 Wei Sun
Wei Sun Changrong Liu1
Changrong Liu1 Zhiyong Li
Zhiyong Li- 1State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
- 2Instrumental Analysis Center, Shanghai Jiao Tong University, Shanghai, China
Sponge-derived actinomycetes represent a significant component of marine actinomycetes. Members of the genus Kocuria are distributed in various habitats such as soil, rhizosphere, clinical specimens, marine sediments, and sponges, however, to date, little is known about the mechanism of their environmental adaptation. Kocuria flava S43 was isolated from a coastal sponge. Phylogenetic analysis revealed that it was closely related to the terrestrial airborne K. flava HO-9041. In this study, to gain insights into the marine adaptation in K. flava S43 we sequenced the draft genome for K. flava S43 by third generation sequencing (TGS) and compared it with those of K. flava HO-9041 and some other Kocuria relatives. Comparative genomics and phylogenetic analyses revealed that K. flava S43 might adapt to the marine environment mainly by increasing the number of the genes linked to potassium homeostasis, resistance to heavy metals and phosphate metabolism, and acquiring the genes associated with electron transport and the genes encoding ATP-binding cassette (ABC) transporter, aquaporin, and thiol/disulfide interchange protein. Notably, gene acquisition was probably a primary mechanism of environmental adaptation in K. flava S43. Furthermore, this study also indicated that the Kocuria isolates from various marine and hyperosmotic environments possessed common genetic basis for environmental adaptation.
Introduction
Marine sponges are well known to be a rich source of diverse actinomycetes such as Streptomyces, Salinispora, Micromonospora, Kocuria, etc. (Taylor et al., 2007). Sponge-derived actinomycetes are distributed randomly in sponge hosts, and they are thought to be taken up from the flowing sea water by filtration and dwell in the mesohyl matrix of sponge hosts (Abdelmohsen et al., 2014). Accordingly, the actinomycetes are endowed with the ability to survive in both nutrient-poor sea water and nutrient-rich sponge mesohyl. They are physiologically or metabolically distinct from their terrestrial counterparts owing to their adaptation to marine environments. A number of isolates grow and differentiate faster on the medium prepared with sea water (Jiang et al., 2007). Some Streptomyces strains strictly require salt for their growth (Khan et al., 2011). A few isolates own several secondary metabolite biosynthesis gene clusters absent in their closely related terrestrial relatives. For instance, sponge-derived Streptomyces sp. GBA 94-10 and PVA 94-07 share 10 identical secondary metabolite gene clusters not present in their terrestrial relative S. albus J1074, most of which are responsible for encoding non-ribosomal peptide synthetases and polyketide synthases (Ian et al., 2014). To date, research interest has been largely focused on the diversity and natural products of marine sponge-derived actinomycetes, however, the mechanisms behind their environmental adaptation are still not well understood. With the rapid development of the second/third next generation sequencing techniques, genomics may provide new insights into the genetic basis for environmental adaptation in sponge-derived actinomycetes.
Over the past decades, great progresses have been made in understanding the genetic basis for marine adaptation in Gram-negative bacteria. The discovery of the sodium-pumping NADH dehydrogenase Nqr (Unemoto and Hayashi, 1993) and the associated genes nqrA-F (Mulkidjanian et al., 2008) provided the genetic link to sodium dependence in Gram-negative marine bacteria. Nqr can create an ionic motive force to generate ATP and drive other cellular processes (Schäfer et al., 2008). The gene acquisition of certain transporters represents another important marine adaptation mechanism. High abundance of ABC branched chain amino acid (BCAA) transporters was found in several marine Roseobacter strains (Moran et al., 2007), and the associated genes were detected in marine metagenomes as well (Morris et al., 2010). BCAAs can be converted into L-glutamate, which would help acidify the basic cytoplasm (Takami et al., 2002). Marine cyanobacterium Synechococcus, with greater capacity to transport Na+ than freshwater species, might adapt to the oligotrophic environment by using more sodium-dependent transporters than a model freshwater cyanobacterium (Palenik et al., 2003). In addition, intracellular accumulation of compatible solutes seems to be an alternative marine adaptation strategy. The marine adaption in Novosphingobium was suggested to be based on the organic osmolyte (ectoine) mechanism that was different from those reported in Gram-negative marine bacteria that exported Na+ via the sodium-pumping NADH dehydrogenase Nqr (Gan et al., 2013).
Initial attempts to provide the evidence of marine adaptation in actinomycetes concentrated in the obligate marine genus Salinispora (Penn and Jensen, 2012). Marine adaptation genes (MAGs) were identified from S. tropica and S. arenicola by comparative genomic analysis, and the loss of a mechanosensitive channel gene mscL was thought to result in the inability of Salinispora strains to grow in low osmotic environment. The finding was later confirmed by genetic complementation of S. tropica with mscL (Bucarey et al., 2012). By the genomic comparison of two sponge-derived Streptomyces albus isolates with their terrestrial relative S. albus J1074, several putative MAGs linked to electron transport and potassium uptake were identified (Ian et al., 2014). In addition, marine-derived Streptomyces subgroup was found to possess some common characteristics of marine adaptation on the basis of comparative genomics (Tian et al., 2016). In specific, marine streptomycetes possessed more functional genes and transporters than other streptomycetes to adapt to the cold, hyperosmosis, oligotrophy, and other marine environments (Tian et al., 2016). Nonetheless, among diverse actinomycete groups, only a few representative members belonging to Salinispora, Streptomyces, Janibacter, Aeromicrobium, Rhodococcus, and one unclassified species “marine actinobacterium PHSC20C1” have been studied on their marine adaptation to date (Penn and Jensen, 2012; Tian et al., 2016). To improve our knowledge of the mechanisms for environmental adaptation in marine actinomycetes, it is significant to investigate a wider range of actinomycete lineages.
Taxonomically, Kocuria belongs to the family Micrococcaceae and comprises a group of coccoid actinomycetes with high environmental adaptability. Kocuria species have been recovered from various habitats, particularly marine environments such as sediments (Bala et al., 2012; Jiang et al., 2015), seawater (Seo et al., 2009), sponges (Abdelmohsen et al., 2014), and corals (Mahmoud and Kalendar, 2016). In our efforts of isolating actinomycetes from sponges collected from South China Sea (Li and Liu, 2006; Sun et al., 2015), Kocuria flava S43 was isolated from a coastal sponge Siphonochalina sp. (Sun et al., 2015), which had 99.7% similarity in 16S rRNA gene sequence with K. flava HO-9041 from the air of Xinjiang, an inland province in China (Zhou et al., 2008). Evidently, the inland airborne K. flava HO-9041 represents a terrestrial microorganism whereas the sponge-derived K. flava S43 is a marine representative. These two sister organisms can serve as a model for gaining insights into marine adaptation in K. flava. The complete genome sequence of K. flava HO-9041 is currently available (Zhou et al., 2016). Furthermore, a few complete or draft genome sequences of marine Kocuria isolates are publicly available, which provided an opportunity to make comparison with K. flava S43 at the genomic level. In this study, de novo genome sequencing for K. flava S43 was performed by TGS, and the genetic basis for marine adaptation in K. flava S43 was revealed by comparative genomic analysis of several Kocuria isolates from various marine and terrestrial environments.
Materials and Methods
Strain and 16S rRNA Gene Phylogeny
K. flava S43 was previously isolated from the tissue of the marine sponge Siphonochalina sp., which was collected from Xincun Harbor (18.42°N, 109.97°E), the South China Sea. The 16S rRNA gene sequence of K. flava S43 was deposited to GenBank under the accession number: JX007971 (Sun et al., 2015).
The type strains of the genus Kocuria were retrieved from RDP database (Release 11.5) by using the hierarchy browser1. Furthermore, literature search for the novel Kocuria species validly published in recent years was performed to update the type strain pool. The 16S rRNA gene sequences of the currently described 26 Kocuria species and K. flava S43 were manually aligned using ClustalX 1.81 (Thompson et al., 1997). Phylogenetic analysis was conducted using the maximum likelihood method (Felsenstein, 1981) provided by the software package MEGA 6.06 (Tamura et al., 2013). The consistency of the tree was verified by bootstrapping (1,000 replicates) for parsimony (Felsenstein, 1985).
Genomic DNA Extraction and Genome Sequencing
A single colony of K. flava S43 was inoculated to a 50-ml centrifuge tube with 10 ml of GYM4 artificial seawater (26.52 g NaCl, 5.228 g MgCl26H2O, 3.305 g MgSO4, 1.141 g CaCl2, 0.725 g KCl, 0.202 g NaHCO3, 0.083 g NaBr, and 1 L distilled water) medium (10 g glucose, 4 g yeast extract, 4 g malt extract, and 1 L artificial seawater) (Schneemann et al., 2010). After incubation at 28°C and 180 rpm for 2 days, the culture was collected by centrifugation and its genomic DNA was extracted using a DNeasy® Blood and Tissue Kit (Qiagen, United States). The integrity, concentration and purity of genomic DNA were detected by Agarose Gel Electrophoresis, Nanodrop and Qubit Fluorimeter. The genome was sequenced by the third generation sequencing (TGS), i.e., the Pacific Biosciences (PacBio) Single Molecule Real-Time (SMRT) sequencing technology. A whole-genome sequencing library was constructed using the SMRTcell Template Prep Kit (PacBio, United States) according to the manufacturer’s protocol, and the library was sequenced by using g-Tube on the PacBio RS II platform (PacBio).
Genome Annotation and Comparative Genomic Analysis
De novo assembly of the sequences was performed using hierarchical genome assembly process (HGAP) version 3.0 (Chin et al., 2013). This whole-genome shotgun project was deposited at DDBJ/EMBL/GenBank under accession number LOMZ00000000. The version described in this paper was the first version, LOMZ01000000. The draft genome sequence of K. flava S43 was uploaded to NCBI and aligned with the deposited genomes using Basic Local Alignment Search Tool2.
The genome sequences of K. flava HO-9041 from inland air, Kocuria sp. SM24M-10 from the mucus of the coral Mussismilia hispida (Palermo et al., 2016), K. indica DSM 25126 from a marine sediment sample (Dastager et al., 2014), K. rhizophila P7-4 from the intestine of the fish Siganus doliatus (Kim et al., 2011) and K. rhizophila DC2201 from a soil sample (Takarada et al., 2008) were downloaded from NCBI3 and served as the reference genomes for subsequent analysis. Genome alignment of K. flava S43 and K. flava HO-9041 was carried out using MAUVE software (Darling et al., 2010). For rapid function-based comparison by the comparative tool in the SEED Viewer (Overbeek et al., 2014), all the six Kocuria genome sequences were annotated by using Rapid Annotation using Subsystem Technology (RAST) version 2.04 (Aziz et al., 2008). Genome Browser was used to visualize gene arrangement and annotation.
The subsystem category distribution pattern was compared between the K. flava S43 and HO-9041 genomes. The subsystems with significant difference in feature counts were marked and further compared at a finer scale. The common genes with marked increase in number in the K. flava S43 genome were screened. Meanwhile, the function-based comparison was performed among the 6 Kocuria genomes, the genes common in at least three marine Kocuria isolates but absent in two terrestrial Kocuria representatives were also screened. All the screened genes were listed as putative MAGs and uploaded to NCBI for protein match using BLASTp. The original RAST annotations were manually checked according to the BLAST hits using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP) (Angiuoli et al., 2008). The genes incorrectly annotated by RAST were excluded.
Phylogenetic Analysis of Genes Potentially Relevant to Environmental Adaptation
The screened candidate genes were subjected to phylogenetic analysis to test for a shared evolutionary history with other bacteria or actinomycetes derived from marine and hyperosmotic environments. The top 15–20 BLASTp hits of each gene were downloaded from the NCBI protein database. Maximum likelihood phylogenies were constructed for each representative gene using the online tool MABL with default settings5 (Dereeper et al., 2008). The genes that were clustered in the clades totally or largely comprising the homologs from the isolates of marine or hyperosmotic sources were kept in the final MAG pool.
Results
Phylogeny and Genome Feature of K. flava S43
Based on 16S rRNA gene sequence analysis, the phylogenetic position of K. flava S43 with respect to currently described Kocuria species was shown in Figure 1. K. flava S43 was clearly positioned within the genus Kocuria and formed a subgroup together with K. flava HO-9041.
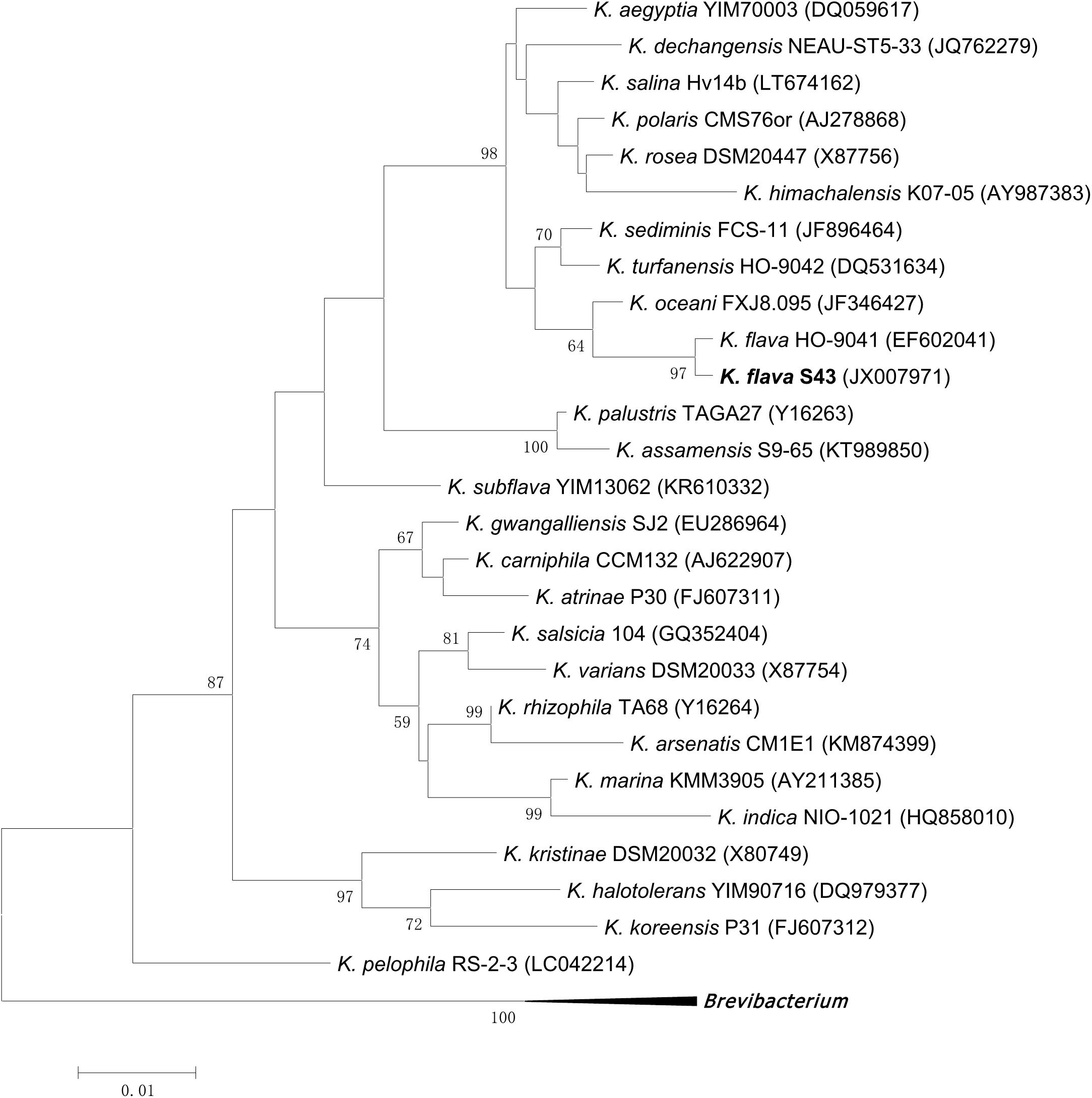
FIGURE 1. Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences of K. flava S43 and all currently described Kocuria species. Brevibacterium iodinum DSM 20626 (X83813), Brevibacterium linens DSM 20425 (X7745), and Brevibacterium casei DSM 20657 (AJ251418) were used as the out-group. Only bootstrap values of being greater than 50% were shown on the tree.

TABLE 1. BLAST match of the K. flava S43 genome against the deposited bacterial genomes.
A total of 99,440 reads of K. flava S43 were obtained, with a mean read length of 16,484 bp, providing 361.84× average reference coverage. The final assembly yielded six contigs, with the largest contig of 3,548,480 bp. The remaining five contigs were from 2,899 to 152,580 bp in length. BLAST matches indicated that the largest contig represented chromosome genome, four contigs were plasmid genomes and the smallest contig could not match any known genome (Table 1).
The genome sequence of K. flava S43 was aligned with that of K. flava HO-9041. This alignment showed a high homology between K. flava S43 and K. flava HO-9041 (Figure 2A), supporting their close phylogenetic relationship as suggested by 16S rRNA gene phylogeny. Both genomes shared a large number of genomic traits, however, various translocations and inversions occurred in the genome of K. flava S43 relative to the reference genome. In addition, it appeared that gene transfer from plasmids to chromosomes happened in both genomes (Figure 2B). In specific, a few genes located in the K. flava S43 plasmid 1 and 2 showed a high homology with those in the K. flava HO-9041 genome and a few genes occurring in the K. flava HO-9041 plasmid 1 showed a high homology with those in the K. flava S43 genome, suggesting potential horizontal gene transfer.
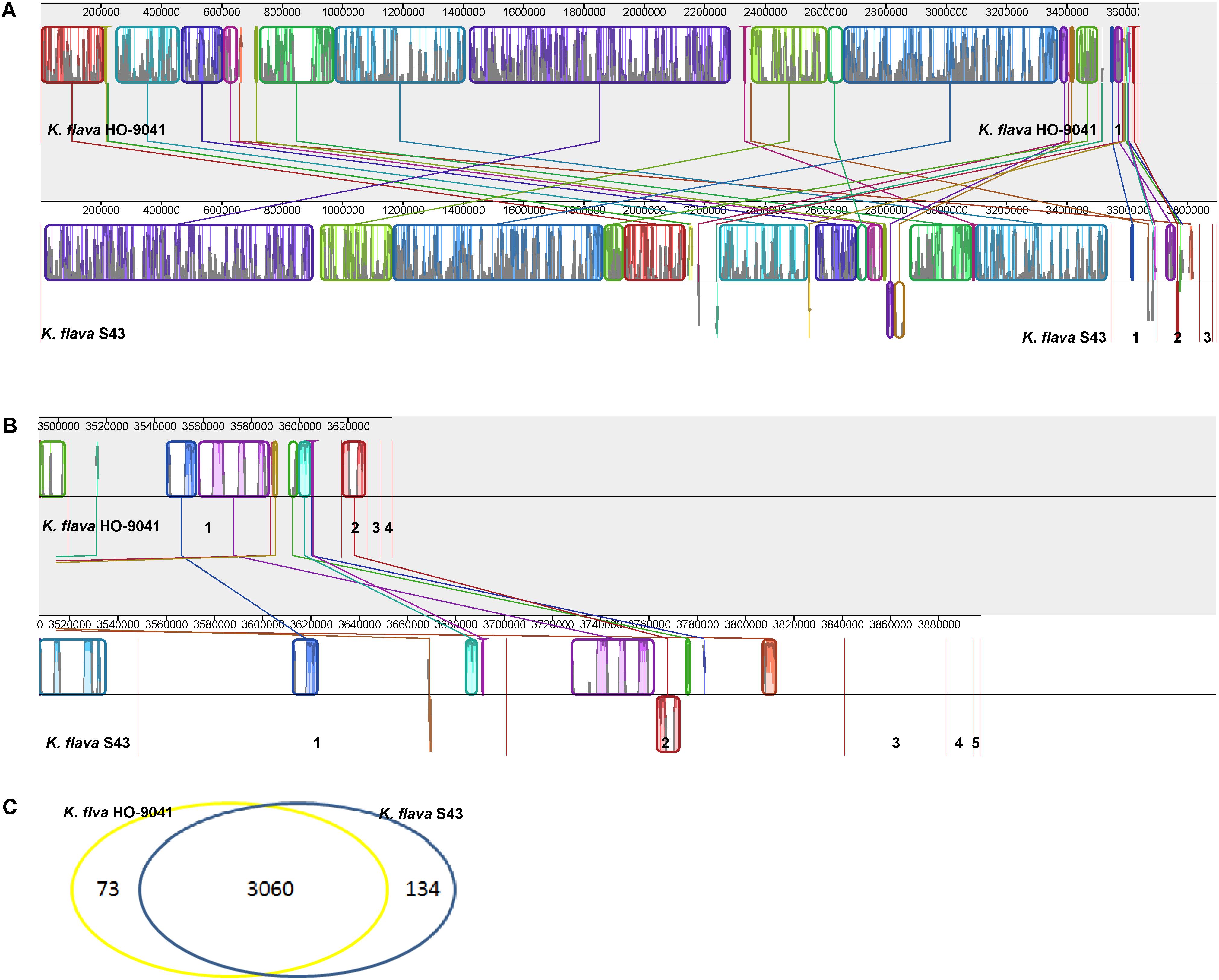
FIGURE 2. MAUVE alignment of the chromosome and plasmid genome sequences of K. flava S43 and K. flava HO-9041. The contigs were separated by red lines. When boxes had the same color, this indicated syntenic regions. Boxes below the horizontal line indicated inverted regions. Rearrangements were shown by colored lines. Scale was in nucleotides. (A) Chromosome with the boundaries marked by strain name. (B) Plasmid with the figures referring to different contigs. (C) Venn diagram showing the unique and common ORFs in 2 K. flava genomes.
Gene annotation of the draft genome sequence yielded 3,494 protein-coding sequences (CDSs), 49 tRNA and 11 rRNA (three 5S rRNA, four 23S rRNA, and four 16S rRNA) genes. In total, 3,194 CDSs, 48 tRNA, and 11 rRNA genes were located in the chromosome genome. The rest of the CDSs and one tRNA gene were distributed in the plasmid genomes.
Genes Potentially Relevant to Marine Adaptation Based on Comparative Genomic Analysis
The comparison of the genomic characteristics between K. flava S43 and K. flava HO-9041 was presented in Table 2. The chromosome genome of K. flava S43 was slightly larger in size than that of K. flava HO-9041, suggesting the draft genome sequence was nearly complete. The same number of tRNA genes occurred in two genomes. Notably, although one copy was incomplete (5S rRNA missing and 23S rRNA partial) due to the draft sequence, the K. flava S43 genome owned one more copy of rRNA operon than the K. flava HO-9041 genome.
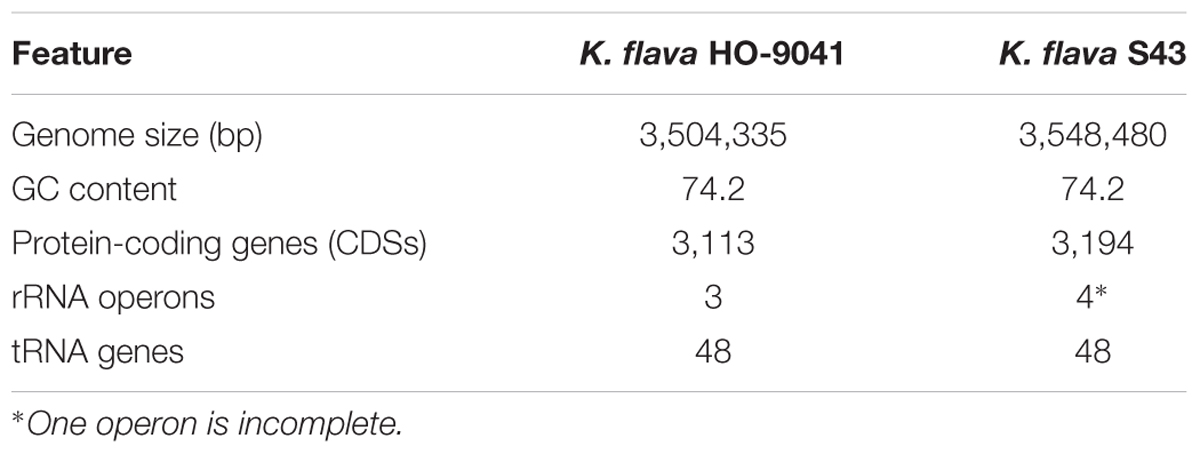
TABLE 2. Genomic characteristics of K. flava HO-9041 and K. flava S43.
A Venn diagram showed the unique and common ORFs in two K. flava genomes (Figure 2C). The functional annotation of the unique ORFs in the K. flava S43 genome was shown in Supplementary Table S1. The comparison of the subsystem distribution between K. flava S43 and K. flava HO-9041 was demonstrated in Table 3. Significant difference was observed in the count of the genes categorized in six subsystems: respiration, potassium metabolism, virulence, disease and defense, phosphorus metabolism, cell wall and capsule, and nitrogen metabolism. The count of the genes related to respiration markedly increased in the K. flava S43 genome, which was due to the occurrence of the genes encoding respiratory complex I, terminal cytochrome d ubiquinol oxidase and the genes associated with the biogenesis of c-type cytochromes. Additional 4 kdpD (osmosensitive K+ channel histidine kinase) genes led to the higher number of genes related to potassium homeostasis in K. flava S43. Similarly, the counts of czcD (cobalt–zinc–cadmium resistance protein) and copD (copper resistance protein D) genes increased in K. flava S43, which were grouped into the virulence, disease, and defense subsystem. In the phosphorus metabolism subsystem, six phoB (phosphate regulon transcriptional regulatory protein) genes occurred in K. flava S43 whereas only three were present in K. flava HO-9041. In addition, the count of the genes categorized in cell wall/capsule and nitrogen metabolism markedly decreased in the K. flava S43 genome. In specific, more than 20 genes related to peptidoglycan biosynthesis were absent in K. flava S43. The number of the genes involved in nitrate and nitrite ammonification and encoding denitrifying reductase were less in K. flava S43 than in K. flava HO-9041.
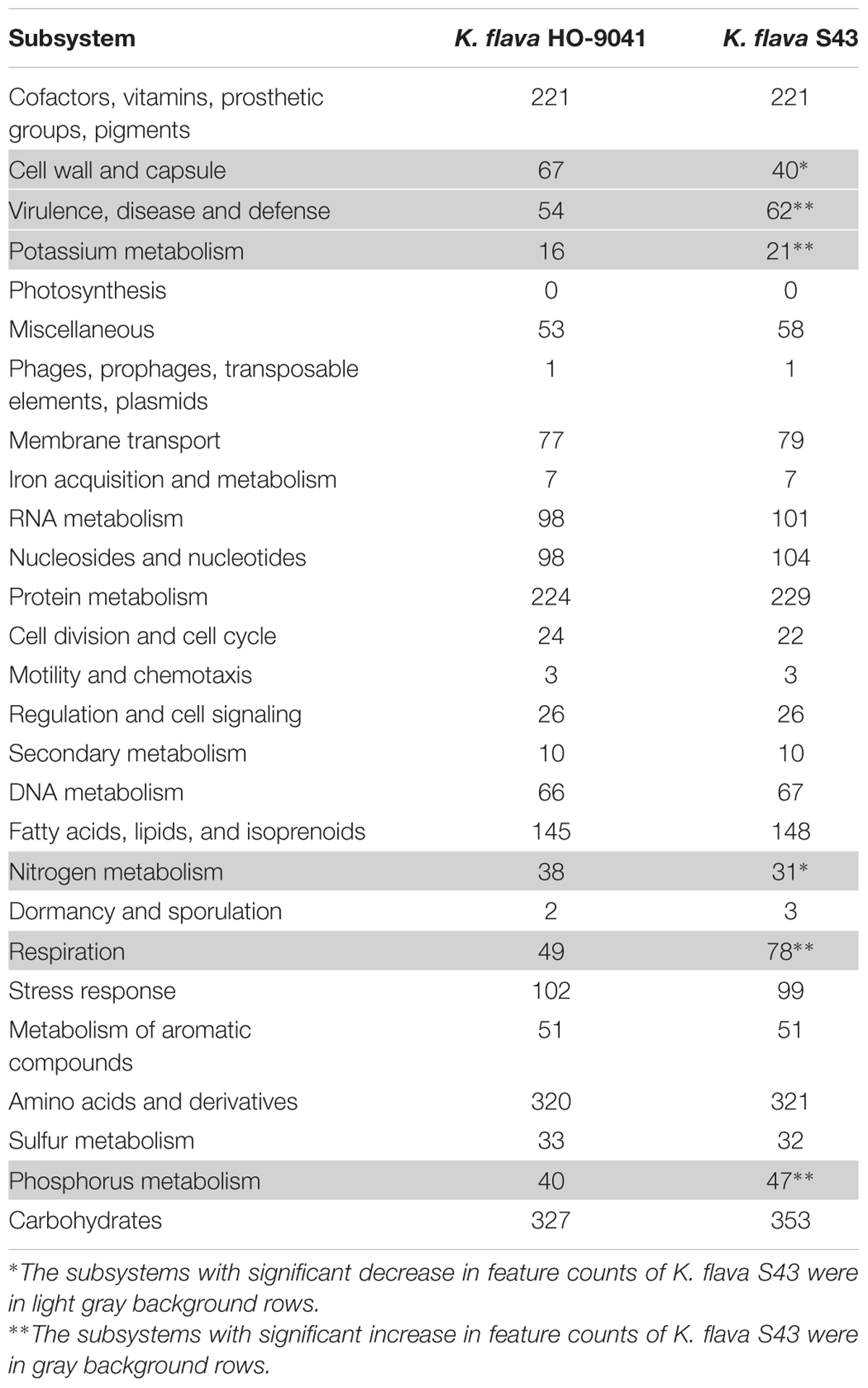
TABLE 3. Subsystem distribution in the K. flava HO-9041 and K. flava S43 genomes.
Phylogenetic analyses of the kdpD, czcD, copD, and phoB genes in K. flava S43 were performed (Figures 3A–D). Apparently, the number increase of these genes was not due to gene duplication but gene gain of K. flava S43 or gene loss of K. flava HO-9041. Notably, each of the phylogenetic trees contained at least one clade totally comprising the homologs from the isolates of marine or hyperosmotic sources, such as Kocuria sp. SM24M-10 from a coral and K. polaris CD08-4 from a celiac disease patient (Chander et al., 2017), suggesting that a total of eight genes (two kdpD, one czcD, three copD, and two phoB genes) in such clades may be MAGs.
A total of 13 genes (nuoA, nuoH, nuoJ, nuoK, nuoL, nuoM, nuoN, cydA, cydB, cydCD, aqpZ, and two dsbA genes) common in at least three marine isolates but absent in two terrestrial representatives were screened as putative MAGs. Seven nuo genes constituted a partial NDH-1 operon, which encoded a part of a proton-pumping NADH dehydrogenase. The nuo genes were previously identified as MAGs in Salinispora (Penn and Jensen, 2012). The organization of the operon in K. flava S43 was consistent with that in S. arenicola CNS-205 (Figure 4A). Three cyd genes in a cluster were responsible for encoding cytochrome d ubiquinol oxidase subunit I, II and an ATP-binding cassette-type transporter, respectively. As one type of terminal respiratory oxidases, cytochrome d ubiquinol oxidase acted as the terminal acceptor of electron transport chains. The arrangement of the genes in the cluster was similar to that in the marine actinobacterium PHSC20C1 genome (Figure 4B). The aqpZ gene was located next to cydCD, which encoded an aquaporin, a specific water channel that facilitated the rapid influx/efflux of water, and the dsbA gene was responsible for encoding a periplasmic thiol/disulfide interchange protein, which was involved in the biogenesis of cytochrome C.
The phylogenies of nuoM (Figure 5A), cydB (Figure 5B), aqpZ (Figure 5C), and dsbA (Figure 5D) indicated the presence of several clades totally or largely comprising the orthologs from the isolates of marine or hyperosmotic sources, suggesting that they shared evolutionary history with certain isolates derived from marine or other hyperosmotic environments.
The distribution and abundance of the MAGs in six Kocuria representatives were shown in Table 4. Besides four marine isolates, two strains from hyperosmotic environments, i.e., K. polaris CD08-4 from a celiac disease patient and K. marina SO9-6 from a copper iron sulfide mineral (Castro et al., 2015), were also included. Apparently, the MAGs were distributed in all or the majority of the analyzed Kocuria isolates though their abundance sometimes varied as the species, indicating that the Kocuria isolates possessed common genetic basis for marine adaptation. The result based on RAST annotation and comparison was consistent with the phylogeny test based on NCBI BLASTp.
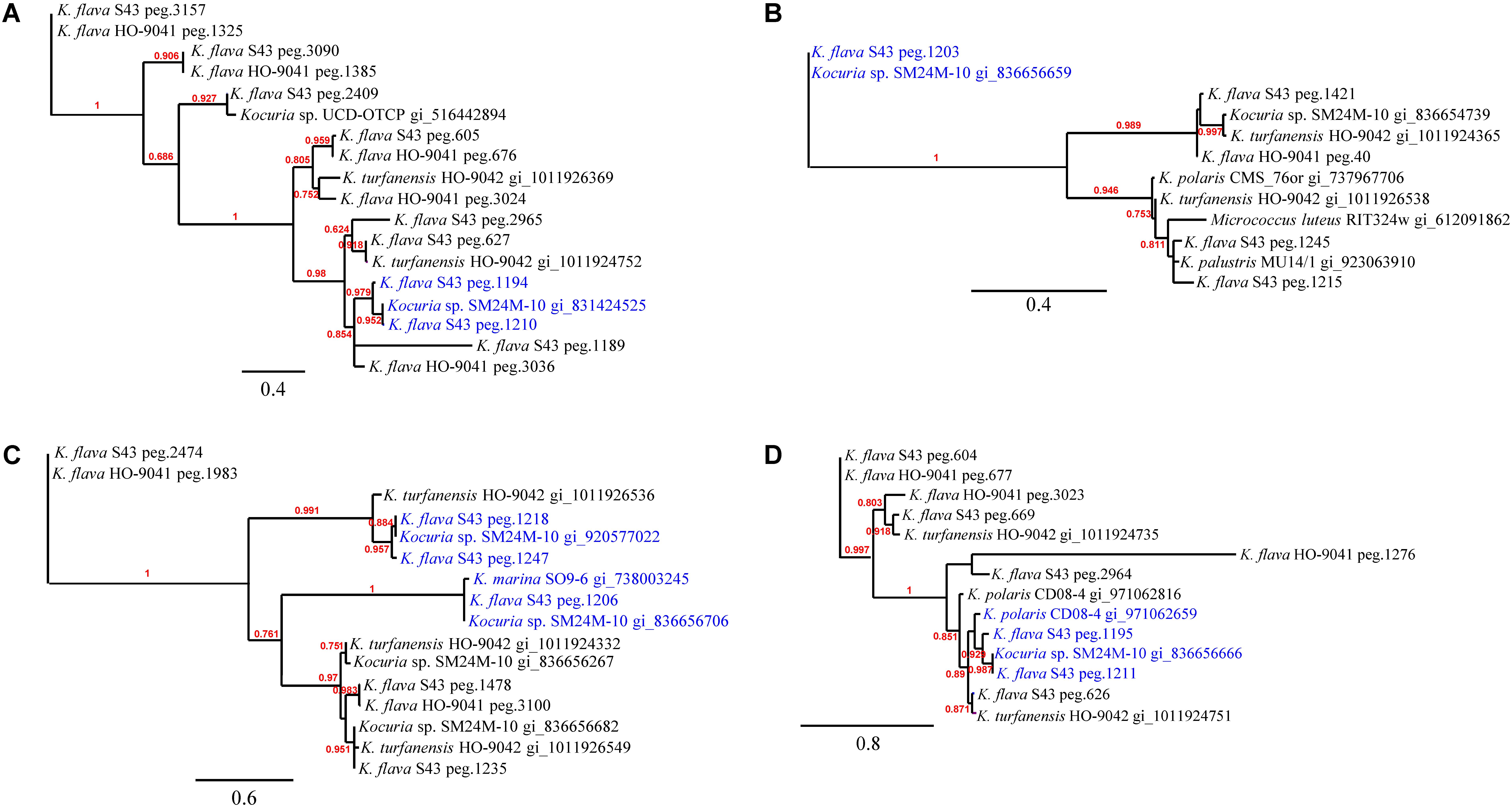
FIGURE 3. The phylogenies of the genes occurring in higher number in the K. flava S43 genome potentially relevant to marine adaptation. (A) kdpD, (B) czcD, (C) copD, (D) phoB. The clades totally comprising the homologs from the isolates of marine or hyperosmotic sources were colored blue. Midpoint rooting was used and likelihood values shown for each node. Only bootstrap values of being greater than 0.5 were shown on the tree. Scale bar represented changes per site.
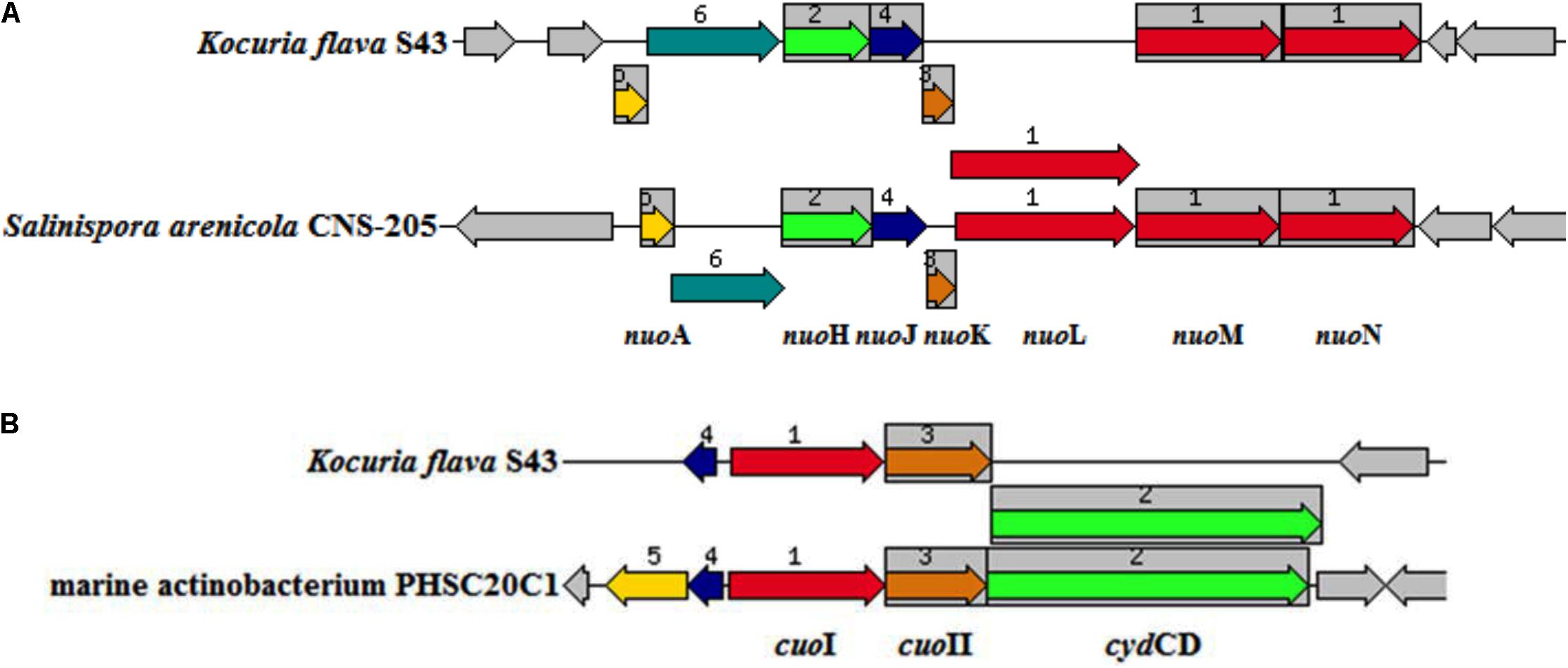
FIGURE 4. The arrangement of the operons potentially relevant to environmental adaptation in K. flava S43 and obligate marine actinomycetes. (A) nuo (NADH-ubiquinone oxidoreductase) operon, (B) cuo (cytochrome ubiquinol oxidase) operon. The chromosomal region of the focus gene (top) was compared with similar organisms. The graphic was centered on the focus gene, which was red and numbered 1. Sets of genes with similar sequence were grouped with the same number and color. Genes whose relative position was conserved were functionally coupled and shared gray background boxes.
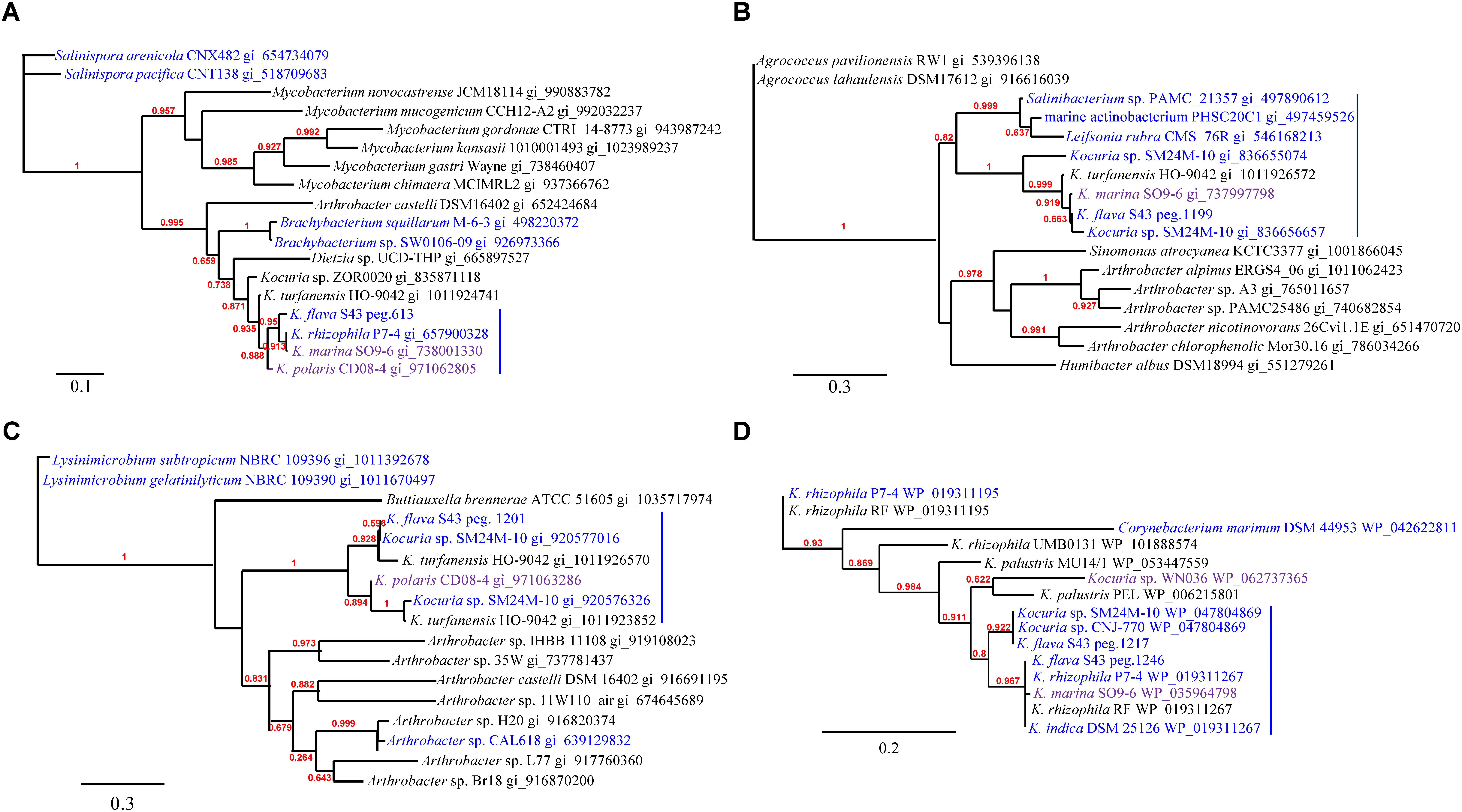
FIGURE 5. The phylogenies of the genes newly acquired in the K. flava S43 genome potentially relevant to environmental adaptation. (A) nuoM, (B) cydB, (C) aqpZ, (D) dsbA. Strains of marine, hyperosmotic, and terrestrial sources were colored blue, purple, and black, respectively. Midpoint rooting was used and likelihood values shown for each node. Only bootstrap values of being greater than 0.5 were shown on the tree. Scale bar represented changes per site.
Discussion
Actinomycetes are widely distributed in marine and terrestrial environments. Besides a few obligate marine lineages such as Salinispora and Marinispora, the majority of the actinomycetes can survive in both marine and terrestrial habitats. Undoubtedly, marine actinomycetes acquire marine adaptive traits relative to their terrestrial counterparts. The genes relevant to marine adaptation can be identified by comparing the genomes of closely related marine and terrestrial microorganisms. Previous comparative genomic analyses of marine actinomycetes were performed mainly at the species or genus level (Penn and Jensen, 2012; Tian et al., 2016). In this study, comparative genomic analysis of Kocuria spp. was carried out at both strain and species level. Several MAGs were finally identified, indicating that comparative genomic analysis at the strain level was efficient and informative.
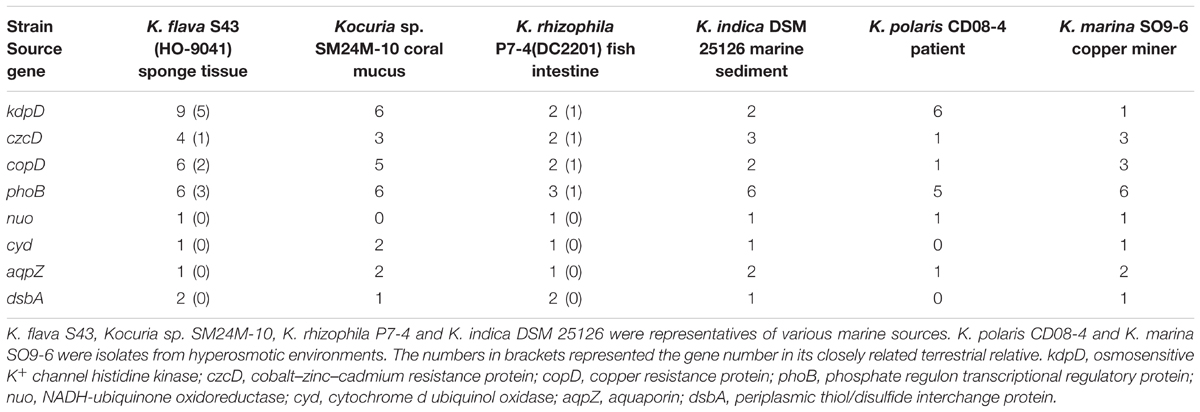
TABLE 4. The abundances of the MAGs in the investigated Kocuria spp. genomes.
Gene gain is a major force driving bacterial evolution. The presence of the MAGs may be attributed to gene gain in marine Kocuria or gene loss in terrestrial Kocuria. In this study, the quantification of gene gain and loss was not performed. However, the gene arrangement in the K. flava S43 genome can provide a clue on gene gain or loss. The identified 21 MAGs are not scattered in the genome but concentrate in two genomic regions. One region consists of the seven nuo genes, and the other centers on the cyd genes. Particularly, a total of 12 genes (kdpD, phoB, cycA, cycB, cycCD, aqpZ, czcD, copD, kdpD, phoB, dsbA, copD) arrange in a cluster. On the upstream and downstream of the cluster, respectively, locates one mobile element, suggesting the gene cluster largely comprising the MAGs is acquired by horizontal gene transfer. Thus, gene acquisition potentially represents a primary mechanism of marine adaptation in K. flava S43.
The MAGs identified in the K. flava S43 genome potentially function in various ways. The respiratory complex I encoded by the nuo operon can create a proton-motive force for the generation of ATP and help maintain a proton gradient in seawater (Tokuda and Unemoto, 1982). The terminal respiratory oxidase encoded by cydA and cydB can act as the terminal acceptor of an electron transport chain and is therefore the key enzyme of respiration, and the ABC transporter encoded by cydCD can export cysteine, which is crucial for redox homeostasis in the periplasm (Pittman et al., 2002). The thiol/disulfide interchange protein encoded by dsbA can oxidize cysteine thiols of apocytochromes c and play a role in the biogenesis of c-type cytochromes (Peek and Taylor, 1992). Considering the high osmolarity in marine environments, the aquaporin AqpZ can provide the osmoregulation by mediating influx/efflux of water (Calamita, 2000), and the sensor kinase KdpD can osmotically regulate the potassium uptake (Jung et al., 1997). Surface seawater is known to contain trace amounts of heavy metals such as cadmium, copper, and zinc, the intracellular accumulation of which is harmful for the microorganisms. The copper resistance protein CopD can maintain the balance of copper by exporting copper and the cobalt–zinc–cadmium resistance protein CzcD can detoxify the periplasm by the export of toxic metal cations (cobalt, zinc, and cadmium) (Nies and Silver, 1995). Given the two-component response regulator PhoB can regulate phosphate assimilation in a sophisticated manner (Marzan and Shimizu, 2011), it probably facilitates the survival of K. flava S43 under phosphate-limiting niche. Taken together, the MAGs can help the marine Kocuria spp. adapt to oligotrophy and hyperosmolarity and resist to heavy metal ions.
The present study reveals that the Kocuria isolates from various marine and hyperosmotic environments have some common genomic features related to environmental adaptation. K. flava S43, Kocuria sp. SM24M-10, K. indica DSM 25126, K. rhizophila P7-4, K. polaris CD08-4, and K. marina SO9-6 were isolated from distinct habitats. The presence of some common MAGs suggests that the Kocuria isolates may adapt to respective niche in the same ways. The abundance of the MAGs in the Kocuria spp. varies as species, which can be attributed to the difference in geographic and environmental factors. It warrants attention that the nuo genes are missing in Kocuria sp. SM24M-10, and both cyd and dsbA are absent in K. polaris CD08-4. This phenomenon can be explained by their close associations with the hosts (coral mucus and patient), which provide nutrient-rich niches for the Kocuria isolates so they may reduce the energy requirement. Furthermore, Kocuria sp. SM24M-10, K. indica DSM 25126 and K. marina SO9-6 have more aqpZ genes than other Kocuria spp., implying the requirement of more aquaporins to regulate osmolarity. Taken together, these results indicate that the MAGs are not essential for the survival in marine environments. However, the presence of the genes potentially improves the environmental adaptability of the Kocuria isolates.
It was proposed that no common genetic basis for marine adaptation existed among different actinobacterial genera (Penn and Jensen, 2012). Our study shows that the MAG pool in marine Kocuria isolates markedly differs from those in Salinispora or marine Streptomyces, further supporting this suggestion. Specifically, over half the MAGs in Kocuria are grouped into electron transport systems, however, in Salinispora channels and pores account for more than half of the MAGs (Penn and Jensen, 2012). K+ transporters (Trk) and betaine/carnitine/choline transporters are highly rich in marine Streptomyces group (Tian et al., 2016). It is noteworthy that although the MAG pool varies significantly in gene content and abundance among different actinomycete genera, the type and function of the MAGs are almost consistent, i.e., respiration/electron transport system, cation and ABC transporters and channels/pores. The results suggest that the functional adaptation of diverse actinomycetes to marine environments is convergent. Previous analyses revealed that the mechanisms of marine adaptation in Salinispora spp. were fundamentally different from those reported for Gram-negative bacteria (Penn and Jensen, 2012). Our finding that the gene content in the MAG pool of Kocuria significantly differs from that of Gram-negative bacteria also supports this conclusion. The role of Nqr in Gram-negative marine bacteria seems to be replaced by that of a proton-pumping NADH dehydrogenase in marine actinomycetes. In contrast to the well-studied Gram-negative bacteria, much fewer researches have focused on Gram-positive bacteria. With increasing Gram-positive bacteria investigated from a genomic view, a clear picture of marine adaptation in Gram-positive bacteria will be obtained.
Conclusion
This study reveals that K. flava S43 might adapt to the marine environment by increasing the number of the genes related to potassium homeostasis, phosphate metabolism and resistance to heavy metals, and acquiring the genes associated with electron transport and the genes encoding ABC transporter, aquaporin, and thiol/disulfide interchange protein. Gene acquisition is potentially a primary mechanism of environmental adaptation in K. flava S43. Furthermore, this study indicates that the Kocuria isolates from various marine and hyperosmotic environments have common genomic characteristics for environmental adaptation. The findings expand our knowledge of the genetic basis for marine adaptation in Kocuria.
Author Contributions
ZL and WS conceived and designed the study. MZ was responsible for sequencing and assembly. WS, CL, and FZ performed data analyses. WS and ZL drafted the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC, 41776138, U1301131, 81102417).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01257/full#supplementary-material
TABLE S1 | The functional annotation of the unique ORFs in the K. flava S43 genome.
Footnotes
- ^http://rdp.cme.msu.edu/
- ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ftp://ftp.ncbi.nih.gov/genomes/genbank/bacteria/Kocuria
- ^http://rast.nmpdr.org/
- ^http://phylogeny.lirmm.fr/phylo_cgi/simple_phylogeny.cgi
References
Abdelmohsen, U. R., Bayer, K., and Hentschel, U. (2014). Diversity, abundance and natural products of marine sponge-associated actinomycetes. Nat. Prod. Rep. 31, 381–399. doi: 10.1039/c3np70111e
Angiuoli, S. V., Gussman, A., Klimke, W., Cochrane, G., Field, D., Garrity, G., et al. (2008). Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 12, 137–141. doi: 10.1089/omi.2008.0017
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bala, M., Kaur, C., Kaur, I., Khan, F., and Mayilraj, S. (2012). Kocuria sediminis sp. nov., isolated from a marine sediment sample. Antonie Van Leeuwenhoek 101, 469–478. doi: 10.1007/s10482-011-9654-2
Bucarey, S. A., Penn, K., Paul, L., Fenical, W., and Jensen, P. R. (2012). Genetic complementation of the obligate marine actinobacterium Salinispora tropica with the large mechanosensitive channel gene mscL rescues cells from osmotic downshock. Appl. Environ. Microbiol. 78, 4175–4182. doi: 10.1128/AEM.00577-12
Calamita, G. (2000). The Escherichia coli aquaporin-Z water channel. Mol. Microbiol. 37, 254–262. doi: 10.1046/j.1365-2958.2000.02016.x
Castro, D. B. A., Pereira, L. B., e Silva, M. V. M., da Silva, B. P., Palermo, B. R. Z., Carlos, C., et al. (2015). High-quality draft genome sequence of Kocuria marina SO9-6, an actinobacterium isolated from a copper mine. Genom. Data 5, 34–35. doi: 10.1016/j.gdata.2015.05.006
Chander, A. M., Kumari, M., Kochhar, R., Dhawan, D. K., Bhadada, S. K., and Mayilraj, S. (2017). Genome sequence of Kocuria polaris strain CD08_4, an isolate from the duodenal mucosa of a celiac disease patient. Genome Announc. 5:e1158-17. doi: 10.1128/genomeA.01158-17
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Dastager, S. G., Tang, S. K., Srinivasan, K., Lee, J. C., and Li, W. J. (2014). Kocuria indica sp. nov., isolated from a sediment sample. Int. J. Syst. Evol. Microbiol. 64, 869–874. doi: 10.1099/ijs.0.052548-0
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36(Suppl. 2), W465–W469. doi: 10.1093/nar/gkn180
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Gan, H. M., Hudson, A. O., Rahman, A. Y. A., Chan, K. G., and Savka, M. A. (2013). Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics 14:431. doi: 10.1186/1471-2164-14-431
Ian, E., Malko, D. B., Sekurova, O. N., Bredholt, H., Rückert, C., Borisova, M. E., et al. (2014). Genomics of sponge-associated Streptomyces spp. closely related to Streptomyces albus J1074: insights into marine adaptation and secondary metabolite biosynthesis potential. PLoS One 9:e96719. doi: 10.1371/journal.pone.0096719
Jiang, S., Sun, W., Chen, M., Dai, S., Zhang, L., Liu, Y., et al. (2007). Diversity of culturable actinobacteria isolated from marine sponge Haliclona sp. Antonie Van Leeuwenhoek 92, 405–416. doi: 10.1007/s10482-007-9169-z
Jiang, Z., Zhang, W. H., Yuan, C. G., Chen, J. Y., Cao, L. X., Park, D. J., et al. (2015). Kocuria subflava sp nov., isolated from marine sediment from the Indian Ocean. Antonie Van Leeuwenhoek 108, 1349–1355. doi: 10.1007/s10482-015-0587-z
Jung, K., Tjaden, B., and Altendorf, K. (1997). Purification, reconstitution, and characterization of KdpD, the turgor sensor of Escherichia coli. J. Biol. Chem. 272, 10847–10852.
Khan, S. T., Komaki, H., Motohashi, K., Kozone, I., Mukai, A., Takagi, M., et al. (2011). Streptomyces associated with a marine sponge Haliclona sp.; biosynthetic genes for secondary metabolites and products. Environ. Microbiol. 13, 391–403. doi: 10.1111/j.1462-2920.2010.02337.x
Kim, W. J., Kim, Y. O., Kim, D. S., Choi, S. H., Kim, D. W., Lee, J. S., et al. (2011). Draft genome sequence of Kocuria rhizophila P7-4. J. Biotechnol. 193, 4286–4287. doi: 10.1128/JB.05334-11
Li, Z. Y., and Liu, Y. (2006). Marine sponge Craniella austrialiensis-associated bacterial diversity revelation based on 16S rDNA library and biologically active actinomycetes screening, phylogenetic analysis. Lett. Appl. Microbiol. 43, 410–416. doi: 10.1111/j.1472-765X.2006.01976.x
Mahmoud, H. M., and Kalendar, A. A. (2016). Coral-associated Actinobacteria: diversity, abundance, and biotechnological potentials. Front. Microbiol. 7:204. doi: 10.3389/fmicb.2016.00204
Marzan, L. W., and Shimizu, K. (2011). Metabolic regulation of Escherichia coli and its phoB and phoR genes knockout mutants under phosphate and nitrogen limitations as well as at acidic condition. Microb. Cell Fact. 10:39. doi: 10.1186/1475-2859-10-39
Moran, M. A., Belas, R., Schell, M. A., Gonzalez, J. M., Sun, F., Sun, S., et al. (2007). Ecological genomics of marine roseobacters. Appl. Environ. Microbiol. 73, 4559–4569. doi: 10.1128/AEM.02580-06
Morris, R. M., Nunn, B. L., Frazar, C., Goodlett, D. R., Ting, Y. S., and Rocap, G. (2010). Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 4, 673–685. doi: 10.1038/ismej.2010.4
Mulkidjanian, A. Y., Dibrov, P., and Galperin, M. Y. (2008). The past and present of sodium energetics: may the sodium-motive force be with you. Biochim. Biophys. Acta 1777, 985–992. doi: 10.1016/j.bbabio.2008.04.028
Nies, D. H., and Silver, S. (1995). Ion efflux systems involved in bacterial metal resistances. J. Ind. Microbiol. 14, 186–199. doi: 10.1007/BF01569902
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Palenik, B., Brahamsha, B., Larimer, F. W., Land, M., Hauser, L., Chain, P., et al. (2003). The genome of a motile marine Synechococcus. Nature 424, 1037–1042. doi: 10.1038/nature01943
Palermo, B. R. Z., Castro, D. B. A., Pereira, L. B., Cauz, A. C. G., Magalhães, B. L., Carlos, C., et al. (2016). Draft genome sequence of Kocuria sp. SM24M-10 isolated from coral mucus. Genom. Data 7, 121–123. doi: 10.1016/j.gdata.2015.12.016
Peek, J. A., and Taylor, R. K. (1992). Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 89, 6210–6214. doi: 10.1073/pnas.89.13.6210
Penn, K., and Jensen, P. R. (2012). Comparative genomics reveals evidence of marine adaptation in Salinispora species. BMC Genomics 13:86. doi: 10.1186/1471-2164-13-86
Pittman, M. S., Corker, H., Wu, G., Binet, M. B., Moir, A. J., and Poole, R. K. (2002). Cysteine is exported from the Escherichia coli cytoplasm by CydDC, an ATP-binding cassette-type transporter required for cytochrome assembly. J. Biol. Chem. 277, 49841–49849. doi: 10.1074/jbc.M205615200
Schäfer, G., Penefsky, H., Kerscher, S., Dröse, S., Zickermann, V., and Brandt, U. (2008). The three families of respiratory NADH dehydrogenases. Results Probl. Cell Differ. 45, 185–222.
Schneemann, I., Nagel, K., Kajahn, I., Labes, A., Wiese, J., and Imhoff, J. F. (2010). Comprehensive investigation of marine Actinobacteria associated with the sponge Halichondria panicea. Appl. Environ. Microbiol. 76, 3702–3714. doi: 10.1128/aem.00780-10
Seo, Y. B., Kim, D. E., Kim, G. D., Kim, H. W., Nam, S. W., Kim, Y. T., et al. (2009). Kocuria gwangalliensis sp. nov., an actinobacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 59, 2769–2772. doi: 10.1099/ijs.0.008482-0
Sun, W., Zhang, F., He, L., Karthik, L., and Li, Z. (2015). Actinomycetes from the South China Sea sponges: isolation, diversity, and potential for aromatic polyketides discovery. Front. Microbiol. 6:1048. doi: 10.3389/fmicb.2015.01048
Takami, H., Takaki, Y., and Uchiyama, I. (2002). Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res. 30, 3927–3935. doi: 10.1093/nar/gkf526
Takarada, H., Sekine, M., Kosugi, H., Matsuo, Y., Fujisawa, T., Omata, S., et al. (2008). Complete genome sequence of the soil actinomycete Kocuria rhizophila. J. Bacteriol. 190, 4139–4146. doi: 10.1128/JB.01853-07
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/MMBR.00040-06
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Tian, X., Zhang, Z., Yang, T., Chen, M., Li, J., Chen, F., et al. (2016). Comparative genomics analysis of Streptomyces species reveals their adaptation to the marine environment and their diversity at the genomic level. Front. Microbiol. 7:998. doi: 10.3389/fmicb.2016.00998
Tokuda, H., and Unemoto, T. (1982). Characterization of the respiration-dependent Na+ pump in the marine bacterium Vibrio alginolyticus. J. Biol. Chem. 257, 10007–10014.
Unemoto, T., and Hayashi, M. (1993). Na+-translocating NADH-quinone reductase of marine and halophilic bacteria. J. Bioenerg. Biomembr. 25, 385–391. doi: 10.1007/BF00762464
Zhou, G., Luo, X., Tang, Y., Zhang, L., Yang, Q., Qiu, Y., et al. (2008). Kocuria flava sp. nov. and Kocuria turfanensis sp. nov., airborne actinobacteria isolated from Xinjiang, China. Int. J. Syst. Evol. Microbiol. 58, 1304–1307. doi: 10.1099/ijs.0.65323-0
Keywords: marine sponge, Kocuria flava, environmental adaptation, comparative genomics, third generation sequencing (TGS)
Citation: Sun W, Liu C, Zhang F, Zhao M and Li Z (2018) Comparative Genomics Provides Insights Into the Marine Adaptation in Sponge-Derived Kocuria flava S43. Front. Microbiol. 9:1257. doi: 10.3389/fmicb.2018.01257
Received: 30 December 2017; Accepted: 24 May 2018;
Published: 08 June 2018.
Edited by:
Wen-Jun Li, Sun Yat-sen University, ChinaReviewed by:
Fei Gan, Pennsylvania State University, United StatesGuangyi Wang, Tianjin University, China
Kui Hong, Wuhan University, China
Copyright © 2018 Sun, Liu, Zhang, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyong Li, enlsaUBzanR1LmVkdS5jbg==