 Roey Angel†
Roey Angel† Maximilian Nepel
Maximilian Nepel Christopher Panhölzl
Christopher Panhölzl Hannes Schmidt
Hannes Schmidt Craig W. Herbold
Craig W. Herbold Stephanie A. Eichorst
Stephanie A. Eichorst Dagmar Woebken*
Dagmar Woebken*- Division of Microbial Ecology, Department of Microbiology and Ecosystem Science, Research Network ‘Chemistry meets Microbiology’, University of Vienna, Vienna, Austria
Diazotrophic microorganisms introduce biologically available nitrogen (N) to the global N cycle through the activity of the nitrogenase enzyme. The genetically conserved dinitrogenase reductase (nifH) gene is phylogenetically distributed across four clusters (I–IV) and is widely used as a marker gene for N2 fixation, permitting investigators to study the genetic diversity of diazotrophs in nature and target potential participants in N2 fixation. To date there have been limited, standardized pipelines for analyzing the nifH functional gene, which is in stark contrast to the 16S rRNA gene. Here we present a bioinformatics pipeline for processing nifH amplicon datasets – NifMAP (“NifH MiSeq Illumina Amplicon Analysis Pipeline”), which as a novel aspect uses Hidden-Markov Models to filter out homologous genes to nifH. By using this pipeline, we evaluated the broadly inclusive primer pairs (Ueda19F–R6, IGK3–DVV, and F2–R6) that target the nifH gene. To evaluate any systematic biases, the nifH gene was amplified with the aforementioned primer pairs in a diverse collection of environmental samples (soils, rhizosphere and roots samples, biological soil crusts and estuarine samples), in addition to a nifH mock community consisting of six phylogenetically diverse members. We noted that all primer pairs co-amplified nifH homologs to varying degrees; up to 90% of the amplicons were nifH homologs with IGK3–DVV in some samples (rhizosphere and roots from tall oat-grass). In regards to specificity, we observed some degree of bias across the primer pairs. For example, primer pair F2–R6 discriminated against cyanobacteria (amongst others), yet captured many sequences from subclusters IIIE and IIIL-N. These aforementioned subclusters were largely missing by the primer pair IGK3–DVV, which also tended to discriminate against Alphaproteobacteria, but amplified sequences within clusters IIIC (affiliated with Clostridia) and clusters IVB and IVC. Primer pair Ueda19F–R6 exhibited the least bias and successfully captured diazotrophs in cluster I and subclusters IIIE, IIIL, IIIM, and IIIN, but tended to discriminate against Firmicutes and subcluster IIIC. Taken together, our newly established bioinformatics pipeline, NifMAP, along with our systematic evaluations of nifH primer pairs permit more robust, high-throughput investigations of diazotrophs in diverse environments.
Introduction
Nitrogen gas (N2) fixing microorganisms (diazotrophs) are one of the most ecologically important functional guilds on Earth, providing the primary natural source for nitrogen to ecosystems through biological N2 fixation (BNF; Fowler et al., 2013). Isotopic evidence suggests that BNF has emerged as early as ca. 3.2 Gyr ago (Stüeken et al., 2015). It is thought to have evolved in an anaerobic archaeon and was later transferred to an aerobic bacterium (Boyd et al., 2015). Considering the time scale of the BNF evolution and the importance of N2 fixation, it is not surprising that the genetic potential to perform N2 fixation (i.e., the nif genes) is found widely among different branches in the phylogenetic trees of archaea and bacteria. It is estimated that 6–15% of all sequenced microbial genomes harbor the minimum number of nif genes to provide them with the genetic capacity to fix N2 (Dos Santos et al., 2012; Boyd et al., 2015), making diazotrophs one of the most diverse functional guilds.
Among all known diazotrophs, N2 fixation is mediated by the nitrogenase enzyme complex along with ca. 20 functional and regulatory genes that are organized in several operons termed together the Nif regulon (Rubio and Ludden, 2002). Of those genes, the nifH gene encoding for the dinitrogenase reductase is considered one of the most genetically conserved genes in the regulon and has been traditionally used as a marker gene for studying the genetic diversity of diazotrophs in nature (Zehr et al., 2003; Gaby and Buckley, 2011). In addition to the canonical MoFe nitrogenase that is shared among all known diazotrophs, some diazotrophs possess in their genome one of the two alternative versions of nitrogenase, which use either VFe (Vnf) or FeFe (Anf) as metal cofactors (Raymond et al., 2004).
Based on phylogenetic analysis, the nifH genes typically form four clusters, termed clusters I–IV (Zehr et al., 2003; Raymond et al., 2004). Most nifH sequences fall into cluster I, which is composed almost entirely of sequences from the canonical MoFe nitrogenase. This cluster typically consists of sequences from Cyanobacteria, Frankia, Proteobacteria, and some are affiliated with Clostridia, Bacilli, and Nitrospirae. In addition, cluster I contains some sequences of the alternative nitrogenase vnfH. Cluster II comprises nearly all sequences of the alternative vnfH nitrogenase as well as all the known sequences of the second alternative nitrogenase, anfH. In addition, the nifH genes of some Archaea also fall into cluster II. Cluster III consists mostly of nifH sequences of anaerobic bacteria and archaea such as methanogens, spirochetes, sulfate reducers, non-sulfur purple bacteria, green sulfur bacteria, acetogens and Clostridia. Cluster IV was considered until recently to only contain “uncharacterized” or “non-functional” nifH sequences, but in 2015 the first isolate of a diazotroph containing an active nitrogenase belonging to cluster IV was cultivated from a termite gut (Zheng et al., 2016). While it has been shown in several pure cultures that most of the known cluster IV nitrogenases do not encode for a protein involved in N2 fixation (e.g., Staples et al., 2007), it is certainly possible that some of the environmental sequences in this cluster do encode for active nitrogenases.
The dinitrogenase reductase gene is found in nearly every environment studied so far (Gaby and Buckley, 2011), and environmental genetic surveys of the nifH gene have produced extensive datasets encompassing several tens of thousands of unique sequences (Gaby and Buckley, 2014; Heller et al., 2014), and novel sequences continue to be discovered. Such PCR-based surveys continue to serve as an important tool for studying the diazotroph diversity and, if the nifH transcripts are targeted (via cDNA), can also reveal transcription patterns in the environment. Yet, the choice of primer pair can have a significant impact on the extent of diversity that is uncovered. Since the first degenerate primers were used to amplify environmental DNA (Kirshtein et al., 1991), several dozens of primer pairs targeting the nifH gene have been designed to serve as group-specific or general nifH primers. In 2012, Gaby and Buckley published an extensive review of all known primers for nifH, and evaluated their performance in silico using a comprehensive database of nifH sequences (Gaby and Buckley, 2012). Through their analysis, several primer pairs were identified for their potential to capture the largest diversity of nifH sequences and were tested for their ability to produce a PCR product from DNA from several diazotrophic strains and two soil samples (Gaby and Buckley, 2012). However, the extent to which these primers are able to capture nifH diversity in environmental samples and their potential preferences of amplification have not been tested. Furthermore, the majority of the nifH sequences in public databases have sequence data between positions 100 and 500 bases (positions are relative to Azotobacter vinelandii), making it challenging to perform in silico coverage estimates of primer pairs flanking this region.
Given the importance of exploring the diazotrophic communities in the environment and the need for bioinformatics pipelines for analysis of the nifH gene, we developed a bioinformatics pipeline for processing nifH amplicon datasets derived from the MiSeq Illumina sequencing platform – NifMAP (“NifH MiSeq Illumina Amplicon Analysis Pipeline”). Using this pipeline, our further goal of this study was to evaluate the performance of general primer sets – selected based on their high in silico coverage for amplifying nifH – via high-throughput sequencing of a mock community and across a diverse collection of environmental samples. We discuss the performance of the tested primer pairs and provide a standard operating procedure for analyzing nifH genes using high-throughput sequencing.
Materials and Methods
Primers Used in This Study
Four forward primers: Nh21F, Ueda19F, F2, and IGK3 and four reverse primers: nifH1, nifH3, R6, and DVV were chosen following the in silico analysis of Gaby and Buckley (2012) (Table 1). The forward primers Nh21F, Ueda19F, and F2 and the reverse primers nifH1, nifH3, and R6 were tested in all nine combinations. In addition, the primer pair IGK3–DVV was tested, as it was suggested as the best performing primer pair in Gaby and Buckley (2012).
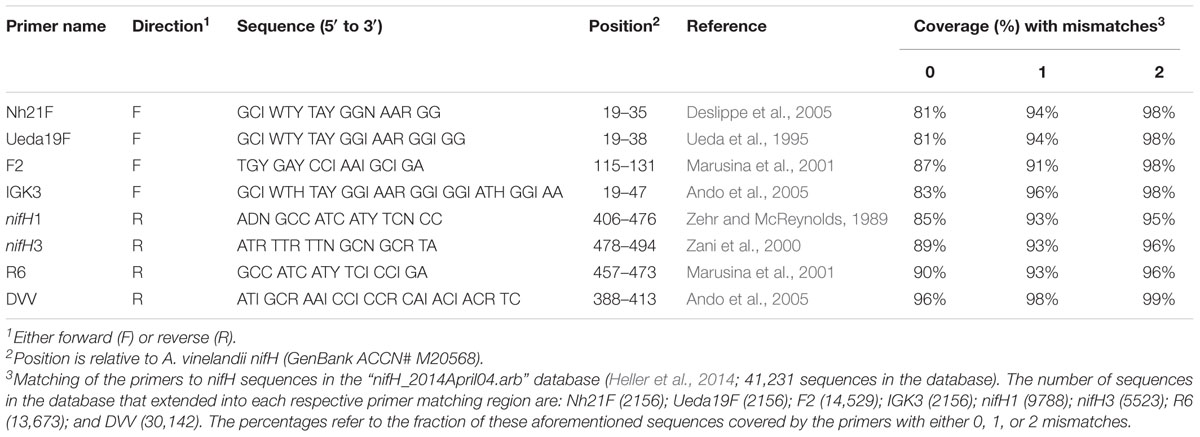
TABLE 1. Summary of the nifH primers used in this study.
Samples Used in This Study
Thirteen different samples were used in this study: soil samples from (a) an Austrian beech forest (Rasche et al., 2011) and (b) an Austrian meadow (Angeringer and Karrer, 2008); (c) rhizosphere and (d) root of Arrhenatherum elatius (tall oat-grass) from an Austrian grassland site (Pötsch et al., 2013); (e) rhizosphere and (f) root of Oryza sativa (wetland rice, grown in the greenhouse on paddy soil from Vercelli, Italy); biological soil crusts (BSCs) from (g) a coastal, sub-arctic crust, Sweden, (h) a temperate crust, Germany, (i) a high Alpine crust, Austria, (j) a semiarid crust, Spain (Büdel et al., 2014) and (k) an arid crust, Israel (Angel and Conrad, 2009); and two estuarine samples from (l) the Great Belt that is separating the North Sea and the Baltic Sea and (m) the Roskilde Fjord, Denmark (Bentzon-Tilia et al., 2015).
DNA Extraction
With the exception of the water samples, DNA was extracted from 0.4 g of soil (or 0.2–0.4 g root and rhizosphere samples) using modification of a standard bead-beating protocol in the presence of a CTAB buffer and phenol, according to a previously published phenol/chloroform-based extraction protocol (Angel, 2012). Following extraction, samples were purified using OneStepTM PCR Inhibitor Removal Kit (Zymo, Irvine, CA, United States). DNA from the water samples were also obtained with a phenol/chloroform-based protocol, as described in Boström et al. (2004). DNA was extracted from biological replicates: the Austrian beech forest (n = 6); Austrian meadow soil (n = 6); BSCs (n = 2/type); root and rhizosphere samples (n = 3/type); and estuarine samples (n = 2).
PCR Amplification and Sequencing
For an initial pre-screening of the different nifH-primer combinations, DNA from the beech forest and meadow soils were used for PCR amplification of the nifH gene fragment, and the PCR products were evaluated using agarose gel electrophoresis. Amplifications were performed in 25 μl volume using the following mixture: 2.5 μl of 10× DreamTaq Green Buffer, 2 mM MgCl2, 0.2 mM of each nucleotide dNTP mixture, 0.08 μg μl-1 of BSA, 0.625 U of DreamTaq Green DNA Polymerase (all from Thermo Fisher Scientific, Waltham, MA, United States) and 0.8 μM of each primer (Thermo Fischer Scientific, Waltham, MA, United States) and 1 μl of DNA template. The primers were designed to include a universal 16 bp head sequence at their 5′ end for subsequent barcoding (Herbold et al., 2015). The following program was used for amplification: 94°C for 5 min followed by 35 cycles of 94°C for 30 s, 52°C for 45 s, and 72°C for 30 s, and a single step of final elongation at 72°C for 10 min. Sequencing of amplified nifH genes was done using multiplexed barcoded amplicon sequencing on an Illumina MiSeq platform (Illumina, San Diego, CA, United States), as described previously (Herbold et al., 2015). First-step PCR amplifications were performed in triplicates of 25 μl each, using the mixture and conditions described above, except that PCRs were done in 25 cycles. Following PCR amplification, samples were purified using ZR-96 DNA Clean-up KitTM (Zymo, Irvine, CA, United States) and 3 μl from the purified sample was used for a second PCR reaction, which was 50 μl in volume and contained the following mixture: 5 μl of 10× DreamTaq Green Buffer, 2 mM MgCl2, 0.2 mM of each nucleotide dNTP mixture, 0.08 μg μl-1 of BSA, 1.25 U of DreamTaq Green DNA Polymerase (all from Thermo Fisher Scientific, Waltham, MA, United States) and 0.4 μM of a barcode primer, which also contained a universal 16 bp head sequence at the 5′ end (Herbold et al., 2015). The following program was used for amplification: 94°C for 5 min followed by 10 cycles of 94°C for 30 s, 52°C for 45 s, and 72°C for 45 s, and a single step of final elongation at 72°C for 10 min. Following this barcoding PCR step, the samples were purified using ZR-96 DNA Clean-up KitTM (Zymo, Irvine, CA, United States), quantified using Quant-iTTM PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States) on a Tecan Safire plate reader (Tecan, Männedorf, Switzerland) and pooled in equimolar amounts of 20 × 109 copies per sample. Library preparation and sequencing services were provided by Microsynth (Balgach, Switzerland). The library was prepared by adaptor ligation and PCR using the TruSeq Nano DNA Library Prep Kit (Illumina, Cat FC-121-4001) according to the TruSeq Nano protocol (Illumina, FC-121-4003), but excluding the fragmentation step. Sequencing was performed on a MiSeq platform (Illumina, San Diego, CA, United States). The MiSeq was run in the 2 × 300 cycle configuration using the MiSeq Reagent kit v3 (Illumina, San Diego, CA, United States). The raw sequence data were deposited into the NCBI Short Read Archive under BioProject accession number PRJNA4 32667.
Construction of Hidden-Markov Models for Filtering Non-nifH Reads and for Aligning nifH Sequences
Non-nifH reads were filtered out using four Hidden-Markov Models (HMMs). One model (hmm_nuc_1160_nifH.hmm) was based on 1160 nucleotide sequences from the nifH_2014April04.arb database (Heller et al., 2014), with sequence data matching the amplification region of Ueda19F–R6 primer pair (the longest amplicon in our test). Three other models (bchX.hmm, chlL-bchL.hmm, and Zehr_2014_1812genomes_nifH_AA.hmm) were based on amino acid sequences. The model Zehr_2014_1812genomes_nifH_AA.hmm was based on 1812 aligned nifH amino acid sequences from sequenced genomes, obtained from the nifH_2014April04.arb database. The model bchX.hmm included 79 amino acid sequences from the NCBI Entrez Protein Cluster chlorophyllide reductase iron protein subunit X (bchX, PCLA_924109; also useful in discriminating against the chromosome partitioning protein (parA) sequences), and chlL-bchL.hmm included 250 amino acid sequences from two light-independent protochlorophyllide reductase iron-sulfur ATP-binding proteins (chlL, CHL00072 and bchL, PCLA_858313, PCLA_3385405). Because bchX and chlL-bchL sequences tend to be more similar to nifH sequences than to each other, an attempt to generate a single HMM covering all three homologs but excluding nifH was unsuccessful. The sequences for the HMMs hmm_nuc_1160_nifH.hmm, bchX.hmm, and chlL-bchL.hmm were aligned using MAFFT (V7.271; Katoh and Standley, 2013), employing the L-INS-i algorithm, and then individual HMMs were built from each multiple sequence alignment using the hmmbuild in HMMER V3.1 (Mistry et al., 2013). For convenience, a single HMM database (nifH_chlL_bchX.hmm) was generated from the three amino-acid based HMMs using command hmmpress in HMMER.
NifMAP – An Automated Pipeline for Analyzing nifH Amplicon Reads
To process the raw MiSeq amplicon reads, we devised the following pipeline (Figure 1): (1) Paired raw MiSeq reads were assembled into contigs using QIIME’s join_paired_ends.py (Caporaso et al., 2012). (2) The merged contigs were filtered against the nucleotide-based HMM (hmm_nuc_1160_nifH.hmm) using hmmsearch command in HMMER. All reads passing through this filtering step were accepted for the next step, irrespective of the model match score. (3) Sequences were chimera-checked and clustered using the UPARSE pipeline (Edgar, 2013). First, contigs were dereplicated with the -derep_fullength command and singleton unique sequences were removed. OTU centroids were then determined with the -cluster_otus command (using 3% radius). Abundances of OTUs were determined by mapping the filtered contigs (prior to dereplication) to OTU centroids using the -usearch_global command (at a 0.97% identity, hereafter referred to as OTU97). (4) Translation into amino acids and (potential) frameshift corrections were done using FrameBot (Wang et al., 2013) against the nifH protein reference set. (5) Homologous genes to the nifH gene (bchX, chlL, bchL, and parA) were filtered out against the HMM nifH_ChlL_bchX.hmm (see above) using the hmmscan command in HMMER. Only OTU representatives that scored highest against the nifH model compared with the bchX and chlL-bchL models were retained. (6) OTU classification and phylogenetic placement: the remaining OTU representatives were classified using BLASTP (Camacho et al., 2009) against the RefSeq database (Pruitt et al., 2005). In addition, OTU representatives were aligned using hmmalign to the HMM Zehr_2014_1812genomes_nifH_AA.hmm and assigned to phylogenetic clusters of nifH using Classification And Regression Trees (CART; Frank et al., 2016). OTU representatives were also placed on a base tree using the Evolutionary Placement Algorithm (EPA) implementation in RAxML (Stamatakis, 2014). The base tree was generated as follows: (1) All entries containing amino acid sequence information for the nifH gene in the nifH_2014April04.arb database were extracted. These included 1971 entries from genomes and 39,258 entries from non-genome sequencing sources. (2) All sequences shorter than 133 AA were filtered out and the remaining sequences were dereplicated. (3) The remaining sequences were clustered at a clustering threshold of 90% identity using CD-HIT (Fu et al., 2012). In addition, the sequences used for constructing the bchX and chlL-bchL HMMs were clustered at a threshold of 80% identity using CD-HIT and merged with the clustered nifH sequences. The combined dataset was then aligned using MAFFT L-INS-i against the aligned collection of 1812 amino acid sequences of genomic origin used for constructing the HMM Zehr_2014_1812genomes_nifH_AA.hmm, in order to maintain an alignment compatible with the nifH_2014April04.arb database. Lastly, a bootstrapped maximum likelihood tree based on a CAT model was reconstructed using RAxML. For placing the OTU representatives on the base tree, the sequences were aligned using MAFFT against the alignment used for constructing the base tree and then added to the base tree using RAxML, employing the EPA. Steps 1 and 3 of the pipeline are identical to the procedures described in Herbold et al. (2015), while steps 2, 4, 5, and 6 are new for this work. The HMMs, base tree and shell script for reproducing steps 2, 4, 5, and 6 are publicly available at https://github.com/roey-angel/NifMAP.
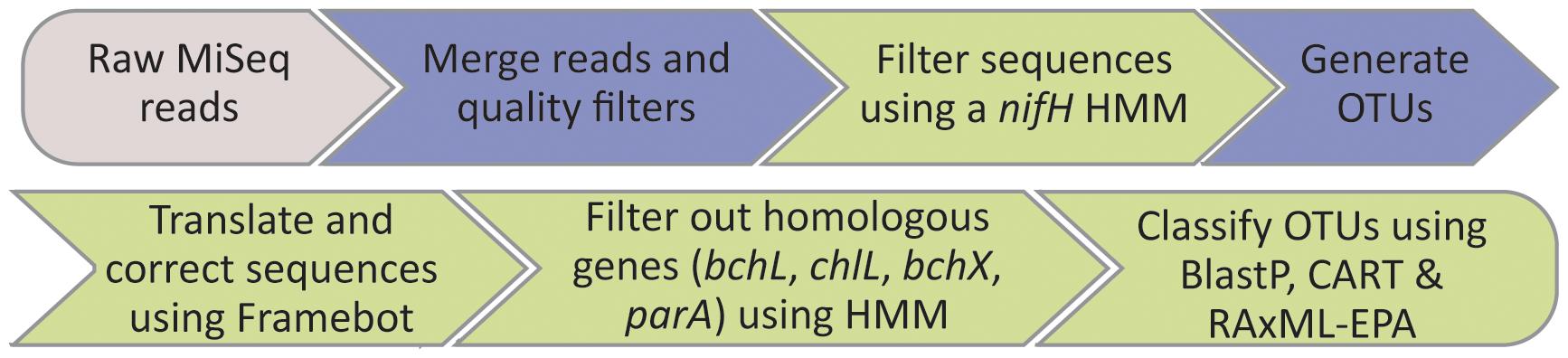
FIGURE 1. Schematic overview of NifMAP – NifH MiSeq Illumina Amplicon Analysis Pipeline. Pipeline begins with raw MiSeq reads from desired sequencing facility (depicted in gray). Steps shaded in blue represent standard processing steps, while steps shaded in green represent steps specific for processing nifH sequences, introduced in this work.
Design and Testing of a nifH Mock Community
A nifH mock community was developed to estimate variations in sequencing quality amongst runs and cross-contamination within a run (Bokulich et al., 2016). This mock community was comprised of DNA from the following six diazotrophic species: Anabaena torulosa (Carm.) Lagerh. (Cyanobacteria) Algae Collection Vienna (ASW 01028), Desulfosporosinus acidophilus strain SJ4T (DSM22704) (Firmicutes) (Alazard et al., 2010), Kosakonia sacchari (Gammaproteobacteria) (in house strain, 16S rRNA gene sequence 99.89% identical (1105 bp) to the 16S rRNA gene sequence of K. sacchari SP1T (Chen et al., 2014), Mesorhizobium loti strain R7A (Alphaproteobacteria) (Kelly et al., 2014), Nostoc microscopicum Vaucher (Cyanobacteria) Algae Collection Vienna (ASW 01020), and Telmatospirillum siberiense 26-4b1T (Alphaproteobacteria) (Hausmann et al., 2018). DNA was extracted using Qiagen DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions except that 200 μl of culture was first transferred to Lysing Matrix A tube and cells were disrupted using bead beating (30 s at 4 m s-1; FastPrep-24, MP Biomedicals, Santa Ana, CA, United States) after adding PBS and AL buffers. DNA was used for PCR amplification of the nifH gene using primers Ueda19F–R6 as described above. PCR products were cloned using TOPO TA Cloning Kit (Thermo Fisher Scientific, Waltham, MA, United States). One clone containing nifH gene fragment from each strain was used for colony PCR amplification using primers M13, flanking the insert region (Thermo Fisher Scientific, Waltham, MA, United States). PCR was performed with the following mixture: 5 μl of 10× DreamTaq Green Buffer, 2 mM MgCl2, 0.2 mM of each nucleotide dNTP mixture, 0.05 μg μl-1 of BSA, 1.25 U of DreamTaq Green DNA Polymerase (all from Thermo Fisher Scientific, Waltham, MA, United States) and 1 μM of each primer and the following conditions for amplification: 94°C for 5 min followed by 30 cycles of 94°C for 60 s, 55°C for 60 s, and 72°C for 60 s, and a single step of final elongation at 72°C for 10 min. Following amplification and purification with ZR-96 DNA Clean-up KitTM (Zymo, Irvine, CA, United States), PCR products were quantified using Quant-iTTM PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States) on a Tecan Safire plate reader (Tecan, Männedorf, Switzerland) and pooled in equal and also tiered proportions. These mixtures were used as PCR templates for MiSeq sequencing. Each mock community mixture was amplified and sequenced in triplicate using primers Ueda19F–R6, IGK3–DVV, and F2–R6 as described above. The information for generating the mock community can be found at the Mockrobiota repository1 under mock-27 and mock-28.
Richness and Diversity Estimates
Richness was estimated using number of observed OTUs, while diversity was estimated using the inverse Simpson metric:
where R represents the number of OTUs in a sample and Pi is the proportional abundance of each OTU. For each sample, both richness and diversity were estimated using a bootstrapped method by iterative subsampling (1000 iterations) to minimum read-depth (after dropping the lowest 15th percentile samples).
Quantitative PCR Assays
Quantitative PCR (qPCR) reactions were performed on a C1000 Touch thermocycler equipped with a CFX96 Real Time System and the data were processed using CFX Manager software (all from Bio-Rad, Hercules, CA, United States). For all reaction plates, serially diluted standards containing known quantities of DNA copies of the target gene (ranging between 4.2 × 101 and 4.2 × 107) were used for establishing quantitative calibration curves. The standard was generated using a cloned fragment of the nifH gene from Didymococcus colitermitum TAV2 (ATCC BAA-2264; Wertz et al., 2012). A SYBR® Green I-based assay for nifH was established as follows: each reaction was 20 μl in volume and contained 10 μl of SYBR Green JumpStart Taq ReadyMix (Bio-Rad, Hercules, CA, United States), 3 mM MgCl2, 0.4 ng μl-1 BSA (Thermo Fisher Scientific, Waltham, MA, United States), 1.4 μM of each primer, and 2 μl of template. The program used was: 95°C for 5 min, followed by 45 cycles of 95°C for 30 s, 52°C for 45 s for annealing, 72°C for 30 s for extension, and 78°C for 10 s for signal acquisition. The reliability of quantification was evaluated using a melting curve from 55 to 95°C. A correction for the nifH copy numbers was done for each sample separately by excluding the fraction of non-nifH sequences (i.e., the proportion of reads that were detected as non-nifH by the pipeline) from the initial values obtained from qPCR. Data were evaluated using ANOVA on log-transformed data.
Results and Discussion
NifMAP – NifH MiSeq Illumina Amplicon Analysis Pipeline
As the majority of amplicon based diversity surveys employ the 16S rRNA gene as a phylogenetic marker, there are standardized analysis pipelines and procedures (e.g., Caporaso et al., 2012; Edgar, 2013; Kozich et al., 2013). This is in stark contrast to amplicon-based surveys targeting functional genes, such as nifH. Some of the challenges limiting the development of standardized pipelines for functional genes include co-amplification of non-target genes due to gene homology (Holmes et al., 1995); amino acid-based analyses making sequencing error (generating insertion and deletion errors) more detrimental; and lack of classification methods or databases. Furthermore as every functional gene is different, the methods oftentimes need to be adjusted in a gene-specific manner (Penton et al., 2013). Several methods have been proposed in the literature to tackle issues such as alignment and filtering (Fish et al., 2013), correct conversion to amino acids (Wang et al., 2013) and classification (Dumont et al., 2014; Frank et al., 2016), yet complete pipelines for analyzing environmental functional genes are still lacking. Recently, Gaby et al. (2017) published a perl-based pipeline for OTU clustering and inference of taxonomic affiliation through BLAST of nifH amplicon data; however, this pipeline did not employ the use of HMMs to filter out homologous genes.
We developed a bioinformatics pipeline for processing nifH reads derived from the MiSeq Illumina sequencing platform called “NifH MiSeq Illumina Amplicon Analysis Pipeline” – NifMAP, which uses HMMs to filter out homologous genes and taxonomically classifies sequences using three different approaches. In the first step, the sequences are filtered against nucleic acids based HMMs to remove non-target, non-homologous sequences. Subsequently, sequences are clustered based on 97% identity to the centroid sequence, translated into amino acids, and corrected for possible frame shifts. The OTU representatives are screened against specific HMMs in order to filter out nifH-homolog sequences (namely bchX, chlL, bchL, or parA). The use of HMMs ensures a highly specific and sensitive method to detect and remove non-target sequences that is orders of magnitude faster than filtering based on taxonomic or phylogenetic affiliation. Finally, the remaining sequences are classified taxonomically to their closest relatives and their phylogenetic cluster (Figure 1). We chose a combined classification approach using several independent methods, providing information on both the closest taxonomic relative of the queried sequence in addition to its placement in a phylogenetic cluster, because the two classification approaches might occasionally disagree (Frank et al., 2016). Our pipeline was evaluated with a nifH mock community and a diverse collection of environmental samples (including roots and rhizosphere samples, BSCs and estuarine water samples).
Evaluating Coverage of nifH Primer Pair Combinations
Across numerous environments, the distribution of the nifH gene has been evaluated using various primer pair combinations (e.g., Ueda et al., 1995; Farnelid et al., 2011; Collavino et al., 2014). Although there have been some in silico investigations evaluating the performance of these primer pairs along with suggestions as to the best combination(s) (Gaby and Buckley, 2012), these primer pairs have yet to be thoroughly evaluated and tested in the wet-lab, especially when considering samples from highly diverse environments such as soils. To that end, we evaluated the performance of 10 different nifH primer combinations. The primer pairs consisted of all possible combinations of three forward primers (Nh21F, Ueda19F, and F2) and three reverse primers (nifH1, nifH3, and R6) (Table 1) as described previously (Gaby and Buckley, 2012), along with the combination IGK3 and DVV. These primers were chosen due to their presumed high coverage (Table 1) and ability to generate amplicons in lengths that are suitable for MiSeq sequencing (200–500 bp). Extracted DNAs from beech forest and meadow soil were initially used in a pilot study to evaluate these combinations. The initial criterion was generation of a correctly sized PCR fragment. Only three primer pairs produced PCR fragments of the correct size: F2–R6 (358 bp), Ueda19F–R6 (394 bp), and IGK3–DVV (454 bp) without unspecific bands (data not shown). Primer pairs F2–R6 and IGK3–DVV have been used in the past for analyzing diazotrophic communities (Gaby and Buckley, 2012), while this was the first time that the Ueda19F–R6 combination was used.
Assessing nifH Primer Pair Coverage Using a Mock Community
In order to assess potential primer biases of these three primer combinations, their performance was tested on constructed nifH mock communities containing six phylogenetically diverse members. Mock communities, i.e., a defined mixture of known microbial strains, have become a standard tool for benchmarking different aspects of high-throughput sequencing techniques, for example, base calling error, variation between indecent runs and cross contamination (Schloss et al., 2011; Bokulich et al., 2016; Singer et al., 2016). In addition, mock communities can be used to assess amplification bias of different primers. Until now nearly all established mock communities were designed for 16S rRNA gene surveys, while there is a growing need to develop mock communities for other (functional) genes.
Two mock communities were generated using six clones containing nifH gene fragments of diazotrophic cultures of even and tiered proportions (Table 2). Prior to pooling the clones, each insert was confirmed to perfectly match the used primers based on Sanger sequencing (data not shown). The nifH genes in each mock community were amplified and sequenced in triplicate using all three primers pairs, in order to assess the ability of the primers to reconstruct the diazotrophic community structure. Across all three primer pairs, sequencing produced 664–7883 reads per sample after quality filtering, which were classified into 6 (primers Ueda19F–R6, and F2–R6) and 7 (IGK3–DVV) OTUs. OTUs 1–6 were confirmed to match the mock community members (Table 2), while OTU 7 was a contaminant identified as a Geobacter sp. and comprised of a single read in two mock community replicates amplified using the primer pair IGK3–DVV (data not shown). Sequencing exhibited a high degree of reproducibility among replicates as indicated by the low standard errors (Table 2), but showed deviations from the expected read distribution in the even and tiered mock communities. Using primers F2–R6, over 90% of the reads were affiliated to M. loti and about 6% were affiliated to K. sacchari, while other members of the mock community (including two cyanobacteria) were nearly not detected. This is despite perfect in silico primer matching to the nifH sequences of the mock community members.
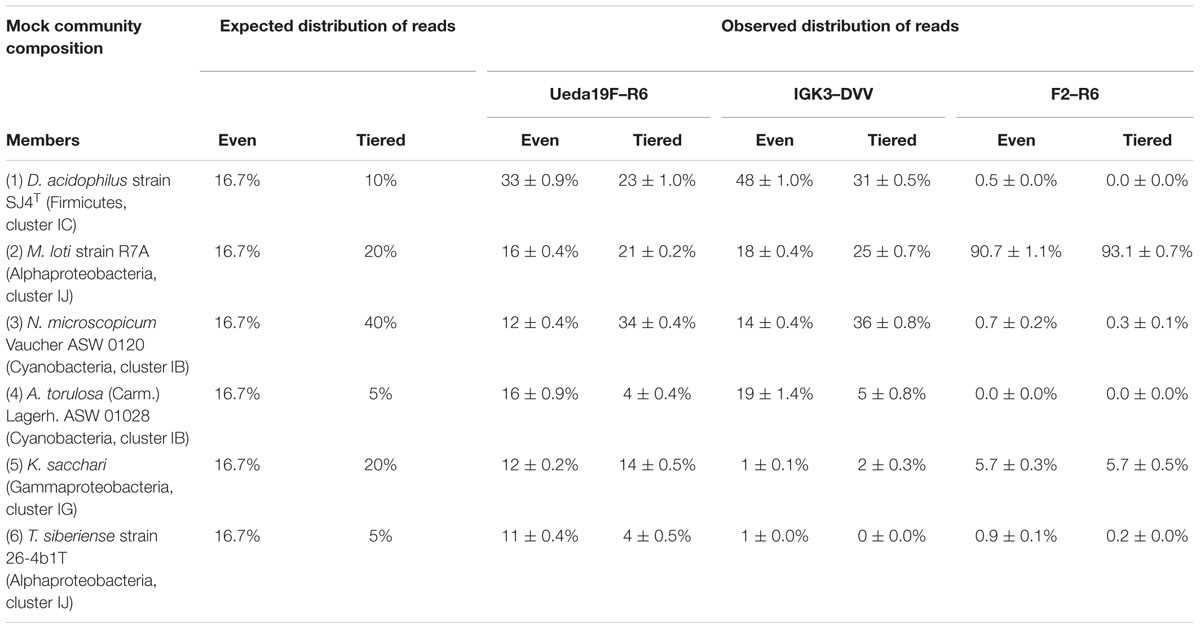
TABLE 2. Relative proportion of the expected and observed reads for an even and tiered nifH gene mock community for the different primer pairs.
In contrast, all mock community members could be recovered using primers Ueda19F–R6 and IGK3–DVV. The bias from the expected distribution (even and tiered) was greater in the IGK3–DVV primers compared to the Ueda19F–R6 primers (Table 2). Specifically, D. acidophilus was consistently overrepresented in both the even and tiered mock communities for both primer pairs, while K. sacchari and T. siberiense were underrepresented. D. acidophilus, K. sacchari, and T. siberiense were particularly biased against using the primer pair IGK3–DVV. Biases in the composition of microbial communities resulting from differential amplification of templates by PCR especially when using degenerate primer pairs, are well documented through high-throughput sequencing of 16S rRNA gene as well as functional gene mock communities (e.g., Caporaso et al., 2011; Schloss et al., 2011; Pinto and Raskin, 2012; Pelikan et al., 2016). As in 16S rRNA gene amplicon sequencing (Schloss et al., 2011), our results therefore stress the need for including mock community samples that are specific to the target gene of interest in sequencing experiments for assessing the extent of primer biases. Furthermore, when mock communities are sequenced along with environmental samples on one sequencing run, it allows verifying the reproducibility of different runs.
Assessing nifH Primer Pair Performance Using Environmental Samples
The specificities of the primer pairs Ueda19F–R6, IGK3–DVV, and F2–R6 were further evaluated by amplicon sequencing of environmental samples. In addition to the above-mentioned temperate soil samples, we extended our investigation to a diverse collection of terrestrial samples (such as BSCs, rhizosphere and root samples) along with two estuarine water samples. A total of 424,234 raw Illumina paired-end reads were generated across these primer pairs: Ueda19F–R6 (156,476 reads), IGK3–DVV (202,300 reads), and F2–R6 (65,458 reads). The raw reads were processed through our automated pipeline NifMAP (see section “Materials and Methods” and Figure 1). After contig assembly, the merged reads were filtered using the nucleic acids based HMM, which filtered out different proportions of reads depending on the used primer combinations. Most reads were removed in the F2–R6 primer pair (ca. 7.5%) followed by Ueda19F–R6 (ca. 2.0%) and IGK3–DVV (ca. 0.3%). Investigating these non-nifH sequences using BLASTN against the Nr database gave no significant matches. The remaining reads were clustered into OTUs with the IGK3–DVV primer pair generating the most reads (89,453) that were clustered into 884 OTU97, followed by Ueda19F–R6 (76,897 reads; 528 OTU97) and F2–R6 (45808 reads; 763 OTU97). OTU representatives were converted to amino acid sequences, but in no case did we observe that frame shift correction was needed. This is in line with previous reports showing a particularly low indel error rate for Illumina MiSeq technology (Schirmer et al., 2015). Next, filtering of the OTU sequence representative via HMMs of homologous genes to nifH showed that all primer pairs and samples produced reads, and consequently OTUs, which were not classified as nifH, but rather as these homologous genes. Most of these homologs were classified as chlL-bchL, followed by bchX, and only a minority of sequences were classified as parA.
While the amplification of homologous genes was observed in all three primer pairs, striking differences amongst the primer pairs were seen in the proportion of reads and OTUs classified as homologs rather than as nifH (Figure 2). Across many environmental samples, the IGK3–DVV combination exhibited the highest proportion of non-specific products with on average 48% non-nifH OTUs and up to 92% non-nifH reads (Figure 2, green bars). The primer pairs F2–R6 and the new tested combination, Ueda19F–R6, amplified less non-nifH sequences across many of the investigated samples, with an average of 7.5 and 13% for primers Ueda19F–R6 and F2–R6, respectively. Our analysis shows not only large differences in the extent of non-specific products amongst the different primer pairs (P < 0.01), but also amongst environmental samples when the same primer pair was used (P = 0.02). The most striking difference was detectable amongst the samples of rice and tall oat-grass. Using primers IGK3–DVV, nearly all reads and OTUs in the rice samples were classified as nifH-related (Figure 2, samples “e, f”), while in the tall oat-grass samples only around 10% of them were identified as nifH-related (Figure 2, samples “c, d”). Similar trends can be seen for the crust and water samples, though to a lesser extent. The problem of co-amplification of non-target homologous or non-homologous genes during PCR is well documented in the literature, particularly when amplifying functional genes (Holmes et al., 1995). Unfortunately, this phenomenon can erroneously inflate diversity estimates and population size when left unnoticed. Our analysis clearly shows that different primer pairs can have striking differences in amplification specificities and that this should be taken into consideration when designing a genetic survey.
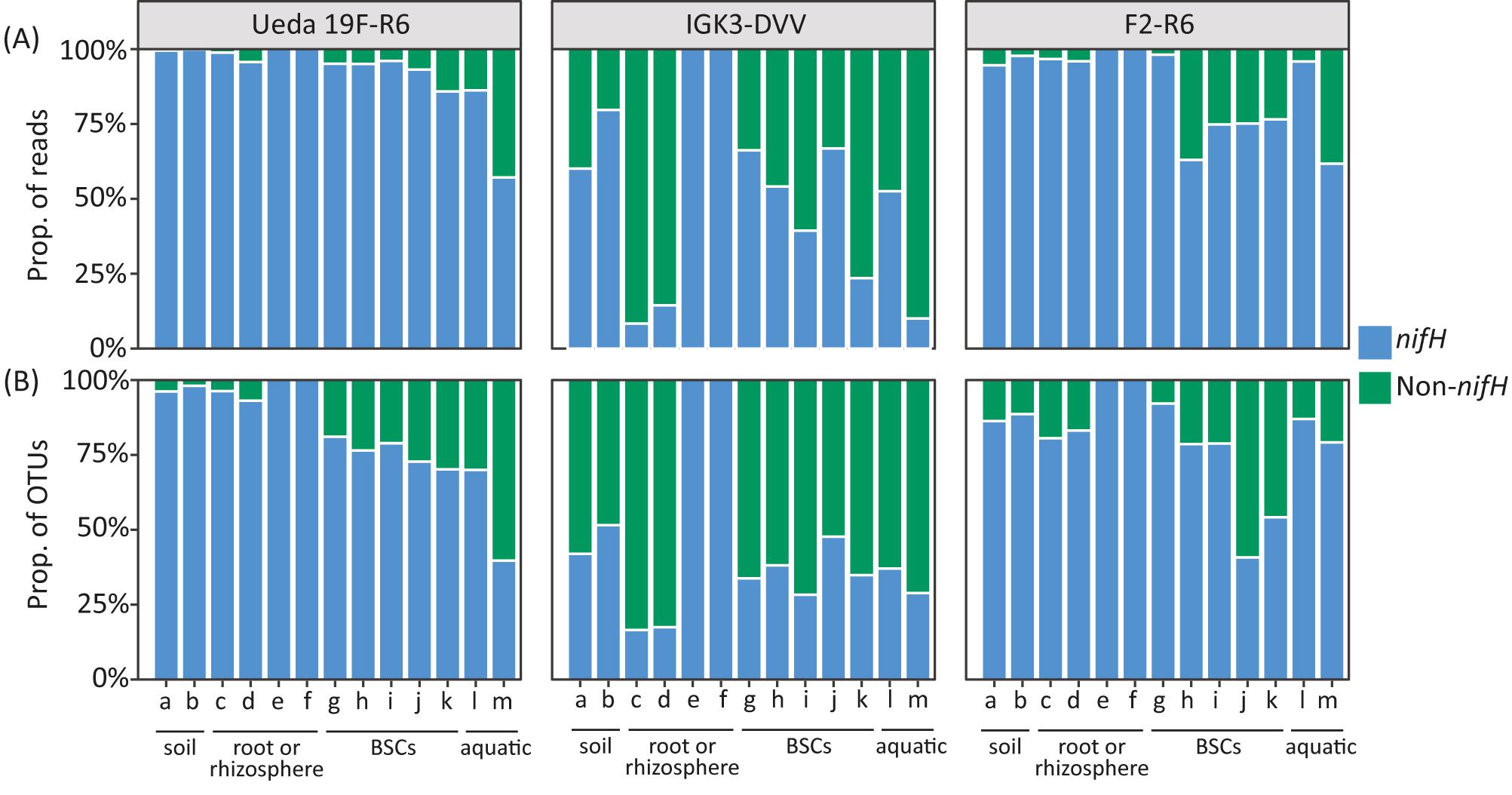
FIGURE 2. Performance of the nifH primer pairs in amplifying nifH and non-nifH (homologous) genes across environmental samples based on proportion of reads (A) and OTUs (B). The environmental samples include: (a) beech forest soil; (b) meadow soil; (c) rhizosphere and (d) root samples of Arrhenatherum elatius; (e) rhizosphere and (f) root samples of Oryza sativa; (g) coastal, sub-arctic biological soil crust (BSC); (h) temperate BSC; (i) high alpine BSC; (j) semiarid BSC; (k) arid BSC; estuarine samples from the (l) Great Belt and (m) Roskilde Fjord. More details on the samples can be found in the Section “Materials and Methods.” Reads or OTUs classified as homologous genes (such as bchX, chlL, bchL, and parA) are summarized as “non-nifH.”
Comparing Alpha-Diversity Metrics Across the Primer Pairs
We calculated the number of observed OTUs (richness estimate) and the inverse Simpson metric (diversity estimate) from each sample type and with the three different primer pairs. On average, similar numbers of nifH OTUs per sample were detected using each of the three primer pairs Ueda19F–R6 (49 ± 3); IGK3–DVV (39 ± 3); and F2–R6 (49 ± 4), while the inverse Simpson index was somewhat lower using primers Ueda19F–R6 (9.5 ± 0.5) compared to the other two primer pairs, IGK3–DVV (14.0 ± 1.5) and F2–R6 (14.0 ± 1.8) (Supplementary Figure S1) indicating no clear advantage of one primer pair in capturing higher richness. A general agreement amongst the primer pairs was seen across the different sample types regarding richness and diversity patters. For example, the beech forest and meadow soils harbored richer and more diverse communities than the crust and water samples. Nevertheless, there were some differences amongst the primer pairs with regards to richness and diversity estimates. Most notably primer pair F2–R6 yielded higher richness and diversity estimates for rice rhizosphere and root samples (Supplementary Figure S1, samples “e, f”), but lower for many of the crust samples (Supplementary Figure S1, samples “g–k”), compared to primer pairs IGK3–DVV and Ueda19F–R6.
Taxonomic Description of the Diazotrophic Community Using Multiple Classification Approaches Across the Primer Pairs
The classification of nifH genes is not an easy task due to a lack of taxonomic affiliation to functional gene sequences (Gaby and Buckley, 2011), poor reflection of the nifH phylogeny to the 16S rRNA gene phylogeny (Zehr et al., 2003), and insufficient information in k-mer frequencies to differentiate amongst the subclusters (Frank et al., 2016) as was also observed with pmoA sequences (Dumont et al., 2014). As such, we propose that a combined classification approach, using several independent methods might be more beneficial for characterizing nifH sequence datasets. Several approaches have been described in the literature for rapid, accurate and biologically meaningful classification of functional gene sequences in general and nifH in particular, including nearest neighbor identification in pairwise alignment using FrameBot (Wang et al., 2013), lowest common ancestor parsing of BLAST bit scores using MEGAN (Huson et al., 2007; Dumont et al., 2014), or CART (Frank et al., 2016). Here, we used a combined approach in our pipeline and describe the taxonomic affiliation of the sequences using closest relative via BLASTP, nifH cluster and subcluster affiliation using CART (Frank et al., 2016), and the EPA (Berger et al., 2011) for phylogenetic tree generation.
A representative from each OTU was classified using the best matching hit in BLASTP (Figure 3), CART (Figure 4), and the EPA for phylogeny (Figure 5). The BLASTP classifies sequences based on their closest relative. This classification yielded an overall agreement amongst the three primer pairs, with mainly members of the Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Cyanobacteria, and Firmicutes populating the diazotrophs across the environments (Figure 3). However, the relative abundance of these taxonomic groups varied amongst the investigated primer pairs. For example, Cyanobacteria were underrepresented by primer pair F2–R6 in comparison to the other two primer pairs (in concordance with the performance of primers F2–R6 amplifying the mock community). Furthermore, the relative abundance of Alphaproteobacteria derived in PCRs with primer pair IGK3–DVV was lower in some samples than in PCRs with the other two primer combinations, and Firmicutes were comparatively underrepresented by primer pair Ueda19F–R6 in some samples (Figure 3). Some taxa (such as the Green Sulfur Bacteria) were completely missing with the IGK3–DVV primer pair, while Euryarchaeota were missing in the Ueda19F–R6 primer pair and at low relative abundance with primer pair F2–R6 (Figure 3).
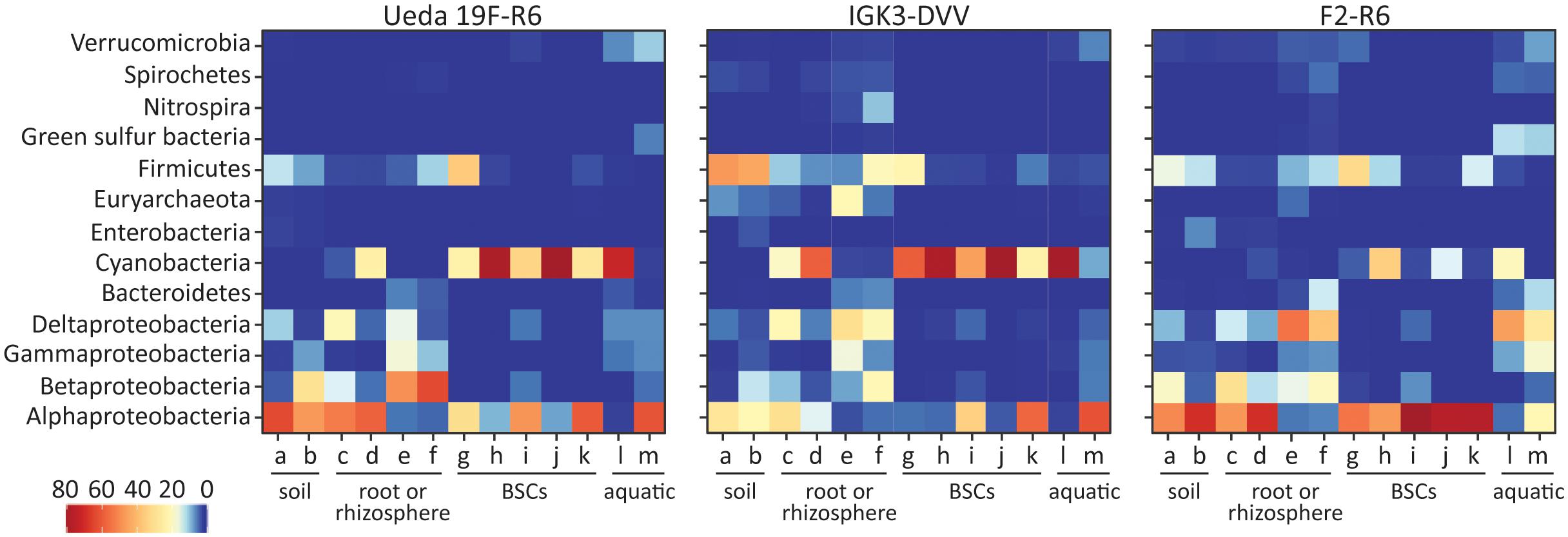
FIGURE 3. Taxonomic classification of nifH sequences based on a BLASTP search for the different primer pairs and environmental samples. Colors of the heatmap indicate the mean relative abundance of the taxonomic classes from each sample. The environmental samples include: (a) beech forest soil; (b) meadow soil; (c) rhizosphere and (d) root samples of Arrhenatherum elatius; (e) rhizosphere and (f) root samples of Oryza sativa; (g) coastal, sub-arctic biological soil crust (BSC); (h) temperate BSC; (i) high alpine BSC; (j) semiarid BSC; (k) arid BSC; estuarine samples from the (l) Great Belt and (m) Roskilde Fjord.
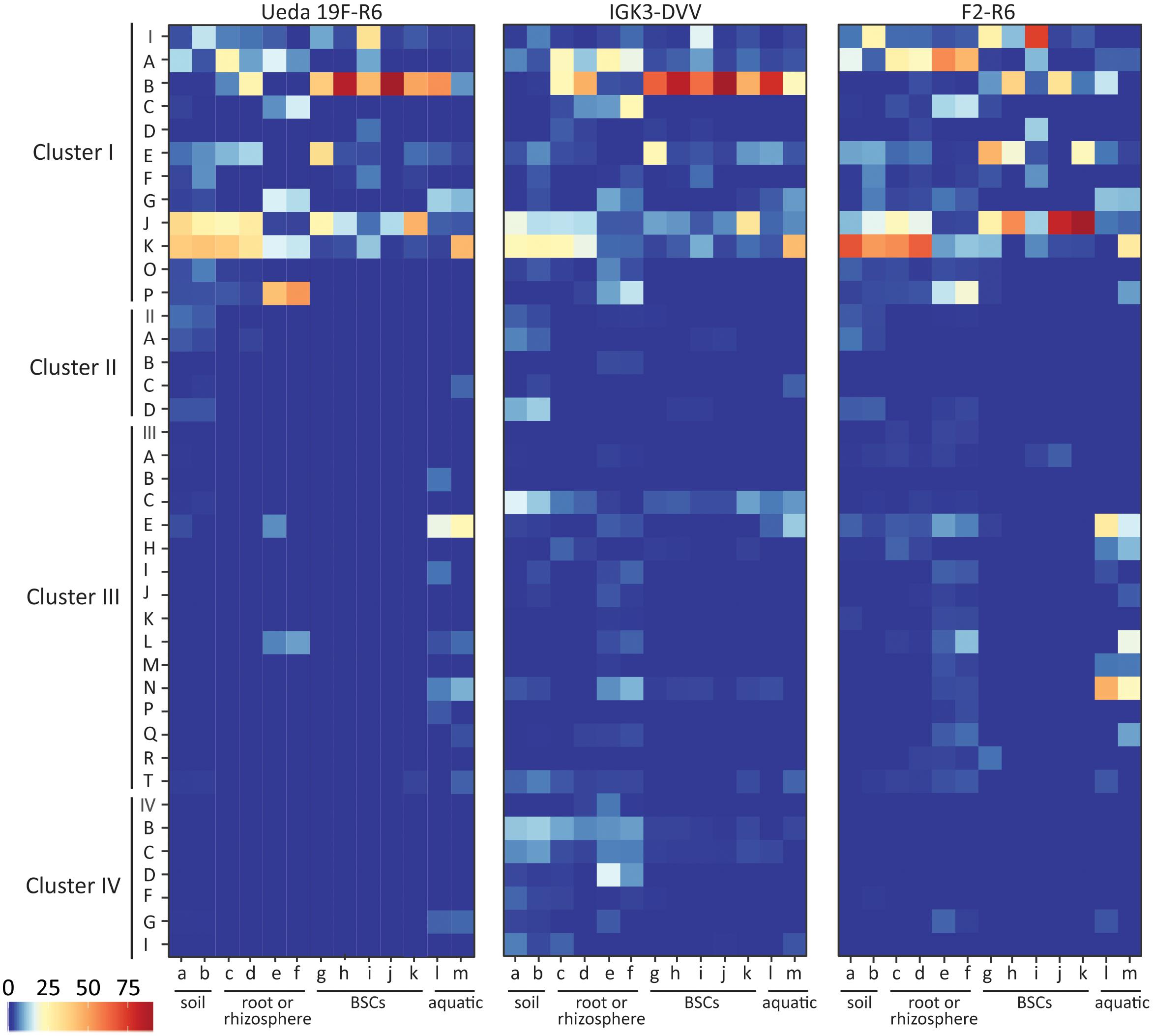
FIGURE 4. Taxonomic classification of nifH sequences based on CART for the different primer pairs and environmental samples. Colors of the heatmap indicate the mean relative abundance of the subclusters in each sample. The environmental samples include: (a) beech forest soil; (b) meadow soil; (c) rhizosphere and (d) root samples of Arrhenatherum elatius; (e) rhizosphere and (f) root samples of Oryza sativa; (g) coastal, sub-arctic biological soil crust (BSC); (h) temperate BSC; (i) high alpine BSC; (j) semiarid BSC; (k) arid BSC; estuarine samples from the (l) Great Belt and (m) Roskilde Fjord.
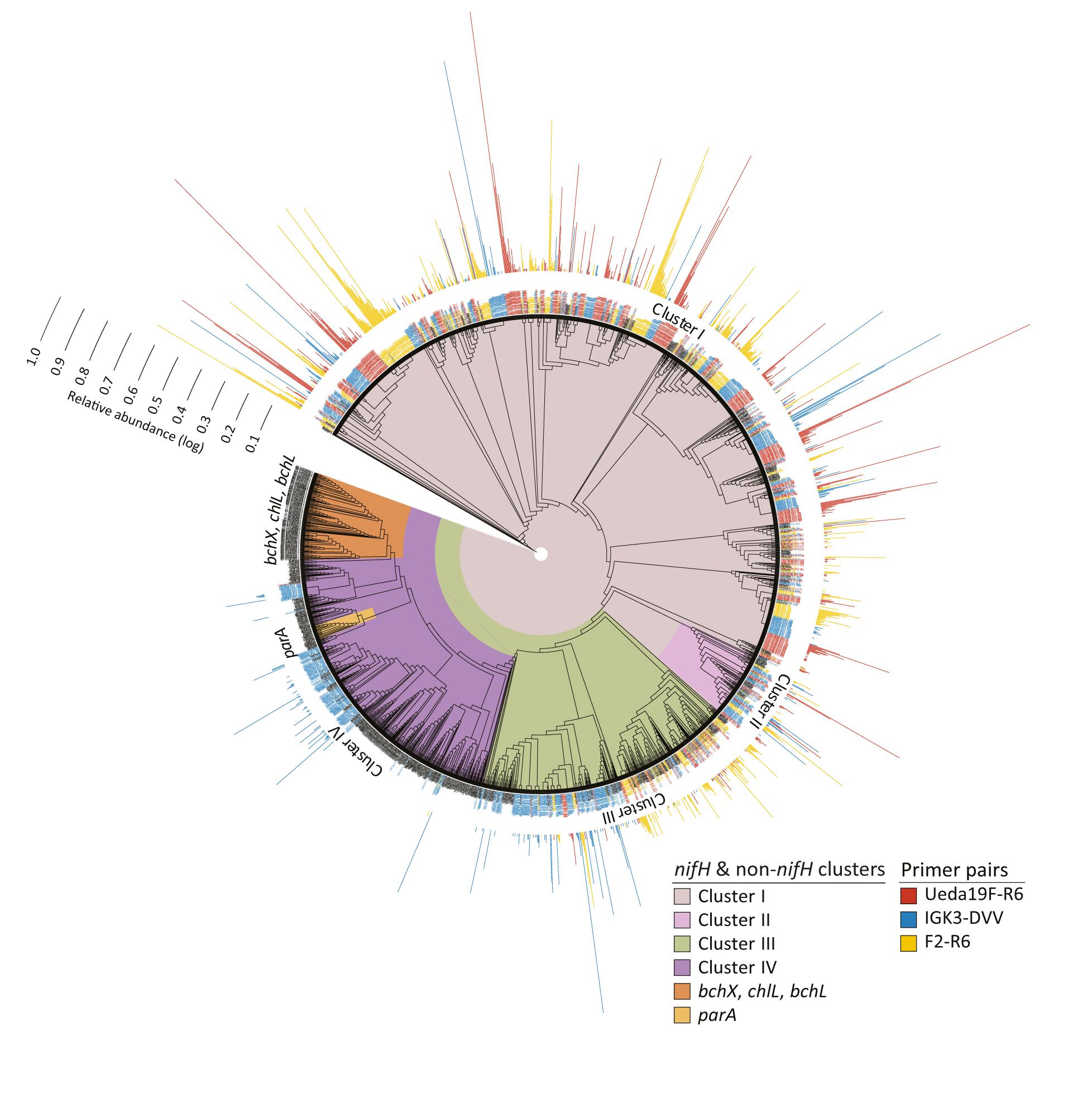
FIGURE 5. Unrooted RAxML-EPA tree of nifH deduced amino acid sequences (with at least 133 positions). The nifH and non-nifH (homologous genes) clusters are represented in different colors. Representative sequences from the “nifH_2014April04.arb” database (Heller et al., 2014) are depicted in black. The phylogenetic placements of OTU representatives from this study for the three primer pairs are depicted at the terminal nodes: sequences obtained with primer pair Ueda19F–R6 are shown in red, IGK3–DVV-derived sequences in blue, and F2–R6-derived sequences in yellow. Outermost bars represent the relative abundance [log(x% + 1)] of each OTU.
The CART classification assigns sequences to four clusters of nifH, termed clusters I–IV with subclusters (Zehr et al., 2003; Raymond et al., 2004). The majority of the sequences amongst all primer pairs were classified into cluster I based on the CART classification (Figure 4). This cluster is known to reflect 16S rRNA-based phylogeny relatively well (Zehr et al., 2003) and in many environments is the largest and most ecologically important diazotroph cluster (Gaby and Buckley, 2011). Within cluster I, differences in the relative abundances of certain defined clusters can be observed, some of which are in agreement with the BLASTP results. For instance, the relative abundance of sequences in cluster IB (affiliated with Cyanobacteria) derived from PCRs with primer pair F2–R6 was lower than from the other two primer combinations. Furthermore, less sequences assigned to cluster IJ and IK (containing Alphaproteobacteria) were derived from primer pair IGK3–DVV than in the other two primer pairs (Figure 4). The same trend was observed when the sequences were analyzed via BLASTP.
As expected, only a minority of the sequences were affiliated with clusters II, III, and IV. All three primer pairs amplified sequences from the same subclusters in clusters II–IV, with some exceptions in clusters III and IV, indicating preferential amplifications of subclusters amongst these primer sets (Figure 4). Most notably the IGK3–DVV primer pair amplified many sequences from clusters IIIC (affiliated with Clostridia) as well as from clusters IVB and IVC. In contrast, primer pairs Ueda19F–R6 and F2–R6 amplified very few sequences from clusters IIIC, IVB and IVC. A close examination of the sequence region matching the R6 primer showed that the primer has indeed two mismatches at the 3′ end of several Clostridia sequences affiliated with cluster IIIC (with residues CT instead of GC).
The phylogenetic placement of OTU representatives suggests that the sequences are in clusters I–IV (Figure 5). It showed a nearly identical coverage of clusters I and II by all three primer pairs, thus indicating that all three primer pairs cover equally well the diazotrophic diversity in these clusters. Although all three primer pairs captured sequences from cluster III, discrepancies within subclusters were noted, especially with the primer pair IGK3–DVV as compared to F2–R6 and Ueda19F–R6. While primers IGK3–DVV captured many sequences in subcluster IIIC, primers F2–R6 and Ueda19F–R6 captured many from subclusters IIIE, IIIL, IIIM, and IIIN, which were largely missed out by primers IGK3–DVV. Cluster IV sequences were nearly only captured by primer pair IGK3–DVV. However, the ecological relevance of cluster IV members is still debatable, as until recently it was considered to be populated only by genes encoding for functions unrelated to N2 fixation (Raymond et al., 2004; Zheng et al., 2016). Moreover this cluster includes several nifH homologs, which might be more abundant than nifH in certain environments. Attempts to use primers that capture sequences from this cluster could inflate datasets with non-target genes.
Suitability of nifH Primers for qPCR Assays
The primer pairs F2–R6, IGK3–DVV, and Ueda19F–R6 were also evaluated for their suitability in qPCR assays. Since using degenerate primers incurs a potential template-depended quantification bias, we increased the primer concentration in these reactions to 1.4 μM. This has been recently shown to minimize the effect of this specific bias (Gaby and Buckley, 2017) and has increased efficiency in our assays (data not shown). High assay efficiencies across all three primer pairs – Ueda19F–R6 (97.4%), IGK3–DVV (92.5%), and F2–R6 (90.5%) – were attained (Supplementary Figure S2), illustrating that these primer sets worked almost equally well and therefore can be used for reliable quantification of nifH genes and transcripts. In a proof-of-principle experiment, we quantified nifH genes in the environmental samples used in this study across all three primer pairs. A caveat when using such quantitative assays is that qPCR works under the underlying assumption that all quantified templates belong to the target gene. As we have demonstrated above, this is not the case for many types of samples when using general nifH-targeting primers. Therefore, we corrected for the proportion of nifH sequences from the total reads based on the sequencing results (Figure 6). Amongst these diverse sample types, the numbers of nifH copies per ng DNA ranged between 1.2 × 101 and 1.8 × 105 with a geometric average of 1.3 × 103. Interestingly, no single primer pair gave consistently higher or lower estimations compared to other primer pairs (P = 0.5, ANOVA test), further reiterating that all three primer sets worked equally well. Yet, significant differences appeared to be sample-type dependent (P < 0.01, ANOVA test). The estimated nifH copies per ng DNA were congruent within a given sample amongst the three primer pairs, considering the precision limitations of qPCR (two- to threefold difference between samples; Hospodsky et al., 2010). However, some exceptions to that observation were notable. For instance, primer pairs F2–R6 and Ueda19F–R6 estimated similar copy numbers in estuarine water samples (Figure 6, samples “l, m”) and the arid crust sample (Figure 6, sample “k”), while primer pair IGK3–DVV estimated an order of magnitude more copies. Furthermore, primers Ueda19F–R6 and IGK3–DVV measured similar copy numbers in the temperate and semiarid BSCs (Figure 6, samples “h, j”) while primers F2–R6 measured an order of magnitude less copies. This latter observation could be explained by the fact that primer pair F2–R6 has the tendency to discriminate against cyanobacteria, which would result in a decreased copy number in these cyanobacteria-dominated samples.
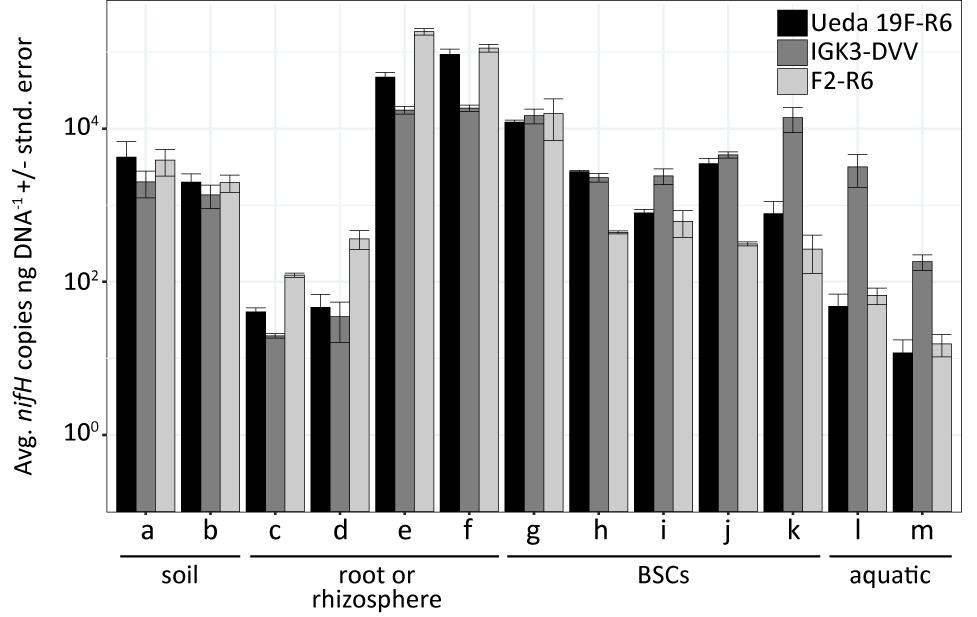
FIGURE 6. Average nifH copies per ng DNA ± standard error across environmental samples based on quantitative PCR using the different primer pairs. The environmental samples include: (a) beech forest soil; (b) meadow soil; (c) rhizosphere and (d) root samples of Arrhenatherum elatius; (e) rhizosphere and (f) root samples of Oryza sativa; (g) coastal, sub-arctic biological soil crust (BSC); (h) temperate BSC; (i) high alpine BSC; (j) semiarid BSC; (k) arid BSC; estuarine samples from the (l) Great Belt and (m) Roskilde Fjord. Numbers of copies were corrected to exclude non-nifH genes that were co-amplified using information obtained from amplicon sequencing of the samples with the specific primer pairs.
Conclusion
We tested the nifH-targeting primer combinations Ueda19F–R6, IGK3–DVV, and F2–R6 for their performance in genetic surveys of diazotrophs, thereby elaborating on the in silico analysis of Gaby and Buckley (2012). All primer pairs had a propensity to co-amplify homologs of the nifH gene at varying proportions, which is common in many studies of functional gene diversity. Most severe, primer combination IGK3–DVV had the largest tendency to co-amplify these sequences (which are most closely related to cluster IV), with some samples yielding up to ca. 90% homolog sequences. Using a pipeline such as our newly established NifMAP, that permits the detection of non-specific co-amplified reads via specifically designed HMMs, proved to be a useful strategy in filtering out such reads.
When analyzing the specificity of these primers through the use of a mock community along with a diverse collection of environmental samples, we observed some degree of bias amongst the primer pairs. As such, care should be taken when choosing primer pairs for diazotrophic diversity investigations. More specifically, primer pair F2–R6 has a propensity to discriminate against cyanobacteria (amongst others) in our mock communities and environmental samples, yet captured many sequences from subclusters IIIE, IIIL, IIIM, and IIIN. These aforementioned subclusters were largely missing by the primer pair IGK3–DVV, which also tended to discriminate against the Alphaproteobacteria, but amplified many sequences within clusters IIIC (affiliated with Clostridia) and clusters IVB and IVC. Primer pair Ueda19F–R6 exhibited the least bias based on our mock community analysis and successfully captured diazotrophs in cluster I as well as in subclusters IIIE, IIIL, IIIM, and IIIN, but discriminated against Firmicutes (based on BLASTP analysis) and subcluster IIIC. Thus, depending on the investigated environmental sample and the aforementioned primer performance, one should choose the appropriate primer combination. Apart from providing useful analysis protocols and standards for the study of environmental diazotrophs, our work highlights some of the important pitfalls and caveats of studying nifH, and potentially other functional genes in the environment. Furthermore, these primer pairs can be used in targeted functional investigations in tandem with activity measurements of N2 fixation (Angel et al., 2018) to better understand the ecophysiology of diazotrophs in the environment and could potentially be used to identify target groups for downstream single-cell analyses (Eichorst et al., 2015; Woebken et al., 2015). We hope this knowledge, along with our newly develop pipeline (NifMAP) will aide investigators to better detect and capture diazotrophs in their respective environment.
Author Contributions
RA, SE, and DW designed the study and participated in writing. RA, CP, and HS extracted DNA and performed the PCRs. MN and HS constructed the mock community. RA, MN, and CH established and tested the analysis pipeline, NifMAP. RA analyzed the data.
Funding
This work was supported by an Austrian Science Fund (FWF) project grant (P25700-B20 to DW and SE). SE was supported by the Austrian Science Fund (FWF) project grant (P26392-B20 to DW and SE). CP and HS were supported by an ERC Starting grant (Grant Agreement No: 636928 to DW) from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program. HS was supported by a Marie Curie Intra European Fellowship within the 7th European Community Framework Program (Grant Agreement No: 628361). MN was funded by a fellowship from the Austrian Academy of Sciences (ÖAW). RA was also supported by the Ministry of Education, Youth and Sports of the Czech Republic – MEYS (projects LM2015075 and EF16_013/0001782).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Lasse Riemann for providing estuarine samples, Gerhard Karrer for granting access to the sampling site at Lainzer Tiergarten, Osnat Gillor and Burkhard Büdel for assistance in obtaining biological soil crust samples, Erich M. Pötsch for his assistance with sampling Arrhenatherum elatius plants at AREC Raumberg-Gumpenstein, Michael Schagerl for providing a culture of A. torulosa and N. microscopicum, Stefanie Wienkoop for providing a culture of M. loti, Bela Hausmann for providing gDNA of Desulfosporosinus acidophilus and Telmatospirillum siberiense, David Seki for his assistance with the nifH mock community and culture of K. sacchari, and Florian Höggerl for his assistance in preparing samples for amplicon sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00703/full#supplementary-material
Footnotes
References
Alazard, D., Joseph, M., Battaglia-Brunet, F., Cayol, J. L., and Ollivier, B. (2010). Desulfosporosinus acidiphilus sp. nov.: a moderately acidophilic sulfate-reducing bacterium isolated from acid mining drainage sediments. Extremophiles 14, 305–312. doi: 10.1007/s00792-010-0309-4
Ando, S., Goto, M., Meunchang, S., Thongra-ar, P., Fujiwara, T., Hayashi, H., et al. (2005). Detection of nifH sequences in sugarcane (Saccharum officinarum l.) and pineapple (Ananas comosus [l.] merr.). Soil Sci. Plant Nutr. 51, 303–308. doi: 10.1111/j.1747-0765.2005.tb00034.x
Angel, R. (2012). Total nucleic acid extraction from soil. Protoc. Exch. doi: 10.1038/protex.2012.046
Angel, R., and Conrad, R. (2009). In situ measurement of methane fluxes and analysis of transcribed particulate methane monooxygenase in desert soils. Environ. Microbiol. 11, 2598–2610. doi: 10.1111/j.1462-2920.2009.01984.x
Angel, R., Panhölzl, C., Gabriel, R., Herbold, C., Wanek, W., Richter, A., et al. (2018). Application of stable-isotope labelling techniques for the detection of active diazotrophs. Environ. Microbiol. 20, 44–61. doi: 10.1111/1462-2920.13954
Angeringer, W., and Karrer, G. (2008). “Preserving species richness in hay meadows of Vienna’s nature conservation area Lainzer Tiergarten,” in University of Natural Resources and Applied Life Sciences, Vienna, Center for Environmental Studies and Nature Conservation (Hrsg.), Preservation of Biocultural Diversity – a Global Issue. Book of Abstracts, ed. U. Pröbstl (Wien: BOKU), 9.
Bentzon-Tilia, M., Traving, S. J., Mantikci, M., Knudsen-Leerbeck, H., Hansen, J. L. S., Markager, S., et al. (2015). Significant N2 fixation by heterotrophs, photoheterotrophs and heterocystous cyanobacteria in two temperate estuaries. ISME J. 9, 273–285. doi: 10.1038/ismej.2014.119
Berger, S. A., Krompass, D., and Stamatakis, A. (2011). Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol. 60, 291–302. doi: 10.1093/sysbio/syr010
Bokulich, N. A., Rideout, J. R., Mercurio, W. G., Shiffer, A., Wolfe, B., Maurice, C. F., et al. (2016). mockrobiota: a public resource for microbiome bioinformatics benchmarking. mSystems 1:e00062-16. doi: 10.1128/mSystems.00062-16
Boström, K. H., Simu, K., Hagström, A., and Riemann, L. (2004). Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol. Oceanogr. Methods 2, 365–373. doi: 10.4319/lom.2004.2.365
Boyd, E. S., Costas, A. M. G., Hamilton, T. L., Mus, F., and Peters, J. W. (2015). Evolution of molybdenum nitrogenase during the transition from anaerobic to aerobic metabolism. J. Bacteriol. 197, 1690–1699. doi: 10.1128/JB.02611-14
Büdel, B., Colesie, C., Green, T. G. A., Grube, M., Suau, R. L., and Loewen-Schneider, K. (2014). Improved appreciation of the functioning and importance of biological soil crusts in Europe: the soil crust international project (SCIN). Biodivers. Conserv. 23, 1639–1658. doi: 10.1007/s10531-014-0645-2
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chen, M., Zhu, B., Lin, L., Yang, L., Li, Y., and An, Q. (2014). Complete genome sequence of Kosakonia sacchari type strain SP1T. Stand. Genome Sci. 9, 1311–1318. doi: 10.4056/sigs.5779977
Collavino, M. M., Tripp, H. J., Frank, I. E., Vidoz, M. L., Calderoli, P. A., Donato, M., et al. (2014). nifH pyrosequencing reveals the potential for location-specific soil chemistry to influence N2-fixing community dynamics. Environ. Microbiol. 16, 3211–3223. doi: 10.1111/1462-2920.12423
Deslippe, J. R., Egger, K. N., and Henry, G. H. R. (2005). Impacts of warming and fertilization on nitrogen-fixing microbial communities in the Canadian High Arctic. FEMS Microbiol. Ecol. 53, 41–50. doi: 10.1016/j.femsec.2004.12.002
Dos Santos, P. C., Fang, Z., Mason, S. W., Setubal, J. C., and Dixon, R. (2012). Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13:162. doi: 10.1186/1471-2164-13-162
Dumont, M. G., Lüke, C., Deng, Y., and Frenzel, P. (2014). Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 5:34. doi: 10.3389/fmicb.2014.00034
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Eichorst, S. A., Strasser, F., Woyke, T., Schintlmeister, A., Wagner, M., and Woebken, D. (2015). Advancements in the application of NanoSIMS and Raman microspectroscopy to investigate the activity of microbial cells in soils. FEMS Microbiol. Ecol. 91:fiv106. doi: 10.1093/femsec/fiv106
Farnelid, H., Andersson, A. F., Bertilsson, S., Al-Soud, W. A., Hansen, L. H., Sørensen, S., et al. (2011). Nitrogenase gene amplicons from global marine surface waters are dominated by genes of non-cyanobacteria. PLoS One 6:e19223. doi: 10.1371/journal.pone.0019223
Fish, J. A., Chai, B., Wang, Q., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2013). FunGene: the functional gene pipeline and repository. Front. Microbiol. 4:291. doi: 10.3389/fmicb.2013.00291
Fowler, D., Coyle, M., Skiba, U., Sutton, M. A., Cape, J. N., Reis, S., et al. (2013). The global nitrogen cycle in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 368:20130164.
Frank, I. E., Turk-Kubo, K. A., and Zehr, J. P. (2016). Rapid annotation of nifH gene sequences using classification and regression trees facilitates environmental functional gene analysis. Environ. Microbiol. Rep. 8, 905–916. doi: 10.1111/1758-2229.12455
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gaby, J. C., and Buckley, D. H. (2011). A global census of nitrogenase diversity. Environ. Microbiol. 13, 1790–1799. doi: 10.1111/j.1462-2920.2011.02488.x
Gaby, J. C., and Buckley, D. H. (2012). A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. doi: 10.1371/journal.pone.0042149
Gaby, J. C., and Buckley, D. H. (2014). A comprehensive aligned nifH gene database: a multipurpose tool for studies of nitrogen-fixing bacteria. Database 2014:bau001. doi: 10.1093/database/bau001
Gaby, J. C., and Buckley, D. H. (2017). The use of degenerate primers in qPCR analysis of functional genes can cause dramatic quantification bias as revealed by investigation of nifH primer performance. Microb. Ecol. 74, 701–708. doi: 10.1007/s00248-017-0968-0
Gaby, J. C., Rishishwar, L., Valderrama-Aguirre, L. C., Green, S. J., and Valderrama-Aguirre, A. (2017). Diazotroph community characterization via a high-throughput nifH amplicon sequencing and analysis pipeline. Appl. Environ. Microbiol. 84:e01512-17. doi: 10.1128/AEM.01512-17
Hausmann, B., Pjevac, P., Schreck, K., Herbold, C. W., Daims, H., Wagner, M., et al. (2018). Draft genome sequence of Telmatospirillum siberiense 26-4b1T, an acidotolerant peatland alphaproteobacterium potentially involved in sulphur cycling. Genome Announc. 6:e01524-17. doi: 10.1128/genomeA.01524-17
Heller, P., Tripp, H. J., Turk-Kubo, K., and Zehr, J. P. (2014). ARBitrator: a software pipeline for on-demand retrieval of auto-curated nifH sequences from GenBank. Bioinformatics 30, 2883–2890. doi: 10.1093/bioinformatics/btu417
Herbold, C. W., Pelikan, C., Kuzyk, O., Hausmann, B., Angel, R., Berry, D., et al. (2015). A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front. Microbiol. 6:731. doi: 10.3389/fmicb.2015.00731
Holmes, A. J., Costello, A., Lidstrom, M. E., and Murrell, J. C. (1995). Evidence that participate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 132, 203–208. doi: 10.1111/j.1574-6968.1995.tb07834.x
Hospodsky, D., Yamamoto, N., and Peccia, J. (2010). Accuracy, precision, and method detection limits of quantitative PCR for airborne bacteria and fungi. Appl. Environ. Microbiol. 76, 7004–7012. doi: 10.1128/AEM.01240-10
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kelly, S., Sullivan, J., Ronson, C., Tian, R., Bräu, L., Munk, C., et al. (2014). Genome sequence of the Lotus spp. Microsymbiont Mesorhizobium loti strain R7A. Stand. Genomic Sci. 9:6. doi: 10.1186/1944-3277-9-6
Kirshtein, J. D., Paerl, H. W., and Zehr, J. (1991). Amplification, cloning, and sequencing of a nifH segment from aquatic microorganisms and natural communities. Appl. Environ. Microbiol. 57, 2645–2650.
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Marusina, A. I., Boulygina, E. S., Kuznetsov, B. B., Tourova, T. P., Kravchenko, I. K., and Gal’chenko, V. F. (2001). A system of oligonucleotide primers for the amplification of nifH genes of different taxonomic groups of prokaryotes. Microbiology 70, 73–78. doi: 10.1023/A:1004849022417
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41:e121. doi: 10.1093/nar/gkt263
Pelikan, C., Herbold, C. W., Hausmann, B., Müller, A. L., Pester, M., and Loy, A. (2016). Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ. Microbiol. 18, 2994–3009. doi: 10.1111/1462-2920.13139
Penton, C. R., Johnson, T. A., Quensen, J. F. I., Iwai, S., Cole, J. R., and Tiedje, J. M. (2013). Functional genes to assess nitrogen cycling and aromatic hydrocarbon degradation: primers and processing matter. Front. Microbiol. 4:279. doi: 10.3389/fmicb.2013.00279
Pinto, A. J., and Raskin, L. (2012). PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7:e43093. doi: 10.1371/journal.pone.0043093
Pötsch, E. M., Resch, R., and Buchgraber, K. (2013). Auswirkung der Düngung auf P-Gehaltswerte im boden und futter sowie P-bilanzen von grünlandflächen. Alpenländisches Exp. 18, 41–50.
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504. doi: 10.1093/nar/gki025
Rasche, F., Knapp, D., Kaiser, C., Koranda, M., Kitzler, B., Zechmeister-Boltenstern, S., et al. (2011). Seasonality and resource availability control bacterial and archaeal communities in soils of a temperate beech forest. ISME J. 5, 389–402. doi: 10.1038/ismej.2010.138
Raymond, J., Siefert, J. L., Staples, C. R., and Blankenship, R. E. (2004). The natural history of nitrogen fixation. Mol. Biol. Evol. 21, 541–554. doi: 10.1093/molbev/msh047
Rubio, L. M., and Ludden, P. W. (2002). CHAPTER 4 - The Gene Products of the nif Regulon A2 - Leigh, G. Jeffery. Nitrogen Fixation at the Millennium. Amsterdam: Elsevier Science, 101–136. doi: 10.1016/B978-044450965-9/50004-5
Schirmer, M., Ijaz, U. Z., D’Amore, R., Hall, N., Sloan, W. T., and Quince, C. (2015). Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 43:e37. doi: 10.1093/nar/gku1341
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. doi: 10.1371/journal.pone.0027310
Singer, E., Andreopoulos, B., Bowers, R. M., Lee, J., Deshpande, S., Chiniquy, J., et al. (2016). Next generation sequencing data of a defined microbial mock community. Sci. Data 3:160081. doi: 10.1038/sdata.2016.81
Stamatakis, A. (2014). RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Staples, C. R., Lahiri, S., Raymond, J., Herbulis, L. V., Mukhophadhyay, B., and Blankenship, R. E. (2007). Expression and association of group IV nitrogenase nifD and nifH homologs in the non-nitrogen-fixing archaeon Methanocaldococcus jannaschii. J. Bacteriol. 189, 7392–7398. doi: 10.1128/JB.00876-07
Stüeken, E. E., Buick, R., Guy, B. M., and Koehler, M. C. (2015). Isotopic evidence for biological nitrogen fixation by molybdenum-nitrogenase from 3.2 Gyr. Nature 520, 666–669. doi: 10.1038/nature14180
Ueda, T., Suga, Y., Yahiro, N., and Matsuguchi, T. (1995). Remarkable N2-fixing bacterial diversity detected in rice roots by molecular evolutionary analysis of nifH gene sequences. J. Bacteriol. 177, 1414–1417. doi: 10.1128/jb.177.5.1414-1417.1995
Wang, Q., Quensen, J. F., Fish, J. A., Lee, T. K., Sun, Y., Tiedje, J. M., et al. (2013). Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using framebot, a new informatics tool. mBio 4:e00592-13. doi: 10.1128/mBio.00592-13
Wertz, J., Kim, E., Breznak, J. A., Schmidt, T. M., and Rodrigues, J. L. M. (2012). Genomic and physiological characterization of the Verrucomicrobia Diplosphaera colitermitum gen. nov., sp. nov., reveals microaerophily and nitrogen fixation genes. Appl. Environ. Microbiol. 78, 1544–1555. doi: 10.1128/AEM.06466-11
Woebken, D., Burow, L. C., Behnam, F., Mayali, X., Schintlmeister, A., and Fleming, E. D. (2015). Revisiting N2 fixation in Guerrero Negro intertidal microbial mats with a functional single-cell approach. ISME J. 9, 485–496. doi: 10.1038/ismej.2014.144
Zani, S., Mellon, M. T., Collier, J. L., and Zehr, J. P. (2000). Expression of nifH genes in natural microbial assemblages in Lake George, New York, detected by reverse transcriptase PCR. Appl. Environ. Microbiol. 66, 3119–3124. doi: 10.1128/AEM.66.7.3119-3124.2000
Zehr, J. P., Jenkins, B. D., Short, S. M., and Steward, G. F. (2003). Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ. Microbiol. 5, 539–554. doi: 10.1046/j.1462-2920.2003.00451.x
Zehr, J. P., and McReynolds, L. A. (1989). Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl. Environ. Microbiol. 55, 2522–2526.
Zheng, H., Dietrich, C., Radek, R., and Brune, A. (2016). Endomicrobium proavitum, the first isolate of Endomicrobia class. nov. (phylum Elusimicrobia) – an ultramicrobacterium with an unusual cell cycle that fixes nitrogen with a Group IV nitrogenase. Environ. Microbiol. 18, 191–204. doi: 10.1111/1462-2920.12960
Keywords: nitrogen fixation, primer evaluation, nifH gene, Illumina amplicon sequencing, NifMAP
Citation: Angel R, Nepel M, Panhölzl C, Schmidt H, Herbold CW, Eichorst SA and Woebken D (2018) Evaluation of Primers Targeting the Diazotroph Functional Gene and Development of NifMAP – A Bioinformatics Pipeline for Analyzing nifH Amplicon Data. Front. Microbiol. 9:703. doi: 10.3389/fmicb.2018.00703
Received: 28 January 2018; Accepted: 27 March 2018;
Published: 30 April 2018.
Edited by:
Marcus A. Horn, Leibniz University of Hanover, GermanyReviewed by:
Oliver Schmidt, University of Bayreuth, GermanyPetri Penttinen, Zhejiang Agriculture and Forestry University, China
Copyright © 2018 Angel, Nepel, Panhölzl, Schmidt, Herbold, Eichorst and Woebken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Woebken, d29lYmtlbkBtaWNyb2JpYWwtZWNvbG9neS5uZXQ=
†Present address: Roey Angel, Soil and Water Research Infrastructure, Biology Centre CAS, České Budějovice, Czechia