 Sarah J. de Jong1
Sarah J. de Jong1 Elias Imahorn2
Elias Imahorn2 Peter Itin2,3Jouni Uitto4,5
Peter Itin2,3Jouni Uitto4,5 Gérard Orth6Emmanuelle Jouanguy1,7,8
Gérard Orth6Emmanuelle Jouanguy1,7,8 Jean-Laurent Casanova1,7,8,9,10*
Jean-Laurent Casanova1,7,8,9,10* Bettina Burger2
Bettina Burger2- 1St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, United States
- 2Department of Biomedicine, University Hospital of Basel, University of Basel, Basel, Switzerland
- 3Department of Dermatology, University Hospital of Basel, Basel, Switzerland
- 4Department of Dermatology and Cutaneous Biology, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, United States
- 5Jefferson Institute of Molecular Medicine, Thomas Jefferson University, Philadelphia, PA, United States
- 6Pasteur Institute, Paris, France
- 7INSERM UMR 1163, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, Necker Hospital for Sick Children, Paris, France
- 8Imagine Institute, Paris Descartes University, Paris, France
- 9Pediatric Hematology and Immunology Unit, Necker Hospital for Sick Children, Paris, France
- 10Howard Hughes Medical Institute, New York, NY, United States
Epidermodysplasia verruciformis (EV) is an autosomal recessive skin disorder with a phenotype conditional on human beta-papillomavirus (beta-HPV) infection. Such infections are common and asymptomatic in the general population, but in individuals with EV, they lead to the development of plane wart-like and red or brownish papules or pityriasis versicolor-like skin lesions, from childhood onwards. Most patients develop non-melanoma skin cancer (NMSC), mostly on areas of UV-exposed skin, from the twenties or thirties onwards. At least half of the cases of typical EV are caused by biallelic loss-of-function mutations of TMC6/EVER1 or TMC8/EVER2. The cellular and molecular basis of disease in TMC/EVER-deficient patients is unknown, but a defect of keratinocyte-intrinsic immunity to beta-HPV is suspected. Indeed, these patients are not susceptible to other infectious diseases and have apparently normal leukocyte development. In contrast, patients with an atypical form of EV due to inborn errors of T-cell immunity invariably develop clinical symptoms of EV in the context of other infectious diseases. The features of the typical and atypical forms of EV thus suggest that the control of beta-HPV infections requires both EVER1/EVER2-dependent keratinocyte-intrinsic immunity and T cell-dependent adaptive immunity.
Introduction
Epidermodysplasia verruciformis (EV) is a rare skin disease characterized by persistent disseminated flat warts and pityriasis versicolor-like lesions, associated with a high risk of non-melanoma skin cancer (NMSC). About 501 patients have been reported worldwide (Burger and Itin, 2014; Imahorn et al., 2017). EV was first described in Lewandowsky and Lutz (1922) as a congenital skin disease. The observation of parental consanguinity and familial cases led to the suggestion that EV might be a genetic disease with autosomal recessive inheritance (Cockayne, 1933; Rajagopalan et al., 1972). This notion was not confirmed until 70 years later, when inactivating mutations of the EVER1/TMC6 and EVER2/TMC8 genes accounting for about 50% of all known cases of EV were identified (Ramoz et al., 2002; Orth, 2006, 2008; Imahorn et al., 2017). Auto- and heteroinoculation experiments performed from 1946 onward showed that the phenotype of EV was dependent on a viral infection (Lutz, 1946; Jablonska and Milewski, 1957). Viral particles were then detected in EV lesions (Ruiter and van Mullem, 1966; Jablonska et al., 1968), and, subsequently, EV-specific human papillomaviruses (HPVs) were identified, some of which, such as HPV-5 in particular, were shown to be associated with the cancers observed in EV patients (reviewed in Orth et al., 1980; Orth, 2006). EV thus results from a genetically determined susceptibility to specific skin-tropic HPVs (Orth, 2006, 2008; Imahorn et al., 2017). These viruses belong to the beta-genus, and are almost ubiquitous and apparently benign in the general population. EV patients are not prone to other viral, bacterial, or fungal infections. NMSC is the only type of cancer presenting a higher incidence in these patients than in the general population (Orth, 2006, 2008). The EVER1 and EVER2 proteins are thought to govern keratinocyte-intrinsic immunity to beta-HPVs. However, patients with cutaneous clinical symptoms of beta-HPV infection and other infectious manifestations caused by profound T-cell defects have been reported. This clinical picture is referred to as “atypical EV." The cumulative findings of almost a century of studies of typical and atypical EV suggest that both keratinocytes and T cells are required for the control of beta-HPVs. We review here the various facets of EV.
Clinical Manifestations and Histopathology of Ev
EV manifests as multiple polymorphic skin lesions, which begin to appear during infancy or early childhood and persist throughout the individual’s life. Flat-topped papular lesions resembling verrucae planae typically develop on the extremities. The lesions may also present as red or red-brownish papules or pityriasis versicolor-like lesions, mostly on the trunk, neck, and face (Lewandowsky and Lutz, 1922). In addition, verruca-like papillomatous or seborrheic keratosis-like lesions may develop. The frequency of verruca vulgaris is no higher in these patients than in the general population. In many patients, isomorphic lesions develop preferentially at sites of traumas (the Koebner phenomenon) (de Oliveira et al., 2003). Some patients have persistent palmar lesions similar to palmar pits and dermatoscopically consistent with plane warts (Imahorn et al., 2017). Over the course of the patient’s lifetime, the phenotype of a particular skin area may change, and lesions may progress to NMSC. There is currently no cure for EV. The development of EV lesions cannot be prevented, but frequent examinations of skin lesions that might develop into skin cancers and appropriate treatment of these lesions are recommended, such as surgical removal and cryotherapy. Patients with typical EV are otherwise healthy and are not particularly susceptible to any other unusually severe infectious diseases. Provided that cancers are treated appropriately, life expectancy is similar to that in the general population; multifocal carcinomas might, however, be fatal if untreated. Histological examinations of lesions typically reveal hyperkeratosis and parakeratosis, mild acanthosis, and the presence of koilocytes, keratinocytes with pale-stained cytoplasm in the upper epidermis associated with high levels of intranuclear viral replication. On hematoxylin-eosin staining, the cytoplasm of the affected cells stains pale blue (so-called “blue cells”) and numerous round basophilic keratohyalin granules are visible. This histological finding is pathognomonic for infections with HPV (Orth, 2006). Possible immune infiltration has not been comprehensively characterized in EV lesions. A number of atypical EV cases have been reported, which will be reviewed below.
Clinical Manifestations of Atypical Ev
Patients with atypical EV have a broader clinical phenotype than those with typical EV. The phenotype of EV, as described above, is undistinguishable between patients with typical and atypical forms. Indeed, the skin lesions develop early in life and are caused by the same beta-HPV subtypes that are found in patients with typical EV (see Table 1). The other clinical phenotypes, mostly infectious and auto-immune, differ between patients (Crequer et al., 2012a,b; Sanal et al., 2012; Stray-Pedersen et al., 2014; Stepensky et al., 2015; Li et al., 2016; Platt et al., 2017; Tahiat et al., 2017). The clinical features of patients with atypical EV are reviewed in Table 1. Some patients have growth retardation or mild developmental delay. Others present auto-immune features, which may be clinically overt and affect various tissues and organs, or covert, with detectable auto-antibodies but no clinical signs. Bacterial infections, including skin abscesses, pneumonia, and ear infections, are frequent. Infections with cutaneous herpes viruses, molluscum, mucocutaneous candidiasis, and non-beta-HPV infections have also been documented. Moreover, some patients display recurrent respiratory and gastrointestinal infections. Cancers, such as Burkitt lymphoma and EBV-lymphoma, have been reported in some patients. Finally, a few patients have been reported to display clinical manifestations of beta- and alpha-HPV co-infections in different lesions (Azzimonti et al., 2005; Borgogna et al., 2014; Landini et al., 2014). These patients display cutaneous alpha- and beta-HPV- (Borgogna et al., 2014) or genital (Landini et al., 2014) alpha-HPV-driven infections, but no broad susceptibility to infections. These cases suggest that covert immunological abnormalities might favor the development of beta- and alpha-HPV-induced cutaneous and mucosal lesions in other individuals. Based on these clinical observations, the molecular and cellular basis of typical and atypical EV phenotype appear to be different. The typical EV phenotype is, in itself, suggestive of a keratinocyte-intrinsic defect, whereas the atypical EV phenotype, with its myriad of infections and auto-immune features, is more consistent with an adaptive T-cell defect.
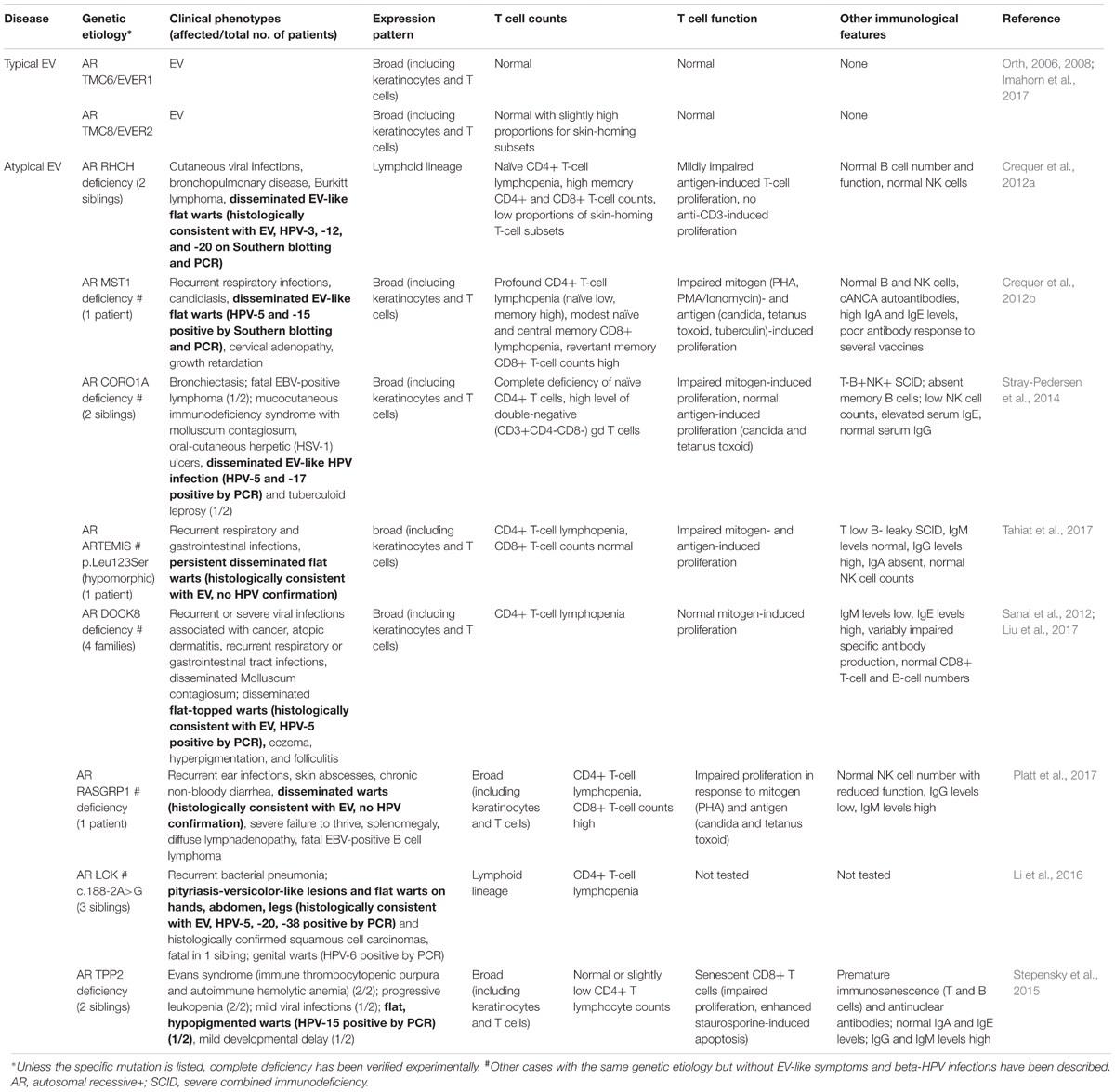
TABLE 1. Immunological parameters of patients with typical and atypical EV.
Skin Cancer
Patients with EV have a higher than normal risk of developing actinic keratosis and NMSC, particularly cutaneous squamous cell carcinoma (cSCC), cSCC in situ (Bowen’s disease) and, to a lesser extent basal cell carcinoma (BCC) (Lewandowsky and Lutz, 1922; Rajagopalan et al., 1972; Orth et al., 1979; Mitsuishi et al., 2008). About two third of EV patients develop NMSC, with an onset during their twenties or thirties (Lewandowsky and Lutz, 1922; Rajagopalan et al., 1972; Orth et al., 1978; Majewski and Jablonska, 1997; de Oliveira et al., 2003). NMSC typically occurs in lesions exposed to the sun, about 20 years after the appearance of the first lesions, suggesting that UV irradiation and HPV are cocarcinogens for the development of this cancer. EV patients should, therefore, pay particular attention to protecting their skin against exposure to the sun. The role of HPVs in carcinogenesis is well established. The first evidence for the oncogenic behavior of HPVs was actually documented in a study of EV (Orth et al., 1980). EV has long served as a human model for studies of viral cutaneous oncogenesis (Jablonska et al., 1972; Casanova and Abel, 2004). This oncogenic behavior of beta-HPVs led to speculations that these viruses might also contribute to the development of cSCC in the general population. However, in cases of cSCC in the general population, less than one viral genome per cell is generally detected; by contrast, viral load is very high in cSCC of EV patients, indicating that different mechanisms of cancer initiation and maintenance are probably at work (Howley and Pfister, 2015). In particular, oncogenic HPV-5 and HPV-8 have been found in EV-associated NMSC, leading to their classification, by the WHO, as possible carcinogens in EV patients (Bouvard et al., 2009). The detection of high loads of HPV-5 E6 and E7 transcripts suggested a role in the cancer induction or maintenance, although the underlying mechanism remains unclear (Orth, 1987). Radiotherapy for cSCC treatment can be effective in the general population, but is counterproductive in EV patients, as the development of more aggressive tumors after treatment has been reported (Rajabi et al., 2014; de Oliveira et al., 2015). Patients should undergo frequent physical examinations, to facilitate the identification of precancerous and cancer lesions as early as possible. However, it has been shown that infection with oncogenic HPVs known to be associated with a high risk of skin cancer is not sufficient, in itself, to cause cancer. Environmental and genetic factors thus contribute to the inter-individual variability of outcome for HPV infection.
Viral Etiology
More than half a century ago, EV was recognized as a virus-induced disease, based on several experimental observations. Inoculation experiments showed EV to be transmissible (Lutz, 1946; Jablonska and Milewski, 1957; Jablonska et al., 1966). The first viral particles were then detected in lesions by electron microscopy (Ruiter and van Mullem, 1966; Jablonska et al., 1968) and, several years later, the first EV-specific HPV types, all from genus beta-HPV, were isolated from EV lesions (Orth et al., 1978; Orth, 1986). EV patients not only have high HPV loads in their lesions, they also have a high serum antibody reactivity to beta-HPVs (Michael et al., 2010). At least 25 different beta-HPV genotypes have been found in patients with EV, and patients are usually infected with multiple EV-HPV types, HPV-5 being the most prevalent genotype (Orth, 2006). HPV-5 is also the most common genotype in cases of malignant conversion (Orth, 1986; Imahorn et al., 2017). Beta-HPVs are also frequently found in the skin of healthy individuals of the general population, but mostly at low copy numbers, and they do not cause clinical disease. This suggests that beta-HPVs are commensals of the skin (Antonsson et al., 2003a,b). However, it remains a matter of debate whether these beta-HPVs are really non-pathogenic in individuals without EV, as some studies have associated EV-HPV seropositivity and viral DNA load in the eyebrow with the risk of cSCC in the general population (Neale et al., 2013; Iannacone et al., 2014). The mechanisms of persistent, asymptomatic EV-HPV infection remain largely unknown. It has been suggested that beta-HPVs lack an essential growth-promoting function, limiting their pathogenicity. Indeed, unlike the alpha- and gamma-HPVs responsible for cutaneous warts, cervical cancers, and laryngeal papillomatosis, beta-HPVs do not possess the E5 or E8 (also reported as E101) open-reading frames typically found in pathogenic non-beta HPVs and shown to be a growth-promoting factor for keratinocytes in vivo (Danos et al., 1983; Giri et al., 1985; Hu et al., 2002; Nonnenmacher et al., 2006; Orth, 2006, 2008). Beta-HPVs can overcome this lack of a growth-promoting function and attain full pathogenicity, only in genetically predisposed individuals, implying that these patients have a specific cutaneous immune defect (reviewed by Orth, 2006, 2008).
Human Genetics of Typical Ev
Typical EV is a Mendelian condition that is transmitted as an autosomal recessive trait and shows complete penetrance in the first decade of life, across different ethnic groups (Orth, 2006). An autosomal recessive mode of inheritance was initially proposed in Cockayne (1933), confirmed in 1974 (Rajagopalan et al., 1972) and subsequently supported by a study of 147 EV cases, in which 11% of patients were the offspring of consanguineous marriages, and 10% came from multiplex families in which 25% of the siblings were affected, with an equal sex ratio (Lutzner, 1978). Shortly after the mapping of two susceptibility loci for EV to chromosomes 17q25 (EV1) and 2p21-p24 (EV2) (Ramoz et al., 1999, 2000), bi-allelic loss-of-function mutations of two adjacent genes termed TMC6 and TMC8, also named EVER1 and EVER2, respectively, located within the EV1 locus, were identified (Ramoz et al., 2002). Eight different mutations of TMC6/EVER1 and 11 mutations of TMC8/EVER2 have been described to date, in a total of 32 patients from 19 families and 5 ethnicities reviewed in Burger and Itin (2014) and Imahorn et al. (2017). These mutations are nonsense, frameshift, or splice-site mutations with full clinical penetrance for EV in homozygotes and compound heterozygotes. The effects of these mutations on mRNA and protein levels have not been investigated in most patients. Nonsense-mediated RNA decay and undetectable TMC8/EVER2 protein have been reported in two patients with premature stop codons (Ramoz et al., 2002; Landini et al., 2012). However, splice-site mutations of TMC8/EVER2 have been identified that have no effect on mRNA levels, despite the detection of aberrantly spliced, shorter transcripts (Miyauchi et al., 2016; Imahorn et al., 2017). Nevertheless, all mutations are assumed to be loss-of-function. By contrast, the missense polymorphisms found in the general population are not predicted to be deleterious and are too common to account for EV. Despite these genetic analyses, more than 40% of the EV families identified to date have no mutations of the genes known to be associated with this disease (Zuo et al., 2006; Arnold et al., 2011; Imahorn et al., 2017) suggesting that high-throughput sequencing studies are required to identify the causal genes in these families.
Human Genetics of Atypical Ev
Inactivating mutations of RHOH (Crequer et al., 2012b), STK4 (encoding the MST1 protein) (Crequer et al., 2012a), CORO1A (Stray-Pedersen et al., 2014), DCLRE1C (encoding the Artemis protein) (Tahiat et al., 2017), DOCK8 (Sanal et al., 2012; Liu et al., 2017), RASGRP1 (Platt et al., 2017), LCK (Li et al., 2016), and TPP2 (Stepensky et al., 2015) have been reported in patients with atypical EV. Interestingly, the clinical penetrance of these genetic disorders for EV lesions is low, as only a few patients present with manifestations of EV. This is at odds with the findings for typical EV, which is a fully penetrant Mendelian trait. A complete loss of function has been reported for all mutations, except those for Artemis (hypomorphic) and Lck (no functional investigation). All these patients suffer from a classic T-cell primary immunodeficiency. Some also display impaired mitogen- and/or antigen-induced T-cell proliferation. NK cells counts are normal in all these diseases, except for CORO1A. Hyper-IgM and hyper-IgE are observed in RASGRP1 and in CORO1A and DOCK8, respectively. Autoimmunity has been reported for STK4 and TPP2 deficiencies. T-cell senescence has been reported for RHOH and TPP2 deficiencies. It remains unclear whether these observations are the cause or consequence of severe, disseminated, chronic, life-long beta-HPV and other recurrent acute and chronic infections in these patients. Few studies have addressed the specific responses of leukocytes, but a lack of autologous T-cell response to HPV-infected keratinocytes has been described (Cooper et al., 1990). Most of these genes are broadly expressed (including in T cells and other types of skin-resident cells), with the exception of RHOH and LCK, which are expressed exclusively in the lymphoid linage (Table 1). An additional contribution of cells other than T lymphocytes, including other types of skin-resident cells, to the pathogenesis of atypical EV cannot, therefore, be ruled out. Nevertheless, the T-cell defects underlying atypical EV are likely to be specific, as most inherited and acquired forms of T-cell deficiency do not cause clinically apparent infections with beta-HPVs.
Leukocyte Immunity
Atypical EV is associated with T-cell deficiency. The only immunological feature common to all known etiologies is CD4+ lymphopenia. More detailed immunological studies have been performed only in RhoH-deficient patients. These patients had impaired tissue-homing T-cells marker expression, with a small decreased in the number of memory skin-homing (CLA+) CD4+ T cells and a large decrease in the number of integrin beta7+ CD4+ T cells. Broad immunological evaluations of patients with typical EV have proved difficult, due to the rarity of the disease, and the data available are therefore highly patchy. TMC6/EVER1 and TMC8/EVER2, which are mutated in patients with typical EV, are widely expressed, including in lymphocytes and the skin (Lazarczyk et al., 2012). Patients with typical EV have normal humoral immunity and antibody responses (Jablonska et al., 1979), their Langerhans cells have normal alloantigen presentation capacity (Majewski and Jablonska, 1992), and they have largely normal NK cell activity (Majewski et al., 1986). They have been reported to display impairments of cellular immunity, including low T-cell counts, CD4+/CD8+ T-cell ratios, a lack of responsiveness to T-cell mitogens, anergy to skin antigens and sensitization to dinitrochlorobenzene (Glinski et al., 1976, 1981; Prawer et al., 1977; Majewski et al., 1986; Majewski and Jablonska, 1992; Majewski et al., 1997; de Oliveira et al., 2003), but these defects were inconsistent and further complicated the assessment of immunological dysfunction in EV patients. All these observations suggest that a role for TMC6/EVER1 and TMC8/EVER2 in skin-intrinsic immunity.
Keratinocyte Immunity
Keratinocytes contribute to protective immunity in the skin in at least three ways. First, they form a physical barrier. Second, they secrete and respond to cytokines, chemokines, and growth factors, participating in leukocyte-mediated operations (Kupper and Fuhlbrigge, 2004). Third, keratinocyte-intrinsic mechanisms are thought to contribute to the control of keratinocyte-tropic viruses. TMC6/EVER1 and TMC8/EVER2 thus provide us a unique opportunity to study the role of keratinocytes in host defense against HPV infection. TMC6/EVER1 and TMC8/EVER2 belong to a large family of highly conserved transmembrane channel-like (TMC) proteins that are expressed in the endoplasmic reticulum, where they act as or modulate the activity of transmembrane channels (Keresztes et al., 2003; Kurima et al., 2003). It has been suggested that these two proteins interact with the zinc transporter ZnT-1, influencing intracellular Zn2+ concentration, and, thus, the activity of the transcription factors, such as AP-1, a key activator in the HPV life cycle and cellular proliferation (Lazarczyk et al., 2008). It has also been suggested that TMC8/EVER2 influences the cellular response to TNF-α by promoting apoptosis rather than NF-κB activation and pro-survival signaling pathways (Gaud et al., 2013; Vuillier et al., 2014). TMC6/EVER1 and TMC8/EVER2 are thus thought to serve as restriction factors for beta-HPVs in keratinocytes, through the limitation of viral replication and gene expression. Studies of TMC6/EVER1 and TMC8/EVER2 have proved technically difficult, partly due to problems raising robust antibodies against these highly hydrophobic proteins, which have 10 and 8 transmembrane domains, respectively. Known molecular functions/mechanism of TMC6/EVER1 and TMC8/EVER2 are reviewed in Orth (2008) and Lazarczyk et al. (2009). The cellular and molecular bases of beta-HPV-driven lesions in TMC6/EVER1 and TMC8/EVER2-deficient patients with EV remain unknown. The genetic defects prevent patients from controlling beta-HPV in keratinocytes (Akgul et al., 2006). The patients are susceptible to skin infections with these particular types of HPV (Boxman et al., 1999). In addition, some patients with severe combined immunodeficiency (SCID) caused by mutations of IL2RG or JAK3 may develop EV. These patients develop EV only after successful bone marrow transplantation, indicating that disease development in these patients is related to a keratinocyte defect (Laffort et al., 2004).
A Classification of Ev Into Two Major Types
In the past, EV patients rarely underwent immunological follow-up, and typical and atypical cases were treated as the same diagnostic entity. This has led to conflicting reports about the impairment of immunity in only some patients. Patients with typical EV probably have a keratinocyte-intrinsic defect allowing beta-HPVs to persist in the skin, leading to the development of clinically apparent lesions. Consistent with this notion, three TMC8/EVER2-deficient EV patients have been reported to display a largely normal partitioning of circulating T cells, possibly even with slightly larger than normal skin-homing T-cell populations (Crequer et al., 2013). These normal numbers of circulating T cells do not exclude the possibility of an impaired recognition of and response to HPV-infected keratinocytes by these cells (Cooper et al., 1990). Once it became clear that these patients had a selective susceptibility to beta-HPV infections and TMC6/EVER1 and TMC8/EVER2 deficiencies had been described as a genetic etiology, EV was identified as a primary immunodeficiency (Notarangelo et al., 2004; Orth, 2008; Casanova, 2015). It has been suggested that the TMC/EVER proteins function as antiviral restriction factors in keratinocytes, and that they control constitutive intrinsic immunity (Orth, 2008). Consistent with this hypothesis, patients with acquired or treatment-induced immunosuppression do not normally develop EV, despite the commensal nature of beta-HPVs (Antonsson et al., 2003a,b). This contrasts with common warts due to alpha-HPVs, which are more common in immunosuppressed individuals (Wieland et al., 2014). Rare cases of EV-like syndromes developing in previously unaffected adults, and known as “acquired EV,” have been described in patients with suppressed cell-mediated immunity. The clinical presentations of these patients and proposed treatments have been reviewed in detail elsewhere (Zampetti et al., 2013; Ovits et al., 2017). The rarity of atypical EV in patients with broad and severe genetic T-cell deficiencies and in immunocompromised individuals suggests that immunosuppression per se is not sufficient for the development of this disease. This is the case for most infectious diseases striking patients with such T-cell disorders, and it suggests that other host and environmental factors are involved in pathogenesis.
Concluding Remarks
In summary, both keratinocytes and T cells probably contribute to immunity to beta-HPV. Genetic defects affecting one or, potentially, both cell types lead to the EV-defining clinical manifestations of beta-HPV infection. All individuals with severe, recurrent, and persistent flat warts and with a diagnosis of typical or atypical EV should be tested for genetic etiologies of typical and atypical EV. The presence and extent of a systemic T-cell defect determines the spectrum of other clinical manifestations of an infectious, autoimmune, or allergic nature. Future studies should provide more conclusive demonstrations of the keratinocyte-intrinsic nature of EVER-dependent immunity to beta-HPVs. The identification of new genes associated with typical and atypical EV will pave the way for analyses of the molecular and cellular bases of EV, including studies of the respective contributions of keratinocytes and T cells to anti-beta-HPV immunity. The highly restricted susceptibility to beta-HPV observed in patients with EV has important clinical and biological implications in the fields of HPV, mucocutaneous immunology, and cell-intrinsic immunity. Elucidation of the genetic and immunological bases of EV will facilitate studies of the virulence mechanisms of other HPVs, including the role of E5 and E8 proteins produced by alpha- and gamma-HPVs, respectively. These studies will also facilitate the development of novel approaches for the diagnosis and management of patients with various conditions due to HPVs.
Author Contributions
SdJ, EI, PI, GO, EJ, J-LC, and BB wrote the manuscript. SdJ, EI, PI, JU, GO, EJ, J-LC, and BB reviewed and edited the manuscript.
Funding
The Laboratory of Human Genetics of Infectious Diseases was supported by grants from the National Institutes of Health (NIH, Grant No: 5 R21 AI107508-02), the French National Cancer Institute (Grant No. 2013-1-PL BIO-11-1), and the French National Research Agency (ANR) under the “Investissements d’Avenir” program (Grant No. ANR-10-IAHU-01). SdJ was supported by the German Research Foundation (Jo 1151-1), funds from the Women & Science Fellowship program at The Rockefeller University, the National Institute of Allergy and Infectious Diseases of the NIH (Grant No. R21AI107508), and the NIH Clinical and Translational Science Award (CTSA) program (Grant No. UL1TR000043).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GS and handling Editor declared their shared affiliation.
Footnotes
References
Akgul, B., Cooke, J. C., and Storey, A. (2006). HPV-associated skin disease. J. Pathol. 208, 165–175. doi: 10.1002/path.1893
Antonsson, A., Erfurt, C., Hazard, K., Holmgren, V., Simon, M., Kataoka, A., et al. (2003a). Prevalence and type spectrum of human papillomaviruses in healthy skin samples collected in three continents. J. Gen. Virol. 84, 1881–1886.
Antonsson, A., Karanfilovska, S., Lindqvist, P. G., and Hansson, B. G. (2003b). General acquisition of human papillomavirus infections of skin occurs in early infancy. J. Clin. Microbiol. 41, 2509–2514.
Arnold, A. W., Burger, B., Kump, E., Rufle, A., Tyring, S. K., Kempf, W., et al. (2011). Homozygosity for the c.917A?T (p.N306l) polymorphism in the EVER2/TMC8 gene of two sisters with epidermodysplasia verruciformis Lewandowsky-Lutz originally described by Wilhelm Lutz. Dermatology 222, 81–86. doi: 10.1159/000322536
Azzimonti, B., Mondini, M., De Andrea, M., Gioia, D., Dianzani, U., Mesturini, R., et al. (2005). CD8+ T-cell lymphocytopenia and lack of EVER mutations in a patient with clinically and virologically typical epidermodysplasia verruciformis. Arch. Dermatol. 141, 1323–1325.
Borgogna, C., Landini, M. M., Lanfredini, S., Doorbar, J., Bouwes Bavinck, J. N., Quint, K. D., et al. (2014). Characterization of skin lesions induced by skin-tropic alpha- and beta-papillomaviruses in a patient with epidermodysplasia verruciformis. Br. J. Dermatol. 171, 1550–1554. doi: 10.1111/bjd.13156
Bouvard, V., Baan, R., Straif, K., Grosse, Y., Secretan, B., El Ghissassi, F., et al. (2009). A review of human carcinogens–Part B: biological agents. Lancet Oncol. 10, 321–322. doi: 10.1016/S1470-2045(09)70096-8
Boxman, I. L., Mulder, L. H., Russell, A., Bouwes Bavinck, J. N., Green, A., and Ter Schegget, J. (1999). Human papillomavirus type 5 is commonly present in immunosuppressed and immunocompetent individuals. Br. J. Dermatol. 141, 246–249. doi: 10.1046/j.1365-2133.1999.02972.x
Burger, B., and Itin, P. H. (2014). Epidermodysplasia verruciformis. Curr. Probl. Dermatol. 45, 123–131. doi: 10.1159/000356068
Casanova, J. L. (2015). Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. U.S.A. 112, E7128–E7137. doi: 10.1073/pnas.1521651112
Casanova, J. L., and Abel, L. (2004). The human model: a genetic dissection of immunity to infection in natural conditions. Nat. Rev. Immunol. 4, 55–66. doi: 10.1038/nri1264
Cockayne, E. (1933). Inherited Abnormalities of the Skin and its Appendages. London: Oxford University Press.
Cooper, K. D., Androphy, E. J., Lowy, D., and Katz, S. I. (1990). Antigen presentation and T-cell activation in epidermodysplasia verruciformis. J. Invest. Dermatol. 94, 769–776. doi: 10.1111/1523-1747.ep12874631
Crequer, A., Picard, C., Patin, E., D’Amico, A., Abhyankar, A., Munzer, M., et al. (2012a). Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One 7:e44010. doi: 10.1371/journal.pone.0044010
Crequer, A., Picard, C., Pedergnana, V., Lim, A., Zhang, S. Y., Abel, L., et al. (2013). EVER2 deficiency is associated with mild T-cell abnormalities. J. Clin. Immunol. 33, 14–21. doi: 10.1007/s10875-012-9749-1
Crequer, A., Troeger, A., Patin, E., Ma, C. S., Picard, C., Pedergnana, V., et al. (2012b). Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J. Clin. Invest. 122, 3239–3247. doi: 10.1172/JCI62949
Danos, O., Engel, L., Chen, E., Yaniv, M., and Howley, P. (1983). Comparative analysis of the human type 1a and bovine type 1 papillomavirus genomes. J. Virol. 46, 557–566.
de Oliveira, W. R., da Cruz Silva, L. L., Neto, C. F., and Tyring, S. (2015). Deleterious effect of radiation therapy on epidermodysplasia verruciformis patients. J. Cutan. Med. Surg. 19, 416–421. doi: 10.1177/1203475415576859
de Oliveira, W. R., Festa Neto, C., Rady, P. L., and Tyring, S. K. (2003). Clinical aspects of epidermodysplasia verruciformis. J. Eur. Acad. Dermatol. Venereol. 17, 394–398. doi: 10.1046/j.1468-3083.2003.00703.x
Gaud, G., Guillemot, D., Jacob, Y., Favre, M., and Vuillier, F. (2013). EVER2 protein binds TRADD to promote TNF-alpha-induced apoptosis. Cell Death Dis. 4:e499. doi: 10.1038/cddis.2013.27
Giri, I., Danos, O., and Yaniv, M. (1985). Genomic structure of the cottontail rabbit (Shope) papillomavirus. Proc. Natl. Acad. Sci. U.S.A. 82, 1580–1584. doi: 10.1073/pnas.82.6.1580
Glinski, W., Jablonska, S., Langner, A., Obalek, S., Haftek, M., and Proniewska, M. (1976). Cell-mediated immunity in epidermodysplasia verruciformis. Dermatologica 153, 218–227. doi: 10.1159/000251060
Glinski, W., Obalek, S., Jablonska, S., and Orth, G. (1981). T cell defect in patients with epidermodysplasia verruciformis due to human papillomavirus type 3 and 5. Dermatologica 162, 141–147. doi: 10.1159/000250262
Howley, P. M., and Pfister, H. J. (2015). Beta genus papillomaviruses and skin cancer. Virology 479–480, 290–296. doi: 10.1016/j.virol.2015.02.004
Hu, J., Han, R., Cladel, N. M., Pickel, M. D., and Christensen, N. D. (2002). Intracutaneous DNA vaccination with the E8 gene of Cottontail Rabbit Papillomavirus induces protective immunity against virus challenge in Rabbits. J. Virol. 76, 6453–6459. doi: 10.1128/JVI.76.13.6453-6459.2002
Iannacone, M. R., Gheit, T., Pfister, H., Giuliano, A. R., Messina, J. L., Fenske, N. A., et al. (2014). Case-control study of genus-beta human papillomaviruses in plucked eyebrow hairs and cutaneous squamous cell carcinoma. Int. J. Cancer 134, 2231–2244. doi: 10.1002/ijc.28552
Imahorn, E., Yuksel, Z., Spoerri, I., Gurel, G., Imhof, C., Saracoglu, Z. N., et al. (2017). Novel TMC8 splice site mutation in epidermodysplasia verruciformis and review of HPV infections in patients with the disease. J. Eur. Acad. Dermatol. Venereol. 31, 1722–1726. doi: 10.1111/jdv.14431
Jablonska, S., Biczysko, W., Jakubowicz, K., and Dabrowski, J. (1968). On the viral etiology of epidermodysplasia verruciformis Lewandowsky-Lutz. Electron microscope studies. Dermatologica 137, 113–125. doi: 10.1159/000254038
Jablonska, S., Dabrowski, J., and Jakubowicz, K. (1972). Epidermodysplasia verruciformis as a model in studies on the role of papovaviruses in oncogenesis. Cancer Res. 32, 583–589.
Jablonska, S., Fabjanska, L., and Formas, I. (1966). On the viral etiology of epidermodysplasia verruciformis. Dermatologica 132, 369–385. doi: 10.1159/000254437
Jablonska, S., and Milewski, B. (1957). Information on epidermodysplasia verruciformis Lewandowsky-Lutz; positive results of auto- and heteroinoculation. Dermatologica 115, 1–22. doi: 10.1159/000255982
Jablonska, S., Orth, G., Jarzabek-Chorzelska, M., Glinski, W., Obalek, S., Rzesa, G., et al. (1979). Twenty-one years of follow-up studies of familial epidermodysplasia verruciformis. Dermatologica 158, 309–327. doi: 10.1159/000250775
Keresztes, G., Mutai, H., and Heller, S. (2003). TMC and EVER genes belong to a larger novel family, the TMC gene family encoding transmembrane proteins. BMC Genomics 4:24. doi: 10.1186/1471-2164-4-24
Kupper, T. S., and Fuhlbrigge, R. C. (2004). Immune surveillance in the skin: mechanisms and clinical consequences. Nat. Rev. Immunol. 4, 211–222. doi: 10.1038/nri1310
Kurima, K., Yang, Y., Sorber, K., and Griffith, A. J. (2003). Characterization of the transmembrane channel-like (TMC) gene family: functional clues from hearing loss and epidermodysplasia verruciformis. Genomics 82, 300–308. doi: 10.1016/S0888-7543(03)00154-X
Laffort, C., Le Deist, F., Favre, M., Caillat-Zucman, S., Radford-Weiss, I., Debre, M., et al. (2004). Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common gammac cytokine receptor subunit or JAK-3 deficiency. Lancet 363, 2051–2054. doi: 10.1016/S0140-6736(04)16457-X
Landini, M. M., Borgogna, C., Peretti, A., Colombo, E., Zavattaro, E., Boldorini, R., et al. (2014). alpha- and beta-papillomavirus infection in a young patient with an unclassified primary T-cell immunodeficiency and multiple mucosal and cutaneous lesions. J. Am. Acad. Dermatol. 71, 108.e1–115.e1. doi: 10.1016/j.jaad.2014.01.859
Landini, M. M., Zavattaro, E., Borgogna, C., Azzimonti, B., De Andrea, M., Colombo, E., et al. (2012). Lack of EVER2 protein in two epidermodysplasia verruciformis patients with skin cancer presenting previously unreported homozygous genetic deletions in the EVER2 gene. J. Invest. Dermatol. 132, 1305–1308. doi: 10.1038/jid.2011.399
Lazarczyk, M., Cassonnet, P., Pons, C., Jacob, Y., and Favre, M. (2009). The EVER proteins as a natural barrier against papillomaviruses: a new insight into the pathogenesis of human papillomavirus infections. Microbiol. Mol. Biol. Rev. 73, 348–370. doi: 10.1128/MMBR.00033-08
Lazarczyk, M., Dalard, C., Hayder, M., Dupre, L., Pignolet, B., Majewski, S., et al. (2012). EVER proteins, key elements of the natural anti-human papillomavirus barrier, are regulated upon T-cell activation. PLoS One 7:e39995. doi: 10.1371/journal.pone.0039995
Lazarczyk, M., Pons, C., Mendoza, J. A., Cassonnet, P., Jacob, Y., and Favre, M. (2008). Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J. Exp. Med. 205, 35–42. doi: 10.1084/jem.20071311
Lewandowsky, F., and Lutz, W. (1922). Ein Fall einer bisher nicht beschriebenen hauterkrankung (Epidermodysplasia verruciformis). Arch. Dermatol. Syphilol. 141, 193–203. doi: 10.1007/BF01938833
Li, S. L., Duo, L. N., Wang, H. J., Dai, W., Zhou, E. H., Xu, Y. N., et al. (2016). Identification of LCK mutation in a family with atypical epidermodysplasia verruciformis with T-cell defects and virus-induced squamous cell carcinoma. Br. J. Dermatol. 175, 1204–1209. doi: 10.1111/bjd.14679
Liu, Y. Q., Zhang, G. L., Mo, X. H., Wang, B., Wu, F., Chen, J., et al. (2017). A novel homozygous DOCK8 mutation associated with unusual coexistence of gross molluscum contagiosum and epidermodysplasia verruciformis in a DOCK8 deficiency patient. J. Eur. Acad. Dermatol. Venereol. 31, e504–e505. doi: 10.1111/jdv.14344
Lutz, W. (1946). A propos de l’épidermodysplasie verruciforme. Dermatologica 92, 30–43. doi: 10.1159/000255805
Lutzner, M. A. (1978). Epidermodysplasia verruciformis. An autosomal recessive disease characterized by viral warts and skin cancer. A model for viral oncogenesis. Bull. Cancer 65, 169–182.
Majewski, S., and Jablonska, S. (1992). Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancers: the role of local immunosurveillance. Am. J. Med. Sci. 304, 174–179. doi: 10.1097/00000441-199209000-00006
Majewski, S., and Jablonska, S. (1997). Skin autografts in epidermodysplasia verruciformis: human papillomavirus-associated cutaneous changes need over 20 years for malignant conversion. Cancer Res. 57, 4214–4216.
Majewski, S., Jablonska, S., and Orth, G. (1997). Epidermodysplasia verruciformis. Immunological and nonimmunological surveillance mechanisms: role in tumor progression. Clin. Dermatol. 15, 321–334. doi: 10.1016/S0738-081X(96)00169-1
Majewski, S., Skopinska-Rozewska, E., Jablonska, S., Wasik, M., Misiewicz, J., and Orth, G. (1986). Partial defects of cell-mediated immunity in patients with epidermodysplasia verruciformis. J. Am. Acad. Dermatol. 15, 966–973. doi: 10.1016/S0190-9622(86)70258-2
Michael, K. M., Waterboer, T., Pfister, H., Gariglio, M., Majewski, S., Favre, M., et al. (2010). Seroreactivity of 38 human papillomavirus types in epidermodysplasia verruciformis patients, relatives, and controls. J. Invest. Dermatol. 130, 841–848. doi: 10.1038/jid.2009.356
Mitsuishi, T., Ohara, K., Suzuki, T., Mochizuki, T., Kaneko, T., and Kawana, S. (2008). Epidermodysplasia verruciformis with keratoacanthoma, Bowen’s disease and squamous cell carcinoma: isolation of high-risk types of HPV 5 and unknown type of human papillomavirus. J. Eur. Acad. Dermatol. Venereol. 22, 1126–1127. doi: 10.1111/j.1468-3083.2007.02526.x
Miyauchi, T., Nomura, T., Suzuki, S., Takeda, M., Shinkuma, S., Arita, K., et al. (2016). Genetic analysis of a novel splice-site mutation in TMC8 reveals the in vivo importance of the transmembrane channel-like domain of TMC8. Br. J. Dermatol. 175, 803–806. doi: 10.1111/bjd.14569
Neale, R. E., Weissenborn, S., Abeni, D., Bavinck, J. N., Euvrard, S., Feltkamp, M. C., et al. (2013). Human papillomavirus load in eyebrow hair follicles and risk of cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 22, 719–727. doi: 10.1158/1055-9965.EPI-12-0917-T
Nonnenmacher, M., Salmon, J., Jacob, Y., Orth, G., and Breitburd, F. (2006). Cottontail rabbit papillomavirus E8 protein is essential for wart formation and provides new insights into viral pathogenesis. J. Virol. 80, 4890–4900. doi: 10.1128/JVI.80.10.4890-4900.2006
Notarangelo, L., Casanova, J. L., Fischer, A., Puck, J., Rosen, F., Seger, R., et al. (2004). Primary immunodeficiency diseases: an update. J. Allergy Clin. Immunol. 114, 677–687. doi: 10.1016/j.jaci.2004.06.044
Orth, G. (1986). Epidermodysplasia verruciformis: a model for understanding the oncogenicity of human papillomaviruses. Ciba Found. Symp. 120, 157–174.
Orth, G. (1987). “Epidermodysplasia verruciformis,” in The Papovaviridae. The Papillomaviruses, Vol. 2, eds P. M. Howley and N. P. Salzman (New York, NY: Plenum Press Inc.), 199–243.
Orth, G. (2006). Genetics of epidermodysplasia verruciformis: insights into host defense against papillomaviruses. Semin. Immunol. 18, 362–374. doi: 10.1016/j.smim.2006.07.008
Orth, G. (2008). Host defenses against human papillomaviruses: lessons from epidermodysplasia verruciformis. Curr. Top. Microbiol. Immunol. 321, 59–83. doi: 10.1007/978-3-540-75203-5_3
Orth, G., Favre, M., Breitburd, F., Croissant, O., Jablonska, M., Obalek, S., et al. (1980). “Epidermodysplasia verruciformis: a model for the role of papillomavirus in human cancer,” in Viruses in Naturally Occurring Cancers, eds M. Essex, G. Todaro, and H. Hausen (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 259.
Orth, G., Jablonska, S., Favre, M., Croissant, O., Jarzabek-Chorzelska, M., and Rzesa, G. (1978). Characterization of two types of human papillomaviruses in lesions of epidermodysplasia verruciformis. Proc. Natl. Acad. Sci. U.S.A. 75, 1537–1541. doi: 10.1073/pnas.75.3.1537
Orth, G., Jablonska, S., Jarzabek-Chorzelska, M., Obalek, S., Rzesa, G., Favre, M., et al. (1979). Characteristics of the lesions and risk of malignant conversion associated with the type of human papillomavirus involved in epidermodysplasia verruciformis. Cancer Res. 39, 1074–1082.
Ovits, C. G., Amin, B. D., and Halverstam, C. (2017). Acquired epidermodysplasia verruciformis and its relationship to immunosuppressive therapy: report of a case and review of the literature. J. Drugs Dermatol. 16, 701–704.
Platt, C. D., Fried, A. J., Hoyos-Bachiloglu, R., Usmani, G. N., Schmidt, B., Whangbo, J., et al. (2017). Combined immunodeficiency with EBV positive B cell lymphoma and epidermodysplasia verruciformis due to a novel homozygous mutation in RASGRP1. Clin. Immunol. 183, 142–144. doi: 10.1016/j.clim.2017.08.007
Prawer, S. E., Pass, F., Vance, J. C., Greenberg, L. J., Yunis, E. J., and Zelickson, A. S. (1977). Depressed immune function in epidermodysplasia verruciformis. Arch. Dermatol. 113, 495–499. doi: 10.1001/archderm.1977.01640040103018
Rajabi, M. T., Ghasemi, H., Safizadeh, M., Jamshidi, S., Asadi-Amoli, F., Abrishami, Y., et al. (2014). Conjunctival squamous cell carcinoma with intraocular invasion after radiotherapy in epidermodysplasia verruciformis. Can. J. Ophthalmol. 49, e43–e46. doi: 10.1016/j.jcjo.2013.12.009
Rajagopalan, K., Bahru, J., Loo, D. S., Tay, C. H., Chin, K. N., and Tan, K. K. (1972). Familial epidermodysplasia verruciformis of Lewandowsky and Lutz. Arch. Dermatol. 105, 73–78. doi: 10.1001/archderm.1972.01620040045008
Ramoz, N., Rueda, L. A., Bouadjar, B., Favre, M., and Orth, G. (1999). A susceptibility locus for epidermodysplasia verruciformis, an abnormal predisposition to infection with the oncogenic human papillomavirus type 5, maps to chromosome 17qter in a region containing a psoriasis locus. J. Invest. Dermatol. 112, 259–263. doi: 10.1046/j.1523-1747.1999.00536.x
Ramoz, N., Rueda, L. A., Bouadjar, B., Montoya, L. S., Orth, G., and Favre, M. (2002). Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 32, 579–581. doi: 10.1038/ng1044
Ramoz, N., Taieb, A., Rueda, L. A., Montoya, L. S., Bouadjar, B., Favre, M., et al. (2000). Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J. Invest. Dermatol. 114, 1148–1153. doi: 10.1046/j.1523-1747.2000.00996.x
Ruiter, M., and van Mullem, P. J. (1966). Demonstration by electronmicroscopy of an intranuclear virus in epidermodysplasia verruciformis. J. Invest. Dermatol. 47, 247–252. doi: 10.1038/jid.1966.137
Sanal, O., Jing, H., Ozgur, T., Ayvaz, D., Strauss-Albee, D. M., Ersoy-Evans, S., et al. (2012). Additional diverse findings expand the clinical presentation of DOCK8 deficiency. J. Clin. Immunol. 32, 698–708. doi: 10.1007/s10875-012-9664-5
Stepensky, P., Rensing-Ehl, A., Gather, R., Revel-Vilk, S., Fischer, U., Nabhani, S., et al. (2015). Early-onset Evans syndrome, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood 125, 753–761. doi: 10.1182/blood-2014-08-593202
Stray-Pedersen, A., Jouanguy, E., Crequer, A., Bertuch, A. A., Brown, B. S., Jhangiani, S. N., et al. (2014). Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. J. Clin. Immunol. 34, 871–890. doi: 10.1007/s10875-014-0074-8
Tahiat, A., Badran, Y. R., Chou, J., Cangemi, B., Lefranc, G., Labgaa, Z. M., et al. (2017). Epidermodysplasia verruciformis as a manifestation of ARTEMIS deficiency in a young adult. J. Allergy Clin. Immunol. 139, 372.e4–375.e4. doi: 10.1016/j.jaci.2016.07.024
Vuillier, F., Gaud, G., Guillemot, D., Commere, P. H., Pons, C., and Favre, M. (2014). Loss of the HPV-infection resistance EVER2 protein impairs NF-kappaB signaling pathways in keratinocytes. PLoS One 9:e89479. doi: 10.1371/journal.pone.0089479
Wieland, U., Kreuter, A., and Pfister, H. (2014). Human papillomavirus and immunosuppression. Curr. Probl. Dermatol. 45, 154–165. doi: 10.1159/000357907
Zampetti, A., Giurdanella, F., Manco, S., Linder, D., Gnarra, M., Guerriero, G., et al. (2013). Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol. Surg. 39, 974–980. doi: 10.1111/dsu.12135
Keywords: epidermodysplasia verruciformis, beta-HPV, TMC/EVER, non-melanoma skin cancer, primary immunodeficiency
Citation: de Jong SJ, Imahorn E, Itin P, Uitto J, Orth G, Jouanguy E, Casanova J-L and Burger B (2018) Epidermodysplasia Verruciformis: Inborn Errors of Immunity to Human Beta-Papillomaviruses. Front. Microbiol. 9:1222. doi: 10.3389/fmicb.2018.01222
Received: 31 January 2018; Accepted: 22 May 2018;
Published: 12 June 2018.
Edited by:
Herbert Johannes Pfister, Universität zu Köln, GermanyReviewed by:
Marisa Gariglio, Università degli Studi del Piemonte Orientale, ItalyGertrud Steger, Universität zu Köln, Germany
Thomas Richard Broker, The University of Alabama at Birmingham, United States
Copyright © 2018 de Jong, Imahorn, Itin, Uitto, Orth, Jouanguy, Casanova and Burger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Laurent Casanova, Y2FzYW5vdmFAcm9ja2VmZWxsZXIuZWR1; amVhbi1sYXVyZW50LmNhc2Fub3ZhQG1haWwucm9ja2VmZWxsZXIuZWR1