 Rong Huang
Rong Huang Ting Li1,2†
Ting Li1,2† Jiajia Ni
Jiajia Ni Yi Gao
Yi Gao- 1Department of Hepatobiliary Surgery II, Guangdong Provincial Research Center of Artificial Organ and Tissue Engineering, Zhujiang Hospital of Southern Medical University, Guangzhou, China
- 2State Key Laboratory of Organ Failure Research, Southern Medical University, Guangzhou, China
- 3Department of Cell Biology, School of Basic Medical Science, Southern Medical University, Guangzhou, China
Gut microbial dysbiosis is correlated with the development of hepatocellular carcinoma (HCC). Therefore, analyzing the changing patterns in gut microbiota during HCC development, especially before HCC occurrence, is essential for the diagnosis and prevention of HCC based on gut microbial composition. However, these changing patterns in HCC are poorly understood, especially considering the sex differences in HCC incidence and mortality. Here, with an aim to determine the relationship between gut microbiota and HCC development in both sexes, and to screen potential microbial biomarkers for HCC diagnosis, we studied the changing patterns in the gut microbiota from mice of both sexes with liver-specific knockout of Tsc1 (LTsc1KO) that spontaneously developed HCC by 9–10 months of age and compared them to the patterns observed in their wide-type Tsc1fl/fl cohorts using high-throughput sequencing. Using the LTsc1KO model, we were able to successfully exclude the continuing influence of diet on the gut microbiota. Based on gut microbial composition, the female LTsc1KO mice exhibited gut microbial disorder earlier than male LTsc1KO mice during the development of HCC. Our findings also indicated that the decrease in the relative abundance of anaerobic bacteria and the increase in the relative abundance of facultative anaerobic bacteria can be used as risk indexes of female HCC, but would be invalid for male HCC. Most of the changes in the gut bacteria were different between female and male LTsc1KO mice. In particular, the increased abundances of Allobaculum, Erysipelotrichaceae, Neisseriaceae, Sutterella, Burkholderiales, and Prevotella species have potential for use as risk indicators of female HCC, and the increased abundances of Paraprevotella, Paraprevotellaceae, and Prevotella can probably be applied as risk indicators of male HCC. These relationships between the gut microbiota and HCC discovered in the present study may serve as a platform for the identification of potential targets for the diagnosis and prevention of HCC in the future.
Introduction
Recent research on gut microbiota has changed our understanding about their role in human diseases and their potential medical impact (Qin et al., 2012). From the perspective of both taxonomic and functional composition, gut microbiota might be linked to and contribute to many complex diseases such as obesity (Turnbaugh et al., 2009; Zhang et al., 2009; Fei and Zhao, 2013), type 2 diabetes (Larsen et al., 2010; Qin et al., 2012), and Crohn’s disease (Manichanh et al., 2006; Joossens et al., 2011).
Due to the anatomic connection, the liver is constantly exposed to microbial products from the gut microbiota (Zhang et al., 2012). Therefore, disruption of gut homeostasis (dysbiosis) is associated with numerous liver diseases (Schnabl, 2013; Xie et al., 2016) such as alcoholic fatty liver disease (Adachi et al., 1995), nonalcoholic fatty liver disease (Li et al., 2003), liver fibrosis (Seki and Schnabl, 2012), and liver cirrhosis (Karakan, 2014; Qin et al., 2014).
Such dysbiosis is also associated with the development of hepatocellular carcinoma (HCC, Zhang et al., 2012), which is the most frequent and aggressive primary tumor of the liver and has limited treatment options (El-Serag and Rudolph, 2007; Sherman, 2010; Shao et al., 2017). Lipopolysaccharide (LPS, i.e., endotoxins) is a major component of the outer membrane of gram-negative bacteria; consequently, an increase in the relative abundance of gram-negative bacteria in the gut microbiota could increase the concentrations of endotoxins in the plasma and the liver (Xie et al., 2016). Several lines of evidence indicate that LPS accumulation contributes to the pathogenesis of HCC by eliciting proinflammatory responses in the liver (Yu et al., 2010).
These data have led to a growing interest in gut microbiota and their metabolites as a new therapeutic target for the prevention, diagnosis, and treatment of metabolic diseases including HCC (Jia et al., 2008; Cani and Delzenne, 2011; Rajpal and Brown, 2013). However, despite considerable progress, most studies have been focused on comparing differences in gut microbiota between patients with HCC and healthy controls. Although differences in phylogenetic and functional compositions have been found, it cannot be determined whether these differences are an inducing factor or a consequence of HCC, only through comparison of the differences between patients with HCC and healthy controls. A better understanding of how variations in the symbiotic supraorganism contribute to HCC risk sustainability during HCC development and progression will point the way to new therapeutic interventions and HCC prevention strategies (Nicholson et al., 2012). Therefore, the phylogenetic and functional compositions of gut microbiota associated with HCC development and progression deserve more attention. More research focused on obtaining detailed information about changes in gut microbial composition and their roles in HCC development and progression is also needed.
Previously, Xie et al. (2016) analyzed fecal microbiota at various pathological stages in the liver and found gut microbiota to be significantly altered in the progression of liver disease. However, the authors did not exclude the continuing influence of streptozotocin-high fat diet (STZ-HFD) on the gut microbiota, despite diet being a major factor that casts and changes the gut microbiota (Faith et al., 2011; Claesson et al., 2012). However, Menon et al. (2012) reported that mice with liver-specific knockout of Tsc1(LTsc1KO) developed spontaneous HCC by 9–10 months of age. In this LTsc1KO model, tumor development was preceded by all of the hallmarks of HCC and was initiated by hepatocyte damage (Menon et al., 2012). This study provided an appropriate model to investigate the causality between HCC and gut microbiota disorder, which excludes the influence of diet and other inducers. We predicted that the gut microbiota from the LTsc1KO mice gradually divided to those from the wild types accompanying their growth and gram-negative bacteria were elevated in the gut microbiota of the LTsc1KO mice. The increase in the abundance of gram-negative bacteria resulted in increased endotoxin levels and triggered liver inflammation and other factors that induce HCC. In the present study, to evaluate the changes occurring in the gut microbiota of LTsc1KO mice with growth, the gut microbiota of LTsc1KO mice and their wide-type Tsc1fl/fl cohorts at different ages were investigated using MiSeq high-throughput sequencing of the 16S rRNA gene. Our data provide insights into the characteristics of gut microbiota related to the development of HCC, provide a paradigm for future studies on the pathophysiological roles of gut microbiota in other relevant diseases, and highlight the potential usefulness of a gut microbiota-based approach for the assessment of individuals at risk for HCC.
Materials and Methods
Animal Studies
Adult LTsc1KO mice and their wide-type Tsc1fl/fl cohorts were purchased from the Jackson Laboratory (Bar Harbor, ME, United States). All the mice were housed in individual stainless-steel cages, provided water ad libitum, and fed a nutritionally replete SPF solid diet that contained ≤100 g of moisture, ≥200 g of crude protein, ≥40 g of crude fat, ≤50 g of crude fiber, and ≤80 g of crude ash per kg. The male and female mice used in this study were propagated using adult LTsc1KO and Tsc1fl/fl mice. The mice were subdivided into five groups according to their age: group 1, age: 68–75 days (seven and six individuals of LTsc1KO and Tsc1fl/fl mice, respectively); group 2, age: 100–133 days (six and six individuals of LTsc1KO and Tsc1fl/fl mice, respectively); group 3, age: 171–172 days (six and five individuals of LTsc1KO and Tsc1fl/fl mice, respectively); group 4, age: 185–191 days (seven and five individuals of LTsc1KO and Tsc1fl/fl mice, respectively); and group 5, age: 222–322 days (six and four individuals of LTsc1KO and Tsc1fl/fl mice, respectively) (Supplementary Table S1). The animal experiments were approved by the Animal Ethics Committee of Southern Medical University under the approval number SYXK(Yue)2011-0074 and performed in accordance with animal ethics guidelines and approved protocols.
Measurement of Physical Indexes and Observation of Liver Histology
The mice were anesthetized using 1% of carbrital and their blood was collected from the heart. The blood was placed for 2 h at room temperature before centrifugation for 3 min at 1370 g for collecting the serum. The serum concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using an animal dry biochemical automatic analyzer (Catalyst One, United States). Liver histological analysis was conducted by dissecting the anesthetized mice and observing sections of paraffin-embedded, formalin-fixed tissues stained with hematoxylin and eosin (Yang et al., 1997).
Fecal Sample Collection and High-Throughput Sequencing
Approximately 0.1 g of fresh fecal pellets was collected from each mouse and stored at -20°C for microbial DNA extraction. Fecal microbial DNA was extracted using a PowerSoilTM DNA isolation kit (MO BIO, United States). DNA concentration and quality were checked using a NanoDrop spectrophotometer (Thermo Fisher Scientific, United States).
The V4-V5 hypervariable region of 16S rRNA gene was amplified and sequenced using the MiSeq system, as described previously (Li et al., 2016; Ni et al., 2017). The sequences were processed and alpha-diversities were calculated using the QIIME Pipeline 1.9.0 with default parameters (Caporaso et al., 2010). Chimeric sequences were identified and removed using the Uchime algorithm before further analysis (Edgar et al., 2011). To assess the repeatability of the MiSeq sequencing in microbiota analysis, the samples were sequenced at least two times. Samples with less than 5000 sequences were removed and then 5000 sequences were randomly sampled from each residual sample to calculate the weighted UniFrac dissimilarity, perform procrustes analysis (Muegge et al., 2011; Rampelli et al., 2013), and employ the unweighted pair group method with arithmetic means (UPGMA). The sequences from the same sample were merged as the sequence dataset of the corresponding sample for further analysis. Subsequently, all the samples were randomly re-sampled to obtain the same number of sequences. The high-quality sequences were clustered into operational taxonomic units (OTUs) at a 97% identity using UPARSE (Edgar, 2013). Taxonomic assignments of each OTU were determined using the RDP classifier (Wang et al., 2007). The metabolic characteristics, ratio of gram-negative bacteria to gram-positive bacteria, and functional profiles of the fecal microbiota were calculated using BugBase (Ward et al., unpublished) and PICRUSt (Langille et al., 2013).
All the DNA datasets have been submitted to the NCBI Sequence Read Archive database under the accession number SRP107826.
Statistical Analysis
Results for each parameter are presented as mean ± standard error (SE) for each group. Non-parametric multivariate analysis of variance (MANOVA) (Anderson, 2001) that was used to test the different significance among three and more groups was conducted using the R vegan package (Dixon, 2003). Canonical correspondence analysis (CCA) was conducted using the R vegan package and ade4 package. The Wilcoxon rank sum test was used to test significant difference between two groups. Tukey’s method was used to perform multiple comparisons of means. P-values ≤ 0.05 were considered statistically significant.
Results
HCC Development of LTsc1KO Mice
Consistent with a previous report (Menon et al., 2012), we found no detectable tumors in any of the LTsc1KO mice in postnatal 6 months. However, Menon et al. (2012) reported that, at this stage (postnatal 6 months), the livers of all the mice had shown various characteristics of liver damage, including increased serum concentrations of the liver enzymes ALT and AST. In contrast, no significant difference was detected in the serum concentrations of AST and ALT between the LTsc1KO and Tsc1fl/fl mice at postnatal 6 months in the present study (Wilcoxon rank sum test, P > 0.05). However, at 10–14 months of age, an increase in the serum concentrations of the liver enzymes ALT and AST was observed in the LTsc1KO mice (Figure 1). Liver histology with hematoxylin and eosin staining showed no significant pathological change in the livers of the 6-month-old LTsc1KO mice. However, HCC was detected at 10 and 14 months of age. No pathological change was detected in the livers of the Tsc1fl/fl mice at any age (Figure 1).
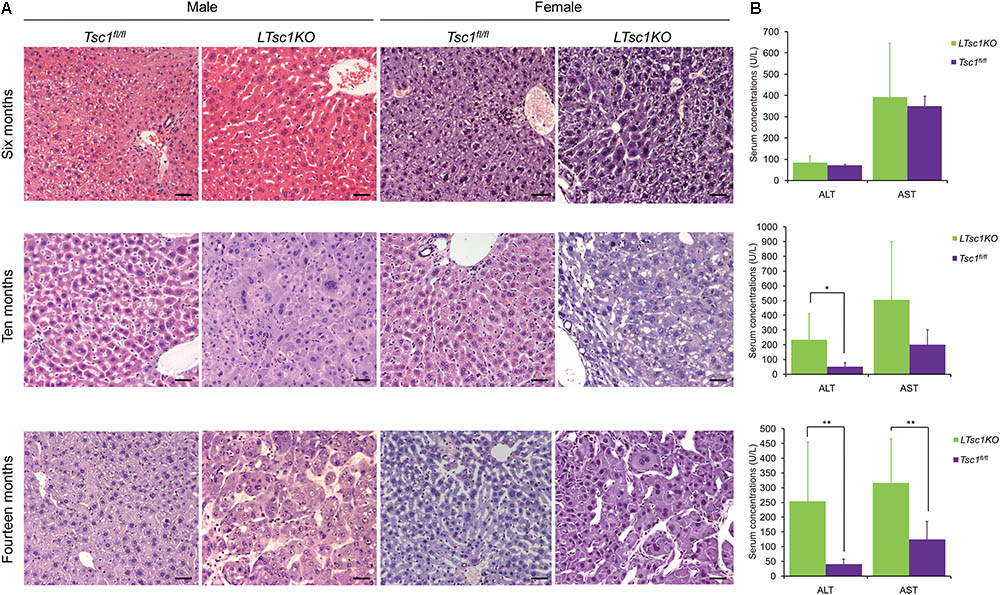
FIGURE 1. Hepatocellular carcinoma (HCC) development in the LTsc1KO mice. (A) Representative hematoxylin and eosin-stained sections of the livers of the Tsc1fl/fl and LTsc1KO mice. (B) Serum concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are presented as the means ± standard error of the mean (SEM). ∗P < 0.05 and ∗∗P < 0.01. Scale bars, 50 μm (×200).
Repeatability of the Results Obtained Using Sequencing Technology to Analyze Fecal Microbiota
We carried out high-throughput sequencing of the 16S rRNA gene in individual fecal DNA samples from 58 mice (26 LTsc1KO mice and 32 Tsc1fl/fl controls, Supplementary Table S1). After removing samples with less than 5000 sequences from single sampling, 54 fecal samples remained and were used to verify the repeatability of the results obtained using the sequencing technology. Despite using only 5,000 sequences of each specimen, MiSeq sequencing still exhibited a forceful repeatability for each sample (Figure 2).
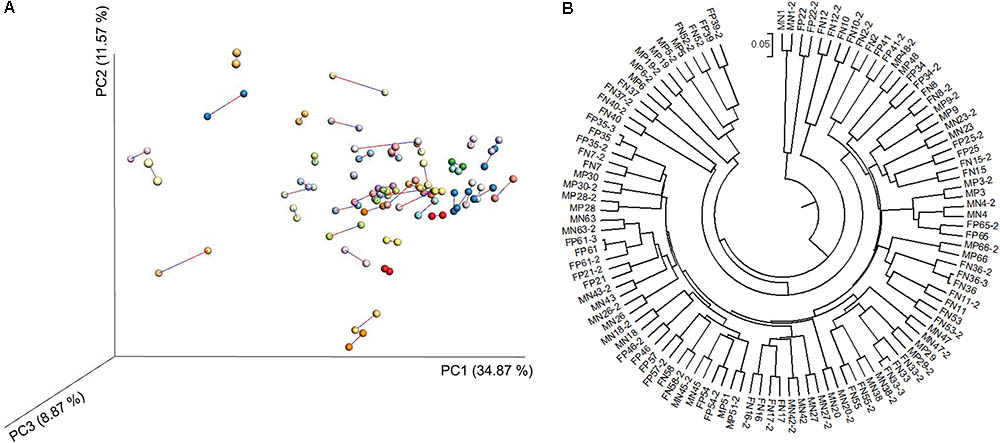
FIGURE 2. Principal coordinate analysis (PCoA) profile of procrustes analysis (A) and unweighted pair group method with arithmetic means (UPGMA) profile (B) based on the weighted UniFrac dissimilarity matrix of fecal microbiota samples from LTsc1KO and Tsc1fl/fl mice. The LTsc1KO mice are a genetic mouse model with liver-specific knockout of the Tsc1 gene, which causes them to develop spontaneous HCC by 9–10 months of age, and the Tsc1fl/fl mice are their wide-type cohorts.
Changes in the Gut Microbiota Between LTsc1KO and Wide-Type Tsc1fl/fl Mice at Different Ages
After merging the sequences from the same sample, a total of 1,703,063 high-quality sequences were obtained. To eliminate the influence of sequencing depth, 15,885 sequences were randomly sampled from each sample for further analysis. A total of 291,336 OTUs from 498 genera were identified by grouping sequences at the 97% similarity level. Although the alpha diversity of the gut microbiota from the LTsc1KO and Tsc1fl/fl mice fluctuated during the experiment (Supplementary Figure S1), no significant difference was detected between the LTsc1KO and Tsc1fl/fl mice.
The sequences were found to belong to 36 phyla with the exception of tiny unclassified sequences (0.019–6.29%). However, only seven phyla — Bacteroidetes, Firmicutes, Proteobacteria, Deferribacteres, Tenericutes, Spirochaetes, and Cyanobacteria — dominated the fecal microbial communities (their relative abundances were more than 1% in at least one sample; Figure 3A). No significant difference was detected between the gut microbiota from the LTsc1KO and Tsc1fl/fl mice of the same age (Figure 3B). However, this result contradicted the results of two previous studies that indicated that, with advancing cirrhosis, there is an increase in the abundance of Proteobacteria, the phylum containing gram-negative families such as Enterobacteriaceae (Chen et al., 2011; Bajaj et al., 2012).
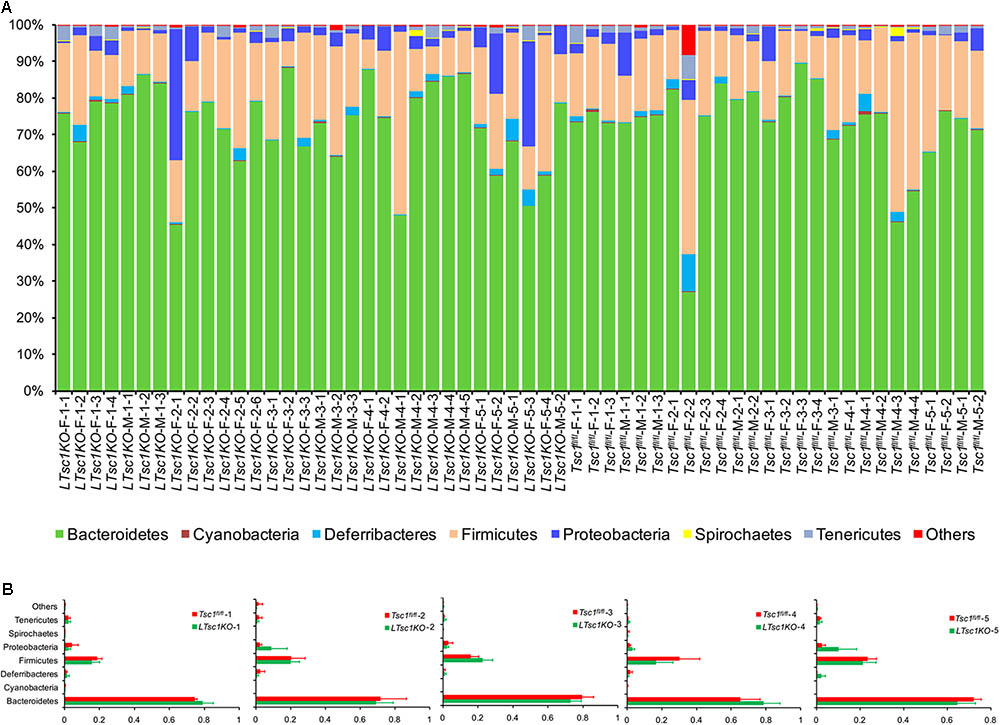
FIGURE 3. Percentage bar diagram (A) and composite bar diagram (B) showing compositions of the dominant phyla in fecal microbiota from LTsc1KO and Tsc1fl/fl mice. LTsc1KO mice are a genetic mouse model with liver-specific knockout of the Tsc1 gene, which causes them to develop spontaneous HCC by 9–10 months of age, and the Tsc1fl/fl mice are their wide-type Tsc1fl/fl cohorts. The first numbers in the sample names indicate the group number. Group 1–5 contained mice of postnatal ages of 68–75, 100–133, 171–172, 185–191, and 222–322 days, respectively. The second numbers in the sample names indicate the individual numbers of the mice in each group.
Aerobic gram-negative bacteria in the gut microbiota have been found to be the major source of serum endotoxin (Riordan and Williams, 2006). Therefore, we compared the relative abundance of aerobic gram-negative bacteria in the fecal microbiota of the LTsc1KO and Tsc1fl/fl mice of different ages. Different changing patterns were detected between female and male mice at different ages (Supplementary Figure S2). The relative abundance of aerobic bacteria in the LTsc1KO mice in group 5 was obviously higher than that in the Tsc1fl/fl mice among the females, but no significant difference was found in the male mice (Supplementary Figure S2A). In contrast, the relative abundance of anaerobic bacteria was obviously decreased in the female LTsc1KO mice in group 3 compared with that in the corresponding female Tsc1fl/fl cohorts. Such obviously decreases were also observed in male LTsc1KO mice in groups 1 and 5 (compared with that in male Tsc1fl/fl mice; Supplementary Figure S2B). An increase in the abundance of facultative anaerobic bacteria in the gut microbiota is commonly considered to be associated with HCC (Zhang et al., 2012). However, we did not find any significant changes in the relative abundance of facultative anaerobic bacteria between the LTsc1KO and Tsc1fl/fl mice in any of the groups, except in group 3, where it was obviously increased in female LTsc1KO mice compared with that in corresponding female Tsc1fl/fl mice (Supplementary Figure S2C). Moreover, no significant difference was detected between the relative abundance of gram-negative or gram-positive bacteria from the LTsc1KO and Tsc1fl/fl mice in any of the groups (Supplementary Figures S2D,E). For LPS metabolism, no significant difference was detected in the relative abundance of the genes that participate in the KEGG pathways of LPS biosynthesis, LPS transport system, or LPS export system between the LTsc1KO and Tsc1fl/fl mice in any of the groups (Supplementary Figures S2F–H). Together, these results implied that gut microbiota of female mice were more sensitive to liver injury than those of male mice. Based on our results, we believe that the decrease in the relative abundance of anaerobic bacteria and an increase in the relative abundance of facultative anaerobic bacteria maybe used as risk indexes for female HCC, but are invalid for male HCC.
Changes in Gut Microbiota Between LTsc1KO and Wide-Type Tsc1fl/fl Mice With the Development of HCC
CCA based on the detected 498 genera showed that age, sex, and genetic (LTsc1KO or wide-type Tsc1fl/fl) differences significantly influenced the gut microbiota of the mice (Figure 4A). In the first two groups (groups 1 and 4), genetic differences did not significantly influence the gut microbiota of the mice. However, in the last three groups, genetic differences significantly influenced the gut microbiota (Figures 4B–F).
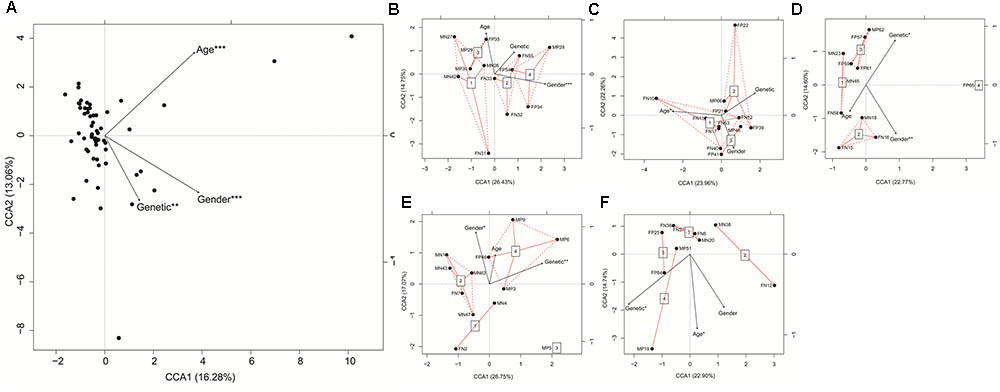
FIGURE 4. Canonical correspondence analysis (CCA) profiles showed influences of age, gender, and genetic differences on gut microbiota of LTsc1KO and wide-type Tsc1fl/fl mice. LTsc1KO mice are a genetic mouse model with liver-specific knockout of the Tsc1 gene, which causes them to develop spontaneous HCC by 9–10 months of age, and Tsc1fl/fl mice are their wide-type Tsc1fl/fl cohorts. The mice were subdivided into five groups according to their age: group 1, age: 68–75 days; group 2, age: 100–133 days; group 3, age: 171–172 days; group 4, age: 185–191 days; and group 5, age: 222–322 days. (A) All samples; (B) Samples in group 1; (C) Samples in group 2; (D) Samples in group 3; (E) Samples in group 4; (F) Samples in group 5. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
From the detected 498 genera, 75 genera dominated the fecal microbial communities (their relative abundances were more than 0.1% in at least one sample, Supplementary Figure S3). Together, these genera account for 99.63 ± 0.26% (mean ± standard deviation) of the relative abundance. Genera with relative abundances less than 0.1% were ignored in the further analysis as their abundances would be greatly influenced randomly by sampling error (Ni et al., 2017).
At the genus level, the relative abundance of a genus of gram-negative bacterium of the S24-7 family showed significant differences between male and female LTsc1KO mice in group 1. In group 2, the relative abundance of gram-positive bacteria belonging to genus Allobaculum in male Tsc1fl/fl mice was significantly higher than that in other cohorts. A genus belonging to the S24-7 family also showed significant difference between male and female LTsc1KO mice in group 2. However, in groups 1 and 2, differences in the gut microbiota between LTsc1KO and wide-type Tsc1fl/fl mice were mostly caused by gender differences (Figure 5). In group 3, no significant difference for any of the genera was detected between the LTsc1KO and wide-type Tsc1fl/fl mice or between genders. After postnatal 185 days, differences in the gut microbiota between the LTsc1KO and wide-type Tsc1fl/fl mice were obvious. Moreover, the changing patterns were different for the female and male mice (Figure 5). For all the genera that showed significant differences in their relative abundance, those from female LTsc1KO mice were significant higher than those from others in group 4. However, no other difference was evident, with the exception of gram-positive Allobaculum that showed significantly lower abundance in female Tsc1fl/fl mice than in male mice. The genera showing increased abundances in gut microbiota from female LTsc1KO mice in group 4 did not maintain these increased abundances in group 5. The relative abundance of three genera from the Paraprevotellaceae family of gram-negative bacteria was significantly higher in the gut microbiota from male LTsc1KO mice than in the other mice (Figure 5).

FIGURE 5. Changes in gut microbiota from LTsc1KO and wide-type Tsc1fl/fl mice at different ages. LTsc1KO mice are a genetic mouse model with liver-specific knockout of the Tsc1 gene, which causes them to develop spontaneous HCC by 9–10 months of age, and Tsc1fl/fl mice are their wide-type Tsc1fl/fl cohorts.
Discussion
From the perspective of both taxonomic and functional composition, gut microbiota might be linked to and contribute to many complex diseases, including HCC. However, despite considerable progress, most studies have been focused on comparing differences in gut microbiota between patients with HCC and healthy controls. Although differences in phylogenetic and functional compositions have been found, comparison of the differences between patients with HCC and healthy controls cannot determine whether the differences are an inducing factor or a consequence of HCC. Previously, Xie et al. (2016) reported that the composition of the gut microbiota changes significantly in mice responding to STZ-HFD, which is highly relevant to human liver disease. However, the authors did not exclude the continuing influence of STZ-HFD on gut microbiota, given that diet is a major factor affecting the gut microbiota (Faith et al., 2011). Using the LTsc1KO model, we were able to successfully exclude the continuing influence of diet on the gut microbiota while describing changes in the gut microbiota during the progression of HCC. Using the LTsc1KO model, we found no difference in gut microbiota between LTsc1KO and Tsc1fl/fl mice at postnatal 6 months. However, after postnatal 6 months, differences in gut microbiota between LTsc1KO and Tsc1fl/fl mice emerged before the physiological and biochemical characteristics of the hosts. In addition, female LTsc1KO mice exhibited gut microbial disorder earlier than male LTsc1KO mice (Figure 5). The relatively few genera showing significant differences between LTsc1KO and Tsc1fl/fl mice in our study also implied that diet probably caused false positive differences between STZ-HFD and control mice in previous studies.
Epidemiological studies have provided compelling evidence for the role of sex in liver cancer etiology and survival (Hartwell et al., 2014; Xie et al., 2017). The incidence of newly diagnosed hepatic cancer is 28,410 cases for males and 10,820 cases for females, a 2.6:1 ratio, according to a United States 2016 cancer statistics report (Siegel et al., 2016). The number of deaths from hepatic cancer in 2016 was 18,280 for males and 8,890 for females, a 2.1:1 ratio (Siegel et al., 2016). HCC accounts for 90% of primary liver cancer (Yeh et al., 2013). Xie et al. (2017) reported that sex-dependent effects of gut microbiota regulate hepatic carcinogenic outcomes in a STZ-HFD-induced nonalcoholic steatohepatitis-HCC murine model. Although Menon et al. (2012) detected similar morbidity in both male and female cohorts, in our study, we found that their gut microbiota showed different changing patterns during the development of HCC (Figure 5). The female LTsc1KO mice exhibited gut microbial disorder earlier than the male LTsc1KO mice did. Most of the changes in the gut bacteria were different between female and male LTsc1KO mice (Figure 5). Moreover, our results also implied that gut microbiota of female mice were more sensitive to liver injury than those of male mice. Even though dysbiosis is considered to promote liver injury and HCC, our results imply that the changes in the gut microbiota probably represented reverse feedback regulation of liver injury and actually prevented the liver injury. This speculation should be verified in future studies.
Previously, Allobaculum has been found to be dominant in the intestines of hamsters fed an AIN-93 M diet, and its abundance increased upon grain sorghum lipid extract supplementation, and this abundance correlated with cholesterol metabolic improvement (Miriam and Buenviaje, 1989). The blood cholesterol-lowering effect of alginate has also been reported, which might be associated with its fermentation properties (Kuda et al., 1997). However, the reason for the increase in the relative abundance of Allobaculum species in the LTsc1KO mice in group 4 is unclear. Except Allobaculum (a gram-positive bacterial genus; An et al., 2013), all the other genera showing significant changes contained gram-negative bacteria, and several pathogenic or disease-related bacteria have been previously reported to belong to these genera (van Ulsen and Tommassen, 2006; Sakon et al., 2008). For instance, Erysipelotrichaceae species have shown changes in their abundance in patients with inflammatory diseases (Kaakoush, 2015). In our study as well, the relative abundance of Erysipelotrichaceae species was found to be significantly increased in the gut microbiota of female LTsc1KO mice in group 4. An increase in the abundance of Sutterella species has also observed in the feces of children with autism spectrum disorder (Wang et al., 2013). Burkholderiales bacteria are especially dangerous for intensive care unit patients and patients with chronic lung diseases (Voronina et al., 2015). Therefore, the increases in the abundance of Allobaculum, Erysipelotrichaceae, Neisseriaceae, Sutterella, Burkholderiales, and Prevotella species might have potential for application as indicators of risk of female HCC. Although Paraprevotellaceae species have been commonly correlated with diet (Fernandes et al., 2014; Li et al., 2015), they have also been reported to be correlated with diseases, especially those caused by Prevotella species (Gharbia et al., 1994; Brook et al., 1995). Therefore, an increased abundance of Parapervotella, Paraprevotellaceae, and Prevotella species can be applied as indicators of risk of male HCC.
An increase in the relative abundance of gram-negative bacteria in the gut microbiota could increase the concentrations of endotoxins in the plasma and the liver (Xie et al., 2016). Several lines of evidence indicate that LPS accumulation contributes to the pathogenesis of HCC by eliciting proinflammatory responses in the liver (Yu et al., 2010). However, our results showed no significant difference was detected between the relative abundance of gram-negative or gram-positive bacteria from the LTsc1KO and Tsc1fl/fl mice in any of the groups (Supplementary Figures S2D,E). For LPS metabolism, no significant difference was detected in the relative abundance of the genes that participate in the KEGG pathways of LPS biosynthesis, LPS transport system, or LPS export system between the LTsc1KO and Tsc1fl/fl mice in any of the groups (Supplementary Figures S2F–H). These results imply that STZ-HFD might be the cause of the relative abundance of gram-negative bacteria and endotoxins, as HFD could significantly increase the relative abundance of gram-negative bacteria. This shows that the LTsc1KO model is an ideal model for studying the relationship between gut microbiota and the development of HCC, which excludes the influence of diet and other inducers.
The gut microbiota is influenced by diverse endogenous and exogenous factors, such as host development stage, diet components, health condition and many as-yet uncharacterized factors (Hooper and Gordon, 2001; Muegge et al., 2011; Ni et al., 2014; Yan et al., 2016; Li et al., 2017). This leads to obvious differences in the microbiota between host individuals or between different habitats (Costello et al., 2009), which in turn increases the difficulty of detecting significant differences. Therefore, many reports broaden the level of significant difference from 0.05 to 0.10 when detecting whether a specific factor significantly influences the microbiota (Ross et al., 2004; Xu et al., 2014; Zijlmans et al., 2015). In addition, although the LTsc1KO model is an ideal model for studying the relationship between gut microbiota and the development of HCC, our study had limitations of sample quantities and some liver biochemical and histological characteristics of the mice with different ages were deficient; moreover, only one sample or sometimes no samples were collected in some sample groups. These might be the reasons why the genera showing increased abundance in the gut microbiota from female LTsc1KO mice in group 4 did not maintain the increased abundance in group 5. Therefore, the causality between the changes in gut microbial composition and the development of HCC needs to be further verifies using germ-free mice. Moreover, the internal mechanisms causing changes in the gut microbiota and the effect of these changes on the development HCC should be study in more detail in the future.
Nevertheless, we detected different changing patterns between female and male LTsc1KO mice of different ages in our study with the development of HCC. Our results also suggest that the decrease in the relative abundance of anaerobic bacteria and the increase in the relative abundance of facultative anaerobic bacteria have potential for use as risk indexes of female HCC. The increased abundances of Allobaculum, Erysipelotrichaceae, Neisseriaceae, Sutterella, Burkholderiales, and Prevotella species might also be applied as risk indicators of female HCC, and for male HCC, increased abundances of Paraprevotella, Paraprevotellaceae, and Prevotella species might be potential risk indicators.
Author Contributions
RH, JN, and YiG designed the experiments. RH, TL, XB, YL, and YaG performed the experiments. RH, JN, and PZ analyzed the data. JN and YiG wrote the paper. All authors reviewed and edited the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [Grant No. 31500417] and the China Postdoctoral Science Foundation [Grant No. 2017M61 2691].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AA and handling Editor declared their shared affiliation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01008/full#supplementary-material
References
Adachi, Y., Moore, L. E., Bradford, B. U., Gao, W., and Thurman, R. G. (1995). Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108, 218–224. doi: 10.1016/0016-5085(95)90027-6
An, C., Kuda, T., Yazaki, T., Takahashi, H., and Kimura, B. (2013). FLX pyrosequencing analysis of the effects of the brown-algal fermentable polysaccharides alginate and laminaran on rat cecal microbiotas. Appl. Environ. Microbiol. 79, 860–866. doi: 10.1128/AEM.02354-12
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46.
Bajaj, J. S., Ridlon, J. M., Hylemon, P. B., Thacker, L. R., Heuman, D. M., Smith, S., et al. (2012). Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G168–G175. doi: 10.1152/ajpgi.00190.2011
Brook, I., Foote, P. A. Jr., and Slotes, J. (1995). Immune response to Fusobacterium nucleatum and Prevotella intermedia in patients with peritonsillar cellulitis and abscress. Clin. Infect. Dis. 20, S220–S221. doi: 10.1093/clinids/20.Supplement_2.S220
Cani, P. D., and Delzenne, N. M. (2011). The gut microbiome as therapeutic target. Pharmacol. Ther. 130, 202–212. doi: 10.1016/j.pharmthera.2011.01.012
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, Y., Yang, F., Lu, H., Wang, B., Chen, Y., Lei, D., et al. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572. doi: 10.1002/hep.24423
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O’Connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi: 10.1038/nature11319
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
El-Serag, H. B., and Rudolph, K. L. (2007). Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576. doi: 10.1053/j.gastro.2007.04.061
Faith, J. J., McNulty, N. P., Rey, F. E., and Gordon, J. E. (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. doi: 10.1126/science.1206025
Fei, N., and Zhao, L. (2013). An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 7, 880–884. doi: 10.1038/ismej.2012.153
Fernandes, K. A., Kittelmann, S., Rogers, C. W., Gee, E. K., Bolwell, C. F., Bermingham, E. N., et al. (2014). Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS One 9:e112846. doi: 10.1371/journal.pone.0112846
Gharbia, S. E., Haapasalo, M., Shah, H. N., Kotiranta, A., Lounatmaa, K., Pearce, M. A., et al. (1994). Characterization of Prevotella intermedia and Prevotella nigrescens isolates from periodontic and endodontic infections. J. Periodontol. 65, 56–61. doi: 10.1902/jop.1994.65.1.56
Hartwell, H. J., Petrosky, K. Y., Fox, J. G., Horseman, N. D., and Rogers, A. B. (2014). Prolactin prevents hepatocellular carcinoma by restricting innate immune activation of c-Myc in mice. Proc. Natl. Acad. Sci. U.S.A. 31, 11455–11460. doi: 10.1073/pnas.1404267111
Hooper, L. V., and Gordon, J. I. (2001). Commensal host-bacterial relationships in the gut. Science 292, 1115–1118. doi: 10.1126/science.1058709
Jia, W., Li, H., Zhao, L., and Nicholson, J. K. (2008). Gut microbiota: a potential new territory for drug targeting. Nat. Rev. Drug Discov. 7, 123–129. doi: 10.1038/nrd2505
Joossens, M., Huys, G., Cnockaert, M., De Preter, V., Verbeke, K., Rutgeerts, P., et al. (2011). Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637. doi: 10.1136/gut.2010.223263
Kaakoush, N. O. (2015). Insights into the role of Erysipelotrichaceae in the human host. Front. Cell Infect. Microbiol. 5:84. doi: 10.3389/fcimb.2015.00084
Karakan, T. (2014). Gut microbiota modulation in cirrhosis: a new frontier in hepatology. Turk. J. Gastroenterol. 25:126. doi: 10.5152/tjg.2014.0007
Kuda, T., Yokoyama, M., and Fujii, T. (1997). Effect of high and low viscous sodium alginates on levels of serum lipids and cecal microbiota in rats. Shokuhin Kagaku Kogaku Kaishi 44, 226–229. doi: 10.3136/nskkk.44.226
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Larsen, N., Vogensen, F. K., van den Berg, F. W., Nielsen, D. S., Andreasen, A. S., Pedersen, B. K., et al. (2010). Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5:e9085. doi: 10.1371/journal.pone.0009085
Li, H., Qu, J., Li, T., Li, J., Lin, Q., and Li, X. (2016). Pika population density is associated with the composition and diversity of gut microbiota. Front. Microbiol. 7:758. doi: 10.3389/fmicb.2016.00758
Li, X., Zhou, L., Yu, Y., Ni, J., Xu, W., and Yan, Q. (2017). Composition of gut microbiota in the gibel carp (Carassius auratus gibelio) varies with host development. Microb. Ecol. 74, 239–249. doi: 10.1007/s00248-016-0924-4
Li, Z., Wright, A. G., Liu, H., Bao, K., Zhang, T., Wang, K., et al. (2015). Bacterial community composition and fermentation patterns in the rumen of Sika deer (Cervus nippon) fed three different diets. Microb. Ecol. 69, 307–318. doi: 10.1007/s00248-014-0497-z
Li, Z., Yang, S., Lin, H., Huang, J., Watkins, P. A., Moser, A. B., et al. (2003). Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 37, 343–350. doi: 10.1053/jhep.2003.50048
Manichanh, C., Rigottier-Gois, L., Bonnaud, E., Gloux, K., Pelletier, E., Frangeul, L., et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Menon, S., Yecies, J. L., Zhang, H. H., Howell, J. J., Nicholatos, J., Harputlugil, E., et al. (2012). Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 5:ra24. doi: 10.1126/scisignal.2002739
Miriam, B., and Buenviaje, M. D. (1989). Quantitative sputum culture and gram stain: pulmonary infection vs. colonization. Philipp. J. Microbiol. Infect. Dis. 18, 28–35.
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
Ni, J., Li, X., He, Z., and Xu, M. (2017). A novel method to determine the minimum number of sequences required for reliable microbial community analysis. J. Microbiol. Methods 139, 196–201. doi: 10.1016/j.mimet.2017.06.006
Ni, J., Yan, Q., Yu, Y., and Zhang, T. (2014). Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol. Ecol. 87, 704–714. doi: 10.1111/1574-6941.12256
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Jia, W., et al. (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Shao, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Rajpal, D. K., and Brown, J. R. (2013). Modulating the human gut microbiome as an emerging therapeutic paradigm. Sci. Prog. 96, 224–236. doi: 10.3184/003685013X13691404141587
Rampelli, S., Candela, M., Turroni, S., Biagi, E., Collino, S., Franceschi, C., et al. (2013). Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging 5, 902–912. doi: 10.18632/aging.100623
Riordan, S. M., and Williams, R. (2006). The intestinal flora and bacterial infection in cirrhosis. J. Hepatol. 45, 744–757. doi: 10.1016/j.jhep.2006.08.001
Ross, D. J., Newton, P. C. D., and Tate, K. R. (2004). Elevated [CO2] effects on herbage production and soil carbon and nitrogen pools and mineralization in a species-rich, grazed pasture on a seasonally dry sand. Plant Soil 260, 183–196. doi: 10.1023/B:PLSO.0000030188.77365.46
Sakon, H., Nagai, F., Morotomi, M., and Tanaka, R. (2008). Sutterella parvirubra sp. nov. and Megamonas funiformis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 58, 970–975. doi: 10.1099/ijs.0.65456-0
Schnabl, B. (2013). Linking intestinal homeostasis and liver disease. Curr. Opin. Gastroenterol. 29, 264–270. doi: 10.1097/MOG.0b013e32835ff948
Seki, E., and Schnabl, B. (2012). Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J. Physiol. 590, 447–458. doi: 10.1113/jphysiol.2011.219691
Shao, Y. Y., Chen, B. B., Ou, D. L., Lin, Z. Z., Hsu, C. H., Wang, M. J., et al. (2017). Lenalidomide as second-line therapy for advanced hepatocellular carcinoma: exploration of biomarkers for treatment efficacy. Aliment. Pharmacol. Ther. 46, 722–730. doi: 10.1111/apt.14270
Sherman, M. (2010). Hepatocellular carcinoma: epidemiology, surveillance, and diagnosis. Semin. Liver Dis. 30, 3–16. doi: 10.1055/s-0030-1247128
Siegel, R. L., Miller, K. D., and Jemal, A. (2016). Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30. doi: 10.3322/caac.21332
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
van Ulsen, P., and Tommassen, J. (2006). Protein secretion and secreted proteins in pathogenic Neisseriaceae. FEMS Microbiol. Rev. 30, 292–319. doi: 10.1111/j.1574-6976.2006.00013.x
Voronina, O. L., Kunda, M. S., Ryzhova, N. N., Aksenova, E. I., Semenov, A. N., Lasareva, A. V., et al. (2015). The variability of the order Burkholderiales representatives in the healthcare units. Biomed Res. Int. 2015:680210. doi: 10.1155/2015/680210
Wang, L., Christophersen, C. T., Sorich, M. J., Gerber, J. P., Angley, M. T., and Conlon, M. A. (2013). Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 4:42. doi: 10.1186/2040-2392-4-42
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Xie, G., Wang, X., Liu, P., Wei, R., Chen, W., Rajani, C., et al. (2016). Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 7, 19355–19366. doi: 10.18632/oncotarget.8466
Xie, G., Wang, X., Zhao, A., Yan, J., Chen, W., Jiang, R., et al. (2017). Sex-dependent effects on gut microbiota regulate hepatic carcinogenic outcomes. Sci. Rep. 7:45232. doi: 10.1038/srep45232
Xu, M., Zhang, Q., Xia, C., Zhong, Y., Sun, G., Guo, J., et al. (2014). Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J. 8, 1932–1944. doi: 10.1038/ismej.2014.42
Yan, Q., Li, J., Yu, Y., Wang, J., He, Z., Van Nostrand, J. D., et al. (2016). Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ. Microbiol. 18, 4739–4754. doi: 10.1111/1462-2920.13365
Yang, S. Q., Lin, H. Z., Lane, M. D., Clemens, M., and Diehl, A. M. (1997). Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. U.S.A. 94, 2557–2562. doi: 10.1073/pnas.94.6.2557
Yeh, Y. T., Chang, C. W., Wei, R. J., and Wang, S. N. (2013). Progesterone and related compounds in hepatocellular carcinoma: basic and clinical aspects. Biomed Res. Int. 2013:290575. doi: 10.1155/2013/290575
Yu, L. X., Yan, H. X., Liu, Q., Yang, W., Wu, H. P., Dong, W., et al. (2010). Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 52, 1322–1333. doi: 10.1002/hep.23845
Zhang, H., DiBaise, J. K., Zuccolo, A., Kudrna, D., Braidotti, M., Yu, Y., et al. (2009). Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. U.S.A. 106, 2365–2370. doi: 10.1073/pnas.0812600106
Zhang, H., Yu, L., Yang, W., Tang, L., Lin, Y., Wu, H., et al. (2012). Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 57, 803–812. doi: 10.1016/j.jhep.2012.06.011
Keywords: biomarker, gut microbiota, hepatocellular carcinoma, sex-based response, Tsc1-knockout mice
Citation: Huang R, Li T, Ni J, Bai X, Gao Y, Li Y, Zhang P and Gong Y (2018) Different Sex-Based Responses of Gut Microbiota During the Development of Hepatocellular Carcinoma in Liver-Specific Tsc1-Knockout Mice. Front. Microbiol. 9:1008. doi: 10.3389/fmicb.2018.01008
Received: 12 December 2017; Accepted: 30 April 2018;
Published: 16 May 2018.
Edited by:
Andrea Masotti, Bambino Gesù Ospedale Pediatrico (IRCCS), ItalyReviewed by:
Alinne Castro, Universidade Católica Dom Bosco, BrazilAnna Alisi, Bambino Gesù Ospedale Pediatrico (IRCCS), Italy
Copyright © 2018 Huang, Li, Ni, Bai, Gao, Li, Zhang and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Gao, gaoyi6146@163.com; drgaoy@126.com
†These authors have contributed equally to this work.