 Josephine Z. Rapp
Josephine Z. Rapp Mar Fernández-Méndez
Mar Fernández-Méndez Christina Bienhold
Christina Bienhold Antje Boetius
Antje Boetius
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 May 2018
Sec. Aquatic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01035
In summer 2012, Arctic sea ice declined to a record minimum and, as a consequence of the melting, large amounts of aggregated ice-algae sank to the seafloor at more than 4,000 m depth. In this study, we assessed the composition, turnover and connectivity of bacterial and microbial eukaryotic communities across Arctic habitats from sea ice, algal aggregates and surface waters to the seafloor. Eukaryotic communities were dominated by diatoms, dinoflagellates and other alveolates in all samples, and showed highest richness and diversity in sea-ice habitats (∼400–500 OTUs). Flavobacteriia and Gammaproteobacteria were the predominant bacterial classes across all investigated Arctic habitats. Bacterial community richness and diversity peaked in deep-sea samples (∼1,700 OTUs). Algal aggregate-associated bacterial communities were mainly recruited from the sea-ice community, and were transported to the seafloor with the sinking ice algae. The algal deposits at the seafloor had a unique community structure, with some shared sequences with both the original sea-ice community (22% OTU overlap), as well as with the deep-sea sediment community (17% OTU overlap). We conclude that ice-algal aggregate export does not only affect carbon export from the surface to the seafloor, but may change microbial community composition in central Arctic habitats with potential effects for benthic ecosystem functioning in the future.
The Arctic Ocean is one of the marine regions most strongly affected by global climate change, with temperatures currently warming two to three times faster than the global average (Overland et al., 2015). As a result, thick multi-year sea ice is being replaced by thinner first-year sea ice, which only lasts for one melt season (Maslanik et al., 2007; Polyakov et al., 2012). An Arctic Ocean free of summer sea ice has been projected before the end of this century (Johannessen et al., 2004; Overland and Wang, 2013; Notz and Stroeve, 2016). First indications for planktonic community changes associated with Arctic change have been described, including a trend toward reduced eukaryote cell sizes with upper ocean freshening (Li et al., 2009) and shifts in species composition associated with increasing water temperature (Nöthig et al., 2015). Sea-ice decline is likely to increase primary production on the Arctic shelves (Carmack and Chapman, 2003; Arrigo et al., 2008), in regions where enough nutrients are supplied (Tremblay and Gagnon, 2009; Arrigo and van Dijken, 2015; Tremblay et al., 2015a). Increasing light availability in and under thinning ice may also enhance ice-algal productivity and under-ice blooms in the deep ice-covered central basins, which have a lower nutrient availability than the shelves (Lalande et al., 2014; Arrigo and van Dijken, 2015; Fernández-Méndez et al., 2015). Furthermore, the extent of melt ponds in summer is increasing (Rösel and Kaleschke, 2012), opening up new habitats for sea-ice biota (Lee et al., 2011). Recent studies have suggested an important, previously underestimated, role of sea-ice algae in primary production and export flux in the central Arctic Ocean (Assmy et al., 2013; Boetius et al., 2013; Fernández-Méndez et al., 2015). Especially diatoms can form extensive blooms in sea ice, melt ponds and at the bottom of ice floes, and constitute the majority of sea-ice associated biomass (Arrigo, 2014 and references therein). Particularly Melosira arctica, which forms long filaments attached to the ice matrix, can build up patchy, but dense accumulations (Melnikov and Bondarchuk, 1987; Fernández-Méndez et al., 2014; Katlein et al., 2014; Poulin et al., 2014). During rapid ice melt, large pulses of these ice algae can be released from the ice (Tamelander et al., 2009) and sink out of the surface ocean, thereby significantly altering the magnitude and composition of organic matter reaching the seafloor (Ambrose et al., 2005; Boetius et al., 2013). Yet, the low number of documented observations of such large ice algae export events and the relatively poor knowledge of spatial and temporal variability of microbial community composition in the central Arctic currently impede predictions about the effects of such environmental changes on biodiversity and ecosystem functioning (Galand et al., 2010; Comeau et al., 2011; Wassmann, 2011; Ghiglione et al., 2012; Thaler and Lovejoy, 2015).
In the Arctic Ocean, in contrast to other ocean environments, temperatures close to the freezing point prevail across sea ice, water column and seafloor habitats, potentially permitting a close vertical connectivity of communities. Seed communities in transient environments such as sea ice are likely derived from directly connected environments (Ackley and Sullivan, 1994), i.e., surface ocean waters and sediments that are incorporated during ice formation on the shallow Arctic shelves (Wegner et al., 2017). In addition, the focused seasonal particulate organic carbon flux from the sea ice and surface ocean to the seafloor may transport viable microbes to the seafloor, where they could contribute to the turnover of carbon and nutrients, and potentially also become members of the benthic communities (Turley and Mackie, 1995; Ruff et al., 2014).
This study aimed at assessing the community composition and turnover of bacterial and eukaryotic microorganisms associated with ice-associated and sinking algal aggregates, as well as their similarity with potential source communities of sea ice, water and deep-sea sediments using next-generation sequencing. We hypothesized (i) that sea-ice algae aggregates select for specific members of eukaryotes and bacteria; (ii) that these originate from sea ice rather than the water column; (iii) that these are exported by rapidly sinking aggregates that transport sea-ice life to the deep sea, but (iv) get quickly overgrown by sediment microbes. Furthermore, this study contributes baseline knowledge of microbial diversity in the central Arctic Ocean in times of rapid sea-ice melt.
Our samples were retrieved from nine sampling locations across the Eurasian Basin of the central Arctic Ocean during RV Polarstern expedition ARK-XXVII/3 (PS80) in August and September 2012 at the end of the productive season (Figure 1). Surface seawater temperatures were between -1.8 to -1.5°C and salinity ranged from 30.3 to 33.2 (Boetius et al., 2013). Nutrient inventories in the surface layer were already very low and nitrate generally limiting (Fernández-Méndez et al., 2015). First-year ice was the dominant ice type during the time of sampling (Supplementary Table S1), and ice thickness varied between 0.7 and 2 m (Fernández-Méndez et al., 2014). Salinity of the total sea-ice surface ranged between 0.1–2.9, and 1.0–3.6 in the bottom layer. Temperatures within surface sea ice varied from -8.5 to 0°C and were between -1.9 and -1.1°C in bottom ice (Hardge et al., 2017). Melt-pond coverage on ice floes varied between stations and ranged from 20 to 50%. Different types of melt ponds were observed, including closed (closed to the underlying seawater), and open ponds (directly connected to the underlying seawater). Some of them had a refrozen surface ice cover. Sea-ice algal aggregates of varying size and color were observed in all different types of ponds (Fernández-Méndez et al., 2014). Seafloor observations revealed the presence of deposited ice-algal aggregates on the sediment surface in the deep sea, with an especially high carbon deposition of 32 and 156 g C m-2 at stations Ice7 and Ice8 (Figure 1), when compared to the remaining stations where values ranged from 0 to 20 g C m-2 (Boetius et al., 2013). The presence of algae deposits at the seafloor often coincided with the presence of opportunistic holothurians of the species Kolga hyalina, which were attracted by the fresh food source (Boetius et al., 2013). Bottom water temperatures in the deep sea were stable at around -0.7°C (Rabe et al., 2013). Direct links to the original data on environmental conditions during sampling can be found via the PANGAEA database entry (Rapp et al., 2017).
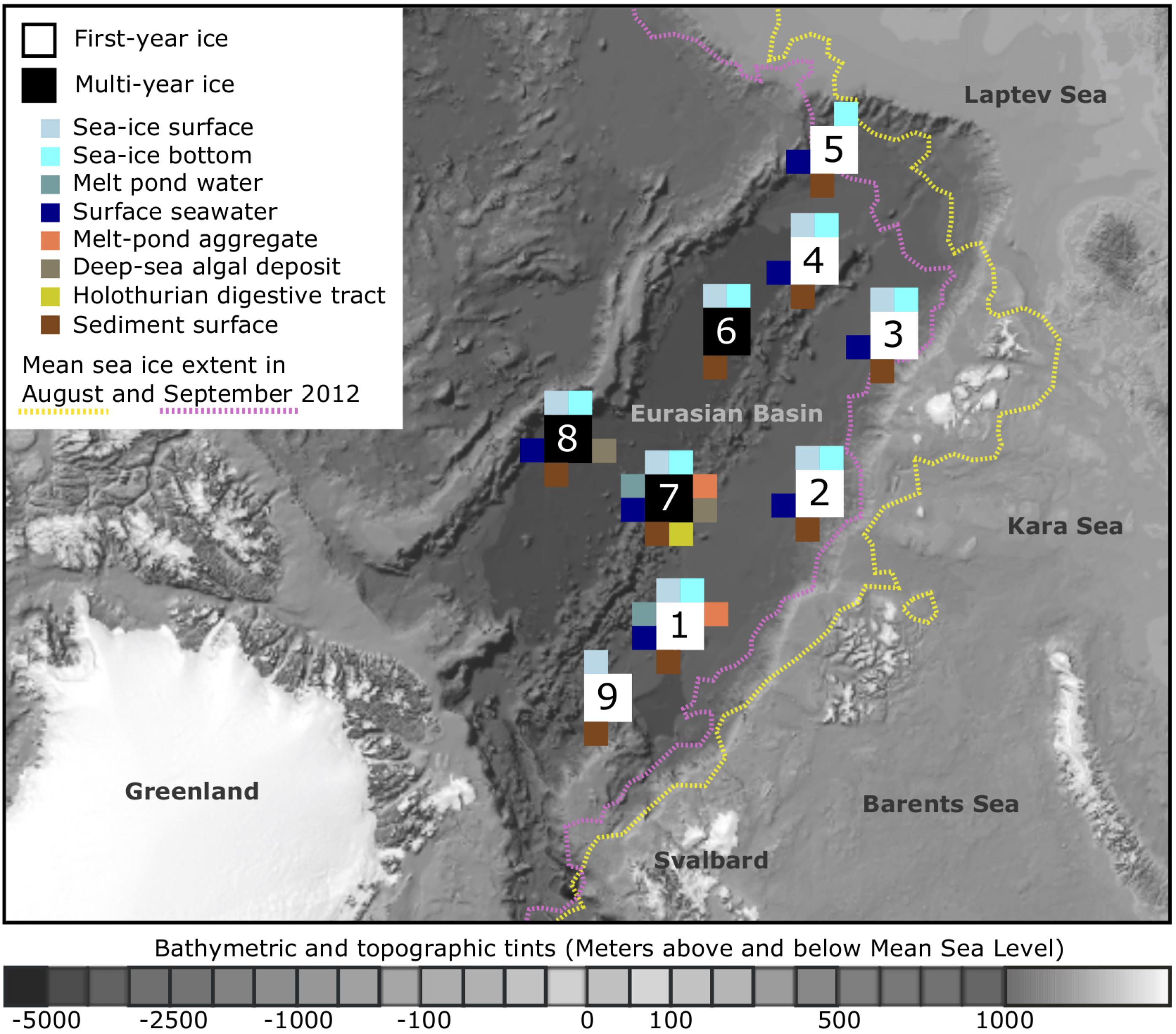
FIGURE 1. Sampling locations and sampled environments. Map of the central Arctic Ocean, depicting sampling locations and mean sea-ice extent during RV Polarstern expedition ARK-XXVII/3 (PS80) in 2012. A total of nine stations were visited from August to the end of September, with the individual sampled environments indicated by color. The bathymetric map was modified from Jakobsson et al. (2012). For further details on sampling location and the conducted analyses see Supplementary Table S1.
Sea-ice algal aggregates and melt pond water were sampled from three distinct melt ponds at stations Ice1 and Ice7 (Figure 1 and Supplementary Table S1) using a manually operated vacuum pump (Model 6132-0010, Nalgene, Penfield, NY, United States). Deposited ice-algal aggregates from the deep-sea floor at stations Ice7 and Ice8 were retrieved using a TV-guided multicorer or a MultiGrab, and individually collected from the sediment surface with sterile plastic pipets or forceps. Sea-ice cores from all stations were taken with an ice corer (9 cm diameter) (Kovacs Enterprise, Roseburg, OR, United States), cut into two equal sections and melted in plastic containers (previously rinsed with ethanol and ultrapure water) on a shaker in the dark at 4°C (Mikkelsen et al., 2008; Rintala et al., 2014). Ice core length ranged from 0.8 to 2.0 m (Supplementary Table S1). Four ice samples were not sectioned: two ice cores from station 9 (one of them from newly formed ice and the other one from very thin ice adjacent to it), as well as two cores from station 7 and 8, which showed a strong brownish coloration (indication of high algal biomass) (Supplementary Table S1). Melting of the ice took around 24 h and samples were immediately processed as soon as the last piece of ice melted, to avoid abrupt changes of temperature in the sample. Surface seawater was sampled either with a peristaltic pump through a hole in the sea ice (Masterflex® E/STM portable sampler, 115 VAC, Oldham, United Kingdom), or with a conductivity–temperature–depth (CTD) rosette sampler (Seabird SBE 911 plus, Bellevue, WA, United States) at 0–2 m depth. Between 0.5 and 2 L of melted sea ice or water were filtered through 0.22 μm polyethersulfone membranes (Millipore® SterivexTM, Merck KGaA, Darmstadt, Germany) with a multichannel peristaltic pump (Model PD 51; Heidolph, Schwabach, Germany). Three parallel deep-sea sediment cores were retrieved from each sampling site by a TV-guided multicorer and subsamples of the uppermost centimeter of each core were pooled per site. At station Ice1, we obtained a total of nine sediment cores, allowing us to prepare three replicate sediment samples (Supplementary Table S1). Holothurians feeding on the deposited algal aggregates were collected using an Agassiz trawl, digestive tracts were dissected and the content stored in plastic syringes. All Sterivex filters and deep-sea samples were stored at -20°C until further processing.
A total of 54 samples were taken for microbial community analysis, 52 of these were subject to bacterial community analysis, and 45 of these samples to eukaryotic community analysis (Supplementary Table S1).
Total community DNA was extracted from half Sterivex filters for sea-ice, melt-pond and seawater samples, from 0.5 g of algae aggregates and holothurian digestive tract content and from 1 g of sediment using the UltraCleanTM Soil DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, CA, United States) following the manufacturer’s instructions for maximum yields, with minor modifications. Instead of using the kit’s enclosed solution S5, DNA extracts were eluted in a final volume of 80 μl TE-buffer (10 mM Tris-Cl, pH 8.0; 1 mM EDTA). By using one standardized extraction kit across all sample types, we aimed to minimize potential biases toward differential extraction yields for individual microbial taxa and thus increase comparability. DNA concentration and purity were determined by fluorescence-based Qubit® quantitation assays (Life Technologies GmbH, Darmstadt, Germany). Amplicon libraries of the bacterial V4-V6 region of the 16S rRNA gene and the eukaryotic V4 region of the 18S rRNA gene were generated according to the protocol recommended by Illumina (16S Metagenomic Sequencing Library Preparation, Part #15044223, Rev. B). For selected sediment, sea-ice algae and deposited algae samples we obtained technical replicates, by preparing several sequencing libraries from the same DNA extract (Supplementary Table S1). For Bacteria we selected the S-D-Bact-0564-a-S-15 and S-∗Univ-1100-a-A-15 primer pair based on a primer evaluation by Klindworth et al. (2013) and for Eukaryota the TAReukFWD1 and TAReukREV3 primers (Stoeck et al., 2010) (Supplementary Information Scripts bac and euk). Libraries were sequenced on an Illumina MiSeq platform in 2x300 cycles paired end runs. Raw paired-end sequences have been submitted to ENA under INSDC accession number PRJEB23005 using the data brokerage service of the German Federation for Biological Data (GFBio, Diepenbroek et al., 2014), in compliance with the Minimal Information about any (X) Sequence (MIxS) standard (Yilmaz et al., 2011). To corroborate the observed bacterial community patterns, we used Automated Ribosomal Intergenic Spacer Analysis in parallel to the tag sequencing approach for a wider set of in total 68 samples (Supplementary Information Material and Methods).
An initial 10,266,909 bacterial and 3,717,438 eukaryotic raw sequence pairs were generated (Supplementary Table S2). We used cutadapt (v. 1.8.1; Martin, 2011) for the removal of primer sequences and a custom awk script to ensure the correct orientation of reads prior to merging (Supplementary Information Scripts bac and euk). For merging forward and reverse reads we used pear (v. 0.9.5; Zhang et al., 2014) and trimmed and quality filtered all sequences using trimmomatic (v. 0.32; Bolger et al., 2014). 2,225,003 bacterial and 2,777,311 eukaryotic merged and quality trimmed sequences were retained after processing (Supplementary Table S2). We reassured correct formatting of the fastq files with bbmap (v. 34.00; B. Bushnell1) before clustering the reads into 613,270 bacterial and 59,853 eukaryotic OTUs by applying a local clustering threshold of d = 1 and the fastidious option in swarm (v. 2.1.1; Mahé et al., 2015). After alignment with the SINA aligner (v. 1.2.10; Pruesse et al., 2012) and classification of the seed sequence of each OTU with the SILVA SSU database release 123 (Quast et al., 2013), we removed all OTUs that were classified as chloroplasts, mitochondria, archaea, or those that could not be classified at domain level from further analysis. OTUs that were classified as bacteria within the eukaryotic dataset and vice versa, were removed as well. Furthermore, we removed all absolute singletons, i.e., OTUs that were only represented by a single sequence across the complete dataset (Supplementary Table S2). By doing so we reduced the effect of artificially inflated diversity potentially introduced through sequencing errors (Tremblay et al., 2015b). Filtering and removal of absolute singletons resulted in a final number of 8,869 bacterial and 7,627 eukaryotic OTUs (Supplementary Table S2). All further analyses were performed with these processed OTU abundance tables (Rapp et al., 2017). The full bioinformatic scripts are provided as supplementary information (Supplementary Information Scripts bac and euk).
We calculated OTU numbers, as well as the Chao1 richness estimate and the inverse Simpson diversity index per sample. To obtain reliable estimates independent of differences in sequencing depth between samples, we subsampled our input OTU tables (Rapp et al., 2017) 100-times to the lowest read number of each dataset (bacteria: 10,068 reads; eukaryotes: 16,372 reads) and used mean values across all iterations. Significant differences in OTU numbers between habitats were determined by analysis of variance using permutation tests as implemented in the function aovp from the lmperm package (Wheeler and Torchiano, 2016). Post hoc pairwise permutation tests were performed using the function pairwisePermutationTest from the rcompanion package (Mangiafico, 2017), and p-values were corrected for multiple testing using the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995). We generated species accumulation curves at different taxonomic resolution to assess the efficiency of our sampling effort to capture the present diversity in the individual environments (Supplementary Figures S1–S4). To assess overall differences in community structure we calculated Bray-Curtis dissimilarity matrices and visualized the results in non-metric multidimensional scaling (NMDS) plots. Due to the high variability in 18S gene copy numbers of microbial eukaryotes, which can vary by at least four orders of magnitude, depending on genome (Prokopowich et al., 2003) and cell (Zhu et al., 2005) size, we additionally assessed community structure using the Jaccard dissimilarity measure for presence/absence of eukaryotic taxa (Supplementary Figure S5). Correlation of both dissimilarity measures was examined by performing a mantel test with 999 permutations (Supplementary Figure S6). Concomitantly, we performed an analysis of similarity (ANOSIM) to test for the significance of differences between groups of samples from different environment types. All analyses were conducted in R (v. 3.1.2; R Core Team, 2014) using the vegan package (Oksanen et al., 2014) and custom R-scripts (Supplementary Information Scripts bac and euk; Gobet et al., 2010). For further community comparisons we chose a conservative presence-absence approach. We aimed to distinguish between abundant, widely dispersed groups of an environment and transient groups, which are present at a subset of locations (Shade and Handelsman, 2012). Therefore, we applied a rule that would count a taxon (i.e., an OTU) as dispersed and abundant, if found in >50% of the samples of a habitat, and represented by at least 100 sequences (Supplementary Tables S3, S4). Next, we determined the proportion of community overlap (i.e., shared groups) between the sampled environments using Jaccard similarities. Further, we defined those OTUs that were shared across sea ice, water column, aggregates, as well as deep-sea surface sediments as generalists. Consequently, habitat-specific OTUs are members of only one environmental community.
We saw indications for higher eukaryotic and bacterial community richness in sea-ice algal aggregates than in the surrounding melt-pond water, and in the case of bacteria, also than in adjacent sea ice (Table 1 and Figure 2). Only 11–12% of the OTUs associated with ice-algae aggregates met our definition of dispersed, abundant community members, i.e., they were present in >50% of aggregate samples and represented by at least 100 sequences. These OTUs represented the majority of sea-ice algae associated sequences (Table 1). Melt-pond waters showed the lowest eukaryote and bacterial OTU richness of the sampled ice habitats, but had a relatively large fraction of abundant bacterial types (23%). Bacterial communities in sea ice also exhibited relatively low richness, while eukaryotic communities showed highest richness in sea-ice samples (Figure 2).
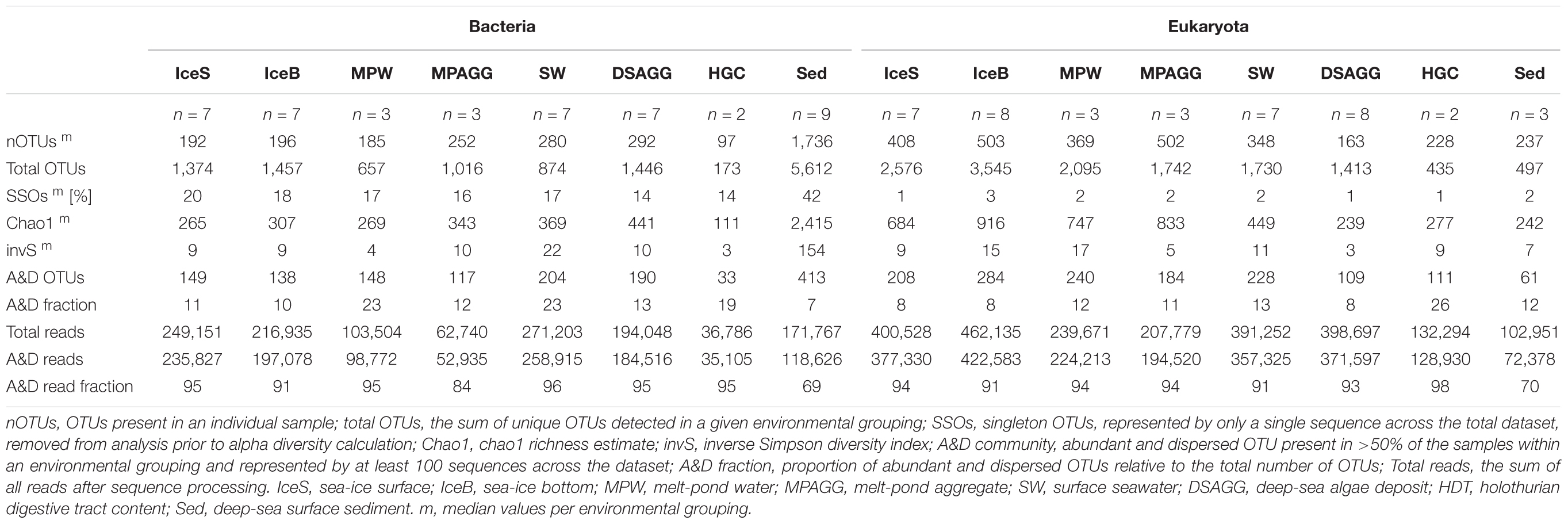
TABLE 1. Community alpha diversity and contribution of abundant and dispersed community members.
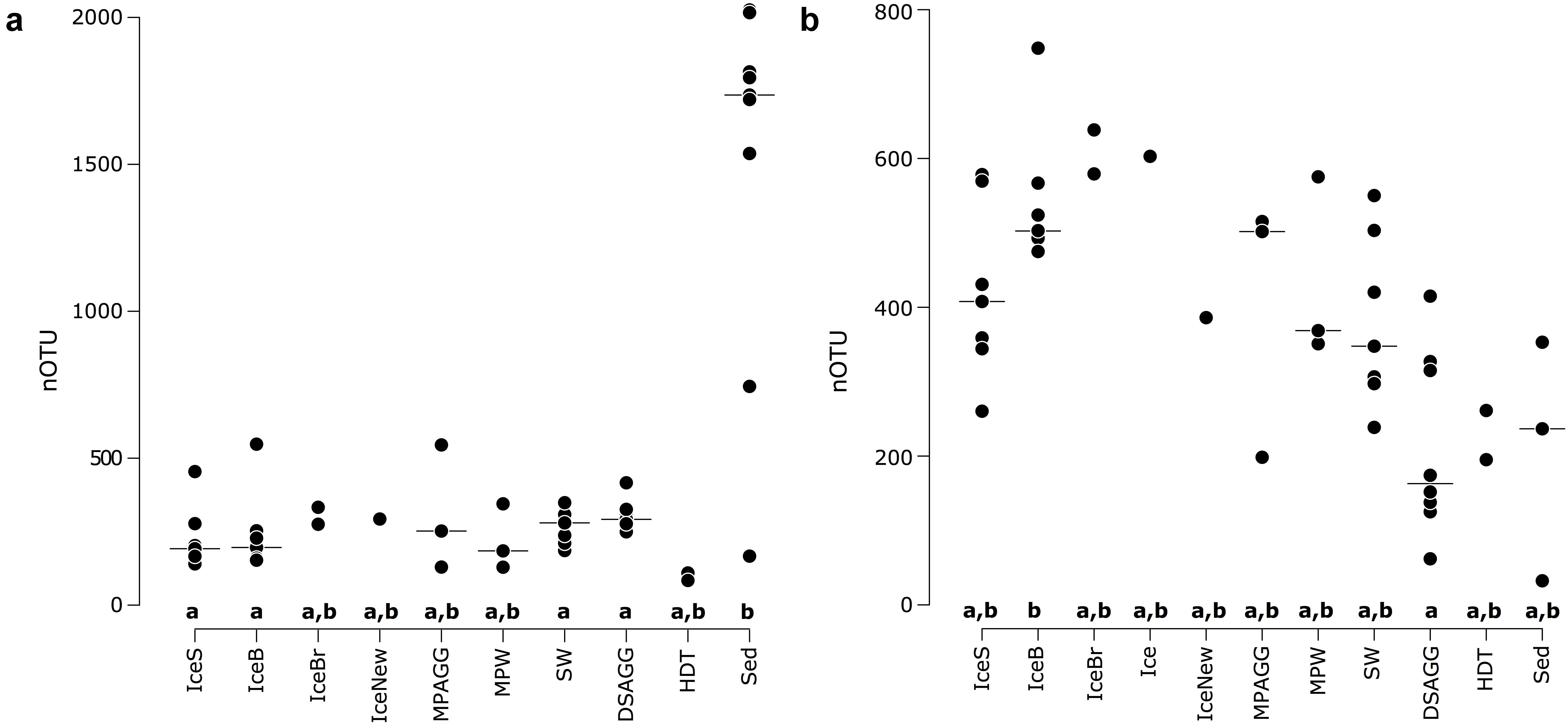
FIGURE 2. Number of OTUs for (a) bacterial communities and (b) eukaryotic communities, with the horizontal black line indicating the median value per environmental grouping. Lower case letters at the bottom of the figure indicate environments that are significantly different from each other based on ANOVA permutation tests at a significance threshold of p < 0.05. IceS, sea-ice surface; IceB, sea-ice bottom; IceBr, brown sea ice; Ice, sea ice; IceNew, freshly formed sea ice; MPAGG, melt-pond aggregate; MPW, melt-pond water; SW, surface seawater; DSAGG, deep-sea algae deposit; HDT, holothurian digestive tract content; Sed, deep-sea surface sediment.
Bacterial communities on sunken ice-algal aggregates deposited at the seafloor (4,400 m water depth) during the summer season were slightly richer than the communities on photosynthetically active aggregates in melt ponds, communities in sea ice and in surface seawater. They still shared 22% of their abundant OTUs with the original ice-algae associated community, but also 17% with the abundant, dispersed sediment bacteria (Figure 3a). We derived the relative contribution of these shared OTUs (Supplementary Table S3) to the total communities detected in deposits and sediments (Rapp et al., 2017). The OTUs shared between sediments and deposits accounted for on average 55% of the bacterial sequences in the algal deposits (up to 79% in individual aggregates), but only for 28% of sequences in the total sediment community. In contrast, eukaryotic communities of the deposited algae showed lowest richness and diversity compared to all other environments (Table 1 and Figure 2). They shared only 29% with the algae-associated abundant types (Figure 3b), and 16% with the abundant sediment eukaryotes. Bacterial richness and diversity was lowest in the digestive tract content of holothurians feeding on the deposited aggregates on the seafloor (Table 1 and Figure 2), and these bacteria were mostly unique to the holothurian digestive tract, with only 4% overlap with the abundant, dispersed types of the sediment community (Figure 3). Deep-sea surface sediments contained a significantly richer and more diverse bacterial community than sea ice, seawater and algae deposits, and showed almost no overlap with water column and sea-ice communities, while eukaryotic richness in sediments was at the lower end of the observed range across all sampled environments (Figure 2).
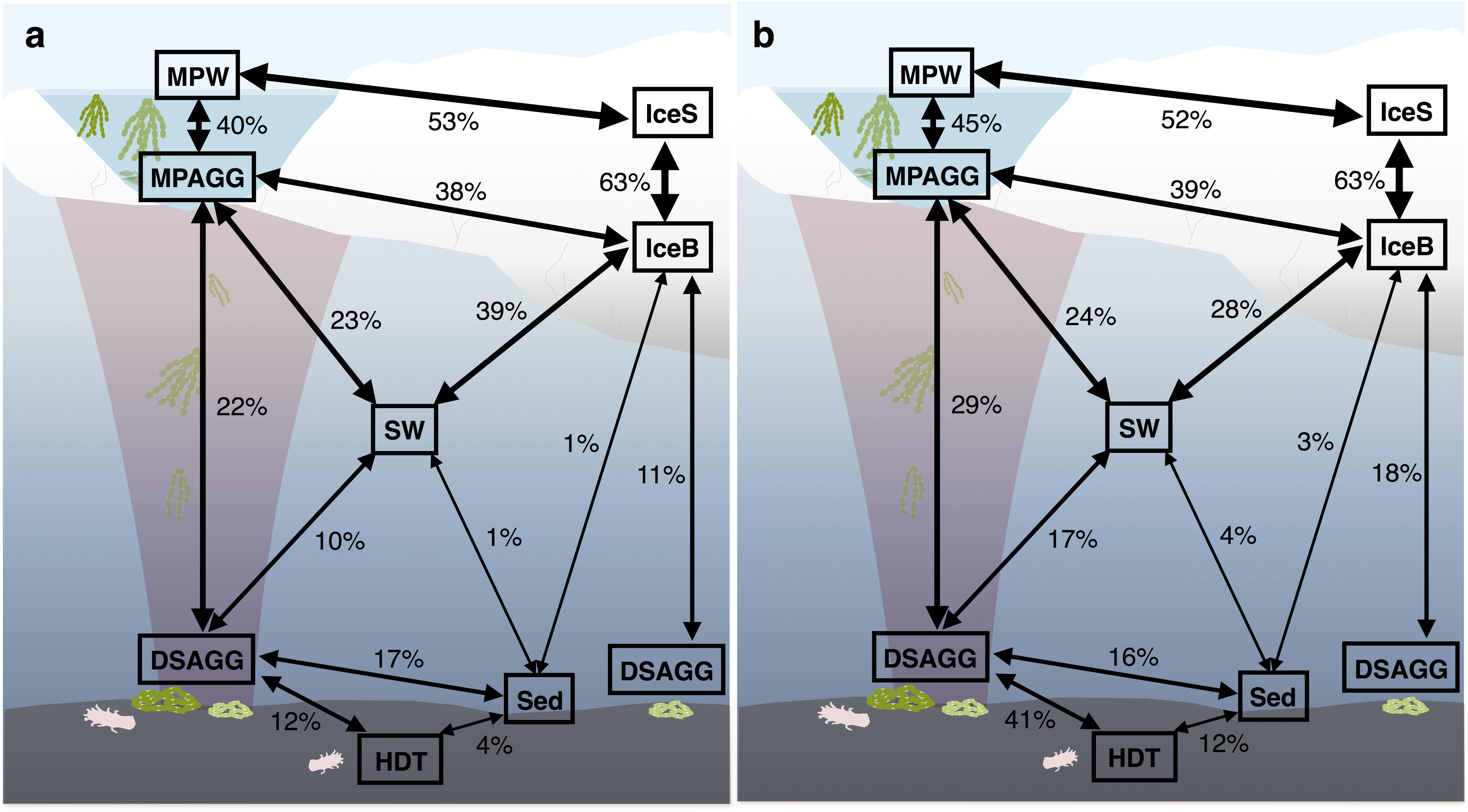
FIGURE 3. Community overlap between environments for (a) bacteria and (b) eukaryotes. Shared proportions are based on presence-absence data for abundant and dispersed community OTUs only, including these OTUs that were present in at least 50% of the samples from one environment and represented by at least 100 sequences in the dataset. IceS, sea-ice surface; IceB, sea-ice bottom; MPAGG, melt-pond aggregate; MPW, melt-pond water; SW, surface seawater; DSAGG, deep-sea algae deposit; HDT, holothurian digestive tract content; Sed, deep-sea surface sediment.
Non-metric multidimensional scaling (NMDS) of Illumina OTUs clustered the samples into five groups according to their habitat (Figure 4 and Supplementary Figure S5), including communities from (i) sea-ice associated environments (sea-ice algae aggregates, melt ponds and sea ice), (ii) surface seawater, (iii) deep-sea sediment, (iv) deposited sea-ice algal aggregates and (v) holothurian digestive tract both for bacteria (ANOSIM: R = 0.86, p = 0.001) and eukaryotes (ANOSIM: R = 0.63, p = 0.001). In both datasets, the communities in sea-ice algal aggregates showed high within-group variation. Notably, eukaryotic community structure in deep-sea sediment and exported ice-algal aggregates showed a much larger dissimilarity than bacterial communities (Figure 4b). Overall, the dissimilarities of bacterial and eukaryotic community structures were significantly correlated across all habitats (r = 0.76; p = 0.001) (Supplementary Figure S7). Bacterial community patterns observed with Illumina tag sequences were supported by analyses of ARISA patterns, which built on a larger set of samples, i.e., from melt pond water, sea-ice algae aggregates and holothurian digestive tract (Supplementary Figure S8); dissimilarity matrices from both methods were significantly correlated (Supplementary Figure S9 and Supplementary Information Results).
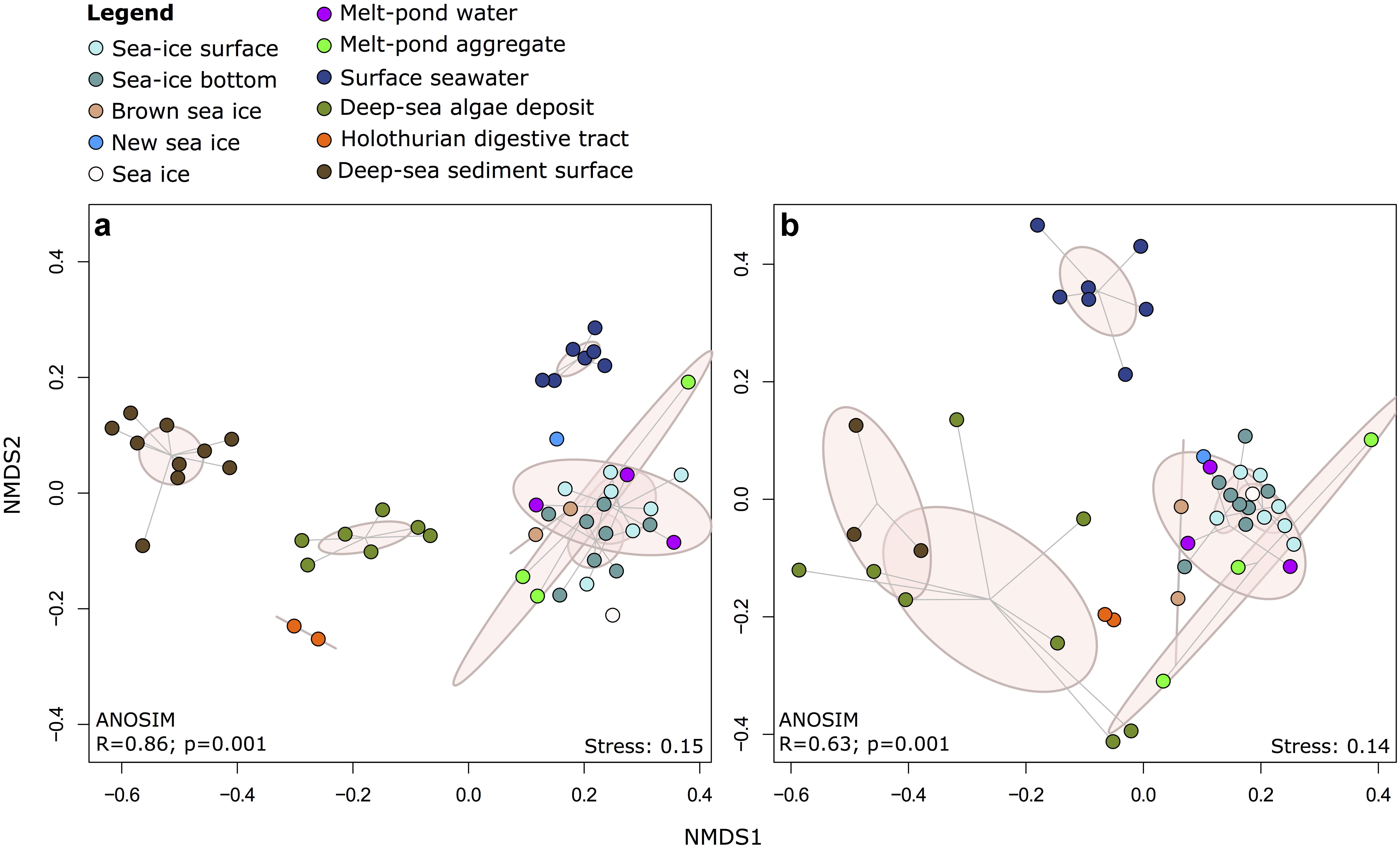
FIGURE 4. Two-dimensional NMDS ordination of community dissimilarities for (a) bacteria and (b) eukaryotes. Dissimilarity matrices and ANOSIM were calculated using the Bray-Curtis dissimilarity measure on the basis of relative abundances of Illumina OTUs. Environments are depicted by color coding and points within each environmental grouping are connected to their group centroid through a spider diagram. Pink ellipses indicate the estimated 95% dispersion limits of each group.
At the time of sampling in late summer, contribution of diatom sequences in surface seawater was minor (Figure 5a). Dinophyceae were the predominant group in surface seawater, but the majority of its representatives could not be classified (Figure 6). One dominant genus was Karlodinium, which was also observed in individual deposited algae aggregates at the seafloor, and in very high sequence abundance in Ice7 deep-sea sediment, where we observed high algae deposition. Eukaryote community composition in sea ice comprised green-brown algae and Dinophyceae, mainly the genus Scrippsiella, members of the Gymnodinium clade and unclassified Suessiaceae in the upper part of the cores, and mostly unclassified diatoms at the bottom of the ice (Figure 6). The genera Pseudo-nitzschia and Melosira exhibited a patchy distribution with low relative abundance (<1%) at all stations except for Ice7. All of these groups were also present in melt-pond waters, in addition to other algae from the Pelagophyceae and a larger contribution of Thecofilosea, including the nanoflagellate Cryothecomonas. In deep-sea sediments, Labyrinthulomycetes, dinoflagellates and other flagellates from the Imbricatea dominated eukaryote communities (Figures 5a, 6). At Ice7 we observed a relatively high contribution of the diatom Chaetoceros, the only diatom detected in the sediment outside of the algal deposits.
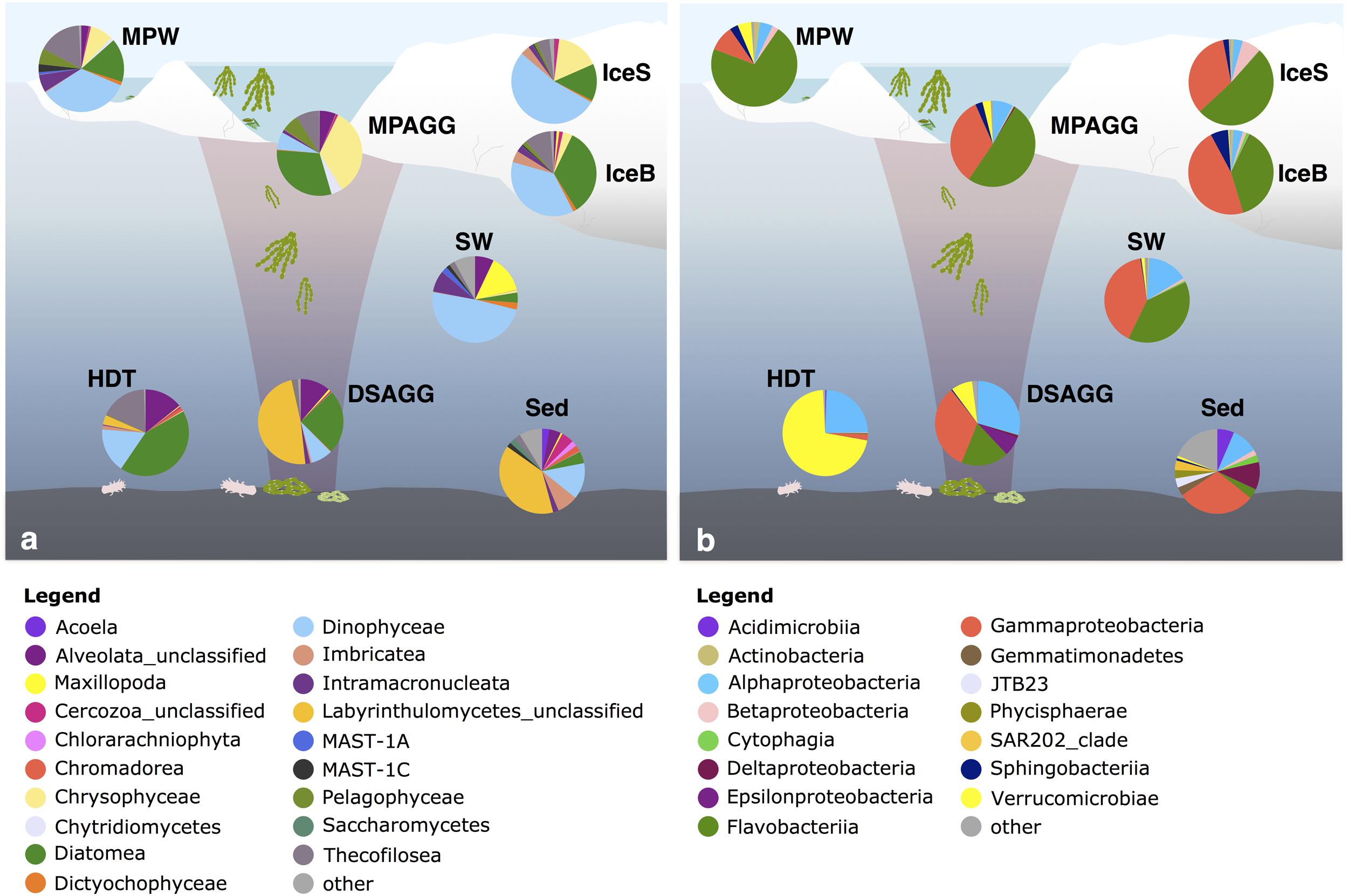
FIGURE 5. Dominant (a) eukaryotic and (b) bacterial classes in the central Arctic Ocean. Shown are average values per environment, displaying classes with a relative sequence abundance of at least 2%. The remaining classes were grouped as ‘other.’ IceS, sea-ice surface; IceB, sea-ice bottom; MPAGG, melt-pond aggregate; MPW, melt-pond water; SW, surface seawater; DSAGG, deep-sea algae deposit; HDT, holothurian digestive tract content; Sed, deep-sea surface sediment. For details on the samples, see Supplementary Table S1.
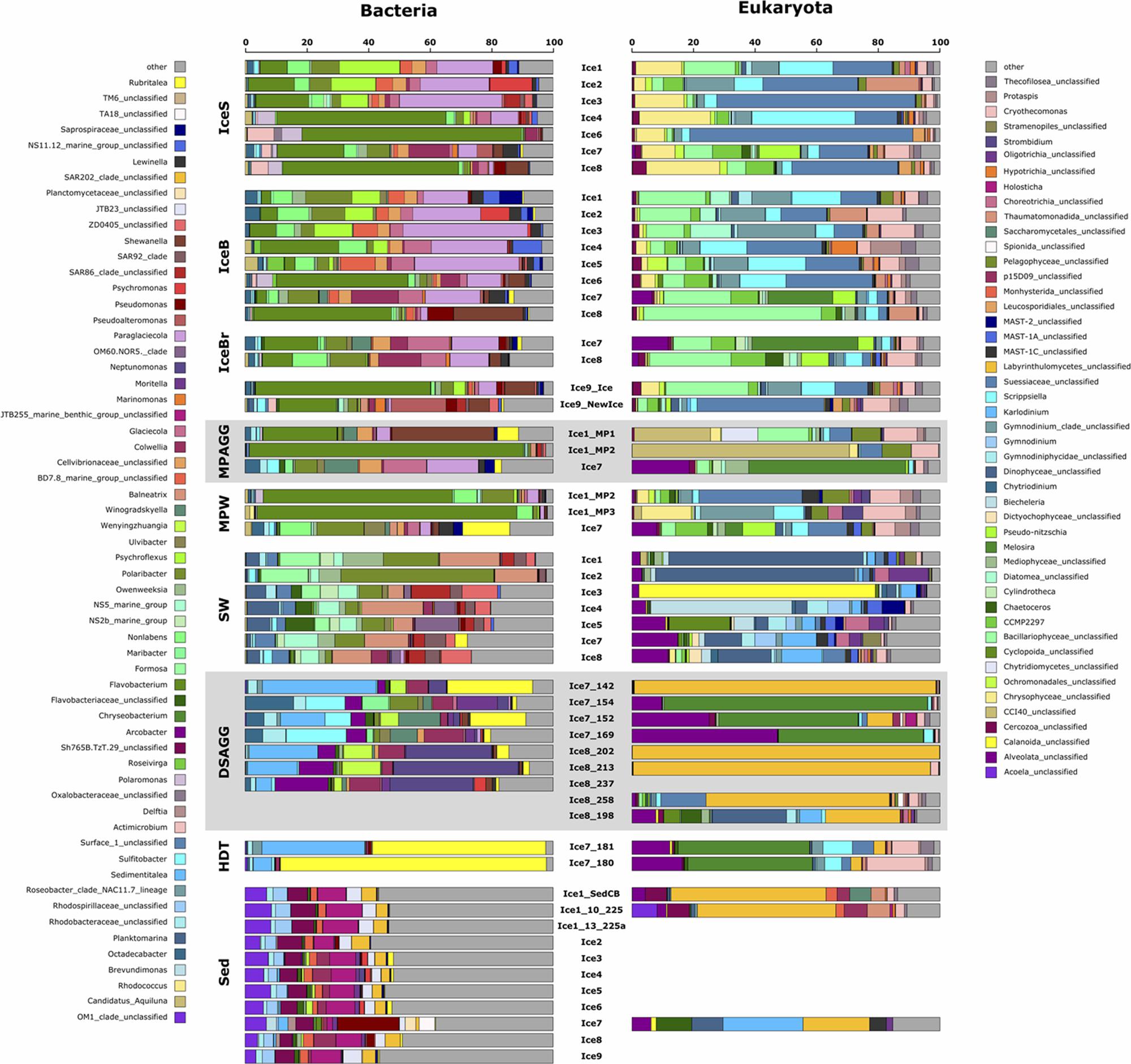
FIGURE 6. Dominant bacterial and eukaryotic genera in a range of environments across the Eurasian basin of the Arctic Ocean. Shown are the five most abundant genera across all samples, based on relative sequence abundance. Samples are grouped by environment and corresponding samples from the bacterial and eukaryotic dataset are plotted next to each other. IceS, sea-ice surface; IceB, sea-ice bottom; IceBr, brownish-colored sea ice; MPAGG, melt-pond aggregate; MPW, melt-pond water; SW, surface seawater; DSAGG, deep-sea algae deposit; HDT, holothurian digestive tract content; Sed, deep-sea surface sediment.
Flavobacteriia, Gamma- and Alphaproteobacteria dominated bacterial communities in sea ice, surface seawater and sediments (Figure 5b), yet with pronounced differences in their predominant representatives at the genus level (Figure 6). In sea ice and melt ponds, Flavobacteriia were mainly represented by the genus Flavobacterium, with reads of this single genus constituting 9–71% of the total community in the ice surface and 3–45% in the bottom of the ice. Also the genera Polaribacter, Psychroflexus, Nonlabens, and Ulvibacter were sequence- abundant in ice. Among these, Polaribacter was the only genus, which was also a dominant flavobacterial member in surface seawater. Here, the genera Owenweeksia, Formosa and the NS5 marine group were the most sequence-abundant Flavobacteriia (Figure 6). Microbial community composition of the deep-sea floor was quite distinct from the water column, and the contribution of Flavobacteriia was markedly reduced (Figure 5b). Most of its representatives in surface sediments could not be classified at genus level and clustered as unclassified Flavobacteriaceae.
Gammaproteobacteria in sea ice were dominated by Paraglaciecola, which displayed a similarly large variability in its contribution as Flavobacterium, ranging from 0 to 33% in sea-ice surface communities and 2–40% in bottom communities (Figure 6). Other important Gammaproteobacteria in sea ice belonged to the genera Colwellia and Glaciecola. In melt-pond waters Gammaproteobacteria representation was lower (Figure 5b), but Paraglaciecola and Glaciecola were also dominant members (Figure 6). In the water column, the most prominent Gammaproteobacteria taxa, Balneatrix, SAR86 clade and ZD0405 clade (Figure 6) showed only minor representation in sea ice, and were absent from the deep-sea samples. The most dominant Gammaproteobacteria in the sediment were members of the JTB255 marine benthic group, and these were restricted to deep-sea samples. Deep-sea surface sediment additionally showed a strong contribution of Deltaproteobacteria and Acidimicrobiia (Figure 5b).
Alphaproteobacteria contributed most to the communities in surface seawater, deep-sea sediment and the digestive tract content of holothurians, while their sequence abundance in sea ice and melt ponds was low (Figure 5b). While the genus Planktomarina and members of the SAR11 Surface1 clade predominated in the water column, Rhodospirillaceae were the most dominant representatives at the seafloor. The bacterial community in the holothurian digestive tract was very different from any other environment (Figure 5b) and almost entirely comprised of the alphaproteobacterial genus Sedimentitalea and the genus Rubritalea from the Verrucomicrobiae (Figure 6).
Based on OTU presence/absence, the microbial community associated with sea-ice algae aggregates showed highest community overlap with both melt-pond water and sea-ice communities. We found lower similarity in composition with the underlying community in surface seawater (Figure 3). Interestingly, the community overlap between ice-algal aggregates associated with melt ponds and the exported ice algae at the seafloor was comparable to or higher than the overlap between aggregates and surface seawater for both bacteria and eukaryotes (Figure 3), despite the >4000 m descent. At the seafloor, deposited ice-algal aggregate communities exhibited similar proportions of community overlap with the surrounding sediment as with the sea-ice bottom and surface seawater. A large overlap was also observed between eukaryotic communities in the deposited ice algae and the digestive tract content of the deep-sea dwelling holothurians (Figure 3b). Sea ice and sediment, as well as surface seawater and sediment communities showed lowest community overlap.
Only three bacterial and seven eukaryotic OTUs in the datasets fit our definition of generalists present across the Arctic habitats. These included members of the genera Colwellia, Oleispira (both Gammaproteobacteria) and Lentimonas (Opitutae) (Supplementary Information Discussion). Eukaryotic generalists included members of the genera Cryothecomonas (Thecofilosea), the NIF-3A7 and NW617 clades (Thecofilosea) and unclassified Dinophyceae, but also the photosynthetic Micromonas (Mamiellophyceae) and unclassified Prasinophytae. We identified a larger number of OTUs in the bacterial and eukaryotic dataset that were shared members between sea-ice algae aggregates in melt ponds, sea ice, water column and deposited algae at the seafloor, but that could not be identified as abundant and dispersed members of the sediment community (Supplementary Tables S3, S4). Overall, a large fraction of community members were also associated with unclassified groups with no closely related cultured representatives, e.g., unclassified Suessiaceae, unclassified Flavobacteriaceae, SAR86 clade and ZD0405 clade, the JTB255 marine benthic group; their ecological role thus remains elusive.
Visual inspection by light microscopy showed a predominance of Melosira in all sampled aggregates retrieved from the sea ice (Fernández-Méndez et al., 2014). Other diatoms observed under the microscope belonged to the pennate genera Nitzschia, Fragilariopsis, and Cylindrotheca (Supplementary Table S1). The ubiquitous presence of the genus Melosira (Coscinodiscophyceae) and of the above diatoms, as well as golden-brown algae (Chrysophyceae) was also revealed by sequencing (Supplementary Table S5). Melosira 16S rRNA gene contribution was <0.01% in the degraded aggregates of station Ice1 composed of empty Melosira frustrules (Supplementary Table S5), but dominated sequences in the fresh, green aggregate of station Ice7 (Figure 6).
Predominant bacterial classes associated with sea-ice algal aggregates were Flavobacteriia and Gammaproteobacteria (Figure 5b). The green aggregate from Ice7 included Glaciecola and Paraglaciecola (Gammaproteobacteria), and Winogradskyella (Flavobacteriia) (Figure 6). The genus Flavobacterium (Flavobacteriia) was the most abundant genus in the highly degraded aggregates from Ice1, where it contributed up to 89% to the total bacterial sequences. Flavobacterium was barely detectable (<0.05%) in the much fresher Melosira aggregate from Ice7 (Supplementary Table S6).
Visual inspection of algal aggregates deposited at the seafloor also identified Melosira as the dominant component (Boetius et al., 2013; Fernández-Méndez et al., 2014). The eukaryotic sequences retrieved from the deposits fell into two groups: those dominated by diatom sequences, mainly Melosira, and unclassified alveolates, and others primarily composed of Labyrinthulomycetes, a group of stramenopile protists (Figure 6). The eukaryotic community in the holothurian digestive tract resembled the sequence composition of deposited algal aggregates, with a high contribution of the diatom Melosira and unclassified alveolates, but also a high contribution of Cryothecomonas and Scrippsiella observed in sea ice (Figure 6 and Supplementary Table S5).
Alpha-, Gammaproteobacteria, and Flavobacteriia dominated all deposited algal aggregates (Figure 5b), and the overlap with the sea-ice aggregates was 22% at the OTU level. Yet, deposit-associated Alphaproteobacteria were dominated by the genera Octadecabacter, Sedimentitalea and Sulfitobacter, which were also detected in sea-ice algae aggregates, but in much lower relative abundances (Figure 6 and Supplementary Table S6). The relative contribution of Gammaproteobacteria was similar in sea-ice algal aggregates and the deposited aggregates at the seafloor, but the predominant genera observed in the deposits, Neptunomonas, Moritella, and Colwellia, differed from the sea-ice aggregates. In contrast, Winogradskyella, the most prominent flavobacterial member associated with the deposits was also well represented in sea-ice aggregates. Overall, the contribution of Flavobacteriia associated with the deposited aggregates was lower than in the sea-ice aggregates. Instead, the contribution of other classes was higher, including Epsilonproteobacteria and Verrucomicrobiae (Figure 5b). Epsilonproteobacteria were entirely represented by the genus Arcobacter, which was not detected in any other environment, except for the surrounding surface sediments. Verrucomicrobiae were represented by the genus Rubritalea, which was also present in sea-ice algae aggregates. The two types of algae deposits that were differentiated by their eukaryotic community composition showed only minor differences in bacterial community composition. Melosira-dominated samples showed higher contributions of the genera Octadecabacter (on average by a factor of 30), Sulfitobacter (on average by a factor of 45), Ulvibacter (on average by a factor of 18) and Winogradskyella (on average by a factor of 21). Labyrinthulomycetes-dominated samples contained a higher proportion of Neptunomonas (on average by a factor of 12; Figure 6).
The Arctic Ocean sea-ice cover of the past 10 years shows minima both in extent as well as in thickness relative to the 1981–2010 mean2. In autumn 2012, at the time of this study, Arctic sea-ice extent reached a record minimum, a loss that has been unprecedented since the beginning of satellite records. The bottom of the relatively thin ice floes had been populated by diatoms, foremost the colonial Melosira arctica forming large filamentous aggregates. Strong melting resulted in a wide-spread export and rapid deposition of sea-ice algal aggregates at the deep-sea floor at around 4,000 m water depth (Boetius et al., 2013). Only few types of benthic fauna, primarily holothurians, were observed to feed on the exported algal material, but a substantial bacterial degradation of the algal deposits was recorded, leading to locally enhanced respiration rates and a depletion of oxygen in and under the algal food falls (Boetius et al., 2013). Other studies have confirmed recent high under-ice productivity and the high contribution of sea-ice algae to export fluxes (Comeau et al., 2013; Lalande et al., 2014; Poulin et al., 2014). Here we compared bacterial and eukaryote community composition on ice-algae aggregates and the surrounding habitats, to understand if the ice-algal aggregates would associate with specific kinds of bacterial groups, if these would be selected from sea ice rather than surrounding waters, and if these communities would get exported to the seafloor. Our analyses did not target archaea, and to our knowledge no data on archaeal community composition is currently available for the Eurasian basin, despite their presence in other Arctic regions (Galand et al., 2009; Comeau et al., 2011), and their potential importance in the biogeochemical cycling of nutrients and carbon (Kirchman et al., 2007). Archaea do not seem to be as tightly linked to phytoplankton biomass, as has been suggested for bacteria (Herfort et al., 2007; Kirchman et al., 2007; Comeau et al., 2011; Wilson et al., 2017), however, their ability to utilize algae-derived organic matter has been demonstrated (Alderkamp et al., 2006; Orsi et al., 2016). As Arctic archaeal communities appear to exhibit high spatio-temporal variability in composition, relative abundance, and heterotrophic activity (Bano et al., 2004; Kirchman et al., 2007; Alonso-Sáez et al., 2008; Galand et al., 2008, 2009; Wilson et al., 2017), future studies of the central Arctic should aim to include archaea in microbial community assessments.
Sea-ice algal aggregates were mostly composed of healthy, green or degraded cells of the diatom Melosira arctica, but also included a variety of other sea-ice diatoms (Boetius et al., 2013). Microscopic analyses and sequence distribution of eukaryotes largely overlapped. Both eukaryotic and bacterial aggregate sequences showed highest community overlap with the surrounding melt-pond water and the sea ice (Figure 3), and the dominant members were also predominant members of the sea-ice and melt-water communities (Figure 5). The algal aggregates selected specifically for Flavobacteriia, particularly the genus Flavobacterium, as well as the gammaproteobacterial Glaciecola and Paraglaciecola (Figure 6). Both are known to be tightly coupled to phytoplankton bloom dynamics (Teeling et al., 2012), and especially members of the Flavobacteriia exhibit the ability to hydrolyze complex polymers, such as polysaccharides found in plant and algal cell walls (Humphry et al., 2001; Knoll et al., 2001; Williams et al., 2013). Also for Glaciecola a key role in the breakdown of organic matter was suggested, and a specialization on diatom-derived material was observed (Beier et al., 2015). Most members of this genus seem to be psychrophilic, and recent experimental work suggests that cold-adapted Glaciecola can be the dominant consumers of algae material in low-temperature environments (von Scheibner et al., 2017). However, bacteria and eukaryotes associated with the ice-algal aggregates both exhibited high beta-diversity, reflecting substantial variations in community structure between individual aggregates. It has been observed before that differently composed phytoplankton blooms may lead to the establishment of distinct bacterial assemblages, due to differences in organic material released by individual algal taxa (Pinhassi et al., 2004; Grossart et al., 2005).
Previous studies observed a dominance of Betaproteobacteria in bacterial melt-pond communities (Brinkmeyer et al., 2004), while we detected a clear predominance of Flavobacteriia (Figure 5b). Betaproteobacteria are known as particle colonizers and were shown to play key roles in the decomposition of aggregates and nutrient cycling, especially in freshwater ecosystems (Knoll et al., 2001; Schweitzer et al., 2001; Lemarchand et al., 2006). However, while Betaproteobacteria were described as early colonizers of particles, Flavobacteriia contribution was shown to increase in the late phase of blooms (Knoll et al., 2001), when they potentially benefit from their capacity to break down refractory material. The high contribution of Flavobacteriia in the melt ponds and sea ice may therefore reflect a late or post bloom state of the system, indicated by the low nutrient inventories during the time of the sampling (Fernández-Méndez et al., 2015). Melt ponds, even when closed, can be directly connected to the surface ocean through a network of brine channels that penetrate the sea-ice matrix, allowing exchange of communities between environments (Boetius et al., 2015). However, the community in melt-pond water differed considerably from that of seawater, even at ice station 7, where the melt pond was open and salinity in the pond was comparable to the underlying water column (Figure 6). Our results, therefore, underpin a recruitment of bacterial groups mainly from sea ice, best adapted to the utilization of ice algal material in the ponds, which differed from the phytoplankton material in the surface ocean. We observed first indications for specific associations between bacterial groups and ice-algae aggregates, with 12 dispersed and abundant bacterial OTUs as exclusive members of ice-algae aggregates and algae deposits (Supplementary Table S3). These comprised mainly Alphaproteobacteria, including several members of the Roseobacter clade, e.g., Sulfitobacter. Examples of both mutualistic and pathogenic interactions have been described for Roseobacter-algae associations, with important implications especially for carbon and sulfur cycling in the environment (Ramanan et al., 2016). Roseobacter are known as rapid colonizers of algae surfaces, where they often outcompete other bacterial groups, probably facilitated by their ability to sense and utilize several compounds released by phytoplankton, including dimethylsulfoniopropionate (Buchan et al., 2014).
Comparative analyses of microbial communities from upper ocean and deep sea showed that these communities barely overlap (Countway et al., 2007; Amaral-Zettler et al., 2010; Zinger et al., 2011; Walsh et al., 2016). Recent studies targeting the local overlap between surface water and surface sediment communities indicated a role of sinking particles and overlying water column properties in structuring benthic bacterial communities (Nagata et al., 2000; Hamdan et al., 2013; Ruff et al., 2014; Lindh et al., 2017). Here we investigated whether the rapidly sinking sea-ice algae deliver their associated bacteria to the deep-sea floor. Melosira filaments differ from other sinking particles in their large size and concomitant fast sinking rate (Alldredge and Gotschalk, 1988; Fernández-Méndez et al., 2014), with aggregates of a diameter of 3 cm potentially reaching the deep-sea floor within a single day after losing buoyancy at the surface (Katlein et al., 2014). We observed algae deposits, which were almost entirely composed of fresh Melosira and alveolates from the sea ice, with intact chloroplasts, presenting a fresh carbon source for benthic communities. These aggregates can potentially transport large numbers of microbial cells from the surface to the seafloor. In the most degraded Melosira aggregates, the dominant sequences belonged to members of the Labyrinthulomycetes (Figure 6), a group of heterotrophic protists that play important roles as saprobes of dead algal material and marine snow (Raghukumar, 2002; Bochdansky et al., 2017). Labyrinthulomycetes display an absorptive mode of nutrition and possess the ability to chemically alter and degrade algal detritus through the production of extracellular enzymes (Bahnweg, 1979; Raghukumar, 2002). Interestingly, in the investigated holothurian digestive tract Melosira sequences dominated largely over those of Labyrinthulomycetes, indicating a preference of the holothurians for fresh deposits (Figure 6).
We detected a large proportion of shared bacterial OTUs (22%) belonging to both the abundant types present on sea-ice algae aggregates and on deposited aggregates at the seafloor (Figure 3a and Supplementary Table S3). They included representatives of the genera Paraglaciecola, Glaciecola (both Gammaproteobacteria), Octadecabacter (Alphaproteobacteria), Psychroserpens and Polaribacter (Flavobacteriia), which have been previously described as psychrophiles, and were observed in sea ice and surface seawater (Bowman et al., 1997, 2012; Gosink et al., 1997; Junge et al., 2002; Brinkmeyer et al., 2003; Han et al., 2014; Hatam et al., 2014). While these OTUs represented 37% of the bacterial sequences associated to sea-ice aggregates and were also abundant members in sea ice, they contributed only 1% of the sequences in the deposits and were absent or represented by a minor fraction in sediments (Rapp et al., 2017). We therefore conclude that they were exported with the aggregates, but overgrown by other bacteria.
A considerable amount of bacteria can be exported to the deep sea attached to particles (Turley and Mackie, 1995), but their fate and biogeochemical roles remain elusive. Indigenous deep-sea bacteria are likely better adapted to in situ temperature and pressure of the deep-sea environment than surface-derived bacteria (Turley and Lochte, 1990; Turkey, 1993; Poremba, 1994; Tamburini et al., 2006, 2013). The results of this study suggest that the deposited algal aggregates were overgrown within a few weeks to months by specific bacterial groups of the surrounding sediment (Figure 6). Accordingly, the bacterial genera of highest relative sequence abundance (i.e., up to 37%) in the deposited ice-algae aggregates consisted of benthic groups that were absent from ice-associated environments and only of minor relative abundance in the surrounding sediment surface (Figure 6 and Supplementary Table S6). Most prominent were members of the alphaproteobacterial Roseobacter clade, i.e., the genus Sedimentitalea, but also the genera Arcobacter (Epsilonproteobacteria) and Neptunomonas (Gammaproteobacteria) (Figure 6). The Roseobacter clade is a physiologically versatile group, known for its capability to utilize a wide range of organic and inorganic compounds and its tight interactions with phytoplankton, important for organic matter decomposition (Buchan et al., 2014 and references therein). Arcobacter has been recorded in a variety of sediment environments, including surface sediment from the Antarctic shelf (Bowman and McCuaig, 2003), the Wadden Sea (Llobet-Brossa et al., 1998) and the deep sea (Thamdrup et al., 2000). Its capacity to attach to surfaces (Assanta et al., 2002), denitrify (Heylen et al., 2006), fix nitrogen (Wirsen et al., 2002), as well as its ability to perform dissimilatory manganese reduction (Thamdrup et al., 2000) and recycle sulfur (Wirsen et al., 2002) indicate a potentially important role of Arcobacter in re-mineralizing nutrients from aggregates. Suboxic patches occurred under the algal aggregates in otherwise fully oxygenated sediments (Boetius et al., 2013), potentially selecting for Arcobacter. Members of the Neptunomonas genus have been isolated from sediments (Zhang et al., 2010), but were also found associated to unicellular eukaryotes (Frommlet et al., 2015) or in close vicinity to whale carcasses (Miyazaki et al., 2008). This genus is known for its capacity to degrade polycyclic aromatic hydrocarbons (Hedlund et al., 1999), indicating an involvement in the breakdown of recalcitrant carbon sources in the deep sea. The selection of rare sediment taxa best adapted to the utilization of the deposited algal detritus could on the long term induce shifts in the indigenous benthic bacterial communities, as has recently been observed in deep-sea surface sediments along the Antarctic Polar Front (Ruff et al., 2014). Overall, the algal deposits changed community composition locally at the deep-sea floor not only by introducing surface ice-bacteria, but also by selecting for specific sediment community members, which resulted in a unique community profile, distinct from any source community (Figures 4a, 6). It remains to be further investigated whether this is a transient feature or whether such events, if reoccurring, will result in a gradual shift of deep-sea sediment communities.
As areas of strong summer ice melt are expanding in the central Arctic, it is expected that abrupt export events may become more frequent in areas covered by seasonal sea ice. This is supported by observations during recent expeditions in late summer 2016, where large areas of the central Arctic seafloor were again covered with Melosira deposits (Soltwedel, 2016; Boetius and Purser, 2017). Consequently, recurrent deposition of sea ice algae at the seafloor could facilitate the establishment of unique microbial assemblages as has been proposed for other discrete resource patches (Grassle and Morse-Porteous, 1987; Durán et al., 2013; Lindh et al., 2017) and would then likely lead to changes in bacterial community structure and in the biogeochemical cycling of carbon and nutrients in surface sediments.
Bacteria in polar ice-associated environments need to be well-equipped to quickly adapt to changes and cope with rapid transitions of seasonal extremes, ranging from high salt concentration in brine inclusions to freshwater salinity in melt ponds (Deming, 2010), and protect themselves against sub-zero temperatures. Several studies showed the production of so-called compatible solutes by some sea-ice bacteria, which they can use as osmo- and cryoprotectants (Methe et al., 2005; Bowman, 2008). These specific adaptations to life around the freezing point of water may also facilitate the exchange of bacteria between habitats, e.g., after ice melt and export of attached bacteria in aggregates. We detected a high number of abundant OTUs shared between sea ice, melt ponds and aggregates, but only a low number of generalist OTUs (Supplementary Information Discussion and Supplementary Table S5), i.e., OTUs present in all sampled environments, despite the relatively uniform temperatures below zero degree.
Hence, this study confirms that sea-ice bacterial and eukaryotic communities are distinct from other Arctic habitats such as seawater and sediment (Figure 4). Consequently, ice dwelling organisms, and especially multi-year ice communities, which are distinct from those of seasonal ice (Leu et al., 2011; Bowman et al., 2012; Boetius et al., 2015; Hatam et al., 2016), may be lost from an Arctic devoid of summer sea ice, with unknown repercussions for productivity, organic matter cycling and other ecological functions. In addition, ice melt may contribute to upper water column freshening, potentially inducing shifts in the bacterioplankton composition, as has been observed for the phytoplankton community (Li et al., 2009). We saw first indications for a bacterial surface seawater community response to massive ice melt in the predominance of the gammaproteobacterial genus Balneatrix, which was previously described to thrive under freshwater conditions (González and Whitman, 2006), and members of the genus Planktomarina of the Roseobacter clade (Figure 6), which was observed to correlate negatively with salinity (Giebel et al., 2011). Other than previous studies on bacterial community composition in the Arctic water column, we did not observe a predominance of the SAR11 clade (Kirchman et al., 2010; Bowman et al., 2012). This may be due to a primer bias (Apprill et al., 2015; Parada et al., 2016), but may also be ascribed to a lower representation of this oligotrophic clade during the post-bloom state of the system at the time of sampling. We observed a significant correlation between bacterial and eukaryote community patterns (Supplementary Figure S7), likely indicating similar drivers of diversification and potentially also biotic interactions, which remain largely unknown for polar organisms to date (Lima-Mendez et al., 2015). Future efforts should therefore aim to integrate bacterial, archaeal and eukaryotic community analyses, and expand the temporal resolution of sampling to better resolve seasonal dynamics.
JR and MF-M designed the study, with contributions by CB and AB. MF-M, CB, and AB collected the samples. JR performed the laboratory molecular and bioinformatic analysis. JR and CB performed the statistical analysis. JR interpreted the data and all authors contributed to the discussion of the results. JR wrote the manuscript, with support and input from all co-authors.
The study was funded by the European Research Council Advanced Investigator grant 294757 to AB and additional funding came from the Max Planck Society. The ship time was funded by AWI grant no. AWI_PS80_01. This publication is Eprint ID 41750 of the Alfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung, Bremerhaven, Germany, and contributes to the Helmholtz PACESII program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the captain and crew of RV Polarstern expedition IceArc (PS80, ARKXXVII/3) for the excellent support in the field. We thank J. Bäger for assistance in the laboratory and H. Tegetmeyer for conducting Illumina sequencing. Further, we thank C. Hassenrück, P. L. Buttigieg, and A. Fernandez-Guerra for providing parts of the bioinformatic script, as well as A. Strache for help with the taxonomic classification of Melosira arctica sequences and K. Metfies for helpful discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01035/full#supplementary-material
Ackley, S. F., and Sullivan, C. W. (1994). Physical controls on the development and characteristics of Antarctic sea ice biological communities- a review and synthesis. Deep Res. Part I 41, 1583–1604. doi: 10.1016/0967-0637(94)90062-0
Alderkamp, A., Sintes, E., and Herndl, G. (2006). Abundance and activity of major groups of prokaryotic plankton in the coastal North Sea during spring and summer. Aquat. Microb. Ecol. 45, 237–246. doi: 10.3354/ame045237
Alldredge, A. L., and Gotschalk, C. (1988). In situ settling behavior of marine snow. Limnol. Oceanogr. 33, 339–351. doi: 10.4319/lo.1988.33.3.0339
Alonso-Sáez, L., Sánchez, O., Gasol, J. M., Balagué, V., and Pedrós-Alio, C. (2008). Winter-to-summer changes in the composition and single-cell activity of near-surface Arctic prokaryotes. Environ. Microbiol. 10, 2444–2454. doi: 10.1111/j.1462-2920.2008.01674.x
Amaral-Zettler, L., Artigas, L. F., Baross, J., Bharathi, P. A. L., Boetius, A., Chandramohan, D., et al. (2010). “A global census of marine microbes,” in Life in the World’s Oceans: Diversity, Distribution, and Abundance, ed. A. D. McIntyre (Oxford: Blackwell Publishing Ltd.), 221–245. doi: 10.1002/9781444325508.ch12
Ambrose, W. G., Von Quillfeldt, C., Clough, L. M., Tilney, P. V. R., and Tucker, T. (2005). The sub-ice algal community in the Chukchi sea: large- and small-scale patterns of abundance based on images from a remotely operated vehicle. Polar Biol. 28, 784–795. doi: 10.1007/s00300-005-0002-8
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. doi: 10.3354/ame01753
Arrigo, K. R. (2014). Sea ice ecosystems. Ann. Rev. Mar. Sci. 6, 439–467. doi: 10.1146/annurev-marine-010213-135103
Arrigo, K. R., van Dijken, G., and Pabi, S. (2008). Impact of a shrinking Arctic ice cover on marine primary production. Geophys. Res. Lett. 35:L19603. doi: 10.1029/2008GL035028
Arrigo, K. R., and van Dijken, G. L. (2015). Continued increases in Arctic Ocean primary production. Prog. Oceanogr. 136, 60–70. doi: 10.1016/j.pocean.2015.05.002
Assanta, M. A., Roy, D., Lemay, M.-J., and Montpetit, D. (2002). Attachment of Arcobacter butzleri, a new waterborne pathogen, to water distribution pipe surfaces. J. Food Prot. 65, 1240–1247. doi: 10.4315/0362-028X-65.8.1240
Assmy, P., Ehn, J. K., Fernández-Méndez, M., Hop, H., Katlein, C., Sundfjord, A., et al. (2013). Floating ice-algal aggregates below melting Arctic sea ice. PLoS One 8:e76599. doi: 10.1371/journal.pone.0076599
Bahnweg, G. (1979). Studies on the physiology of Thraustochytriales. I. Carbon nutrition of Thraustochytrium spp., Schizochytrium sp., Japonochytrium sp., Ulkenia spp. and Labyrinthuloides spp. Veröff. Inst. Meeresforsch. Bremerh. 17, 269–274.
Bano, N., Ruffin, S., Ransom, B., and Hollibaugh, J. T. (2004). Phylogenetic composition of Arctic Ocean archaeal assemblages and comparison with Antarctic assemblages. Appl. Environ. Microbiol. 70, 781–789. doi: 10.1128/AEM.70.2.781-789.2004
Beier, S., Rivers, A. R., Moran, M. A., and Obernosterer, I. (2015). The transcriptional response of prokaryotes to phytoplankton-derived dissolved organic matter in seawater. Environ. Microbiol. 17, 3466–3480. doi: 10.1111/1462-2920.12434
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.2307/2346101
Bochdansky, A. B., Clouse, M. A., and Herndl, G. J. (2017). Eukaryotic microbes, principally fungi and labyrinthulomycetes, dominate biomass on bathypelagic marine snow. ISME J. 11, 362–373. doi: 10.1038/ismej.2016.113
Boetius, A., Albrecht, S., Bakker, K., Bienhold, C., Felden, J., Fernández-Méndez, M., et al. (2013). Export of algal biomass from the melting Arctic sea ice. Science 339, 1430–1432. doi: 10.1126/science.1231346
Boetius, A., Anesio, A. M., Deming, J. W., Mikucki, J., and Rapp, J. Z. (2015). Microbial ecology of the cryosphere: sea ice and glacial habitats. Nat. Rev. Microbiol. 13, 677–690. doi: 10.1038/nrmicro3522
Boetius, A., and Purser, A. (2017). The expedition PS101 of the research vessel POLARSTERN to the Arctic Ocean in 2016. Ber. Polar Meeresforsch. 706, 1–236.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowman, J. P. (2008). “Genomic analysis of psychrophilic prokaryotes,” in Psychrophiles: From Biodiversity to Biotechnology, eds R. Margesin, F. Schinner, J.-C. Marx, and C. Gerday (Berlin: Springer), 265–284. doi: 10.1007/978-3-540-74335-4_16
Bowman, J. P., McCammon, S. A., Brown, J. L., Nichols, P. D., and McMeekin, T. A. (1997). Psychroserpens burtonensis gen. nov., sp. nov., and Gelidibacter algens gen. nov., sp. nov., psychrophilic bacteria isolated from Antarctic lacustrine and sea ice habitats. Int. J. Syst. Bacteriol. 47, 670–677. doi: 10.1099/00207713-47-3-670
Bowman, J. P., and McCuaig, R. D. (2003). Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl. Environ. Microbiol. 69, 2463–2483. doi: 10.1128/AEM.69.5.2463-2483.2003
Bowman, J. S., Rasmussen, S., Blom, N., Deming, J. W., Rysgaard, S., and Sicheritz-Ponten, T. (2012). Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene. ISME J. 6, 11–20. doi: 10.1038/ismej.2011.76
Brinkmeyer, R., Glöckner, F.-O., Helmke, E., and Amann, R. (2004). Predominance of ß-proteobacteria in summer melt pools on Arctic pack ice. Limnol. Oceanogr. 49, 1013–1021. doi: 10.4319/lo.2004.49.4.1013
Brinkmeyer, R., Knittel, K., Ju, J., Weyland, H., Amann, R., Helmke, E., et al. (2003). Diversity and structure of bacterial communities in Arctic versus Antarctic pack ice. Appl. Environ. Microbiol. 69, 6610–6619. doi: 10.1128/AEM.69.11.6610-6619.2003
Buchan, A., LeCleir, G. R., Gulvik, C. A., and Gonzalez, J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12, 686–698. doi: 10.1038/nrmicro3326
Carmack, E., and Chapman, D. C. (2003). Wind-driven shelf/basin exchange on an Arctic shelf: the joint roles of ice cover extent and shelf-break bathymetry. Geophys. Res. Lett. 30:1778. doi: 10.1029/2003GL017526
Comeau, A. M., Li, W. K. W., Tremblay, J. -É., Carmack, E. C., and Lovejoy, C. (2011). Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS One 6:e27492. doi: 10.1371/journal.pone.0027492
Comeau, A. M., Philippe, B., Thaler, M., Gosselin, M., Poulin, M., and Lovejoy, C. (2013). Protists in Arctic drift and land-fast sea ice. J. Phycol. 49, 229–240. doi: 10.1111/jpy.12026
Countway, P. D., Gast, R. J., Dennett, M. R., Savai, P., Rose, J. M., and Caron, D. A. (2007). Distinct protistan assemblages characterize the euphotic zone and deep sea (2500 m) of the western North Atlantic (Sargasso Sea and Gulf Stream). Environ. Microbiol. 9, 1219–1232. doi: 10.1111/j.1462-2920.2007.01243.x
Deming, J. W. (2010). “Sea ice bacteria and viruses,” in Sea Ice - An Introduction to its Physics, Chemistry, Biology and Geology, eds D. N. Thomas and G. S. Dieckmann (Oxford: Blackwell Science Ltd.), 247–282.
Diepenbroek, M., Glöckner, F. O., Grobe, P., Güntsch, A., Huber, R., König-Ries, B., et al. (2014). “Towards an integrated biodiversity and ecological research data management and archiving platform: the German federation for the curation of biological data (GFBio),” in Proceedings of the Informatik 2014 – Big Data Komplexität Meistern. GI-Edition: Lecture Notes in Informatics (LNI), eds E. Plödereder, L. Grunske, E. Schneider, and D. Ull (Bonn: Gesellschaft für Informatik eV), 1711–1724.
Durán, R., Canals, M., Lastras, G., Micallef, A., Amblas, D., Pedrosa-Pàmies, R., et al. (2013). Sediment dynamics and post-glacial evolution of the continental shelf around the Blanes submarine canyon head (NW Mediterranean). Prog. Oceanogr. 118, 28–46. doi: 10.1016/j.pocean.2013.07.031
Fernández-Méndez, M., Katlein, C., Rabe, B., Nicolaus, M., Peeken, I., Bakker, K., et al. (2015). Photosynthetic production in the central Arctic Ocean during the record sea-ice minimum in 2012. Biogeosciences 12, 3525–3549. doi: 10.5194/bg-12-3525-2015
Fernández-Méndez, M., Wenzhöfer, F., Peeken, I., Sørensen, H. L., Glud, R. N., and Boetius, A. (2014). Composition, buoyancy regulation and fate of ice algal aggregates in the central Arctic Ocean. PLoS One 9:e107452. doi: 10.1371/journal.pone.0107452
Frommlet, J., Guimarães, B., Sousa, L., Serôdio, J., and Alves, A. (2015). Neptunomonas phycophila sp. nov. isolated from a culture of Symbiodinium sp., a dinoflagellate symbiont of the sea anemone Aiptasia tagetes. Int. J. Syst. Evol. Microbiol. 65, 915–919. doi: 10.1099/ijs.0.000039
Galand, P. E., Casamayor, E. O., Kirchman, D. L., Potvin, M., and Lovejoy, C. (2009). Unique archaeal assemblages in the Arctic Ocean unveiled by massively parallel tag sequencing. ISME J. 3, 860–869. doi: 10.1038/ismej.2009.23
Galand, P. E., Lovejoy, C., Gv, Q. C., and Vincent, W. F. (2008). Microbial community diversity and heterotrophic production in a coastal Arctic ecosystem: a stamukhi lake and its source waters. Limnol. Oceangr. 53,813–823. doi: 10.4319/lo.2008.53.2.0813
Galand, P. E., Potvin, M., Casamayor, E. O., and Lovejoy, C. (2010). Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J. 4, 564–576. doi: 10.1038/ismej.2009.134
Ghiglione, J.-F., Galand, P. E., Pommier, T., Pedrós-Alió, C., Maas, E. W., Bakker, K., et al. (2012). Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 109, 17633–17638. doi: 10.1073/pnas.1208160109
Giebel, H.-A., Kalhoefer, D., Lemke, A., Thole, S., Gahl-Janssen, R., Simon, M., et al. (2011). Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 5, 8–19. doi: 10.1038/ismej.2010.87
Gobet, A., Quince, C., and Ramette, A. (2010). Multivariate cutoff level analysis (MultiCoLA) of large community data sets. Nucleic Acids Res. 38:e155. doi: 10.1093/nar/gkq545
González, J., and Whitman, W. (2006). “Oceanospirillum and related genera,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 887–915. doi: 10.1007/0-387-30746-x
Gosink, J. J., Herwig, R. P., and Staley, J. T. (1997). Octadecabacter arcticus gen. nov., sp. nov., and O. antarcticus, sp. nov., nonpigmented, psychrophilic gas vacuolate bacteria from polar sea ice and water. Syst. Appl. Microbiol. 20, 356–365. doi: 10.1016/S0723-2020(97)80003-3
Grassle, J. F., and Morse-Porteous, L. S. (1987). Macrofaunal colonization of disturbed deep-sea environments and the structure of deep-sea benthic communities. Deep Sea Res. Part A Oceanogr. Res. Pap. 34, 1911–1950. doi: 10.1016/0198-0149(87)90091-4
Grossart, H. P., Levold, F., Allgaier, M., Simon, M., and Brinkhoff, T. (2005). Marine diatom species harbour distinct bacterial communities. Environ. Microbiol. 7, 860–873. doi: 10.1111/j.1462-2920.2005.00759.x
Hamdan, L. J., Coffin, R. B., Sikaroodi, M., Greinert, J., Treude, T., and Gillevet, P. M. (2013). Ocean currents shape the microbiome of Arctic marine sediments. ISME J. 7, 685–696. doi: 10.1038/ismej.2012.143
Han, D., Kang, I., Ha, H. K., Kim, H. C., Kim, O. S., Lee, B. Y., et al. (2014). Bacterial communities of surface mixed layer in the pacific sector of the western Arctic Ocean during sea-ice melting. PLoS One 9:e86887. doi: 10.1371/journal.pone.0086887
Hardge, K., Peeken, I., Neuhaus, S., Lange, B. A., Stock, A., Stoeck, T., et al. (2017). The importance of sea ice for exchange of habitat-specific protist communities in the Central Arctic Ocean. J. Mar. Syst. 165, 124–138. doi: 10.1016/j.jmarsys.2016.10.004
Hatam, I., Charchuk, R., Lange, B., Beckers, J., Haas, C., and Lanoil, B. (2014). Distinct bacterial assemblages reside at different depths in Arctic multiyear sea ice. FEMS Microbiol. Ecol. 90, 115–125. doi: 10.1111/1574-6941.12377
Hatam, I., Lange, B., Beckers, J., Haas, C., and Lanoil, B. (2016). Bacterial communities from Arctic seasonal sea ice are more compositionally variable than those from multi-year sea ice. ISME J. 10, 2543–2552. doi: 10.1038/ismej.2016.4
Hedlund, B. P., Geiselbrecht, A. D., Bair, T. J., and Staley, J. T. (1999). Polycyclic aromatic hydrocarbon degradation by a new marine bacterium, Neptunomonas naphthovorans gen. nov., sp. nov. Appl. Environ. Microbiol. 65, 251–259.
Herfort, L., Schouten, S., Abbas, B., Veldhuis, M. J. W., Coolen, M. J. L., Wuchter, C., et al. (2007). Variations in spatial and temporal distribution of Archaea in the North Sea in relation to environmental variables. FEMS Microbiol. Ecol. 62, 242–257. doi: 10.1111/j.1574-6941.2007.00397.x
Heylen, K., Vanparys, B., Wittebolle, L., Verstraete, W., Boon, N., and De Vos, P. (2006). Cultivation of denitrifying bacteria: optimization of isolation conditions and diversity study. Appl. Environ. Microbiol. 72, 2637–2643. doi: 10.1128/AEM.72.4.2637-2643.2006
Humphry, D. R., George, A., Black, G. W., and Cummings, S. P. (2001). Flavobacterium frigidarium sp. nov., an aerobic, psychrophilic, xylanolytic and laminarinolytic bacterium from Antarctica. Int. J. Syst. Evol. Microbiol. 51, 1235–1243. doi: 10.1099/00207713-51-4-1235
Jakobsson, M., Mayer, L., Coakley, B., Dowdeswell, J. A., Forbes, S., Fridman, B., et al. (2012). The international bathymetric chart of the Arctic Ocean (IBCAO) version 3.0. Geophys. Res. Lett. 39:L12609. doi: 10.1029/2012GL052219
Johannessen, O. M., Bengtsson, L., Miles, M. W., Kuzmina, S. I., Semenov, V. A., Alekseev, G. V., et al. (2004). Arctic climate change: observed and modelled temperature and sea-ice variability. Tellus Ser. A Dyn. Meteorol. Oceanogr. 56, 328–341. doi: 10.1111/j.1600-0870.2004.00060.x
Junge, K., Imhoff, F., Staley, T., and Deming, J. W. (2002). Phylogenetic diversity of numerically important Arctic sea-ice bacteria cultured at subzero temperature. Microb. Ecol. 43, 315–328. doi: 10.1007/s00248-001-1026-4
Katlein, C., Fernández-Méndez, M., Wenzhöfer, F., and Nicolaus, M. (2014). Distribution of algal aggregates under summer sea ice in the Central Arctic. Polar Biol. 38, 719–731. doi: 10.1007/s00300-014-1634-3
Kirchman, D. L., Cottrell, M. T., and Lovejoy, C. (2010). The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 12, 1132–1143. doi: 10.1111/j.1462-2920.2010.02154.x
Kirchman, D. L., Elifantz, H., Dittel, A. I., Malmstrom, R. R., and Cottrell, M. T. (2007). Standing stocks and activity of Archaea and Bacteria in the western Arctic Ocean. Limnol. Oceanogr. 52, 495–507. doi: 10.4319/lo.2007.52.2.0495
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Knoll, S., Zwisler, W., and Simon, M. (2001). Bacterial colonization of early stages of limnetic diatom microaggregates. Aquat. Microb. Ecol. 25, 141–150. doi: 10.3354/ame025141
Lalande, C., Nöthig, E. M., Somavilla, R., Bauerfeind, E., Shevchenko, V., and Okolodkov, Y. (2014). Variability in under-ice export fluxes of biogenic matter in the Arctic Ocean. Glob. Biogeochem. Cycles 28, 571–583. doi: 10.1002/2013GB004735
Lee, S. H., McRoy, C. P., Joo, H. M., Gradinger, R., Cui, X. H., Yun, M. S., et al. (2011). Holes in progressively thinning Arctic sea ice lead to new ice algae habitat. Oceanography 24, 302–308. doi: 10.5670/oceanog.2011.81
Lemarchand, C., Jardillier, L., Carrias, J. F., Richardot, M., Debroas, D., Sime-Ngando, T., et al. (2006). Community composition and activity of prokaryotes associated to detrital particles in two contrasting lake ecosystems. FEMS Microbiol. Ecol. 57, 442–451. doi: 10.1111/j.1574-6941.2006.00131.x
Leu, E., Søreide, J. E., Hessen, D. O., Falk-Petersen, S., and Berge, J. (2011). Consequences of changing sea-ice cover for primary and secondary producers in the European Arctic shelf seas: timing, quantity, and quality. Prog. Oceanogr. 90, 18–32. doi: 10.1016/j.pocean.2011.02.004
Li, W. K., McLaughlin, F. A., Lovejoy, C., and Carmack, E. C. (2009). Smallest algae thrive as the Arctic Ocean freshens. Science 326, 539–539. doi: 10.1126/science.1179798
Lima-Mendez, G., Faust, K., Henry, N., Decelle, J., Colin, S., Carcillo, F., et al. (2015). Determinants of community structure in the global plankton interactome. Science 348:1262073. doi: 10.1126/science.1262073
Lindh, M. V., Maillot, B. M., Shulse, C. N., Gooday, A. J., Amon, D. J., Smith, C. R., et al. (2017). From the surface to the deep-sea: bacterial distributions across polymetallic nodule fields in the clarion-clipperton zone of the Pacific Ocean. Front. Microbiol. 8:1696. doi: 10.3389/fmicb.2017.01696
Llobet-Brossa, E., Rossello-Mora, R., and Amann, R. (1998). Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64, 2691–2696.
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2015). Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ 3:e1420. doi: 10.7717/peerj.1420
Mangiafico, S. (2017). rcompanion: Functions to Support Extension Education Program Evaluation - R Package Version 1.11.1. Available at: https://cran.r-project.org/package=rcompanion [accessed February 14, 2018].
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Maslanik, J. A., Fowler, C., Stroeve, J., Drobot, S., Zwally, J., Yi, D., et al. (2007). A younger, thinner Arctic ice cover: increased potential for rapid, extensive sea-ice loss. Geophys. Res. Lett. 34:L24501. doi: 10.1029/2007GL032043
Melnikov, I. A., and Bondarchuk, L. L. (1987). Ecology of mass accumulations of colonial diatom algae under drifting Arctic ice. Oceanology 27, 233–236. doi: 10.1594/PANGAEA.756627
Methe, B. A., Nelson, K. E., Deming, J. W., Momen, B., Melamud, E., Zhang, X., et al. (2005). The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. U.S.A. 102, 10913–10918. doi: 10.1073/pnas.0504766102
Mikkelsen, D. M., Rysgaard, S., and Glud, R. N. (2008). Microalgal composition and primary production in Arctic sea ice: a seasonal study from Kobbefjord (Kangerluarsunnguaq). West Greenland. Mar. Ecol. Prog. Ser. 368, 65–74. doi: 10.3354/meps07627
Miyazaki, M., Nogi, Y., Fujiwara, Y., and Kawato, M. (2008). Neptunomonas japonica sp. nov., an Osedax japonicus symbiont-like bacterium isolated from sediment adjacent to sperm whale carcasses off Kagoshima, Japan. Int. J. Syst. Evol. Microbiol. 58, 866–871. doi: 10.1099/ijs.0.65509-0
Nagata, T., Fukuda, H., Fukuda, R., and Koike, I. (2000). Bacterioplankton distribution and production in deep Pacific waters: large-scale geographic variations and possible coupling with sinking particle fluxes. Limnol. Oceanogr. 45, 426–435. doi: 10.4319/lo.2000.45.2.0426
Nöthig, E.-M., Bracher, A., Engel, A., Metfies, K., Niehoff, B., Peeken, I., et al. (2015). Summertime plankton ecology in Fram Strait — a compilation of long- and short-term observations. Polar Res. 34:23349. doi: 10.3402/polar.v34.23349
Notz, D., and Stroeve, J. (2016). Observed Arctic sea-ice loss directly follows anthropogenic CO2 emission. Science 354, 747–750. doi: 10.1126/science.aag2345
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., et al. (2014). vegan: Community Ecology Package - R Package Version 2.2-0. Available at: http://cran.r-project.org/package=vegan
Orsi, W. D., Smith, J. M., Liu, S., Liu, Z., Sakamoto, C. M., Wilken, S., et al. (2016). Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J. 10, 2158–2173. doi: 10.1038/ismej.2016.20
Overland, J., Hanna, E., Hanssen-Bauer, I., Kim, S.-J., Walsh, J. E., Wang, M., et al. (2015). Surface Air Temperature. Arctic Report Card 2015.
Overland, J. E., and Wang, M. (2013). When will the summer Arctic be nearly sea ice free? Geophys. Res. Lett. 40, 2097–2101. doi: 10.1002/grl.50316
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Pinhassi, J., Sala, M. M., Havskum, H., Peters, F., Guadayol,Ò., Malits, A., et al. (2004). Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 70, 6753–6766. doi: 10.1128/AEM.70.11.6753-6766.2004
Polyakov, I. V., Walsh, J. E., and Kwok, R. (2012). Recent changes of Arctic multiyear sea ice coverage and the likely causes. Bull. Am. Meteorol. Soc. 93, 145–151. doi: 10.1175/BAMS-D-11-00070.1
Poremba, K. (1994). Simulated degradation of phytodetritus in deep- sea sediments of the NE Atlantic (47° N, 19° W). Mar. Ecol. Prog. Ser. 105, 291–299. doi: 10.3354/meps105291
Poulin, M., Underwood, G. J. C., and Michel, C. (2014). Sub-ice colonial Melosira arctica in Arctic first-year ice. Diatom Res. 29, 213–221. doi: 10.1080/0269249X.2013.877085
Prokopowich, C. D., Gregory, T. R., and Crease, T. J. (2003). The correlation between rDNA copy number and genome size in eukaryotes. Genome 46, 48–50. doi: 10.1139/g02-103
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28,1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2014). R: A Language and Environment for Statistical Computing. Available at: http://www.r-project.org/
Rabe, B., Wisotzki, A., Rettig, S., Somavilla Cabrillo, R., and Sander, H. (2013). Physical Oceanography Measured on Water Bottle Samples During Polarstern Cruise ARK-XXVII/3. Bremerhaven: Alfred Wegener Institute, Helmholtz Center for Polar and Marine Research. doi: 10.1594/PANGAEA.819452
Raghukumar, S. (2002). Ecology of the marine protists, the Labyrinthulomycetes (Thraustochytrids and Labyrinthulids). Eur. J. Protistol. 38, 127–145. doi: 10.1078/0932-4739-00832
Ramanan, R., Kim, B.-H., Cho, D.-H., Oh, H.-M., and Kim, H.-S. (2016). Algae–bacteria interactions: evolution, ecology and emerging applications. Biotechnol. Adv. 34, 14–29. doi: 10.1016/J.BIOTECHADV.2015.12.003
Rapp, J. Z., Fernández-Méndez, M., Bienhold, C., and Boetius, A. (2017). Bacterial and eukaryotic operational taxonomic units (OTU) in sea ice, water and deep-sea sediment samples of the Central Arctic collected during POLARSTERN cruise ARK-XXVII/3 (IceArc) in 2012. PANGAEA. doi: 10.1594/PANGAEA.882580
Rintala, J. M., Piiparinen, J., Blomster, J., Majaneva, M., Müller, S., Uusikivi, J., et al. (2014). Fast direct melting of brackish sea-ice samples results in biologically more accurate results than slow buffered melting. Polar Biol. 37, 1811–1822. doi: 10.1007/s00300-014-1563-1
Rösel, A., and Kaleschke, L. (2012). Exceptional melt pond occurrence in the years 2007 and 2011 on the Arctic sea ice revealed from MODIS satellite data. J. Geophys. Res. Oceans 117:C05018. doi: 10.1029/2011JC007869
Ruff, E. S., Probandt, D., Zinkann, A.-C. C., Iversen, M. H., Klaas, C., Würzberg, L., et al. (2014). Indications for algae-degrading benthic microbial communities in deep-sea sediments along the Antarctic Polar Front. Deep Sea Res. Part II Top. Stud. Oceanogr. 108, 6–16. doi: 10.1016/j.dsr2.2014.05.011
Schweitzer, B., Huber, I., Amann, R., Ludwig, W., and Simon, M. (2001). α- and β-Proteobacteria control the consumption and release of amino acids on lake snow aggregates. Appl. Environ. Microbiol. 67, 632–645. doi: 10.1128/AEM.67.2.632-645.2001
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Soltwedel, T. (2016). The expeditions PS99.1 and PS99.2 of the research vessel POLARSTERN to the Fram strait in 2016. Ber. Polar Meeresforsch. 704:107.
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Tamburini, C., Boutrif, M., Garel, M., Colwell, R. R., and Deming, J. W. (2013). Prokaryotic responses to hydrostatic pressure in the ocean - a review. Environ. Microbiol. 15, 1262–1274. doi: 10.1111/1462-2920.12084
Tamburini, C., Garcin, J., Grégori, G., Leblanc, K., Rimmelin, P., and Kirchman, D. L. (2006). Pressure effects on surface Mediterranean prokaryotes and biogenic silica dissolution during a diatom sinking experiment. Aquat. Microb. Ecol. 43, 267–276. doi: 10.3354/ame043267
Tamelander, T., Reigstad, M., Hop, H., and Ratkova, T. (2009). Ice algal assemblages and vertical export of organic matter from sea ice in the Barents Sea and Nansen Basin (Arctic Ocean). Polar Biol. 32, 1261–1273. doi: 10.1007/s00300-009-0622-5
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Thaler, M., and Lovejoy, C. (2015). Biogeography of heterotrophic flagellate populations indicates the presence of generalist and specialist taxa in the Arctic Ocean. Appl. Environ. Microbiol. 81, 2137–2148. doi: 10.1128/AEM.02737-14
Thamdrup, B., Rosselló-Mora, R., and Amann, R. (2000). Microbial manganese and sulfate reduction in Black Sea shelf sediments. Appl. Environ. Microbiol. 66, 2888–2897. doi: 10.1128/AEM.66.7.2888-2897.2000
Tremblay, J. É., Anderson, L. G., Matrai, P., Coupel, P., Bélanger, S., Michel, C., et al. (2015a). Global and regional drivers of nutrient supply, primary production and CO2 drawdown in the changing Arctic Ocean. Prog. Oceanogr. 139, 171–196. doi: 10.1016/j.pocean.2015.08.009
Tremblay, J., Singh, K., Fern, A., Kirton, E. S., He, S., Woyke, T., et al. (2015b). Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 6:771. doi: 10.3389/fmicb.2015.00771
Tremblay, J. -É., and Gagnon, J. (2009). “The effects of irradiance and nutrient supply on the productivity of Arctic waters: a perspective on climate change,” in Proceedings of the NATO Advanced Research Workshop on Influence of Climate Change on the Changing Arctic: Influence of Climate Change on the Changing Arctic and Sub-Arctic Conditions, Liège, 73–93. doi: 10.1007/978-1-4020-9460-6_7
Turkey, C. M. (1993). The effect of pressure on leucine and thymidine incorporation by free-living bacteria and by bacteria attached to sinking oceanic particles. Deep Res. Part I 40, 2193–2206. doi: 10.1016/0967-0637(93)90098-N
Turley, C. M., and Lochte, K. (1990). Microbial response to the input of fresh detritus to the deep-sea bed. Glob. Planet. Change 3, 3–23. doi: 10.1016/0921-8181(90)90052-E
Turley, C. M., and Mackie, P. J. (1995). Bacterial and cyanobacterial flux to the deep NE Atlantic on sedimenting particles. Deep Res. Part I 42, 1453–1474. doi: 10.1016/0967-0637(95)00056-C
von Scheibner, M., Sommer, U., and Jürgens, K. (2017). Tight coupling of Glaciecola spp. and diatoms during cold-water phytoplankton spring blooms. Front. Microbiol. 8:27. doi: 10.3389/fmicb.2017.00027
Walsh, E. A., Kirkpatrick, J. B., Rutherford, S. D., Smith, D. C., Sogin, M., and D’Hondt, S. (2016). Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 10, 979–989. doi: 10.1038/ismej.2015.175
Wassmann, P. (2011). Arctic marine ecosystems in an era of rapid climate change. Prog. Oceanogr. 90, 1–17. doi: 10.1016/j.pocean.2011.02.002
Wegner, C., Wittbrodt, K., Hölemann, J. A., Janout, M. A., Krumpen, T., Selyuzhenok, V., et al. (2017). Sediment entrainment into sea ice and transport in the Transpolar Drift: a case study from the Laptev Sea in winter 2011/2012. Cont. Shelf Res. 141, 1–10. doi: 10.1016/j.csr.2017.04.010
Wheeler, B., and Torchiano, M. (2016). lmPerm: Permutation Tests for Linear Models - R Package Version 2.1.0. Available at: https://cran.r-project.org/package=lmPerm [accessed February 14, 2018].
Williams, T. J., Wilkins, D., Long, E., Evans, F., DeMaere, M. Z., Raftery, M. J., et al. (2013). The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317. doi: 10.1111/1462-2920.12017
Wilson, B., Müller, O., Nordmann, E.-L., Seuthe, L., Bratbak, G., and Øvreås, L. (2017). Changes in marine prokaryote composition with season and depth over an Arctic polar year. Front. Mar. Sci. 4:95. doi: 10.3389/fmars.2017.00095
Wirsen, C. O., Sievert, S. M., Cavanaugh, C. M., Molyneaux, S. J., Ahmad, A., Taylor, L. T., et al. (2002). Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl. Environ. Microbiol. 68, 316–325. doi: 10.1128/AEM.68.1.316-325.2002
Yilmaz, P., Kottmann, R., Field, D., Knight, R., Cole, J. R., Amaral-Zettler, L., et al. (2011). Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420. doi: 10.1038/nbt.1823
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Zhang, X.-Y., Zhang, Y.-J., Yu, Y., Li, H.-J., Gao, Z.-M., Chen, X.-L., et al. (2010). Neptunomonas antarctica sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 60, 1958–1961. doi: 10.1099/ijs.0.017756-0
Zhu, F., Massana, R., Not, F., Marie, D., and Vaulot, D. (2005). Mapping of picoeukaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol. Ecol. 52, 79–92. doi: 10.1016/j.femsec.2004.10.006
Keywords: sea-ice algae, deep-sea sediment, Illumina tag sequencing, microbial eukaryotes, sea-ice decline, microbial ecology, bacterial diversity
Citation: Rapp JZ, Fernández-Méndez M, Bienhold C and Boetius A (2018) Effects of Ice-Algal Aggregate Export on the Connectivity of Bacterial Communities in the Central Arctic Ocean. Front. Microbiol. 9:1035. doi: 10.3389/fmicb.2018.01035
Received: 25 February 2018; Accepted: 01 May 2018;
Published: 18 May 2018.
Edited by:
George S. Bullerjahn, Bowling Green State University, United StatesReviewed by:
Trinity L. Hamilton, University of Minnesota Twin Cities, United StatesCopyright © 2018 Rapp, Fernández-Méndez, Bienhold and Boetius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josephine Z. Rapp, am9zZXBoaW5lLnJhcHBAYXdpLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.