 Claudia Ibacache-Quiroga
Claudia Ibacache-Quiroga Juan C. Oliveros
Juan C. Oliveros Alejandro Couce3
Alejandro Couce3 Jesus Blázquez
Jesus Blázquez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 March 2018
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00427
Antibiotic resistance is a major concern in public health worldwide, thus there is much interest in characterizing the mutational pathways through which susceptible bacteria evolve resistance. Here we use experimental evolution to explore the mutational pathways toward aminoglycoside resistance, using gentamicin as a model, under low and high mutation supply rates. Our results show that both normo and hypermutable strains of Escherichia coli are able to develop resistance to drug dosages > 1,000-fold higher than the minimal inhibitory concentration for their ancestors. Interestingly, such level of resistance was often associated with changes in susceptibility to other antibiotics, most prominently with increased resistance to fosfomycin. Whole-genome sequencing revealed that all resistant derivatives presented diverse mutations in five common genetic elements: fhuA, fusA and the atpIBEFHAGDC, cyoABCDE, and potABCD operons. Despite the large number of mutations acquired, hypermutable strains did not pay, apparently, fitness cost. In contrast to recent studies, we found that the mutation supply rate mainly affected the speed (tempo) but not the pattern (mode) of evolution: both backgrounds acquired the mutations in the same order, although the hypermutator strain did it faster. This observation is compatible with the adaptive landscape for high-level gentamicin resistance being relatively smooth, with few local maxima; which might be a common feature among antibiotics for which resistance involves multiple loci.
Darwinian evolution, through mutation, recombination, and horizontal transfer, enables bacteria to adapt to widely different environmental conditions (Lan and Reeves, 1996; Rosenberg, 2001; Didelot and Maiden, 2010). One of the main examples of bacterial evolution is antibiotic resistance (Woodford and Ellington, 2007), which has become a healthcare issue worldwide and produces more than 25,000 deaths per year only in Europe. While resistance to certain antibiotics requires only one mutation (Jin and Gross, 1988; Leavis et al., 2006), the development of clinically relevant resistance levels to many compounds typically involves the accumulation of multiple mutations (El'Garch et al., 2007; Toprak et al., 2011). Much experimental work is being devoted to catalog these mutations, as well as to understand how they combine to produce high-level resistance. Nowadays, there have been identified bacterial strains resistant to every clinically available class of antibiotic, including aminoglycosides (Davies and Davies, 2010).
In recent years, the combination of experimental evolution with next-generation sequencing has afforded the means to explore the mutational pathways through which bacteria can acquire high-level resistance (Pál et al., 2015; Lukacišinová and Bollenbach, 2017). One important insight has been the realization that non-additive interactions among mutations can create adaptive landscapes that are rugged, that is, with divergent mutational pathways that lead to different local fitness peaks (de Visser and Krug, 2014). Adaptive dynamics under these circumstances can be complex, as illustrated by the apparent paradox that populations with a small mutation supply, although limited in their adaptation speed, may have more chances of finding global fitness optima than populations with large ones (Rozen et al., 2008; Handel and Rozen, 2009). This is because large mutation supplies increase the likelihood of substituting the most beneficial first-step mutations, which sometimes can lead toward an evolutionary dead-end. In turn, by substituting the best mutations less often, populations with smaller mutation supplies show more stochastic trajectories, which allows them to explore sub-optimal paths; incidentally reaching more distant, perhaps globally optimal, fitness peaks. This phenomenon has been recently confirmed in an experimental model of beta-lactam resistance evolution (Salverda et al., 2017).
In this work we sought to characterize the mutational pathways toward resistance to gentamicin, an aminoglycoside antibiotic to which high-level resistance is known to require mutations in multiple loci (Garneau-Tsodikova and Labby, 2016). It is important to highlight here that we have used the concept of operational definition of antibiotic resistance, based on the pairwise comparison of a parental strain with a mutant strain. Therefore, a strain is considered resistant if it has a higher MIC value for the studied compound than its parental wild-type strain (Martinez et al., 2015). Apart from evaluating how readily gentamicin resistance can develop, we aimed at exploring the impact of the mutation supply rate on the evolutionary dynamics. To this end, we conducted experiments with two variants of the common Escherichia coli MG1655 strain: a normomutator and its ΔdnaQ derivative, the hypermutator mutant with the highest known mutation rate and the broadest mutational spectrum (Scheuermann et al., 1983).
Aminoglycosides are an important family of broad-spectrum antibiotics that inhibit protein synthesis by binding to the 30S ribosomal subunit, thus generating truncated peptides and inhibiting tRNA translocation. The first step in aminoglycoside uptake involves reversible ionic binding to the cell surface, while the second and third phases are energy dependent (Jana and Deb, 2006). Thus, mutations on ribosomal components, translation machinery and electron transport chain have been identified as resistant determinants in bacteria, affecting the antibiotic target and uptake (Garneau-Tsodikova and Labby, 2016). Mutations in different genes are known to contribute to aminoglycoside resistance in gram-negative bacteria; like, for example, in fusA, galU, rplY, rplF, and genes involved in energy metabolism such as nqr, nuo, and cyo operons (Ahmad et al., 1980; Mogre et al., 2014; Wang et al., 2015). Independent mutations in these genes are associated to slight decreases in aminoglycoside susceptibility, although they can have a cumulative effect on aminoglycoside resistance and, therefore, multiple mutations are needed to reach higher-level resistance (El'Garch et al., 2007). Nevertheless, the main mechanism for gentamicin resistance in clinical isolates is related to the expression of aminoglycoside-modifying enzymes, involving acyltransferases, methyltransferases, and nucleotidyltransferases (Ramirez and Tolmasky, 2010).
Mutants with greatly elevated mutation rates, commonly referred to as hypermutators, are often isolated in clinical settings and in evolution experiments (Sniegowski et al., 1997; Oliver et al., 2000). Despite mutations typically being deleterious or neutral (Andersson and Levin, 1999), an elevated mutation rate has been shown to represent an advantage when adapting to new environments under strong selective pressures, by increasing the mutational supply rate of beneficial mutations (Taddei et al., 1997). The prevalence of hypermutators has also been linked to the fact that a higher mutation rate may favor the acquisition of compensatory mutations that alleviate the resistance fitness cost (Perron et al., 2010). Moreover, a larger mutation supply may guarantee that mutations conferring advantage in a subsequent environment are present in the population, even despite a sub-optimal fitness value under the current conditions (Couce et al., 2016).
Our experimental design consisted of propagating normo and hypermutable bacterial populations in chemostats in which the concentration of gentamicin was doubled every 3 days. The experiment was finished when the concentration of antibiotic exceeded the minimal inhibitory concentration (MIC) for the ancestors by more than a 1,000-fold factor. Chemostats allow varying just the antibiotic concentration while maintaining constant environmental conditions (Monod, 1950), many of which are known to affect aminoglycoside susceptibility (Tresse et al., 1995; Xiong et al., 1996) (e.g., pH, oxygen levels, or temperature). Moreover, chemostats keep bacteria in the exponential phase of growth, a situation that maximizes both the generation of mutations and the uptake of the drug, thus ensuring the best circumstances to select for mutations that confer resistance. At the end of the experiment, to gain insight into the genetic basis of adaptation, end-point isolates were subjected to diverse phenotypic assays, which guided the choice of candidates for whole-genome sequencing.
Strains used in this study are listed in Supplementary Table S1. Strains derived from MG1655 were grown in M9 minimal medium supplemented with glucose [0.2% (w/v)], liquid or agar plates [1.5% (w/v)]. Unless otherwise indicated, cultures were incubated at 37°C for 48 h. MG1655 ΔdnaQ was constructed according to Datsenko and Wanner (Datsenko and Wanner, 2000) and ΔdnaQ::Kan was amplified from strain JW0205 (Baba et al., 2006). When needed for strain selection and plasmid propagation, antibiotics were added at the following concentrations: ampicillin (AMP) at 100 μg/ml, chloramphenicol (CHL) at 50 μg/ml, and kanamycin (KAN) at 50 μg/ml. For cloning and propagation of pGE-derivatives, E. coli CC118 λ-pir was used (de Lorenzo and Timmis, 1994; Herring et al., 2003).
Plasmids used in this study are listed in Supplementary Table S2. Primers were synthetized by Sigma Genosys and are listed in Supplementary Table S3. Gene amplification was performed with Expand High Fidelity PCR System (Roche).
Construction of mutant strains of E. coli MG1655 was based on the expression of I-SceI endonuclease (Sniegowski et al., 1997). Selected genes were amplified from the evolved strain CIM8M using oligonucleotides listed in Supplementary Table S3 and cloned into pGE vector (KANR). Mutants construction was performed as described in Piñero-Lambea et al. (2015).
Complementation assays were carried out using vectors (Supplementary Table S2) from the ASKA collection for all selected strains (Kitagawa et al., 2005).
Prior to the evolutionary process, a wild-type E. coli MG1655 strain and its ΔdnaQ derivative were adapted to M9 minimal medium by serial passages into fresh media every 24 h during 7 days, equivalent to 56 generations, approximately. This previous adaptation was done to diminish the putative confounding effects of mutations not directly related with resistance to gentamicin. Bacterial evolution was performed in chemostats with M9 minimal medium with glucose as only carbon and energy source and increasing concentrations of gentamicin. For each evolutionary process, four chemostats containing 40 ml of culture medium supplemented with gentamicin at 0.03 μg/ml, equivalent to 0.125-fold their minimal inhibitory concentrations (MIC), were inoculated with 400 μl of an overnight culture of the ancestor. Chemostats were continuously fed with fresh culture medium with gentamicin at a dilution rate of 0.24 h−1, and maintained at 37°C and aeration of 1.2 l/h. Gentamicin concentration in culture media was doubled every 72 h until it reached 256 μg/ml. Samples from every stage of the evolutionary process were taken and cryopreserved at −80°C, with glycerol at a final concentration of 20% (v/v). Samples from the last stage of the evolution process were plated on M9 minimal medium agar with glucose and gentamicin (256 μg/ml). After 72 h of incubation, 22 MG1655-derivatives and 38 MG1655 ΔdnaQ-derivatives were isolated and cryopreserved for further characterization. Selection of these colonies was based on colony size and morphology.
Antimicrobial susceptibility profiles were established by evaluating MICs of different antimicrobials in M9 minimal medium supplemented with glucose at 0.2% (w/v), through micro-broth dilution method. The evaluated compounds were amikacin (AMK), ampicillin (AMP), chloramphenicol (CHL), colistin (COL), ciprofloxacin (CIP), fosfomycin (FOS), gentamicin (GEN), kanamycin (KAN), tetracycline (TET), rifampicin (RIF), trimethoprim (TRM), and ethidium bromide (EtB).
For strains derived from wild-type MG1655, the mutant frequency was determined on plates of M9 minimal medium with glucose containing 40 μg/ml of nalidixic acid, while rifampicin at 50 μg/ml was used for ΔdnaQ-derivatives. For both ancestors, overnight cultures were plated on media with antibiotic. Viable cells were measured by plating 103 cells from each overnight culture on M9 minimal medium supplemented with glucose and incubated under the same conditions. Mutant frequency was estimated by the ratio between colony-forming units (CFU) on rifampicin or nalidixic acid and total viable CFU. Frequency was evaluated in triplicates for each strain.
Bacterial susceptibility to hydrogen peroxide was measured on plates with M9 minimal medium agar with glucose, by measuring growth inhibition. Bacterial cells from overnight cultures were inoculated into top agar and exposed to 26 μmol of hydrogen peroxide. The halo of growth inhibition was measured in millimeters (mm) after 48 h. Susceptibility to hydrogen peroxide was evaluated in triplicates for each strain.
The ability of bacterial strains to use different organic compounds as sole energy and carbon source was evaluated using Biolog'S GN2 system, according to manufacturer's instructions. Biochemical characterization was performed using API 20E strips (Biomerieux), according to manufacturer'S instructions. Both systems were incubated at 37°C for 72 h.
Bacterial growth was evaluated in presence and absence of gentamicin (64 μg/ml). Ninety-six-well plates were filled with 200 μl of minimal medium and inoculated with 103 cells per well. Growth curves were performed in a Tecan Infinite M200 spectrophotometer with incubation, at 37°C, with linear shaking, for 48 h. Growth rates were determined using GrowthRates program (Hall et al., 2014). Growth curves were evaluated in triplicates for each strain.
Whole genome sequencing of all selected strains and their ancestors was carried out using the pair-end protocol in an Illumina HiSeq2000 Sequencer. More than 1,000,000 sequences, between 35 - 82 bp, were obtained per sample, and their quality was analyzed through FASTAQC. Sequences were aligned with BWA (Li and Durbin, 2009), using “bwa aln” and “bwa sampe” commands. Genome sequence of MG1655 was obtained from Genebank Database (NCBI): https://www.ncbi.nlm.nih.gov/nuccore/U00096.3. Alignment files were transformed to BAM format, sorted and indexed through samtools (Li et al., 2009). snpEff (Cingolani et al., 2012) was used for the annotation of gene polymorphisms. For mutations in upstream and downstream regions, 200 bp were considered. Results were filtered and analyzed in an interactive manner using a visualizer developed by the Computational Genomic Service of the National Center for Biotechnology (CNB-CSIC) in Madrid, Spain. Polymorphisms were considered relevant when they reached a minimal quality of QC > 30 and a biological impact of “High” or “Moderate.” The sequences obtained in this study have been deposited in the NCBI-SRA database (https://www.ncbi.nlm.nih.gov/sra) under the accession number SRP128878.
To determine the evolutionary trajectories of the selected genes, samples from chemostats were taken at 0.5, 2.0, 4.0, and 8.0 μg/ml of gentamicin, and plated on M9 minimal medium agar with glucose. Two single colonies were selected from each chemostat. Genes of interest were amplified from the selected strains, sequenced, and analyzed in order to determine if gene mutations were present at the selected evolutionary steps.
Statistical analysis was carried out with StatPlus:mac LE (AnalystSoft Inc., www.analystsoft.com). For all analyses, the level of significance was set at 95% (p-value < 0.05).
To minimize the confounding effects of mutations related to medium adaptation, both MG1655 and its ΔdnaQ derivative were adapted to M9 minimal medium without antibiotic, during 56 generations prior to the evolutionary process. Evolution to gentamicin resistance was carried out in chemostats under increasing concentrations of this antibiotic. Four parallel cultures of MG1655 and MG1655 ΔdnaQ, with MIC of gentamicin of 0.25 μg/ml in both cases, were evolved until antibiotic concentration reached 256 μg/ml. In total, 60 E. coli strains resistant to gentamicin were isolated (Supplementary Table S4): 22 derived from MG1655 (denominated CIM) and 38 from MG1655 ΔdnaQ (denominated CIQ), all of them presenting gentamicin MIC ≥256 μg/ml. These strains presented different colony sizes and morphology, including the presence of small colony variants especially in MG1655-derivatives. Thus, the 60 E. coli strains selected in this study represent the different colony phenotypes observed in each sample. Clones were named according to the author's name (CI), their ancestor (M for MG1655-derivatives and Q for ΔdnaQ), the replicate from which they were isolated (N°) and the isolated colony (letter).
All isolates were characterized for their antibiotic susceptibility profiles (Supplementary Tables S5, S6). All strains decreased their sensibility to fosfomycin. Increased susceptibility to chloramphenicol was also observed in 68% of strains. Interestingly, only MG1655-derivatives presented an increased resistance to rifampicin. From all 60 strains, 4 MG1655-derivatives (CIM5H, CIM5N, CIM8C, CIM8M), and 4 ΔdnaQ-derivatives (CIQ1E, CIQ1G, CIQ2J, CIQ4J) were selected for further studies. These strains were selected accordingly to their antibiotic-resistance profile in order to represent the different phenotypes observed among all characterized strains from each replicate. All MG1655-derivatives showed an increase in gentamicin resistance (≥2,000-fold) as well as rifampicin and fosfomycin resistance (Tables 1, 2). In particular, CIM5H presented an increase in chloramphenicol (16-fold) and ethidium bromide (≥4-fold) susceptibility; CIM5N showed an increase in chloramphenicol (4-fold) and a decrease in trimethoprim susceptibility (4-fold); CIM8C presented a decrease in chloramphenicol susceptibility (4-fold); CIM8M presented a decrease in chloramphenicol susceptibility (≥32-fold) and an increase in trimethoprim susceptibility (4-fold). Regarding the MG1655 ΔdnaQ derivatives, all strains showed increased fosfomycin resistance and were selected according to the following parameters: CIQ1E was selected due to its increase in chloramphenicol and tetracycline susceptibility (8-fold); CIQ1G showed an increase in its susceptibility to chloramphenicol (16-fold), trimethoprim (8-fold) and rifampicin (≥4-fold), and a 16-fold increase in resistance to ethidium bromide; CIQ2J presented increased resistance to ampicillin (4-fold) and a decrease to chloramphenicol (4-fold); CIQ4J showed increased susceptibility to ciprofloxacin (2-fold) and rifampicin (≥4-fold).
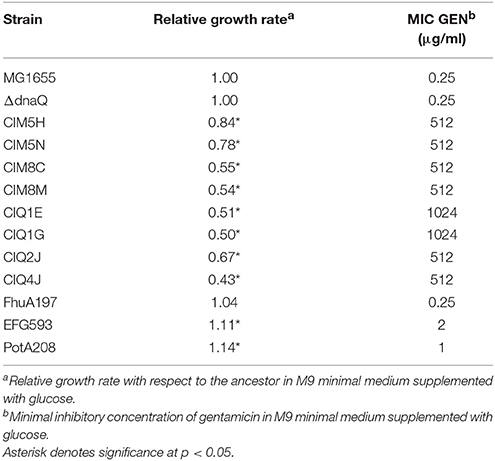
Table 1. Antibiotic susceptibility profile and relative growth rates of the selected gentamicin-resistant strain.
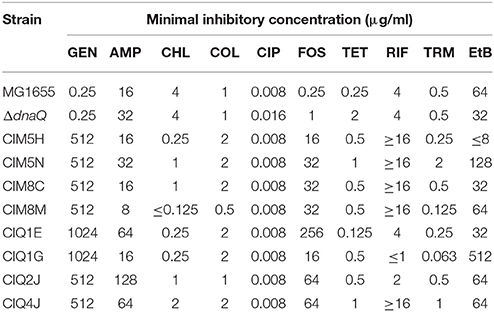
Table 2. Phenotypic characterization of the strains derived from this study.
Fitness cost of gentamicin resistance was observed for all evaluated strains according to growth curves, metabolic and enzymatic profiles. All isolates presented lower growth rates relative to their ancestor, showing a significant reduction of this parameter in all strains (p-value < 0.05, one-sample t-test) (Table 1). No significant differences in relative growth rates were observed between MG1655-derivatives and ΔdnaQ-derivatives (p-value < 0.05, two-samples t-test). All strains presented a reduced ability to use different organic compound as a sole energy and carbon source (Supplementary Table S7). Changes on their enzymatic activities were also observed (Supplementary Table S8). Interestingly, even though hypermutation has been associated with an increased deleterious burden (Giraud et al., 2001), dnaQ-derivatives did not displayed a particularly large reduction in growth rate or metabolic capacity compared to MG1655-derivatives with respect to their ancestors (77.7 vs. 72.8% reduction, p-value < 0.05, Fisher's exact test). On the contrary, even when mutations in electron transfer chain and exposure to aminoglycosides have been associated with the development of small colony variants (SCV) (McNamara and Proctor, 2000), only two (CIM8C and CIM8M) out of eight strains showed smaller colony sizes (data not shown). Of note, these strains were the only ones to maintain the normomutator phenotype after the evolutionary process, in line with previous suggestions that mutators could keep their evolutionary advantage through substituting secondary mutations that compensate costly phenotypes (Perron et al., 2010).
The effect of adaptation on the spontaneous mutant frequencies of MG1655 and MG1655 ΔdnaQ was analyzed. The MG1655 evolved strains CIM5H and CIM5N showed mutant frequencies two orders of magnitude higher than their ancestor MG1655 (Figure 1A). Genome sequence analysis of CIM5H and CIM5N revealed a H162Q substitution in dnaQ, affecting a functional residue, likely reducing its proofreading activity (Cisneros et al., 2009). Note that this change is unrelated with the ΔdnaQ derivative, ruling out the possibility of a cross-contamination from the MG1655 ΔdnaQ strain. On the other hand, the evolved ΔdnaQ derivative CIQ4J showed a frequency of spontaneous mutants one order of magnitude higher than that of its ancestor (Figure 1B). CIQ4J showed an I695S substitution in MutS, a key enzyme of the Mismatch Repair System (Junop et al., 2003). This is a semi-conserved residue located in a highly conserved region of the protein. The addition of a mutation on genes of the mismatch repair pathway (MMR) on a dnaQ background has shown to generate a slight increase in mutant frequency (Schaaper, 1993), thus this mutation on mutS may be involved in the activity of MutS and in the increase of mutant frequency in CIQ4J.
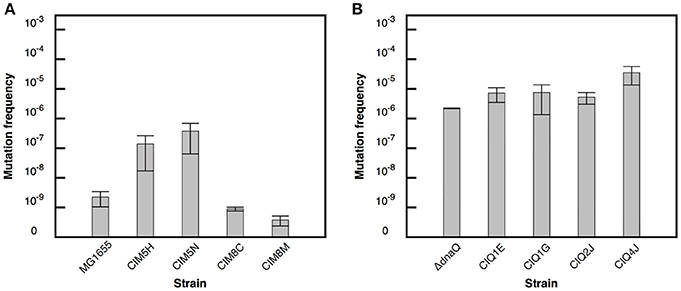
Figure 1. Mutation frequency estimates for the evolved strains. Frequency of nalidixic acid resistant mutants in the evolved strains derived from MG1655 (A). Frequency of rifampicin resistant mutants in the evolved strains derived from ΔdnaQ (B). Error bars correspond to the standard deviation of three independent replicates.
In order to identify the genetic modifications arisen during the evolutionary process related to gentamicin resistance, whole genome sequencing and bioinformatic analysis of all selected strains and their ancestors were performed. Agreeing with their ancestors' phenotype, ΔdnaQ-derived strains presented a higher number of mutations, being CIQ1G the strain with more genetic modifications (Table 3). After the evolutionary process, strains derived from MG1655 showed two different mutations profiles. While CIM8C and CIM8M retained the normomutator phenotype, showing 29 and 22 mutations, respectively, CIM5H and CIM5N developed a mutator phenotype, gaining 345 and 636 genetic modifications, respectively (Table 3).
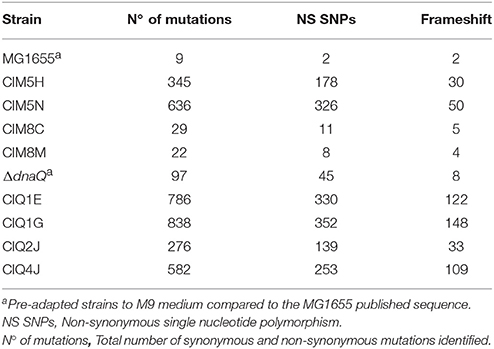
Table 3. Number of mutations in the gentamicin-resistant strains.
Despite the effort in pre-adapting both backgrounds to the medium without antibiotic, it remains the possibility that some of the observed mutations were linked to adaptation to the environment. For that reason, we focused on mutational targets for which previous knowledge rendered reasonable to expect some involvement with gentamycin susceptibility. All eight evolved strains shared non-synonymous mutations on five common candidate genetic elements: fhuA and fusA genes and atpIBEFHAGDC, cyoABCDE, and potABCD operons (Table 4). In Table 5 we show that, indeed, most of these mutations are associated with increases in resistance. Isolate CIM8M was selected as model for further studies in order to evaluate the putative effect of the acquired mutations on gentamicin resistance, due to its high-level resistance to gentamicin (MIC of 512 μg/ml) and lower number of mutations on its genome. Beside its mutations in the five common genetic elements, strain CIM8M presented a SNP in csrA, affecting the fifth residue (T5I) and a H526Y substitution in RpoB, which is related to its resistance to rifampicin. CsrA is a small RNA-binding protein that acts as a global regulator in E. coli, playing a key role in central carbon metabolism (Wei et al., 2000). It is also involved in biofilm formation and flagellum biosynthesis (Wei et al., 2001; Jackson et al., 2002).
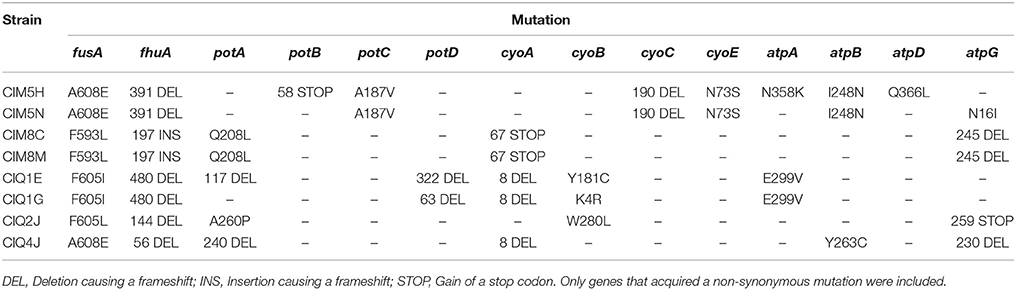
Table 4. Mutations in common genetic elements of strains resistant to gentamicin.
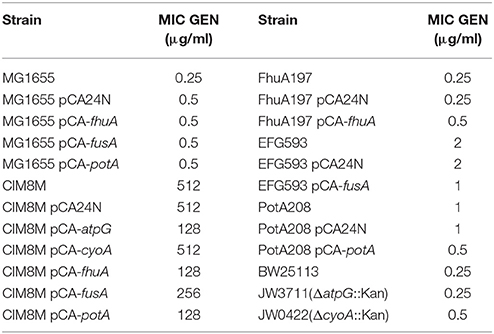
Table 5. Susceptibility to gentamicin of strains used in this study.
Related to the five common genetic elements, strain CIM8M presented mutations on atpG, cyoA, fhuA, fusA, and potA. In the case of atpG, cyoA, and fhuA, mutations generated frameshifts in residues 245, 68, and 197, respectively, while F593L and Q208L substitutions were identified in fusA and potA, respectively (Table 4).
fhuA encodes a ferrichrome transporter (Hantke and Braun, 1975) and its mutation in CIM8M, as well as in all evaluated strains, generated a frameshift on the amino acid sequence. In CIM8M the insertion of a nucleotide in position 197 generated a frameshift (Endriss et al., 2003). This mutation did not generate variations in the gentamicin susceptibility of the strain FhuA197, a MG1655-derivative with a nucleotide insertion at residue 197 of fhuA (constructed as indicated in Materials and Methods). Complementation of CIM8M with the wild-type fhuA gene reduced gentamicin MIC from 512 to 128 μg/ml (Table 5). No fitness cost was observed for FhuA197 (Table 2), nevertheless, the complementation indeed decreased the duplication time (Table 6), suggesting that in this genetic background it has a detrimental effect on bacterial fitness. Anyway, a positive effect of the expression of fhuA in multicopy on growth rate cannot be discarded.
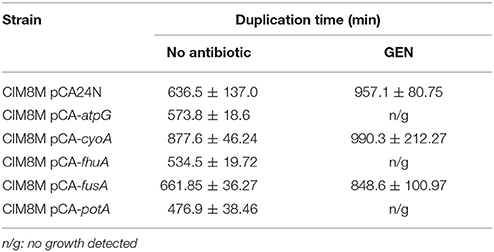
Table 6. Growth rate of CIM8M complemented with the wild type genotype of selected genes in presence and absence of gentamicin (128 μg/ml).
fusA encodes the Elongation Factor G (EF-G) and mutations were mapped to domains IV or V of the protein (Table 4), which are essential for its activity (Savelsbergh et al., 2000). CIM8M complementation with wild-type fusA produced a slight but consistent decrease on the MIC of gentamicin from 512 to 256 μg/ml (Table 5), and a bacterial growth improvement in absence of gentamicin (Supplementary Figure S1). The substitution F593L in fusA (strain EFG593) generated an 8-fold decrease in bacterial susceptibility to gentamicin respect MG1655 (Table 5), without any fitness cost (Table 2). However, complementation with wild-type fusA increased growth rate in the CIM8M strain (Supplementary Figure S1), suggesting that even if FusAF593L has no effect on fitness in a wild-type background, it is detrimental in the presence of other mutations.
We also identified alterations on electron transport chain, specifically on the atp and cyo operons. Even though electron transport chain has been mainly related to the aminoglycoside uptake due to the importance of membrane potential for this process (Damper and Epstein, 1981), it has been recently described that aminoglycosides accelerate respiration, potentiating their lethal effect on bacteria (Lobritz et al., 2015). CIM8M complementation with the wild-type atpG gene reduced gentamicin MIC (from 512 to 128 μg/ml) and duplication time in absence of gentamicin (Table 6). Complementation with the wild-type cyoA did not generate changes on its MIC of gentamicin, while a decrease in growth rate was observed (Table 6). Despite all efforts, it was not possible to generate single mutants on atpG and cyoA reproducing those on CIM8M. For that reason we resorted to use gene knockout strains from the KEIO collection (Baba et al., 2006), JW3711 (ΔatpG::Km) and JW0422 (ΔcyoA::Km), to evaluate the effect of these genotypes on gentamicin resistance. The cyoA deletion caused a 2-fold decrease in gentamicin susceptibility (Table 5). In contrast to previous studies where atpG deletion caused an increase in gentamicin susceptibility (Liu et al., 2010), we failed to observe any effect over resistance. On the other hand, complementation of CIM8M with the wild-type atpG gene reduced its resistance to gentamicin and decreased its duplication time (Tables 5, 6). Because aminoglycoside uptake is related to growth rate in absence of the antibiotic (Muir et al., 1984), this genetic modification could generate a decreased uptake. The difference in gentamicin susceptibility between atpG deletion in JW3711 and atpG complementation in CIM8M may be related to the characteristics of the mutation. It has been described that only certain genetic changes in atpG generate decreased aminoglycoside susceptibility (Mogre et al., 2014). While JW3711 has a complete deletion of the protein, CIM8M presents a frameshift in position 245.
The fifth mutation identified in all resistant strains affected the potABCD operon, encoding the spermidine-preferential uptake system PotABCD (Igarashi and Kashiwagi, 1999). Polyamines are involved in several cellular processes such as energy metabolism, oxidative stress tolerance, biofilm formation, and iron transport, as well as in gene expression of an oligopeptide transporter OppA (Tkachenko and Nesterova, 2003; Yoshida et al., 2004; Karatan et al., 2005; Patel et al., 2006; Tkachenko et al., 2012). PotAQ208L identified in CIM8M affected the ATPase subunit of the transporter (Igarashi and Kashiwagi, 1996), generating a 4-fold increase in gentamicin MIC (Table 5), without fitness cost (Table 2). It is worth noting that both transporters, OppA and PotABCD, have been described to be able to uptake some aminoglycosides into bacterial cell (Holtje, 1978; Acosta et al., 2000), suggesting that PotAQ208L may generate an altered gentamicin intake. In CIM8M, complementation with wild-type potA reduced gentamicin MIC 4-fold (Table 5). Also, we observed a reduction in the cellular growth in absence of this antibiotic (Supplementary Figure S1).
Besides gentamicin resistance, CIM8M presented increases in both kanamycin and amikacin MICs (Supplementary Table S9), indicating that its mechanism of resistance is shared with other aminoglycosides.
Evolutionary trajectories of the common resistance determinants were determined by analyzing the time of apparition of mutations along the experiment. For this purpose, two single colonies were randomly selected from chemostat populations at different gentamicin concentrations. Only clones from populations obtained at 0.5, 2.0, 4.0, and 8.0 μg/ml gentamicin were selected, as isolates from higher gentamicin concentrations showed high-level gentamicin resistance. For instance, gentamycin MIC of isolates from 8.0 μg/ml ranged from 16 to >128 μg/ml (data not shown). We chose five loci (atpG, cyoA, fhuA, fusA, and potA) as representative of the five common genetic elements that were repeatedly targeted by selection to be subjected to PCR amplification and Sanger sequencing. Results show a sequential and cumulative appearance of mutations in both backgrounds, MG1655 and its hyper-mutator derivative (Figure 2; Supplementary Table S10). The first locus to present alterations was fusA (observed first, in both backgrounds, at 0.5 μg/ml). Secondly, we found mutations in cyoA and potA, observed earlier in the hypermutator than in the normomutator (0.5 vs. 4 μg/ml). Next, we detected mutations in fhuA, also appearing earlier in the hypermutator than in the normomutator (2 vs. 8 μg/ml). The last mutations to be added mapped onto atpG, being present in both backgrounds from a concentration of 8 μg/ml onwards.
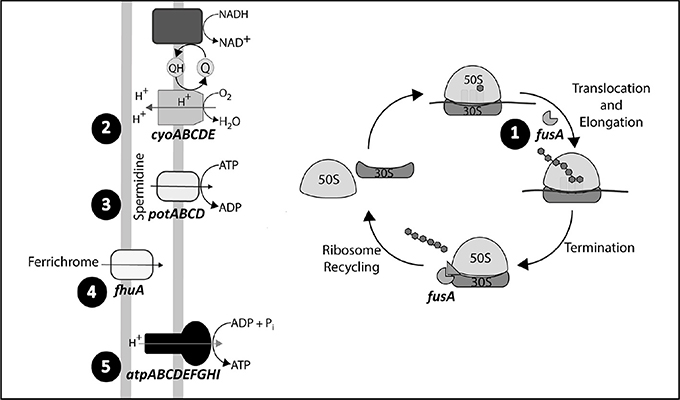
Figure 2. Mutational trajectories. Order of appearance of mutations in the selected genetic elements: fhuA, fusA, and atp, cyo, and potABCD operons. Mutations in fusA where identified from a gentamicin concentration of 0.5 μg/ml in MG1655 and its ΔdnaQ derivative, while mutations in the cyo and potABCD operons where detected from an antibiotic concentration of 4 and 0.5 μg/ml in MG1655 and ΔdnaQ, respectively. Genetic modifications in fhuA were identified from a gentamicin concentration of 8 and 2 μg/ml in MG1655 and ΔdnaQ, respectively, while mutations on the atp operon were detected from a gentamicin concentration of 8 μg/ml in both ancestors.
The effect of gentamicin over oxidative stress has been widely studied and discussed during the past years. While several studies have established that these antibiotics generate increased concentration of reactive oxygen species (ROS) as a consequence of antibiotic-stress and that this oxidative stress contributes to the bactericidal effect of aminoglycosides (Zhao and Drlica, 2014), other studies indicate that bactericidal effect of aminoglycosides is unrelated to oxidative stress (Keren et al., 2013; Liu and Imlay, 2013). On the other hand, Ezraty et al. (2013) established that iron concentration is related to gentamicin susceptibility in a Fenton-independent manner, where iron depletion reduces the activity of respiratory Complex I and therefore reduces proton motive force generation (essential for aminoglycoside uptake). Consequently, iron depletion reduces aminoglycosides entry and bacterial susceptibility to them (Ezraty et al., 2013). In order to evaluate if gentamicin resistance is related to a reduction in oxidative stress, the susceptibility of gentamicin-resistant strains to hydrogen peroxide was evaluated. Hydrogen peroxide, as well as increased intracellular concentration of iron, induces oxidative stress due to Fenton's reaction, where ferrous iron is reduced by superoxide, generating hydroxyl radicals. Therefore, an increase in hydrogen peroxide or iron concentration may lead to oxidative stress. Unexpectedly, all resistant strains were more susceptible to hydrogen peroxide than their ancestors (p-value < 0.05, two-samples t-test) (Figure 3), suggesting that their mechanisms of resistance are not related to enhanced tolerance to ROS. Differences in the effect of aminoglycoside over oxidative stress may be related to differences in experimental approaches and cell growth status. Wang et al. recently established that several genes involved in oxidative stress protection are under RpoS regulation in late exponential and early stationary phase of growth, showing that aminoglycosides induce oxidative stress in this phase of growth and that RpoS is directly related to oxidative stress protection (Wang et al., 2014). Because these strains were evolved in continuous cultures, maintaining the exponential phase of growth, the effect of gentamicin on oxidative stress may have not been significant in this condition and, therefore, the mechanism of resistance of these strains does not involve decreased susceptibility to ROS.
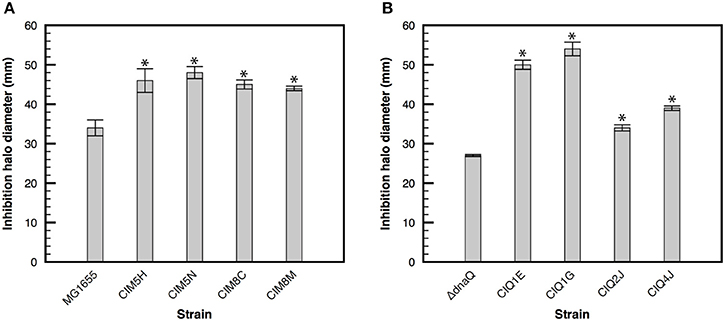
Figure 3. Susceptibility to hydrogen peroxide of evolved strains. Bacterial susceptibility to hydrogen peroxide was determined with disks containing 26 μmoles of hydrogen peroxide in M9 minimal medium supplemented with glucose. Halo of inhibition in the presence of hydrogen peroxide of the evolved strains derived from MG1655 (A) and its ΔdnaQ derivative (B). Asterisk denotes significance at p < 0.05.
In order to evaluate if the evolution to gentamicin resistance altered the susceptibility to other antibiotics, the MICs of nine antimicrobial compounds were determined (Supplementary Tables S5, S6). All selected strains developed fosfomycin resistance, but whole genome sequencing revealed that they did not present mutations on genes previously related to this phenotype (Nilsson et al., 2003; Takahata et al., 2010; Couce et al., 2012). Nevertheless, 6 and 4 out of 8 strains were unable to grow with glucose-6-phosphate and glycerol-3-phosphate, respectively, as sole energy and carbon source (Supplementary Table S7), suggesting that a defect in fosfomycin uptake may be involved in their resistance. On the other hand, to evaluate whether gentamicin resistance genetic determinants were related to this phenotype, the MIC of fosfomycin was determined for CIM8M and its derivatives complemented with the wild-type genes (Supplementary Table S11). Except for a weak reduction with fhuA, complementation with wild-type genes did not modified fosfomycin resistance. Further studies are required to elucidate the genetic basis of this indirectly acquired resistance.
Besides fosfomycin resistance, all studied strains derived from MG1655, but not from its ΔdnaQ derivative, developed rifampicin resistance due to a H526Y substitution in RpoB (Cavusoglu et al., 2006; Ma et al., 2006). The H526Y mutation is known to cause temperature-sensitive growth and decreased termination efficiency at Rho-dependent and independent transcriptional terminators in vivo and in vitro (Jin and Gross, 1989), increasing the transcription elongation rate and decreasing pausing in vitro (McDowell et al., 1994). Changes in rpoB have been related to changes in the expression of hundreds of genes and large fitness advantages (Rodríguez-Verdugo et al., 2016). The importance of this mutation, if any, on the adaptation to gentamicin remains to be studied. Its absence from the genomes of ΔdnaQ-derivatives merits also a brief consideration. A simple possibility is that the wild-type mutational spectrum is particularly prone to produce the CAC → TAC substitution that underlies mutation H526Y, although this was not found in previous studies (Couce et al., 2013). Another, more speculative possibility is that many of the neutral or slightly deleterious mutations that the mutator has accumulated become strongly deleterious due to the pleiotropic effects associated to changes in the RNA polymerase (Rodríguez-Verdugo et al., 2016), precluding its selection. Interestingly, the ΔdnaQ-derivative CIQ4J displays an increased rifampicin resistance, yet without any mutation in rpoB. This is an intriguing result that deserves further studies to stablish the molecular basis of this resistance mechanism.
On the contrary, all selected strains increased their susceptibility to chloramphenicol (Table 1). This finding agrees with previous reports that indicate that aminoglycoside resistance is related to an increased susceptibility to chloramphenicol (Imamovic and Sommer, 2013; Lázár et al., 2013; Lobritz et al., 2015). It has been proposed that this phenomenon could be related to alterations in electron transport chain and thus a reduction in the activity of efflux pumps that export chloramphenicol but not aminoglycosides, like AcrAB-TolC (Nishino and Yamaguchi, 2001). The effect of single mutations on previously selected genes related to gentamicin resistance over chloramphenicol susceptibility was evaluated through MIC of the single gene mutants (Supplementary Table S12). FhuA197 and EFG593 were the only strains that decreased 2-fold their MIC of chloramphenicol, while atpG deletion increased it 2-fold, suggesting that, at least in an individual manner, these mutations do not explain CIM8M increase in chloramphenicol susceptibility from 4 to ≤ 0.25 μg/ml. Liu et al. determined the contribution of gene deletions in E. coli over antibiotic sensibility, including chloramphenicol (Liu et al., 2010). However, no mutations on genes described by Lui et al. were detected in CIM8M. An effect of the other mutations (synonymous, intergenic, etc.) found in this and other strains on gene regulation cannot be discarded.
In this work, we studied the patterns of gentamicin resistance development in E. coli under high and low mutation supply rates. For this purpose, a normomutator (MG1655) and a hypermutator (MG1655 ΔdnaQ) strain were exposed to increasing concentrations of gentamicin in chemostats. We found that both genetic backgrounds were capable of evolving resistance to antibiotic concentrations >1,000 higher than the ancestral MIC. All sequenced gentamicin-resistant strains carried mutations on five common loci: the cytochrome bo3 oxidase (cyoABCDE operon), the ATP synthase (atpIBEFHAGDC operon), the PotABCD transporter (potABCD operon), the ferrichrome transporter (fhuA), and the Elongation Factor G (fusA). These targets can be grouped according to at least two putative resistance mechanisms: inhibition of gentamicin uptake (cyoABCDE, atpIBEFHAGDC, and potABCD) and alteration of the antibiotic binding to the ribosome (fusA). The mechanistic basis of fhuA-based resistance remains to be elucidated.
The first group of loci is related to aminoglycoside uptake. This process requires a threshold membrane potential (Mates et al., 1982), thus modifications in electron transport chain may lead to decreased antibiotic uptake (Miller et al., 1980; Bryan and Kwan, 1983). Cytochrome mutations have been widely associated with aminoglycoside resistance in E. coli, Pseudomonas aeruginosa, Listeria monocytogenes, and Staphylococcus aureus (Miller et al., 1980; Schurek et al., 2008; Kastbjerg et al., 2014; Lázár et al., 2014). We found that cyoA mutations contribute to a small extent to the resistance phenotype and that this impact on resistance is lost after new mutations arise, suggesting that its contribution is only important at low antibiotic concentrations. Regarding atpG mutations, its effect on gentamicin resistance seems to be related to structural changes in the γ-subunit of the ATP synthase rather than to the loss of its functionality, agreeing with Mogre et al. who described that only certain gene deletions generate reduced aminoglycoside susceptibility (Mogre et al., 2014). These structural modifications may cause a proton leakage, reducing membrane potential, and gentamicin uptake (Humbert and Altendorf, 1989). Even though the main mechanism of entry of these antibiotics into the cell is a three-phase mechanism that requires membrane potential, it has been described that other membrane proteins can transport aminoglycoside into the cell (Holtje, 1978; Acosta et al., 2000). This is the case of the PotABCD transporter, the preferential uptake system for the polyamine spermidine, which is able to transport also streptomycin (Holtje, 1978). Despite potA mutations being observed in aminoglycoside-resistant strains of E. coli in prior studies (Lázár et al., 2014), this work provides further evidence that potA non-synonymous SNP contributes to a resistance phenotype. Of note, the culture medium in this study did not contain polyamines, discarding that the effect of potA mutations on gentamicin resistance is linked to a reduction in polyamine concentration and an altered gene expression of the genetic elements regulated by these compounds.
Functionally speaking, the second group of mutations is related to the translation. Mutations in fusA have been associated with relevant resistance levels in E. coli, P. aeruginosa, and L. monocytogenes (Hou et al., 1994; Wang et al., 2015; Feng et al., 2016). Despite these reports, the fact that FusAF593L alters only mildly gentamicin susceptibility in the strain EFG593 and the rise of this mutation at early in our experiment, suggests that mutations in this locus contribute to gentamicin resistance mainly at low concentrations. As advanced above, the effect of fhuA mutation alone on gentamicin resistance could not be identified. Despite the mutation in FhuA197 having no impact on resistance, its complementation in the strain CIM8M reduces gentamicin MIC, suggesting that its effect depends on the genetic background. Overall, the effect of each of the five mutations over gentamicin susceptibility depends to different degrees on the genetic background. Interestingly, the development of resistance in the model strain CIM8M did not involve mutations in genetic determinants of gentamicin resistance or their regulatory regions (Soo et al., 2011; Krahn et al., 2012; Lau et al., 2015).
In agreement with Lázár et al. our results show that the development of aminoglycoside resistance does not generate extensive multi-drug resistance (Lázár et al., 2014). We did find, however, a strong association of gentamicin and fosfomycin resistance. Although the genetic basis of this cross-resistance remains to be elucidated, it may imply a threat for the usage of fosfomycin, an old antibiotic that has regained clinical interest. In general, more work is needed to understand how indirect selection influences the dynamics of resistance in clinical settings.
Here we documented an example of parallel evolution, in which selection targeted the same genetic elements in independent replicate populations (Stern, 2013). Similar observations have been reported in diverse organisms, including virus (Wichman et al., 1999), bacteria (Woods et al., 2006), and yeast (Gerstein et al., 2012). Some degree of parallelism is understandably common insofar most studies typically start from a single genotype and involve directional selective pressures: parallelism might simply reflect that the space of possible solutions is very limited (Lee and Marx, 2013), or that strong mutational biases repeatedly drive populations toward a particular solution (Stoltzfus and McCandlish, 2017). To gain further insight into the shape of the underlying adaptive landscape, researchers have used different experimental approaches. Multiple fitness peaks can be revealed by massively increasing the number of replicates (Tenaillon et al., 2012), blocking the major adaptive pathways through genetic manipulation (Lind et al., 2015), using strains with strong and contrasting mutational biases (Couce et al., 2015), or by studying evolutionary trajectories under different mutational supplies (Rozen et al., 2008). This last approach was followed by a recent study, similar to ours, in which bacterial populations carrying the beta-lactamase TEM1 were exposed to increasing concentrations of a drug to which the wild-type allele displayed low activity (Salverda et al., 2017). By altering the population size, the effect of the mutation supply on the evolutionary trajectories was explicitly tested. Their main result is that larger populations, following the adaptive pathways with the steeper slope, were repeatedly trapped in a local maximum; while a fraction of the smaller populations managed to find a secondary, higher fitness peak. In contrast, we observed that populations with orders-of-magnitude differences in mutation supply rate followed roughly the same adaptive pathways, albeit at a different pace. This observation suggests that local maxima, if present, are not readily accessible in our experiment. A possible explanation for these differences may be found in the fact that our system involves the combination of mutations in several independent loci, whereas in Salverda et al. all adaptation was restricted to a single locus. Intra-locus epistasis is thought to be generally more frequent than inter-gene epistasis (Watson et al., 2006), partly due to the strong stability-activity trade-offs exhibited by many proteins (Thomas et al., 2010). Overall, our work raises the possibility that antibiotics for which resistance involves multiple loci exhibit relatively smooth fitness landscapes, which would cause evolutionary outcomes to be more repeatable and less sensitive to population bottlenecks. More experiments with other antibiotics need to be conducted to establish the generality of this pattern.
Our results show that high-level resistance to gentamicin can be readily achieved through the accumulation of multiple mutations with moderate to low effects on susceptibility. We shall stress, however, that caution must be taken in extrapolating these findings to clinical settings. Hospital environments are very complex, with multiple antibiotics and biocides at different concentrations challenging bacterial populations at different times. In contrast, our experimental setting has been purposely designed to be simple and homogeneous. Inclusion of multiple antimicrobial agents and fluctuating concentrations will be necessary in future studies to constitute a more realistic account of clinical settings.
CI-Q designed and performed experiments, analyzed data, and wrote the manuscript; JO performed bioinformatic analysis; AC contributed to the experimental design, statistical analysis, interpretation of the results and writing the manuscript. JB designed the work, analyzed data, and wrote the manuscript. All authors approved the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank people from Victor de Lorenzo and José Luis Martínez groups for helping with some experiments. Supported by Plan Nacional de I+D+i 2013-2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía, Industria y Competitividad, Spanish Network for Research in Infectious Diseases (REIPI RD16/0016/0009)—cofinanced by European Development Regional Fund A way to achieve Europe, Operative program Intelligent Growth 2014-2020 and grant FIS PI13/00063 from Instituto de Salud Carlos III. CI-Q was supported by grant Becas Chile Doctorado en el Extranjero (72110933), Consejo Nacional de Investigación Científica y Tecnológica, Chilean Government, and CIDI-DIUV 4/2016.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00427/full#supplementary-material
Acosta, M. B., Ferreira, R. C., Padilla, G., Ferreira, L. C., and Costa, S. O. (2000). Altered expression of oligopeptide-binding protein (OppA) and aminoglycoside resistance in laboratory and clinical Escherichia coli strains. J. Med. Microbiol. 49, 409–413. doi: 10.1099/0022-1317-49-5-409
Ahmad, M. H., Rechenmacher, A., and Böck, A. (1980). Interaction between aminoglycoside uptake and ribosomal resistance mutations. Antimicrob. Agents Chemother. 18, 798–806. doi: 10.1128/AAC.18.5.798
Andersson, D. I., and Levin, B. R. (1999). The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2, 489–493. doi: 10.1016/S1369-5274(99)00005-3
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006), Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006 0008. doi: 10.1038/msb4100050
Bryan, L. E., and Kwan, S. (1983). Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob. Agents Chemother. 23, 835–845. doi: 10.1128/AAC.23.6.835
Cavusoglu, C., Turhan, A., Akinci, P., and Soyler, I. (2006). Evaluation of the Genotype MTBDR assay for rapid detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis isolates. J. Clin. Microbiol. 44, 2338–2342. doi: 10.1128/JCM.00425-06
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Cisneros, G. A., Perera, L., Schaaper, R. M., Pedersen, L. C., London, R. E., Pedersen, L. G., et al. (2009). Reaction mechanism of the epsilon subunit of E. coli DNA polymerase III: insights into active site metal coordination and catalytically significant residues. J. Am. Chem. Soc. 131, 1550–1556. doi: 10.1021/ja8082818
Couce, A., Briales, A., Rodríguez-Rojas, A., Costas, C., Pascual, A., and Blázquez, J. (2012). Genomewide overexpression screen for fosfomycin resistance in Escherichia coli: MurA confers clinical resistance at low fitness cost. Antimicrob. Agents Chemother. 56, 2767–2769. doi: 10.1128/AAC.06122-11
Couce, A., Guelfo, J. R., and Blázquez, J. (2013). Mutational spectrum drives the rise of mutator bacteria. PLoS Genet. 9:e1003167. doi: 10.1371/journal.pgen.1003167
Couce, A., Rodríguez-Rojas, A., and Blázquez, J. (2015). Bypass of genetic constraints during mutator evolution to antibiotic resistance. Proc. Biol. Sci. 282:20142698. doi: 10.1098/rspb.2014.2698
Couce, A., Rodríguez-Rojas, A., and Blázquez, J. (2016). Determinants of genetic diversity of spontaneous drug resistance in bacteria. Genetics 203, 1369–1380. doi: 10.1534/genetics.115.185355
Damper, P. D., and Epstein, W. (1981). Role of the membrane potential in bacterial resistance to aminoglycoside antibiotics. Antimicrob. Agents Chemother. 20, 803–808. doi: 10.1128/AAC.20.6.803
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Davies, J., and Davies, D. (2010). Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433. doi: 10.1128/MMBR.00016-10
de Lorenzo, V., and Timmis, K. N. (1994). Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235, 386–405. doi: 10.1016/0076-6879(94)35157-0
de Visser, J. A., and Krug, J. (2014). Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. doi: 10.1038/nrg3744
Didelot, X., and Maiden, M. C. (2010). Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322. doi: 10.1016/j.tim.2010.04.002
Endriss, F., Braun, M., Killmann, H., and Braun, V. (2003). Mutant analysis of the Escherichia coli FhuA protein reveals sites of FhuA activity. J. Bacteriol. 185, 4683–4692. doi: 10.1128/JB.185.16.4683-4692.2003
Ezraty, B., Vergnes, A., Banzhaf, M., Duverger, Y., Huguenot, A., Brochado, A. R., et al. (2013). Fe-S cluster biosynthesis controls uptake of aminoglycosides in a ROS-less death pathway. Science 340, 1583–1587. doi: 10.1126/science.1238328
El'Garch, F., Jeannot, K., Hocquet, D., Llanes-Barakat, C., and Plesiat, P. (2007). Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 51, 1016–1021. doi: 10.1128/AAC.00704-06
Feng, Y., Jonker, M. J., Moustakas, I., Brul, S., and Ter Kuile, B. H. (2016). Dynamics of mutations during development of resistance by Pseudomonas aeruginosa against five antibiotics. Antimicrob. Agents Chemother. 60, 4229–4236. doi: 10.1128/AAC.00434-16
Garneau-Tsodikova, S., and Labby, K. J. (2016). Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. Medchemcomm 7, 11–27. doi: 10.1039/C5MD00344J
Gerstein, A. C., Lo, D. S., and Otto, S. P. (2012). Parallel genetic changes and nonparallel gene-environment interactions characterize the evolution of drug resistance in yeast. Genetics 192, 241–252. doi: 10.1534/genetics.112.142620
Giraud, A., Radman, M., Matic, I., and Taddei, F. (2001). The rise and fall of mutator bacteria. Curr. Opin. Microbiol. 4, 582–585. doi: 10.1016/S1369-5274(00)00254-X
Hall, B. G., Acar, H., Nandipati, A., and Barlow, M. (2014). Growth rates made easy. Mol. Biol. Evol. 31, 232–238. doi: 10.1093/molbev/mst187
Handel, A., and Rozen, D. E. (2009). The impact of population size on the evolution of asexual microbes on smooth versus rugged fitness landscapes. BMC Evol. Biol. 9:236. doi: 10.1186/1471-2148-9-236
Hantke, K., and Braun, V. (1975). Membrane receptor dependent iron transport in Escherichia coli. FEBS Lett. 49, 301–305. doi: 10.1016/0014-5793(75)80771-X
Herring, C. D., Glasner, J. D., and Blattner, F. R. (2003). Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311, 153–163. doi: 10.1016/S0378-1119(03)00585-7
Holtje, J. V. (1978). Streptomycin uptake via an inducible polyamine transport system in Escherichia coli. Eur. J. Biochem. 86, 345–351. doi: 10.1111/j.1432-1033.1978.tb12316.x
Hou, Y., Lin, Y. P., Sharer, J. D., and March, P. E. (1994). In vivo selection of conditional-lethal mutations in the gene encoding elongation factor G of Escherichia coli. J. Bacteriol. 176, 123–129. doi: 10.1128/jb.176.1.123-129.1994
Humbert, R., and Altendorf, K. (1989). Defective gamma subunit of ATP synthase (F1F0) from Escherichia coli leads to resistance to aminoglycoside antibiotics. J. Bacteriol. 171, 1435–1444. doi: 10.1128/jb.171.3.1435-1444.1989
Igarashi, K., and Kashiwagi, K. (1996). Polyamine transport in Escherichia coli. Amino Acids 10, 83–97. doi: 10.1007/BF00806095
Igarashi, K., and Kashiwagi, K. (1999). Polyamine transport in bacteria and yeast. Biochem. J. 344, Pt 3 633–642.
Imamovic, L., and Sommer, M. O. (2013). Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med. 5, 204ra132. doi: 10.1126/scitranslmed.3006609
Jackson, D. W., Suzuki, K., Oakford, L., Simecka, J. W., Hart, M. E., and Romeo, T. (2002). Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J. Bacteriol. 184, 290–301. doi: 10.1128/JB.184.1.290-301.2002
Jana, S., and Deb, J. K. (2006). Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 70, 140–150. doi: 10.1007/s00253-005-0279-0
Jin, D. J., and Gross, C. A. (1988). Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 202, 45–58. doi: 10.1016/0022-2836(88)90517-7
Jin, D. J., and Gross, C. A. (1989). Three rpoBC mutations that suppress the termination defects of rho mutants also affect the functions of nusA mutants. Mol. Gen. Genet. 216, 269–275. doi: 10.1007/BF00334365
Junop, M. S., Yang, W., Funchain, P., Clendenin, W., and Miller, J. H. (2003). In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair 2, 387–405. doi: 10.1016/S1568-7864(02)00245-8
Karatan, E., Duncan, T. R., and Watnick, P. I. (2005). NspS, a predicted polyamine sensor, mediates activation of Vibrio cholerae biofilm formation by norspermidine. J. Bacteriol. 187, 7434–7443. doi: 10.1128/JB.187.21.7434-7443.2005
Kastbjerg, V. G., Hein-Kristensen, L., and Gram, L. (2014). Triclosan-induced aminoglycoside-tolerant Listeria monocytogenes isolates can appear as small-colony variants. Antimicrob. Agents Chemother. 58, 3124–3132. doi: 10.1128/AAC.02266-13
Keren, I., Wu, Y., Inocencio, J., Mulcahy, L. R., and Lewis, K. (2013). Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339, 1213–1216. doi: 10.1126/science.1232688
Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., et al. (2005). Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299. doi: 10.1093/dnares/dsi012
Krahn, T., Gilmour, C., Tilak, J., Fraud, S., Kerr, N., Lau, C. H., et al. (2012). Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56, 5591–5602. doi: 10.1128/AAC.01446-12
Lan, R., and Reeves, P. R. (1996). Gene transfer is a major factor in bacterial evolution. Mol. Biol. Evol. 13, 47–55. doi: 10.1093/oxfordjournals.molbev.a025569
Lau, C. H., Krahn, T., Gilmour, C., Mullen, E., and Poole, K. (2015). AmgRS-mediated envelope stress-inducible expression of the mexXY multidrug efflux operon of Pseudomonas aeruginosa. Microbiologyopen 4, 121–135. doi: 10.1002/mbo3.226
Lázár, V., Nagy, I., Spohn, R., Csörgo, B., Györkei, Á., Nyerges, Á., et al. (2014). Genome-wide analysis captures the determinants of the antibiotic cross-resistance interaction network. Nat. Commun. 5, 4352. doi: 10.1038/ncomms5352
Lázár, V., Pal Singh, G., Spohn, R., Nagy, I., Horváth, B., Hrtyan, M., et al. (2013). Bacterial evolution of antibiotic hypersensitivity. Mol. Syst. Biol. 9:700. doi: 10.1038/msb.2013.57
Leavis, H. L., Willems, R. J., Top, J., and Bonten, M. J. (2006). High-level ciprofloxacin resistance from point mutations in gyrA and parC confined to global hospital-adapted clonal lineage CC17 of Enterococcus faecium. J. Clin. Microbiol. 44, 1059–1064. doi: 10.1128/JCM.44.3.1059-1064.2006
Lee, M. C., and Marx, C. J. (2013). Synchronous waves of failed soft sweeps in the laboratory: remarkably rampant clonal interference of alleles at a single locus. Genetics 193, 943–952. doi: 10.1534/genetics.112.148502
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lind, P. A., Farr, A. D., and Rainey, P. B. (2015). Experimental evolution reveals hidden diversity in evolutionary pathways. eLife 4:e07074. doi: 10.7554/eLife.07074
Liu, A., Tran, L., Becket, E., Lee, K., Chinn, L., Park, E., et al. (2010). Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob. Agents Chemother. 54, 1393–1403. doi: 10.1128/AAC.00906-09
Liu, Y., and Imlay, J. A. (2013). Cell death from antibiotics without the involvement of reactive oxygen species. Science 339, 1210–1213. doi: 10.1126/science.1232751
Lobritz, M. A., Belenky, P., Porter, C. B., Gutierrez, A., Yang, J. H., Schwarz, E. G., et al. (2015). Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 8173–8180. doi: 10.1073/pnas.1509743112
Lukacišinová, M., and Bollenbach, T. (2017). Toward a quantitative understanding of antibiotic resistance evolution. Curr. Opin. Biotechnol. 46, 90–97. doi: 10.1016/j.copbio.2017.02.013
Ma, X., Wang, H., Deng, Y., Liu, Z., Xu, Y., Pan, X., et al. (2006). rpoB Gene mutations and molecular characterization of rifampin-resistant Mycobacterium tuberculosis isolates from Shandong Province, China. J. Clin. Microbiol. 44, 3409–3412. doi: 10.1128/JCM.00515-06
Martinez, J. L., Coque, T. M., and Baquero, F. (2015). What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 13, 116–123. doi: 10.1038/nrmicro3399
Mates, S. M., Eisenberg, E. S., Mandel, L. J., Patel, L., Kaback, H. R., and Miller, M. H. (1982). Membrane potential and gentamicin uptake in Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 79, 6693–6697. doi: 10.1073/pnas.79.21.6693
McDowell, J. C., Roberts, J. W., Jin, D. J., and Gross, C. (1994). Determination of intrinsic transcription termination efficiency by RNA polymerase elongation rate. Science 266, 822–825. doi: 10.1126/science.7526463
McNamara, P. J., and Proctor, R. A. (2000). Staphylococcus aureus small colony variants, electron transport and persistent infections. Int. J. Antimicrob. Agents 14, 117–122. doi: 10.1016/S0924-8579(99)00170-3
Miller, M. H., Edberg, S. C., Mandel, L. J., Behar, C. F., and Steigbigel, N. H. (1980). Gentamicin uptake in wild-type and aminoglycoside-resistant small-colony mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 18, 722–729. doi: 10.1128/AAC.18.5.722
Mogre, A., Sengupta, T., Veetil, R. T., Ravi, P., and Seshasayee, A. S. (2014). Genomic analysis reveals distinct concentration-dependent evolutionary trajectories for antibiotic resistance in Escherichia coli. DNA Res. 21, 711–726. doi: 10.1093/dnares/dsu032
Monod, J. (1950). La technique de culture continue, theorie et applications. Ann. Inst. Pasteur 79, 390–410.
Muir, M. E., van Heeswyck, R. S., and Wallace, B. J. (1984). Effect of growth rate on streptomycin accumulation by Escherichia coli and Bacillus megaterium. J. Gen. Microbiol. 130, 2015–2022. doi: 10.1099/00221287-130-8-2015
Nilsson, A. I., Berg, O. G., Aspevall, O., Kahlmeter, G., and Andersson, D. I. (2003). Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob. Agents Chemother. 47, 2850–2858. doi: 10.1128/AAC.47.9.2850-2858.2003
Nishino, K., and Yamaguchi, A. (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001
Oliver, A., Cantón, R., Campo, P., Baquero, F., and Blázquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1254. doi: 10.1126/science.288.5469.1251
Pál, C., Papp, B., and Lázár, V. (2015). Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 23, 401–407. doi: 10.1016/j.tim.2015.02.009
Patel, C. N., Wortham, B. W., Lines, J. L., Fetherston, J. D., Perry, R. D., and Oliveira, M. A. (2006). Polyamines are essential for the formation of plague biofilm. J. Bacteriol. 188, 2355–2363. doi: 10.1128/JB.188.7.2355-2363.2006
Perron, G. G., Hall, A. R., and Buckling, A. (2010). Hypermutability and compensatory adaptation in antibiotic-resistant bacteria. Am. Nat. 176, 303–311. doi: 10.1086/655217
Piñero-Lambea, C., Bodelón, G., Fernández-Periáñez, R., Cuesta, A. M., Álvarez-Vallina, L., and Fernández, L. Á. (2015). Programming controlled adhesion of E. coli to Target surfaces, cells, and tumors with synthetic adhesins. ACS Synth. Biol. 4, 463–473. doi: 10.1021/sb500252a
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Rodríguez-Verdugo, A., Tenaillon, O., and Gaut, B. S. (2016). First-Step mutations during adaptation restore the expression of hundreds of genes. Mol. Biol. Evol. 33, 25–39. doi: 10.1093/molbev/msv228
Rosenberg, S. M. (2001). Evolving responsively: adaptive mutation. Nat. Rev. Genet. 2, 504–515. doi: 10.1038/35080556
Rozen, D. E., Habets, M. G., Handel, A., and de Visser, J. A. (2008). Heterogeneous adaptive trajectories of small populations on complex fitness landscapes. PLoS ONE 3:e1715. doi: 10.1371/journal.pone.0001715
Salverda, L. M., Koomen, J., Koopmanschap, B., Zwart, M. P., and de Visser, J. A. G. M. (2017). Adaptive benefits from small mutation supplies in an antibiotic resistance enzyme. Proc. Natl. Acad. Sci. U.S.A. 114, 12773–12778. doi: 10.1073/pnas.1712999114
Savelsbergh, A., Matassova, N. B., Rodnina, M. V., and Wintermeyer, W. (2000). Role of domains 4 and 5 in elongation factor G functions on the ribosome. J. Mol. Biol. 300, 951–961. doi: 10.1006/jmbi.2000.3886
Schaaper, R. M. (1993). Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 268, 23762–23765.
Scheuermann, R., Tam, S., Burgers, P. M., Lu, C., and Echols, H. (1983). Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: a fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. U.S.A. 80, 7085–7089. doi: 10.1073/pnas.80.23.7085
Schurek, K. N., Marr, A. K., Taylor, P. K., Wiegand, I., Semenec, L., Khaira, B. K., et al. (2008). Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52, 4213–4219. doi: 10.1128/AAC.00507-08
Sniegowski, P. D., Gerrish, P. J., and Lenski, R. E. (1997). Evolution of high mutation rates in experimental populations of E. coli. Nature 387, 703–705. doi: 10.1038/42701
Soo, V. W., Hanson-Manful, P., and Patrick, W. M. (2011). Artificial gene amplification reveals an abundance of promiscuous resistance determinants in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 1484–1489. doi: 10.1073/pnas.1012108108
Stern, D. L. (2013). The genetic causes of convergent evolution. Nat. Rev. Genet. 14, 751–764. doi: 10.1038/nrg3483
Stoltzfus, A., and McCandlish, D. M. (2017). Mutational biases influence parallel adaptation. Mol. Biol. Evol. 34, 2163–2172. doi: 10.1093/molbev/msx180
Taddei, F., Radman, M., Maynard-Smith, J., Toupance, B., Gouyon, P. H., and Godelle, B. (1997). Role of mutator alleles in adaptive evolution. Nature 387, 700–702. doi: 10.1038/42696
Takahata, S., Ida, T., Hiraishi, T., Sakakibara, S., Maebashi, K., Terada, S., et al. (2010). Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 35, 333–337. doi: 10.1016/j.ijantimicag.2009.11.011
Tenaillon, O., Rodríguez-Verdugo, A., Gaut, R. L., McDonald, P., Bennett, A. F., Long, A. D., et al. (2012). The molecular diversity of adaptive convergence. Science 335, 457–461. doi: 10.1126/science.1212986
Thomas, V. L., McReynolds, A. C., and Shoichet, B. K. (2010). Structural bases for stability-function tradeoffs in antibiotic resistance. J. Mol. Biol. 396, 47–59. doi: 10.1016/j.jmb.2009.11.005
Tkachenko, A. G., Akhova, A. V., Shumkov, M. S., and Nesterova, L. Y. (2012). Polyamines reduce oxidative stress in Escherichia coli cells exposed to bactericidal antibiotics. Res. Microbiol. 163, 83–91. doi: 10.1016/j.resmic.2011.10.009
Tkachenko, A. G., and Nesterova, L. Y. (2003). Polyamines as modulators of gene expression under oxidative stress in Escherichia coli. Biochemistry Mosc. 68, 850–856. doi: 10.1023/A:1025790729797
Toprak, E., Veres, A., Michel, J. B., Chait, R., Hartl, D. L., and Kishony, R. (2011). Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 44, 101–105. doi: 10.1038/ng.1034
Tresse, O., Jouenne, T., and Junter, G. A. (1995). The role of oxygen limitation in the resistance of agar-entrapped, sessile-like Escherichia coli to aminoglycoside and beta-lactam antibiotics. J. Antimicrob. Chemother. 36, 521–526. doi: 10.1093/jac/36.3.521
Wang, D., Dorosky, R. J., Han, C. S., Lo, C. C., Dichosa, A. E., Chain, P. S., et al. (2015). Adaptation genomics of a small-colony variant in a Pseudomonas chlororaphis 30-84 biofilm. Appl. Environ. Microbiol. 81, 890–899. doi: 10.1128/AEM.02617-14
Wang, J. H., Singh, R., Benoit, M., Keyhan, M., Sylvester, M., Hsieh, M., et al. (2014). Sigma S-dependent antioxidant defense protects stationary-phase Escherichia coli against the bactericidal antibiotic gentamicin. Antimicrob. Agents Chemother. 58, 5964–5975. doi: 10.1128/AAC.03683-14
Watson, R. A., Weinreich, D. M., and Wakeley, J. (2006). Effects of intra-gene fitness interactions on the benefit of sexual recombination. Biochem. Soc. Trans. 34, 560–561. doi: 10.1042/BST0340560
Wei, B. L., Brun-Zinkernagel, A. M., Simecka, J. W., Prüss, B. M., Babitzke, P., and Romeo, T. (2001). Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol. Microbiol. 40, 245–256. doi: 10.1046/j.1365-2958.2001.02380.x
Wei, B., Shin, S., LaPorte, D., Wolfe, A. J., and Romeo, T. (2000). Global regulatory mutations in csrA and rpoS cause severe central carbon stress in Escherichia coli in the presence of acetate. J. Bacteriol. 182, 1632–1640. doi: 10.1128/JB.182.6.1632-1640.2000
Wichman, H. A., Badgett, M. R., Scott, L. A., Boulianne, C. M., and Bull, J. J. (1999). Different trajectories of parallel evolution during viral adaptation. Science 285, 422–424. doi: 10.1126/science.285.5426.422
Woodford, N., and Ellington, M. J. (2007). The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 13, 5–18. doi: 10.1111/j.1469-0691.2006.01492.x
Woods, R., Schneider, D., Winkworth, C. L., Riley, M. A., and Lenski, R. E. (2006). Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 103, 9107–9112. doi: 10.1073/pnas.0602917103
Xiong, Y. Q., Caillon, J., Drugeon, H., Potel, G., and Baron, D. (1996). Influence of pH on adaptive resistance of Pseudomonas aeruginosa to aminoglycosides and their postantibiotic effects. Antimicrob. Agents Chemother. 40, 35–39.
Yoshida, M., Kashiwagi, K., Shigemasa, A., Taniguchi, S., Yamamoto, K., Makinoshima, H., et al. (2004). A unifying model for the role of polyamines in bacterial cell growth, the polyamine modulon. J. Biol. Chem. 279, 46008–46013. doi: 10.1074/jbc.M404393200
Keywords: antibiotic resistance, evolution, mutation rate, aminoglycosides, Escherichia coli
Citation: Ibacache-Quiroga C, Oliveros JC, Couce A and Blázquez J (2018) Parallel Evolution of High-Level Aminoglycoside Resistance in Escherichia coli Under Low and High Mutation Supply Rates. Front. Microbiol. 9:427. doi: 10.3389/fmicb.2018.00427
Received: 11 September 2017; Accepted: 22 February 2018;
Published: 19 March 2018.
Edited by:
Patrícia Poeta, University of Trás-os-Montes and Alto Douro, PortugalReviewed by:
Valeria Souza, Universidad Nacional Autónoma de México, MexicoCopyright © 2018 Ibacache-Quiroga, Oliveros, Couce and Blázquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Ibacache-Quiroga, Y2xhdWRpYS5pYmFjYWNoZUB1di5jbA==
Jesus Blázquez, YmxhenF1ZXpAY25iLmNzaWMuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.