth in a zebrafish model of MABSC infection (Dupont et al., 2016). An interesting property of MmpL3 inhibitors first revealed in M. tuberculosis is their ability to synergize with a number of other antimycobacterial drugs or drug candidates including rifampicin, bedaquiline, clofazimine, and β-lactams (Li et al., 2017). Our preliminary studies with IC25 (see Table 1) in MABSC similarly point to the existence of a synergistic interaction between this MmpL3 inhibitor and clofazimine in MABSC (Table S1).
Collectively, these results highlight the therapeutic potential of MmpL3 inhibitors in the treatment of NTM infections and provide a strong incentive to develop these compounds into new generation antimycobacterial drugs as their inclusion in anti-NTM drug regimens has the potential to lead to the faster and more efficient clearance of NTM from infected tissues.
MmpL3: A Promiscuous Mycobacterial Target
The reason why so many chemical scaffolds kill M. tuberculosis and NTM through the inhibition of MmpL3 remains incompletely understood. MmpL3 belongs to the Resistance, Nodulation and Division (RND) superfamily of transporters that requires the transmembrane electrochemical proton gradient for activity. The observation that the most common resistance mutations identified in both M. tuberculosis and MABSC tend to map to a transmembrane region of MmpL3 overlapping with functional residues required for proton translocation or proton-driven conformational changes in the transporter has led to the hypothesis that inhibitors might target the proton relay site of MmpL3 (Belardinelli et al., 2016). MmpL3 inhibitors are typically lipophilic (logP ~2.6–7.0) and many suffer from poor aqueous solubility which likely favors their concentration in the inner membrane where MmpL3 is located. This property and the extreme vulnerability of MmpL3 (Li et al., 2016) that may allow inhibitors with relatively weak binding affinity to the transporter to still inhibit enough of its activity to cause growth arrest, could explain the bias of phenotypic screens toward small hydrophobic inhibitors of MmpL3. The exquisite vulnerability of MmpL3 may further mask potential secondary targets of the inhibitors as illustrated by THPP derivatives that were found to target another essential mycolic acid-related protein in M. tuberculosis (Cox et al., 2016) and compounds such as SQ109, BM212, and some THPPs that show activity against non-replicating M. tuberculosis bacilli, a property typically not shared by other MmpL3 inhibitors Li W. et al., 2014). The fact that the hydrophobicity of ICs, THPPs, SQ109 analogs and urea derivatives is a key driver of their efficacy provides further support to the notion that the concentration of MmpL3 inhibitors in the phospholipid bilayer plays a key role in their activity (Biava et al., 2005, 2007; Onajole et al., 2010, 2013; Brown et al., 2011; Scherman et al., 2012; Kondreddi et al., 2013; North et al., 2013; Li K. et al., 2014; Poce et al., 2016; Stec et al., 2016; Franz et al., 2017; Kozikowski et al., 2017).
A second mechanism through which high rates of apparent MmpL3 inhibitors may arise from phenotypic screens was proposed after it was found that unspecific uncouplers such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or the K+ ionophore, valinomycin, both abolished MmpL3 activity in M. tuberculosis and M. smegmatis (Li W. et al., 2014). This finding indicated that any compound with the ability to dissipate the proton motive force may indirectly inhibit MmpL3 activity with immediate consequences on mycobacterial growth and viability. Accordingly, and most likely explaining the relatively broad spectrum of activity of some of these compounds including against bacteria devoid of MmpL3 homolog, inhibitors such as the 1,2-ethylenediamine SQ109, the adamantyl urea AU1235 and the 1,5-diarylpyrrole derivative BM212 were found to impact to some degree the membrane potential, the electrochemical proton gradient or both components of the proton motive force of mycobacterial cells (Li K. et al., 2014; Li W. et al., 2014; Feng et al., 2015; Foss et al., 2016). This unspecific activity, however, was later disputed in the case of BM212 and this compound proposed to directly inhibit MmpL3 on the basis of its demonstrated binding to the purified MmpL3 protein from M. smegmatis (Xu et al., 2017).
In conclusion, both direct and indirect mechanisms can lead to MmpL3 inhibition in treated mycobacterial cells and contribute to the promiscuity of the target. While not mutually exclusive, a precise understanding of how these two mechanism(s) play out for each inhibitor to eventually abolish mycolic acid export will require a detailed analysis of how each of them interacts with the transporter and affects the energy metabolism of the bacterium.
NTM vs. M. Tuberculosis Efficacy
From the MIC data presented in Table 1 and previous studies (Biava et al., 1999, 2007; Biava, 2002; Li K. et al., 2014; Franz et al., 2017; Kozikowski et al., 2017; Low et al., 2017; De Groote et al., in revision), it is obvious that the overall activity of MmpL3 inhibitors against NTM, particularly MAC, is less than that observed against M. tuberculosis. While the structural diversity of the chemotypes found to inhibit MmpL3 is very broad, spanning from compounds such as BM212 and THPP1, which are large (for MmpL3 inhibitors) multicyclic compounds, to SQ109 which is an ethylene diamine originally designed as an ethambutol analog, the majority of MmpL3 inhibitors reported to date have come from two other classes that contain the same pharmacophore which are the ureas (e.g., AU1235) and indole-2-carboxamides. The pharmacophore for these classes of MmpL3 inhibitors are two hydrogen bond donors and one hydrogen bond acceptor in the center of the molecule and one bulky lipophilic aliphatic ring (adamantyl, cyclooctyl, cycloheptyl, or substituted cyclohexyl groups) and one aromatic ring on either side of the core. For structure activity relationships, generally, as lipophilicity is increased on either the bulky aliphatic ring (typically through ring expansion or addition of methylene or methyl groups) or aromatic ring (typically through addition of halogens or methyl groups), anti-NTM activity is improved.
Since a number of factors could account for the overall better activity of MmpL3 inhibitors against M. tuberculosis than NTM, including species-specific variations in the structure of MmpL3 orthologs, increased drug efflux/degradation/modification in NTM relative to M. tuberculosis, or reduced compound penetration in NTM, we first sought to compare the MIC of the inhibitors presented in Table 1 against M. smegmatis recombinant strains expressing different MmpL3 orthologs. To this end, mmpL3 from M. tuberculosis, M. smegmatis and M. abscessus subsp. abscessus were expressed from the same expression plasmid in the background of a M. smegmatis mutant whose endogenous mmpL3 gene was deleted by allelic replacement (MsmgΔmmpL3) (Belardinelli et al., 2016). Expressing all orthologs from the same promoter in the same M. smegmatis strain abolished any potential differences in compound uptake, modification and efflux allowing for a direct comparison of the effect of the inhibitors against the three MmpL3 proteins. Importantly, all three mmpL3 orthologs were expressed at comparable levels in this recombinant system (Figure S1). The results of these comparative MIC studies clearly indicated that the MICs of the inhibitors against the different M. smegmatis strains generally reflected their MICs against the Mycobacterium species from which the rescue mmpL3 ortholog originated (Table 1). The structure of MmpL3 thus appears to be the main driver of the susceptibility of each Mycobacterium species to these inhibitors.
MmpL3 Modeling
To investigate the structural basis for the different susceptibilities of MmpL3 orthologs to various classes of inhibitors, the I-TASSER server (Yang et al., 2015) was used for automated full-length 3D structure prediction of MmpL3 transporters from M. tuberculosis H37Rv, M. abscessus ATCC 19977, M. smegmatis mc2155, and M. avium 104. The top predicted structure for each MmpL3 transporter corresponded to a C-score of >-1.5 suggesting a correct fold. All MmpL3 orthologs resemble each other (root mean square differences based C-alpha atoms < 0.4Å) and had as closest target the crystal structure of the Burkholderia multivorans hopanoid transporter HpnN (PDB 5khnB). Comparison of the predicted structures with that of HpnN yielded a high TM-score >0.8 and low RMSD <2.0 between residues that were structurally aligned by TM-align (Zhang, 2008). The superposition of all three NTM MmpL3 orthologs onto MmpL3 from M. tuberculosis H37Rv shows very similar spatial overlap of the C-alpha positions of essential residues identified in the reference MmpL3 transporter (Belardinelli et al., 2016) that can be seen in Figure 1A. Each of the predicted structures contained 12 TM helices.
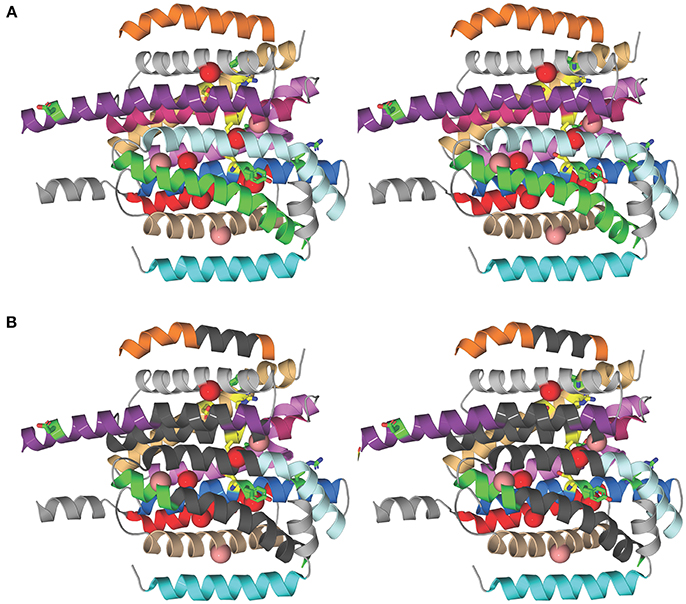
Figure 1. Structural comparison of NTM and M. tuberculosis MmpL3 transporters. (A) Stereo model showing the transmembrane (TM) helices of the M. tuberculosis MmpL3 subunit structure as predicted by I-TASSER. The TM helices are color-coded to improve visibility. From top to bottom, TMS-7 (orange), TMS-9 (gray), TMS-8 (violet purple), TMS-10 (pink), TMS-12 (light orange), TMS-11 (violet), TMS-5 (pale cyan), TMS-4 (marine), TMS-6 (green), TMS-2 (red), TMS-3 (wheat), and TMS-1 (cyan). The TM helices encompass residues whose mutations resulted in significant reduction in transport activity, shown in green, and residues 251, 288, 640, 641, 710, and 715 whose mutation abolished transport activity, colored yellow (Belardinelli et al., 2016). The positions of frequently encountered resistance mutations to one or more MmpL3 inhibitor series are shown as red and salmon spheres centered on the Cα atom of the native MmpL3 residue (Belardinelli et al., 2016). These residues map to TM helices. Resistance mutations also producing a significant reduction in growth are shown in red and those that slightly attenuated growth are colored salmon. (B) Stereo model as in (A) showing regions of the TM helices where the majority of residues are not conserved between MmpL3 orthologs (dark gray). Most of the dissimilar residues represent semi-conservative and non-conservative mutations (see Figure S2). Several of these regions map vicinal to the functional residues and mutations that induce resistance.
We next aligned the amino acid sequences of the MmpL3 orthologs among each other using PSI/TM-Coffee (Chang et al., 2012; Figure S2). Essential functional residues identified in M. tuberculosis MmpL3 (namely, residues: 251, 288, 640, 641, 710, and 715; boxed in green in Figure S2; Belardinelli et al., 2016) are conserved and are all located in the central regions of the 12 TM helical bundle that is thought to be involved in proton translocation.
We then searched for sequence dissimilarities among the TM helices given the lipophilicity of the MmpL3 inhibitors. The stereo model in Figure 1B shows regions of the transmembrane helices in dark gray where the majority of amino acid residues are not conserved between MmpL3 orthologs and which span the central regions of the 12 TM helical bundle. Most of the dissimilar residues represent semi-conservative and non-conservative mutations that are shown as red boxes on the sequence alignment shown in Figure S2. Several of these regions map vicinal to the functional residues and mutations that induce resistance. The shape differences in the geometries of the hydrophobic and polar side chains alters the packing of the 12 TM helices and is likely to concomitantly modify their dynamical behavior important for transport activity and inhibitor binding. Given that the inhibitors are partitioned into the lipid bilayer, from a thermodynamic perspective, their propensity to interact with the TM helices will further depend on two factors: their ability to interact preferentially with the hydrophobic side chains of the TM helices and their ability to form polar interactions with either backbone or polar side chains. It follows that both the differential helical packing modifying the binding loci of the inhibitors and the nature of the side chains of the TM helices probably account for the ortholog-dependent activity of MmpL3 inhibitors.
Future Directions
There is an unmet medical need for the development of new bactericidal agents to treat pulmonary NTM infections. The novel classes of bactericidal MmpL3 inhibitors that have been reported in the last few years, some of which have demonstrated activity against M. tuberculosis and MABSC in vivo, highlight the therapeutic potential of this transporter in tuberculous and nontuberculous mycobacteria and provide much needed translational opportunities for the treatment of NTM infections. Future research is expected to gain further insight into the structure of MmpL3 and its variations across Mycobacterium species in order to leverage the emerging structure-activity relationship information now available for some of these compound series (Brown et al., 2011; Scherman et al., 2012; North et al., 2013; Li K. et al., 2014; Poce et al., 2016, 2018; Stec et al., 2016; Franz et al., 2017; Kozikowski et al., 2017). Also, critical to the further development of these inhibitors will be the availability of a simple, non-radioactive, and relatively high-throughput assay to screen optimized analogs with increased activity against MmpL3. Currently available cell-free and whole cell-based assays (e.g., Grzegorzewicz et al., 2012; Li W. et al., 2014; Li et al., 2016; Xu et al., 2017) indeed lack the simplicity of use and/or specificity required to rapidly screen such analogs. The development of such assays is currently the object of intense efforts in our laboratories.
Author Contributions
EN, JS, and MJ conceived the project, analyzed the data, and wrote the manuscript. WL, PH, WT, and VC generated and characterized the M. smegmatis recombinant strains, and carried out the MIC determinations and checkerboard assays. AP synthesized inhibitors, contributed to the preparation of Table 1 and analyzed SAR data. AY performed the MmpL3 modeling studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (AI116525) (to MJ and EN) and a grant from the Bill and Melinda Gates Foundation (OPP1181207) (to MJ and JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We are grateful to the Global Alliance for TB Drug Development for the provision of NITD-304 and NITD-349.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01547/full#supplementary-material
References
Belardinelli, J. M., Yazidi, A., Yang, L., Fabre, L., Li, W., Jacques, B., et al. (2016). Structure-Function profile of MmpL3, the essential mycolic acid transporter from Mycobacterium tuberculosis. ACS Infect. Dis. 2, 702–713. doi: 10.1021/acsinfecdis.6b00095
Biava, M. (2002). BM 212 and its derivatives as a new class of antimycobacterial active agents. Curr. Med. Chem. 9, 1859–1869. doi: 10.2174/0929867023368953
Biava, M., Fioravanti, R., Porretta, G. C., Deidda, D., Maullu, C., and Pompei, R. (1999). New pyrrole derivatives as antimycobacterial agents analogs of BM212. Bioorg. Med. Chem. Lett. 9, 2983–2988.
Biava, M., Porretta, G. C., and Manetti, F. (2007). New derivatives of BM212: A class of antimycobacterial compounds based on the pyrrole ring as a scaffold. Mini-Rev. Med. Chem. 7, 65–78. doi: 10.2174/138955707779317786
Biava, M., Porretta, G. C., Poce, G., Deidda, D., Pompei, R., Tafi, A., et al. (2005). Antimycobacterial compounds. Optimization of the BM 212 structure, the lead compound for a new pyrrole derivative class. Bioorg. Med. Chem. 13, 1221–1230. doi: 10.1016/j.bmc.2004.11.018
Brown, J. R., North, E. J., Hurdle, J. G., Morisseau, C., Scarborough, J. S., Sun, D., et al. (2011). The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg. Med. Chem. 19, 5585–5595. doi: 10.1016/j.bmc.2011.07.034
Brown-Elliott, B. A., Nash, K. A., and Wallace, R. J. Jr. (2012). Antimicrobial susceptibility testing, drug resistance mechanisms, and therapy of infections with nontuberculous mycobacteria. Clin. Microbiol. Rev. 25, 545–582. doi: 10.1128/CMR.05030-11
Bryant, J. M., Grogono, D. M., Rodriguez-Rincon, D., Everall, I., Brown, K. P., Moreno, P., et al. (2016). Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 354, 751–757. doi: 10.1126/science.aaf8156
Chang, J. M., Di Tommaso, P., Taly, J. F., and Notredame, C. (2012). Accurate multiple sequence alignment of transmembrane proteins with PSI-Coffee. BMC Bioinformatics 13(Suppl. 4):S1. doi: 10.1186/1471-2105-13-S4-S1
Cox, J. A., Abrahams, K. A., Alemparte, C., Ghidelli-Disse, S., Rullas, J., Angulo-Barturen, I., et al. (2016). THPP target assignment reveals EchA6 as an essential fatty acid shuttle in mycobacteria. Nat. Microbiol. 1:15006. doi: 10.1038/nmicrobiol.2015.6
Degiacomi, G., Benjak, A., Madacki, J., Boldrin, F., Provvedi, R., Palu, G., et al. (2017). Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci. Rep. 7:43495. doi: 10.1038/srep43495
Dupont, C., Viljoen, A., Dubar, F., Blaise, M., Bernut, A., Pawlik, A., et al. (2016). A new piperidinol derivative targeting mycolic acid transport in Mycobacterium abscessus. Mol. Microbiol. 101, 515–529. doi: 10.1111/mmi.13406
Feng, X., Zhu, W., Schurig-Briccio, L. A., Lindert, S., Shoen, C., Hitchings, R., et al. (2015). Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. U.S.A. 112, E7073–E7082. doi: 10.1073/pnas.1521988112
Floto, R. A., Olivier, K. N., Saiman, L., Daley, C. L., Herrmann, J. L., Nick, J. A., et al. (2016). US Cystic fibrosis foundation and european cystic fibrosis society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis: executive summary. Thorax 71, 88–90. doi: 10.1136/thoraxjnl-2015-207983
Foss, M. H., Pou, S., Davidson, P. M., Dunaj, J. L., Winter, R. W., Pou, S., et al. (2016). Diphenylether-Modified 1,2-Diamines with improved drug properties for development against Mycobacterium tuberculosis. ACS Infect. Dis. 2, 500–508. doi: 10.1021/acsinfecdis.6b00052
Franz, N. D., Belardinelli, J. M., Kaminski, M. A., Dunn, L. C., Calado Nogueira de Moura, V., Blaha, M. A., et al. (2017). Design, synthesis and evaluation of indole-2-carboxamides with pan anti-mycobacterial activity. Bioorg. Med. Chem. 25, 3746–3755. doi: 10.1016/j.bmc.2017.05.015
Grzegorzewicz, A. E., Pham, H., Gundi, V. A., Scherman, M. S., North, E. J., Hess, T., et al. (2012). Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341. doi: 10.1038/nchembio.794
Jarand, J., Levin, A., Zhang, L., Huitt, G., Mitchell, J. D., and Daley, C. L. (2011). Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin. Infect. Dis. 52, 565–571. doi: 10.1093/cid/ciq237
Kondreddi, R. R., Jiricek, J., Rao, S. P., Lakshminarayana, S. B., Camacho, L. R., Rao, R., et al. (2013). Design, synthesis, and biological evaluation of Indole-2-carboxamides: a promising class of antituberculosis agents. J. Med. Chem. 56, 8849–8859. doi: 10.1021/jm4012774
Kozikowski, A. P., Onajole, O. K., Stec, J., Dupont, C., Viljoen, A., Richard, M., et al. (2017). Targeting mycolic acid transport by Indole-2-carboxamides for the treatment of Mycobacterium abscessus infections. J. Med. Chem. 60, 5876–5888. doi: 10.1021/acs.jmedchem.7b00582
La Rosa, V., Poce, G., Canseco, J. O., Buroni, S., Pasca, M. R., Biava, M., et al. (2012). MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 56, 324–331. doi: 10.1128/AAC.05270-11
Li, K., Schurig-Briccio, L. A., Feng, X., Upadhyay, A., Pujari, V., Lechartier, B., et al. (2014). Multi-target drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 57, 3126–3139. doi: 10.1021/jm500131s
Li, W., Obregon-Henao, A., Wallach, J. B., North, E. J., Lee, R. E., Gonzalez-Juarrero, M., et al. (2016). Therapeutic potential of the Mycobacterium tuberculosis mycolic acid transporter, MmpL3. Antimicrob. Agents Chemother. 60, 5198–5207. doi: 10.1128/AAC.00826-16
Li, W., Sanchez-Hidalgo, A., Jones, V., de Moura, V. C., North, E. J., and Jackson, M. (2017). Synergistic interactions of mmpl3 inhibitors with antitubercular compounds in vitro. Antimicrob. Agents Chemother. 61:e02399-e02316. doi: 10.1128/AAC.02399-16
Li, W., Upadhyay, A., Fontes, F. L., North, E. J., Wang, Y., Crans, D. C., et al. (2014). Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6413–6423. doi: 10.1128/AAC.03229-14
Low, J. L., Wu, M. L., Aziz, D. B., Laleu, B., and Dick, T. (2017). Screening of TB actives for activity against nontuberculous mycobacteria delivers high hit rates. Front. Microbiol. 8:1539. doi: 10.3389/fmicb.2017.01539
Lun, S., Guo, H., Onajole, O. K., Pieroni, M., Gunosewoyo, H., Chen, G., et al. (2013). Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat. Commun. 4:2907. doi: 10.1038/ncomms3907
Martin, A., Camacho, M., Portaels, F., and Palomino, J.-C. (2003). Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 47, 3616–3619. doi: 10.1128/AAC.47.11.3616-3619.2003
Martiniano, S. L., Nick, J. A., and Daley, C. L. (2016). Nontuberculous mycobacterial infections in cystic fibrosis. Clin. Chest. Med. 37, 83–96. doi: 10.1016/j.ccm.2015.11.001
Maurer, F. P., Bruderer, V. L., Ritter, C., Castelberg, C., Bloemberg, G. V., and Bottger, E. C. (2014). Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob. Agents Chemother. 58, 3828–3836. doi: 10.1128/AAC.02448-14
North, E. J., Scherman, M. S., Bruhn, D. F., Scarborough, J. S., Maddox, M. M., Jones, V., et al. (2013). Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg. Med. Chem. 21, 2587–2599. doi: 10.1016/j.bmc.2013.02.028
Onajole, O. K., Govender, P., van Helden, P. D., Kruger, H. G., Maguire, G. E., Wiid, I., et al. (2010). Synthesis and evaluation of SQ109 analogues as potential anti-tuberculosis candidates. Eur. J. Med. Chem. 45, 2075–2079. doi: 10.1016/j.ejmech.2010.01.046
Onajole, O. K., Pieroni, M., Tipparaju, S. K., Lun, S., Stec, J., Chen, G., et al. (2013). Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. J. Med. Chem. 56, 4093–4103 doi: 10.1021/jm4003878
Park, I. K., and Olivier, K. N. (2015). Nontuberculous mycobacteria in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin. Respir. Crit. Care Med. 36, 217–224. doi: 10.1055/s-0035-1546751
Parkins, M. D., and Floto, R. A. (2015). Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J. Cyst. Fibros 14, 293–304. doi: 10.1016/j.jcf.2015.03.012
Poce, G., Cocozza, M., Alfonso, S., Consalvi, S., Venditti, G., Fernandez-Menendez, R., et al. (2018). In vivo potent BM635 analogue with improved drug-like properties. Eur. J. Med. Chem. 145, 539–550. doi: 10.1016/j.ejmech.2017.12.075
Poce, G., Consalvi, S., and Biava, M. (2016). MmpL3 inhibitors: diverse chemical scaffolds inhibit the same target. Mini-Rev. Med. Chem. 16, 1274–1283. doi: 10.2174/1389557516666160118105319
Rao, S. P., Lakshminarayana, S. B., Kondreddi, R. R., Herve, M., Camacho, L. R., Bifani, P., et al. (2013). Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 5:214ra168. doi: 10.1126/scitranslmed.3007355
Remuinan, M. J., Perez-Herran, E., Rullas, J., Alemparte, C., Martinez-Hoyos, M., Dow, D. J., et al. (2013). Tetrahydropyrazolo[1,5-a]Pyrimidine-3-Carboxamide and N-Benzyl-6',7'-Dihydrospiro[Piperidine-4,4'-Thieno[3,2-c] Pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLoS ONE 8:e60933. doi: 10.1371/journal.pone.0060933
Sacksteder, K. A., Protopopova, M., Barry, C. E. III., Andries, K., and Nacy, C. A. (2012). Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 7, 823–837. doi: 10.2217/fmb.12.56
Scherman, M. S., North, E. J., Jones, V., Hess, T. N., Grzegorzewicz, A. E., Kasagami, T., et al. (2012). Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability. Bioorg. Med. Chem. 20, 3255–3262. doi: 10.1016/j.bmc.2012.03.058
Stec, J., Onajole, O. K., Lun, S., Guo, H., Merenbloom, B., Vistoli, G., et al. (2016). Indole-2-carboxamide-based MmpL3 inhibitors show exceptional antitubercular activity in an animal model of tuberculosis infection. J. Med. Chem. 59, 6232–6247. doi: 10.1021/acs.jmedchem.6b00415
Tahlan, K., Wilson, R., Kastrinsky, D. B., Arora, K., Nair, V., Fischer, E., et al. (2012). SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809. doi: 10.1128/AAC.05708-11
Xu, Z., Meshcheryakov, V. A., Poce, G., and Chng, S. S. (2017). MmpL3 is the flippase or mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 114, 7993–7998. doi: 10.1073/pnas.1700062114
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yokokawa, F., Wang, G., Chan, W. L., Ang, S. H., Wong, J., Ma, I., et al. (2013). Discovery of tetrahydropyrazolopyrimidine carboxamide derivatives as potent and orally active antitubercular agents. ACS Med. Chem. Lett. 4, 451–455. doi: 10.1021/ml400071a
Keywords: Mycobacterium abscessus, nontuberculous mycobacteria, MmpL3, mycolic acids, drug development
Citation: Li W, Yazidi A, Pandya AN, Hegde P, Tong W, Calado Nogueira de Moura V, North EJ, Sygusch J and Jackson M (2018) MmpL3 as a Target for the Treatment of Drug-Resistant Nontuberculous Mycobacterial Infections. Front. Microbiol. 9:1547. doi: 10.3389/fmicb.2018.01547
Received: 18 May 2018; Accepted: 21 June 2018;
Published: 10 July 2018.
Edited by:
Thomas Dick, Rutgers, The State University of New Jersey, Newark, United StatesReviewed by:
Shashank Gupta, Brown University, United StatesKapil Tahlan, Memorial University of Newfoundland, Canada
Copyright © 2018 Li, Yazidi, Pandya, Hegde, Tong, Calado Nogueira de Moura, North, Sygusch and Jackson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: E. Jeffrey North, amVmZnJleW5vcnRoQGNyZWlnaHRvbi5lZHU=
Jurgen Sygusch, anVyZ2VuLnN5Z3VzY2hAdW1vbnRyZWFsLmNh
Mary Jackson, bWFyeS5qYWNrc29uQGNvbG9zdGF0ZS5lZHU=
†Co-first authors.