 Huihui Fu
Huihui Fu Peng Jiang1,2,3*
Peng Jiang1,2,3* Chunhui Wu
Chunhui Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 02 July 2018
Sec. Aquatic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01458
Algae-bacteria associations occurred widely in marine habitats, however, contributions of bacteria to macroalgal blooming were almost unknown. In this study, a potential endophytic strain SI-3 was isolated from Ulva prolifera, the causative species for the world's largest green tide in the Yellow Sea, following a strict bleaching treatment to eliminate epiphytes. The genomic sequence of SI-3 was determined in size of 4.8 Mb and SI-3 was found to be mostly closed to Pseudomonas stutzeri. To evaluate the characteristics of SI-3 as a potential endophyte, the genomes of SI-3 and other 20 P. stutzeri strains were compared. We found that SI-3 had more strain-specific genes than most of the 20 P. stutzeri strains. Clusters of Orthologous Groups (COGs) analysis revealed that SI-3 had a higher proportion of genes assigned to transcriptional regulation and signal transduction compared with the 20 P. stutzeri strains, including four rhizosphere bacteria, indicating a complicated interaction network between SI-3 and its host. P. stutzeri is renowned for its metabolic versatility in aromatic compounds degradation. However, significant gene loss was observed in several aromatic compounds degradation pathways in SI-3, which may be an evolutional adaptation that developed upon association with its host. KEGG analysis revealed that dissimilatory nitrate reduction to ammonium (DNRA) and denitrification, two competing dissimilatory nitrate reduction pathways, co-occurred in the genome of SI-3, like most of the other 20 P. stutzeri strains. We speculated that DNRA of SI-3 may contribute a competitive advantage in nitrogen acquisition of U. prolifera by conserving nitrogen in NH4+ form, as in the case of microalgae bloom. Collectively, these data suggest that Pseudomonas sp. strain SI-3 was a suitable candidate for investigation of the algae-bacteria interaction with U. prolifera and the ecological impacts on algal blooming.
Substance exchange is considered the fundation of the partnership between algae and associated bacteria (Dittami et al., 2014; Cooper and Smith, 2015; Kouzuma and Watanabe, 2015). Algae, as the primary producer in freshwater and marine environments, provide dissolved organic nutrients to phycosphere heterotrophic bacteria (Field et al., 1998; Kouzuma and Watanabe, 2015). Complementarily, associated bacteria contribute to growth, morphogenesis, spore germination and colonization of algae by fixing nitrogen, as well as releasing minerals, vitamins, auxins, and quorum sensing signaling molecules (Joint et al., 2002; Croft et al., 2005; Marshall et al., 2006; Weinberger et al., 2007; Goecke et al., 2010; Foster et al., 2011; Singh and Reddy, 2014, 2015). Algae-associated bacteria have the potential to stimulate the growth of algae, similar to plant growth-promoting rhizobacteria. In addition, algae-associated bacteria can confer the competitive advantages to their host. Although limited in study, it has been suggested that associated bacteria promote algae bloom in the interaction between diatom and bacteria (Doucette, 1995).
Most, if not all, algae live with associated bacteria (Delbridge et al., 2004; Dittami et al., 2014). It generally accepted that algae exudates, such as polysaccharides, amino acids, proteoglycans or glycoproteins, form a phycosphere and influence the community structure of algae-associated bacteria (Myklestad, 1995; Sapp et al., 2007; Sison-Mangus et al., 2014). 16S rDNA sequencing and denaturing gradient gel electrophoresis (DGGE) fingerprinting have revealed that algae-associated bacteria communities are highly distinct from those of planktonic communities (Burke et al., 2011a; Amin et al., 2012; Goecke et al., 2013). Furthermore, algae-associated bacteria community presents host specificity. For example, pyrosequencing of 16S rDNA genes revealed that three species of the diatom genus Pseudo-nitzschia have different bacterial community compositions (Sison-Mangus et al., 2014). According to the spatial distribution of algae-associated bacteria, they can be divided into epiphyte and endophyte. Burke et al. (2011b) revealed the epiphytic bacterial community structure of Ulva australis varied according to space and time. However, the endophytic bacteria of algae are more closely associated withtheir host. For example, the endophytic bacterial communities of Bryopsis were found to be well-defined, even though samples were collected several hundred kilometers apart (Hollants et al., 2011). Additionally, the relative stability of endophytic bacterial communities of algae was successfully used to trace the invasive Caulerpa racemosa in Mediterranean to Australian range (Aires et al., 2013).
Ulva prolifera is the only dominant alga that causes successive green tides in the Yellow Sea, China (Zhao et al., 2013; Li et al., 2016), inducing harmful ecological impacts and economic losses. The community structures of U. prolifera associated bacteria have also been investigated to obtain information regarding the cause and influence of the world's largest green tide, but these studies were limited to communities from thalli surface and environmental water during the algal blooming (Guo et al., 2011; Liu et al., 2011). Moreover, the endophytic bacteria of U. prolifera have never been studied. In the present study, we obtained potential endophytes by treating the thalli of U. prolifera with ethanol plus bleach for sterilization. This method has been extensively used to eliminate epiphytes of algae and higher plants (Coombs and Franco, 2003; Kientz et al., 2011; Aires et al., 2012; Baoune et al., 2018). Pseudomonas sp. strain SI-3, which is mostly closed to P. stutzeri, was isolated from rich medium in which the homogenate of pretreated thalli was plated, indicating that strain SI-3 was probably an endophyte of U. prolifera.
Pseudomonas is a diverse genus that is known to occupy a wide range of niches and metabolic versatility. As a remarkable member of the Pseudomonas genus, P. stutzeri has received particular attention for its ability to conduct denitrification, degradation of aromatic compounds, and nitrogen-fixation (Lalucat et al., 2006). Some P. stutzeri strains even associate with plants endophytically or in the rhizosphere, stimulating plant growth or protecting plants against pathogens (Yan et al., 2008; Shen et al., 2013). In this study, the genome of strain SI-3 was sequenced and compared with that of 20 other P. stutzeri strains from diverse environments. Comparative genomic analysis revealed distinct characteristics of strain SI-3. Overall, the availability of genome sequence of strain SI-3 and comparative genomics results suggest that Pseudomonas sp. strain SI-3 is a suitable candidate to further investigation of algae-bacteria interaction with seaweed host U. prolifera.
The protoplast of U. prolifera was prepared according to Wu et al. (accepted). Complete cells and protoplast of U. prolifera were observed by optical microscope to determine if endophytic bacteria exist. The external bacteria of thalli of U. prolifera were removed by ethanol plus bleach treatment according to Aires et al. (2012). Briefly, U. prolifera samples were placed in 99% ethanol for 1 min and transferred to 3% bleach solution for 5 min, then immersed in 99% ethanol for about 30 s. The effect of epiphytes removal of the treated sample was detected by scanning electron microscopy (SEM, S-3400N, Hitachi, Tokyo, Japan) using untreated sample as a control. The endophytic bacteria of U. prolifera were released by grinding thoroughly, then cultured on 2216E agar medium and cultured at 28oC. For identification, PCR amplification was conducted using universal bacterial 16S rRNA primers 8F and 1492R. Strain SI-3, which was identified by 16S rDNA sequencing to be most closely related to P. stutzeri, was used for genome sequencing.
The genome of strain SI-3 was sequenced using PacBio RS II system and assembled using HGAP assembler. rRNAs and tRNAs were predicted using Barranp 0.4.2 and tRNAscan-SE v1.3.1, respectively. Protein coding sequences were predicted using Glimmer 3.02 and annotated by BLASTp alignment (BLAST 2.2.28+) with the Non-redundant (Nr), string and GO databases. Gene islands were predicted using IslandPATH-DIMOB v1.0.0 and SIGI-HMM 4.0. Carbohydrate-active enzymes (CAZymes) were functional annotated using similarity searches against the CAZy database.
The complete nucleotide genome sequence of Pseudomonas sp. strain SI-3 has been deposited in GenBank under accession no. CP026511.
We selected 20 P. stutzeri strains, including all 10 completely sequenced strains by far and 10 of 14 strains with genomes that were assembled into <100 contigs. The isolation environments of the 20 P. stutzeri strains were diverse, including seawater and marine sediment, rhizosphere, contaminated soil and water, and clinical specimens (Table 1) (Yan et al., 2008; Chen et al., 2011; Yu et al., 2011; Brunet-Galmés et al., 2012; Busquets et al., 2012, 2013; Li A. et al., 2012; Li X. et al., 2012; Chauhan et al., 2013; Grigoryeva et al., 2013; Smith et al., 2014; Hirose et al., 2015; Hu et al., 2015; Iyer and Damania, 2016; Wu et al., 2017).
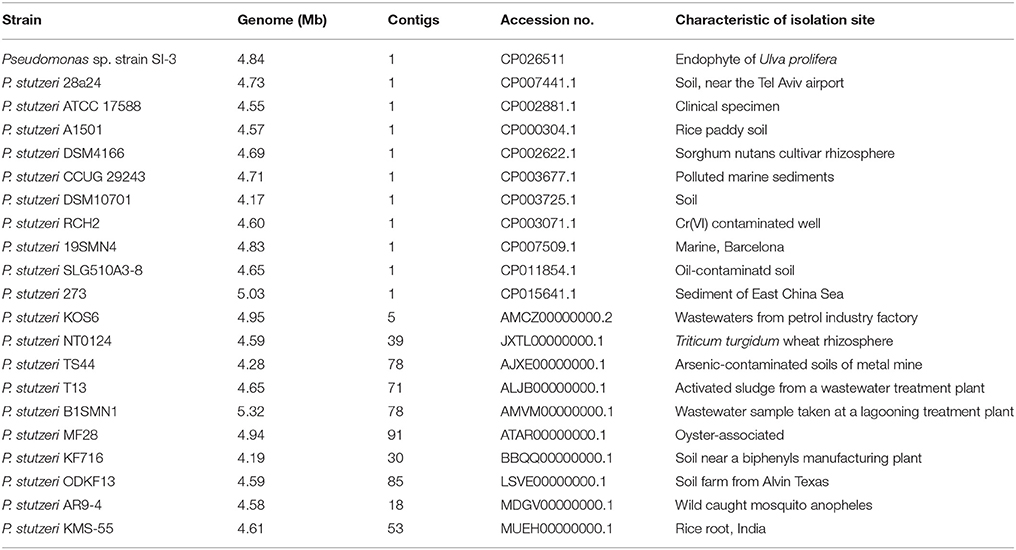
Table 1. General information of Pseudomonas sp. strain SI-3 and 20 P. stutzeri strains.
The genome sequence of SI-3 was compared with those of the 20 P. stutzeri above. Homologous genes were obtained by aligning the amino acids or nucleic acids sequences of all 21 strains using Ortho MCL v2.0.3 with an E-value <10−5. Conserved and strain-specific genes were identified by aligning the whole genome sequences using MUSCLE v3.7. The maximum likelihood (ML) phylogenetic tree based on the amino acids sequences of housekeeping gene rpoD which were aligned by MUSCLE v3.7, was constructed using MEGA 5. RAxML was used to construct the ML phylogenetic tree of concatenated amino acid sequences of single-copy orthologous conserved genes of 21 strains, after alignment using MUSCLE v3.7. For Clusters of Orthologous Groups (COGs) analysis, we used BLASTp (BLAST 2.2.28+) alignment against the string database (v9.05) to obtain the COG annotations of genes, then clustered them into different COG catagories. Metabolic pathway annotation was conducted using BLASTp (blastx/blastp 2.2.28+) against the Kyoto Encyclopedia of Genes and Genomes (KEGG) gene database.
Seaweed-associated bacteria and their interactions with the hosts have attracted the attention of many researchers worldwide. Studies have revealed green macroalgae such as Caulerpa and Bryopsis contain endophytic bacteria (Dawes and Lohr, 1978; Delbridge et al., 2004; Hollants et al., 2011, 2013; Aires et al., 2013). However, the endophytic bacteria of Ulva, the cosmopolitan green macroalgae, remain poorly studied. We found numerous highly moving bacteria when observing the thalli of U. prolifera by optical microscope (see the Supplementary Video 1). In addition, the existence of highly moving bacteria was observed in the protoplast of U. prolifera using enzymatic digestion (Wu et al., accepted) as well by optical microscope (see the Supplementary Video 2). This is the first time the potential endophytic bacteria of U. prolifera have been visualized.
To isolate potential endophytic bacteria of U. prolifera, we eliminated the epiphytic bacteria using the ethanol plus bleach sterilization. In this combination, ethanol serves as a detergent/solvent to break down the phospholipid bilayer and promote the invasion of bleach. Moreover, bleach eliminates the epiphytes as a strong oxidant. This method has been used to efficiently eliminate epiphytic bacteria and chloroplastidial DNA of green alga Caulerpa taxifolia (Aires et al., 2012), remove epiphytic bacteria to extract bioactive compounds originating from macroalgae (Kientz et al., 2011), and isolate endophytic actinobacteria from roots of wheat and other plants grown in petroleum contaminated soil (Coombs and Franco, 2003; Baoune et al., 2018). The effect of epiphytes removal of U. prolifera was observed by SEM using the untreated sample as a control. No bacteria were observed on the surface of the treated sample (Figure 1), suggesting the efficient removal of epiphytic bacteria. This result intensively implied that the isolates we obtained from the treated thalli were endophytes.

Figure 1. The epiphytes removal effect of ethanol plus bleach sterilization using SEM. (A) Untreated U. prolifera. (B) Ehtanol plus bleach treated U. prolfiera.
The homogenate of treated thalli was cultured and a few of colonies appeared, among which some hard, dry, and coral-like colonies resembled P. stutzeri morphologically (Lalucat et al., 2006). Phylogenetic analysis of the 16S rDNA sequence of one isolate, designated as SI-3, showed 99% sequence identity to about 20 P. stutzeri strains. P. stutzeri species possess a high degree of genotypic heterogeneity, so they are easily confused during phylogenetic classification (Wolterink et al., 2002; Romanenko et al., 2005; Cladera et al., 2006; Lalucat et al., 2006). Since the systematically phylogenetic identification of strain SI-3 has not yet been completed, we identified it as Pseudomonas sp. strain SI-3, and this strain had been repeatedly obtained from bloomed U. prolifera samples in different years. P. stutzeri has received particular attention because of its metabolic versatility, such as the ability to fix nitrogen, as well as to conduct denitrification and degradation of aromatic compounds. In addition, some P. stutzeri strains form intimate relationships with plants. For example, P. stutzeri A1501 is capable of associating with or colonizing the higher plant and promotes its growth, and has been used as crop inoculants in China (You et al., 1995; Rediers et al., 2003).
The genome of Pseudomonas sp. strain SI-3 was sequenced using PacBio RS II system. A total 117394 reads were assembled into one single contig using HGAP assembler. SI-3 had a 60.22%-GC circular chromosome of 4,838.607 bp, with no extrachromosomal elements such as plasmid (Figure 2A). Nine rRNAs and 58 tRNAs were predicted using Barranp 0.4.2 and tRNAscan-SE v1.3.1, respectively. In total, 4,563 protein-coding sequences (CDSs) comprising approximately 88.1% of the genome were predicted using Glimmer 3.02. COG analysis assigned all genes into different functional categories, about 10% of which are functionally unknown (Figure 2B). Using IslandPATH-DIMOB v1.0.0 and SIGI-HMM 4.0, 68 genomic islands were predicted at least by one method in the genome of SI-3 (Figure 2C).
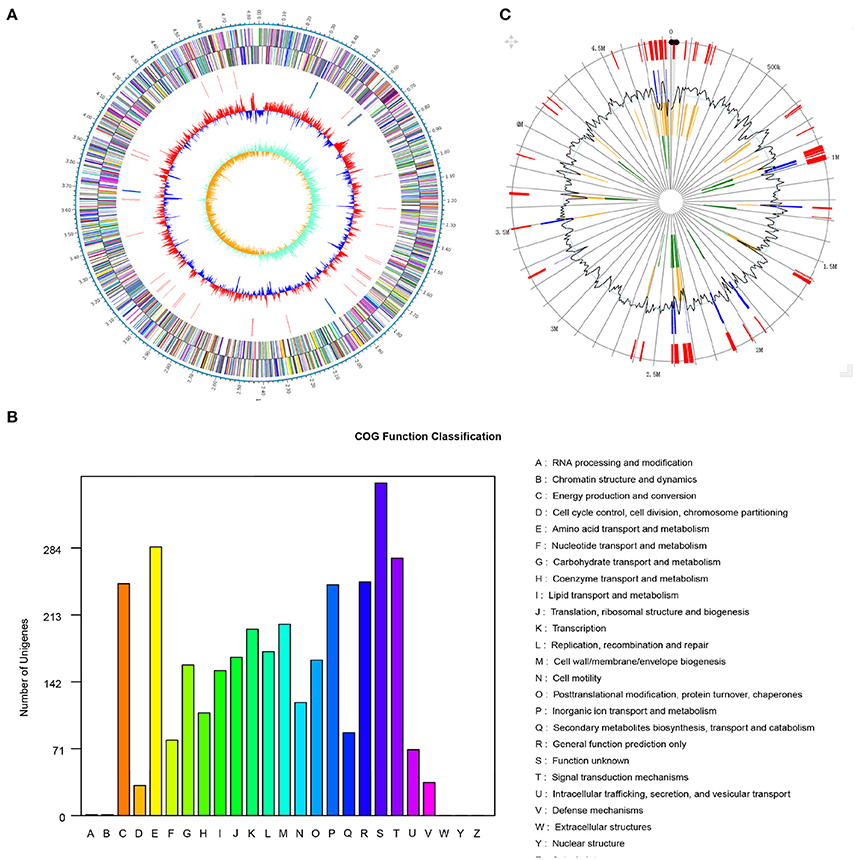
Figure 2. Genome features of Pseudomonas sp. strain SI-3. (A) Graphical map of chromosome of Pseudomonas sp. strain SI-3. The outer scale is marked in 0.1 Mb. From the outside to the center of each circle: Circle 1 and 2, genes encoded on forward and reverse strands, respectively. Coding sequences are colored by COG categories. Circle 3, RNA genes. Circle 4, GC content. Circle 5, GC skew (G–C/G+C). (B) COG function classification of SI-3. The ordinate axis indicates the gene numbers in each COG functional category. (C) Genomic islands predicted by two methods. Blue lines present the genomic islands predicted by IslandPATH-DIMOB method. Yellow lines present the genomic islands predicted by SIGI-HMM method. Red lines present the integrated genomic islands predicted by two methods. The second circle indicates the GC content.
We compared SI-3 with the 20 P. stutzeri strains in Table 1, including all 10 strains that have been completely sequenced to dateand 10 of 14 strains for which their genomes were assembled into <100 contigs. The amino acid sequence of housekeeping gene rpoD, which is a qualified representative of the whole genomes for different P. stutzeri isolates (Yamamoto et al., 2000; Cladera et al., 2004) was selected as a genetic marker to construct a ML phylogenetic tree (Figure 3A). Moremover, a ML phylogenetic tree was established based on 2,291 concatenated amino acid sequences of single-copy orthologous genes from all strains (Figure 3B). Both trees shared a similar topological relationship and showed that SI-3 was mostly closed to P. stutzeri 273, which was isolated from sediment in a green-tide affected sea area (Wu et al., 2017), then clustered with 28a24 and MF28, which is a marine animal oyster-associated strain.
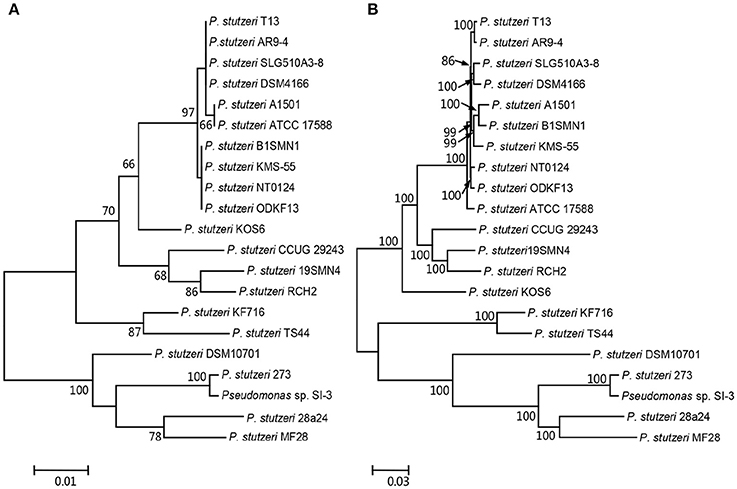
Figure 3. Phylogenetic trees of Pseudomonas sp. strain SI-3 and 20 other P. stutzeri strains. The Maximum Likelihood (ML) phylogeny trees were constructed based on amino acid sequences of housekeeping gene rpoD (A) and concatenated amino acid sequences of single copy orthologous genes (B). The topology of the tree was tested with 1,000 bootstrap replications and bootstrap values were shown near the horizontal branches of the trees.
The pan- and core-genome of strain SI-3 and the 20 P. stutzeri strains was analyzed to reveal unique genes of SI-3. We identified a pan-genome of 6,468 genes and a core-genome of 2,216 genes using 21 genomes (Figure 4). Taking into consideration of the average gene numbers of 4,282 for the 21 strains, the 2,216 genes of core-genome represent approximately 52% of the total genome. In other words, half of the genomic regions are conserved. P. stutzeri strains B1SMN1 and KOS6, and strain SI-3 were ranked highest based on the presence of 705, 378, and 332 strain-specific genes, respectively (Figure 4). For B1SMN1 and KOS6, which were both isolated from wastewater samples (Table 1), quite a few of their strain-specific genes encodes transporters, chemotaxis proteins, secretion systems, motility related proteins, and transcriptional regulators, which can contribute to the survival under extreme conditions. Thirteen of the strain-specific genes of strain SI-3, encoded kinases, transcriptional regulators, sensor proteins, chemotaxis proteins, and GGDEF-domain containing proteins, which are indicative of the need to respond to changes in the host environment (Burke et al., 2011a). Fifteen transposase genes are presented within or around the SI-3 specific genes regions, while 71 strain-specific genes are located in genomic islands, indicating the strain-specific genes of SI-3 may beexogenous. Moreover, up to 82% of the strain-specific genes of SI-3 are function-unknown, which may contribute to its survival and adaptation to unique environment.
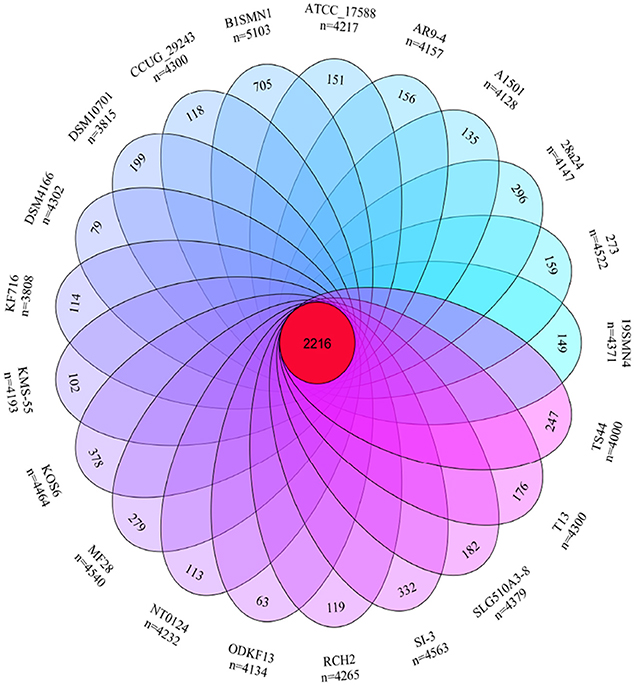
Figure 4. Venn diagram showing the pan- and core-genome of Pseudomonas sp. strain SI-3 and 20 P. stutzeri strains. The center red circle represented the core-genome. The strain name and total gene number of each ellipse presented were marked outside. The specific genes number of each strain was marked in corresponding ellipse.
We compared the functional distribution of all genes from 21 strains based on COG database. Strain SI-3 had 198 genes assigned to transcription (COG K) category, occupying 5.81% of the genes of all COG categories. This was the highest level among the 20 P. stutzeri strains, including the closely related strains 273 and 28a24 (Figure 5A). Transcriptional regulators were overrepresented in COG K category, which regulated transcription involved in attachment and colonization, quorum sensing, motility, and uptake of elements (Kovacikova and Skorupski, 1999; Cao et al., 2001; Fillat, 2014; Taw et al., 2015), indicating a complicated regulatory network within SI-3. In addition, genes enriched in signal transduction mechanism category (COG T) in SI-3 occupied 8.01% of the total COG categories genes, which was significantly higher than that of the other 20 P. stutzeri strains (Figure 5B). There were many homologs of the histidine kinase and GGDEF and EAL domain proteins, which were known to be involved in osmoregulation, chemotaxis, multidrug export, motility, and biofilm formation (Foynes et al., 2000; Nagakubo et al., 2002; Simm et al., 2004; Baker et al., 2006; Jenal and Malone, 2006; Yoshida et al., 2007), and these processes were involved in the colonization steps and interactions with other community members or the host. It is widely accepted that the host and its metabolites affect the gene regulation of associated bacteria (Mark et al., 2005; Pothier et al., 2007), and that the high proportion of transduction regulators and signal transducers could be indicative of the need of strain SI-3 to respond to changes in the host environment (Burke et al., 2011a). Notably, genes contributing to transcription and signal transduction mechanism in P. stutzeri strains A1501, DSM4166, NT0124, and KSM-55, which were isolated from rhizosphere of higher plants, were not as abundant as that in strain SI-3. We speculated that, as a potential endophytic bacterium, strain SI-3 may establish a more intimate and stable relationship with its host, than rhizosphere bacteria, therefore, they are prone to evolution of more complicated regulation and signal transduction networks to adapt to theire host environment. Collectively, these data indicate that strain SI-3 has evolved strain-specific features, which may contribute to its adaptability in the green macroalga U. prolifera.
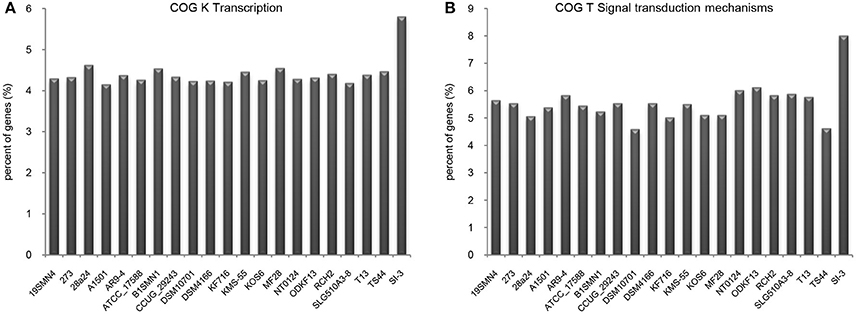
Figure 5. Enhanced transcription and signal transduction network in Pseudomonas sp. strain SI-3. The proportions of genes assigned into COG K (A) and COG T (B) categories were normalized to the total gene number of all COG categories in each strain.
It is well known that genes work with other genes in a well-organized cooperative relationship rather than functioning individually. To evaluate their biological functions systematically, the overall metabolic pathways of all 21 strains were analyzed according to KEGG analysis. The gene number of each KEGG pathway of strain SI-3 was normalized against the average gene number of the corresponding KEGG pathway of all 21 strains. All KEGG pathways of SI-3 were ranked based on relative gene contents and the gene numbers of most KEGG pathways of SI-3 were close to the averages. Specifically, pathways with relative gene contents lower than 0.8 and higher than 1.2 were presented considering the significant differences compared with the averages (Figure 6). Coincidentally, 8 of 11 metabolic pathways with the relative values lower than 0.8 were involved in degradation of benzene ring containing compounds, indicating the compromised ability of SI-3 to degrade aromatic compounds. The greatest gene loss was observed in pathways of xylene degradation (PATH: ko00622), toluene degradation (PATH: ko00623), fluorobenzoate degradation (PATH: ko00364), and degradation of aromatic compounds (PATH: ko01220). Strain SI-3 was found to be closely related to P. stutzeri, which is well known for its metabolic versatility in aromatic compounds degradation (Baggi et al., 1987; Kozlovsky et al., 1993; DiLecce et al., 1997; Dijk et al., 2003; Lalucat et al., 2006), however, there was no xylene degradation pathway in SI-3, indicating the defect of SI-3 in xylene degradation. In addition, the homologs of enzymes catalyzing the initial steps of xylene degradation were absent in strains 28a24 and MF28, which were isolated from soil and oyster, respectively, and closely clustered with SI-3 (Figure 3). In contrast, as the most phylogenetic closely related strain of SI-3, P. stutzeri 273 was isolated from a sediment sample collected in the green-tide affected sea area, but the complete set of xylene degradation enzymes was conserved. P. stutzeri strains 19SMN4, B1SMN1, CCUG_29243, KF716, KOS6, and TS44 had homologs catalyzing the xylene degradation intermediate methylbenzoate to methylcaltechol (Figure 7A). For the toluene degradation pathway, strain SI-3, 28a24 and MF28 all lacked the enzymes needed to catalyze toluene and its intermediates degradation. Strains KF716, KOS6, and TS44, which were isolated from contaminated environments (Table 1) all contained enzymes responsible for degradation of toluene via 2-hydroxytoluene to 3-methylcatechol. Based on KEGG analysis, strain 273 was able to degrade toluene to benzoate. However, other strains could not degrade toluene, but conserved enzymes catalyzing 4-methylcatechol, the intermediate of toluene-4-sulfonate degradation pathway (Figure 7B). Significant gene loss in fluorobenzoate degradation pathway was observed in strain SI-3, as well as P. stutzeri strains 28a24, KOS6, and MF28, all of which lacked the homologs of genes involved in fluorobenzoate degradation (Figure 7C). Hence, the number of SI-3 genes involved in the degradation of aromatic compounds degradation (PATH: ko01220) pathway was rather low (Figure 7D) because the number of genes involved in degradation of multiple benzene ring containing compounds was below average. Collectively, these data, demonstrated that P. stutzeri strains isolated from contaminated environments were likely to conserve more pollutant degradation genes, while strain SI-3, a potential endophytic bacterium of U. prolifera, lost the majority of genes involved in aromatic compounds degradation. Interestingly, Zhang et al. (2017) demonstrated that sterilized host U. prolifera was capable of efficiently removing polycyclic aromatic hydrocarbons. In contrast, enrichment of aromatic compounds biodegradation pathways was found in the rhizoplane microbiome by metagenomic sequencing of root microbes of foxtail millet (Jin et al., 2017). Therefore, we speculated that the bacterial spatial distribution and host capacity of aromatic compounds degradation could shape the adaptation features of the plant-associated bacterial genome.
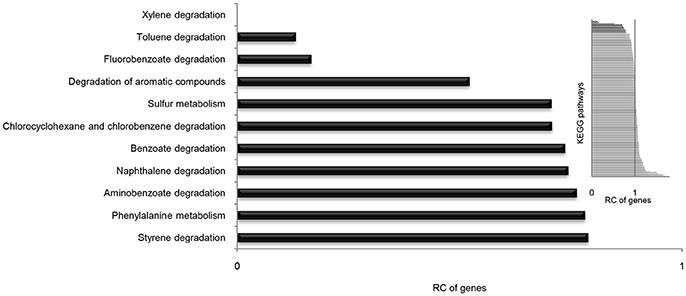
Figure 6. Relative contents (RCs) of gene numbers of KEGG metabolic pathways in Pseudomonas sp. strain SI-3. The average gene numbers for each metabolic pathway in strain SI-3 and 20 P. stutzeri strains were set to 1, after which the gene numbers of corresponding KEGG metabolic pathways of SI-3 were normalized. All pathways of SI-3 were ranked based on the RC of genes. The pathways with RCs between 0.8 and 1.2 were omitted to make the results clear. The embedded panel in the upper right was the entire profile of all pathways of SI-3.
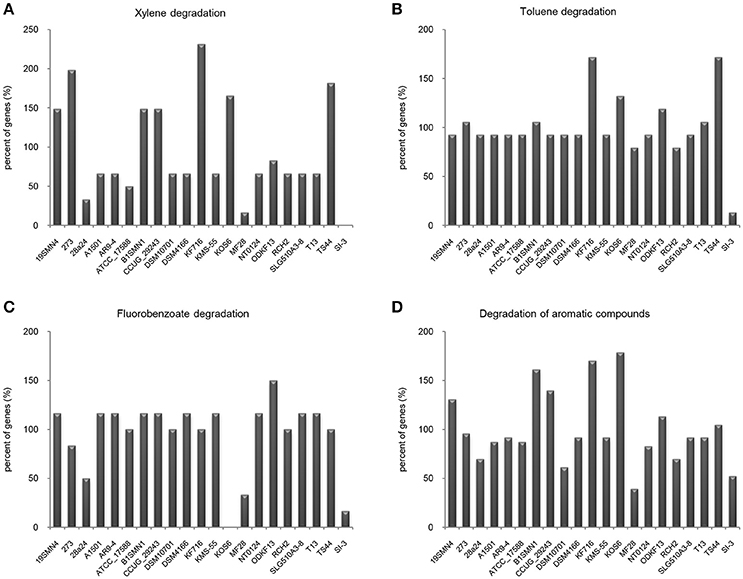
Figure 7. Loss of genes involved in aromatic compounds degradation pathways in Pseudomonas sp. strain SI-3. The gene numbers in each strain were normalized to the average gene numbers of SI-3 and 20 P. stutzeri strains in xylene degradation (A), toluene degradation (B), fluorobenzoate degradation (C), and degradation aromatic compounds (D) pathways.
Pseudomonas stutzeri is renowned for its specific metabolic properties in nitrogen metabolism, such as nitrogen fixation and denitrification. Since SI-3 was phylogenetically close to P. stutzeri, we evaluated the nitrogen metabolism pathways of SI-3 based on KEGG analysis. There were no homologs of nif genes, the nitrogen fixation cluster, in strain SI-3. Homologs of nitrogen fixation genes were only identified in P. stutzeri strains A1501, DSM4166, KMS55, B1SMN1, and KOS6, among which the first three were all isolated from rhizosphere (Table 1). A complete set of denitrification genes were found in the genome of SI-3, as well as all of the investigated P. stutzeri strains except for strain 28a24. Moreover, genes of dissmilatory nitrate reduction to ammonium (DNRA), a competing dissimilatory nitrate reduction pathway of denitrification, were identified in all strains but 28a24. DNRA and denitrification determine fates of nitrate, with the former conserving nitrogen in a more stable and biological preferred form, while the latter serves as a nitrogen sink by turning nitrate into gaseous products. The importance of DNRA in many ecosystems has been ignored until recently (Silver et al., 2001; Rütting et al., 2011). It was proposed that DNRA mechanism by providing could explain the successive bloom of Texas Brown Tide caused by monospecific microalga Aureomonas lagunensis (An and Gardner, 2002). However, the role of DNRA in the bloom of macroalgae has never been considered and studied. Additionally, the interspecific ecological success of U. prolifera has been shown to be partially a result of its higher uptake rate of nitrogen (Luo et al., 2012), and is the preferred nitrogen form (Guo et al., 2017). Since the nitrate concentration of the Yellow Sea presented increasing trend in the last few decades while the concentration of fluctuated (Li et al., 2015, 2017), we wondered whether coupling of DNRA from associated bacteria with the high nitrogen uptake rate would confer U. prolifera a competitive advantages over other seaweeds in the Yellow Sea against other seaweeds. Based on the results of the present study, the role of bacterial DNRA in the algae-bacteria interaction of U. prolifera is worthy of further investigation.
Substance exchange is essential to maintenance of an algae-bacteria interaction. Pseudomonas strains can use a wide range of nutrients (Clarke, 1982). We identified 118 CAZymes encoded in genome of strain SI-3, including 12 α-amylases (GH13) and five cellulases (GH5) and one β-1,4-xylosidase (GH39), whose substrates were constituents of polysaccharide of U. prolifera (Ray, 2006; Lahaye and Robic, 2007). In addition, SI-3 contained 218 genes involved in transport of diverse substrates, such as amino acids, sugars, organic acids, lipopolysaccharide, and biopolymers and 88 genes encoding peptidases or proteases, indicating the capacity of SI-3 to degrade plant-derived compounds. Apart from obtaining organic matter from their hosts, algae-associated bacteria can provide micronutrients in return. For example, iron is essential for most organisms, but often limited in marine environment. The availability of iron regulates the nitrate acquisition in Ulva (Viaroli et al., 2005) and utilization of xenosiderophores, high-affinity ferric iron chelators, is a common advantage for cells (Miethke and Marahiel, 2007). Strain SI-3 possessed genes encoding non-ribosomal peptides synthetases involved in the biogenesis of enterobactin, vibrobactin, bacillibactin, and myxochelin precursor according to the analysis against KEGG database, which may contribute to maintenance of its association with U. prolifera.
Toxic metals contamination in the margin sea are receiving more and more attention, and numerous studies have shown green macroalgae, such as U. linza and U. prolifera, are able to absorb and accumulate toxic metals (Jiang et al., 2013). Strain SI-3 contained two different copper resistance systems, cus and cop (Cha and Cooksey, 1991; Franke et al., 2003), the arsenic-resistance genes arsC andarsRB, nickel-resistance proteins and multiple toxic metal efflux and transporters. These genes may help SI-3 adapt to high metal niches of its host. In addition to defense against toxic metals, SI-3 also contained many genes involved in defense against oxidative stress, such as oxidative stress response regulator OxyR, catalase and peroxidase. Macroalgae can employ oxidative bursts, in which rapid activation of reactive oxygen species occurs, to defend against pathogens (Goecke et al., 2010). Obviously, the survival and stable existence of endophytic bacteria depend on the ability of these genes to cope with oxidative bursts from their host.
Pseudomonas sp. strain SI-3, which was phylogenetically close to P. stutzeri, was isolated from U. prolifera, the only dominant alga of the world's largest green tide. Here, we reported the complete genome sequence of strain SI-3 and revealed its unique genomic characteristic as a potential endophyte based on comparison with 20 P. stutzeri strains. Stain SI-3 contained more strain-specific genes than most of the others, which may have facilitated its adaptationto the host environment. The remarkably high proportion of genes assigned to transcriptional regulation and signal transduction functional categories indicated the extraordinary ability of SI-3 to respond to its host environment. Loss of genes associated with aromatic compounds degradationwas observed in SI-3, which may be an evolutionary adaptation when associate with its host. Our results revealed the co-existence of DNRA and denitrification, two competing dissimilatory nitrate reduction pathways, in strain SI-3, which was similar to most of the P. stutzeri strains it was compared with. The role of bacterial DNRA in algae-bacteria interaction during algal blooming process is noteworthy. Moreover, SI-3 was found to have many genomic traits that probably contributed to maintenance of its algae-bacteria interaction with U. prolifera, such as the potential to transport and degrade plant-derived compounds, providing micronutrient to the host and resistance to toxic metals and oxidative stress. Therefore, we suggested that Pseudomonas sp. strain SI-3 was a suitable candidate for investigation of the algae-bacteria interaction with macroalgae U. prolifera.
HF designed the study, performed the analysis and interpreted the results. PJ, JZ, and CW contributed to data processing. HF and PJ wrote the manuscript.
This research was supported by National Natural Science Foundation of China (31700049, 41776153), China Post-doctoral Science Foundation (2017M610451), Scientific & Technological Innovation Project financially supported by Qingdao National Laboratory for Marine Science and Technology (2016ASKJ02-1), National Post doctoral Program for Innovative Talents (BX201600167), Post doctoral Application Research Program funded by Qingdao (2016189), Open Fund of Qingdao National Laboratory for Marine Science and Technology (QNLM2016ORP0303), and Strategic Priority Research Program of the Chinese Academy of Sciences (XDA11020304).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01458/full#supplementary-material
Supplementary Video 1. Highly moving bacteria in the thalli of U. prolifera observed under optical microscope magnified by 600X. The representative cells in which bacteria existed were indicated by red arrows.
Supplementary Video 2. Highly moving bacteria in protoplast of U. prolifera observed under optical microscope magnified by 600X. Several highly moving bacteria were limited within the spherical shape of protoplast all the time. The larger spherical moving particle inside of the protoplast was pushed passively by the bacteria around it.
Aires, T., Marbà, N., Serrao, E. A., Duarte, C. M., and Arnaud-Haond, S. (2012). Selective elimination of chloroplastidial DNA for metagenomics of bacteria associated with the green alga Caulerpa taxifolia (Bryopsidophyceae). J. Phycol. 48, 483–490. doi: 10.1111/j.1529-8817.2012.01124.x
Aires, T., Serrao, E. A., Kendrick, G., Duarte, C. M., and Arnaud-Haond, S. (2013). Invasion is a community affair: clandestine followers in the bacterial community associated to green algae, Caulerpa racemosa, track the invasion source. PLoS ONE 8:e68429. doi: 10.1371/journal.pone.0068429
Amin, S. A., Parker, M. S., and Armbrust, E. V. (2012). Interactions between diatoms and bacteria. Microbiol. Mol. Biol. Rev. 76, 667–684. doi: 10.1128/MMBR.00007-12
An, S., and Gardner, W. S. (2002). Dissimilatory nitrate reduction to ammonium (DNRA) as a nitrogen link, versus denitrification as a sink in a shallow estuary (Laguna Madre/Baffin Bay, Texas). Mar. Ecol. Prog. Ser. 237, 41–50. doi: 10.3354/meps237041
Baggi, G., Barbieri, P., Galli, E., and Tollari, S. (1987). Isolation of a Pseudomonas stutzeri strain that degrades o-xylene. Appl. Environ. Microbiol. 53, 2129–2132.
Baker, M. D., Wolanin, P. M., and Stock, J. B. (2006). Signal transduction in bacterial chemotaxis. Bioessays 28, 9–22. doi: 10.1002/bies.20343
Baoune, H., Ould El Hadj-Khelil, A., Pucci, G., Sineli, P., Loucif, L., and Polti, M. A. (2018). Petroleum degradation by endophytic Streptomyces spp. isolated from plants grown in contaminated soil of southern Algeria. Ecotox. Environ. Safe. 147, 602–609. doi: 10.1016/j.ecoenv.2017.09.013
Brunet-Galmés, I., Busquets, A., Peña, A., Gomila, M., Nogales, B., García-Valdés, E., et al. (2012). Complete genome sequence of the naphthalene-degrading bacterium Pseudomonas stutzeri AN10 (CCUG 29243). J. Bacteriol. 194, 6642–6643. doi: 10.1128/JB.01753-12
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S., and Thomas, T. (2011a). Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U.S.A. 108, 14288–14293. doi: 10.1073/pnas.1101591108
Burke, C., Thomas, T., Lewis, M., Steinberg, P., and Kjelleberg, S. (2011b). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600. doi: 10.1038/ismej.2010.164
Busquets, A., Peña, A., Gomila, M., Bosch, R., Nogales, B., García-Valdés, E., et al. (2012). Genome sequence of Pseudomonas stutzeri strain JM300 (DSM 10701), a soil isolate and model organism for natural transformation. J. Bacteriol. 194, 5477–5478. doi: 10.1128/JB.01257-12
Busquets, A., Peña, A., Gomila, M., Mayol, J., Bosch, R., Nogales, B., et al. (2013). Draft genome sequence of Pseudomonas stutzeri strain B1SMN1, a nitrogen-fixing and naphthalene-degrading strain isolated from wastewater. Genome Announc. 1:e00584-3. doi: 10.1128/genomeA.00584-13
Cao, H., Krishnan, G., Goumnerov, B., Tsongalis, J., Tompkins, R., and Rahme, L. G. (2001). A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc. Natl. Acad. Sci. U.S.A. 98, 14613–14618. doi: 10.1073/pnas.251465298
Cha, J. S., and Cooksey, D. A. (1991). Copper resistance in Pseudomonas syringae mediated by periplasmic and outer membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 88, 8915–8919. doi: 10.1073/pnas.88.20.8915
Chauhan, A., Green, S., Pathak, A., Thomas, J., and Venkatramanan, R. (2013). Whole-genome sequences of five oyster-associated bacteria show potential for crude oil hydrocarbon degradation. Genome Announc. 1:e00802-13. doi: 10.1128/genomeA.00802-13
Chen, M., Yan, Y., Zhang, W., Lu, W., Wang, J., Ping, S., et al. (2011). Complete genome sequence of the type strain Pseudomonas stutzeri CGMCC 1.1803. J. Bacteriol. 193, 6095–6095. doi: 10.1128/JB.06061-11
Cladera, A. M., Bennasar, A., Barceló, M., Lalucat, J., and García-Valdés, E. (2004). Comparative genetic diversity of Pseudomonas stutzeri genomovars, clonal structure, and phylogeny of the species. J. Bacteriol. 186, 5239–5248. doi: 10.1128/JB.186.16.5239–5248.2004
Cladera, A. M., Sepúlveda-Torres, L. C., Valens-Vadell, M., Meyer, J. M., Lalucat, J., and García-Valdés, E. (2006). A detailed phenotypic and genotypic description of Pseudomonas strain OX1. Syst. Appl. Microbiol. 29, 422–430. doi: 10.1016/j.syapm.2005.11.007
Clarke, P. H. (1982). The metabolic versitility of Pseudomonads. Antonie Van Leeuwenhoek 48, 105–130.
Coombs, J. T., and Franco, C. M. M. (2003). Isolation and identification of actinobacteria from surface-sterilized wheat roots. Appl. Environ. Microbiol. 69, 5603–5608. doi: 10.1128/aem.69.9.5603-5608.2003
Cooper, M. B., and Smith, A. G. (2015). Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr. Opin. Plant Biol. 26, 147–153. doi: 10.1016/j.pbi.2015.07.003
Croft, M. T., Lawrence, A. D., Raux-Deery, E., Warren, M. J., and Smith, A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
Dawes, C. J., and Lohr, C. A. (1978). Cytoplasmic organization and endosymbiotic bacteria in the growing points of Caulerpa prolifera. Rev. Algol. 13, 309–314.
Delbridge, L., Coulburn, J., Fagerberg, W., and Tisa, L. S. (2004). Community profiles of bacterial endosymbionts in four species of Caulerpa. Symb. 37, 335–344.
Dijk, J. A., Stams, A. J., Schraa, G., Ballerstedt, H., de Bont, J. A., and Gerritse, J. (2003). Anaerobic oxidation of 2-chloroethanol under denitrifying conditions by Pseudomonas stutzeri strain JJ. Appl. Microbiol. Biotech. 63, 68–74. doi: 10.1007/s00253-003-1346-z
DiLecce, C., Accarino, M., Bolognese, F., Galli, E., and Barbieri, P. (1997). Isolation and metabolic characterization of a Pseudomonas stutzeri mutant able to grow on the three isomers of xylene. Appl. Environ. Microbiol. 63, 3279–3281.
Dittami, S. M., Eveillard, D., and Tonon, T. (2014). A metabolic approach to study algal–bacterial interactions in changing environments. Mol. Ecol. 23, 1656–1660. doi: 10.1111/mec.12670
Doucette, G. J. (1995). Interactions between bacteria and harmful algae: a review. Nat. Toxins 3, 65–74. doi: 10.1002/nt.2620030202
Field, C., Behrenfeld, M. J., Randerson, J. T., and Falkowski, P. (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281, 237–240. doi: 10.1126/science.281.5374.237
Fillat, M. F. (2014). The FUR (ferric uptake regulator) superfamily: diversity and versatility of key transcriptional regulators. Arch. Biochem. Biophys. 546, 41–52. doi: 10.1016/j.abb.2014.01.029
Foster, R. A., Kuypers, M. M., Vagner, T., Paerl, R. W., Musat, N., and Zehr, J. P. (2011). Nitrogen fixation and transfer in open ocean diatom-cyanobacterial symbioses. ISME J. 5, 1484–1493. doi: 10.1038/ismej.2011.26
Foynes, S., Dorrell, N., Ward, S. J., Stabler, R. A., McColm, A. A., Rycroft, A. N., et al. (2000). Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 68, 2016–2023. doi: 10.1128/IAI.68.4.2016-2023.2000
Franke, S., Grass, G., Rensing, C., and Nies, D. H. (2003). Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J. Bacteriol. 185, 3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003
Goecke, F., Labes, A., Wiese, J., and Imhoff, J. F. (2010). Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 409, 267–300. doi: 10.3354/meps08607
Goecke, F., Thiel, V., Wiese, J., Labes, A., and Imhoff, J. F. (2013). Algae as an important environment for bacteria – phylogenetic relationships among new bacterial species isolated from algae. Phycologia 52, 14–24. doi: 10.2216/12–24.1.s1
Grigoryeva, T. V., Laikov, A. V., Naumova, R. P., Manolov, A. I., Larin, A. K., Karpova, I. Y., et al. (2013). Draft genome of the nitrogen-fixing bacterium Pseudomonas stutzeri strain KOS6 isolated from industrial hydrocarbon sludge. Genome Announc. 1:e00072-12. doi: 10.1128/genomeA.00072-12
Guo, C., Li, F., Jiang, P., Liu, Z., and Qin, S. (2011). Bacterial diversity in surface water of the Yellow Sea during and after a green alga tide in 2008. Chin. J. Oceanol. Limnol. 29, 1147–1154. doi: 10.1007/s00343-011-0264-7
Guo, Y., Wang, H., Wu, C., Fu, H., and Jiang, P. (2017). Cloning and characterization of nitrate reductase gene in Ulva prolifera (Ulvophyceae, Chlorophyta). J. Phycol. 53, 1035–1043. doi: 10.1111/jpy.12556
Hirose, J., Yamazoe, A., Hosoyama, A., Kimura, N., Suenaga, H., Watanabe, T., et al. (2015). Draft genome sequence of the polychlorinated biphenyl-degrading bacterium Pseudomonas stutzeri KF716 (NBRC 110668). Genome Announc. 3:e01215. doi: 10.1128/genomeA.01215-15
Hollants, J., Leliaert, F., Verbruggen, H., Willems, A., and De-Clerck, O. (2013). Permanent residents or temporary lodgers: characterizing intracellular bacterial communities in the siphonous green alga Bryopsis. Proc. R. Soc. Lond. B 280:20122659. doi: 10.1098/rspb.2012.2659
Hollants, J., Leroux, O., Leliaert, F., Decleyre, H., De Clerck, O., and Willems, A. (2011). Who is in there? Exploration of endophytic bacteria within the siphonous green seaweed Bryopsis (Bryopsidales, Chlorophyta). PLoS ONE 6:e26458. doi: 10.1371/journal.pone.0026458
Hu, B., Nie, Y., Geng, S., and Wu, X. (2015). Complete genome sequence of the petroleum-emulsifying bacterium Pseudomonas stutzeri SLG510A3-8. J. Biotech. 211, 1–2. doi: 10.1016/j.jbiotec.2015.06.421
Iyer, R., and Damania, A. (2016). Draft genome sequence of Pseudomonas stutzeri ODKF13, isolated from farmland soil in Alvin, Texas. Genome Announc. 4:e00293-16. doi: 10.1128/genomeA.00293-16
Jenal, U., and Malone, J. (2006). Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 40, 385–407. doi: 10.1146/annurev.genet.40.110405.090423
Jiang, H., Gao, B., Li, W., Zhu, M., Zheng, C., Zheng, Q., et al. (2013). Physiological and biochemical responses of Ulva prolifera and Ulva linza to cadmium stress. Sci. World J. 2013:289537. doi: 10.1155/2013/289537
Jin, T., Wang, Y., Huang, Y., Xu, J., Zhang, P., Wang, N., et al. (2017). Taxonomic structure and functional association of foxtail millet root microbiome. Gigascience 6, 1–12. doi: 10.1093/gigascience/gix089
Joint, I., Tait, K., Callow, M. E., Callow, J. A., Milton, D., Williams, P., et al. (2002). Cell-to-cell communication across the prokaryote-eukaryote boundary. Science 298:1207. doi: 10.1126/science.1077075
Kientz, B., Thabard, M., Cragg, S. M., Pope, J., and Hellio, C. (2011). A new method for removing microflora from macroalgal surfaces: an important step for natural product discovery. Bot. Mar. 54, 457–469. doi: 10.1515/BOT.2011.053
Kouzuma, A., and Watanabe, K. (2015). Exploring the potential of algae/bacteria interactions. Curr. Opin. Biotech. 33, 125–129. doi: 10.1016/j.copbio.2015.02.007
Kovacikova, G., and Skorupski, K. (1999). A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181, 4250–4256.
Kozlovsky, S. A., Zaitsev, G. M., Kunc, F., Gabriel, J., and Boronin, A. M. (1993). Degradation of 2-chlorobenzoic and 2,5-dichlorobenzoic acids in pure culture by Pseudomonas stutzeri. Folia. Microbiol. 38, 371–375.
Lahaye, M., and Robic, A. (2007). Structure and functional properties of ulvan, a polyasccaride from green seaweeds. Biomacromolecules 8, 1765–1774. doi: 10.1021/bm061185q
Lalucat, J., Bennasar, A., Bosch, R., García-Valdés, E., and Palleroni, N. J. (2006). Biology of Pseudomonas stutzeri. Microbiol. Mol. Biol. Rev. 70, 510–547. doi: 10.1128/MMBR.00047-05
Li, A., Gai, Z., Cui, D., Ma, F., Yang, J., Zhang, X., et al. (2012). Genome sequence of a highly efficient aerobic denitrifying bacterium, Pseudomonas stutzeri T13. J. Bacteriol. 194:5720. doi: 10.1128/JB.01376-12
Li, H., Zhang, C., Han, X., and Shi, X. (2015). Changes in concentrations of oxygen, dissolved nitrogen, phosphate, and silicate in the southern Yellow Sea, 1980–2012: Sources and seaward gradients. Estuar. Coast. Shelf Sci. 163, 44–55. doi: 10.1016/j.ecss.2014.12.013
Li, H., Zhang, Y., Han, X., Shi, X., Rivkin, R. B., and Legendre, L. (2016). Growth responses of Ulva prolifera to inorganic and organic nutrients: implications for macroalgal blooms in the southern Yellow Sea, China. Sci. Rep. 6:26498. doi: 10.1038/srep26498
Li, H., Zhang, Y., Tang, H., Shi, X., Rivkin, R. B., and Legendre, L. (2017). Spatiotemporal variations of inorganic nutrients along the Jiangsu coast, China, and the occurrence of macroalgal blooms (green tides) in the southern Yellow Sea. Harmful Algae 63, 164–172. doi: 10.1016/j.hal.2017.02.006
Li, X., Gong, J., Hu, Y., Cai, L., Johnstone, L., Grass, G., et al. (2012). Genome sequence of the moderately halotolerant, arsenite-oxidizing bacterium Pseudomonas stutzeri TS44. J. Bacteriol. 194, 4473–4474. doi: 10.1128/JB.00907-12
Liu, M., Dong, Y., Zhao, Y., Zhang, G., Zhang, W., and Xiao, T. (2011). Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay, China. World J. Microbiol. Biotech. 27, 1703–1712. doi: 10.1007/s11274-010-0627-9
Luo, M., Liu, F., and Xu, Z. (2012). Growth and nutrient uptake capacity of two co-occurring species, Ulva prolifera and Ulva linza. Aquat. Bot. 100, 18–24. doi: 10.1016/j.aquabot.2012.03.006
Mark, G. L., Dow, J. M., Kiely, P. D., Higgins, H., Haynes, J., Baysse, C., et al. (2005). Transcriptome profiling of bacterial responses to root exudates identifies genes involved in microbe-plant interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 17454–17459. doi: 10.1073/pnas.0506407102
Marshall, K., Joint, I., Callow, M. E., and Callow, J. A. (2006). Effect of marine bacterial isolates on the growth and morphology of axenic plantlets of the green alga Ulva linza. Microbial Ecol. 52, 302–310. doi: 10.1128/MMBR.00012-07
Miethke, M., and Marahiel, M. A. (2007). Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451. doi: 10.1007/s00248-006-9060-x
Myklestad, S. M. (1995). Release of extracellular products by phytoplankton with special emphasis on polysaccharides. Sci. Tot. Environ. 165, 155–164. doi: 10.1016/0048-9697(95)04549-G
Nagakubo, S., Nishino, K., Hirata, T., and Yamaguchi, A. (2002). The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J. Bacteriol. 184, 4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002
Pothier, J. F., Wisniewski-Dye, F., Weiss-Gayet, M., Moenne-Loccoz, Y., and Prigent-Combaret, C. (2007). Promoter-trap identification of wheat seed extract-induced genes in the plant-growth-promoting rhizobacterium Azospirillum brasilense Sp245. Microbiology 153, 3608–3622. doi: 10.1099/mic.0.2007/009381-0
Ray, B. (2006). Polysaccharides from Enteromorpha compressa: isolation, purification and structural features. Carbohyd. Polymer. 66, 408–416. doi: 10.1016/j.carbpol.2006.03.027
Rediers, H., Bonnecarrère, V., Rainey, P. B., Hamonts, K., Vanderleyden, J., and De Mot, R. (2003). Development and application of a dapB-based in vivo expression technology system to study colonization of rice by the endophytic nitrogen-fixing bacterium Pseudomonas stutzeri A15. Appl. Environ. Microbiol. 69, 6864–6874. doi: 10.1128/AEM.69.11.2003
Romanenko, L. A., Uchino, M., Falsen, E., Lysenko, A. M., Zhukova, N. V., and Mikhailov, V. V. (2005). Pseudomonas xanthomarina sp. nov., a novel bacterium isolated from marine ascidian. J. Gen. Appl. Microbiol. 51, 65–71. doi: 10.2323/jgam.51.65
Rütting, T., Huygens, D., Staelens, J., Muller, C., and Boeckx, P. (2011). Advances in 15N-tracing experiments: new labelling and data analysis approaches. Biochem. Soc. Trans. 39, 279–283. doi: 10.1042/BST0390279
Sapp, M., Schwaderer, A. S., Wiltshire, K. H., Hoppe, H. G., Gerdts, G., and Wichels, A. (2007). Species-specific bacterial communities in the phycosphere of microalgae? Microbial. Ecol. 53, 683–699. doi: 10.1007/s00248-006-9162-5
Shen, X., Hu, H., Peng, H., Wang, W., and Zhang, X. (2013). Comparative genomic analysis of four representative plant growth-promoting rhizobacteria in Pseudomonas. BMC Genomics 14:271. doi: 10.1186/1471-2164-14-271
Silver, W. L., Herman, D. J., and Firestone, M. K. (2001). Dissimilatory nitrate reduction to ammonium in upland tropical forest soils. Ecology 82, 2410–2416. doi: 10.1890/0012-9658(2001)082[2410:DNRTAI]2.0.CO;2
Simm, R., Morr, M., Kader, A., Nimtz, M., and Römling, U. (2004). GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 53, 1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x
Singh, R. P., and Reddy, C. R. (2014). Seaweed-microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88, 213–230. doi: 10.1111/1574-6941.12297
Singh, R. P., and Reddy, C. R. (2015). Unraveling the functions of the macroalgal microbiome. Fron. Microbiol. 6:1488. doi: 10.3389/fmicb.2015.01488
Sison-Mangus, M. P., Jiang, S., Tran, K. N., and Kudela, R. M. (2014). Host-specific adaptation governs the interaction of the marine diatom, Pseudo-nitzschia and their microbiota. ISME J. 8, 63–76. doi: 10.1038/ismej.2013.138
Smith, B. A., Dougherty, K. M., and Baltrus, D. A. (2014). Complete genome sequence of the highly transformable Pseudomonas stutzeri strain 28a24. Genome Announc. 2:e00543-14. doi: 10.1128/genomeA.00543-14
Taw, M. N., Lee, H. I., Lee, S. H., and Chang, W. S. (2015). Characterization of MocR, a GntR-like transcriptional regulator, in Bradyrhizobium japonicum: its impact on motility, biofilm formation, and soybean nodulation. J. Microbiol. 53, 518–525. doi: 10.1007/s12275-015-5313-z
Viaroli, P., Bartoli, M., Azzoni, R., Giordani, G., Mucchino, C., Naldi, M., et al. (2005). Nutrient and iron limitation to Ulva blooms in a eutrophic coastal lagoon (Sacca di Goro, Italy). Hydrobiologia 550, 57–71. doi: 10.1007/s10750-005-4363-3
Weinberger, F., Beltran, J., Correa, J. A., Lion, U., Pohnert, G., Kumar, N., et al. (2007). Spore release in Acrochaetium sp. (Rhodophyta) is bacterially controlled. J. Phycol. 43, 235–241. doi: 10.1111/j.1529-8817.2007.00329.x
Wolterink, A. F. W. M., Jonker, A. B., Kengen, S. W. M., and Stams, A. J. M. (2002). Pseudomonas chloritidismutans sp. nov., a non-denitrifying, chlorate-reducing bacterium. Int. J. Syst. Evol. Microbiol. 52, 2183–2190. doi: 10.1099/ijs.0.02102-0
Wu, C., Jiang, P., Zhao, J., and Fu, H. (accepted). High efficiency of protoplast preparation for artifically cultured Ulva prolifera (Ulvophyceae, Chlorophyta). Chin. J. Oceanol. Limnol.
Wu, S., Zheng, R., Sha, Z., and Sun, C. (2017). Genome sequence of Pseudomonas stutzeri 273 and identification of the exopolysaccharide EPS273 biosynthesis locus. Mar. Drugs 15:218. doi: 10.3390/md15070218
Yamamoto, S., Kasai, H., Arnold, D. L., Jackson, R. W., Vivian, A., and Harayama, S. (2000). Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 146, 2385–2394. doi: 10.1099/00221287-146-10-2385
Yan, Y., Yang, J., Dou, Y., Chen, M., Ping, S., Peng, J., et al. (2008). Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proc. Natl. Acad. Sci. U.S.A. 105, 7564–7569. doi: 10.1073/pnas.0801093105
Yoshida, T., Phadtare, S., and Inouye, M. (2007). Functional and structural characterization of EnvZ, an osmosensing histidine kinase of E. coli. Method Enzymol. 423, 184–202. doi: 10.1016/S0076-6879(07)23008-3
You, C., Lin, M., Fang, X., and Song, W. (1995). Attachment of Alcaligenes to rice roots. Soil. Biol. Biochem. 27, 463–466.
Yu, H., Yuan, M., Lu, W., Yang, J., Dai, S., Li, Q., et al. (2011). Complete genome sequence of the nitrogen-fixing and rhizosphere-associated bacterium Pseudomonas stutzeri strain DSM4166. J. Bacteriol. 193, 3422–3423. doi: 10.1128/JB.05039-11
Zhang, C., Lu, J., Wu, J., and Luo, Y. (2017). Removal of phenanthrene from coastal waters by green tide algae Ulva prolifera. Sci. Total Environ. 609, 1322–1328. doi: 10.1016/j.scitotenv.2017.07.187
Keywords: Pseudomonas, genome sequence, Comparative genomics, Ulva prolifera, algae-associated bacteria
Citation: Fu H, Jiang P, Zhao J and Wu C (2018) Comparative Genomics of Pseudomonas sp. Strain SI-3 Associated With Macroalga Ulva prolifera, the Causative Species for Green Tide in the Yellow Sea. Front. Microbiol. 9:1458. doi: 10.3389/fmicb.2018.01458
Received: 11 February 2018; Accepted: 12 June 2018;
Published: 02 July 2018.
Edited by:
Raquel Peixoto, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Rui Zhang, Xiamen University, ChinaCopyright © 2018 Fu, Jiang, Zhao and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Jiang, amlhbmdwZW5nQHFkaW8uYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.