Abstract
The coinage “pan-genome” was first introduced dating back to 2005, and was used to elaborate the entire gene repertoire of any given species. Core genome consists of genes shared by all bacterial strains studied and is considered to encode essential functions associated with species’ basic biology and phenotypes, yet its relatedness with bacterial lifestyle of the species remains elusive. We performed the pan-genome analysis of sulfur-oxidizing acidophile Acidithiobacillus thiooxidans as a case study to highlight species’ core genome and its relevance with autotrophic lifestyle of bacterial species. The mathematical modeling based on bacterial genomes of A. thiooxidans species, including a novel strain ZBY isolated from Zambian copper mine plus eight other recognized strains, was attempted to extrapolate the expansion of its pan-genome, suggesting that A. thiooxidans pan-genome is closed. Further investigation revealed a common set of genes, many of which were assigned to metabolic profiles, notably with respect to energy metabolism, amino acid metabolism, and carbohydrate metabolism. The predicted metabolic profiles of A. thiooxidans were characterized by the fixation of inorganic carbon, assimilation of nitrogen compounds, and aerobic oxidation of various sulfur species. Notably, several hydrogenase (H2ase)-like genes dispersed in core genome might represent the novel classes due to the potential functional disparities, despite being closely related homologous genes that code for H2ase. Overall, the findings shed light on the distinguishing features of A. thiooxidans genomes on a global scale, and extend the understanding of its conserved core genome pertaining to autotrophic lifestyle.
Introduction
The development of next-generation sequencing technologies has engendered the public databases inundated with plentiful sequenced genomes (Vernikos et al., 2015). Using the gene content of microbial genomes, large-scale comparative genomics has advanced our understanding of extensive intra-species diversity (Pallen and Wren, 2007; Zhang et al., 2016a). To address the question of how many genomes were needed to fully characterize a bacterial species, the best approximation was to introduce the concept of pan-genome (“pan,” derived from Greek word “παν,” meaning “whole”; Medini et al., 2005; Tettelin et al., 2005), which was first coined to define the global gene repertoire across all strains of individual species. Core genome consists of genes shared by all strains of any given species, and probably encodes functions necessary for its fundamental biological processes and phenotypes (Medini et al., 2005; Tettelin et al., 2008). And the remaining is dispensable genome that contains genes present in some but not all the strains studied and strain-specific genes, and probably contributes to the species’ diversity (Tettelin et al., 2008). Pan-genome analyses provide a framework not only to estimate the genomic diversity of a species using the datasets at hand, but also to predict, via extrapolation, the extension of bacterial pan-genome (Vernikos et al., 2015). Mathematical modeling based on bacterial pan-genomes indicated that in the species with an open pan-genome, novel genes would be added to the gene repertoire of a species with new genomes sequenced. In the species having the closed pan-genome, however, novel genomes sequenced might not provide additional new genes to expand the pan-genome (Vernikos et al., 2015). An open pan-genome hints that species has multiple ways of exchanging genetic material to extend the microbial gene pool. For a closed pan-genome, in contrast, species’ genome is more conserved due to a low capacity to acquire alien genes (Medini et al., 2005; Tettelin et al., 2005), indicating a more genome stability. And in fact, in both open and closed pan-genome, the essence of a species pertains to core genome, which is responsible for the basic biology and major phenotypic traits. Thus, it is of interest to investigate the genetic attributes of core genome that encodes for all possible lifestyles.
Members of the genus Acidithiobacillus (Kelly and Wood, 2000) are Gram-negative and rod-shaped bacteria, which are affiliated to the class Acidithiobacillia (Williams and Kelly, 2013). As of 2018, considerable efforts have been made to delineate several recognized species belonging to Acidithiobacillus genus: Acidithiobacillus thiooxidans (Waksman and Joffe, 1922), Acidithiobacillus ferrooxidans (Temple and Colmer, 1951), Acidithiobacillus caldus (Hallberg and Lindström, 1994), Acidithiobacillus. albertensis (Kelly and Wood, 2000), Acidithiobacillus ferrivorans (Hallberg et al., 2010), Acidithiobacillus ferridurans (Hedrich and Johnson, 2013a), and Acidithiobacillus ferriphilus (Falagán and Johnson, 2016). Very recently, a comprehensive phylogenetic analysis has been performed to elucidate the hierarchical relationships among an extensive set of Acidithiobacillus strains using molecular systematics approaches (Nuñez et al., 2017). It has been widely acknowledged that these microorganisms ubiquitously occur in both pristine ecological niches (e.g., acid rock drainage and sulfur springs) and acidic settings of anthropogenic origins (e.g., acid mine drainage and bioleaching operations; Valdés et al., 2008; You et al., 2011; Bonnefoy and Holmes, 2012; Chen et al., 2013; Talla et al., 2014; Yin et al., 2014a; Nuñez et al., 2017). Except for the scientific merits as model extreme acidophiles, the consortium of Acidithiobacillus isolates has been successfully exploited in the bio-hydrometallurgical operations for metal extraction from sulfur-bearing minerals (Johnson, 2014; Nuñez et al., 2016). Due to their capabilities of dissimilatory oxidation of elemental sulfur and a wide range of reduced inorganic sulfur compounds (RISCs), Acidithiobacillus species are believed to play a crucial role in the biogeochemical cycle of sulfur in the living habitats (Zhang et al., 2016c).
Acidithiobacillus thiooxidans (formerly Thiobacillus thiooxidans), a validated species of Acidithiobacillus genus, has been traditionally studied dating back to 1920s (Waksman and Joffe, 1922). It is a sulfide-oxidizing autotroph, which obtains energy derived from sulfur oxidation to support its autotrophic growth. In the last decade, numerous studies on sulfur oxidation of A. thiooxidans have been conducted mainly based on genome-wide analyses (Valdes et al., 2011; Travisany et al., 2014; Yin et al., 2014a,b; Zhang et al., 2015). Also, stoichiometric modeling has been attempted to construct the potential metabolic pathway of sulfur oxidation (Bobadilla Fazzini et al., 2013). In contrast to members of A. ferrooxidans and A. ferrivorans, A. thiooxidans isolates have no ability to aerobically oxidize the ferrous iron. In addition, hydrogen utilization was experimentally observed in several Acidithiobacillus organisms including A. ferrooxidans, A. ferridurans, and A. caldus, but not in A. thiooxidans and A. ferrivorans (Hedrich and Johnson, 2013b). Much of recent efforts have expanded the scope of genetic traits and bioenergetic pathways within A. thiooxidans strains.
In this study, nine draft genomes of A. thiooxidans strains (Valdes et al., 2011; Travisany et al., 2014; Yin et al., 2014b; Zhang et al., 2016a) were included. The geographic origins of these nine strains were previously reported in our earlier study (Zhang et al., 2016a), such as copper mine, Kimmeridge clay, and coal heap drainage worldwide. Comparisons of A. thiooxidans genomes have been made to gain a deeper appreciation of hereditary variation of A. thiooxidans isolates (Travisany et al., 2014; Zhang et al., 2016a). However, relatively few comparative analyses have focused on its conserved genome potentially related to the autotrophic lifestyle. Here a novel strain ZBY isolated from Zambian copper mine tailings was phylogenetically affiliated to A. thiooxidans species, and its genome was released to the GenBank database. Using the newly sequenced genome plus other recognized genomes of A. thiooxidans strains, pan-genome analysis presents the elucidation of their gene repertoire, metabolic profiles, and functional features. This work provides a coherent picture of genomic traits of A. thiooxidans isolates, and sheds light on the potential relevance between a large conserved common genes and autotrophic lifestyle of A. thiooxidans species.
Materials and Methods
Sample Collection, Strain Isolation, and Bacterial Culture
Samples were collected from bioleaching heaps at Chambishi copper mine in Zambia. Mineral samples were repeatedly flushed with distilled water (pH 2.0), and the solution was then filtered using a 0.22-μm pore-size filter membrane as previously described (Zhang et al., 2016d). Gradient dilution was implemented to isolate the targeted microorganisms according to their respective growth conditions. Before inoculation, elemental sulfur (boiling sterilized, 10 g/L) was added in liquid 9K basic culture medium. Bacterial strain was grown at 30°C in liquid medium on a shaking table at 170 rpm, as previously described by Yin et al. (2014a). After sequential dilution, pure isolate, designated ZBY, was aerobically cultivated at optimized conditions as above.
Genomic DNA Extraction, Genome Sequencing, and Assembly
We collected bacterial cells at the stationary phase by centrifugation (12,000 g) for 20 min at 4°C. Total genomic DNA was extracted using TIANamp Bacteria DNA Kit (TIANGEN, Beijing, China), and the DNA quality was then assessed using a NanoDrop® ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, United States). The Illumina MiSeq platform (Illumina, CA, United States) was applied for whole-genome sequencing. We constructed the shotgun library with an average DNA insert size of 300 bp, which was used for 2 × 150 bp paired-end sequencing. After sequencing, all raw reads were screened using NGS QC Toolkit v2.3.1 (Patel and Jain, 2012) with the following parameters: cut-off read length for high-quality (HQ), 70%; cut-off quality score, 20. After quality control, the HQ sequences were de novo assembled using the Velvet program (Zerbino and Birney, 2008) with various k-mers. The best assembly was then used to generate unigene sequences by the iAssembler program (Zheng et al., 2011). GC content, N50, and N90 sequence length of genome assembly were calculated using the script “N50Stat.pl” within NGS QC Toolkit, and its completeness was evaluated using the CheckM program (Parks et al., 2015).
Analysis of Phylogeny and Taxonomy
In order to establish how this strain was related to other Acidithiobacillus strains, we used the 16S rRNA sequences to construct a phylogeny. High sequence identities (≥97%) between newly sequenced strain and other recognized Acidithiobacillus strains preliminarily suggested their phylogenetic relationships. Accordingly, 16S rRNA gene sequences of Acidithiobacillus spp. deposited in GenBank database were acquired from individual genomes according to the approach as above. Multiple sequence alignment based on 16S rRNA sequences from various strains including newly sequenced strain was performed using a ClustalX v1.81. The ClustalX result was then used to construct the maximum likelihood tree implementing MEGA v5.05 (Tamura et al., 2011). Herein, the Tamura–Nei model of nucleotide substitution was applied for phylogenetic reconstruction. To evaluate clade support, we constructed a bootstrap analysis with 1,000 replicates. Furthermore, a genome-based phylogeny of Acidithiobacillus spp. was constructed using the online platform CVTree3 with K-tuple length 12 (Zuo and Hao, 2015). Here Leptospirillum ferrooxidans C2-3 was included as an out-group.
Comparisons of average nucleotide identity (ANI) based on BLAST algorithm (ANIb; Goris et al., 2007) and MUMmer algorithm (ANIm; Kurtz et al., 2004), as well as tetranucleotide frequency correlation coefficient (TETRA, Teeling et al., 2004) were further conducted using the software JSpecies v1.2.1 (Richter and Rossellómóra, 2009) to infer the phylogenetic relationships between pairs of bacterial genomes. For ANI calculation, default parameters were applied as follows: sequence identity cut-off, 30%; alignment cut-off, 70%; and query length, 1,020 bp. General features of the selected genomes are shown in Supplementary Table S1. A software HemI (Deng et al., 2014) was used to visualize the calculation results.
Pan-Genome Analysis
In our study, nine of A. thiooxidans strains including new sequenced strain were selected for pan-genome analysis (Supplementary Table S2). Draft genome of ATCC 19377 was excluded in this part due to the low sequencing depth. We performed the BLASTP all-versus-all comparisons of entire amino acid sequences between pairs of A. thiooxidans strains. BLAST results with a tabular format (m8) were subsequently used to identify the orthologs among all of A. thiooxidans strains using the Pan-genome Ortholog Clustering Tool (PanOCT) v3.18 program (Fouts et al., 2012) with E-value cut-off of 1e-5, sequence identity cut-off of 50%, and match length cut-off of 65 bp. In this procedure, mobile genetic elements were removed from the datasets prior to the determination of orthologous genes as described previously (Snipen and Ussery, 2010). The results were then manually inspected and corrected.
Mathematical Modeling of A. thiooxidans Pan-Genome
The sizes of core genome and novel genes of A. thiooxidans species depend on how many genomes are taken into consideration. To extrapolate the sizes of core-genome and pan-genome, the number of core and strain-specific genes were used to fit the exponential decaying curve Fc = 𝜀cexp(-n/τc) + Ω and Fs = 𝜀sexp(-n/τs) + tg(𝜃), respectively. In this formula, n is the number of strains and 𝜀c, 𝜀s, τc, τs, Ω, and tg(𝜃) are fitting parameters. Ω represents the size of core-genome and tg(𝜃) is the growth rate of pan-genome when n →∞ (Tettelin et al., 2005). Subsequently, we implemented the power law Ps = κn-α (Tettelin et al., 2008) to determine whether the A. thiooxidans pan-genome is open (α ≤ 1) or closed (α > 1). Here, κ and α are fitting parameters. To evaluate the normality of the number of common and new genes, normality test was carried out by function “shapiro.test” in the package R v3.3.3 (R Core Team, 2000). In consideration of the data non-normality (Supplementary Figure S1), medians were used to estimate the average number of common and new genes when the nth genome was added. Parameter fitting were performed by using function “nls” with least-squares algorithm. The fitting results were summarized by function “summary.” Visualization for fitting curves were carried out by “ggplot2” package.
Structural and Functional Analyses of Bacterial Genomes
NCBI prokaryotic genome annotation pipeline was applied to gene prediction and functional annotation of bacterial genome. Protein-coding sequences (CDSs) assigned to core genome and dispensable genome within strain ZBY were extracted using in-house script, and aligned against the specialized database, such as the extended Clusters of Orthologous Groups (COG; Franceschini et al., 2013); the results were screened according to the highest hit coverage value. Insertion sequences (ISs) and transposases were detected using the online platform ISfinder with an E-value cutoff set to 1e-5 (Siguier et al., 2006). The tRNAscan-SE program (Lowe and Eddy, 1997) was used for the identification of tRNA. Finally, BLASTN-based whole genome comparison was performed and visualized using the Circos software (Krzywinski et al., 2009) to exhibit the architecture and gene repertoire of A. thiooxidans pan-genome.
KEGG Automatic Annotation Server (KAAS; Moriya et al., 2007) was employed to infer the predicted metabolic profiles of core genome by sequence alignment against the manually curated KEGG GENES database. We gathered the amino acid sequences of interest for further analyses. Multiple sequence alignment was performed using the software DNAMAN v7.0.2, and phylogenetic tree based on the targeted proteins from various species was constructed using the MEGA v5.05. Additionally, pairwise comparisons of putative gene clusters within A. thiooxidans genomes were visualized using the software EasyFig v2.1 (Sullivan et al., 2011).
Availability of Supporting Data
The datasets supporting the findings of this study are available in NCBI repository. The Whole Genome Shotgun project regarding the newly sequenced strain ZBY has been deposited at DDBJ/EMBL/GenBank under the accession number LZYI00000000. The version described in this paper is version LZYI01000000. All additional datasets generated and/or analyzed during the current study are included within the additional files of this article.
Results
General Genome Features of New Sequenced ZBY
HQ reads generated from quality control were used to genome assembly. As a result, the draft genome contains 3,793,418 base pairs (bps) distributed in 277 contigs and its GC content is 53.17%. The lengths of maximum and minimum contigs are 155,963 and 225 bp, respectively. Genome completeness evaluated by CheckM indicated that the genome assembly was near complete (99.34%, Table 1). Draft genome of novel strain ZBY was predicted to comprise 3,713 CDSs, 117 tRNA that cover all the 20 amino acids, and 134 ISs. Further alignment against the NCBI-nr database revealed that only 2,029 (54.65%) CDSs were assigned to putative proteins with known functions, while 1,684 (45.35%) CDSs were annotated as hypothetical proteins. Functional analysis based on COG classification showed that there were a total of 2,743 (73.88%) significant BLAST hits, in which 2,741 (73.82%) CDSs were assigned to the COG categories. The four most abundant functional categories within the ZBY genome were “replication, recombination, and repair (L),” “cell wall/membrane/envelope biogenesis (M),” “energy production and conversion (C),” and “amino acid transport and metabolism (E)” (Supplementary Table S3). Furthermore, COG annotation suggested that a plenty of poorly characterized sequences (20.74%) were dispersed in the draft genome of strain ZBY, making the potential roles unclear.
Table 1
| Property | Value |
|---|---|
| Coverage | 91× |
| Number of contigs | 277 |
| Total bases | 3,793,418 |
| Completeness (%) | 99.34 |
| Min sequence length (bp) | 225 |
| Max sequence length (bp) | 155,963 |
| N50 length (bp) | 32,632 |
| N90 length (bp) | 6,269 |
| GC content (%) | 53.17 |
| Total number of CDSs | 3,713 |
| tRNAs | 117 |
| IS elements | 134 |
| CDSs with known function | 2,029 (54.65%) |
| Hypothetical proteins | 1,684 (45.35%) |
| CDSs with COG hits | 2,743 (73.88%) |
| CDSs with COGs | 2,741 (73.82%) |
General features of draft genome of newly sequenced strain ZBY.
Phylogeny and Taxonomy of Novel Strain
Strategy for phylogenetic construction was employed to assess the relationships between newly sequenced strain originally isolated from Zambian copper mine and other known Acidithiobacillus spp. deposited in public database. A dendrogram based on 16S rRNA gene sequences showed that the novel strain was clustered on a distinct branch within the identified A. thiooxidans species, but was clearly distinguished from other Acidithiobacillus isolates including A. ferrooxidans, A. ferrivorans, and A. caldus (unpublished data). Accordingly, we preliminary inferred that strain ZBY in this study was likely to be assigned to A. thiooxidans species. Since species identification at the 16S rRNA gene level has its limitation, a genome-based phylogeny was further conducted implementing the CVTree3 web server (Figure 1). In addition, ANIb and ANIm, and TETRA were further determined to infer the phylogenetic relationships between strain ZBY and other Acidithiobacillus strains (Supplementary Figure S2). High values of ANIb (≥97.27%), ANIm (≥97.88%), and TETRA (≥0.995) between pairs indicated that strain ZBY was very closely related to all recognized A. thiooxidans strains. The results combined with whole-genome-based phylogeny strongly supported the prior notion of 16S rRNA sequence-based phylogenetic tree. Taken together, we inferred that strain ZBY was phylogenetically affiliated with A. thiooxidans species.
FIGURE 1
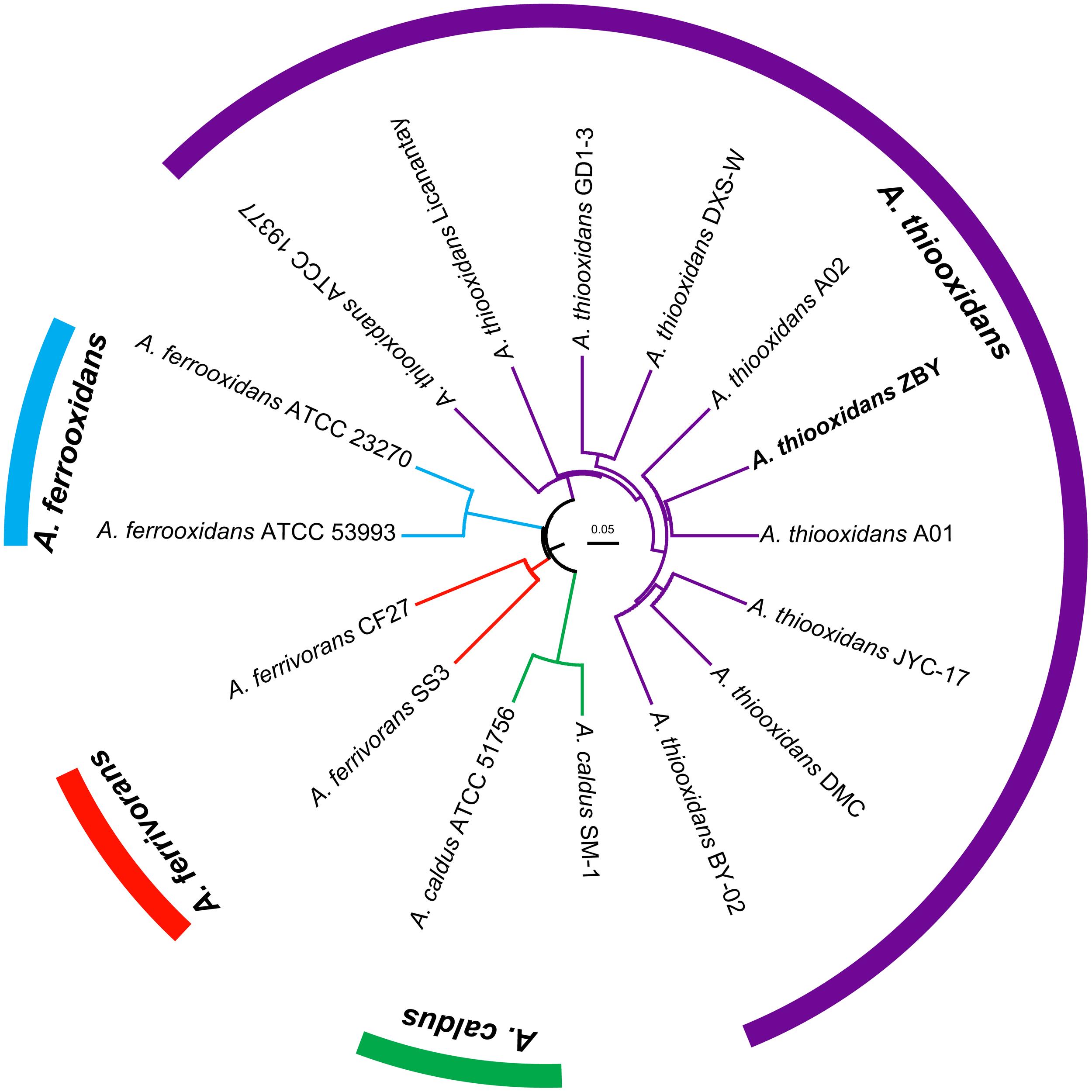
Whole-genome-based phylogeny depicting the phylogenetic relationships amongst bacterial strains of Acidithiobacillus spp. CVTree3 with a composition vector approach was applied and Leptospirillum ferrooxidans C2-3 was used as an out-group.
Pan-Genome Analysis of A. thiooxidans Strains
BLASTN-based whole genome comparisons were performed to identify the core and dispensable genomes among these A. thiooxidans strains. Circular plot depicting genome structure was visualized using the software Circos (Figure 2). Pan-genome analysis based on nine A. thiooxidans strains was conducted, showing that there were a total of 5,999 genes, in which 1,994 were identified to be shared genes (core genome; Figure 2). Further inspection showed that the percentages of shared genes accounting for each genome vastly varied, ranging from 52.0% (DXS-W) to 54.8% (A02). The non-core genes (1,719) within A. thiooxidans ZBY were assigned to dispensable genome, which contains genes shared by a subset of A. thiooxidans genomes and strain-specific genes. Interestingly, relatively few strain-specific genes (23) unique in strain ZBY were predicted compared to that in strain Licanantay (923). COG classification showed that the most abundant strain-specific genes in these strains were assigned to COG categories [L], while was in line with previous studies (Travisany et al., 2014; Zhang et al., 2016a).
FIGURE 2
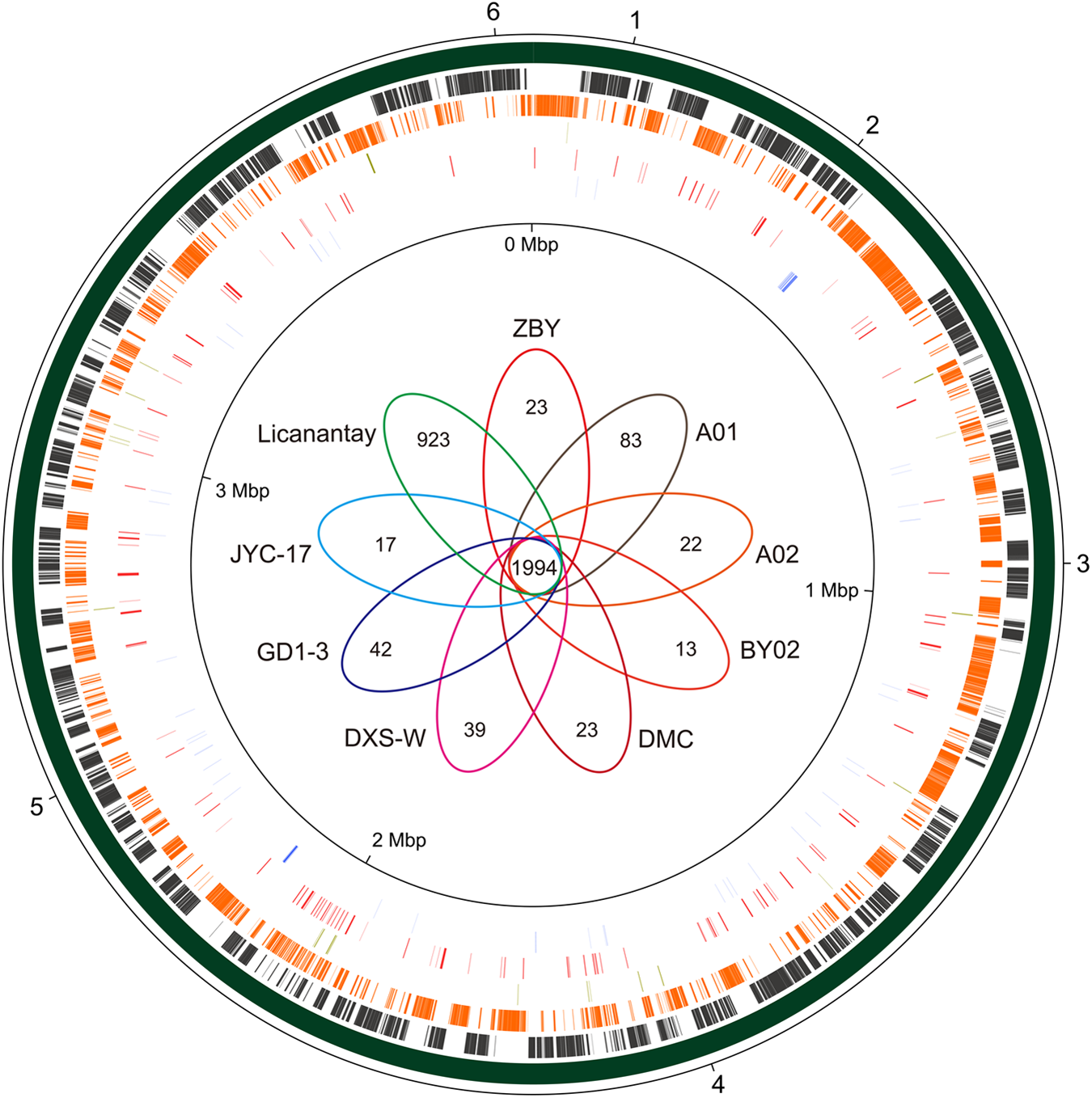
Circular representation of A. thiooxidans pan-genome. BLASTN-based whole genome comparisons of A. thiooxidans strains were performed and visualized using Circos. Here the newly sequenced strain ZBY was used as a reference. The predicted core genome based on nine strains of A. thiooxidans was shown in the second ring. Moving inward, dispensable genome and ZBY-specific genes were indicated in the following two rings (more details for the strategy of strain selection are shown in section “Materials and Methods”). In addition, transposases and tRNA were shown on the fifth and sixth circles. Venn diagram depicting the orthologous and strain-specific CDSs of A. thiooxidans strains was shown in the figure center. Several genomic regions of interest were numbered with a pan-genome locus (PL) of 1–6, including nirBD/nasA (1), carboxysome-associated gene cluster (2), hydrogenase-like gene cluster 2 (3), narGHJI (4), hydrogenase-like gene cluster 1 (5), and coxLMS (6). PL 1–6 were linked to Figure 5 and Supplementary Figures S4A–D, S5, respectively.
Mathematical Extrapolation for A. thiooxidans Pan-Genome
As stated by Tettelin et al. (2005), one or two bacterial genomes per species might provide insufficient information to understand its genetic variability. Vernikos et al. (2015) also recommended that at least five genomes were necessary for mathematical extrapolation. Accordingly, strains A01 (Yin et al., 2014b), Licanantay (Travisany et al., 2014), A02, BY-02, DMC, DXS-W, GD1-3, JYC-17 (Zhang et al., 2016a), and the novel strain ZBY, which was sequenced as part of our present study, were chosen for comparison survey. Genome sizes of these nine strains varied from 3.72 (A02) to 3.95 Mbp (DXS-W; Supplementary Table S2). The completeness values of ≥98.72% supported the mathematical modeling of A. thiooxidans pan-genome.
To predict the pan-genome size of A. thiooxidans species, the number of specific genes with each new sequenced strain was calculated. Mathematical extrapolation of nine genomic data indicated that an average of 690 novel genes were added when a second genome was sequenced (Supplementary Figure S1). The new genes decayed with new sequenced genome, and quickly converge to a non-zero asymptotic value of 28 (Figure 3A). Heaps’ law model revealed that the increase of new genes became harder as sampling proceeded (α = 4.9 ± 0.4, estimate ± SE; Supplementary Table S4), suggesting that A. thiooxidans pan-genome was predicted to be closed. In addition, the number of shared genes exponentially decayed with the sequential addition of each new genome (Figure 3B). The extrapolated curve indicated that the number of core genes reached a constant number (1,994 ± 118) when the ninth genome was added.
FIGURE 3
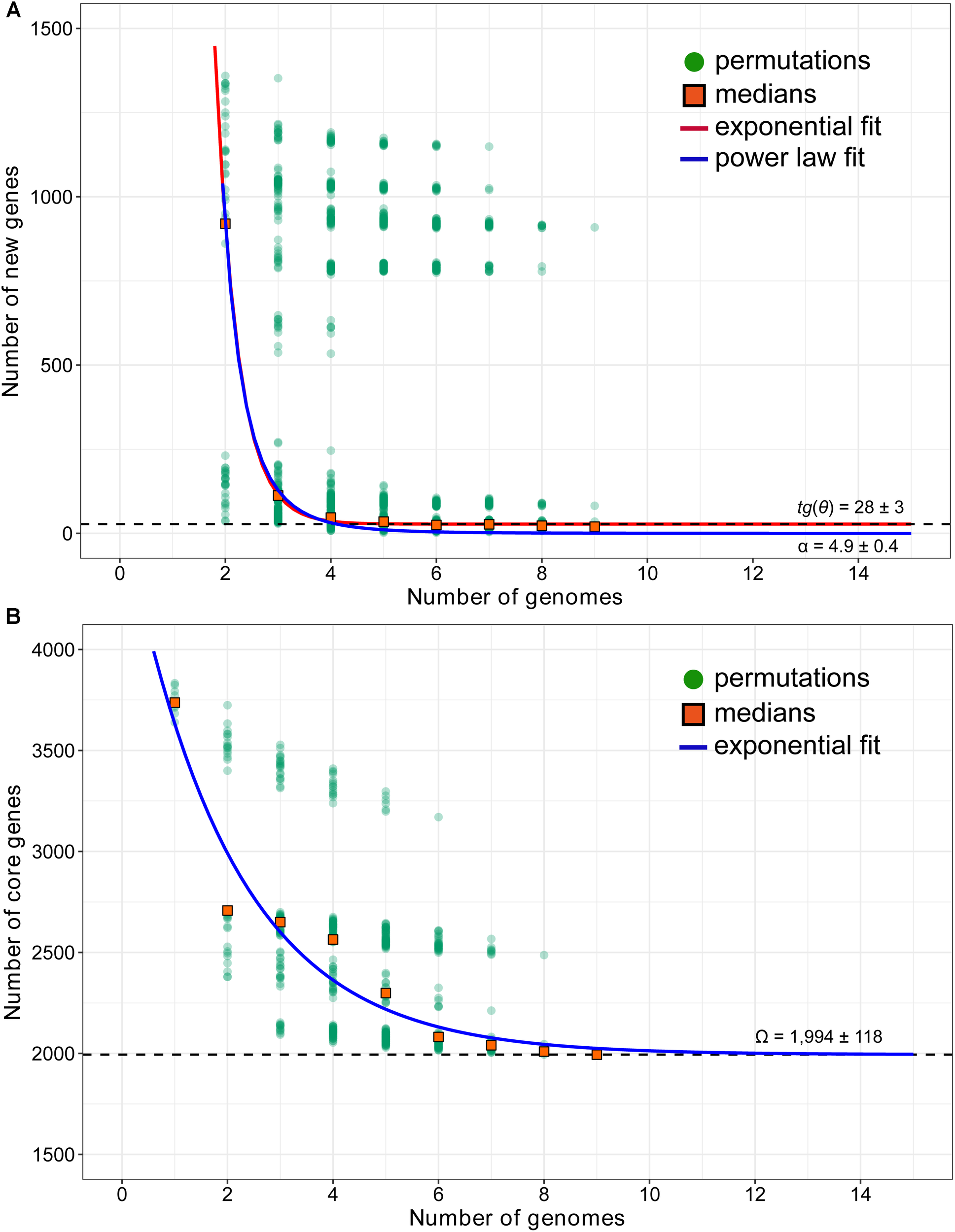
Mathematical extrapolation estimating the new genes (A) and core genome (B) of A. thiooxidans species, based on these sequenced genomes of bacterial strains (except for ATCC 19377). In the new genes extrapolation, new genes were counted by the growth of the gene pool of (n – 1) strains when they were added. For each n, there are N2 = n⋅N1 observations. Orange squares are the medians of such values presented in n strains. The curve in main window represents the least-squares fit of Fs = 𝜀sexp(–n/τs) + tg(𝜃). The optimal fitting was output with adjust R-square = 0.9996 for 𝜀s = 93,320 ± 16,520, τs = 0.43 ± 0.04, and tg(𝜃) = 28 ± 3. The dashed line displays the extrapolated growth rate of pan-genome tg(𝜃). The curve in child window represents the growth curve of pan-genome as the function P(n). In addition, the blue curve is least-squares fit of the power law Ps = κn-α to medians. A threshold parameter (α) is used to distinguish whether the A. thiooxidans pan-genome is open (α ≤ 1) or closed (α > 1); as for core gnome extrapolation, the number of core genes shared by n of strains was plotted. For each n, there are N1 = S!/[(n – 1)!⋅(S – n)!] observations, where S is the numbers of strains. Orange squares are the medians of such values presented in n strains. The blue curve represents the fit least-squares of Fc = 𝜀cexp(–n/τc) + Ω. The optimal fitting was output with adjust R-square = 0.9912 for 𝜀c = 2,690 ± 421, τc = 2.0 ± 0.54, and Ω = 1,994 ± 118. The dashed line displays the extrapolated size of core genomes Ω.
Functional Classification of the Core and Dispensable Genomes
Functional classification analysis of core and dispensable genomes was carried out by comparison against the extended COG database (Supplementary Figure S3). As expected, relatively high proportion of genes dispersed in the core genome (31.13%) were predicted to be involved in metabolic profiles compared to that in dispensable genome (11.43%). In particular, functional categories [C] (energy production and conversion; 7.17%), [E] (amino acid transport and metabolism; 6.24%), and [G] (carbohydrate transport and metabolism; 4.16%) were abundant in core genome, highlighting the importance of essential genes for basic activities of bacterial strains. In contrast, more genes within dispensable genome were assigned to COG categories [L](replication, recombination, and repair; 8.28%) and [U] (intracellular trafficking, secretion, and vesicular transport; 3.85%) in comparison with that in core genome (4.21% and 1.75%, respectively). In addition, vast genes were predicted to encode hypothetical proteins with unidentified functions, which still need to be further explored.
Linking Core Genome to the Inferred Metabolic Traits
Identification of predicted metabolic profiles of A. thiooxidans genomes was conducted, providing further insights into metabolic traits of these microorganisms. KAAS was applied to investigate the metabolic potentials of all strains (Supplementary Table S5). A large number of core genes were assigned to carbohydrate metabolism (172), amino acid metabolism (142), and energy metabolism (133); this result was consistent with COG classification. In the following sections, we focus on central metabolism of A. thiooxidans strains.
Central Carbon Metabolism
The assimilation of carbon source derived from external environments is essential for organisms. Similar to many other autotrophic acidophiles (Valdés et al., 2008; Ullrich et al., 2016; Zhang et al., 2016a,b,e), A. thiooxidans isolates have the ability to fix atmospheric CO2 via the classical Calvin–Benson–Bassham (CBB) cycle (Figure 4). For carbon fixation, ribulose-1,5-biphosphate carboxylase/oxygenase (Rubisco) directing the regeneration of ribulose-1,5-bisphosphate was commonly recognized as an crucial enzyme in CBB cycle(Zhang et al., 2016c,e). All A. thiooxidans strains were predicted to harbor a carboxysome-associated gene cluster, which potentially encodes Type I Rubisco large and small subunits, carboxysome shell carbonic anhydrase, several copies of carboxysome shell proteins and carbon dioxide-concentrating proteins (Supplementary Figure S4A).
FIGURE 4
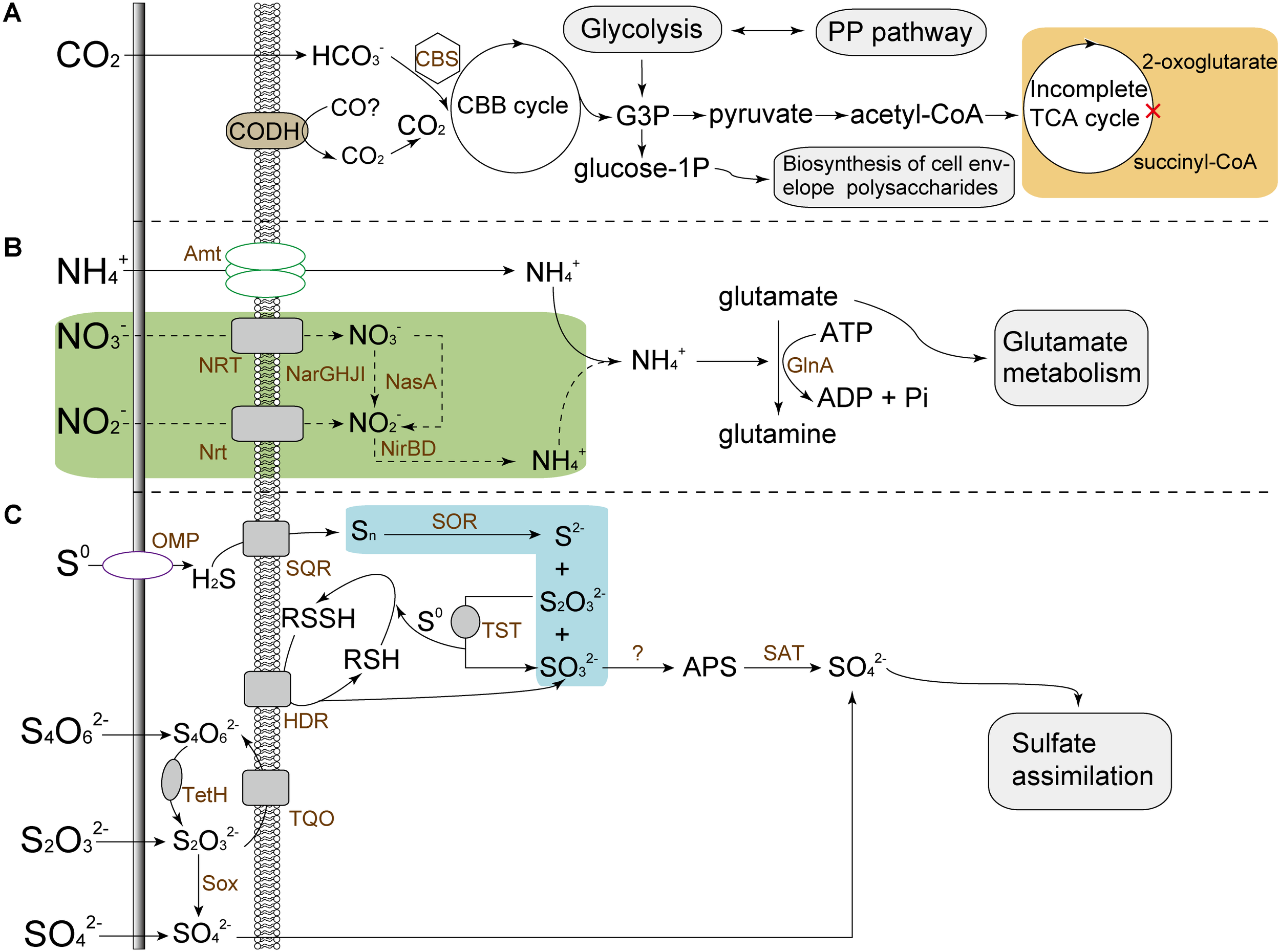
Predicted models for central metabolisms of A. thiooxidans strains, including carbon assimilation (A), nitrogen metabolism (B), and sulfur oxidation (C). In the incomplete TCA cycle (citrate cycle), 2-oxoglutarate dehydrogenase that catalyzes the transformation of 2-oxoglutarate to succinyl-CoA was predicted to be absent (orange area), which were also previously reported in some other studies (Zhang et al., 2016a,e). The nitrate reductase (NarGHJI) and nitrite reductase (NirBD) pertaining to dissimilatory nitrate reduction, and putative assimilatory nitrate reductase catalytic subunit (NasA) associated to assimilatory nitrate reduction were predicted to be present in A. thiooxidans species except for ATCC 19377 (green area). For strain ATCC 19377, ammonium was required as an alternative nitrogen source. Unlike other A. thiooxidans strains, a key enzyme sulfur oxygenase reductase (SOR) involved in sulfur oxidation was absent in draft genome of strain ATCC 19377 probably due to the low sequencing depth (blue area). CBS, carboxysome; PP pathway, pentose phosphate pathway; CODH, carbon-monoxide dehydrogenase; CBB cycle, Calvin–Benson–Bassham cycle; G3P, glyceraldehyde-3-phosphate; Amt, ammonia transporter; Nrt, nitrate/nitrite transporter; NRT, nitrate transporter; NarGHJI, nitrate reductase; NirBD, nitrite reductase; NasA, assimilatory nitrate reductase catalytic subunit; GlnA, glutamine synthetase; OMP, outer membrane protein; SQR, sulfide quinone oxidoreductase; TQO, thiosulfate:quinone oxidoreductase; TetH, tetrathionate hydrolase; Sox, sulfur oxidizing protein; TST, thiosulfate sulfurtransferase; HDR, heterodisulfide reductase.
Glyceraldehyde-3-phosphate (G3P), one of important metabolic intermediates in the process of carbon fixation, was predicted to be involved in the formation of certain precursors for the biosynthesis of amino acids, fatty acids, and polysaccharides via central metabolism. A. thiooxidans genomes were predicted to harbor genes involved in amino sugar and nucleotide sugar metabolism (ko00520; Supplementary Table S5). Of note, glucose-6-phosphate and glucose-1-phosphate converted by G3P might be involved in the conversion of precursors for the biosynthesis of cell envelope polysaccharides (Supplementary Table S6), as previously described by Ullrich et al. (2016). Additionally, a three-gene cluster (coxLMS) encoding putative carbon-monoxide dehydrogenase (EC: 1.2.7.4) were identified in A. thiooxidans genomes (Supplementary Figure S4B).
Nitrogen Assimilation
Unlike A. ferrooxidans (Valdés et al., 2008), no similar components of nitrogenase complex were identified in A. thiooxidans isolates. Comparisons of the metabolic profiles of all A. thiooxidans strains revealed that these microorganisms except for ATCC 19377 harbor genes for dissimilatory nitrate reduction, observed as nirBD and narGHJI (Supplementary Figures S4C, D, respectively). As for assimilatory nitrate reduction, gene nasA encoding putative nitrate reductase (NADH) catalytic subunit was found in all A. thiooxidans strains except for ATCC 19377. Besides, the subunit NasB that transfers electrons derived from NADH to nitrate (Lin and Stewart, 1997) was absent in A. thiooxidans genomes, thereby making the electron donor unclear. In addition, genes coding for nitrate and/or nitrite transporter were absent in A. thiooxidans ATCC 19377.
All A. thiooxidans strains were predicted to harbor the ammonium transporters to take up the external ammonia, and to assimilate the latter into central metabolic pathways by a series of enzymes, such as glutamine synthetase and glutamate synthase (Figure 4).
Sulfur Oxidation and Electron Transfer
According to COG class assignment, numerous genes pertaining to the core genome of A. thiooxidans species were predicted to be related to energy production and conversion (COG category [C]; Supplementary Figure S3). Oxidation of various sulfur species (e.g., tetrathionate and elemental sulfur) was a well-studied characteristic of A. thiooxidans strains (Yin et al., 2014a; Zhang et al., 2016a). In this study, the core genome was replete with enzymes involved in sulfur oxidation, such as sulfide quinone oxidoreductase, tetrathionate hydrolase, thiosulfate:quinone oxidoreductase, sulfur oxidizing protein system, and sulfur oxygenase reductase (Figure 4). However, the sor gene was absent in A. thiooxidans ATCC 19377, possibly due to the low-coverage genome sequencing (Zhang et al., 2016e). Also, a number of genes within core genome of A. thiooxidans species were predicted to be involved in electron transport chain, including cydAB, cyoABCD, and nuoABCDEFGHIJKLMN (Supplementary Table S7).
Hydrogenase-Like in A. thiooxidans Strains?
Numerous genes involved in energy metabolism were predicted to be distributed in the core genome of A. thiooxidans. Notably, several hydrogenase-like genes were observed in all sequenced A. thiooxidans genomes (sections 3 and 5 in Figure 2). In light of previous report showing that A. thiooxidans strains could not grow chemolithotrophically on hydrogen (Hedrich and Johnson, 2013b), the genotypic traits appear to be inconsistent with their phenotypes. To address this issue, the following sections focus on two specific genomic regions containing hydrogenase-like genes.
Hydrogenases (H2ases) directing the interconversion of molecular hydrogen (Vignais et al., 2001; Jenney and Adams, 2008; Lim et al., 2010) are widely distributed in all domains of life. They are believed to play a central role in the biological production as well as hydrogen utilization. According to the classification of H2ases (Vignais et al., 2001), three H2ases in all A. thiooxidans strains seem to be assigned to the putative membrane-bound H2 evolving [NiFe]-H2ases (group 4). A six-gene cluster was predicted to encode the H2ases complex, which consists of two H2ases, two NADH-ubiquinone oxidoreductases, a NADH dehydrogenase, and a formate dehydrogenase. Further inspection showed that these genes were organized into a bipartite fdh–mhy cluster, including three fdh genes coding for formate dehydrogenase and three mhy genes coding for membrane-bound H2ases (Figure 5A). To evaluate the phylogenetic relationship of H2ases, the large subunits encoded by mhyB genes from various species were selected for further analysis. A phylogeny demonstrated that these targeted H2ases were obviously separated into three clusters (Figure 5B), in which clusters I and II were assigned to groups 4b and 4a, respectively, as previously described by Lim et al. (2010). Notably, H2ases in Acidithiobacillus spp. (cluster III) were significantly distinct from those in other species, probably indicating a novel enzyme complex. In support of this designation, DNAMAN program was employed to perform the multiple sequence alignment. As depicted in Figure 5C, two conserved motifs (SGD[TS][TS][AV][GA]Y and NLSYSGHDL) were identified in all Acidithiobacillus spp.
FIGURE 5
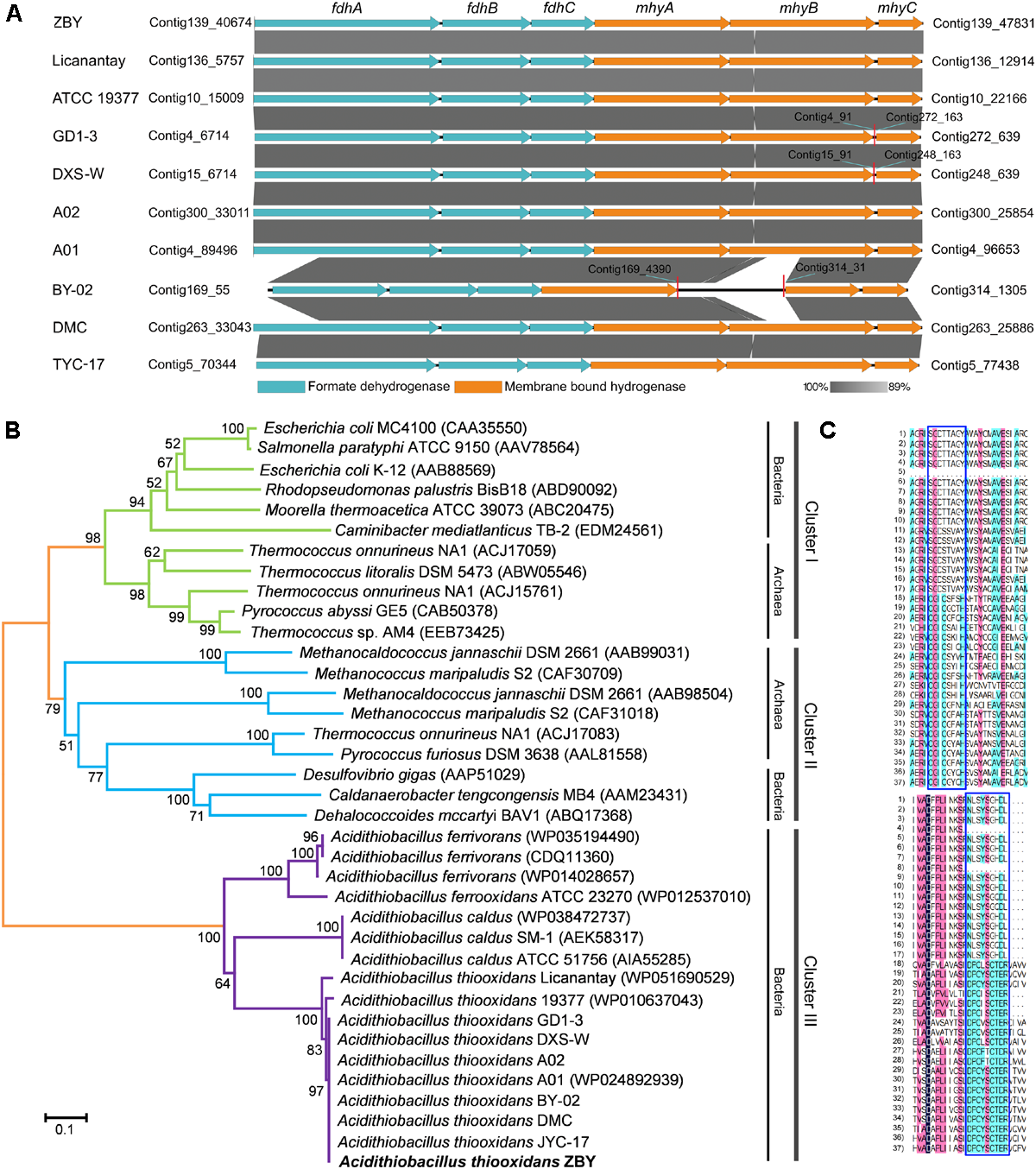
Identification of the putative formate hydrogenlyase in A. thiooxidans strains. (A) Visualization for the putative fdh–mhy gene cluster of A. thiooxidans strains using the software EasyFig. (B) Phylogenetic tree showing the relationships between Acidithiobacillus spp. and other organisms having the group 4 [NiFe]-hydrogenases. The tree was based on the large subunits of putative hydrogenases from Acidithiobacillus spp. plus various large subunits of recognized hydrogenases from different organisms including Bacteria and Archaea, and was generated and visualized using the MEGA. The neighbor-joining tree was constructed with a bootstrap analysis implementing 1,000 replicates. (C) Sequence representations of conserved motifs (in the blue boxes) from various organisms.
Another gene cluster potentially encoding formate dehydrogenase, H2ase, NADH dehydrogenase, two copies of NADH-ubiquinone oxidoreductase, and H2ase was also identified in A. thiooxidans genomes. These six genes were assigned as new labels (orf1–orf6), and compared and visualized using the software EasyFig (Supplementary Figure S5A). To further analyze the possible function of these candidate genes, the corresponding amino acid sequences were extracted for the identification of conserved domains. The results confused us as some conserved domains were involved in the [NiFe]-H2ases, whereas others were associated with the NADH-ubiquinone oxidoreductase (Supplementary Figure S5B). In order to identify their putative functional properties, amino acid sequences from A. thiooxidans ZBY were aligned to other complex, including H2ase Group III and IV and NADH-ubiquinone oxidoreductase. Results showed that orf4 and orf5 was potentially annotated as the large and small subunits of both Group III and IV [NiFe]-H2ases, respectively (Supplementary Figure S5C). According to the conserved regions of [NiFe]-H2ase groups (Vignais et al., 2001), however, no L1 and L2 signature was identified in these large subunits (unpublished).
Discussion
In our mathematical model, a threshold parameter (α > 1) indicated that the pan-genome of A. thiooxidans species was closed (Figure 3 and Supplementary Table S4). Some microbial species with a closed pan-genome, such as Bacillus anthracis and Mycobacterium tuberculosis, inhabit the isolated environments with limited access to the existing global microbial gene pool, suggesting the genome stability and relatively conserved genome (Medini et al., 2005). Aside from core genome, the remaining dispensable genes contribute to species’ genome diversity. In other microorganisms, studies have discussed issues related to the correlation between the dispensable genome and genetic variation (Lindsay et al., 2012; Graña-Miraglia et al., 2017). In comparison with microbial species harboring an open pan-genome, such as Leptospirillum ferriphilum (Zhang et al., 2018), the size of A. thiooxidans pan-genome would reach a plateau as a certain number of genomes were sampled. For species with a closed pan-genome, additional sequenced genomes would not expand the species’ pan-genome, showing a relatively limited gene content diversity (Tettelin et al., 2008).
Clusters of Orthologous Groups classification revealed that relatively more core genes within A. thiooxidans were assigned to COG categories [C], [E], and [M] compared to dispensable genes (Supplementary Figure S3). The COG classes [C] and [E] are involved in metabolisms of energy and amino acids, and COG class [M] is associated with the biogenesis of cell wall/membrane/envelope. Accordingly, we inferred that efficient uptake of energy and nutrients from acidic environments was essential for the autotrophic lifestyle of acidophile A. thiooxidans, and specialized cellular structures might provide a selective advantage to adapt to the harsh econiches. Further inspection showed that genes associated with COG class [L] in dispensable genome were more than that in core genome (Supplementary Figure S3). We therefore interpret this as an indication that the large proportion of genes involved in COG category [L] might endow A. thiooxidans with adaptive strategies to respond to the adverse environments, as high concentrations of toxic substances such as heavy metal elements in acidic bioleaching operations tend to cause DNA injury (Silver and Phung, 1996). Once cell damage occurs, the rescue system may play a critical role in DNA repair. The findings presented here were in line with the previous studies (Travisany et al., 2014; Zhang et al., 2016a), highlighting an adaptive advantage in individual biomining environments. Taken together, our results showed that dispensable genome might provide microorganisms with functions that were essential to niche adaptation (Tettelin et al., 2008).
Unlike other acidophilic bacteria such as Sulfobacillus spp. that utilize both inorganic and organic forms of carbon (Guo et al., 2014; Justice et al., 2014; Zhang et al., 2017) and Leptospirillum spp. that assimilate carbon via the reductive tricarboxylic acid cycle (Levicán et al., 2008; Zhang et al., 2016c), A. thiooxidans was known to be autotrophic carbon fixer that assimilates carbon via classical CBB cycle. In a gene cluster related to carboxysome, one gene was predicted to encode a novel carbonic anhydrase (CA; 𝜀-class CA, Zhang et al., 2016e). The carboxysome-associated carbonic anhydrase elevates the concentration of carbon dioxide near the Rubisco by the conversion of accumulated cytosolic bicarbonate into CO2 (Price et al., 2004), thereby suggesting a more efficient CO2 fixation.
With respect to nitrogen assimilation, in general, microorganisms-mediated inorganic nitrogen metabolisms are commonly involved in the uptake of atmospheric nitrogen, ammonium, nitrite, and nitrate (Arrigo, 2005). The closely related A. ferrooxidans was reported to harbor the diazotrophic lifestyle (Valdés et al., 2008; Zhang et al., 2016c). By contrast, the dissimilatory and assimilatory nitrate reduction in A. thiooxidans species (except for ATCC 19377) could be an alternative pathway to acquire nitrogen source. Additionally, genes associated to nitrate and/or nitrite transporter were absent in strain ATCC 19377. Although genes/gene clusters pertaining to dissimilatory and assimilatory nitrate reduction were identified in a number of A. thiooxidans strains, we could not determine whether these genes/gene clusters were introduced by horizontal gene transfer (HGT), since no mobile genetic element, such as transposases, integrase, and tRNA gene, was found in the neighborhood of these observed genomic regions. In other words, we inferred that the absence of the corresponding genes/gene clusters in strain ATCC 19377 was more likely caused by gene loss rather than HGT. In other organisms, many studies have focused on genome reduction in the condition of increased biological fitness and efficient utilization of limited nutrients especially in oligotrophic ecosystems (Boon et al., 2014; Giovannoni et al., 2014; Martínez-Cano et al., 2015; Zhang et al., 2017). Given that the concentrations of nitrogen resources (such as ammonium, nitrate, and nitrite) are very limited in many acidic eco-environments (Parro et al., 2007; Chen et al., 2015), the genome reduction might provide a selective advantage that drives the abandonment of expensive genes, supporting the effective acquisition of limited nutrients. Therefore, the abandonment of these dispensable genes might increase the cellular economization, reference to a streamlining hypothesis (Mira et al., 2001; Dufresne et al., 2005; D’Souza et al., 2014). In addition, the results revealed that not all A. thiooxidans strains in the microbial communities should reduce their genome sizes as it is of importance to maintain a functional diversity (Giovannoni et al., 2014). Taken together, our findings supported that similar to gene gain such as HGT, gene loss was the equally important driving force that contributes to the microbial genome evolution. Further analysis of potential DNA donor-recipient is an interesting prospect to investigate the genome evolution by accessing the microbial gene pool of the whole community.
In general, A. thiooxidans species was known as the sulfur oxidizer, which has a sulfur-dependent chemolithoautotrophic lifestyle. All of the strains utilize energy and electrons derived from aerobic oxidation of various sulfur species for assimilation of inorganic carbon and other anabolic processes (Yin et al., 2014a; Zhang et al., 2016a,c). Electrons from elemental sulfur and various sulfur compounds are transferred via the quinol pool (i) either to bd-type or bo3-type terminal oxidases, (ii) or to NADH-ubiquinone oxidoreductase (complex I; Yin et al., 2014a). Unlike nitrogen metabolism, in which some genes have undergone the gene loss, genes pertaining to sulfur oxidation were predicted to be dispersed in the core genome of A. thiooxidans species, suggesting that these conserved genes might evolve from a common ancestor and were more stable to be responsible for the sulfur oxidation of A. thiooxidans species.
The closely related A. ferrooxidans (formerly Thiobacillus ferrooxidans) not only utilizes the ferrous iron as the electron donor, but also gains energy generated from the oxidation of RISC, hydrogen, and formate to support its growth (Drobner et al., 1990; Pronk et al., 1991; Valdés et al., 2008). Hedrich and Johnson (2013b) further revealed that three species of Acidithiobacillus (A. ferrooxidans, A. caldus, and A. ferridurans) that oxidize ferrous iron or sulfur as electron donors could also grow on hydrogen. In their study, however, efforts to reveal the hydrogen utilization of two A. thiooxidans strains have been failed, thereby making it questionable whether this species could or could not metabolize hydrogen. Considering the inconsistency between experimental evidence and genomic analyses, we thus inferred that the presence of these hydrogenase-like in A. thiooxidans strains might not necessarily imply that the corresponding proteins could perform the predicted function as they might play other roles in bacterial lifestyle. In the following sections, further analyses were attempted to explain this inconsistency.
In the hyperthermophilic archaeon Thermococcus litoralis, an eight-gene cluster (fdhAB–mhyCDEFGH) was reported to encode the formate hydrogenlyase (formate dehydrogenase-coupled H2ase, FDH–MHY) complex, which supports microbial non-syntrophic growth on formate and metabolizes the hydrogen (Takács et al., 2008). The FDH–MHY complex consists of a formate dehydrogenase and a membrane-bound [NiFe]-H2ase which is considered as the member of H2-evolving, energy-conserving, membrane-associated H2ases. In addition, in silico genomic analysis of Thermococcus onnurineus showed the presence of a tripartite fdh–mfh–mnh cluster encoding for formate dehydrogenase, multimeric membrane-bound H2ases, and cation/proton antiporter, which was classified as the subgroup 4b of [NiFe]-H2ase (Kim et al., 2010; Lim et al., 2010). Further analysis showed that a pair of highly conserved hydrogen and nickel-binding motifs (C[GS][ILV]C[AGNS]xxH and [DE][PL]Cx[AGST]Cx[DE][RL], “x” represents any amino acid) were identified in the large subunits of H2ases (Wu and Mandrand, 1993; Lim et al., 2010). Although two conserved domains were predicted in all putative group 4 H2ases of A. thiooxidans strains used in our study, they were quite distinct from the conserved motifs in catalytic large subunits of other known Group 4 [NiFe]-H2ases, as some variant residues were identified in the former. The similar results were also reported in several other acidophiles (Yelton et al., 2013; Justice et al., 2014). In their studies, the putative group 4 H2ases were thought to perform some other function due to the absence of the binding site motifs that were normally found in [NiFe]-H2ases and were considered to be necessary for H2 formation (Wu and Mandrand, 1993; Lim et al., 2010).
Previous studies revealed the identical order and organization, and sequence similarities of certain essential subunits of [NiFe]-H2ases with subunits of energy-conserving NADH-ubiquinone oxidoreductase (also called respiratory complex I, Albracht et al., 1997; Hedderich, 2004). However, the reaction catalyzed by multi-subunit membrane-bound [NiFe]-H2ases was significantly different from the reaction catalyzed by complex I (EC: 1.6.5.3, Hedderich, 2004). The former is responsible for the interconversion of hydrogen, whereas the latter catalyzes the electron transfer from NADH to ubiquinone (Albracht et al., 1997; Friedrich and Scheide, 2000). In light of the six subunits (NuoB, C, D, H, I, and L) common to complex I and membrane-bound [NiFe]-H2ases, Friedrich and Scheide (2000) proposed a hypothesis that the complex enzyme consisting of the peripheral subunits (NuoB, C, D, and I) and the membraneous subunits (Nuo H and L) might be the common ancestor of complex I and multi-subunit membrane-bound H2ases. On the basis of the aforementioned evolutionary relationship of complex I and membrane-bound [NiFe]-H2ases, we inferred that the enzyme complexes in A. thiooxidans strains might evolve from an ancestral enzyme of complex I and recognized [NiFe]-H2ases, but have some other unclear functional roles.
Conclusion
Pan-genome analysis presented here expands our current understanding of genetic characteristics of A. thiooxidans isolates. Mathematical extrapolation revealed a closed pan-genome of A. thiooxidans species. Comparisons of gene repertoire, especially metabolic profile, of A. thiooxidans strains provide new insights into their autotrophic lifestyle, which strengthen our recognition that A. thiooxidans is an obligately chemolithoautotrophic acidophile capable of assimilating the atmospheric CO2 and inorganic forms of nitrogen, acquiring energy from aerobic oxidation of various sulfur species.
Statements
Author contributions
XZ drafted and wrote the manuscript, and performed the pan-genome analysis. ZL carried out the mathematical modeling. GW, FY, and XL participated in the discussion.
Funding
This work was supported by the National Natural Science Foundation of China (81502787, 81773393, and 31570113) and Innovation driven Program of Central South University (20170027010004).
Acknowledgments
We thank the National Center for Biotechnology Information for providing the genomic sequences of Acidithiobacillus thiooxidans strains A01, Licanantay, A02, BY-02, DMC, DXS-W, GD1-3, JYC-17, and ATCC 19377.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01255/full#supplementary-material
References
1
AlbrachtS. P. J.MarietteA.de JongP. (1997). Bovine-heart NADH: ubiquinone oxidoreductase is a monomer with 8 Fe-S clusters and 2 FMN groups.Biochim. Biophys. Acta131892–106. 10.1016/S0005-2728(96)00153-3
2
ArrigoK. R. (2005). Marine microorganisms and global nutrient cycles.Nature437349–355. 10.1038/nature04159
3
Bobadilla FazziniR. A.CortésM. P.PadillaL.MaturanaD.BudinichM.MaassA.et al (2013). Stoichiometric modeling of oxidation of reduced inorganic sulfur compounds (Riscs) in Acidithiobacillus thiooxidans.Biotechnol. Bioeng.1102242–2251. 10.1002/bit.24875
4
BonnefoyV.HolmesD. S. (2012). Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments.Environ. Microbiol.141597–1611. 10.1111/j.1462-2920.2011.02626.x
5
BoonE.MeehanC. J.WhiddenC.WongD. H. J.LangilleM. G. I.BeikoR. G. (2014). Interactions in the microbiome: communities of organisms and communities of genes.FEMS Microbiol. Rev.3890–118. 10.1111/1574-6976.12035
6
ChenL.HuM.HuangL.HuaZ.KuangJ.LiS.et al (2015). Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage.ISME J.91579–1592. 10.1038/ismej.2014.245
7
ChenL. X.LiJ. T.ChenY. T.HuangL. N.HuaZ. S.HuM.et al (2013). Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings.Environ. Microbiol.152431–2444. 10.1111/1462-2920.12114
8
DengW.WangY.LiuZ.ChengH.XueY. (2014). HemI: a toolkit for illustrating heatmaps.PLoS One9:e111988. 10.1371/journal.pone.0111988
9
DrobnerE.HuberH.StetterK. O. (1990). Thiobacillus ferrooxidans, a facultative hydrogen oxidizer.Appl. Environ. Microbiol.562922–2923.
10
D’SouzaG.WaschinaS.PandeS.BohlK.KaletaC.KostC. (2014). Less is more: selective advantages can explain the prevalent loss of biosynthetic genes in bacteria.Evolution682559–2570. 10.1111/evo.12468
11
DufresneA.GarczarekL.PartenskyF. (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote.Genome Biol.6:R14. 10.1186/gb-2005-6-2-r14
12
FalagánC.JohnsonD. B. (2016). Acidithiobacillus ferriphilus sp. nov., a facultatively anaerobic iron- and sulfur-metabolizing extreme acidophile.Int. J. Syst. Evol. Microbiol.66206–211. 10.1099/ijsem.0.000698
13
FoutsD. E.BrinkacL.BeckE.InmanJ.SuttonG. (2012). PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species.Nucleic Acids Res.40:e172. 10.1093/nar/gks757
14
FranceschiniA.SzklarczykD.FrankildS.KuhnM.SimonovicM.RothA.et al (2013). STRING v9. 1: protein-protein interaction networks, with increased coverage and integration.Nucleic Acids Res.41D808–D815. 10.1093/nar/gks1094
15
FriedrichT.ScheideD. (2000). The respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases.FEBS Lett.4791–5. 10.1016/S0014-5793(00)01867-6
16
GiovannoniS. J.ThrashJ. C.TempertonB. (2014). Implications of streamlining theory for microbial ecology.ISME J.81553–1565. 10.1038/ismej.2014.60
17
GorisJ.KonstantinidisK. T.KlappenbachJ. A.CoenyeT.VandammeP.TiedjeJ. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities.Int. J. Syst. Evol. Microbiol.5781–91. 10.1099/ijs.0.64483-0
18
Graña-MiragliaL.LozanoL. F.VelázquezC.Volkow-FernándezP.Pérez-OsegueraÁCevallosM. A. (2017). Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen.Front. Microbiol.8:1817. 10.3389/fmicb.2017.01817
19
GuoX.YinH.LiangY.HuQ.ZhouX.XiaoY.et al (2014). Comparative genome analysis reveals metabolic versatility and environmental adaptations of Sulfobacillus thermosulfidooxidans strain ST.PLoS One9:e99417. 10.1371/journal.pone.0099417
20
HallbergK. B.González-TorilE.JohnsonD. B. (2010). Acidithiobacillus ferrivorans, sp. nov.; facultatively anaerobic, psychrotolerant iron-, and sulfur-oxidizing acidophiles isolated from metal mine-impacted environments.Extremophiles149–19. 10.1007/s00792-009-0282-y
21
HallbergK. B.LindströmE. B. (1994). Characterization of Thiobacillus caldus sp. nov., a moderately thermophilic acidophile.Microbiology1403451–3456. 10.1099/13500872-140-12-3451
22
HedderichR. (2004). Energy-converting [NiFe] hydrogenases from archaea and extremophiles: ancestors of complex I.J. Bioenerg. Biomembr.3665–75. 10.1023/B:JOBB.0000019599.43969.33
23
HedrichS.JohnsonD. B. (2013a). Acidithiobacillus ferridurans sp. nov., an acidophilic iron-, sulfur-and hydrogen-metabolizing chemolithotrophic gammaproteobacterium.Int. J. Syst. Evol. Microbiol.634018–4025. 10.1099/ijs.0.049759-0
24
HedrichS.JohnsonD. B. (2013b). Aerobic and anaerobic oxidation of hydrogen by acidophilic bacteria.FEMS Microbiol. Lett.34940–45. 10.1111/1574-6968.12290
25
JenneyF. E.AdamsM. W. W. (2008). Hydrogenases of the model hyperthermophiles.Ann. N.Y. Acad. Sci.1125252–266. 10.1196/annals.1419.013
26
JohnsonD. B. (2014). Biomining-biotechnologies for extracting and recovering metals from ores and waste materials.Curr. Opin. Biotechnol3024–31. 10.1016/j.copbio.2014.04.008
27
JusticeN. B.NormanA.BrownC. T.SinghA.ThomasB. C.BanfieldJ. F. (2014). Comparison of environmental and isolate Sulfobacillus genomes reveals diverse carbon, sulfur, nitrogen, and hydrogen metabolisms.BMC Genomics15:1107. 10.1186/1471-2164-15-1107
28
KellyD. P.WoodA. P. (2000). Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov.Int. J. Syst. Evol. Microbiol.50511–516. 10.1099/00207713-50-2-511
29
KimY. J.LeeH. S.KimE. S.BaeS. S.LimJ. K.MatsumiR.et al (2010). Formate-driven growth coupled with H2 production.Nature467352–355. 10.1038/nature09375
30
KrzywinskiM.ScheinJ.BirolİConnorsJ.GascoyneR.HorsmanD. (2009). Circos: an information aesthetic for comparative genomics.Genome Res.191639–1645. 10.1101/gr.092759.109
31
KurtzS.PhillippyA.DelcherA. L.SmootM.ShumwayM.AntonescuC.et al (2004). Versatile and open software for comparing large genomes.Genome Biol.5:R12. 10.1186/gb-2004-5-2-r12
32
LevicánG.UgaldeJ. A.EhrenfeldN.MaassA.ParadaP. (2008). Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations.BMC Genomics9:581. 10.1186/1471-2164-9-581
33
LimJ. K.KangS. G.LebedinskyA. V.LeeJ.LeeH. S. (2010). Identification of a novel class of membrane-bound [NiFe]-hydrogenases in Thermococcus onnurineus NA1 by in silico analysis.Appl. Environ. Microbiol.766286–6289. 10.1128/AEM.00123-10
34
LinJ. T.StewartV. (1997). Nitrate assimilation by bacteria.Adv. Microb. Physiol.391–30. 10.1016/S0065-2911(08)60014-4
35
LindsayJ. A.KnightG. M.BuddE. L.McCarthyA. J. (2012). Shuffling of mobile genetic elements (MGEs) in successful healthcare-associated MRSA (HA-MRSA).Mob. Genet. Elements2239–243. 10.4161/mge.22085
36
LoweT. M.EddyS. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence.Nucleic Acids Res.25955–964. 10.1093/nar/25.5.0955
37
Martínez-CanoD. J.Reyes-PrietoM.Martínez-RomeroE.Partida-MartínezL. P.LatorreA.MoyaA.et al (2015). Evolution of small prokaryotic genomes.Front. Microbiol.5:742. 10.3389/fmicb.2014.00742
38
MediniD.DonatiC.TettelinH.MasignaniV.RappuoliR. (2005). The microbial pan-genome.Curr. Opin. Genet. Dev.15589–594. 10.1016/j.gde.2005.09.006
39
MiraA.OchmanH.MoranN. A. (2001). Deletional bias and the evolution of bacterial genomes.Trends Genet.17589–596. 10.1016/S0168-9525(01)02447-7
40
MoriyaY.ItohM.OkudaS.YoshizawaA. C.KanehisaM. (2007). KAAS: an automatic genome annotation and pathway reconstruction server.Nucleic Acids Res.35W182–W185. 10.1093/nar/gkm321
41
NuñezH.CovarrubiasP. C.Moya-BeltránA.IssottaF.AtavalesJ.AcuñaL. G.et al (2016). Detection, identification and typing of Acidithiobacillus species and strains: a review.Res. Microbiol.167555–567. 10.1016/j.resmic.2016.05.006
42
NuñezH.Moya-BeltránA.CovarrubiasP. C.IssottaF.CárdenasJ. P.GonzálezM.et al (2017). Molecular systematics of the genus Acidithiobacillus: insights into the phylogenetic structure and diversification of the taxon.Front. Microbiol.8:30. 10.3389/fmicb.2017.00030
43
PallenM. J.WrenB. W. (2007). Bacterial pathogenomics.Nature449835–842. 10.1038/nature06248
44
ParksD. H.ImelfortM.SkennertonC. T.HugenholtzP.TysonG. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes.Genome Res.251043–1055. 10.1101/gr.186072.114
45
ParroV.Moreno-PazM.González-TorilE. (2007). Analysis of environmental transcriptomes by DNA microarrays.Environ. Microbiol.9453–464. 10.1111/j.1462-2920.2006.01162.x
46
PatelR. K.JainM. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data.PLoS One7:e30619. 10.1371/journal.pone.0030619
47
PriceG. D.WoodgerF. J.BadgerM. R.HowittS. M.TuckerL. (2004). Identification of a SulP-type bicarbonate transporter in marine cyanobacteria.Proc. Natl. Acad. Sci. U.S.A.10118228–18233. 10.1073/pnas.0405211101
48
PronkJ. T.MeijerW. M.HazeuW.van DijkenJ. P.BosP.KuenenJ. G. (1991). Growth of Thiobacillus ferrooxidans on formic acid.Appl. Environ. Microbiol.572057–2062.
49
RichterM.RossellómóraR. (2009). Shifting the genomic gold standard for the prokaryotic species definition.Proc. Natl. Acad. Sci. U.S.A.10619126–19131. 10.1073/pnas.0906412106
50
SiguierP.PerochonJ.LestradeL.MahillonJ.ChandlerM. (2006). ISfinder: the reference centre for bacterial insertion sequences.Nucleic Acids Res.34D32–D36. 10.1093/nar/gkj014
51
SilverS.PhungL. T. (1996). Bacterial heavy metal resistance: new surprises.Annu. Rev. Microbiol.50753–789. 10.1146/annurev.micro.50.1.753
52
SnipenL.UsseryD. W. (2010). Standard operating procedure for computing pangenome trees.Stand. Genomic Sci.2135–141. 10.4056/sigs.38923
53
SullivanM. J.PettyN. K.BeatsonS. A. (2011). Easyfig: a genome comparison visualizer.Bioinformatics271009–1010. 10.1093/bioinformatics/btr039
54
TakácsM.TóthA.BogosB.VargaA.RákhelyG.KovácsK. L. (2008). Formate hydrogenlyase in the hyperthermophilic archaeon, Thermococcus litoralis.BMC Microbiol.8:88. 10.1186/1471-2180-8-88
55
TallaE.HedrichS.MangenotS.JiB.JohnsonD. B.BarbeV.et al (2014). Insights into the pathways of iron-and sulfur-oxidation, and biofilm formation from the chemolithotrophic acidophile Acidithiobacillus ferrivorans CF27.Res. Microbiol.165753–760. 10.1016/j.resmic.2014.08.002
56
TamuraK.PetersonD.PetersonN.StecherG.NeiM.KumarS. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.Mol. Biol. Evol.282731–2739. 10.1093/molbev/msr121
57
R Core Team (2000). R Language Definition.ViennaR foundation for statistical computing.
58
TeelingH.MeyerdierksA.BauerM.AmannR.GlöcknerF. O. (2004). Application of tetranucleotide frequencies for the assignment of genomic fragments.Environ. Microbiol.6938–947. 10.1111/j.1462-2920.2004.00624.x
59
TempleK. L.ColmerA. R. (1951). The autotrophic oxidation of iron by a new bacterium: Thiobacillus ferrooxidans.J. Bacteriol.62605–611.
60
TettelinH.MasignaniV.CieslewiczM. J.DonatiC.MediniD.WardN. L.et al (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ”pan-genome”.Proc. Natl. Acad. Sci. U.S.A.10213950–13955. 10.1073/pnas.0506758102
61
TettelinH.RileyD.CattutoC.MediniD. (2008). Comparative genomics: the bacterial pan-genome.Curr. Opin. Microbiol.11472–477. 10.1016/j.mib.2008.09.006
62
TravisanyD.CortésM. P.LatorreM.Di GenovaA.BudinichM.Bobadilla-FazziniR. A.et al (2014). A new genome of Acidithiobacillus thiooxidans provides insights into adaptation to a bioleaching environment.Res. Microbiol.165743–752. 10.1016/j.resmic.2014.08.004
63
UllrichS. R.GonzálezC.PoehleinA.TischlerJ. S.DanielR.SchlömannM.et al (2016). Gene loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile ”Ferrovum”.Front. Microbiol.7:797. 10.3389/fmicb.2016.00797
64
ValdesJ.OssandonF.QuatriniR.DopsonM.HolmesD. S. (2011). Draft genome sequence of the extremely acidophilic biomining bacterium Acidithiobacillus thiooxidans ATCC 19377 provides insights into the evolution of the Acidithiobacillus genus.J. Bacteriol.1937003–7004. 10.1128/JB.06281-11
65
ValdésJ.PedrosoI.QuatriniR.DodsonR. J.TettelinH.BlakeR.et al (2008). Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications.BMC Genomics9:597. 10.1186/1471-2164-9-597
66
VernikosG.MediniD.RileyD. R.TettelinH. (2015). Ten years of pan-genome analyses.Curr. Opin. Microbiol.23148–154. 10.1016/j.mib.2014.11.016
67
VignaisP. M.BilloudB.MeyerJ. (2001). Classification and phylogeny of hydrogenases.FEMS Microbiol. Rev.25455–501. 10.1111/j.1574-6976.2001.tb00587.x
68
WaksmanS. A.JoffeJ. S. (1922). Microorganisms concerned in the oxidation of sulfur in the soil. II. Thiobacillus thiooxidans, a new sulfur-oxidizing organism isolated from the soil.J. Bacteriol.7239–256.
69
WilliamsK. P.KellyD. P. (2013). Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria.Int. J. Syst. Evol. Microbiol.632901–2906. 10.1099/ijs.0.049270-0
70
WuL. F.MandrandM. A. (1993). Microbial hydrogenases: primary structure, classification, signatures and phylogeny.FEMS Microbiol. Rev.10243–269. 10.1111/j.1574-6968.1993.tb05870.x
71
YeltonA. P.ComolliL. R.JusticeN. B.CastelleC.DenefV. J.ThomasB. C.et al (2013). Comparative genomics in acid mine drainage biofilm communities reveals metabolic and structural differentiation of co-occurring archaea.BMC Genomics14:485. 10.1186/1471-2164-14-485
72
YinH.ZhangX.LiX.HeZ.LiangY.GuoX.et al (2014a). Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans.BMC Microbiol.14:179. 10.1186/1471-2180-14-179
73
YinH.ZhangX.LiangY.XiaoY.NiuJ.LiuX. (2014b). Draft genome sequence of the extremophile Acidithiobacillus thiooxidans A01, isolated from the wastewater of a coal dump.Genome Announc.2e00222–1410.1128/genomeA.00222-14
74
YouX. Y.GuoX.ZhengH. J.ZhangM. J.LiuL. J.ZhuY. Q.et al (2011). Unraveling the Acidithiobacillus caldus complete genome and its central metabolisms for carbon assimilation.J. Genet Genomics38243–252. 10.1016/j.jgg.2011.04.006
75
ZerbinoD. R.BirneyE. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs.Genome Res.18821–829. 10.1101/gr.074492.107
76
ZhangX.FengX.TaoJ.MaL.XiaoY.LiangY.et al (2016a). Comparative genomics of the extreme acidophile Acidithiobacillus thiooxidans reveals intraspecific divergence and niche adaptation.Int. J. Mol. Sci.17:1355. 10.3390/ijms17081355
77
ZhangX.LiuX.HeQ.DongW.ZhangX.FanF.et al (2016b). Gene turnover contributes to the evolutionary adaptation of Acidithiobacillus caldus: insights from comparative genomics.Front. Microbiol.7:1960. 10.3389/fmicb.2016.01960
78
ZhangX.LiuX.LiangY.FanF.ZhangX.YinH. (2016c). Metabolic diversity and adaptive mechanisms of iron- and/or sulfur-oxidizing autotrophic acidophiles in extremely acidic environments.Environ. Microbiol. Rep.8738–751. 10.1111/1758-2229.12435
79
ZhangX.NiuJ.LiangY.LiuX.YinH. (2016d). Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap.BMC Genet.17:21. 10.1186/s12863-016-0330-4
80
ZhangX.SheS.DongW.NiuJ.XiaoY.LiangY.et al (2016e). Comparative genomics unravels metabolic differences at species and/or strain level and extremely acidic environmental adaptation of ten bacteria belonging to the genus Acidithiobacillus.Syst. Appl. Microbiol.39493–502. 10.1016/j.syapm.2016.08.007
81
ZhangX.LiuX.LiangY.GuoX.XiaoY.MaL.et al (2017). Adaptive evolution of extreme acidophile Sulfobacillus thermosulfidooxidans potentially driven by horizontal gene transfer and gene loss.Appl. Environ. Microbiol.83e3098–e3016. 10.1128/AEM.03098-16
82
ZhangX.LiuX.YangF.ChenL. (2018). Pan-genome analysis links the hereditary variation of Leptospirillum ferriphilum with its evolutionary adaptation.Front. Microbiol.9:577. 10.3389/fmicb.2018.00577
83
ZhangX.YinH.LiangY.QiuG.LiuX. (2015). Theoretical model of the structure and the reaction mechanisms of sulfur oxygenase reductase in Acidithiobacillus thiooxidans.Adv. Mater. Res.113067–70. 10.4028/www.scientific.net/AMR.1130.67
84
ZhengY.ZhaoL.GaoJ.FeiZ. (2011). iAssembler: a package for de novo assembly of Roche-454/Sanger transcriptome sequences.BMC Bioinformatics12:453. 10.1186/1471-2105-12-453
85
ZuoG.HaoB. (2015). CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy.Genomics Proteomics Bioinformatics13321–331. 10.1016/j.gpb.2015.08.004
Summary
Keywords
Acidithiobacillus thiooxidans, core genome, mathematical modeling, bacterial lifestyle, metabolic profiles
Citation
Zhang X, Liu Z, Wei G, Yang F and Liu X (2018) In Silico Genome-Wide Analysis Reveals the Potential Links Between Core Genome of Acidithiobacillus thiooxidans and Its Autotrophic Lifestyle. Front. Microbiol. 9:1255. doi: 10.3389/fmicb.2018.01255
Received
04 December 2017
Accepted
24 May 2018
Published
08 June 2018
Volume
9 - 2018
Edited by
Peng Luo, South China Sea Institute of Oceanology (CAS), China
Reviewed by
Santiago Castillo Ramírez, Universidad Nacional Autónoma de México, Mexico; Debmalya Barh, Universidade Federal de Minas Gerais, Brazil
Updates
Copyright
© 2018 Zhang, Liu, Wei, Yang and Liu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Yang, phfyang@csu.edu.cn Xueduan Liu, xueduanliu@yahoo.com
This article was submitted to Evolutionary and Genomic Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.