 Sebastian Reichert1
Sebastian Reichert1 Patrick Ebner1
Patrick Ebner1 Eve-Julie Bonetti2Arif Luqman1Mulugeta Nega1
Eve-Julie Bonetti2Arif Luqman1Mulugeta Nega1 Jacques Schrenzel2Cathrin Spröer3
Jacques Schrenzel2Cathrin Spröer3 Boyke Bunk3
Boyke Bunk3 Jörg Overmann3
Jörg Overmann3 Peter Sass4
Peter Sass4 Patrice François2
Patrice François2 Friedrich Götz1*
Friedrich Götz1*- 1Microbial Genetics, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Tübingen, Germany
- 2Genomic Research Laboratory, Division of Infectious Diseases, Geneva University Hospital, Geneva, Switzerland
- 3Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany
- 4Microbial Bioactive Compounds, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Tübingen, Germany
In this study we addressed the question how a mevalonate (MVA)-auxotrophic Staphylococcus aureusΔmvaS mutant can revert to prototrophy. This mutant couldn’t grow in the absence of MVA. However, after a long lag-phase of 4–6 days the mutant adapted from auxotrophic to prototrophic phenotype. During that time, it acquired two point mutations: One mutation in the coding region of the regulator gene spx, which resulted in an amino acid exchange that decreased Spx function. The other mutation in the upstream-element within the core-promoter of the mevalonolactone lactonase gene drp35. This mutation led to an increased expression of drp35. In repeated experiments the mutations always occurred in spx and drp35 and in the same order. The first detectable mutation appeared in spx and allowed slight growth; the second mutation, in drp35, increased growth further. Phenotypical characterizations of the mutant showed that small amounts of the lipid-carrier undecaprenol are synthesized, despite the lack of mvaS. The growth of the adapted clone, ΔmvaSad, indicates that the mutations reawake a rescue bypass. We think that this bypass enters the MVA pathway at the stage of MVA, because blocking the pathway downstream of MVA led to growth arrest of the mutant. In addition, the lactonase Drp35 is able to convert mevalonolactone to MVA. Summarized, we describe here a mutation-based two-step adaptation process that allows resuscitation of growth of the ΔmvaS mutant.
Introduction
The very large and diverse class of isoprenoids is composed of 1000s of different organic compounds. Representatives can be found in all living organisms, where they fulfill essential roles. They are involved in the electron transport during aerobic respiration (quinones), the cell wall synthesis (bactoprenols), the photosynthesis (carotenoids), membrane stabilization, protein translation and degradation, gene transcription and many more (Holstein and Hohl, 2004). All isoprenoids share a common basic precursor, the five-carbon molecule IPP and its isomer DMAPP. There are two alternative routes for the synthesis of IPP: the classical MVA pathway with mevalonic acid as an intermediary product, and the MEP pathway, also known as deoxyxylulose 5-phosphate (DXP) pathway or non-MVAP (Eisenreich et al., 1996; Schwender et al., 1996). In general, green algae and most eubacteria use the MEP pathway (Lange et al., 2000), whereas mammals, yeast, archaea and some Gram-positive cocci synthesize IPP via the MVA pathway (Boucher and Doolittle, 2000; Wilding et al., 2000). Some organisms such as plants and a few bacterial species contain both pathways.
Staphylococci either use the one or the other pathway dependent on their preferred habitat. Staphylococcus species that use the MVA pathway are often associated with humans and primates, whereas the MEP pathway is predominantly found in species linked to companion animals, livestock, and wildlife (Misic et al., 2016). Recently, Staphylococcus sciuri strain ATCC 29059 was found to possess the complete sets of genes for both pathways (Christo-Foroux et al., 2017). Both pathways are also present in Listeria monocytogenes and some Streptomycetes (Begley et al., 2004; Heuston et al., 2012).
The MVA pathway, which is used by Staphylococcus aureus for IPP biosynthesis, starts with the acetylation of acetoacetyl-CoA to Hydroxy-3-methylglutaryl-CoA (HMG-CoA) catalyzed by the HMG-CoA synthase (MvaS) (Ferguson et al., 1959). HMG-CoA is then reduced by the HMG-CoA reductase (MvaA) to MVA (Durr and Rudney, 1960). This part is termed the upper MVA pathway. The lower part of the MVA pathway consists of the double phosphorylation of MVA to mevalonate-5-pyrophosphate by two kinases (MvaK1 and MvaK2) (Tchen, 1958; Tada and Lynen, 1961) and the final decarboxylation and dehydration to IPP and DMAPP by the mevalonate decarboxylase (MvaD) (Bloch et al., 1959). The reaction cascade is shown in Supplementary Figure S1A for better understanding.
Because of the importance of isoprenoids during different cellular processes the biosynthesis of IPP is essential for all living organisms.
In S. aureus, for example, repressing the expression of each of the MVA pathway genes drastically reduced growth, and a temperature-sensitive mvaA mutant was unable to grow at high temperatures (Balibar et al., 2009; Matsumoto et al., 2016).
In an earlier study we created a deletion mutant (ΔmvaS) in S. aureus and could show that this mutant was unable to grow in the absence of MVA in the medium, meaning that the mutant was auxotrophic for MVA (Yu et al., 2013). Surprisingly, after prolonged cultivation we obtained stable ΔmvaS variants that were able to grow without MVA, which suggests unknown mechanisms for compensating undecaprenol synthesis without MVA in S. aureus. This work demonstrated how flexible and adaptable bacteria are in order to survive under nutrient depletion.
Now, we investigated the adaptation process during the prolonged lag-phase in which the MVA auxotrophic ΔmvaS becomes prototrophic. We hypothesized that the adaptation is the result of mutations or gene amplifications, because long-term adaptation of bacteria to a certain environment is often based on such genetic variations (Wray, 2007; Andersson and Hughes, 2009). We could show that two sequential point mutations occurred in the regulator gene spx and the MVA lactonase drp35. Although the adapted mutant, ΔmvaSad, became prototrophic, its phenotypic characterization implies a weakened cell wall biosynthesis and increased osmotic stress susceptibility.
Materials and Methods
Bacterial Strains, Plasmids and Growth Conditions
All the bacterial strains used in this study are listed in Supplementary Table S1. S. aureus HG001 (Herbert et al., 2010) was used as parent strain. Escherichia coli DC10B (Monk et al., 2012) was used as a cloning host for the shuttle vectors pBASE6 (Geiger et al., 2012), pPTtuf (Popella et al., 2016) and pRAB11 (Helle et al., 2011). Their derivatives were constructed using Gibson assembly (Gibson et al., 2009). E. coli cells were grown at 37°C in basic medium [BM, 1% (w/v) casein peptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl, 0.1% (w/v) K2HPO4, 0.1% (w/v) glucose, pH 7.2], S. aureus cells were grown at 37°C in tryptic soy broth (TSB, Fluka) or BM. E. coli cultures were supplemented with ampicillin (100 μg/ml), when appropriate. In case of S. aureus chloramphenicol (10 μg/ml, pBASE6, pRAB11) or tetracycline (25 μg/ml, pPTtuf) was added.
Construction of Plasmids, Knock-Out and Knock-In Mutants
Oligonucleotides and plasmids are listed in Supplementary Tables S1, S2. The construction of the knock-in (drp35c-41t, spxT11I and drp35c-41t/spxT11I) and knockout mutants (Δspx, ΔmvaSadΔmvaA) in HG001 and ΔmvaS was performed using the plasmid pBASE6 and allelic replacement as described in (Bae and Schneewind, 2006). Briefly, around 1 kb of the upstream and downstream regions were amplified and ligated into the BglII-site of pBASE6. As template, chromosomal DNA of HG001 or ΔmvaSad was used. The knock-in plasmids were constructed by amplifying the mutated site together with roughly 1 kb of each flanking region and ligating it into the BglII-site of pBASE6. Over-expression plasmids were constructed by amplifying the desired gene together with a Shine-Dalgarno sequence (AGGAGGT) and ligating it into the BglII-site of pRAB11 or the EcoRI-site of pPTtuf. The resulting plasmids pBASE_drp35-KO, pBASE_spx-KO, pBASE_mvaA-KO, pBASE_drp35c-41t-KI, pBASE_spxT11I-KI, pPTtuf_drp35-strep and pRAB11_spxDD were transformed into the appropriate strains by electroporation.
Selection of Adapted ΔmvaS Mutants
Adapted mvaS mutants (ΔmvaSad) were selected as described before (Yu et al., 2013) with some modifications. Briefly, S. aureus ΔmvaS was incubated in TSB, supplemented with 500 μM (±)-MVL (Sigma–Aldrich), and grown overnight. Afterward, ΔmvaS was inoculated into fresh TSB without MVL to an OD578 of 0.005 to keep the influence of residual MVL as low as possible. This culture was incubated for several days at 37°C under aerobic conditions and the OD578 was monitored every 24 h. As soon as the culture reached the stationary growth phase a fresh culture was inoculated, and it was streaked on tryptic soy agar (TSA) plates to get single colonies. After several days of incubation one of the biggest colonies was taken, streaked again and used for all upcoming experiments.
Growth Studies
Growth experiments with the ΔmvaS knock-in mutants were done by incubating the cells in TSB, supplemented with 500 μM (±)-MVL (TSBMV A), overnight. Cultures were then diluted to an OD578 of 0.01 and further serially diluted 1:10. Finally, 10 μl of each dilution was dropped on a TSA plate and incubated at 37°C for 5 days.
Super-Resolution Fluorescence Microscopy
Cells were grown to the mid-exponential growth phase and washed with PBS. The pellets were then resuspended in PBS and incubated with BODIPYTM FL Vancomycin (0.25 μg/ml, Invitrogen) for cell wall staining and FM5-95 (7 μg/ml, Molecular Probes) for membrane staining for 10 min at 37°C. To remove unbound dye cells were washed twice in PBS and finally resuspended in PBS. For fluorescence microscopy, bacteria were mounted on microscope slides covered with a thin film of 2% agarose dissolved in PBS. Fluorescence micrographs were obtained using a Zeiss Axio Observer Z1 LSM 800 equipped with Airyscan detector and C Plan-Apo 63x/1.4 Oil DIC objective (Zeiss). Image acquisition and analysis were performed via ZEN 2.3 image analysis software package (Zeiss).
RNA-Isolation for Microarray
RNA for microarray was isolated from bacterial cultures of HG001 and its ΔmvaSad mutant during mid- and late exponential growth phase using the acid guanidinium thiocyanate-phenol-chloroform extraction method described in Schuster and Bertram (2014). DNA was removed by DNaseI digestion as described in Fischer et al. (2011). Pools of 5 μg total RNA for each condition were reverse-transcribed using SuperScript II (Invitrogen, Basel, Switzerland).
Microarray Manufacturing and Microarray Design
The microarray was manufactured by in situ synthesis of 60-base-long oligonucleotide probes (Agilent, Palo Alto, CA, United States), selected as previously described (Charbonnier et al., 2005). The microarray consists in a 15′600 glass slide covering > 95% of all open reading frames (ORFs) annotated in strains NCTC8325, UAMS-1 and SA564 as well as Newman, including their respective plasmids.
Preparation of Labeled Nucleic Acids for Expression Microarrays
Total RNA was purified from the strains HG001 and ΔmvaSad from two independent cultures. After additional DNase treatment, the absence of remaining DNA traces was confirmed by quantitative PCR with an assay specific for 16S rRNA (Sassi et al., 2015). Batches of 5 μg of total S. aureus RNA were labeled by Cy3-dCTP using SuperScript II (Invitrogen, Basel, Switzerland) following the manufacturer’s instructions. Labeled products were then purified onto QiaQuick columns (Qiagen). Purified genomic DNA from the different sequenced strains used for the design of the microarray was extracted (DNeasy; Qiagen), labeled with Cy5 dCTP using the Klenow fragment of DNA polymerase I (BioPrime, Invitrogen, Carlsbad, CA, United States), and used for the normalization process (Talaat et al., 2002). Cy5-labeled DNA (500 ng) and a Cy3-labeled cDNA mixture were diluted in 50 μl of Agilent hybridization buffer and hybridized at a temperature of 60°C for 17 h in a dedicated hybridization oven (Robbins Scientific, Sunnyvale, CA, United States). Slides were washed, dried under nitrogen flow, and scanned (Agilent, Palo Alto, CA, United States) using 100% photon multiplier tube power for both wavelengths.
Microarray Analysis
Fluorescence intensities were extracted using Feature Extraction software (version 9; Agilent). Local background-subtracted signals were corrected for unequal dye incorporation or unequal load of the labeled product. The algorithm consisted of a rank consistency filter and a curve fit using the default LOWESS (locally weighted linear regression) method. Data consisting of three independent biological experiments were expressed as log 10 ratios and analyzed using GeneSpring, version 8.0 (Silicon Genetics, Redwood City, CA, United States). A filter was applied to select oligonucleotides mapping ORFs in the HG001 genome, yielding approximately 95% coverage. Statistical significance of differentially expressed genes was calculated by analysis of variance (Pohl et al., 2009) using GeneSpring, including the Benjamini and Hochberg false discovery rate correction of 5% (p-value cutoff, 0.05) and an arbitrary cutoff of twofold for expression ratios.
Microarray Data Accession Number
The complete microarray data set has been posted on the Gene Expression Omnibus database1 under accession number GSEXXXX for the platform design and GPL10537 for the original data set.
Reverse Transcription-PCR
Reverse Transcription-PCR (RT-PCR) experiments were carried out using the OneTaq One-step RT-PCR kit (New England BioLabs, Frankfurt am Main, Germany). Fifty nanogram of RNA was used as template for each reaction. As positive control the housekeeping gene pykA was amplified and the OneTaq DNA Polymerase was taken for the no-RT negative control. Primers used for RT-PCR are listed in Supplementary Table S2. Gene expression was quantified by measuring the strength of the agarose gel bands after incubation in ethidium bromide using ImageJ software.
Purification and Analysis of Peptidoglycan
Peptidoglycan was isolated from wild type HG001 and the adapted mutant ΔmvaSad as described earlier (de Jonge et al., 1992). Briefly, cells grown up to mid-exponential growth phase were harvested by centrifugation, boiled with 5% SDS for 30 min and broken with glass beads. Broken cells were washed SDS free and resuspended in 100 mM Tris-HCl (pH 7.2) containing 20 mM MgCl2 and treated with 10 μg/ml DNAse and 50 μg/ml RNAse for 2 h and subsequently with 100 μg/ml trypsin, 37°C overnight. To remove wall teichoic acid, the PG preparations were incubated with 48% hydrofluoric acid (HFA) for 24 h at 4°C while stirred after washing with water. PG was harvested by centrifugation and washed several times with water until HFA was completely removed and lyophilized. Lyophilized PG was resuspended in 25 mM sodium phosphate buffer (pH 6.8) to a final OD578 of 5.0, digested with mutanolysin overnight at 37°C, reduced with sodium borohydride and analyzed by HPLC as described earlier (Nega et al., 2015).
Extraction, Analysis and Quantification of Bactoprenol
C55-isoprenoids were extracted with methanol/chloroform/PBS as described in (Barreteau et al., 2009). Cells were harvested during the exponential growth phase and KOH was used to convert undecaprenyl-pyrophosphate (C55-PP) to undecaprenyl-phosphate (C55-P) (Kato et al., 1999). The analysis of the C55-isoprenoids was performed by reverse-phase HPLC as described (Barreteau et al., 2009) with one major difference. A gradient from 95% buffer A (95% methanol, 5% 2-propanol, 10 mM phosphoric acid) to 100% buffer B (70% methanol, 30% 2-propanol, 10 mM phosphoric acid) in 50 min instead of an isocratic run was developed. The flow rate as well as the column temperature were kept constant at 0.6 ml/min and 30°C, respectively. Undecaprenyl-phosphate was detected at 210 nm. A calibration curve with different amounts of commercial Undecaprenyl-MPDA (monophosphate diammonium) (Larodan, Sweden), which were treated in the same way as the samples, was used to quantify undecaprenol. The amount was projected to nmol of undecaprenol per gram of cell mass dry weight. The KOH-treatment allowed the quantification of C55-PP and C55-P in one peak.
Filter Disk Diffusion Assay
To determine the sensitivity of bacterial strains to diamide, an inducer for disulfide stress, and H2O2 filter disk diffusion assays were performed. Overnight cultures were diluted to an OD578 of 0.1 and streaked onto TSA with a cotton swap. Filter disks were prepared by pipetting 20 μl of diamide (50 mM) or H2O2 (10 mM) onto the disks, which were then put on the agar plates. The diameter of the zone of inhibition (ZOI) were measured after the incubation at 37°C for 18 h using the software ImageJ.
Purification of Drp35
An overnight culture of S. aureus HG001, harboring the plasmid pPTtuf_drp35-strep, which constitutively expresses a C-terminal strep-tagged Drp35 protein, was diluted to an OD578 of 0.1 with fresh BM and incubated for at least 4 h at 37°C. The cells were harvested by centrifugation (7000 ×g, 10 min), washed with phosphate-buffered saline (PBS), and broken down by glass beads using a FastPrep FP120 instrument (MP Biomedicals). To remove cell debris the solution was centrifuged (4°C, 12,000 rpm, 10 min) and the supernatant was subjected to the purification procedure using Strep-Tactin Superflow resin (IBA, Goettingen, Germany) described by the manufacturer. During the whole procedure buffers lacking EDTA were used. After elution the purified protein was dialyzed against 20 mM Tris/HCl, pH 8.0 overnight at 4°C. Protein concentration was determined using the BCATM Protein Assay Kit (Thermo Scientific).
Drp35 Activity
Acid production by the hydrolysis of non-aromatic lactones was monitored by a colorimetric assay at 558 nm with correction at 475 nm at 37°C (Draganov et al., 2005). The assay was performed in a Tecan infinite® M200 Microplate Reader, each well contained 2 mM HEPES, pH 8.0, 1 mM CaCl2, 0.004% phenol red, 0.005% bovine serum albumin, 1 mM substrate and 5 – 10 μl purified protein. Spontaneous hydrolysis of the substrate was corrected for by substituting the enzyme by the same volume of 20 mM Tris/HCl, pH 8.0. A calibration curve with different amounts of HCl was used to calculate the rate of hydrolysis (Billecke et al., 1999).
The rate of hydrolysis for MVL was also tested for whole protein extracts of different strains. To do so, cells of the early exponential growth phase were harvested and washed twice with 20 mM Tris/HCl, pH 8.0. Afterward, cells were disrupted with glass beads, centrifuged and the supernatant was sterile filtered. Twenty microliter of each cell extract was used for the activity assay.
Galleria mellonella Infection Model
Larvae of Galleria mellonella were infected as described previously (Ebner et al., 2016) with few modifications. Bacterial cells of an overnight culture were washed twice with PBS (140 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4) and adjusted to a cell concentration of 5 × 108/ml in PBS. The injection volume was 10 μl, which is equal to 5 × 106 cells, per larvae. The larvae were infected with cells of S. aureus HG001 and HG001ΔmvaSad. In total 30 larvae, separated in three different experiments, were used for each strain. PBS served as a control. After injection, larvae were incubated at 37°C, and dead larvae were counted every 24 h.
Statistical Analysis
Multiple comparisons were analyzed by either ordinary one-way ANOVA or RM one-way ANOVA with Bonferroni post-test. The log-rank (Mantel-Cox) test was used to analyze the infection model. Statistical analyses were performed with GraphPad Prism software, with significance defined as p < 0.05. n represents independent biological replicates.
Results
Growth Behavior of the Mevalonate-Prototrophic S. aureusΔmvaS Mutant
Recently we showed that deletion of the mvaS gene, encoding the hydroxymethylglutaryl-coenzyme A (HMG-CoA) synthase, leads to auxotrophy for MVA; however, growth of the mutant could be restored by the addition of MVA or MVL to the medium (Figure 1A). Surprisingly, after prolonged cultivation for about 4–6 days, the ΔmvaS mutant started to grow but reached only an OD578 of about 2.5. When samples of this culture were plated on TSA small and large colonies were observed after several days (Figure 1B). After inoculation in fresh TSB, the cells were able to grow without a prolonged lag-phase; and when plated on TSA it formed uniformly large colonies (Figure 1C). We named these stable variants ΔmvaSad, for adapted ΔmvaS. Although, ΔmvaSad grew without lag-phase the growth was significantly retarded and they reached only of the OD578 of the parent strain (Figure 1A). Also, in contrast to the parent strain, colonies of ΔmvaSad were white suggesting that they were not able to produce the orange staphyloxanthin.
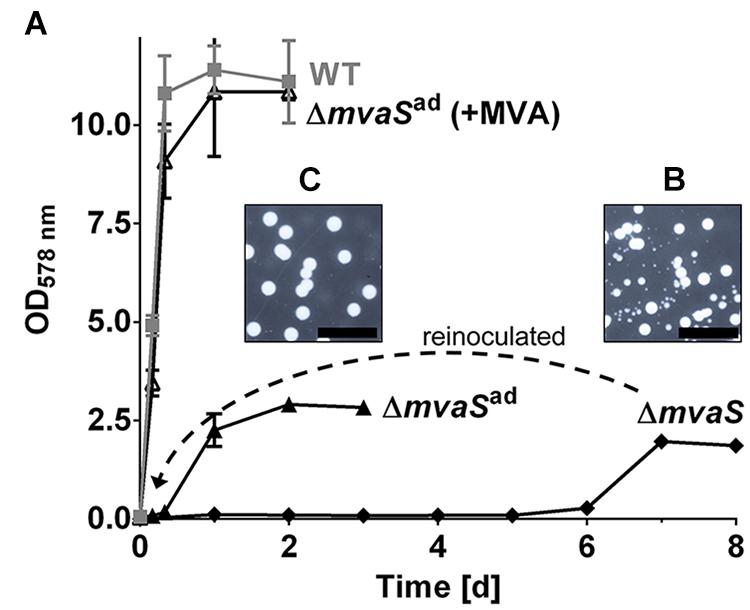
FIGURE 1. The mvaS deletion mutant starts to grow after a prolonged lag-phase of several days. (A) Growth curve of HG001, its isogenic mvaS deletion mutant and the adapted mvaS mutant (mvaSad) in the absence and presence of MVA. (B,C) Growth on agar plates of ΔmvaS (B) and ΔmvaSad (C) after their cultures reached the stationary phase, incubated for 7 days. For all graphs, except of ΔmvaS, each data point is the mean value ± SD; n = 3 for WT and ΔmvaSad, n = 2 for ΔmvaSad (+MVA). The graph of ΔmvaS represents one adaptation trial.
Whole Genome Analysis Revealed Two Important Point Mutations
We hypothesized that adaptation to MVA-prototrophy in ΔmvaSad is due to mutations, why we performed whole genome analysis.
The genomes of the parent strain HG001, the non-adapted ΔmvaS mutant and two independently adapted ΔmvaSad clones from different experiments were sequenced. Isolation of the chromosomal DNA and the sequencing of the genomes were performed by the DSMZ (Braunschweig, Germany) and analyzed for single nucleotide polymorphisms (SNPs). The long-read sequencing technique PacBio RS II with an average read length of 10 kb was used (Rhoads and Au, 2015). For error correction Illumina sequencing was performed. In both ΔmvaSad clones two interesting SNPs could be identified (Table 1). One SNP was located in the promoter region of drp35 (SAOUHSC_03023), which encodes a lactonase-like protein with yet unknown substrate specificity and function. The ΔmvaSad 1-clone carried a base exchange from cytosine to thymine 41 nucleotides upstream of the predicted transcription start site (TSS) (c-41t). The ΔmvaSad 2-clone carried adenine instead of guanine 52 nucleotides upstream of TSS (g-52a) (Figure 2A and Table 1).

TABLE 1. Mutations found by whole genome sequencing.
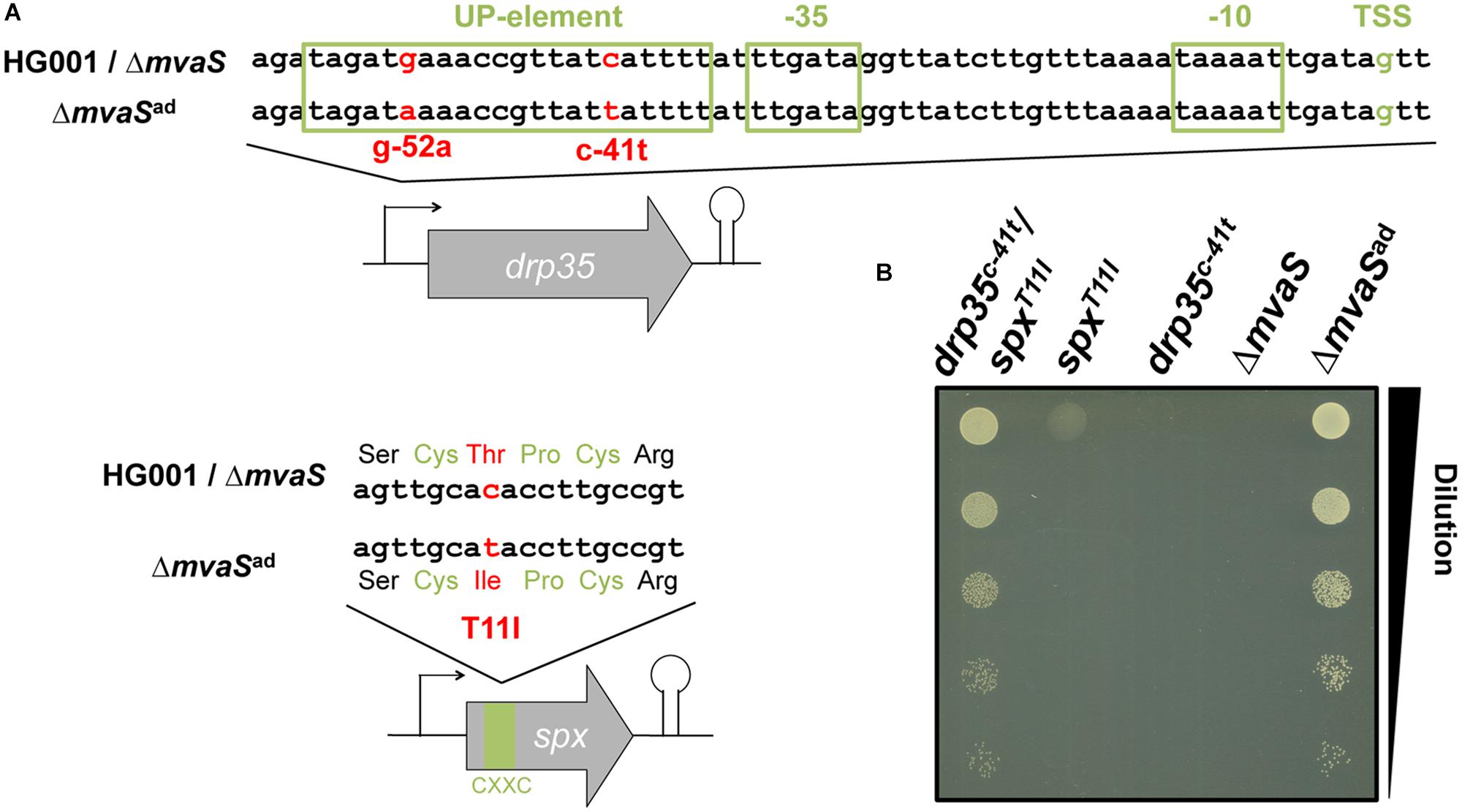
FIGURE 2. Two point mutations are involved in the adaptation of ΔmvaSad. (A) Location of the point mutations c-41t in the promoter of the lactonase gene drp35 and T11I in the C-X-X-C motif of the transcriptional regulator Spx. (B) Culture drop test on TS agar plates after back-cloning of drp35c-41t and spxT11I separately and together into the non-adapted mvaS mutant. Cultures were diluted to an OD578 of 0.1, serially diluted 1:10 and dropped on an agar plate, which was incubated aerobically at 37°C.
Both ΔmvaSad 1- and 2-clones carried a point mutation in the coding region of spx (SAOUHSC_00934), which encodes a transcriptional regulator that is involved in the global stress response (Pamp et al., 2006). The SNPs were located at nucleotide position 32 of the coding region where cytosine was exchanged by thymine (c32t). This mutation caused an amino acid exchange in the protein at position 11 from threonine to isoleucine (T11I). This exchange is located within the C-X-X-C motif (amino acids 10–13), which regulates the activity of the protein via the oxidation and reduction of the cysteine residues (Nakano et al., 2005). In ΔmvaSad 1, we additionally found a SNP (N352D) in PBP1 (penicillin binding protein 1) and in ΔmvaSad 2 a silent mutation in phoR. Since these two mutations are only present in one mutant they do not contribute to the adaptation and were therefore not considered in further experiments.
Both Mutations Are Necessary for the Adaptation of ΔmvaSad to MVA-Prototrophy
To confirm the importance of the two mutations in the adaptation of ΔmvaSad to MVA-prototrophy both mutated genes of ΔmvaSad 1, drp35c-41t and spxT11I, were introduced separately as well as combined into the non-adapted ΔmvaS mutant. The created strains were named ΔmvaS-drp35c-41t, ΔmvaS-spxT11I, and ΔmvaS-drp35c-41t/spxT11I. In a serial dilution (1:10) the clones were tested for growth on TSA in the absence of MVA (Figure 2B). The ΔmvaS clone, which carried both mutations (ΔmvaS-drp35c-41t/spxT11I), and ΔmvaSad grew similar. The ΔmvaS clone, that carries only spxT11I, showed only very little growth indicated by the faint spot at the highest cell concentration. The non-adapted ΔmvaS mutant as well as ΔmvaS-drp35c-41t were not able to grow. This result indicates that both mutations are necessary for the adaptation of ΔmvaSad to MVA prototrophy.
Additionally Isolated ΔmvaSad Clones Also Showed Mutations in spx and drp35
To investigate whether the development of the mutations in spx and drp35 is reproducible we repeated the isolation of adapted ΔmvaSad clones additional five times. In all experiments the adapted clones formed colonies with different size on TSA plates (Figure 1B). All tested large colonies revealed SNPs in both genes, spx and drp35. The identified SNPs were located 40 to 53 nucleotides upstream of the predicted TSS of drp35, and in the codons 11 or 14 of spx, which always led to an amino acid exchange (Table 2).

TABLE 2. Mutations in individually adapted mutants.
As shown in Figure 1B we also observed small colonies when the ΔmvaS mutants grew up after the long lag-phase. We wondered whether the small colonies were also mutated. Indeed, all the small colonies from five independent experiments carried a SNP in the spx gene but not in drp35 (Table 2). The results prove that the adaptation of ΔmvaS to ΔmvaSad occurs stepwise via two successive mutations, the first occurring in spx and the second in the drp35 gene.
Phenotypic Characterization
For phenotypic characterizations and to elucidate the effect of the mutations on cell metabolism, SNPs were also cloned into the wild type, resulting in the strains HG001-drp35c-41t, HG001-spxT11I and HG001-drp35c-41t/spxT11I. In addition, a spx deletion mutant (Δspx) was constructed.
The Growth Rate Was Delayed in ΔmvaSad
The doubling time of the single mutant, ΔmvaS-spxT11I was 29-times longer compared to the parent strain (Table 3). This shows that the ΔmvaS-spxT11I mutant is able to grow but that the growth is severely impaired which is also indicated by the tiny colonies (Figure 1B). The adapted clone ΔmvaSad grew much better, due to the additional mutation in drp35. The doubling time was “only” 8-times higher compared to the parent strain and colonies on TSA were much larger (Figures 1B,C). The single mutant ΔmvaS-drp35c-41t was unable to grow in the absence of MVA, (Figure 2B). This result indicates that the mutation in spx occurs first during the adaptation and that a mutation occurring first in drp35 cannot resuscitate growth.

TABLE 3. Growth rate of S. aureus strains.
In HG001 the mutation drp35c-41t did not influence growth. However, spxT11I slightly increased the generation time 1.1-fold in both HG001-spxT11I and HG001-drp35c-41t/spxT11I (Table 3). However, complete deletion of spx reduced growth 1.8-fold. The strain ΔmvaS-drp35c-41t/spxT11I showed a similar doubling time than ΔmvaSad. Further, both mutations had no influence on staphyloxanthin production. All knock-in strains in HG001 as well as Δspx showed the typical orange color, whereas ΔmvaSad and ΔmvaS-drp35c-41t/spxT11I strains were white (Supplementary Figure S2), suggesting that the missing staphyloxanthin is due to the disrupted MVA pathway and not a result of the mutations. This makes sense as staphyloxanthin biosynthesis affords isoprenoid precursors (Pelz et al., 2005).
Morphological Changes in ΔmvaSad
Already in bright field (BF) imaging it’s visible that the cells of the adapted ΔmvaSad clone were enlarged cocci and look swollen in contrast to the parent strain HG001 or ΔmvaSad grown in the presence of MVA (Figure 3A). Fluorescence staining with BODIPYTM FL Vancomycin revealed that less vancomycin was bound in the cross wall of ΔmvaSad and spots with increased dye accumulation were visible. This suggests that less PG was present and that cross wall is largely deficient of uncross-linked PG. Membrane staining with FM5-95 revealed a weaker staining and dot-like structures (Figure 3A). In the merged image, membrane- and PG-stained areas do not really overlap. Overall, the microscopic studies suggest that ΔmvaSad has a severe deficiency in PG synthesis and that it is osmotic fragile as indicated by the blown-up cells.
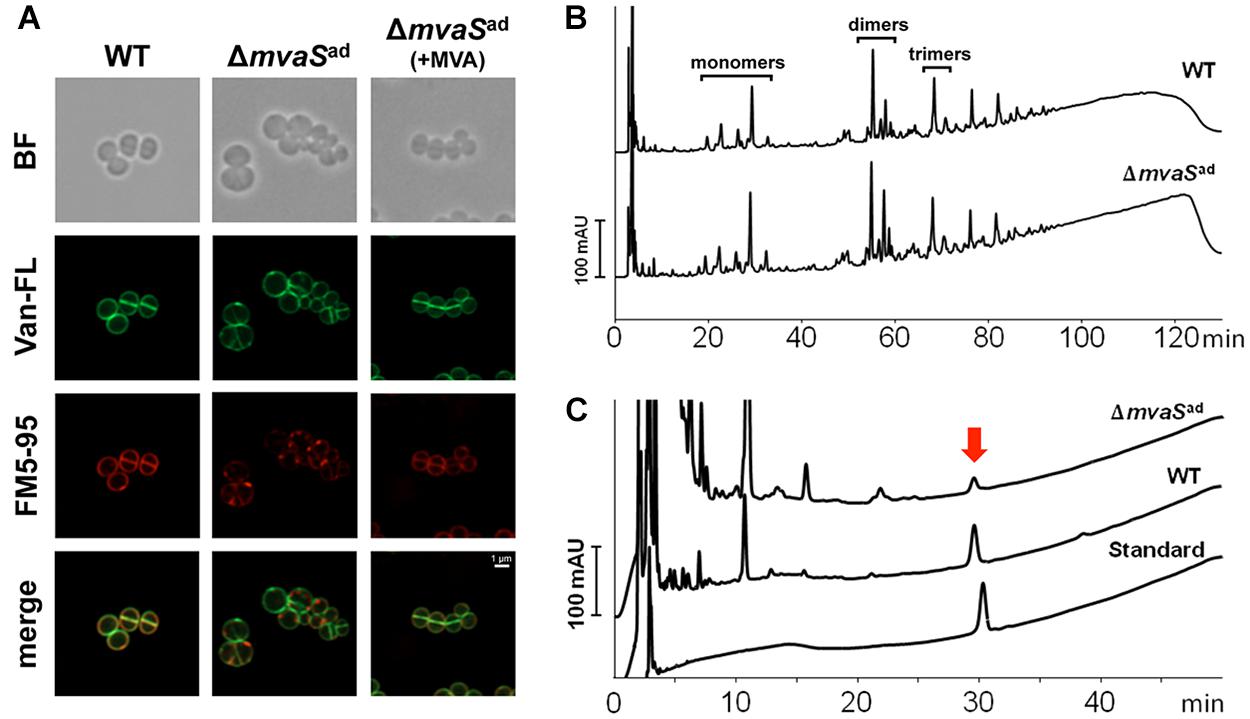
FIGURE 3. Comparison of the cell envelop of HG001 and ΔmvaSad. (A) Fluorescence microscopy of HG001 and ΔmvaSad. Cell membranes were stained with FM5-95 and PG was stained with Vancomycin, BODIPYTM FL Conjugate (Van-FL). BF, bright field. (B) PG pattern of HG001 and ΔmvaSad analyzed by RP-HPLC. v was isolated and digested with mutanolysin prior to analysis. (C) Amount of Undecaprenyl-phosphate (MW: 847.3) in HG001 and ΔmvaSad cells determined by HPLC analysis. The amount was calculated to 287 ± 39 and 16.1 ± 1.7 nmol per gram of cell mass dry weight for HG001 and ΔmvaSad, respectively. Each value is the mean ± SD of three individual experiments (n = 3). Undecaprenyl-monophosphate diammonium (MW: 881.4) was used as standard.
The two mutations seem not to be the reason for the irregular morphology and cell wall staining. The strains HG001-drp35c-41t, HG001-spxT11I and HG001-drp35c-41t/spxT11I showed normal cell size and cell wall staining. Only the strains ΔmvaSad and ΔmvaS-drp35c-41t/spxT11I showed the abnormal morphology (Supplementary Figure S3).
Peptidoglycan Structure Was Not Altered in ΔmvaSad but the Content of Bactoprenol Was Very Low
Because of the abnormal cell size and the irregular cell wall staining in ΔmvaSad we were interested whether the PG composition differed. PG was isolated from mid-exponential growth phase of ΔmvaSad and the parent strain HG001 and adjusted to the same OD578. The HPLC-profile of the mono-, di-, tri- and larger oligomers were not significantly altered in ΔmvaSad compared to the parent strain (Figure 3B).
As the primary structure of PG was apparently not altered we investigated whether undecaprenol, a follow product of MVA, could be formed by ΔmvaSad. Undecaprenol is part of Lipid II and is needed to translocate PG precursors across the membrane. To answer this question, we extracted lipids from whole cells and the chloroform extract was analyzed for the presence of undecaprenol by RP-HPLC. We analyzed the phosphorylated undecaprenyl-phosphate (C55-P) and undecaprenyl-diphosphate (C55-PP). Chemical dephosphorylation with KOH, which converts C55-PP into C55-P, allowed the analysis and quantification of both forms in one peak. Commercial undecaprenyl-MPDA was used as a standard. The untreated standard showed one single and well-defined peak with a retention time of ∼ 30 min (Figure 3C). The chloroform extract of the parent strain showed that most of the substances eluted during the first 5 min and that C55-P could be detected very well. The small shift of the peak of the standard can be explained by the slightly higher molecular mass (881 g/mol compared to 847 g/mol). Interestingly, the same peak could also be found in ΔmvaSad cells, but the amount was 18-fold less compared to the parent strain (Figure 3C). The amount of phosphorylated undecaprenol in the parent strain was 282 nmol/g cell dry weight, while for ΔmvaSad it was only 16.1 nmol/g cell dry weight (Table 4). This result shows that ΔmvaSad can synthesize undecaprenol, whose synthesis normally needs IPP as precursor. Cloning of drp35c-41t or spxT11I into HG001 resulted in an increase in undecaprenol to 461 and 426 nmol/g cell dry weight, respectively. The strain ΔmvaS-drp35c-41t/spxT11I produced similar undecaprenol amounts as ΔmvaSad.

TABLE 4. Amount of undecaprenol in different strains.
Both Mutations in spx and drp35 Enhance drp35 Expression
The mutation in the promoter of drp35 suggested an impact on gene expression. Also, we wondered whether the mutation in the transcriptional regulator gene spx influenced drp35-expression. In a semi-quantitative RT-PCR approach the expression of drp35 in the exponential growth phase was determined in several strains. Relative to the parent strain, drp35 was upregulated 1.7-fold in the knock-in mutant HG001-drp35c-41t, and 1.3-fold in HG001-spxT11I (Figure 4A). The combination of both mutations (drp35c-41t/spxT11I) further increased drp35 expression to 1.8-fold. The deletion of the spx gene resulted in a 2.4-fold higher drp35 expression compared to the parent strain, whereas the over-expression of a proteolysis resistant Spx variant (pRAB11_spxDD, last two amino acids are aspartate) (Nakano et al., 2005; Wang et al., 2010) reduced the expression of drp35 to 0.5-fold. In ΔmvaSad drp35 was 3.8-fold higher expressed (Figure 4A).
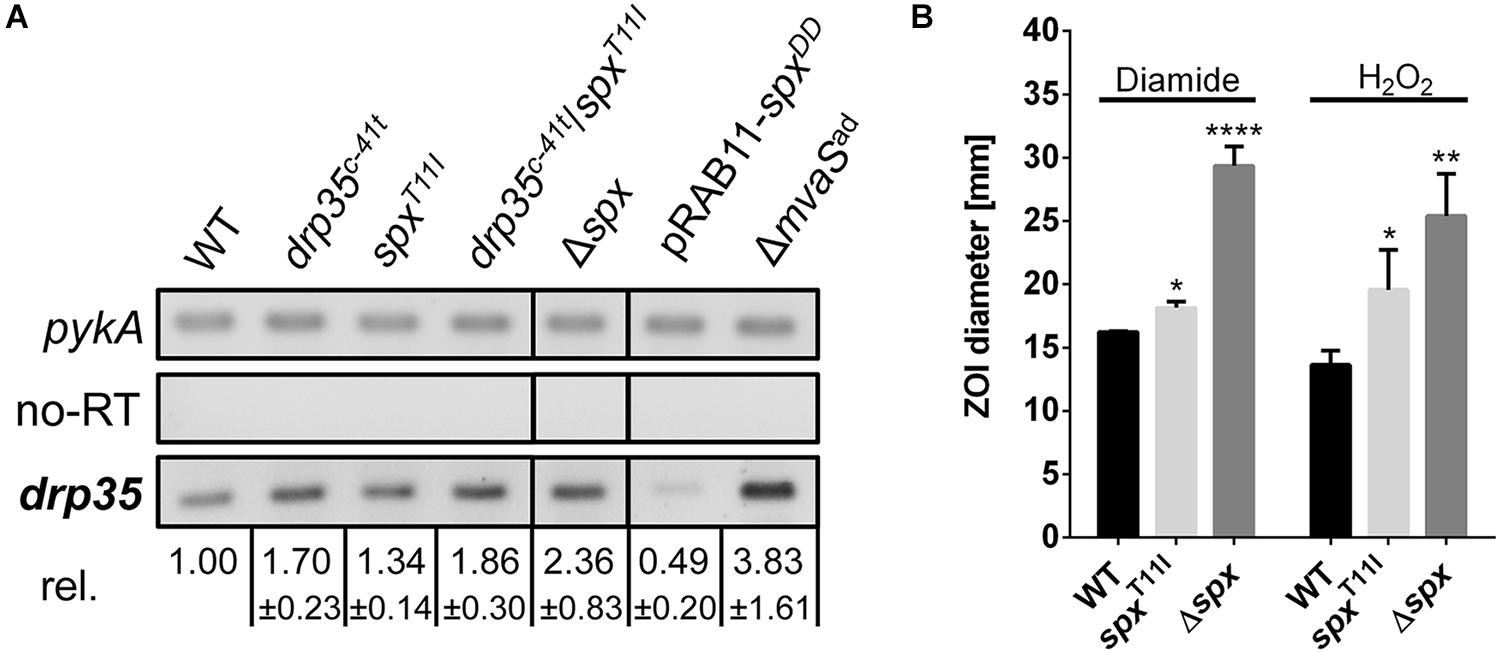
FIGURE 4. The point mutations spxT11I and drp35c-41t reduce the activity of Spx. (A) Expression level of drp35 in HG001, its isogenic spx deletion mutant, its knock-in mutants drp35c-41t, spxT11I and drp35c-41t/spxT11I, and during overexpression of a proteolysis resistant Spx variant (pRAB11_spxDD). Mean values ± SD are from two individual experiments. (B) Sensitivity of HG001, its isogenic spx mutant and HG001-spxT11I toward diamide and H2O2 determined by a filter disk diffusion assay. The ZOI values for diamide were 16.24 ± 0.05 mm (HG001), 18.16 ± 0.39 mm (HG001-spxT11I), and 29.36 ± 1.24 mm (HG001Δspx). For H2O2 the values were 13.64 ± 0.92 (HG001), 19.58 ± 2.58 mm (HG001-spxT11I), and 25.40 ± 2.72 mm (Δspx). Each value is the mean ± SD of three individual experiments (n = 3). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.001 and ordinary one-way ANOVA for (B).
Comparative Transcriptome Analysis by Microarray Approach
By transcriptome analysis we tried to find out which genes in ΔmvaSad were differently expressed compared to the parent strain HG001. For that RNA was isolated from cells in the mid (t1) and late-exponential growth phase (t2). In total 181 genes were more than 2.0-fold differentially expressed at least at one of the time points. Fifty four genes were up-regulated in the adapted ΔmvaSad, whereas 127 genes were down-regulated at t1 and/or t2 (Supplementary Tables S3, S4). Among the upregulated genes were the capsule biosynthesis genes and genes belonging to the cell wall stress stimulon (SAOUHSC_00560, vraX, SAOUHSC_00639, SAOUHSC_01173, SAOUHSC_02596, cwrA, and drp35) (Kuroda et al., 2003; Utaida et al., 2003). Among the down-regulated genes were those involved in nucleotide and amino acid metabolism, in regulation and particularly phage-related genes. It was remarkable that we didn’t find changes in the expression of the remaining genes of the MVAP or genes related to the synthesis of isoprenoids. The only exception was the upregulation of the glycosyl-4,4′-diaponeurosporenoate acyltransferase gene, which encodes the last enzyme in staphyloxanthin biosynthesis (Pelz et al., 2005). The lactonase-encoding drp35 gene was upregulated 3.2- (t1) and 2.4-fold (t2), respectively.
The Point Mutation in spx Decreased Spx Regulator Activity
Since Spx plays a key role during disulfide and oxidative stress (Nakano et al., 2003a), the sensitivity toward diamide and H2O2 was investigated. Filter disk diffusion assays with diamide and H2O2 were performed and the sensitivity was compared by measuring the diameter of the ZOI. Compared to the parent strain HG001, Δspx was, as expected, much more sensitive toward both substances. Also HG001-spxT11I was significantly more sensitive, but not as sensitive as the deletion mutant (Figure 4B). These results, together with the results from the growth analysis and RT-PCR approach, show that the T11I-mutation in Spx reduces the activity of the regulator, but does not abolish it completely; the results also show that spx is an important regulator gene as its deletion has a severe impact on growth.
Drp35 Is a Lactonase That Converts Mevalonolactone to Mevalonate
Drp35 is annotated as a lactonase and it has been shown that it can use the aromatic lactones dihydrocoumarine and 2-coumaranone as substrates (Morikawa et al., 2005). Using a colorimetric activity assay with purified strep-tagged Drp35 we found that MVL was rapidly hydrolyzed with a rate of 478 μmol/min/mg of protein. (Table 5 and Supplementary Figure S4). The conversion rates of other tested lactones were at least 100-fold lower compared to MVL (Table 5). For some lactones we didn’t see any activity. This result suggests MVL as the physiological substrate of the lactonase Drp35.
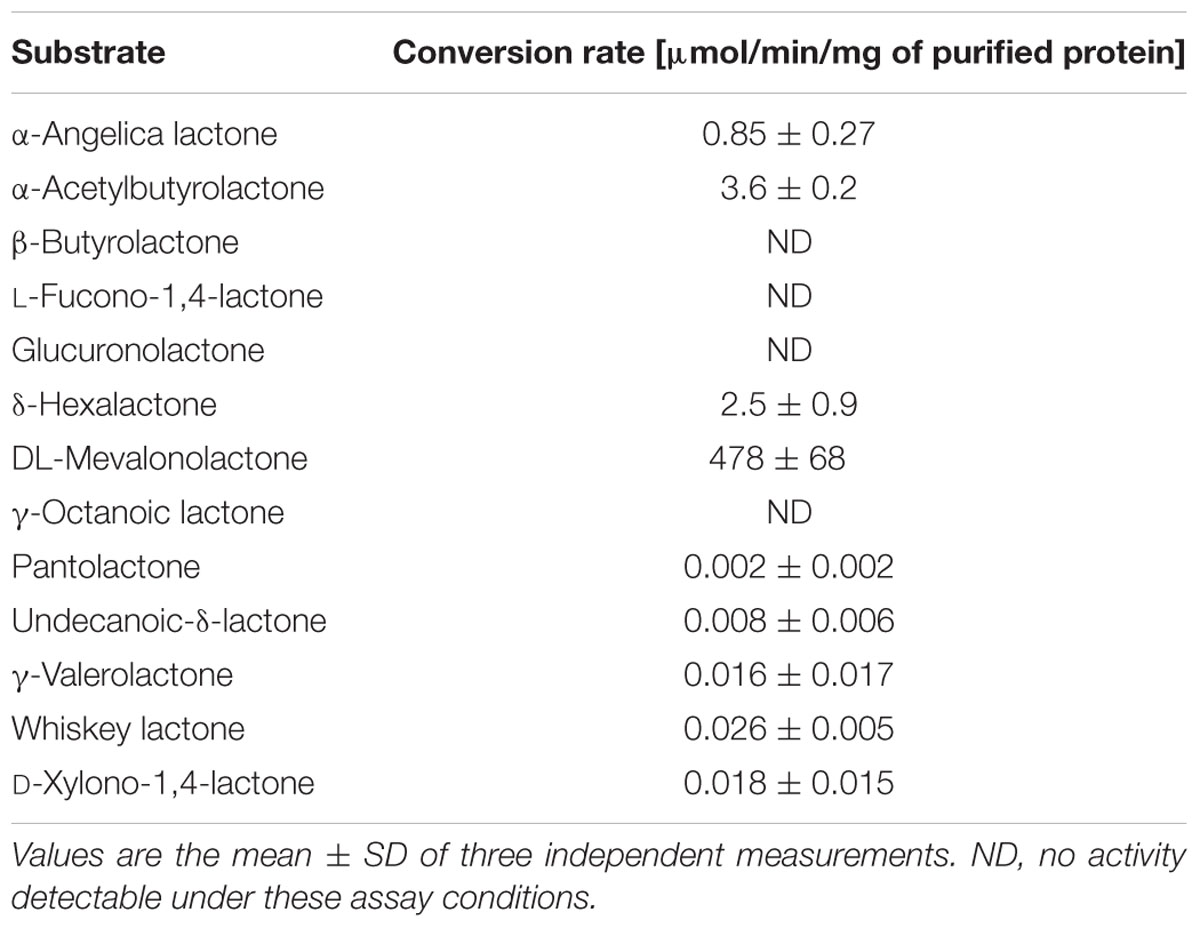
TABLE 5. Conversion rate of different lactones.
We also compared the mevalonolactonase activity in cell extracts of parent and mutant strains. Compared to the parent strain, HG001, the cell extract of strain HG001-drp35c-41t showed a 10-fold higher hydrolysis rate, and in the double mutant, HG001-drp35c-41t/spxT11I, the activity was even 14-fold higher compared to the parent strain (Table 6). The mevalonolactonase activity of cell extracts of strain HG001-spxT11I was slightly increased (1.6-fold) compared to HG001. Cell extracts of the adapted mutant, ΔmvaSad, and the complemented non-adapted mutant, ΔmvaS-drp35c-41t/spxT11I, had a 18- and 10-fold, respectively, higher activity than the parent strain HG001; indicating that the mutation in drp35c-41t contributes more to lactonase activity than the mutation in spxT11I.
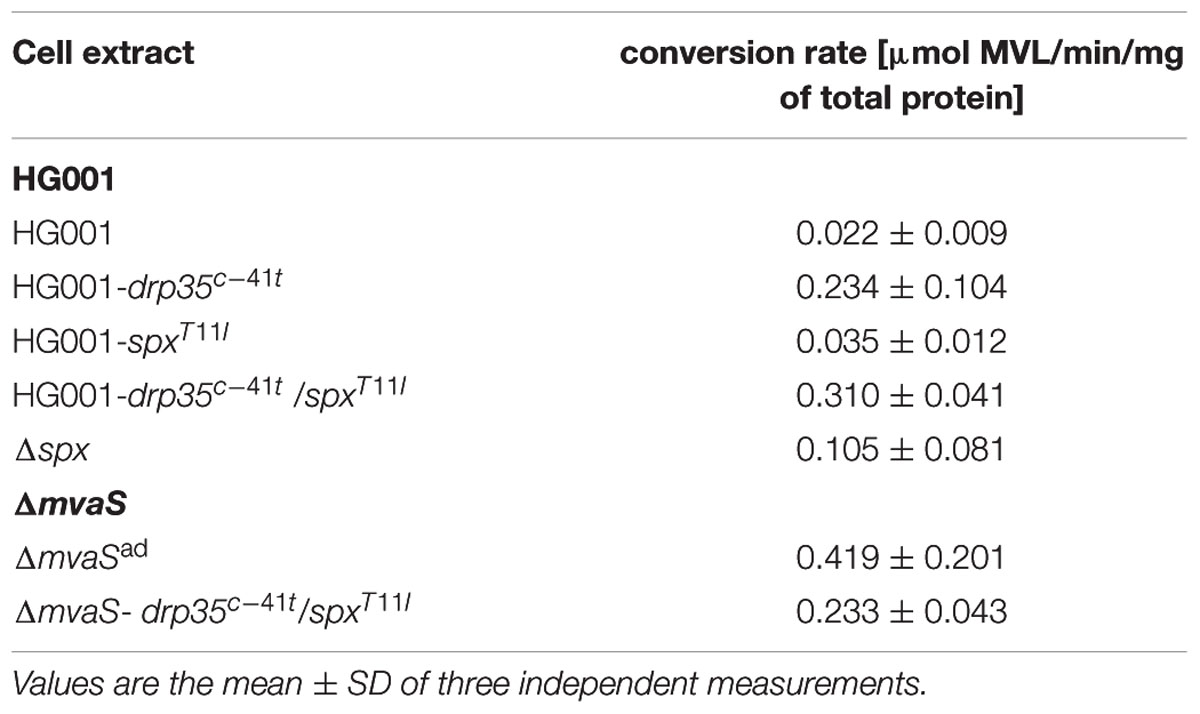
TABLE 6. Hydrolysis rate of MVL by cell extracts.
Growth of ΔmvaSad Is Inhibited by 6-Fluoromevalonate
To investigate whether the MVAP, downstream of MvaS, is still functional, we grew the ΔmvaSad in the presence of FMV. FMV is an inhibitor of the diphosphomevalonate decarboxylase MvaD, which is the last enzyme in the MVAP. In the presence of FMV ΔmvaSad was not able to grow, indicating that the lower MVAP is still needed in the mutant (Figure 5).
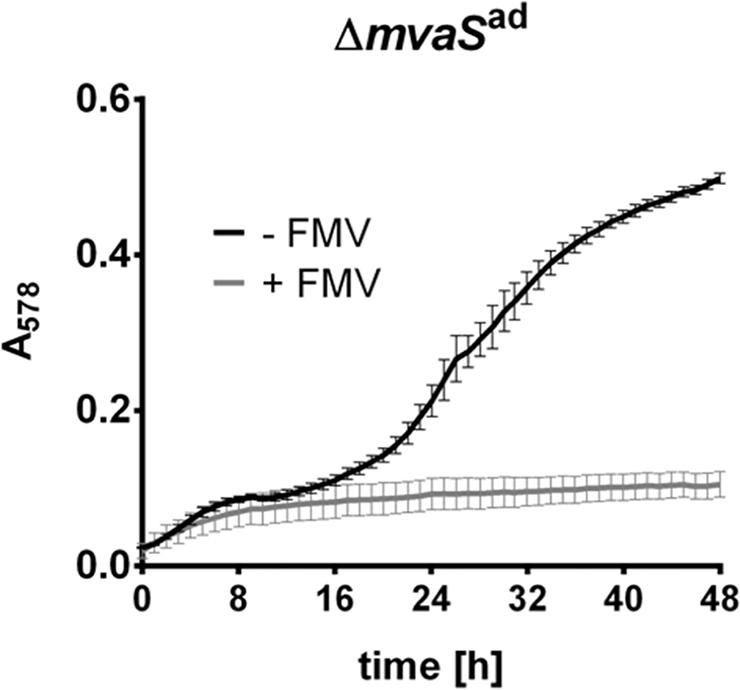
FIGURE 5. Growth of ΔmvaSad is inhibited by 6-Fluoromevalonate. Growth of ΔmvaSad in the presence or absence of 6-Fluoromevalonate (FMV, inhibitor of diphosphomevalonate decarboxylase MvaD). Cells were grown in the presence of 5% DMSO and 150 μg/ml FMV or only 5% DMSO (control). Growth was followed in a Tecan Infinite M200 microplate reader at 37°C with shaking intervals for 48 h. Absorbance at 578 nm was measured every hour. Each data point represents the mean value ± SD of three independent experiments (n = 3).
Virulence of ΔmvaSad Is Decreased
To test whether the adapted mutant is pathogenic the hemolytic activity as well as the ability to cause infections was tested. A drop test on sheep agar plates showed that ΔmvaSad still lysed erythrocytes (Figure 6A). However the appearance of the hemolytic zone was different. The parent strain HG001 showed only a clear halo, whereas ΔmvaSad showed in addition a turbid halo surrounding the clear halo. Wide clear zones are indicative for α-hemolysin and turbid halos for β-hemolysin activity (Adhikari and Novick, 2008). The strains HG001-drp35c-41t, HG001-drp35c-41t/spxT11I and ΔmvaS-drp35c-41t/spxT11I also produced the outer turbid zone, whereas HG001-spxT11I and Δspx did not. Unexpectedly, this result suggests that the expression of ß-hemolysin is somehow affected by the overexpression of drp35 and not by the reduced activity of SpxT11I.
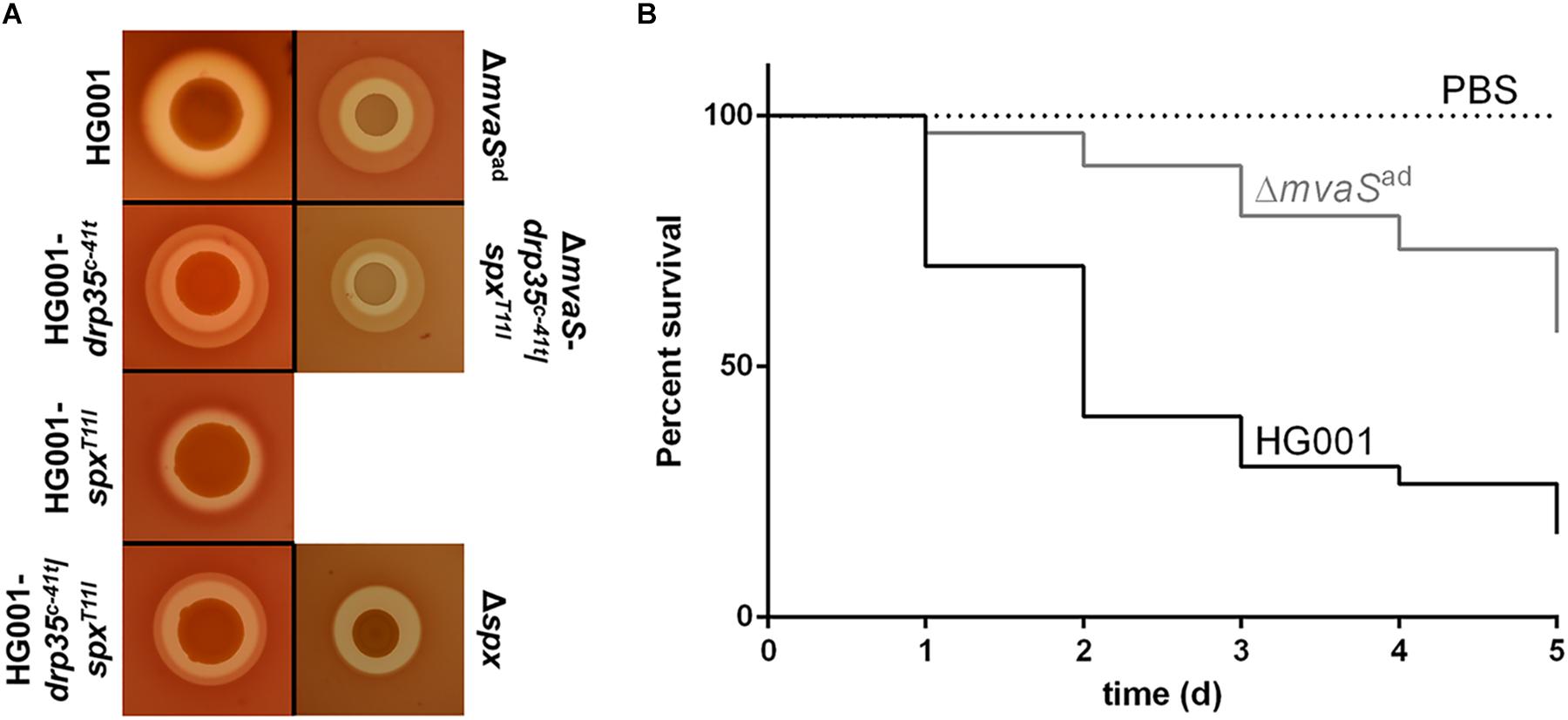
FIGURE 6. The adapted mutant shows differences in the hemolytic activity and reduced virulence. (A) Overnight cultures of HG001, HG001-drp35c-41t, HG001-spxT11I, HG001-drp35c-41t/spxT11I, Δspx, ΔmvaSad, and ΔmvaS--drp35c-41t/spxT11I, were diluted to OD578 1 and dropped on sheep blood agar plates. Plates were incubated at 37°C for 72 h. (B) Larvae of Galleria mellonella were infected with 5 × 106 cells of HG001 and ΔmvaSad. For each strain 30 larvae separated into three individual experiments were used. Larvae were incubated at 37°C for 5 days and survival was checked every 24 h. The virulence of ΔmvaSad is significantly reduced with p < 0.0001, by log-rank (Mantel-Cox) test.
The Galleria mellonella infection model showed that the virulence of ΔmvaSad is significantly reduced compared to the parent strain (Figure 6B). Larvae infected with the parent strain showed a survival rate of only 17% (5 out of 30 larvae), with more than 50% dead larvae within the first 2 days. The survival rate of larvae infected with the adapted mutant amounted to 57% (17 out of 30). In the first 2 days only 10% percent of the larvae died.
Discussion
The synthesis of isoprenoids is essential in all living organisms. In S. aureus the MVAP is the only route to synthesize the universal isoprenoid-precursor isopentenyl-pyrophosphate, which makes the MVA pathway a potential target for antimicrobial compounds. Recently, we created a ΔmvaS deletion mutant in S. aureus and, as expected, the mutant was unable to grow in medium lacking MVA (Yu et al., 2013). However, to our surprise, after prolonged cultivation for 4–6 days the mutant started to grow.
Here, we addressed the question what adaptation processes took place in the prolonged lag-phase that allowed the ΔmvaS mutant to grow. As the ΔmvaS mutant has adapted from auxotrophic to prototrophic phenotype we referred the adapted mutant as ΔmvaSad. Comparative genome sequencing of the parent strain HG001 and the adapted mutant ΔmvaSad revealed, that the mutant aquired two point mutations in different genes, namely spx and drp35.
Only Specific Mutations Lead to Mevalonate Prototrophy in ΔmvaSad
We wondered whether the mutational adaptation of ΔmvaS to MVA-prototrophy in ΔmvaSad could be repeated. Therefore, we carried out the adaptation procedure in total seven times. In all cases we found the mutations in a close range of spx and drp35 (Table 2). This indicates that only a few specific mutations lead to the adaptation of the mutant.
This resembles on the selection of antibiotic resistant mutants such as ciprofloxacin or rifampicin (RIF). Ciprofloxacin and other quinolones target the A subunit of DNA gyrase and selection of spontaneous single-step resistance mutants of E. coli and other bacteria were mostly gyrA mutations (Hooper et al., 1987). Resistance to RIF is nearly always due to a mutation in the β-subunit of bacterial RNA polymerase (RNAP) (Goldstein, 2014). The difference between these single-step resistance mutants involved in antibiotic resistance to the adaptation of ΔmvaSad to MVA-prototrophy is that in our case two mutations, instead of only one mutation, are necessary.
The Adaptation of ΔmvaS to Mevalonate Prototrophy Is Based on Two Sequential Mutations in Different Genes
The question was whether the mutations in spx and drp35 occurred in a special order or simultaneously. A simultaneous mutation is completely unlikely as the frequency would be extremely low (multiplication of the frequency of each single mutation). So the question remained which mutation appears first or if it is a random event. When ΔmvaS started to grow after the lag-phase of 4–6 days we observed always a mixture of very small and larger colonies on agar plates (Figure 1B). All the small colonies we investigated carried only one mutation, namely in spx. All the large colonies carried the double mutations in both spx and drp35 (Figures 1B,C and Table 2). We never found colonies with a mutation in drp35 only. This result indicates that the first mutation is in the regulator gene spx following the second mutation in drp35. The mutations in this order make sense, because, the spxT11I mutation allows already slight growth, which facilitates the development of the second mutation. Growth is necessary for the second mutation, as mutations occur mainly during DNA replication (Luria and Delbruck, 1943; Foster, 2004).
The Mutation spxT11I Reduced the Regulator Activity of Spx
Spx is described as a global transcriptional regulator in S. aureus, which is mainly active under stress conditions, but also contributes to cell fitness under non-stress conditions (Pamp et al., 2006). The Bacillus subtilis specific Spx does not bind directly to DNA, but influences gene transcription by interacting with the C-terminal domain of the α-subunit (αCTD) of the RNA Polymerase, a common target for transcriptional activators (Nakano et al., 2003b). The point mutation in spx is located in the coding region and caused an amino acid exchange from threonine to isoleucine at position 11 (T11I). The amino acid substitution is located in the so-called C-X-X-C motif, which mediates enzyme activity by the reduction of the cysteine residues. We assume that the change in this motif affects the general activity of Spx, in both positive and negative way. To see whether oxidative stress tolerance is affected, we introduced the mutation in the parent strain, HG001-spxT11I. Indeed, this mutation made HG001-spxT11I more sensitive against oxidative stress triggered by the thiol-reactive chemical diamide and by H2O2. Deletion of spx further increased the sensitivity, indicating, that the activity of SpxT11I is reduced but not completely abolished.
Spx is a negative regulator of drp35 expression, as verified by several observations: In HG001-spxT11Idrp35 was 1.7-fold up-regulated and 2.4-fold in Δspx. When spx was over-expressed in HG001 (pRAB11spxDD) drp35 expression was repressed. Furthermore, the value for mevalonolactonase activity of strain HG001-spxT11I was in between the values of Δspx and HG001. All our results suggest that the activity of SpxT11I is not abolished, but severely alters the regulator function. But why this reduced activity is beneficial for the mutant is still not completely clear.
Drp35 Is Overexpressed and Shows Mevalonolactone Lactonase Activity
The point mutation of drp35c-41t in ΔmvaSad was located at position -41, which is in the so-called UP (upstream)-element of the -35-region of the core-promoter. Comparative transcriptome analysis showed that drp35 is up-regulated two to three times in ΔmvaSad, indicating that this mutation increased drp35 expression, which could be confirmed by RT-PCR. UP-elements are in general AT-rich and stimulate gene transcription (Petho et al., 1986; Estrem et al., 1998). In drp35c-41t and the other adapted mutants, the AT-content of the UP-element is further increased and we assume that this is the reason of the transcriptional up-regulation of drp35c-41t. Activity tests showed that the hydrolysis of MVL to MVA can be carried out by Drp35 much more efficient than the hydrolysis of other substrates. This indicates that MVL might be the physiological substrate of the lactonase Drp35. The higher MVL hydrolyzing activity of cell extracts from the adapted mutant and strains harboring the mutations showed that the up-regulation of drp35 results in a higher protein amount in the cells.
Also, we showed that the adapted mutant ΔmvaSad can produce a small amount of undecaprenol, which is essential for cell wall biosynthesis. Both mutations seem to influence undecaprenol biosynthesis, as cloning of drp35c-41t or spxT11I into HG001 increased the undecaprenol level.
Only the Upper Part of the MVA Pathway Is Bypassed
In general, a decreased activity of the transcriptional regulator Spx and the overproduction of the lactonase Drp35 is needed to regain cell proliferation of ΔmvaSad. However, the growth of ΔmvaSad is slow and does not reach the level of growth when the medium contains MVA. The cell wall appears to be fragile and disordered and the blown-up cells indicate that the cells cannot withstand osmotic stress. Nevertheless, the mutations in drp35c-41t and spxT11I seem to allow a limited bypass of the upper MVA pathway to MVA. Bypassing essential metabolic pathways is not unusual in bacteria. For example, glutamate and proline auxotrophic mutants of B. subtilis can revert to prototrophy by suppressor mutations or amplification of specific genomic loci (Zaprasis et al., 2014; Dormeyer et al., 2017).
There are several reasons why we think that MVA is the bypass product. First, we investigated whether genes downstream of mvaS could be deleted in ΔmvaSad. We were able to delete mvaA without affecting the growth (Supplementary Figure S5) but not the mvaK1 gene, suggesting that enzymes downstream of MVA are essential and that there is not a completely different pathway, which was activated (see Supplementary Figure S1A). Second, ΔmvaSad is not able to grow in the presence of FMV (inhibitor of MvaD). Furthermore, when examining the location of drp35 in various staphylococcal genomes we found that in some species, like S. xylosus, drp35 is positioned directly next to mvaS and mvaA. In S. sciuri strain FDAARGOS_285 it is located directly upstream and in the same orientation of the mvaK1/D/K2 operon indicating an involvement of Drp35 in the MVA pathway (Supplementary Figure S1B and Supplementary Table S5). Only in S. aureus and some other species it is dislocated. Finally, it is remarkable that Drp35 shows the highest lactonase activity with MVL.
The upper MVA pathway, in particular MvaA, is considered as a potential target for the development of new antibiotics (Wilding et al., 2000). Therefore, we were interested if the adapted mutant, who seems to have a bypass for the upper MVA pathway, is still pathogenic. If so, the theory of targeting MvaA would be questionable, because challenging S. aureus with such an antibiotic would probably lead to the emergence of resistant and virulent mutants. However, in a Galleria mellonella infection model it turned out that the virulence of the adapted mutant is greatly reduced, which might be due to the missing staphyloxanthin and the more fragile cell wall. Additionally, the growth on sheep blood agar showed that drp35c-41t influenced hemolysin production.
Altogether, we showed here that a MVA auxotrophic mvaS mutant can revert to prototrophy by acquiring two point mutations in the genes spx and drp35. The mutations led to a decreased regulator activity of Spx and to an increased expression of the lactonase Drp35. Based on these mutations the adapted mutant, ΔmvaSad, seems to be able to synthesize sufficient MVA to produce small amounts of the lipid carrier undecaprenol. However, the amount of produced MVA is not high enough to allow a fast and proper cell wall synthesis or to synthesize staphyloxanthin. The adapted ΔmvaSad mutant appears to have a rescue pathway that allows cell proliferation in the absence of the upper MVA pathway. We could not uncover this bypass completely. Nevertheless this study illustrates once again how adaptive the bacterial genome is. Like a lifeless creature a subpopulation is able to rise again by genetic adaptation. Interestingly, the adaptation includes always the two point mutations in spx and drp35 and always in the order spx first and drp35 second, a classical case of adaptive mutation.
Author Contributions
FG and SR designed the study. SR, PE, AL, MN, JS, JO, PS, PF, and FG designed the experiments. SR performed most of the experiments. E-JB and PF performed transcriptome analysis. MN performed PG analysis. BB and CS performed whole genome sequencing and analysis and PS performed fluorescence microscopy. PE contributed to proofreading. FG and SR wrote the manuscript.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG: GRK1708: “Molecular principles of bacterial survival strategies”, SFB/Transregio 34, SFB 766); and DZIF, Deutsches Zentrum für Infektionsforschung.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FC and handling Editor declared their shared affiliation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01539/full#supplementary-material
Abbreviations
DMAPP, dimethylallyl pyrophosphate; FMV, 6-Fluoromevalonate; IPP, isopentenyl pyrophosphate; MEP, 2C-methyl-D-erythritol 4-phosphate pathway; MVA, mevalonate; MVL, mevalonolactone; MVAP, mevalonate pathway; PG, peptidoglycan.
Footnotes
References
Adhikari, R. P., and Novick, R. P. (2008). Regulatory organization of the staphylococcal sae locus. Microbiology 154(Pt 3), 949–959. doi: 10.1099/mic.0.2007/012245-0
Andersson, D. I., and Hughes, D. (2009). Gene amplification and adaptive evolution in bacteria. Annu. Rev. Genet. 43, 167–195. doi: 10.1146/annurev-genet-102108-134805
Bae, T., and Schneewind, O. (2006). Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63. doi: 10.1016/j.plasmid.2005.05.005
Balibar, C. J., Shen, X., and Tao, J. (2009). The mevalonate pathway of Staphylococcus aureus. J. Bacteriol. 191, 851–861. doi: 10.1128/JB.01357-08
Barreteau, H., Magnet, S., El Ghachi, M., Touze, T., Arthur, M., Mengin-Lecreulx, D., et al. (2009). Quantitative high-performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 213–220. doi: 10.1016/j.jchromb.2008.12.010
Begley, M., Gahan, C. G., Kollas, A. K., Hintz, M., Hill, C., Jomaa, H., et al. (2004). The interplay between classical and alternative isoprenoid biosynthesis controls gammadelta T cell bioactivity of Listeria monocytogenes. FEBS Lett. 561, 99–104. doi: 10.1016/S0014-5793(04)00131-0
Billecke, S. S., Primo-Parmo, S. L., Dunlop, C. S., Doorn, J. A., La Du, B. N., and Broomfield, C. A. (1999). Characterization of a soluble mouse liver enzyme capable of hydrolyzing diisopropyl phosphorofluoridate. Chem. Biol. Interact. 119-120, 251–256. doi: 10.1016/S0009-2797(99)00034-4
Bloch, K., Chaykin, S., Phillips, A. H., and De Waard, A. (1959). Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J. Biol. Chem. 234, 2595–2604.
Boucher, Y., and Doolittle, W. F. (2000). The role of lateral gene transfer in the evolution of isoprenoid biosynthesis pathways. Mol. Microbiol. 37, 703–716. doi: 10.1046/j.1365-2958.2000.02004.x
Charbonnier, Y., Gettler, B., Francois, P., Bento, M., Renzoni, A., Vaudaux, P., et al. (2005). A generic approach for the design of whole-genome oligoarrays, validated for genomotyping, deletion mapping and gene expression analysis on Staphylococcus aureus. BMC Genomics 6:95. doi: 10.1186/1471-2164-6-95
Christo-Foroux, E., Vallaeys, T., Loux, V., Dassa, E., Deutscher, J., Wandersman, C., et al. (2017). Manual and expert annotation of the nearly complete genome sequence of Staphylococcus sciuri strain ATCC 29059: a reference for the oxidase-positive staphylococci that supports the atypical phenotypic features of the species group. Syst. Appl. Microbiol. 40, 401–410. doi: 10.1016/j.syapm.2017.07.002
de Jonge, B. L., Chang, Y. S., Gage, D., and Tomasz, A. (1992). Peptidoglycan composition in heterogeneous Tn551 mutants of a methicillin-resistant Staphylococcus aureus strain. J. Biol. Chem. 267, 11255–11259.
Dormeyer, M., Lubke, A. L., Muller, P., Lentes, S., Reuss, D. R., Thurmer, A., et al. (2017). Hierarchical mutational events compensate for glutamate auxotrophy of a Bacillus subtilis gltC mutant. Environ. Microbiol. Rep. 9, 279–289. doi: 10.1111/1758-2229.12531
Draganov, D. I., Teiber, J. F., Speelman, A., Osawa, Y., Sunahara, R., and La Du, B. N. (2005). Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 46, 1239–1247. doi: 10.1194/jlr.M400511-JLR200
Durr, I. F., and Rudney, H. (1960). The reduction of beta-hydroxy-beta-methyl-glutaryl coenzyme A to mevalonic acid. J. Biol. Chem. 235, 2572–2578.
Ebner, P., Rinker, J., Nguyen, M. T., Popella, P., Nega, M., Luqman, A., et al. (2016). Excreted cytoplasmic proteins contribute to pathogenicity in Staphylococcus aureus. Infect. Immun. 84, 1672–1681. doi: 10.1128/IAI.00138-16
Eisenreich, W., Menhard, B., Hylands, P. J., Zenk, M. H., and Bacher, A. (1996). Studies on the biosynthesis of taxol: the taxane carbon skeleton is not of mevalonoid origin. Proc. Natl. Acad. Sci. U.S.A. 93, 6431–6436. doi: 10.1073/pnas.93.13.6431
Estrem, S. T., Gaal, T., Ross, W., and Gourse, R. L. (1998). Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. U.S.A. 95, 9761–9766. doi: 10.1073/pnas.95.17.9761
Ferguson, J. J., Durr, I. F., and Rudney, H. (1959). The biosynthesis of mevalonic acid. Proc. Natl. Acad. Sci. U.S.A. 45, 499–504. doi: 10.1073/pnas.45.4.499
Fischer, A., Yang, S. J., Bayer, A. S., Vaezzadeh, A. R., Herzig, S., Stenz, L., et al. (2011). Daptomycin resistance mechanisms in clinically derived Staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J. Antimicrob. Chemother. 66, 1696–1711. doi: 10.1093/jac/dkr195
Foster, P. L. (2004). Adaptive mutation in Escherichia coli. J. Bacteriol. 186, 4846–4852. doi: 10.1128/JB.186.15.4846-4852.2004
Geiger, T., Francois, P., Liebeke, M., Fraunholz, M., Goerke, C., Krismer, B., et al. (2012). The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 8:e1003016. doi: 10.1371/journal.ppat.1003016
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. III, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Goldstein, B. P. (2014). Resistance to rifampicin: a review. J. Antibiot. (Tokyo) 67, 625–630. doi: 10.1038/ja.2014.107
Helle, L., Kull, M., Mayer, S., Marincola, G., Zelder, M. E., Goerke, C., et al. (2011). Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology 157(Pt 12), 3314–3323. doi: 10.1099/mic.0.052548-0
Herbert, S., Ziebandt, A. K., Ohlsen, K., Schafer, T., Hecker, M., Albrecht, D., et al. (2010). Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78, 2877–2889. doi: 10.1128/IAI.00088-10
Heuston, S., Begley, M., Gahan, C. G., and Hill, C. (2012). Isoprenoid biosynthesis in bacterial pathogens. Microbiology 158(Pt 6), 1389–1401. doi: 10.1099/mic.0.051599-0
Holstein, S. A., and Hohl, R. J. (2004). Isoprenoids: remarkable diversity of form and function. Lipids 39, 293–309. doi: 10.1007/s11745-004-1233-3
Hooper, D. C., Wolfson, J. S., Ng, E. Y., and Swartz, M. N. (1987). Mechanisms of action of and resistance to ciprofloxacin. Am. J. Med. 82, 12–20.
Kato, J., Fujisaki, S., Nakajima, K., Nishimura, Y., Sato, M., and Nakano, A. (1999). The Escherichia coli homologue of yeast RER2, a key enzyme of dolichol synthesis, is essential for carrier lipid formation in bacterial cell wall synthesis. J. Bacteriol. 181, 2733–2738.
Kuroda, M., Kuroda, H., Oshima, T., Takeuchi, F., Mori, H., and Hiramatsu, K. (2003). Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49, 807–821. doi: 10.1046/j.1365-2958.2003.03599.x
Lange, B. M., Rujan, T., Martin, W., and Croteau, R. (2000). Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc. Natl. Acad. Sci. U.S.A. 97, 13172–13177. doi: 10.1073/pnas.240454797
Luria, S. E., and Delbruck, M. (1943). Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511.
Matsumoto, Y., Yasukawa, J., Ishii, M., Hayashi, Y., Miyazaki, S., and Sekimizu, K. (2016). A critical role of mevalonate for peptidoglycan synthesis in Staphylococcus aureus. Sci. Rep. 6:22894. doi: 10.1038/srep22894
Misic, A. M., Cain, C. L., Morris, D. O., Rankin, S. C., and Beiting, D. P. (2016). Divergent isoprenoid biosynthesis pathways in Staphylococcus species constitute a drug target for treating infections in companion animals. mSphere 1:e00258-16. doi: 10.1128/mSphere.00258-16
Monk, I. R., Shah, I. M., Xu, M., Tan, M. W., and Foster, T. J. (2012). Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 3:e00277-11. doi: 10.1128/mBio.00277-11
Morikawa, K., Hidaka, T., Murakami, H., Hayashi, H., and Ohta, T. (2005). Staphylococcal Drp35 is the functional counterpart of the eukaryotic PONs. FEMS Microbiol. Lett. 249, 185–190. doi: 10.1016/j.femsle.2005.06.038
Nakano, S., Erwin, K. N., Ralle, M., and Zuber, P. (2005). Redox-sensitive transcriptional control by a thiol/disulphide switch in the global regulator. Spx. Mol. Microbiol. 55, 498–510. doi: 10.1111/j.1365-2958.2004.04395.x
Nakano, S., Kuster-Schock, E., Grossman, A. D., and Zuber, P. (2003a). Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 100, 13603–13608. doi: 10.1073/pnas.2235180100
Nakano, S., Nakano, M. M., Zhang, Y., Leelakriangsak, M., and Zuber, P. (2003b). A regulatory protein that interferes with activator-stimulated transcription in bacteria. Proc. Natl. Acad. Sci. U.S.A. 100, 4233–4238. doi: 10.1073/pnas.0637648100
Nega, M., Dube, L., Kull, M., Ziebandt, A. K., Ebner, P., Albrecht, D., et al. (2015). Secretome analysis revealed adaptive and non-adaptive responses of the Staphylococcus carnosus femB mutant. Proteomics 15, 1268–1279. doi: 10.1002/pmic.201400343
Pamp, S. J., Frees, D., Engelmann, S., Hecker, M., and Ingmer, H. (2006). Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. J. Bacteriol. 188, 4861–4870. doi: 10.1128/JB.00194-06
Pelz, A., Wieland, K. P., Putzbach, K., Hentschel, P., Albert, K., and Götz, F. (2005). Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem. 280, 32493–32498. doi: 10.1074/jbc.M505070200
Petho, A., Belter, J., Boros, I., and Venetianer, P. (1986). The role of upstream sequences in determining the strength of an rRNA promoter of E.coli. Biochim. Biophys. Acta 866, 37–43. doi: 10.1016/0167-4781(86)90098-9
Pohl, K., Francois, P., Stenz, L., Schlink, F., Geiger, T., Herbert, S., et al. (2009). CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J. Bacteriol. 191, 2953–2963. doi: 10.1128/JB.01492-08
Popella, P., Krauss, S., Ebner, P., Nega, M., Deibert, J., and Götz, F. (2016). VraH is the third component of the Staphylococcus aureus VraDEH system involved in gallidermin and daptomycin resistance and pathogenicity. Antimicrob. Agents Chemother. 60, 2391–2401. doi: 10.1128/AAC.02865-15
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Sassi, M., Sharma, D., Brinsmade, S. R., Felden, B., and Augagneur, Y. (2015). Genome sequence of the clinical isolate Staphylococcus aureus subsp. aureus Strain UAMS-1. Genome Announc. 3:e01584-14. doi: 10.1128/genomeA.01584-14
Schuster, C. F., and Bertram, R. (2014). Fluorescence based primer extension technique to determine transcriptional starting points and cleavage sites of RNases in vivo. J. Vis. Exp. 92:e52134. doi: 10.3791/52134
Schwender, J., Seemann, M., Lichtenthaler, H. K., and Rohmer, M. (1996). Biosynthesis of isoprenoids (carotenoids, sterols, prenyl side-chains of chlorophylls and plastoquinone) via a novel pyruvate/glyceraldehyde 3-phosphate non-mevalonate pathway in the green alga Scenedesmus obliquus. Biochem. J. 316(Pt 1), 73–80. doi: 10.1042/bj3160073
Tada, M., and Lynen, F. (1961). [On the biosynthesis of terpenes. XIV. On the determination of phosphomevalonic acid kinase and pyrophosphomevalonic acid decarboxylase in cell extracts]. J. Biochem. 49, 758–764. doi: 10.1093/oxfordjournals.jbchem.a127368
Talaat, A. M., Howard, S. T., Hale, W., Lyons, R., Garner, H., and Johnston, S. A. (2002). Genomic DNA standards for gene expression profiling in Mycobacterium tuberculosis. Nucleic Acids Res. 30:e104. doi: 10.1093/nar/gnf103
Utaida, S., Dunman, P. M., Macapagal, D., Murphy, E., Projan, S. J., Singh, V. K., et al. (2003). Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149(Pt 10), 2719–2732. doi: 10.1099/mic.0.26426-0
Wang, C., Fan, J., Niu, C., Wang, C., Villaruz, A. E., Otto, M., et al. (2010). Role of spx in biofilm formation of Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 59, 152–160. doi: 10.1111/j.1574-695X.2010.00673.x
Wilding, E. I., Brown, J. R., Bryant, A. P., Chalker, A. F., Holmes, D. J., Ingraham, K. A., et al. (2000). Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J. Bacteriol. 182, 4319–4327. doi: 10.1128/JB.182.15.4319-4327.2000
Wray, G. A. (2007). The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216. doi: 10.1038/nrg2063
Yu, W., Leibig, M., Schafer, T., Bertram, R., Ohlsen, K., and Götz, F. (2013). The mevalonate auxotrophic mutant of Staphylococcus aureus can adapt to mevalonate depletion. Antimicrob. Agents Chemother. 57, 5710–5713. doi: 10.1128/AAC.00726-13
Zaprasis, A., Hoffmann, T., Wunsche, G., Florez, L. A., Stulke, J., and Bremer, E. (2014). Mutational activation of the RocR activator and of a cryptic rocDEF promoter bypass loss of the initial steps of proline biosynthesis in Bacillus subtilis. Environ. Microbiol. 16, 701–717. doi: 10.1111/1462-2920.12193
Keywords: adaptation, mutation, mevalonate pathway, isoprenoids, lactonase, Drp35, Spx
Citation: Reichert S, Ebner P, Bonetti E-J, Luqman A, Nega M, Schrenzel J, Spröer C, Bunk B, Overmann J, Sass P, François P and Götz F (2018) Genetic Adaptation of a Mevalonate Pathway Deficient Mutant in Staphylococcus aureus. Front. Microbiol. 9:1539. doi: 10.3389/fmicb.2018.01539
Received: 24 April 2018; Accepted: 20 June 2018;
Published: 12 July 2018.
Edited by:
Jörg Stülke, Georg-August-Universität Göttingen, GermanyReviewed by:
Fabian Moritz Commichau, Georg-August-Universität Göttingen, GermanyVolker F. Wendisch, Bielefeld University, Germany
Copyright © 2018 Reichert, Ebner, Bonetti, Luqman, Nega, Schrenzel, Spröer, Bunk, Overmann, Sass, Francçois and Götz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedrich Götz, ZnJpZWRyaWNoLmdvZXR6QHVuaS10dWViaW5nZW4uZGU=