 Jednipit Borthong1
Jednipit Borthong1 Ryosuke Omori1,2
Ryosuke Omori1,2 Chihiro Sugimoto3,4
Chihiro Sugimoto3,4 Orasa Suthienkul5
Orasa Suthienkul5 Ryo Nakao6†
Ryo Nakao6† Kimihito Ito1,4,5*†
Kimihito Ito1,4,5*†- 1Division of Bioinformatics, Research Center for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 2Precursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Kawaguchi, Japan
- 3Division of Collaboration and Education, Research Center for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 4Global Institute for Collaborative Research and Education, Hokkaido University, Sapporo, Japan
- 5Faculty of Public Health, Thammasat University, Rangsit Campus, Pathumthani, Thailand
- 6Laboratory of Parasitology, Graduate School of Veterinary Medicine, Hokkaido University, Sapporo, Japan
Metagenomic analysis has become a powerful tool to analyze bacterial communities in environmental samples. However, the detection of a specific bacterial species using metagenomic analysis remains difficult due to false positive detections of sequences shared between different bacterial species. In this study, 16S rRNA amplicon and shotgun metagenomic analyses were conducted on samples collected along a stream and ponds in the campus of Hokkaido University. We compared different database search methods for bacterial detection by focusing on Legionella pneumophila. In this study, we used L. pneumophila-specific nested PCR as a gold standard to evaluate the results of the metagenomic analysis. Comparison with the results from L. pneumophila-specific nested PCR indicated that a blastn search of shotgun reads against the NCBI-NT database led to false positive results and had problems with specificity. We also found that a blastn search of shotgun reads against a database of the catalase-peroxidase (katB) gene detected L. pneumophila with the highest area under the receiver operating characteristic curve among the tested search methods; indicating that a blastn search against the katB gene database had better diagnostic ability than searches against other databases. Our results suggest that sequence searches targeting long genes specifically associated with the bacterial species of interest is a prerequisite to detecting the bacterial species in environmental samples using metagenomic analyses.
Introduction
Metagenomic analysis has become a powerful tool for analyzing bacterial communities in environmental samples. In metagenomic analyses, genetic materials in samples are analyzed directly by next generation sequencing (NGS) (Thomas et al., 2012). In contrast to single gene amplification techniques such as PCR-based assays, metagenomic analysis can detect genomic fragments of thousands of bacteria in a single NGS run (Caporaso et al., 2012). Metagenomic approaches have been used to investigate the bacterial population structure in a variety of samples, including environmental (Daniel, 2005; Sogin et al., 2006; Breitbart et al., 2009), food (Ercolini, 2013), and clinical samples (Cho and Blaser, 2012). The price of NGS platforms and their running costs are decreasing (Lecuit and Eloit, 2014; Muir et al., 2016), increasing the opportunity for application in metagenomic analysis (Garrido-Cardenas and Manzano-Agugliaro, 2017).
Several studies have evaluated the diagnostic potential of metagenomic analysis in clinical settings. Nakamura et al. detected genomic fragments of Campylobacter jejuni from the fecal sample of a diarrheal patient using metagenomic analysis (Nakamura et al., 2008). During an outbreak of acute respiratory distress syndrome in Germany in 2013, Fischer et al. (2014) used metagenomic analysis on patient bronchoalveolar lavage samples to confirm that Chlamydia psittaci was the causative agent of the outbreak. Ortiz-Alcantara et al. (2016) conducted metagenomics analysis using cerebrospinal fluid of a pediatric patient with meningitis to identify the causative agent as Psychrobacter sp. Kujiraoka et al. (2017) showed that metagenomic analysis was useful for rapid bacterial diagnosis of acute cholecystitis. These studies suggest that metagenomic analysis can be used for the diagnosis of infectious diseases when routine methods fail to detect pathogens.
Early detection of potential pathogens in the environment is one of the most important strategies to prevent waterborne and foodborne infectious diseases (Pandey et al., 2014). There are two major approaches in pathogen detection with metagenomic analysis. The first approach, 16S rRNA metagenomic analysis, uses conserved and variable regions in the bacterial 16S rRNA gene to study the taxonomy of bacteria in samples (Janda and Abbott, 2007). 16S rRNA metagenomic analysis has been used to detect pathogens in water and food in numerous studies. Ibekwe et al. detected potential pathogens from the genera Aeromonas, Clostridium, Bacillus, Pseudomonas, and Treponema in water samples collected from the Middle Santa Ana River (Ibekwe et al., 2013). Ye and Zhang detected pathogens from wastewater treatment plants in China, United States, Canada, and Singapore; finding all samples contaminated with Aeromonas and Clostridium (Ye and Zhang, 2011). Mukherjee et al. (2016) investigated the bacterial diversity in water supplies from rural areas in Haiti and found human pathogens such as Aeromonas, Bacillus, Clostridium, and Yersinia in a high proportion of bacterial communities. Several studies applied 16S rRNA analysis to check pathogen contamination in drinking water (Shi et al., 2013; Huang et al., 2014; Pinto et al., 2016; Oh et al., 2018) and vegetables (Leonard et al., 2015; Kim et al., 2018). 16S rRNA metagenomic analysis uses PCR, and the results are affected by this amplification step. Problems due to differences in the copy number of 16S rRNA gene in a genome of bacteria (Vetrovsky and Baldrian, 2013) and chimeric sequences in PCR products (Haas et al., 2011) may arise. No single hypervariable region can be used to differentiate between bacteria (Chakravorty et al., 2007), and closely related bacteria cannot be differentiated (Weinstock, 2012).
The second approach is shotgun metagenomic analysis. By using random primers, DNA fragments can be captured from any part of the bacterial genome (Sharpton, 2014). Since the bacterial genome contains sequences specific to a bacterial species, there is a possibility to increase the specificity of pathogen detection. Several studies have applied whole genome metagenomics to the detection of potential pathogens in the environment. Lu et al. (2015) compared bacterial populations in water before and after processing in a sewage treatment system, and they found that most pathogenic bacteria were eliminated after the treatment. Nordahl Petersen et al. (2015) investigated toilet waste from airplanes using metagenomics and detected Salmonella enterica and Clostridium difficile from the waste after international flights. Several other studies have used whole genome metagenomics to investigate pathogenic bacteria in water samples collected from wastewater treatment (Cai and Zhang, 2013; Ibarbalz et al., 2016), drinking water and drink water systems (Gomez-Alvarez et al., 2012; Chao et al., 2013; Otten et al., 2016), and freshwater (Van Rossum et al., 2015; Mohiuddin et al., 2017). Whole genome metagenomics are used for food safety (Walsh et al., 2017) and investigation of the food production chain (Yang et al., 2016). These studies show the potential usefulness of metagenomic analyses in detecting pathogenic bacteria in environmental samples. Shotgun metagenomics can get narrower sequence coverage than 16S rRNA analysis (Angiuoli et al., 2011). The bacterial diversities analyzed by shotgun metagenomics depend on the method of DNA extraction and/or sequencing protocol (Morgan et al., 2010) and can also capture the host’s genetic material (Kuczynski et al., 2011).
Taxonomic classification is a bioinformatics procedure to infer the population structure of microorganisms based on genomic information obtained from samples, and several computational methods have been developed so far (Lindgreen et al., 2016). The lowest common ancestor (LCA) algorithm implemented in MEGAN assigns sequence reads to taxa on taxonomical trees based on blastn search results of reads against given databases (Huson et al., 2007). Kraken (Wood and Salzberg, 2014), CLARK (Ounit et al., 2015), and One Codex (Minot et al., 2015) use the differences in k-mer distributions among taxa to assign reads to nodes in the taxonomic tree. MetaPhlAn2 uses pre-defined sets of clade-specific marker sequences and classifies reads using reference mapping onto marker sequences (Truong et al., 2015). MGmapper uses alignment scores from reference mappings of reads to reference sequences in a database (Petersen et al., 2017). RDP (Cole et al., 2005) and SILVA (Quast et al., 2013) are specialized to analyze 16S rRNA amplicon reads and determine the taxa of reads according to sequence similarity of the 16S rRNA genes.
Despite recent advancements in sequencing technologies and classification algorithms, several studies using metagenomic analyses have exposed important issues associated with sensitivity and specificity. Loman et al. (2013) reported false negative detections of Shiga-Toxigenic Escherichia coli O104:H4 in the diagnosis of diarrheal patients using metagenomic analysis. Several groups found that bacterial populations identified by 16S rRNA metagenomics and those by shotgun metagenomics were not always consistent with one another (Shah et al., 2011; Clooney et al., 2016). These results suggest that sensitivity and/or specificity of the two methods are different depending on the bacterial species. It is also known that metagenomic analyses generate different results depending on the taxonomical classification algorithms (Clooney et al., 2016) and reference databases (Miller et al., 2013) used.
Legionella pneumophila is the causative agent of Legionnaire’s diseases. This pathogenic bacterium is ubiquitous in natural aquatic environments such as ponds, lakes, rivers, and estuaries (Fliermans et al., 1981). L. pneumophila can be also found in man-made water reservoirs, such as cooling towers (Turetgen et al., 2005), spas (Benkel et al., 2000), and water distribution systems (Stout et al., 1985). Inhalation of water aerosols is the primary cause of transmission to humans, and human-to-human transmission is rare (Correia et al., 2016).
The standard methods of detecting L. pneumophila in water samples are the culture-based and PCR-based methods. The cultured-based method uses centrifugation, filtration, heat and acid treatments, selective media, and antibiotics (Atlas et al., 1995). This method can be used to enumerate the total population of L. pneumophila in samples. The nested PCR and real-time PCR are alternative assays for the detection of L. pneumophila. These PCR-based methods use the primer sequences of the genes specific to L. pneumophila. The 5S rRNA (Mahbubani et al., 1990), 16S rRNA (Cloud et al., 2000; Buchbinder et al., 2002), dotA (Yanez et al., 2005), and mip (Mahbubani et al., 1990; Catalan et al., 1994) are examples of target genes for the detection of L. pneumophila.
Several metagenomic studies detected Legionella spp. and L. pneumophila in water samples (Cai and Zhang, 2013; Delafont et al., 2013; Lu et al., 2015; Mohiuddin et al., 2017). Pereira et al. (2017) conducted 16S rRNA metagenomic analysis and detected six different Legionella spp. in freshwater samples. Peabody et al. (2017) investigated Legionella spp. in water samples from seven different places for a year. They found that L. pneumophila was the most abundant at all sampling sites (Peabody et al., 2017).
Sequence-based typing (SBT) and core genome multilocus sequence typing (cgMLST) are used for outbreak investigation of Legionnaires’ disease (Gaia et al., 2003; Moran-Gilad et al., 2015). Both methods use nucleotide sequences at seven alleles on the genome of L. pneumophila to determine sequence type (Gaia et al., 2005; Ratzow et al., 2007). Whole genome sequencing (WGS) has become a tool for differentiation among L. pneumophila (Reuter et al., 2013; Graham et al., 2014; Levesque et al., 2014; McAdam et al., 2014). WGS maps the NGS reads onto the reference sequences and analyzes single nucleotide polymorphism in the genome. The cgMLST uses more than 1,500 loci in core genes of L. pneumophila (Moran-Gilad et al., 2015; Burckhardt et al., 2016; Petzold et al., 2017).
The aim of this study is to compare different database search methods for detecting L. pneumophila in metagenomic analyses. Using water samples collected from a stream and ponds in the campus of Hokkaido University, 16S rRNA and shotgun metagenomic analyses were conducted. In this study, we used L. pneumophila-specific nested PCR as a gold standard to evaluate the results of the metagenomic analysis.
Materials and Methods
Water Samples
Ten water samples were collected in the Sapporo campus of Hokkaido University on October 16th, 2012. Eight samples were obtained from different points along the Sakushukotoni stream (HKU_A, HKU_B, HKU_C, HKU_E, HKU_F, HKU_G, HKU_H, and HKU_I), one sample was collected from Ohno Pond (HKU_D), and another sample was collected from Hyotan Pond (HKU_J) (Figure 1). Two liters of water were collected from the water surface using sterilized containers (Pope and Patel, 2008; Tekera et al., 2011; Silva et al., 2012). The samples were transferred to a laboratory of the Research Center for Zoonosis Control in Hokkaido University for further analysis.
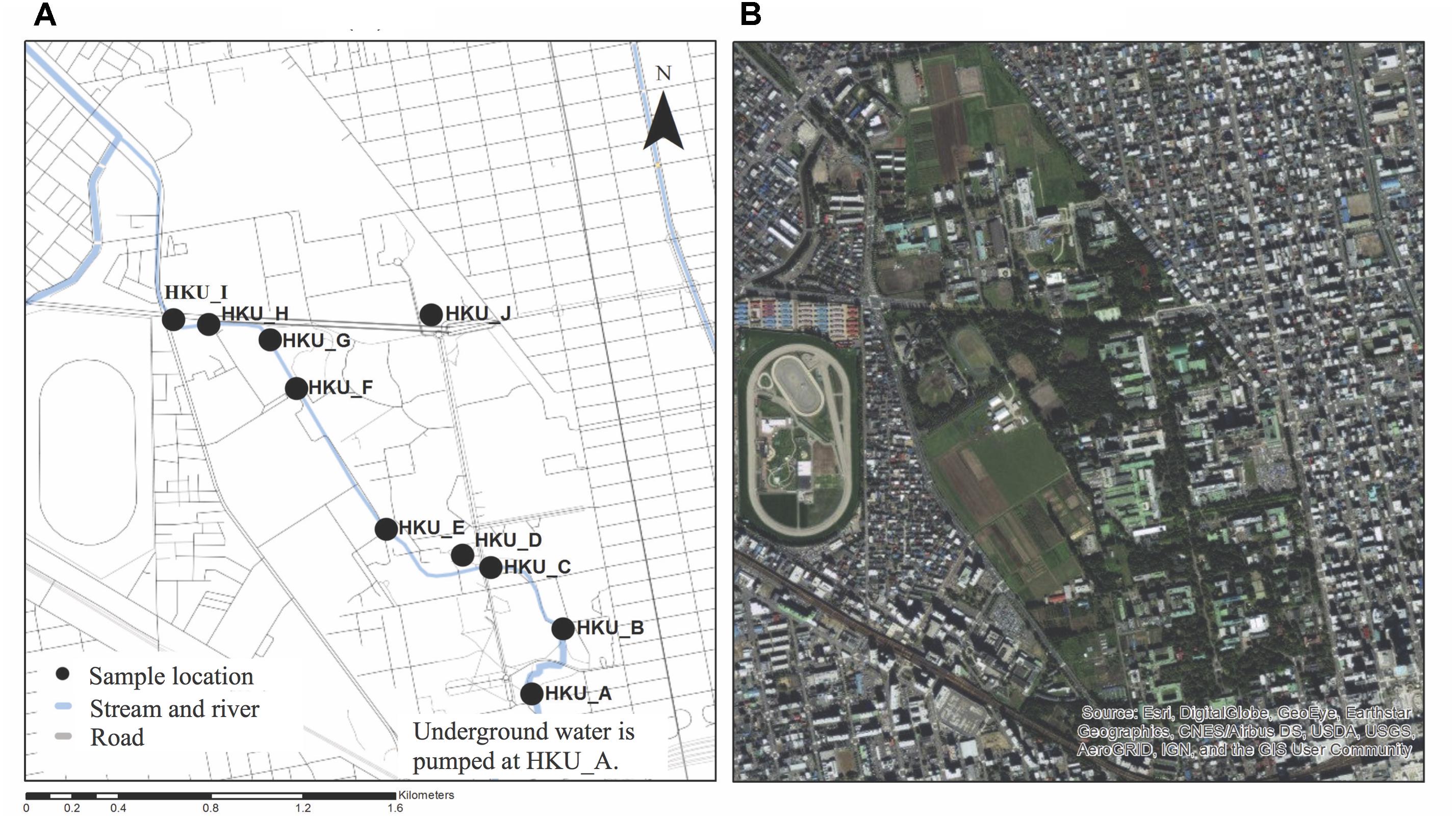
FIGURE 1. Locations of sample collection. Water samples were collected from Sakushukotoni stream (HKU_A, HKU_B, HKU_C, HKU_E, HKU_F, HKU_G, HKU_H, and HKU_I), Ohno Pond (HKU_D), and Hyotan Pond (HKU_J). (A) A map from OpenStreetmap. (B) A satellite image from Google Earth.
Bacterial Concentration and DNA Extraction
Bacteria in the water samples were concentrated using a standard membrane filtration technique with four different pore sizes; 100, 10, 5, and 0.22 μm (Millipore, Tokyo, Japan). The filtrates of 0.22 μm-membrane were used to extract DNA using a PowerWater® DNA Isolation Kit (Mo Bio Laboratories, Inc., Carlsbad, CA, United States). DNA concentration was determined using a QubitTM fluorometer (Invitrogen, Tokyo, Japan).
Detection of Legionella spp. and L. pneumophila Using Nested PCR
SBT and cgMLST are common methods to genotype L. pneumophila isolates (Gaia et al., 2003; Moran-Gilad et al., 2015). In this study, we used L. pneumophila-specific nested PCR as a gold standard to evaluate the results of the metagenomic analysis. Legionella genus-specific nested PCR was conducted amplifying 16S rRNA genes using the outer primers Leg120v and Leg1023r (Buchbinder et al., 2002) and inner primers JFP and JRP (Cloud et al., 2000). L. pneumophila-specific nested PCR was conducted amplifying macrophage infectivity potentiator surface protein (mip) genes using the outer primers Lmip920 and Limp1548 (Mahbubani et al., 1990) and inner primers Lmip976 and Lmip1427 (Catalan et al., 1994). All PCR reactions were performed using Tks Gflex DNA Polymerase (TaKaRa Bio Inc., Shiga, Japan). The amplified PCR products were analyzed using agarose gel electrophoresis and visualized with a UV transilluminator. The amplicons of mip PCR were subjected to Sanger sequencing analysis. The obtained sequences were aligned using ClustalW (Larkin et al., 2007), and p-distances among sequences were calculated by MEGA6 (Tamura et al., 2013).
Illumina Sequencing for Shotgun Metagenomic Analysis
The Illumina MiSeq platform was used for shotgun metagenomic analysis. The sequencing libraries were prepared with a Nextera XT DNA Sample Prep Kit (Illumina, San Diego, CA, United States). Libraries from each sample were tagged with multiplexing barcodes for analysis in one run. The final concentration of the purified libraries was normalized to 4 nM and the pooled libraries were sequenced with a MiSeq Reagent Kit v3 (Illumina). The resulting sequence data were made available at the DNA Data Bank of Japan (DDBJ) with an accession number of DRA006698. The barcoding sequences were removed using CLC Genomic Workbench software 8.0 (CLC bio, Tokyo, Japan). The resulting clean reads were used as shotgun reads for further analysis.
GS Junior Sequencing for 16S rRNA Amplicon Analysis
The GS Junior Titanium System (Roche, Basel, Switzerland) was used for 16S rRNA amplicon analysis. The 16S rRNA library was prepared as described in the previous study (Qiu et al., 2014). The resulting sequence data were made available at the DDBJ with an accession number of DRA006697. Barcoding sequences were removed as described above and reads shorter than 250 bp were also removed using CLC Genomic Workbench software. Potential chimera sequences were removed using Chimera.Slayer (Haas et al., 2011).
Taxonomic Classification of Reads From Shotgun Metagenomic and 16S rRNA Amplicon Analyses Using MEGAN
A blastn search (Altschul et al., 1990) and MEGAN (Huson et al., 2016) were used for taxonomic classification of the reads. For each sample, shotgun reads were aligned against the NCBI-NT database using blastn with a cut off value of 1e-04. Then, the blastn results were analyzed using the naïve LCA algorithm of MEGAN with parameters of min score = 50.0, max expected = 0.01, top percent = 10.0, min support percent = 0.001, and min support = 1. The proportions of bacterial genera (or species) were calculated using the numbers of reads classified to the genus (or species) divided by the numbers of reads classified as bacteria. Numbers of reads mapped to each bacterial genus in each sample were subjected to principal component analysis (PCA) using the prcomp command in R (R Core Team, 2016). The numbers of reads identified as L. pneumophila were collected after taxonomical classification. The reads generated from the 454 GS Junior Titanium System were aligned against the NCBI-16SMicrobial-NT database using blastn with a cut off value of 1e-04. The taxonomic classification and downstream analysis were conducted as mentioned above.
Detection of L. pneumophila Using Kraken and CLARK
In addition to the analysis with MEGAN, we tested two k-mer-based taxonomic classification algorithms, Kraken (Wood and Salzberg, 2014) and CLARK (Ounit et al., 2015). For the Kraken analysis, the reference sequences (RefSeq) of bacteria, archaea, and viruses were downloaded from the Kraken webpage, and a standard Kraken database was constructed. Shotgun reads were aligned and classified to the bacterial taxonomy using Kraken v1.0 with default parameters. For CLARK, only the RefSeq of bacteria were obtained from the CLARK webpage, and they were used to construct a bacterial database. Shotgun reads of each sample were aligned and classified to the bacterial taxonomy using CLARK v1.2.3.2 with default parameters. In both analyses, the numbers of reads identified as L. pneumophila were collected after taxonomic classification.
Detection of L. pneumophila Using Blastn Against VFDB
Nucleotide sequences of virulence factor genes were downloaded from the Virulence Factor Gene Database (VFDB) (Chen et al., 2005). A VFDB blast database was constructed using the ‘makeblastdb’ command in the blast package. Shotgun reads were aligned against the database using blastn with a cut off value of 1e-04. Blastn results with multiple hits from the same query to different regions of the same reference sequence were removed, except one. The proportions of L. pneumophila hits were calculated by dividing the number of reads classified to L. pneumophila by the number of reads classified to bacteria.
Detection of L. pneumophila Using Blastn Against mip Gene
A nucleotide sequence of the mip gene from L. pneumophila subsp. philadelphia str. Philadelphia 1 (NC_002942.5) was download from NCBI, and a mip blast database was constructed using this sequence. Shotgun reads from each sample were aligned to this database using a blastn search with a cut off value of 1e-04, and the numbers of hit reads were collected.
Detection of L. pneumophila Using Blastn Against a Custom VFDB
Based on the results of a blastn search of shotgun reads against a VFDB blast database, virulence factor genes (n = 9) associated with L. pneumophila were identified. For each virulence factor gene, its protein sequences of L. pneumophila subsp. philadelphia str. Philadelphia 1 were downloaded from NCBI. These protein sequences are CcmC (YP_094893.1), CcmF (YP_094896.1), DotA (YP_096691.1), IcmO (YP_094490.1), KatB (YP_096397.1), LvhB10 (YP_095278.1), PilT (YP_096029.1), GTP pyrophosphokinase (YP_095486.1), and superoxide dismutase (YP_096960.1). Nucleotide sequences encoding these nine proteins were collected using the tblastn search at NCBI (4,267, 4,526, 483, 707, 2,686, 5,506, 5,000, 5,000, and 5,000 sequences were obtained for ccmC, ccmF, dotA, icmO, lvhB10, katB, pilT, relA, and sodB, respectively) and a custom VFDB blast database was constructed. A blastn search of shotgun reads against the custom VFDB was performed, and the numbers of reads identified as L. pneumophila were obtained using the naïve LCA algorithm in MEGAN.
Comparison of Database Search Methods for L. pneumophila Detection
The area under the curve (AUC) of receiver operating characteristic curve (ROC) was used to compare the results of different database search methods in the detection of L. pneumophila. We considered that the results of L. pneumophila-specific nested PCR were correct. The true positive rate and false positive rate (1 – specificity) of each database search method were calculated, and area under curves were determined using the AUC package (Ballings and Van den Poel, 2013) in R.
Results
Detection of Legionella spp. and L. pneumophila Using Nested PCR
Legionella spp. was detected in all samples by Legionella genus-specific nested PCR (Figure 2A). The amplification of the mip gene by L. pneumophila-specific nested PCR was observed in only three samples; HKU_G, HKU_H, and HKU_I (Figure 2B). These results suggested that 7 samples, except for HKU_G, HKU_H, and HKU_I, contained Legionella spp. not classified as L. pneumophila. The pairwise distances among the mip gene sequences from amplified samples and positive control were within a range of 0.018 – 0.030, indicating that there was no cross contamination from the positive control during the PCR process. Therefore, we concluded that the samples HKU_G, HKU_H, and HKU_I were contaminated with L. pneumophila.
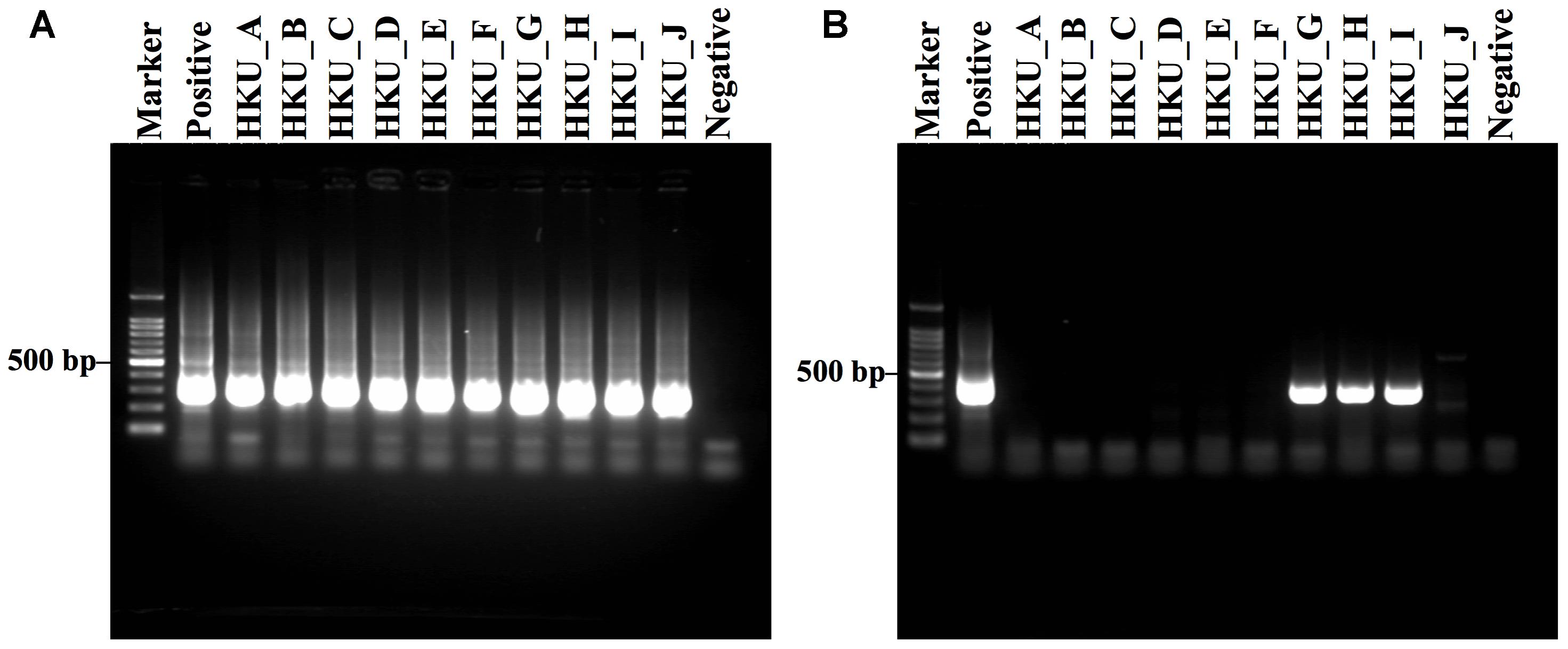
FIGURE 2. Gel electrophoresis of DNA amplification by Legionella genus-specific and Legionella pneumophila-specific nested PCRs. (A) Amplification results of Legionella genus-specific and (B) L. pneumophila-specific nested PCRs are shown. Lane Marker: 100 bp DNA marker; lane Positive: L. pneumophila; lanes HKU_A – HKU_C and HKU_E – HKU_I: DNA from water samples of Sakushokotoni stream; lane HKU_D: DNA from water samples of Ohno Pond; lane HKU_J: DNA from water samples of Hyotan Pond; and lane Negative: distilled water (no DNA).
Next Generation Sequencing
Next generation sequencing was conducted using Illumina MiSeq and GS Junior Titanium System, from which a total of 51,162,136 and 353,913 reads were obtained, respectively (Table 1). The average lengths of bacterial reads obtained from 16S rRNA amplicon analysis were within a range of 453.3 – 473.4 bp, whereas average lengths of bacterial reads obtained from Miseq were within a range of 288.8 – 293.1 bp.
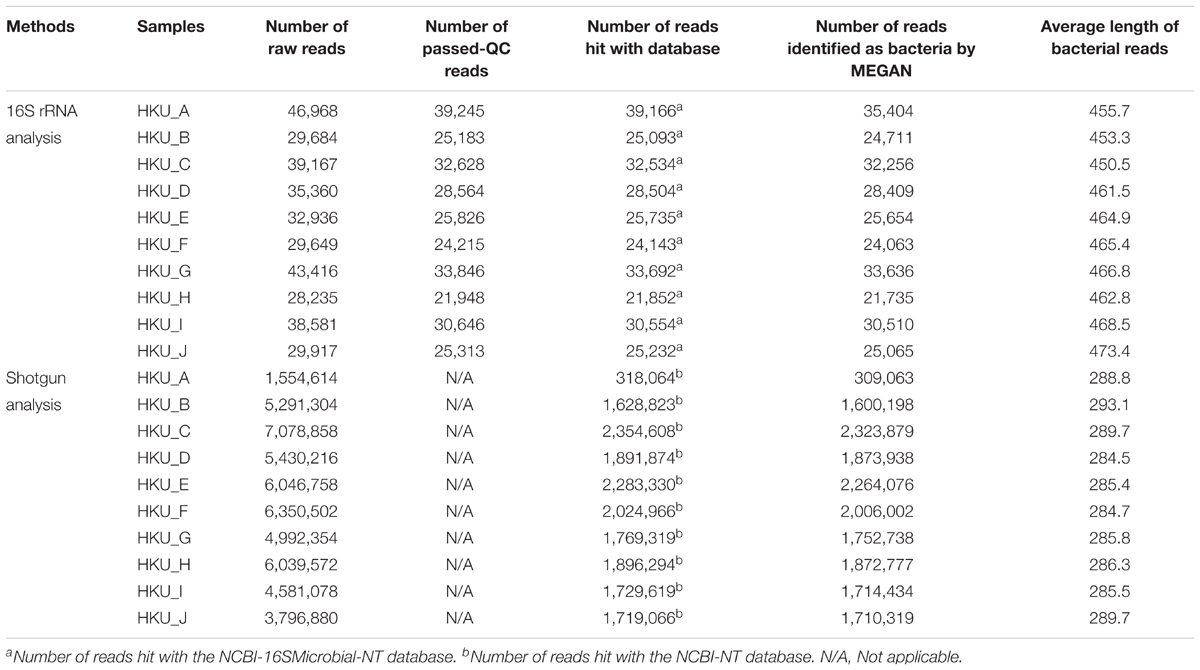
TABLE 1. Summary of next generation sequencing reads.
Bacterial Communities Inferred From 16S rRNA Amplicon and Shotgun Metagenomic Analyses
16S rRNA amplicon and shotgun sequence reads were subjected to a blastn search against NCBI-16SMicrobial-NT and NCBI-NT databases, respectively. The proportions of bacterial genera inferred using the naïve LCA algorithm of MEGAN are shown in Figures 3A,B for 16S rRNA amplicon and shotgun reads, respectively. More than 75% of reads generated by GS Junior Titanium System were identified as having bacterial origins, whereas 19.9 – 45.0% of Illumina reads were identified as having bacterial origins. A total of 977 bacterial genera were detected from 16S rRNA amplicon analysis, while a total of 897 bacterial genera were found in shotgun metagenomic analysis. The PCA suggested that the bacterial communities in samples were divided into three groups in both 16S rRNA amplicon and shotgun metagenomic analyses (Figures 3C,D).
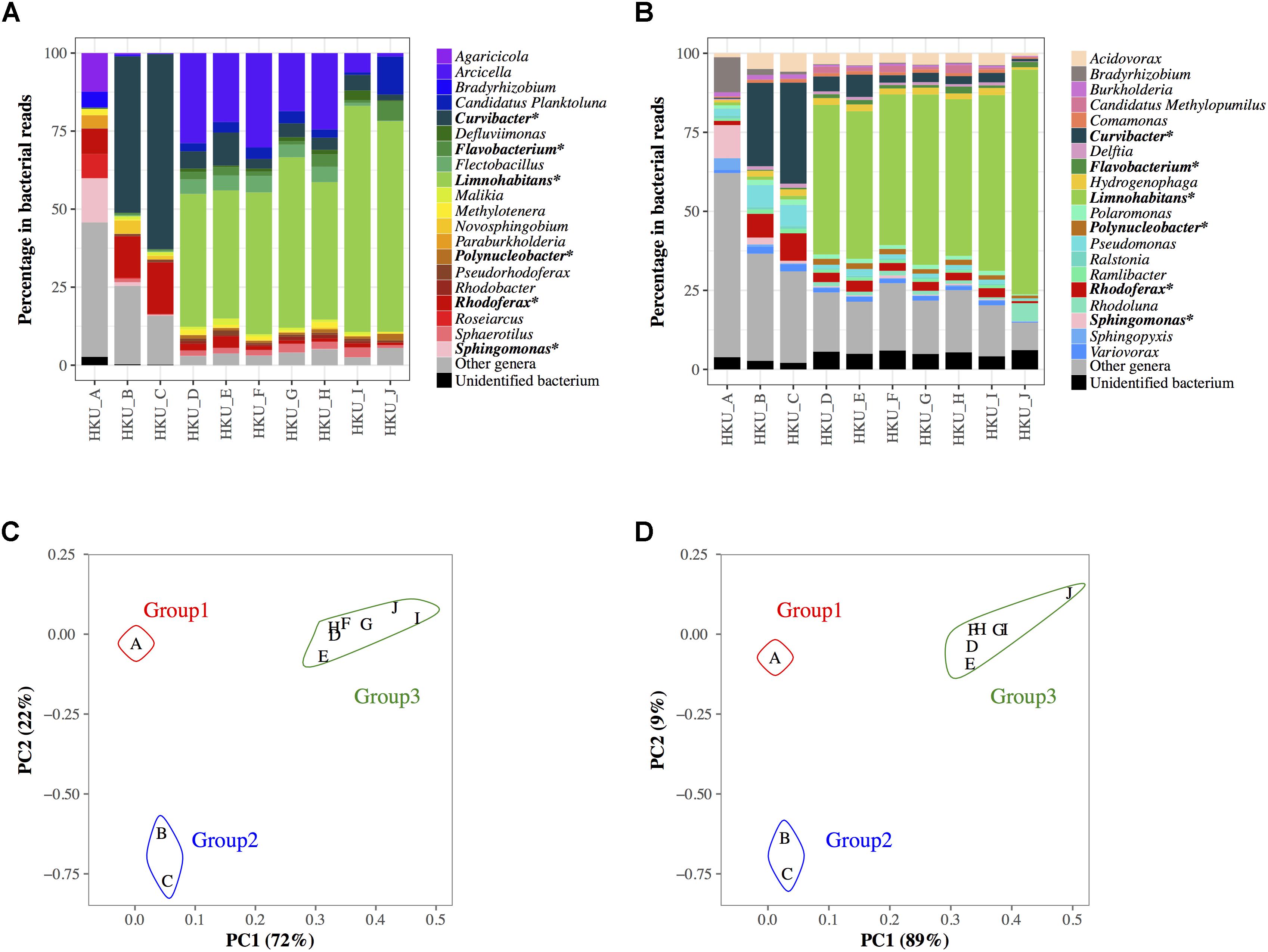
FIGURE 3. Bacterial communities at genus level in water samples determined by the lowest common ancestor algorithm in MEGAN. (A) Bacterial communities based on the results of a blastn search of 16S rRNA amplicon reads against nucleotide sequences from the NCBI-16SMicrobial-NT database. (B) Bacterial communities based on the results of the blastn search of shotgun sequencing reads against nucleotide sequences from the NCBI-NT database. Colored bars represent the top 20 abundant genera in all samples. Reads from other minor genera are represented in gray, and the reads with unidentified genera are represented in black. The genera ranked in top 20 in both 16S rRNA amplicon and shotgun metagenomic analyses are indicated with asterisks. The results of principal component analysis of the bacterial communities using 16S rRNA reads and shotgun reads are shown in (C,D), respectively.
Some genera showed similar proportions of reads between 16S rRNA amplicon and shotgun metagenomic analyses, whereas others did not. For example, more than 10% of reads were identified as Sphingomonas in both 16S rRNA amplicon and shotgun metagenomic analyses (14.1 and 10.4%, respectively) in group 1 (HKU_A). Shotgun metagenomic analysis identified Pseudomonas (6.6 – 7.0%) in group 2, but this genus was not found in the top 20 genera in the 16S rRNA amplicon analysis. The highest portion of a bacterium in group 3 was Limnohabitans in both the 16S rRNA amplicon and shotgun metagenomic analyses (41.0 – 72.4% and 47.3 – 71.1%, respectively). In contrast, the 16S rRNA amplicon analysis identified a moderate number of reads from Arcicella (1.1 – 30.2%) in group 3, but this abundant genus was not listed in the top 20 genera of the shotgun metagenomic analysis. Supplementary Table 1 shows the number of bacterial reads of species classified by MEGAN with NCBI-NT database. Supplementary Figure 1 presents potential pathogens at the species level identified by shotgun reads.
Detection of L. pneumophila Using MEGAN, Kraken, CLARK, VFDB, and mip Gene
To investigate the sensitivity and specificity of different database search methods in the detection of L. pneumophila, we compared the results of each method with that of L. pneumophila-specific nested PCR (Table 2). Although the nested PCR amplified sequence of the mip gene of L. pneumophila in three samples, blastn searches of shotgun reads could not detect any reads encoding the mip gene (Table 2 and Figure 4E). In contrast, MEGAN with NCBI-NT database, Kraken and CLARK with RefSeq database detected a moderate number of L. pneumophila sequences in all samples (Table 2). MEGAN, Kraken, and CLARK identified the highest proportion of L. pneumophila reads in HKU_A (Figures 4A–C) even though HKU_A was negative by L. pneumophila-specific nested PCR assay. On the other hand, the use of VFDB detected no L. pneumophila read in HKU_A, and a relatively higher proportion of L. pneumophila reads in HKU_G, HKU_H, and HKU_I (Figure 4D), which were positive by nested PCR (Figure 2B). VFDB hits contained 19 virulence factor genes (Supplementary Table 2). Blastn searches of detected sequences against NCBI-NT indicated that 10 virulence factor genes were derived from other bacterial species. Finally, 9 virulence factor genes (ccmC, ccmF, dotA, icmO, lvhB10, katB, pilT, relA, and sodB) were identified as L. pneumophila origin (Supplementary Table 3).
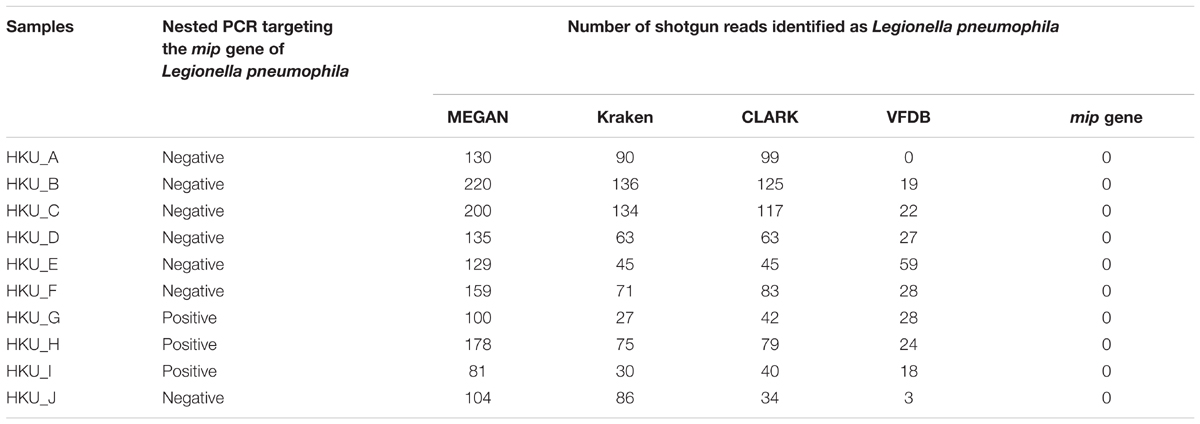
TABLE 2. Number of shotgun reads identified as Legionella pneumophila by MEGAN, Kraken, CLARK, VFDB, and mip gene.
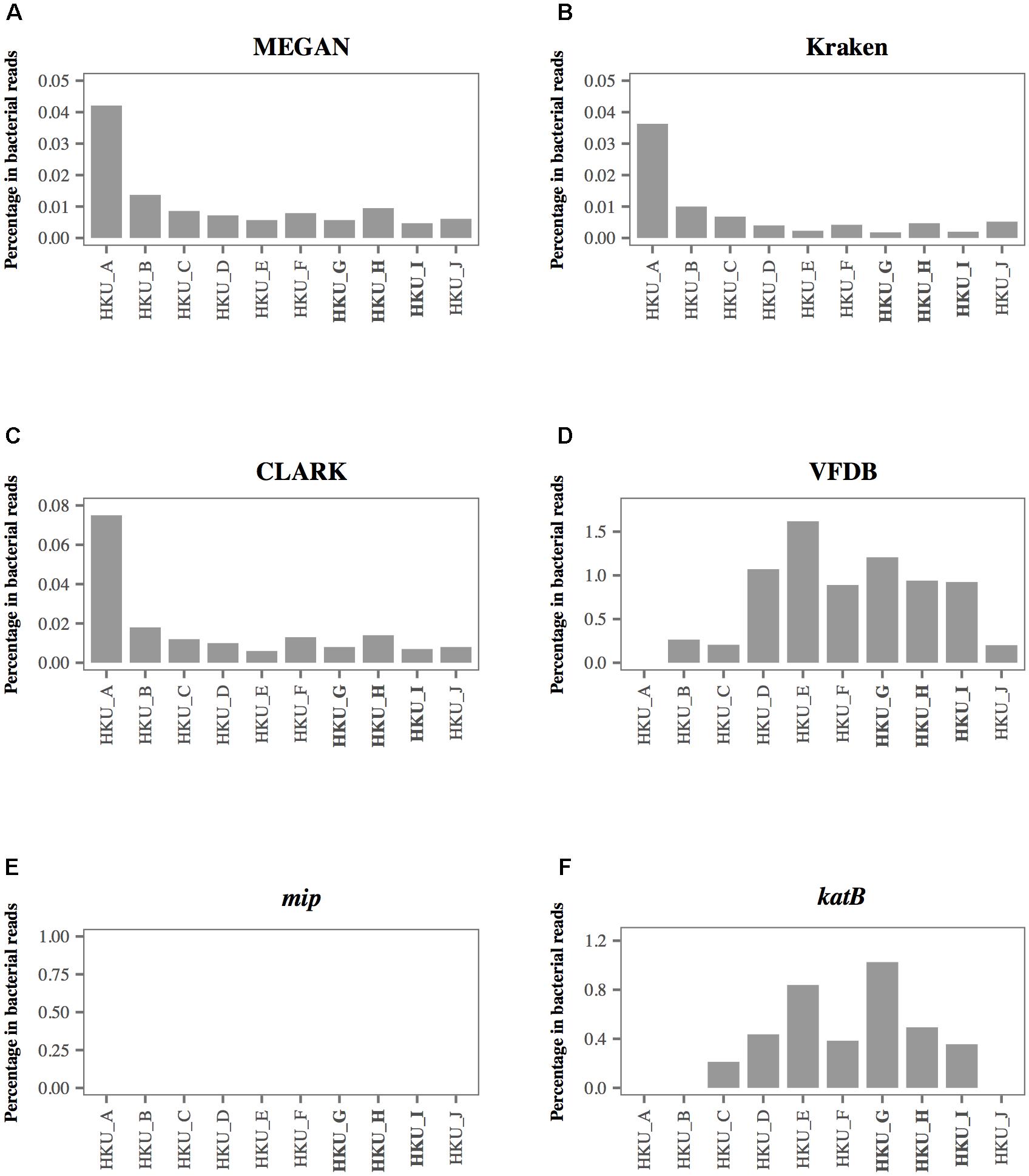
FIGURE 4. The percentage of shotgun reads identified as Legionella pneumophila using 6 different database-based search methods. Proportion of shotgun sequence reads identified as L. pneumophila by (A) MEGAN with the NCBI-NT database, (B) Kraken with RefSeq archaea, bacteria, and viruses, (C) CLARK with RefSeq archaea and bacteria, (D) VFDB, (E) mip gene, and (F) katB gene. The bold labels indicate L. pneumophila-positive samples using nested PCR.
Detection of L. pneumophila Using a Custom VFDB
We further investigated the detection ability of the method using 9 virulence factor genes detected by the VFDB as a database. For each virulence factor gene, we collected related nucleotide sequences from its protein sequence using a tblastn search and constructed a custom VFDB. Table 3 shows the number of shotgun reads identified as virulence factor genes associated with L. pneumophila. Among 9 genes we tested, the blastn search of shotgun reads against the katB gene of L. pneumophila showed the best agreement with the results of nested PCR.
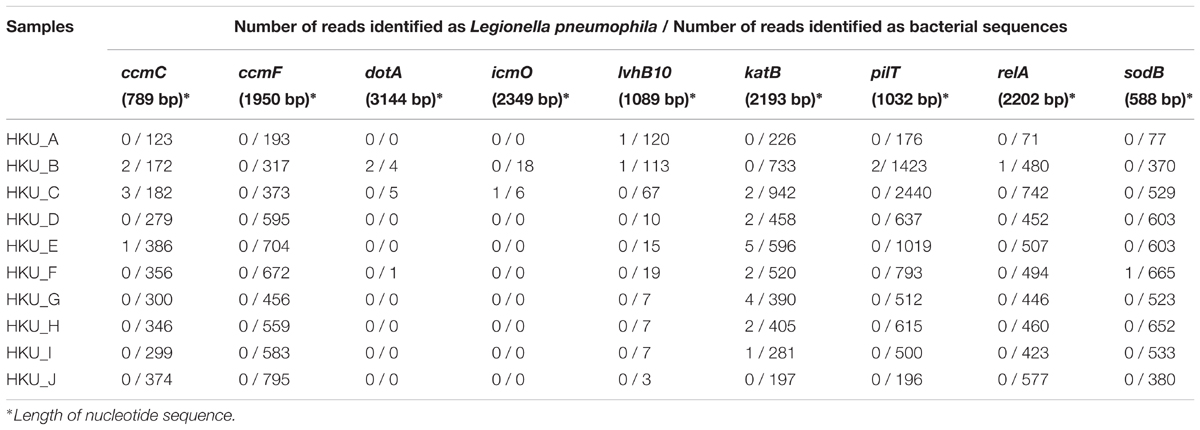
TABLE 3. Number of shotgun reads identified as Legionella pneumophila using a blastn search against custom databases of virulence factor genes associated with Legionella pneumophila.
Diagnostic Ability of L. pneumophila Using a katB Gene
Figure 4 presents the percentage of L. pneumophila-associated reads identified by 6 different database search methods. Among the 6 database search methods we tested, the blastn search against the katB gene showed the best agreement with the results of nested PCR. The highest percentage of shotgun reads identified as L. pneumophila origin was observed in HKU_G. The non-bacterial reads were classified as archaea, fungi, and metazoan reads. None of the reads identified as L. pneumophila was found in HKU_A, HKU_B, and HKU_J (Figure 4F).
The AUC of database search methods demonstrated that the detection of L. pneumophila using the katB gene had the highest AUC at 0.8095 (Figure 5D). Other database search methods such as MEGAN with NCBI-NT, Kraken and CLARK with RefSeq database had AUC values with a range between 0.2142 and 0.3095; lower than that using katB gene (Figures 5A,B). The database search method using the VFDB database had AUC value at 0.7619 (Figure 5C). These results indicate that the blastn search against the katB gene database had higher diagnostic capability than searches against databases containing whole genome sequences of L. pneumophila.
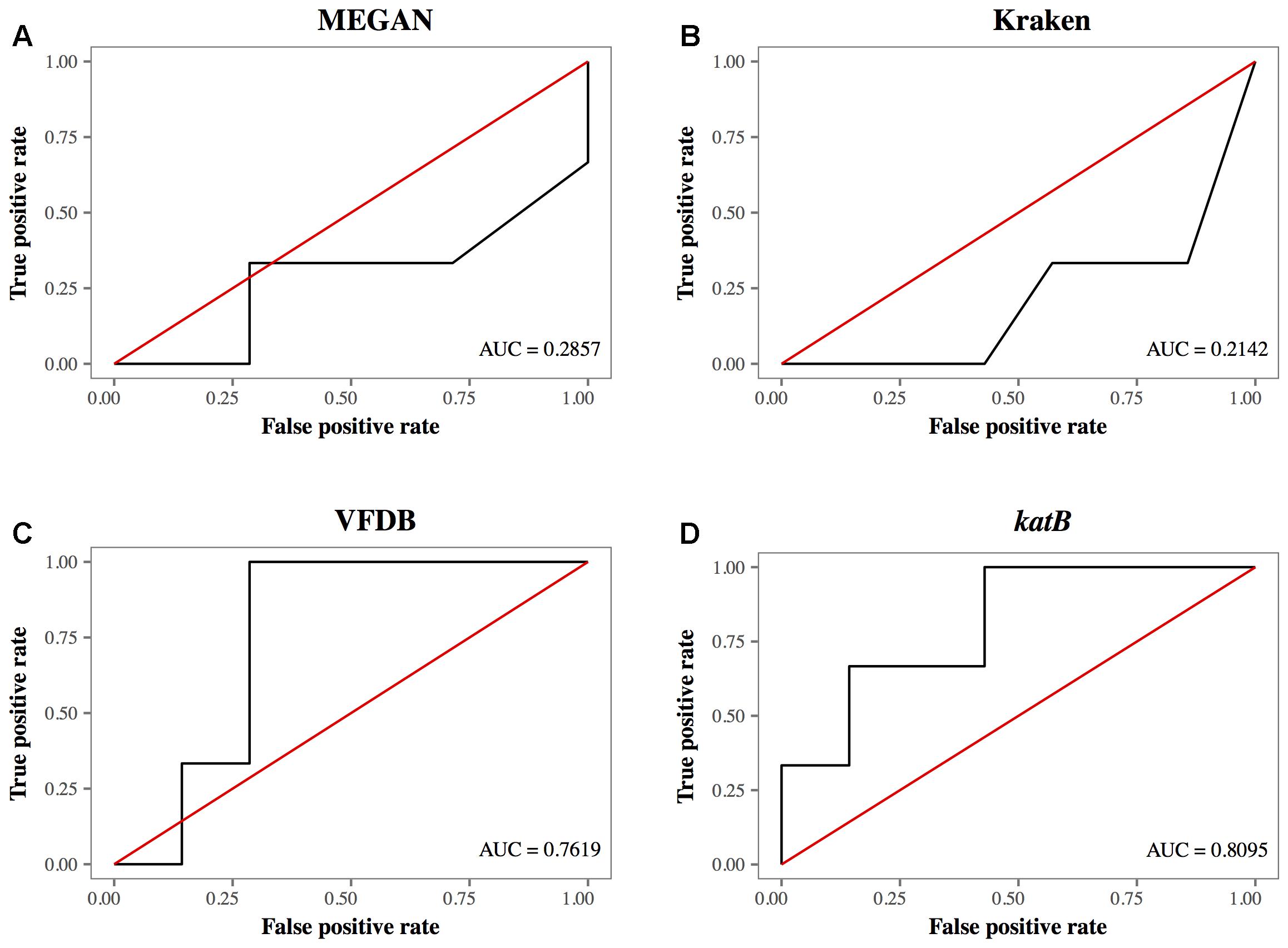
FIGURE 5. The receiver operating characteristic curves for different database search methods. (A) MEGAN with NCBI-NT database, (B) Kraken with RefSeq archaea, bacteria, and viruses, (C) VFDB, and (D) katB gene. The red line is the reference line indicating the test without diagnostic benefit, i.e., random diagnosis.
Discussion
In this study, we conducted metagenomic analyses using water samples collected from a stream and ponds in the campus of Hokkaido University. By focusing on L. pneumophila, we evaluated different database search methods in detecting a specific bacterium in water samples by validating their detection results with those of nested PCR assay. We found that a blastn search of shotgun reads against the NCBI-NT database led to false positive detection and had a potential problem in specificity. Our results indicated that the blastn search against the genes of species-specific virulence factors had better agreement with the results of L. pneumophila-specific nested PCR.
The population structures inferred by 16S rRNA amplicon analysis and those by shotgun metagenomic analysis showed different bacterial communities even at the genus level (Figures 3A,B). On the other hand, PCA using 16S rRNA amplicon and shotgun metagenomic analyses clustered the samples in a similar way (Figures 3C,D). These results indicated that both 16S rRNA amplicon and shotgun metagenomic analyses captured the similarity in population structures among samples, but sensitivity and/or specificity of the two methods were different depending on bacterial genera.
The nested PCR assay detected L. pneumophila DNA in only three out of ten water samples (Figure 2B). In contrast, MEGAN with NCBI-NT database, Kraken and CLARK with the RefSeq database detected a moderate number of L. pneumophila sequences in the shotgun reads from all samples (Table 2). Furthermore, MEGAN with NCBI-NT, Kraken, and CLARK with RefSeq database detected a larger number of L. pneumophila sequences in PCR-negative samples such as HKU_B and HKU_C than in PCR-positive samples including HKU_G, HKU_H, and HKU_I (Table 2). Since the sensitivity of nested PCR assay with the employed primer sets is known to be 10 fg or 10 CFU per ml (Nintasen et al., 2007), the inconsistency is probably attributed to false positive detections due to the low specificity of these database search methods in detecting L. pneumophila.
The NCBI-NT and RefSeq databases contain whole genome sequences of L. pneumophila. The sequences of some of the bacterial genomic regions, for example the loci encoding housekeeping genes, are conserved among closely related bacterial species. The wrong assignment of the reads from such conserved genomic loci may be a possible cause of the false positive detection with MEGAN with NCBI-NT, Kraken and CLARK with RefSeq databases. In fact, the number of reads assigned to L. pneumophila were strongly correlated with the number of reads assigned to other species in genus Legionella with a Pearson correlation coefficient of 0.98 and a p-value of 10-6 (Supplementary Figure 2). A large fraction of reads assigned to L. pneumophila in HKU_A may be attributed to wrong assignment of reads from other abundant species in genus Legionella (Figures 4A–C).
The ROC plot analysis showed that detection using the katB gene had the largest AUC, indicating that the method was the best among the database search methods we tested (Figure 5). The katB gene can be found in several bacterial species, but nucleotide sequences of katB are divergent among different bacterial species (Supplementary Figure 3). This would be the reason for the high diagnostic ability of the method using the katB gene. The mip gene is a genetic marker for detecting L. pneumophila using PCR-based assay (Cianciotto et al., 1989). However, the shotgun reads did not contain a DNA fragment of the mip gene (Table 2 and Figure 4E). The nucleotide length of the mip gene is 702 bp, while the length of a katB gene is 2,163 bp. The read depth of certain genes in shotgun metagenomic sequencing is proportional to the length of the gene. We speculate that the length of the mip gene might affect the absence of the gene in the metagenomic sequencing data. Despite dotA (3,144 bp) having more nucleotides than the katB gene, the number of reads identified as L. pneumophila using the dotA gene is smaller than that using the katB gene (Table 3). It is known that dotA determines the serogroup of L. pneumophila (Ko et al., 2003). There is a possibility that the L. pneumophila present in our samples belong to different serogroups from L. pneumophila subsp. philadelphia str. Philadelphia 1, which is the reference sequence we used for the tblastn search to collect nucleotide sequences.
The nested PCR using specific primers to amplify a mip gene detected L. pneumophila in only three samples; HKU_G, HKU_H, and HKU_I (Figure 2B). L. pneumophila can be found in natural water supplies (Mahbubani et al., 1990), and there is no report of outbreaks of L. pneumophila in the university campus. Since sampling the sites of HKU_G, HKU_H, and HKU_I are near a primeval forest conserved by the university, the pathogen has probably existed naturally and is not associated with the emergence of Legionnaires’ disease.
Although the detection of L. pneumophila using PCR-based methods is relatively rapid and sensitive, it is necessary to know the sequences of the target bacteria in advance. Conversely, a shotgun metagenomic approach does not require sequence information and thus is potentially useful in the detection of new and/or unexpected organisms. High throughput is another advantage of the metagenomic approach in that the method can detect multiple organisms in a single run. In fact, several studies have demonstrated the usefulness of metagenomic analysis in water science. Gomez-Alvarez et al. (2012) used metagenomics to investigate microbial populations in drinking water and found that Legionella like-genes were abundant in free-chlorine-treated drinking water. Metagenomic analysis showed potential risk of Mycobacterium tuberculosis-like in water samples from wastewater treatment plants (Cai and Zhang, 2013). Several studies have detected bacterial genes related to antibiotic resistance in water samples (Zhang et al., 2011; Durso et al., 2012; Wang et al., 2013). Pereira et al. (2017) proposed a novel approach to increase the sensitivity of Legionella detection in metagenomics. These studies are examples of possible directions for future application of metagenomics in detecting pathogens in water.
Our study has a limitation due to a lack of information for L. pneumophila in our water samples. The conventional method could be used to enumerate the number of L. pneumophila in a water sample. Based on the sensitivity the L. pneumophila-specific nested PCR (Nintasen et al., 2007), the number of L. pneumophila were estimated as at least 10 CFU/ml. Another limitation of this study was the number of reads generated by Miseq. Hiseq can produce a larger number of sequence reads with deeper coverage. In this sense, we might increase the sensitivity of detection of L. pneumophila by using Hiseq. At the same time, however, the length of reads from Hiseq are 100 – 150 bp, shorter than that of Miseq, which produces 300 bp. In this sense, specificity of detection might decrease if we used Hiseq. The number and the length of sequence reads are a tradeoff as well as sensitivity and specificity. These tradeoffs should be considered when conducting shotgun metagenomic analysis to detect pathogens in water samples. The one of our future work is the evaluation of detection limit of L. pneumophila in water samples using metagenomic analysis. Comparison of results among culture-based method, quantitative RT PCR, and metagenomic analysis can be used to discuss the detection limit of L. pneumophila in water samples.
In the present study, we compared the different database search methods for detecting L. pneumophila using metagenomic analyses. We used L. pneumophila-specific nested PCR as a gold standard and found that a blastn search against a katB gene database detected L. pneumophila with the highest area under the ROC among the tested search methods. Our study suggests that sequence searches targeting a long gene specifically associated with a bacterial species of interest has better diagnostic potential using current NGS technologies.
Author Contributions
JB, RN, and KI designed the study. JB, RN, and CS conducted sampling and next generation sequencing. OS provided the positive control DNA of L. pneumophila. JB analyzed the data. JB, RO, and KI designed the statistical analysis and wrote the paper.
Funding
This work was supported by the Program for Leading Graduate Schools from the Ministry of Education, Culture, Sports, Science and Technology, Japan (http://www.mext.go.jp/) and CREST and PRESTO programs from Japan Science and Technology Agency (http://www.jst.go.jp/). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01272/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Angiuoli, S. V., White, J. R., Matalka, M., White, O., and Fricke, W. F. (2011). Resources and costs for microbial sequence analysis evaluated using virtual machines and cloud computing. PLoS One 6:e26624. doi: 10.1371/journal.pone.0026624
Atlas, R. M., Williams, J. F., and Huntington, M. K. (1995). Legionella contamination of dental-unit waters. Appl. Environ. Microbiol. 61, 1208–1213.
Ballings, M., and Van den Poel, D. (2013). Threshold Independent Performance Mesures for Probabilistic Classifiers. Available at: http://cran.r-project.org/web/packages/AUC/AUC.pdf
Benkel, D. H., McClure, E. M., Woolard, D., Rullan, J. V., Miller, G. B. Jr., Jenkins, S. R., et al. (2000). Outbreak of Legionnaires’ disease associated with a display whirlpool spa. Int. J. Epidemiol. 29, 1092–1098. doi: 10.1093/ije/29.6.1092
Breitbart, M., Hoare, A., Nitti, A., Siefert, J., Haynes, M., Dinsdale, E., et al. (2009). Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Cienegas, Mexico. Environ. Microbiol. 11, 16–34. doi: 10.1111/j.1462-2920.2008.01725.x
Buchbinder, S., Trebesius, K., and Heesemann, J. (2002). Evaluation of detection of Legionella spp. in water samples by fluorescence in situ hybridization, PCR amplification and bacterial culture. Int. J. Med. Microbiol. 292, 241–245. doi: 10.1078/1438-4221-00213
Burckhardt, F., Brion, A., Lahm, J., Koch, H. U., Prior, K., Petzold, M., et al. (2016). Confirming Legionnaires’ disease outbreak by genome-based method, Germany, 2012. Emerg. Infect. Dis. 22, 1303–1304. doi: 10.3201/eid2207.151738
Cai, L., and Zhang, T. (2013). Detecting human bacterial pathogens in wastewater treatment plants by a high-throughput shotgun sequencing technique. Environ. Sci. Technol. 47, 5433–5441. doi: 10.1021/es400275r
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Catalan, V., Moreno, C., Dasi, M. A., Munoz, C., and Apraiz, D. (1994). Nested polymerase chain reaction for detection of Legionella pneumophila in water. Res. Microbiol. 145, 603–610. doi: 10.1016/0923-2508(94)90077-9
Chakravorty, S., Helb, D., Burday, M., Connell, N., and Alland, D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339. doi: 10.1016/j.mimet.2007.02.005
Chao, Y., Ma, L., Yang, Y., Ju, F., Zhang, X. X., Wu, W. M., et al. (2013). Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment. Sci. Rep. 3:3550. doi: 10.1038/srep03550
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. doi: 10.1038/nrg3182
Cianciotto, N. P., Eisenstein, B. I., Mody, C. H., Toews, G. B., and Engleberg, N. C. (1989). A Legionella pneumophila gene encoding a species-specific surface protein potentiates initiation of intracellular infection. Infect. Immun. 57, 1255–1262.
Clooney, A. G., Fouhy, F., Sleator, R. D., O’ Driscoll, A., Stanton, C., Cotter, P. D., et al. (2016). Comparing apples and oranges?: next generation sequencing and its impact on microbiome analysis. PLoS One 11:e0148028. doi: 10.1371/journal.pone.0148028
Cloud, J. L., Carroll, K. C., Pixton, P., Erali, M., and Hillyard, D. R. (2000). Detection of Legionella species in respiratory specimens using PCR with sequencing confirmation. J. Clin. Microbiol. 38, 1709–1712.
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam, S. A., Mcgarrell, D. M., et al. (2005). The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33, D294–D296. doi: 10.1093/nar/gki038
Correia, A. M., Ferreira, J. S., Borges, V., Nunes, A., Gomes, B., Capucho, R., et al. (2016). Probable person-to-person transmission of Legionnaires’ disease. N. Engl. J. Med. 374, 497–498. doi: 10.1056/NEJMc1505356
Daniel, R. (2005). The metagenomics of soil. Nat. Rev. Microbiol. 3, 470–478. doi: 10.1038/nrmicro1160
Delafont, V., Brouke, A., Bouchon, D., Moulin, L., and Hechard, Y. (2013). Microbiome of free-living amoebae isolated from drinking water. Water Res. 47, 6958–6965. doi: 10.1016/j.watres.2013.07.047
Durso, L. M., Miller, D. N., and Wienhold, B. J. (2012). Distribution and quantification of antibiotic resistant genes and bacteria across agricultural and non-agricultural metagenomes. PLoS One 7:e48325. doi: 10.1371/journal.pone.0048325
Ercolini, D. (2013). High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 79, 3148–3155. doi: 10.1128/AEM.00256-13
Fischer, N., Rohde, H., Indenbirken, D., Gunther, T., Reumann, K., Lutgehetmann, M., et al. (2014). Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia. Emerg. Infect. Dis. 20, 1072–1075. doi: 10.3201/eid2006.131526
Fliermans, C. B., Cherry, W. B., Orrison, L. H., Smith, S. J., Tison, D. L., and Pope, D. H. (1981). Ecological distribution of Legionella pneumophila. Appl. Environ. Microbiol. 41, 9–16.
Gaia, V., Fry, N. K., Afshar, B., Luck, P. C., Meugnier, H., Etienne, J., et al. (2005). Consensus sequence-based scheme for epidemiological typing of clinical and environmental isolates of Legionella pneumophila. J. Clin. Microbiol. 43, 2047–2052. doi: 10.1128/JCM.43.5.2047-2052.2005
Gaia, V., Fry, N. K., Harrison, T. G., and Peduzzi, R. (2003). Sequence-based typing of Legionella pneumophila serogroup 1 offers the potential for true portability in legionellosis outbreak investigation. J. Clin. Microbiol. 41, 2932–2939. doi: 10.1128/JCM.41.7.2932-2939.2003
Garrido-Cardenas, J. A., and Manzano-Agugliaro, F. (2017). The metagenomics worldwide research. Curr. Genet. 63, 819–829. doi: 10.1007/s00294-017-0693-8
Gomez-Alvarez, V., Revetta, R. P., and Santo Domingo, J. W. (2012). Metagenomic analyses of drinking water receiving different disinfection treatments. Appl. Environ. Microbiol. 78, 6095–6102. doi: 10.1128/AEM.01018-12
Graham, R. M., Doyle, C. J., and Jennison, A. V. (2014). Real-time investigation of a Legionella pneumophila outbreak using whole genome sequencing. Epidemiol. Infect. 142, 2347–2351. doi: 10.1017/S0950268814000375
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Huang, K., Zhang, X. X., Shi, P., Wu, B., and Ren, H. (2014). A comprehensive insight into bacterial virulence in drinking water using 454 pyrosequencing and Illumina high-throughput sequencing. Ecotoxicol. Environ. Saf. 109, 15–21. doi: 10.1016/j.ecoenv.2014.07.029
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Huson, D. H., Beier, S., Flade, I., Gorska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition - Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Ibarbalz, F. M., Orellana, E., Figuerola, E. L., and Erijman, L. (2016). Shotgun metagenomic profiles have a high capacity to discriminate samples of activated sludge according to wastewater type. Appl. Environ. Microbiol. 82, 5186–5196. doi: 10.1128/AEM.00916-16
Ibekwe, A. M., Leddy, M., and Murinda, S. E. (2013). Potential human pathogenic bacteria in a mixed urban watershed as revealed by pyrosequencing. PLoS One 8:e79490. doi: 10.1371/journal.pone.0079490
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Kim, D., Hong, S., Kim, Y. T., Ryu, S., Kim, H. B., and Lee, J. H. (2018). Metagenomic approach to identifying foodborne pathogens on Chinese cabbage. J. Microbiol. Biotechnol. 28, 227–235. doi: 10.4014/jmb.1710.10021
Ko, K. S., Hong, S. K., Lee, H. K., Park, M. Y., and Kook, Y. H. (2003). Molecular evolution of the dotA gene in Legionella pneumophila. J. Bacteriol. 185, 6269–6277. doi: 10.1128/JB.185.21.6269-6277.2003
Kuczynski, J., Lauber, C. L., Walters, W. A., Parfrey, L. W., Clemente, J. C., Gevers, D., et al. (2011). Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 13, 47–58. doi: 10.1038/nrg3129
Kujiraoka, M., Kuroda, M., Asai, K., Sekizuka, T., Kato, K., Watanabe, M., et al. (2017). Comprehensive diagnosis of bacterial infection associated with Acute Cholecystitis using metagenomic approach. Front. Microbiol. 8:685. doi: 10.3389/fmicb.2017.00685
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lecuit, M., and Eloit, M. (2014). The diagnosis of infectious diseases by whole genome next generation sequencing: a new era is opening. Front. Cell. Infect. Microbiol. 4:25. doi: 10.3389/fcimb.2014.00025
Leonard, S. R., Mammel, M. K., Lacher, D. W., and Elkins, C. A. (2015). Application of metagenomic sequencing to food safety: detection of Shiga Toxin-producing Escherichia coli on fresh bagged spinach. Appl. Environ. Microbiol. 81, 8183–8191. doi: 10.1128/AEM.02601-15
Levesque, S., Plante, P. L., Mendis, N., Cantin, P., Marchand, G., Charest, H., et al. (2014). Genomic characterization of a large outbreak of Legionella pneumophila serogroup 1 strains in Quebec City, 2012. PLoS One 9:e103852. doi: 10.1371/journal.pone.0103852
Lindgreen, S., Adair, K. L., and Gardner, P. P. (2016). An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 6:19233. doi: 10.1038/srep19233
Loman, N. J., Constantinidou, C., Christner, M., Rohde, H., Chan, J. Z., Quick, J., et al. (2013). A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309, 1502–1510. doi: 10.1001/jama.2013.3231
Lu, X., Zhang, X. X., Wang, Z., Huang, K., Wang, Y., Liang, W., et al. (2015). Bacterial pathogens and community composition in advanced sewage treatment systems revealed by metagenomics analysis based on high-throughput sequencing. PLoS One 10:e0125549. doi: 10.1371/journal.pone.0125549
Mahbubani, M. H., Bej, A. K., Miller, R., Haff, L., Dicesare, J., and Atlas, R. M. (1990). Detection of Legionella with polymerase chain reaction and gene probe methods. Mol. Cell. Probes 4, 175–187. doi: 10.1016/0890-8508(90)90051-Z
McAdam, P. R., Vander Broek, C. W., Lindsay, D. S., Ward, M. J., Hanson, M. F., Gillies, M., et al. (2014). Gene flow in environmental Legionella pneumophila leads to genetic and pathogenic heterogeneity within a Legionnaires’ disease outbreak. Genome Biol. 15:504. doi: 10.1186/PREACCEPT-1675723368141690
Miller, R. R., Montoya, V., Gardy, J. L., Patrick, D. M., and Tang, P. (2013). Metagenomics for pathogen detection in public health. Genome Med. 5:81. doi: 10.1186/gm485
Minot, S. S., Krumm, N., and Greenfield, N. B. (2015). One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. Available at: https://onecodex.com/
Mohiuddin, M. M., Salama, Y., Schellhorn, H. E., and Golding, G. B. (2017). Shotgun metagenomic sequencing reveals freshwater beach sands as reservoir of bacterial pathogens. Water Res. 115, 360–369. doi: 10.1016/j.watres.2017.02.057
Moran-Gilad, J., Prior, K., Yakunin, E., Harrison, T. G., Underwood, A., Lazarovitch, T., et al. (2015). Design and application of a core genome multilocus sequence typing scheme for investigation of Legionnaires’ disease incidents. Euro Surveill. 20:21186. doi: 10.2807/1560-7917.ES2015.20.28.21186
Morgan, J. L., Darling, A. E., and Eisen, J. A. (2010). Metagenomic sequencing of an in vitro-simulated microbial community. PLoS One 5:e10209. doi: 10.1371/journal.pone.0010209
Muir, P., Li, S., Lou, S., Wang, D., Spakowicz, D. J., Salichos, L., et al. (2016). The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol. 17:53. doi: 10.1186/s13059-016-0917-0
Mukherjee, N., Bartelli, D., Patra, C., Chauhan, B. V., Dowd, S. E., and Banerjee, P. (2016). Microbial diversity of source and point-of-use water in rural Haiti - A pyrosequencing-based metagenomic survey. PLoS One 11:e0167353. doi: 10.1371/journal.pone.0167353
Nakamura, S., Maeda, N., Miron, I. M., Yoh, M., Izutsu, K., Kataoka, C., et al. (2008). Metagenomic diagnosis of bacterial infections. Emerg. Infect. Dis. 14, 1784–1786. doi: 10.3201/eid1411.080589
Nintasen, R., Utrarachkij, F., Siripanichgon, K., Bhumiratana, A., Suzuki, Y., and Suthienkul, O. (2007). Enhancement of Legionella pneumophila culture isolation from microenvironments by macrophage infectivity potentiator (mip) gene-specific nested polymerase chain reaction. Microbiol. Immunol. 51, 777–785. doi: 10.1111/j.1348-0421.2007.tb03967.x
Nordahl Petersen, T., Rasmussen, S., Hasman, H., Caroe, C., Baelum, J., Schultz, A. C., et al. (2015). Meta-genomic analysis of toilet waste from long distance flights; a step towards global surveillance of infectious diseases and antimicrobial resistance. Sci. Rep. 5:11444. doi: 10.1038/srep11444
Oh, S., Hammes, F., and Liu, W. T. (2018). Metagenomic characterization of biofilter microbial communities in a full-scale drinking water treatment plant. Water Res. 128, 278–285. doi: 10.1016/j.watres.2017.10.054
Ortiz-Alcantara, J. M., Segura-Candelas, J. M., Garces-Ayala, F., Gonzalez-Duran, E., Rodriguez-Castillo, A., Alcantara-Perez, P., et al. (2016). Fatal Psychrobacter sp. infection in a pediatric patient with meningitis identified by metagenomic next-generation sequencing in cerebrospinal fluid. Arch. Microbiol. 198, 129–135. doi: 10.1007/s00203-015-1168-2
Otten, T. G., Graham, J. L., Harris, T. D., and Dreher, T. W. (2016). Elucidation of taste- and odor-producing bacteria and toxigenic cyanobacteria in a midwestern drinking water supply reservoir by shotgun metagenomic analysis. Appl. Environ. Microbiol. 82, 5410–5420. doi: 10.1128/AEM.01334-16
Ounit, R., Wanamaker, S., Close, T. J., and Lonardi, S. (2015). CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics 16:236. doi: 10.1186/s12864-015-1419-2
Pandey, P. K., Kass, P. H., Soupir, M. L., Biswas, S., and Singh, V. P. (2014). Contamination of water resources by pathogenic bacteria. AMB Express 4:51. doi: 10.1186/s13568-014-0051-x
Peabody, M. A., Caravas, J. A., Morrison, S. S., Mercante, J. W., Prystajecky, N. A., Raphael, B. H., et al. (2017). Characterization of Legionella species from watersheds in British Columbia, Canada. mSphere 2:e00246-17. doi: 10.1128/mSphere.00246-17
Pereira, R. P., Peplies, J., Brettar, I., and Hofle, M. G. (2017). Development of a genus-specific next generation sequencing approach for sensitive and quantitative determination of the Legionella microbiome in freshwater systems. BMC Microbiol. 17:79. doi: 10.1186/s12866-017-0987-5
Petersen, T. N., Lukjancenko, O., Thomsen, M. C. F., Maddalena Sperotto, M., Lund, O., Moller Aarestrup, F., et al. (2017). MGmapper: reference based mapping and taxonomy annotation of metagenomics sequence reads. PLoS One 12:e0176469. doi: 10.1371/journal.pone.0176469
Petzold, M., Prior, K., Moran-Gilad, J., Harmsen, D., and Luck, C. (2017). Epidemiological information is key when interpreting whole genome sequence data - lessons learned from a large Legionella pneumophila outbreak in Warstein, Germany, 2013. Euro Surveill. 22:17-00137. doi: 10.2807/1560-7917.ES.2017.22.45.17-00137
Pinto, A. J., Marcus, D. N., Ijaz, U. Z., Bautista-De Lose Santos, Q. M., Dick, G. J., and Raskin, L. (2016). Metagenomic evidence for the presence of Comammox Nitrospira-like bacteria in a drinking water system. mSphere 1:e00054-15 doi: 10.1128/mSphere.00054-15
Pope, P. B., and Patel, B. K. (2008). Metagenomic analysis of a freshwater toxic cyanobacteria bloom. FEMS Microbiol. Ecol. 64, 9–27. doi: 10.1111/j.1574-6941.2008.00448.x
Qiu, Y., Nakao, R., Ohnuma, A., Kawamori, F., and Sugimoto, C. (2014). Microbial population analysis of the salivary glands of ticks; a possible strategy for the surveillance of bacterial pathogens. PLoS One 9:e103961. doi: 10.1371/journal.pone.0103961
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2016). R : A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ratzow, S., Gaia, V., Helbig, J. H., Fry, N. K., and Luck, P. C. (2007). Addition of neuA, the gene encoding N-acylneuraminate cytidylyl transferase, increases the discriminatory ability of the consensus sequence-based scheme for typing Legionella pneumophila serogroup 1 strains. J. Clin. Microbiol. 45, 1965–1968. doi: 10.1128/JCM.00261-07
Reuter, S., Harrison, T. G., Koser, C. U., Ellington, M. J., Smith, G. P., Parkhill, J., et al. (2013). A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open 3:e002175 doi: 10.1136/bmjopen-2012-002175
Shah, N., Tang, H., Doak, T. G., and Ye, Y. (2011). Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac. Symp. Biocomput. 165–176. doi: 10.1142/9789814335058_0018
Sharpton, T. J. (2014). An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 5:209. doi: 10.3389/fpls.2014.00209
Shi, P., Jia, S., Zhang, X. X., Zhang, T., Cheng, S., and Li, A. (2013). Metagenomic insights into chlorination effects on microbial antibiotic resistance in drinking water. Water Res. 47, 111–120. doi: 10.1016/j.watres.2012.09.046
Silva, C. C., Hayden, H., Sawbridge, T., Mele, P., Kruger, R. H., Rodrigues, M. V., et al. (2012). Phylogenetic and functional diversity of metagenomic libraries of phenol degrading sludge from petroleum refinery wastewater treatment system. AMB Express 2:18. doi: 10.1186/2191-0855-2-18
Sogin, M. L., Morrison, H. G., Huber, J. A., Mark Welch, D., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored ”rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Stout, J. E., Yu, V. L., and Best, M. G. (1985). Ecology of Legionella pneumophila within water distribution systems. Appl. Environ. Microbiol. 49, 221–228.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tekera, M., Lotter, A., Oliver, J., Jonker, N., and Venter, S. (2011). Metagenomic analysis of bacterial diveristy of Siloam hot water spring, Limpopo, South Africa. Afr. J. Biotechnol. 10, 18005–18012.
Thomas, T., Gilbert, J., and Meyer, F. (2012). Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2:3. doi: 10.1186/2042-5783-2-3
Truong, D. T., Franzosa, E. A., Tickle, T. L., Scholz, M., Weingart, G., Pasolli, E., et al. (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. doi: 10.1038/nmeth.3589
Turetgen, I., Sungur, E. I., and Cotuk, A. (2005). Enumeration of Legionella pneumophila in cooling tower water systems. Environ. Monit. Assess. 100, 53–58. doi: 10.1007/s10661-005-7058-3
Van Rossum, T., Peabody, M. A., Uyaguari-Diaz, M. I., Cronin, K. I., Chan, M., Slobodan, J. R., et al. (2015). Year-long metagenomic study of river microbiomes across land use and water quality. Front. Microbiol. 6:1405. doi: 10.3389/fmicb.2015.01405
Vetrovsky, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Walsh, A. M., Crispie, F., Daari, K., O’sullivan, O., Martin, J. C., Arthur, C. T., et al. (2017). Strain-level metagenomic analysis of the fermented dairy beverage nunu highlights potential food safety risks. Appl. Environ. Microbiol. 83:e01144-17. doi: 10.1128/AEM.01144-17
Wang, Z., Zhang, X. X., Huang, K., Miao, Y., Shi, P., Liu, B., et al. (2013). Metagenomic profiling of antibiotic resistance genes and mobile genetic elements in a tannery wastewater treatment plant. PLoS One 8:e76079. doi: 10.1371/journal.pone.0076079
Weinstock, G. M. (2012). Genomic approaches to studying the human microbiota. Nature 489, 250–256. doi: 10.1038/nature11553
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Yanez, M. A., Carrasco-Serrano, C., Barbera, V. M., and Catalan, V. (2005). Quantitative detection of Legionella pneumophila in water samples by immunomagnetic purification and real-time PCR amplification of the dotA gene. Appl. Environ. Microbiol. 71, 3433–3441. doi: 10.1128/AEM.71.7.3433-3441.2005
Yang, X., Noyes, N. R., Doster, E., Martin, J. N., Linke, L. M., Magnuson, R. J., et al. (2016). Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within the microbiome of the beef production chain. Appl. Environ. Microbiol. 82, 2433–2443. doi: 10.1128/AEM.00078-16
Ye, L., and Zhang, T. (2011). Pathogenic bacteria in sewage treatment plants as revealed by 454 pyrosequencing. Environ. Sci. Technol. 45, 7173–7179. doi: 10.1021/es201045e
Keywords: water-borne diseases, metagenomic analysis, bacteria, detection, receiver operating characteristic curve, Legionella pneumophila
Citation: Borthong J, Omori R, Sugimoto C, Suthienkul O, Nakao R and Ito K (2018) Comparison of Database Search Methods for the Detection of Legionella pneumophila in Water Samples Using Metagenomic Analysis. Front. Microbiol. 9:1272. doi: 10.3389/fmicb.2018.01272
Received: 03 December 2017; Accepted: 24 May 2018;
Published: 19 June 2018.
Edited by:
Jacob Moran-Gilad, Ben-Gurion University of the Negev, IsraelReviewed by:
Silke Maria Peter, Universität Tübingen, GermanyNatacha Couto, University Medical Center Groningen, Netherlands
Copyright © 2018 Borthong, Omori, Sugimoto, Suthienkul, Nakao and Ito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimihito Ito, aXRva0BjemMuaG9rdWRhaS5hYy5qcA==
†These authors have contributed equally to this work.