
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 April 2018
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00830
This article is part of the Research Topic Innovative Approaches to Virus Diagnosis and Control View all 27 articles
 Mamoru Oshiki1*
Mamoru Oshiki1* Takayuki Miura2Shinobu Kazama3Takahiro Segawa4
Takayuki Miura2Shinobu Kazama3Takahiro Segawa4 Satoshi Ishii5Masashi Hatamoto6Takashi Yamaguchi7Kengo Kubota8Akinori Iguchi9Tadashi Tagawa1Tsutomu Okubo1Shigeki Uemura1Hideki Harada10Naohiro Kobayashi1Nobuo Araki1Daisuke Sano8
Satoshi Ishii5Masashi Hatamoto6Takashi Yamaguchi7Kengo Kubota8Akinori Iguchi9Tadashi Tagawa1Tsutomu Okubo1Shigeki Uemura1Hideki Harada10Naohiro Kobayashi1Nobuo Araki1Daisuke Sano8Detection and genotyping of pathogenic RNA viruses in human and environmental samples are useful for monitoring the circulation and prevalence of these pathogens, whereas a conventional PCR assay followed by Sanger sequencing is time-consuming and laborious. The present study aimed to develop a high-throughput detection-and-genotyping tool for 11 human RNA viruses [Aichi virus; astrovirus; enterovirus; norovirus genogroup I (GI), GII, and GIV; hepatitis A virus; hepatitis E virus; rotavirus; sapovirus; and human parechovirus] using a microfluidic device and next-generation sequencer. Microfluidic nested PCR was carried out on a 48.48 Access Array chip, and the amplicons were recovered and used for MiSeq sequencing (Illumina, Tokyo, Japan); genotyping was conducted by homology searching and phylogenetic analysis of the obtained sequence reads. The detection limit of the 11 tested viruses ranged from 100 to 103 copies/μL in cDNA sample, corresponding to 101–104 copies/mL-sewage, 105–108 copies/g-human feces, and 102–105 copies/g-digestive tissues of oyster. The developed assay was successfully applied for simultaneous detection and genotyping of RNA viruses to samples of human feces, sewage, and artificially contaminated oysters. Microfluidic nested PCR followed by MiSeq sequencing enables efficient tracking of the fate of multiple RNA viruses in various environments, which is essential for a better understanding of the circulation of human pathogenic RNA viruses in the human population.
Various species of human RNA viruses, including Aichi virus (AiV), astrovirus (AstV), enterovirus (EV), hepatitis A virus (HAV), hepatitis E virus (HEV), norovirus (NoV), rotavirus (RV), sapovirus (SaV), and human parechovirus (HPeV), have been recognized as causative agents of waterborne viral gastroenteritis and hepatitis (Bosch, 1998; Harvala et al., 2008; Sinclair et al., 2009; Pham et al., 2010; Svraka et al., 2010; Drexler et al., 2011). The above mentioned RNA virus species includes multiple (sub)genogroups. For example, NoV is composed of seven genogroups, and each genogroup is further subdivided into genotypes (Vinjé, 2015). These RNA viruses circulate in aquatic environments and the human population (Iwai et al., 2009; Sano et al., 2011; Hata et al., 2015; Miura et al., 2016; Pu et al., 2016). For instance, the viruses exist at high concentration in feces of patients and are released into sewage system (Lee et al., 2007; Drexler et al., 2011). Sewage effluents still contain the viruses at certain level (Miura et al., 2015; Kobayashi et al., 2017), which contaminate natural water resources. The released viruses can be accumulated in shellfishes such as oyster, and outbreak occurs by eating the contaminated shellfish (Le Guyader et al., 1994; Ueki et al., 2005; Nenonen et al., 2008). For a better understanding of the circulation and prevalence of RNA viruses and for identification of a contamination source, detection and genotyping of multiple viruses are essential (Kazama et al., 2017).
Detection and genotyping of human pathogenic RNA viruses have usually been carried out by reverse transcription (RT)-PCR followed by Sanger sequencing (Nasri et al., 2007; Stene-Johansen et al., 2007; Kitajima et al., 2010) because of the high sensitivity, specificity, and reliability of identification of contaminating viruses. Nonetheless, clinical samples (e.g., stool samples) (Román et al., 2003; Iturriza Gómara et al., 2008) and environmental water samples such as river water (Ishii et al., 2014) and wastewater (Miura et al., 2015; Kobayashi et al., 2017) contain various species of human pathogenic RNA viruses, and the conventional PCR assay is labor intensive because many PCR runs are required for detection and genotyping of multiple RNA viruses. Additionally, nested PCR in which the amplicon from the first round of PCR undergoes the second round PCR as a template DNA is often conducted to enhance sensitivity and specificity of detection and genotyping of an RNA virus (Le Guyader et al., 1994; Fischer et al., 2003; Aw et al., 2009); this approach further increases the amount of labor. Moreover, recombination of viral genomes has resulted in emergence of viral variants containing a genome deriving from different genotypes (Eden et al., 2013). As for NoV GII, new GII genotype 2 (GII.2) epidemic strains containing open-reading frame 1 (ORF1) and ORF 2–3 regions of GII.5 and GII.2, respectively, were recently identified (Motomura et al., 2016). In this case, the use of multiple oligonucleotide primer sets targeting different regions of the viral genome is recommended for reliable genotyping, while this practice is even more laborious.
Apart from the PCR-based assay, shotgun sequencing analysis using a next-generation sequencer (NGS) was recently developed for the detection and identification of a human pathogenic RNA virus in human feces and wastewater samples (Batty et al., 2013; Hasing et al., 2016). Abundance of the sequence reads corresponding to NoV in stool samples was, however, only 0.01–1.9% of all sequence reads (Hasing et al., 2016), requiring massive sequencing efforts for detection of the human pathogenic RNA viral population in the sample. This situation increases labor and sequencing costs; therefore, only a limited number of samples can be analyzed this way.
The present authors have developed high-throughput quantitative PCR (qPCR) techniques involving a microfluidic chip for detection and quantification of pathogenic microbes (Ishii et al., 2013, 2014). In the previous microfluidic qPCR (MFqPCR) analyses, which used a 96.96 Dynamic Array chip (Fluidigm, Tokyo, Japan), up to 9,216 independent PCRs could be carried out simultaneously in nanoliter reaction chambers on a microfluidic chip, and the MFqPCR enables simultaneous quantification of 13 viruses in up to 92 samples by means of 13 sets of oligonucleotide primers and TaqMan probes: i.e., adenovirus as a DNA virus and AiV, AstV, EV, NoV GI, NoV GII, NoV GIV, HAV, HEV, RV, SaV, mengovirus, and murine NoV as RNA viruses. Sensitivity and specificity of MFqPCR are comparable with those of a conventional qPCR assay (Ishii et al., 2014), and the MFqPCR has provided a convenient and efficient platform for PCR detection of RNA viruses in water environments (Kobayashi et al., 2017). Moreover, the use of a different type of microfluidic chip, specially designed for the recovery of PCR amplicons (i.e., a 48.48 access array chip, Fluidigm, Tokyo, Japan), allows for the recovery of amplicons after microfluidic PCR, and for their subsequent sequencing using NGS. This approach has been successfully applied in human leukocyte antigen typing (Lange et al., 2014), in genotyping of enterohemorrhagic Escherichia coli (Ison et al., 2016), and in the investigation of the genetic diversity of multiple functional genes related to the nitrogen cycle (Oshiki et al., 2018), but has not yet been used for the genotyping of human pathogenic viruses. The use of microfluidic PCR in combination with amplicon sequencing is a promising approach for simultaneous high-throughput detection and genotyping of multiple viral species in multiple samples, the performance of which (in terms of sensitivity, specificity, and applicability to different types of samples) remains to be investigated.
Accordingly, the aim of this study was to develop a high-throughput tool for detection and genotyping of 11 human pathogenic RNA viruses by microfluidic nested PCR followed by MiSeq amplicon sequencing (MFnPCR–MiSeq; Figure 1). The following 11 human pathogenic RNA viruses were targeted in this study: AiV, AstV, EV, NoV GI, NoV GII, NoV GIV, HAV, HEV, RV, SaV, and HPeV. Notably, the oligonucleotide primers used in our previous MFqPCR assay (Ishii et al., 2014) could not be used to analyze the 11 human pathogenic RNA viruses listed above, as amplicon sequence lengths (ranging from 87 to 172 bp, 113 bp on average) were too short to allow for reliable genotyping. Therefore, 34 sets of newly selected oligonucleotide primers (Supplementary Table S1) were used in the present study. After validation of sensitivity and specificity, MFnPCR–MiSeq was applied to sewage and human fecal samples collected from patients. To the best of our knowledge, this is the first study combining microfluidic PCR and next-generation sequencing techniques for detection and genotyping of multiple human RNA viruses in environments.
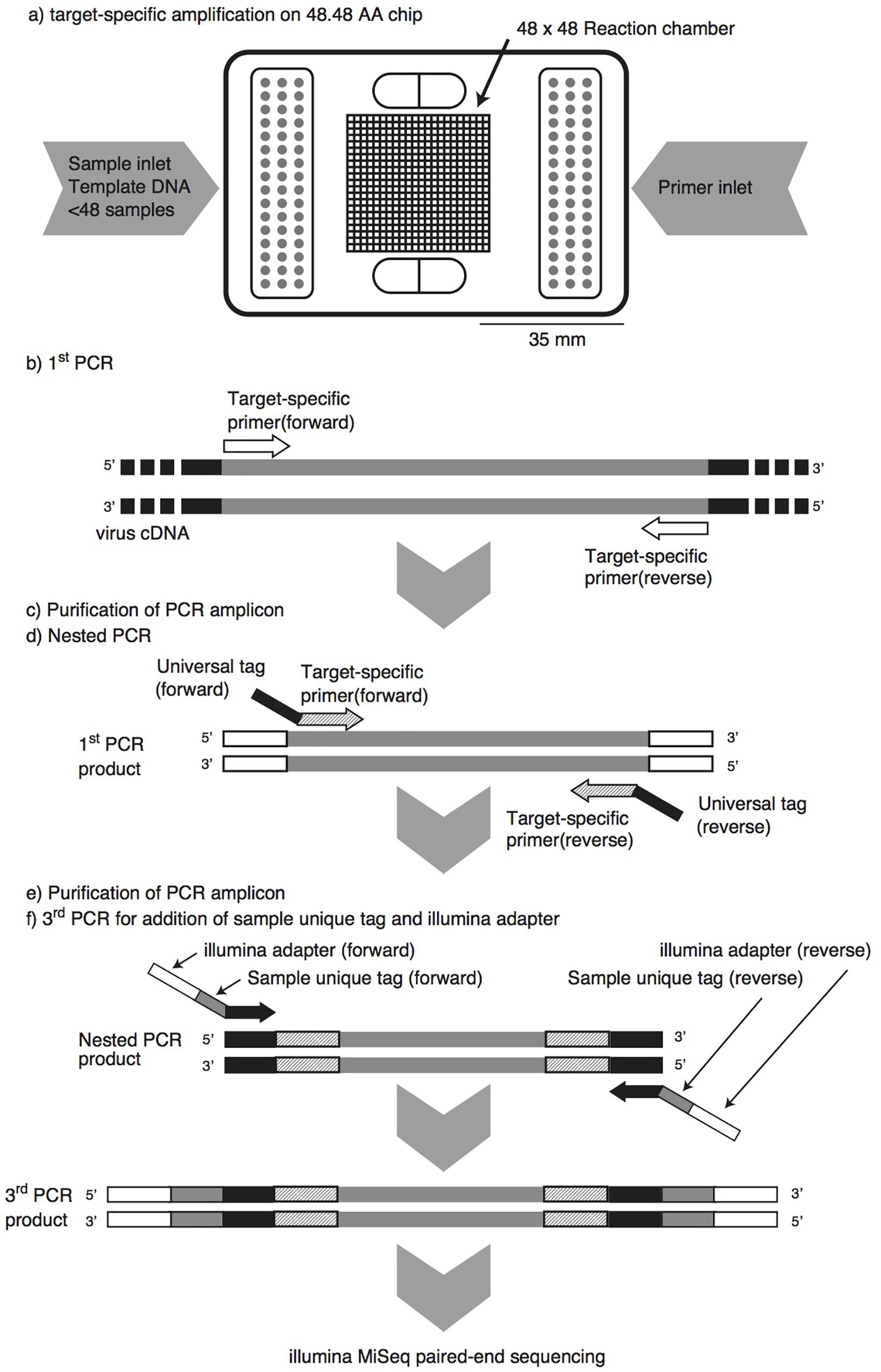
FIGURE 1. Pipeline of microfluidic nested PCR followed by MiSeq amplicon sequencing (MFnPCR–MiSeq). Purification of PCR amplicons was carried out with the NucleoSpin Gel and PCR Clean-up Kit. Amplification by the first-round and nested PCR was performed on a 48.48 Access Array (AA) chip in a BioMark HD reader, while the third PCR for addition of a sample-specific tag and Illumina adapter was run in 96-well PCR plates on a conventional thermal cycler.
Sensitivity of MFnPCR–MiSeq was examined in a mixture of plasmid DNA and viral cDNA (Table 1). As for NoV GI, NoV GII, NoV GIV, HAV, HEV, and HPeV, plasmids containing the target region were constructed or prepared as previously described (Ishii et al., 2014). The target regions were obtained from the following reference strains: Norwalk (nucleotide region 5271–5770), Lordsdale (4998–5497), Saint-Cloud (681–1180), HM175 (2151–3440), Burma (5147–6646), and Harris (2101–3250). DNA concentrations were determined by means of the Qubit dsDNA BR Assay Kit on a Qubit 3.0 Fluorometer (Thermo Fisher Scientific K.K., Yokohama, Japan). As for AiV, AstV, EV, RV, and SaV, viral RNA was extracted from cell culture or fecal samples, cDNA was synthesized, and copy numbers were determined by conventional qPCR (Ishii et al., 2014). The cDNA of AiV strain N1277/91 was kindly provided by Dr. Hiroyuki Katayama (The University of Tokyo, Japan). Poliovirus 1 strain Sabin is courtesy of Dr. Kazuyoshi Yano (Tokyo Metropolitan Institute of Public Health, Japan) and served as the reference strain of EV. RV strain RRV was kindly provided by Prof. Osamu Nakagomi (Nagasaki University, Japan). AstV and SaV were obtained from feces and were a generous gift from Dr. You Ueki (Miyagi Prefectural Institute of Public Health and Environment, Japan). The six plasmids and five synthesized cDNAs were mixed at equimolar concentrations and serially diluted from 100 to 104 copies/μL.
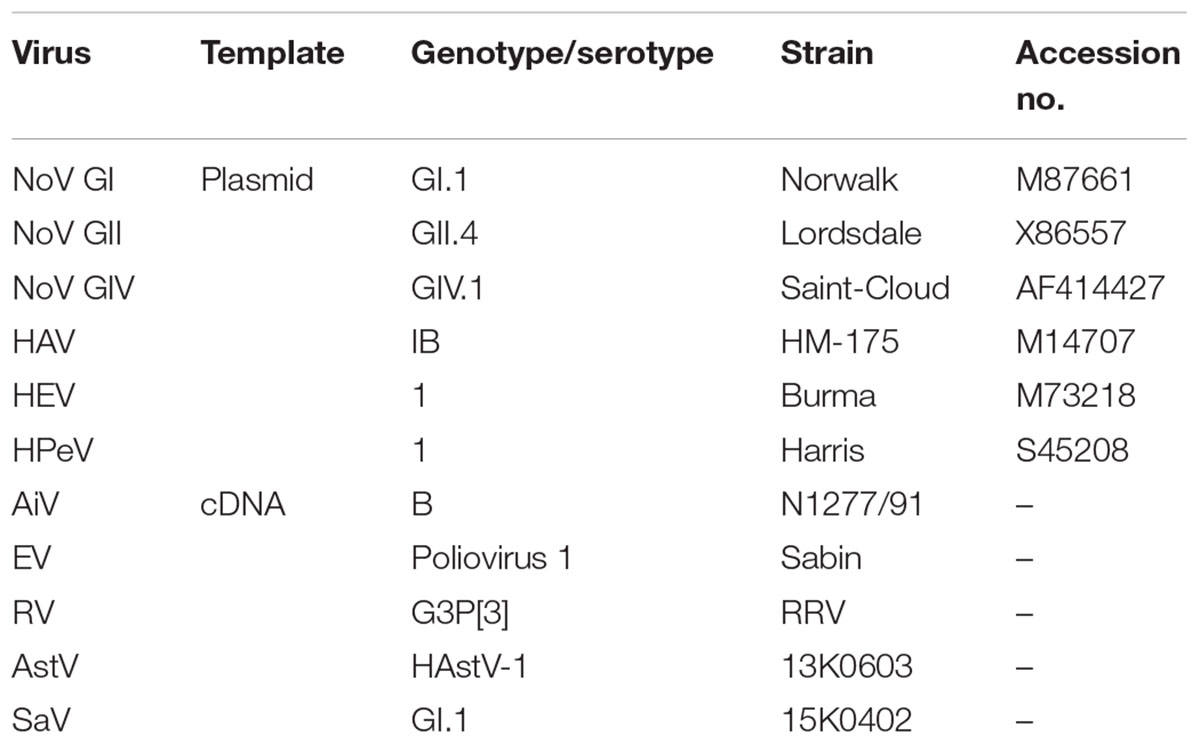
TABLE 1. Plasmid DNAs and viral cDNAs serving as positive controls for detection by microfluidic nested PCR followed by MiSeq amplicon sequencing.
Live oysters were purchased directly from a producer in Miyagi Prefecture, Japan, and were artificially contaminated by placing in seawater seeded with a known concentration of a fecal suspension containing NoV GII genotype 4 (GII.4) Sydney 2012 or GII.17 Kawasaki 2014 strain (107 copies/mL) for 24 h with aeration. Digestive tissues were excised from 15 contaminated oysters and chopped up to obtain a homogenate. Viral RNA was extracted from 2 g of the digestive-tissue homogenate according to the method from ISO with some modifications. Briefly, 100 μL of the murine NoV process control and 2 mL of a 100 μg/mL proteinase K solution (Cat. No. P2308, Sigma-Aldrich, Tokyo, Japan) were added to the 2 g of the digestive-tissue homogenate. The sample was incubated at 60°C for 15 min on a rotator (32 rpm) and then centrifuged at 3,000 ×g for 5 min. Approximately 2.5 mL of the resulting supernatant was collected and 500 μL was used for RNA extraction with the NucliSENS Kit (bioMérieux, Tokyo, Japan).
Eight fecal samples that have tested positive for NoV GI or GII were kindly provided by Dr. You Ueki (Miyagi Prefectural Institute of Public Health and Environment, Japan). The samples were collected from patients with acute gastroenteritis through routine infectious disease surveillance in Miyagi Prefecture, Japan, during the National Epidemiological Surveillance of Infectious Diseases from March 2014 to March 2015. General practitioners obtained oral consent for sample collection and pathogen screening from patients. Screening of pathogens was performed as a part of health services provided by Miyagi Prefectural Institute of Public Health and Environment. Consequently, human ethics approval and informed consent were not required for the present study. All samples were anonymized and there is no possibility of personal identification. The collected fecal samples were diluted in phosphate-buffered saline (pH 7) to obtain a 10% suspension and centrifuged at 10,000 ×g for 10 min. The resulting supernatants were stored at -80°C until processing, and viral RNA was extracted using the NucliSENS kit.
These samples were collected at two domestic wastewater treatment plants in Niigata and Miyagi, Japan. Regarding the sewage samples collected from the wastewater treatment plant in Niigata (the population served by the sewerage system and daily flow rate, respectively: 150,000 people and 120,000 m3/d), the present authors have previously determined virus concentrations by MFqPCR, and these data are shown in Supplementary Table S2 (Kobayashi et al., 2017). The sewage samples were 24-h composite samples (1 L/h) that were collected from May 2014 to January 2015.
The other sewage sample was collected at a wastewater treatment plant (10,000 and 4,000 m3/d: population served by the sewerage system and daily flow rate, respectively) in Miyagi Prefecture in June 2015. One liter of the effluent of the first sedimentation tank was collected as a grab sample. The collected sewage sample was immediately transported to our laboratory on ice and subjected to concentration and recovery of virus particles by the polyethylene glycol precipitation method (Kazama et al., 2016). The virus concentrate was stored at -80°C until processing, and viral RNA was extracted by means of the QIAamp Viral RNA Mini Kit (Qiagen, Tokyo, Japan).
This procedure was carried out using the PrimeScript RT Reagent Kit with a random hexamer primer (Takara Bio, Shiga, Japan), as previously described (Ishii et al., 2014).
The 34 sets of oligonucleotide primers shown in Supplementary Table S1 were used in the present study for genotyping of the following 11 viruses: AiV, AstV, EV, HAV, HEV, NoV GI, NoV GII, NoV GIV, RV, SaV, and HPeV. Two primer sets were newly designed and employed in the present study for the detection of EV, i.e., primers EV1F_66 and EV1R_552 for the first round of PCR and primers EV1F-66 and Ev2R_460 for seminested PCR (see Supplementary Text). The forward and reverse PCR primers used for the nested PCR amplification contained the following Illumina tag sequences at the 5′-end: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′, respectively. The sequence length of PCR amplicons without Illumina tag sequences ranges from 180 to 760 bp (341 bp on average).
Microfluidic PCR was run in triplicate on a BioMark HD reader with a 48.48 AccessArray chip (Fluidigm, Tokyo, Japan). The 20× primer solutions and presample master mix were dispensed into the 48.48 Access Array chip and mixed by an IFC controller AX (Fluidigm, Tokyo, Japan) according to the manufacturer’s instructions. The 20× primer solutions contained the 1× Access Array loading reagent (Fluidigm, Tokyo, Japan) and 4 μM each forward and reverse primers. The presample master mix (5 μL) contained 1× FastStart HiFi reaction buffer (Roche Diagnostics, Tokyo, Japan), 1× Access Array loading reagent, 4.5 mM MgCl2, 5% (v/v) dimethyl sulfoxide, 0.2 mM each dNTP, 0.05 U of FastStart HiFi Enzyme Blend (Roche Diagnostics), 1× EvaGreen (Biotium, CA, United States), 0.5× ROX, and 1 μL of template DNA (ca. 10 ng). Microfluidic PCR was performed under the following thermal conditions: 50°C for 120 s, 70°C for 20 min, 95°C for 10 min, followed by 10 cycles of program A (95°C for 15 s, 48°C for 30 s, and 72°C for 1 min), 2 cycles of program B (95°C for 15 s, 80°C for 30 s, 48°C for 30 s, and 72°C for 1 min), 8 cycles of program A, 2 cycles of program B, 10 cycles of program A, 2 cycles of program B, and the final extension at 72°C for 3 min. The detection of PCR products involved monitoring of the fluorescence intensity of the EvaGreen dye. The threshold cycle (CT) was defined as the cycle at which the EvaGreen signal became significantly greater than the background level. After the microfluidic PCR, the amplicons were recovered with the Access Array Harvest Reagent (Fluidigm, Tokyo, Japan) and the IFC controller AX.
For microfluidic nested-PCR amplification, the recovered 48 amplicons (approx. 10 μL) were purified using NucleoSpin Gel and the PCR Clean-up Kit (Takara Bio, Shiga, Japan). The purified DNAs were eluted with 15 μL of the supplied elution buffer. Microfluidic nested PCR was run with the purified DNAs as a template, and the amplicons were recovered from the chip as described above. The 48 nested-PCR products were purified by means of the NucleoSpin Gel and PCR Clean-up Kit and subjected to the following sequencing library preparation. A sample without viral cDNA (i.e., pure water) was also subjected to the above microfluidic first-round and nested PCR in parallel as a negative control.
The purified nested-PCR products were tagged with sample-specific index sequences at their 5’-end by PCR via the index primers supplied in Nextera XT index Kit v2 Set A (Illumina, Tokyo, Japan). The PCR mixture (20 μL) contained 1× KAPA HiFi HS ReadyMix (Nippon Genetics, Tokyo, Japan), 2 μL of forward and reverse index primers, 1 μL of the purified PCR products, and 5 μL of distilled water. The PCR was conducted under the following cycling conditions: 95°C for 3 min; 15 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and 72°C for 5 min. After agarose gel electrophoresis, gel lanes containing the amplicons of 200–1,000 bp were excised and purified using the NucleoSpin Gel and PCR Clean-up Kit. Tagged amplicons were pooled and analyzed in a MiSeq paired-end sequencing reaction with the v3 reagent kit (Illumina, Tokyo, Japan).
These analyses were performed as follows in the MacQIIME 1.9.1 (Caporaso et al., 2010) and USEARCH 9.2 (Edgar, 2010) software packages. The merging of forward and reverse reads obtained from the MiSeq paired-end sequencing and the quality filtering of the merged reads were conducted via MacQIIME scripts: join_paired_ends.py and split_library.py with a minimum quality score parameter of 25 and a minimum sequence length of 70 bp.
The quality-filtered reads were demultiplexed by sample and clustered into operational taxonomic units (OTUs) by the usearch scripts. Specifically, the reads were dereplicated (-derep_fulllength –sizeout) and sorted, and singletons were removed (-sortbysize–minsize 2); then, the reads were clustered (-cluster_otus), checked for chimeric sequences (-uchime_denovo), and mapped back to OTUs at 97% identity (-usearch_global–strand both–id 0.97). When relative abundance of the OTU in total sequence reads of each sample was <0.1%, the OTU was eliminated and not subjected to the further analysis. The reads representing each OTU were subjected to a blastn search against the nt database, and virus reads were thus identified. For phylogenetic analysis, the virus reads were processed in ClustalW; bootstrapped phylogenetic trees were then constructed by the neighbor-joining method with 1,000 bootstrap replications in the MEGA7 software (Kumar et al., 2016). The genetic distances were calculated by the Kimura two-parameter method. Among NoV reads, genotypes and variants were identified using the NoV Genotyping Tool Version 1.0 (Kroneman et al., 2011) before the phylogenetic analysis.
Nucleotide sequence data from the human feces, sewage, and contaminated oysters were deposited in the DDBJ/EMBL/GenBank databases under the accession number DRA005567.
The mixture of plasmid DNA and viral cDNA was serially diluted from 104 to 100 copies/μL and then subjected to MFnPCR–MiSeq. As shown in Figure 2, all the tested viruses could be detected simultaneously when concentrations were higher than 103 copies/μL. PCR amplicons obtained from each dilution series were recovered and subjected to MiSeq sequencing. Blast analysis of sequence reads revealed that the recovered amplicons derived from the plasmid DNA and viral cDNA serving as templates. When the detection limit was defined as 10 sequence reads per sample, each tested virus was detectable in the following dilution series (copies/μL): 102 for AiV, 102 for AstV, 101 for EV, 101 for NoV GI, 102 for NoV GII, 102 for NoV GIV, 101 for HAV, 100 for HEV, 102 for RV, 103 for SaV, and 102 for HPeV. By considering dilution factors resulting from the processes of virus concentration, RNA extraction, and cDNA synthesis, the detection limits were calculated to be 101–104 copies/mL for sewage, 105–108 copies/g-human feces, and 102–105 copies/g-digestive tissues of oysters. Those detection limits were not dependent on the amplicon size (180–760 bp without Illumina tag sequences) (Supplementary Table S1) but on the PCR primer set. For instance, microfluidic nested PCR amplification was performed with four primer sets for detection of AiV, and PCR amplification was detectable only in the assay containing primers Sense 1 (6261) and 6779 in the first-round PCR and primers Sense 2 and Antisense 2 in the nested PCR. Likewise, multiple PCR primer sets were used for the detection of EV, RV, and SaV. The following PCR primer sets gave the highest sensitivity: Ev1F_66 and Ev2R_460 for EV; R3 and R2 for RV; and SaV1245Rfwd and SVR-DS5 for SaV. This means that the amplification efficiency of each primer set is more critical than amplicon size for the detection of target viruses.
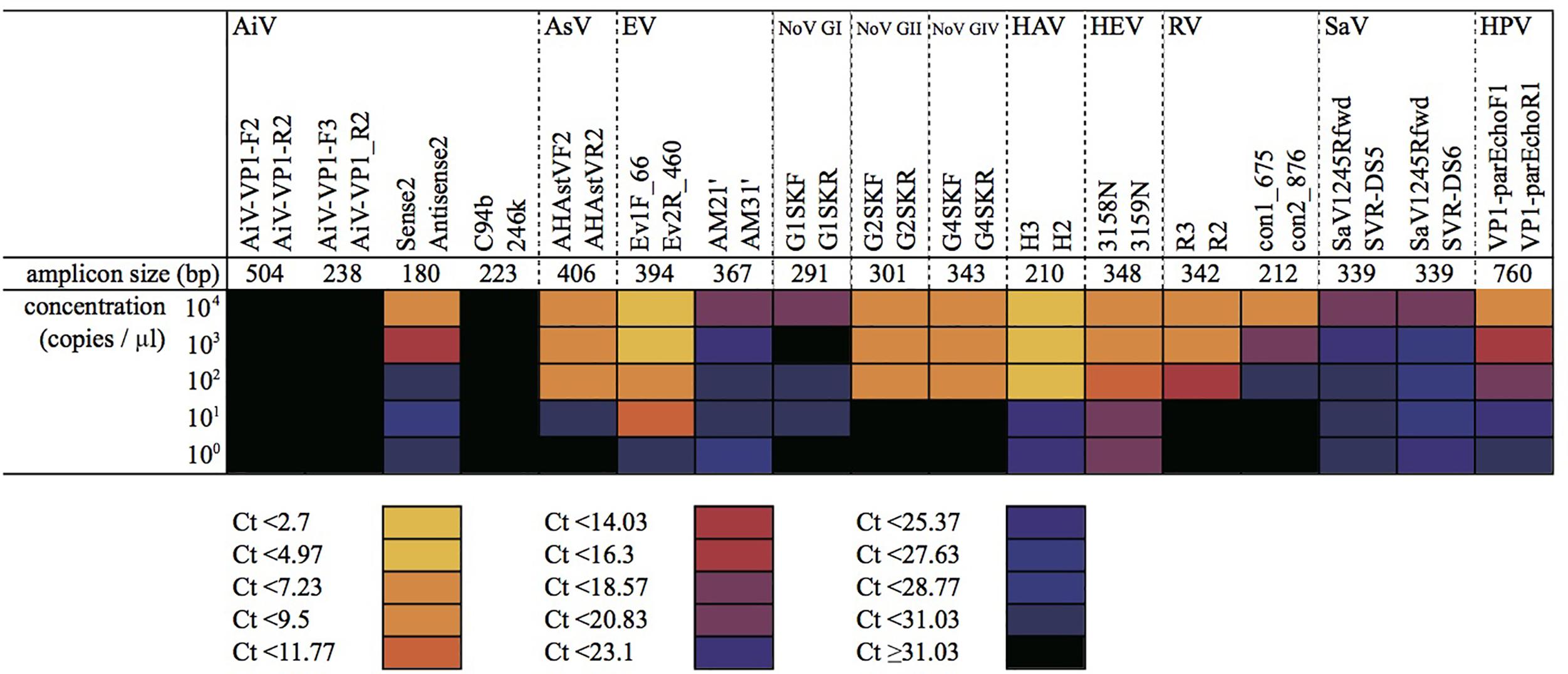
FIGURE 2. Detection of serially diluted virus samples by microfluidic nested PCR followed by MiSeq amplicon sequencing. Plasmid DNA and cDNA samples containing target regions of each virus were mixed at equimolar concentrations and diluted to 104–100 copies/μL. The mixtures were subjected to the first round of MF-PCR, and the amplicon was collected and used for MFnPCR with 20 oligonucleotide primer sets for detection of 11 viruses (i.e., AiV; AsV; EV; NoV GI, GII, and GIV; HAV; HEV; RV; SaV; and HPV). CT values determined in the nested MF PCR are shown as a heat map. Amplicon size represents the size of a PCR amplicon without an Illumina tag sequence.
To validate the genotyping assay based on MFnPCR–MiSeq, viral RNA was extracted from two oyster samples (B005 and B006) in which the known NoV GII strains (i.e., NoV GII.4 Sydney 2012 and GII.17 Kawasaki 2014) were intentionally bioaccumulated, and the synthesized cDNA was subjected to MFnPCR–MiSeq. More than 73,115 sequence reads were identical to those of the artificially added NoV GII (Figure 3), thus, indicating the reliability of the genotyping assay by MFnPCR–MiSeq.
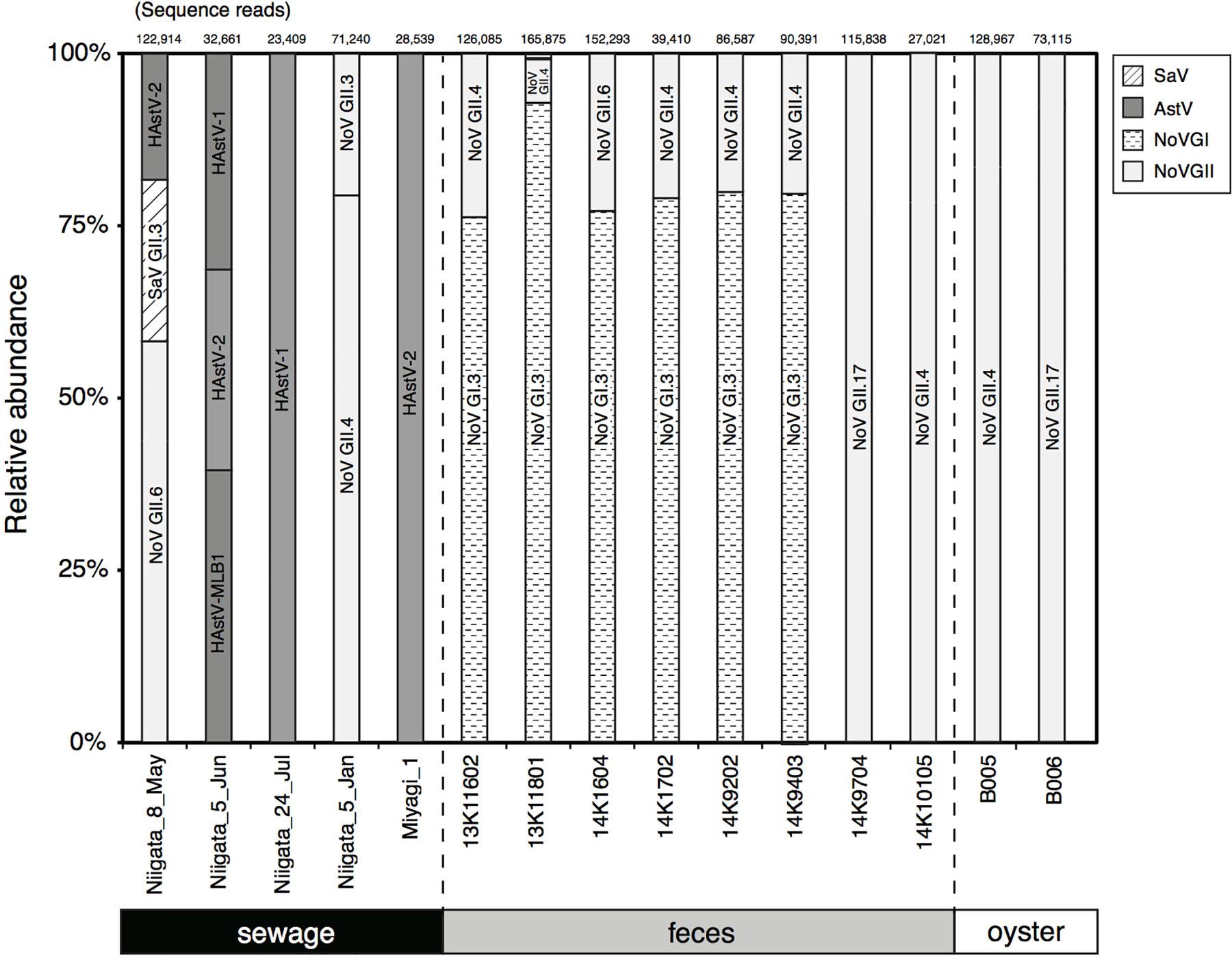
FIGURE 3. Detection and typing of viruses in samples of human feces, sewage, and contaminated oysters. The number of reads obtained by MiSeq sequencing is shown at the top of the panel, and relative abundance of the sequence reads affiliated with a specific genotype of viruses is shown in the figure. The identified genotypes are indicated in the bars.
MFnPCR–MiSeq was applied to eight human fecal and five sewage samples. Coinfection with strains NoV GI and NoV GII was identified in six fecal samples (Figure 3; 13K11602, 13K11801, 14K1604, 14K1702, 14K9202, and 14K9403). Genotyping of the detected NoV GI and GII was conducted by phylogenetic analysis of the MiSeq reads. All the sequence reads of NoV GI were found to be affiliated with GI.3 (Figure 3), whose sequence reads ended up in two phylogenetic clades (Supplementary Figure S1). The sequence reads from feces 13K11602 and 14K9202 were affiliated with clade I, while the other reads clustered in clade II. NoV GI.3 clade I and II contained the nucleotide sequence identified in the sewage sample from Nanning city, China, in 2011 (KM246914) and the sequence identified in a stool sample from Chinese adult outpatients in Beijing in 2007 (Accession No.: GQ856471), respectively. As for NoV GII, GII.4 (Supplementary Figure S2) was detected in fecal samples except for samples 14K1604 and 14K9704. GII.6 and GII.17 were identified in samples 14K1604 (Supplementary Figure S3) and 14K9704 (Supplementary Figure S4), respectively.
In the tested sewage samples collected at wastewater treatment plants in Niigata and Miyagi (designated as Niigata and Miyagi in Figure 3, respectively), NoV GII.3 (Supplementary Figure S5), NoV GII.4 (Supplementary Figure S2), NoV GII.6 (Supplementary Figure S3), SaV GII.3 (Supplementary Figure S6), and HAstV-1 (human AstV type 1), HAstV-2 (Supplementary Figure S7), and HAstV-MLB1 (Supplementary Figure S8) were detected (Figure 3). In the tested Niigata samples, concentrations of AiV (5.2 × 100 to 1.7 × 101 copies/μL), NoV GI (1.2 × 100 to 1.7 × 101 copies/μL), and EV (<7.8 × 101 copies/μL) as determined by the MFqPCR assay (Kobayashi et al., 2017) were lower than the detection limit of MFnPCR–MiSeq as previously mentioned; therefore, those viruses were not detectable. On the other hand, SaV in samples Niigata_5_Jun, Niigata_24_Jul, and Niigata_5_Jan was not detectable although the concentrations were higher than 4.8 × 105 copies/μL (Supplementary Table S2). As for the AstV in the Niigata samples, the dominant genotype changed with time, i.e., AstV was detected from May to July 2014, and the dominant genotype changed from HAstV-2 to HAstV-1.
Detection and genotyping of human pathogenic RNA viruses are essential for the identification of contamination sources, and for better understanding the circulation of those pathogens in the aquatic environments and human population. Here, we propose the MFnPCR–MiSeq assay, in which 11 RNA viruses are detected and quantified simultaneously on a microfluidic chip, and then genotyped by sequencing the recovered PCR amplicons. A single-tube multiplex PCR assay with subsequent agarose gel electrophoresis was previously developed to screen the RNA virus genotypes in fecal samples, but this assay has a number of limitations (Yan et al., 2003; Rohayem et al., 2004; Khamrin et al., 2011; Zeng et al., 2014). Firstly, primer sets yielding PCR amplicons with similar sequence lengths are not suitable because separation of such amplicons on an agarose gel is generally difficult. Secondly, nonspecific amplification can result in false positive detection of a virus. Finally, while analysis by cloning and Sanger sequencing is the accepted gold standard for genotyping of viruses (Nasri et al., 2007; Stene-Johansen et al., 2007; Kitajima et al., 2010), low-abundance viruses are likely to be overlooked if analysis has been carried out with an insufficient number of cloned DNA copies. In contrast, viruses were identified here by MFnPCR–MiSeq based on nucleic acid sequence similarity (and not on amplicon length), and sequence reads derived from nonspecific amplification were eliminated in blastn analysis. Our MiSeq sequencer yielded >20,000 reads per sample, allowing for the identification of low-abundance viruses (e.g., NoV GII.4 in the 13K11801 fecal sample).
Aside from PCR-based approaches, including our MFnPCR–MiSeq technique, shotgun metagenomic approaches have previously been used to detect human pathogenic viruses in sewage (Cantalupo et al., 2011; Ng et al., 2012), activated sludge (Tamaki et al., 2012; Bibby and Peccia, 2013), and reclaimed water (Rosario et al., 2009). A main advantage of metagenomic approaches is that they avoid bias introduced by PCR amplification of cDNA; i.e., PCR-based approaches require the use of oligonucleotide primers, with the consequence that viral populations with primer–template mismatches are overlooked. On the contrary, the population of human pathogenic viruses in environmental samples is generally small. When stool samples from patients with NoV infections were analyzed, even after enrichment of viral RNA by depletion of bacterial rRNA, the NoV reads accounted for only 0.01–1.9% of all sequence reads (Hasing et al., 2016). Therefore, metagenomic approaches require a massive sequencing effort per sample, which cannot be applied to multiple samples. Additionally, genotyping requires sequence information from specific genomic regions, such as the capsid N/S region of the NoV genome, and the probability of recovering sequence reads spanning those regions is low, especially when the targeted human pathogenic viruses form a low proportion of the total viral population. Shotgun metagenomic approaches should preferentially be used to study genetic diversity of whole viral populations. MFnPCR–MiSeq, on the contrary, enables us to perform reliable targeted genotyping of human pathogenic viruses with reasonable sensitivity (i.e., 101–104 copies/mL for sewage, 105–108 copies/g-human feces, and 102–105 copies/g-digestive tissues of oysters).
Notably, selection of appropriate oligonucleotide primers was critical for MFnPCR–MiSeq detection sensitivity because of variations in the amplification efficiency for each primer set, and also the possibility of primer–template mismatches resulting in false-negative detection of target viruses. In the present study, we could not detect SaV in the samples Niigata_5_Jun, Niigata_24_Jul, and Niigata_5_Jan even though the concentrations (4.8 × 105 copies/μL) were higher than our method’s detection limit (103 copies/μL for SaV). This result was probably a consequence of mismatches between the PCR primers and viral cDNA. SaV in the tested sewage samples was detectable by primers SaV1F, SaV5F, and SaV1245R, as well as the TaqMan probes, SaV124TP and SaV5TP, used in the previous assay version (MFqPCR), but was undetectable by the oligonucleotide primers selected in the present study (Supplementary Table S1). In addition to amplification efficiency and the possibility of primer–template mismatches, the length of the PCR amplicon should also be considered during selection of oligonucleotide primers for MFnPCR–MiSeq assay. This is due to the upper read-length limit restriction imposed by the Illumina MiSeq sequencer, which is typically 300 bp × 2 in a MiSeq paired-end sequencing reaction performed using the v3 reagent kit. Due to this limitation, we amplified and analyzed the capsid N/S region (∼350 nt) instead of the complete ORF2 (VP1) sequence (∼2,000 nt) of the NoV genome. Capsid N/S region sequences from environmental samples have provided important insights into NoV genotypes circulating in human society (Kazama et al., 2016), while classification of NoVs based on complete ORF2 (VP1) sequences (∼2,000 nt) has been proposed (Kroneman et al., 2013).
Source tracking of contaminating viruses requires investigation of a large number of samples. In clinical surveillance studies, for example, detection and genotyping of pathogenic viruses were carried out using more than 100 fecal samples (Román et al., 2003; Kageyama et al., 2004). Furthermore, NoV GII detection and genotyping was performed on 28 subsamples of frozen strawberries to identify the source of a contamination that caused a foodborne outbreak of gastroenteritis in Germany in 2012 (Mäde et al., 2013; Bernard et al., 2014). MFnPCR–MiSeq enables simultaneous detection and genotyping of 11 RNA viruses in up to 48 samples at once, making this a powerful tool for source tracking of human pathogenic RNA viruses. Additionally, as a microfluidic PCR technique has been applied for the detection and quantification of DNA viruses as well as RNA viruses (Ishii et al., 2014), with suitable modifications, our MFnPCR–MiSeq method can also be applied to detect DNA viruses. Moreover, recent advances in DNA sequencing techniques have accelerated the accumulation of viral sequence data, which has led to the development of novel PCR primer sets (Hata et al., 2015). These novel primer sets can be included in MFnPCR–MiSeq, along with the primers tested in the present study. MFnPCR–MiSeq should allow for further expansion of our understanding of the emergence and circulation of human pathogenic RNA viruses, and also DNA viruses, in natural environments, and human populations.
MO, TM, and SK performed the sample collection, MFnPCR–MiSeq analysis, and downstream bioinformatics analyses. All the authors wrote the manuscript, and reviewed and approved the final manuscript.
This work was supported by JSPS KAKENHI grant numbers 15K18141, 16H04442, and 16H02371 to SK, NA, and TY; Program for the Strategic Promotion of International Cooperation to Accelerate Innovation in Developing Countries of the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT); National Institute of Polar Research (NIPR) through General Collaboration Project No. 27-19; and Core Research for Evolutionary Science and Technology (CREST) from the Japan Science and Technology Agency (JST).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Ayumi Akiyoshi (National Institute of Polar Research, Japan) for the technical assistance. We also express our appreciation to Dr. Hiroyuki Katayama (The University of Tokyo, Japan), Dr. Kazuyoshi Yano (Tokyo Metropolitan Institute of Public Health, Japan), Prof. Osamu Nakagomi (Nagasaki University, Japan), and Dr. You Ueki (Miyagi Prefectural Institute of Public Health and Environment, Japan) for kindly providing viruses or viral cDNAs.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00830/full#supplementary-material
Aw, T. G., Gin, K. Y. H., Oon, L. L. E., Chen, E. X., and Woo, C. H. (2009). Prevalence and genotypes of human noroviruses in tropical urban surface waters and clinical samples in Singapore. Appl. Environ. Microbiol. 75, 4984–4992. doi: 10.1128/AEM.00489-09
Batty, E. M., Wong, T. H., Trebes, A., Argoud, K., Attar, M., Buck, D., et al. (2013). A modified RNA-Seq approach for whole genome sequencing of RNA viruses from fecal and blood samples. PLoS One 10:e66129. doi: 10.1371/journal.pone.0066129
Bernard, H., Faber, M., Wilking, H., Haller, S., Höhle, M., Schielke, A., et al. (2014). Large multistate outbreak of norovirus gastroenteritis associated with frozen strawberries, Germany, 2012. Euro Surveil. 19:20719. doi: 10.2807/1560-7917.ES2014.19.8.20719
Bibby, K., and Peccia, J. (2013). Identification of viral pathogen diversity in sewage sludge by metagenome analysis. Environ. Sci. Technol. 47, 1945–1951. doi: 10.1021/es305181x
Bosch, A. (1998). Human enteric viruses in the water environment: a minireview. Int. Microbiol. 1, 191–196.
Cantalupo, P. G., Calgua, B., Zhao, G., Hundesa, A., Wier, A. D., Katz, J. P., et al. (2011). Raw sewage harbors diverse viral populations. mBio 2:e180-11. doi: 10.1128/mBio.00180-11
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F., Costello, E. K., et al. (2010). QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Drexler, J. F., Baumgarte, S., de Souza Luna, L. K., Eschbach-Bludau, M., Lukashev, A. N., and Drosten, C. (2011). Aichi virus shedding in high concentrations in patients with acute diarrhea. Emerg. Infect. Dis. 17, 1544–1548. doi: 10.3201/eid1708.101556
Eden, J. S., Tanaka, M. M., Boni, M. F., Rawlinson, W. D., and White, P. A. (2013). Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 87, 6270–6282. doi: 10.1128/JVI.03464-12
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Fischer, T. K., Page, N. A., Griffin, D. D., Eugen-Olsen, J., Pedersen, A. G., Valentiner-Branth, P., et al. (2003). Characterization of incompletely typed rotavirus strains from Guinea-Bissau: identification of G8 and G9 types and a high frequency of mixed infections. Virology 311, 125–133. doi: 10.1016/S0042-6822(03)00153-3
Harvala, H., Robertson, I., McWilliam Leitch, E. C., Benschop, K., Wolthers, K. C., Templeton, K., et al. (2008). Epidemiology and clinical associations of human parechovirus respiratory infections. J. Clin. Microbiol. 46, 3446–3453. doi: 10.1128/JCM.01207-08
Hasing, M. E., Hazes, B., Lee, B. E., Preiksaitis, J. K., and Pang, X. L. (2016). A next generation sequencing-based method to study the intra-host genetic diversity of norovirus in patients with acute and chronic infection. BMC Genomics 17:480. doi: 10.1186/s12864-016-2831-y
Hata, A., Katayama, H., Kitajima, M., and Furumai, H. (2015). Wastewater analysis indicates that genetically diverse astroviruses, including strains belonging to novel clades MLB and VA, are circulating within Japanese populations. Appl. Environ. Microbiol. 81, 4932–4939. doi: 10.1128/AEM.00563-15
Ishii, S., Kitamura, G., Segawa, T., Kobayashi, A., Miura, T., Sano, D., et al. (2014). Microfluidic quantitative PCR for simultaneous quantification of multiple viruses in environmental water sample. Appl. Environ. Microbiol. 80, 7505–7511. doi: 10.1128/AEM.02578-14
Ishii, S., Segawa, T., and Okabe, S. (2013). Simultaneous quantification of multiple food- and waterborne pathogens by use of microfluidic quantitative PCR. Appl. Environ. Microbiol. 79, 2891–2898. doi: 10.1128/AEM.00205-13
Ison, S. A., Delannoy, S., Bugarel, M., Nagaraja, T. G., Renter, D. G., den Bakker, H. C., et al. (2016). Targeted amplicon sequencing for single-nucleotide-polymorphism genotyping of attaching and effacing Escherichia coli O26:H11 cattle strains via a high-throughput library preparation technique. Appl. Environ. Microbiol. 82, 640–649. doi: 10.1128/AEM.03182-15
Iturriza Gómara, M., Simpson, R., Perault, A. M., Redpath, C., Lorgelly, P., Joshi, D., et al. (2008). Structured surveillance of infantile gastroenteritis in East Anglia, UK: incidence of infection with common viral gastroenteric pathogens. Epidemiol. Infect. 136, 23–33. doi: 10.1017/S0950268807008059
Iwai, M., Hasegawa, S., Obara, M., Nakamura, K., Horimoto, E., Takizawa, T., et al. (2009). Continuous presence of noroviruses and sapoviruses in raw sewage reflects infections among inhabitants of Toyama, Japan (2006 to 2008). Appl. Environ. Microbiol. 75, 1264–1270. doi: 10.1128/AEM.01166-08
Kageyama, T., Shinohara, M., Uchida, K., Fukushi, S., Hoshino, F. B., Kojima, S., et al. (2004). Coexistence of multiple genotypes, including newly identified genotypes, in outbreaks of gastroenteritis due to norovirus in Japan. J. Clin. Microbiol. 42, 2988–2995. doi: 10.1128/JCM.42.7.2988-2995.2004
Kazama, S., Masago, Y., Tohma, K., Souma, N., Imagawa, T., Suzuki, A., et al. (2016). Temporal dynamics of norovirus determined through monitoring of municipal wastewater by pyrosequencing and virological surveillance of gastroenteritis cases. Water Res. 92, 244–253. doi: 10.1016/j.watres.2015.10.024
Kazama, S., Miura, T., Masago, Y., Konta, Y., Tohma, K., Manaka, T., et al. (2017). Environmental surveillance of norovirus genogroups I and II for sensitive detection of epidemic variants. Appl. Environ. Microbiol. 83:e3406-16. doi: 10.1128/AEM.03406-16
Khamrin, P., Okame, M., Thongprachum, A., Nantachit, N., Nishimura, S., Okitsu, S., et al. (2011). A single-tube multiplex PCR for rapid detection in feces of 10 viruses causing diarrhea. J. Virol. Methods 173, 390–393. doi: 10.1016/j.jviromet.2011.02.012
Kitajima, M., Oka, T., Haramoto, E., Takeda, N., Katayama, K., and Katayama, H. (2010). Seasonal distribution and genetic diversity of genogroups I, II, and IV noroviruses in the Tamagawa River, Japan. Environ. Sci. Technol. 44, 7116–7122. doi: 10.1021/es100346a
Kobayashi, N., Oshiki, M., Ito, T., Segawa, T., Hatamoto, M., Kato, T., et al. (2017). Removal of human pathogenic viruses in a down-flow hanging sponge (DHS) reactor treating municipal wastewater and health risks associated with utilization of the effluent for agricultural irrigation. Water Res. 110, 389–398. doi: 10.1016/j.watres.2016.10.054
Kroneman, A., Vega, E., Vennema, H., Vinje, J., White, P. A., Hansman, G., et al. (2013). Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 158, 2059–2068. doi: 10.1007/s00705-013-1708-5
Kroneman, A., Vennema, H., Deforche, K., Avoort, H. V. D., Peñaranda, S., Oberste, M. S., et al. (2011). An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51, 121–125. doi: 10.1016/j.jcv.2011.03.006
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lange, V., Böhme, I., Hofmann, J., Lang, K., Sauter, J., Schöne, B., et al. (2014). Cost-efficient high-throughput HLA typing by MiSeq amplicon sequencing. BMC Genomics 15:63. doi: 10.1186/1471-2164-15-63
Le Guyader, F., Dubois, E., Menard, D., and Pommepuy, M. (1994). Detection of hepatitis A virus, rotavirus, and Enterovirus in naturally contaminated shellfish and sediment by reverse transcription-seminested PCR. Appl. Environ. Microbiol. 60, 3665–3671.
Lee, N., Chan, M. C. W., Wong, B., Choi, K. W., Sin, W., Lui, G., et al. (2007). Feral viral concentration and diarrhoea in norovirus gastroenteritis. Emerg. Inf. Dis. 13, 1399–1401. doi: 10.3201/eid1309.061535
Mäde, D., Trübner, K., Neubert, E., Höhne, M., and Johne, R. (2013). Detection and typing of norovirus from frozen strawberries involved in a large-scale gastroenteritis outbreak in Germany. Food Environ. Virol. 5, 162–168. doi: 10.1007/s12560-013-9118-0
Miura, T., Lhomme, S., Saux, J. C. L., Mehaute, P. L., Guillois, Y., Couturier, E., et al. (2016). Detection of hepatitis E virus in sewage after an outbreak on a French island. Food Environ. Virol. 8, 194–199. doi: 10.1007/s12560-016-9241-9
Miura, T., Okabe, S., Nakahara, Y., and Sano, D. (2015). Removal properties of human enteric viruses in a pilot-scale membrane bioreactor (MBR) process. Water Res. 75, 282–291. doi: 10.1016/j.watres.2015.02.046
Motomura, K., Boonchan, M., Noda, M., Tanaka, T., Takeda, N., and Norovirus Surveillance Group of Japan. (2016). Norovirus epidemics caused by new GII.2 chimera viruses in 2012-2014 in Japan. Infect. Genet. Evol. 42, 49–52. doi: 10.1016/j.meegid.2016.04.026
Nasri, D., Bouslama, L., Omar, S., Saoudin, H., Bourlet, T., Aouni, M., et al. (2007). Typing of human Enterovirus by partial sequencing of VP2. J. Clin. Microbiol. 45, 2370–2379. doi: 10.1128/JCM.00093-07
Nenonen, N. P., Hannoun, C., Horal, P., Hernroth, B., and Bergström, T. (2008). Tracing of norovirus outbreak strains in mussels collected near sewage effluents. Appl. Environ. Microbiol. 74, 2544–2549. doi: 10.1128/AEM.02477-07
Ng, T. F., Marine, R., Wang, C., Simmonds, P., Kapusinszky, B., Bodhidatta, L., et al. (2012). High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J. Virol. 86, 12161–12175. doi: 10.1128/JVI.00869-12
Oshiki, M., Segawa, T., and Ishii, S. (2018). Nitrogen cycle evaluation (NiCE) chip for the simultaneous analysis of multiple N-cycle associated genes. Appl. Environ. Microbiol. doi: 10.1128/AEM.02615-17 [Epub ahead of print].
Pham, N. T. K., Trinh, Q. D., Khamrin, P., Maneekarn, N., Shimizu, H., Okitsu, S., et al. (2010). Diversity of human parechoviruses isolated from stool samples collected from Thai children with acute gastroenteritis. J. Clin. Microbiol. 48, 115–119. doi: 10.1128/JCM.01015-09
Pu, J., Kazama, S., Miura, T., Azraini, N. D., Konta, Y., Ito, H., et al. (2016). Pyrosequencing analysis of norovirus genogroup II distribution in sewage and oysters: first detection of gii.17 kawasaki 2014 in oysters. Food Environ. Virol. 8, 310–312. doi: 10.1007/s12560-016-9261-5
Rohayem, J., Berger, S., Juretzek, T., Herchenröder, O., Mogel, M., Poppe, M., et al. (2004). A simple and rapid single-step multiplex RT-PCR to detect Norovirus, Astrovirus and Adenovirus in clinical stool samples. J. Virol. Methods 118, 49–59. doi: 10.1016/j.jviromet.2004.01.016
Román, E., Wilhelmi, I., Colomina, J., Villar, J., Cilleruelo, M. L., Nebreda, V., et al. (2003). Acute viral gastroenteritis: proportion and clinical relevance of multiple infections in Spanish children. J. Med. Microbiol. 52, 435–440. doi: 10.1099/jmm.0.05079-0
Rosario, K., Nilsson, C., Lim, Y. W., Ruan, Y., and Breitbart, M. (2009). Metagenomic analysis of viruses in reclaimed water. Environ. Microbiol. 11, 2806–2820. doi: 10.1111/j.1462-2920.2009.01964.x
Sano, D., Pérez-Sautu, U., Guix, S., Pintó, R. M., Miura, T., Okabe, S., et al. (2011). Quantification and genotyping of human sapoviruses in the Llobregat river catchment, Spain. Appl. Environ. Microbiol. 77, 1111–1114. doi: 10.1128/AEM.01721-10
Sinclair, R. G., Jones, E. L., and Gerba, C. P. (2009). Viruses in recreational water-borne disease outbreaks: a review. J. Appl. Microbiol. 107, 1769–1780. doi: 10.1111/j.1365-2672.2009.04367.x
Stene-Johansen, K., Tjon, G., Schreier, E., Bremer, V., Bruisten, S., Ngui, S. L., et al. (2007). Molecular epidemiological studies show that hepatitis A virus is endemic among active homosexual men in Europe. J. Med. Virol. 79, 356–365. doi: 10.1002/jmv.20781
Svraka, S., Vennema, H., van der Veer, B., Hedlund, K. O., Thorhagen, M., Siebenga, J., et al. (2010). Epidemiology and genotype analysis of emerging sapovirus-associated infections across Europe. J. Clin. Microbiol. 48, 2191–2198. doi: 10.1128/JCM.02427-09
Tamaki, H., Zhang, R., Angly, F. E., Nakamura, S., Hong, P. Y., Yasunaga, T., et al. (2012). Metagenomic analysis of DNA viruses in a wastewater treatment plant in tropical climate. Environ. Microbiol. 14, 441–452. doi: 10.1111/j.1462-2920.2011.02630.x
Ueki, Y., Sano, D., Watanabe, T., Akiyama, K., and Omura, T. (2005). Norovirus pathway in water environment estimated by genetic analysis of strains from patients of gastroenteritis, sewage, treated wastewater, river water and oysters. Water Res. 39, 4271–4280. doi: 10.1016/j.watres.2005.06.035
Vinjé, J. (2015). Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 53, 373–381. doi: 10.1128/JCM.01535-14
Yan, H., Yagyu, F., Okitsu, S., Nishio, O., and Ushijima, H. (2003). Detection of norovirus (GI, GII), Sapovirus and astrovirus in fecal samples using reverse transcription single-round multiplex PCR. J. Virol. Methods 114, 37–44. doi: 10.1016/j.jviromet.2003.08.009
Keywords: human pathogenic viruses, high-throughput detection and genotyping, microfluidic PCR, MiSeq sequencing, sewage, human feces, oyster
Citation: Oshiki M, Miura T, Kazama S, Segawa T, Ishii S, Hatamoto M, Yamaguchi T, Kubota K, Iguchi A, Tagawa T, Okubo T, Uemura S, Harada H, Kobayashi N, Araki N and Sano D (2018) Microfluidic PCR Amplification and MiSeq Amplicon Sequencing Techniques for High-Throughput Detection and Genotyping of Human Pathogenic RNA Viruses in Human Feces, Sewage, and Oysters. Front. Microbiol. 9:830. doi: 10.3389/fmicb.2018.00830
Received: 04 January 2018; Accepted: 11 April 2018;
Published: 27 April 2018.
Edited by:
Heiko Ziebell, Julius Kühn-Institut, GermanyReviewed by:
Hirokazu Kimura, Gumma Paz College, JapanCopyright © 2018 Oshiki, Miura, Kazama, Segawa, Ishii, Hatamoto, Yamaguchi, Kubota, Iguchi, Tagawa, Okubo, Uemura, Harada, Kobayashi, Araki and Sano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mamoru Oshiki, b3NoaWtpQG5hZ2Fva2EtY3QuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.