 Josselin Bodilis1,2*
Josselin Bodilis1,2* Elodie Denet1
Elodie Denet1 Elisabeth Brothier1Arnault Graindorge1†
Elisabeth Brothier1Arnault Graindorge1† Sabine Favre-Bonté1
Sabine Favre-Bonté1 Sylvie Nazaret1
Sylvie Nazaret1- 1Research Group on Environmental Multi-Resistance and Efflux Pump, INRA 1418, UMR CNRS 5557, Laboratoire Ecologie Microbienne, Ecole Nationale Vétérinaire de Lyon, Université de Lyon 1, Villeurbanne, France
- 2EA 4312 Laboratoire de Microbiologie Signaux et Microenvironnement, Université de Rouen, Mont-Saint-Aignan, France
The Burkholderia cenocepacia epidemic ET12 lineage belongs to the genomovar IIIA including the reference strain J2315, a highly transmissible epidemic B. cenocepacia lineage. Members of this lineage are able to cause lung infections in immunocompromised and cystic fibrosis patients. In this study, we describe the genome of F01, an environmental B. cenocepacia strain isolated from soil in Burkina Faso that is, to our knowledge, the most closely related strain to this epidemic lineage. A comparative genomic analysis was performed on this new isolate, in association with five clinical and one environmental B. cenocepacia strains whose genomes were previously sequenced. Antibiotic resistances, virulence phenotype, and genomic contents were compared and discussed with an emphasis on virulent and antibiotic determinants. Surprisingly, no significant differences in antibiotic resistance and virulence were found between clinical and environmental strains, while the most important genomic differences were related to the number of prophages identified in their genomes. The ET12 lineage strains showed a noticeable greater number of prophages (partial or full-length), especially compared to the phylogenetically related environmental F01 strain (i.e., 5–6 and 3 prophages, respectively). Data obtained suggest possible involvements of prophages in the clinical success of opportunistic pathogens.
Introduction
The Burkholderia cepacia complex (Bcc) is a group of opportunistic pathogens that cause lung infections in immunocompromised and cystic fibrosis (CF) patients. The Bcc (originally described as Pseudomonas cepacia) comprises 20 taxonomically related species frequently isolated in water or soil, including plant-associated environments (Ramette et al., 2005; De Smet et al., 2015). Burkholderia multivorans and B. cenocepacia (formerly Genomovar II and III, respectively) are the most clinically frequent members of Bcc, representing from 50% up to 80% of the infections caused by this complex (Mahenthiralingam et al., 2002; Holden et al., 2009; Peeters et al., 2017). Unlike other opportunistic pathogens isolated from CF patients (e.g., Haemophilus influenzae or P. aeruginosa), members of the Bcc could lead to the “cepacia syndrome.” This syndrome is characterized by a rapid clinical deterioration due to necrotizing pneumonia or sepsis, resulting in early death (Isles et al., 1984; Gibson et al., 2003). Moreover, several Bcc species have been shown to be transmissible among CF patients and are able to cause epidemic outbreaks (Biddick et al., 2003).
Given that isolate can result of variable virulence traits depending on the infected patient (Isles et al., 1984; Govan et al., 1993), the determinants of the cepacia syndrome are complex. Indeed, many virulence factors have been highlighted in B. cenocepacia (Drevinek and Mahenthiralingam, 2010; Leitao et al., 2010; Bazzini et al., 2011a; Schwager et al., 2013). Briefly, expression of the cable pilus (cblA), as well as a 22 kDa adhesion, are required for the initial adhesion of the bacteria to the surface of lung epithelial cells (McClean and Callaghan, 2009). Moreover, a functional flagella and lipase are necessary for cell invasion (Tomich et al., 2002; Mullen et al., 2007), and several other virulence factors enhance the B. cenocepacia pathogenicity, like LPS, metalloprotease, siderophores or secretion systems (Leitao et al., 2010; McKeon et al., 2010). Cell-cell communication by quorum sensing (QS), have been described in B. cenocepacia to coordinate expression of these different virulence factors, most of them being necessary for the virulence and also for the normal physiology of B. cenocepacia (Leitao et al., 2010).
Clinical strains of Bcc exhibit high levels of antibiotic resistance that participates to the mortality of the CF patients (Foweraker, 2009). Multiple mechanisms were described as responsible for this drug resistance, including the production of modifying or degrading enzymes, antibiotic target alterations, cell permeability, and drug efflux. In addition, Bcc strains are able to form biofilm on various biotic and abiotic surfaces, increasing their drug tolerance (Coenye, 2010). Because high level of resistance to most of antibiotics (β-lactams, polymyxins, and aminoglycosides) is now commonly found in Bcc strains, treatment of B. cenocepacia by triple antibiotic combination has been proposed (Aaron et al., 2000). Among the antibiotic resistance mechanisms of greatest concern, the RND (Resistance-Nodulation-Cell Division) pumps are particularly problematic because they catalyze the active efflux of many chemically different molecules and, consequently, participate in the multi-drug resistance (MDR) phenotype (Li et al., 2015). No fewer than 15 RND efflux pumps have been described in B. cenocepacia J2315, and their roles in antibiotic resistance was demonstrated for at least 5 of them (Bazzini et al., 2011b; Buroni et al., 2014).
DNA–DNA hybridization and phylogenetic analysis of the recA gene revealed the presence of four distinct recA lineages within B. cenocepacia, i.e., genomovar IIIA to IIID (Vandamme et al., 2003). Even though no clear distinction has been established between clinical and environmental Bcc strains on the basis of phenotypic or genotypic criteria (Bevivino et al., 2002; Peeters et al., 2017), B. cenocepacia strains isolated from CF patients seem to be more invasive and more virulent than environmental strains (Pirone et al., 2008; Bevivino et al., 2012). Moreover, B. cenocepacia IIIA strains were rarely found outside of human infections in comparison to other B. cenocepacia and the Bcc (Baldwin et al., 2007). In particular, the B. cenocepacia epidemic ET12 lineage, which belongs to the genomovar IIIA and includes the reference strain J2315 (also known as LMG 16656T), is a highly transmissible epidemic B. cenocepacia lineage that emerged from the Canada and spread to Europe during the 1990s (Holden et al., 2009). In this study, we describe the genome of F01, a new environmental B. cenocepacia strain isolated from soil in Burkina Faso. This environmental strain presents the same recA sequence as J2315, and is, to our knowledge, the most closely related environmental strain to the epidemic ET12 lineage. A comparative genomic analysis was performed on the genome of this environmental strain, together with available genomes of five clinical and one environmental B. cenocepacia (i.e., J2315, BC7, K56-2, H111, MCO-3, and AU1054 strains). Phylogenetic relationships between these seven B. cenocepacia strains were investigated. In addition, antibiotic resistances, virulence phenotype and genomic contents were compared and discussed.
Materials and Methods
Bacterial Strains and Sampling Sites
The B. cenocepacia strain F01 was isolated form an agricultural soil in the periphery of Ouagadougou in Burkina Faso (sub-sahelian climate). The soil was sampled in a field planted with sorghum in June 2008, 6 months after the crop harvest (Youenou et al., 2016). Characterization of the strain F01 according to the Bcc was performed as described in Mahenthiralingam et al. (2000) by amplification of 1043 bp of the recA gene with BCR1 and BCR2 primers.
Six previously sequenced B. cenocepacia strains from clinical and environmental origins were included in the study (Table 1). The genomes were obtained from NCBI website1 and the strains were kindly provided by Dr. Goldberg and Dr. Varga. Briefly, the B. cenocepacia stains J2315 (Holden et al., 2009), BC7 and K56-2 (Varga et al., 2013) are members of the highly transmissible genomovar IIIA (ET12 lineage) and were all isolated from CF patients. The B. cenocepacia strain H111 is also a member of the genomovar IIIA isolated from CF patient, but this strain is not included in the ET12 lineage (Carlier et al., 2014). The B. cenocepacia strain AU1054 is a member of the genomovar IIIB isolated from a CF patient (Chen et al., 2001). The B. cenocepacia strain MCO-3 is a member of the genomovar IIIB isolated from the rhizosphere of maize (A. Baldwin, unpublished).
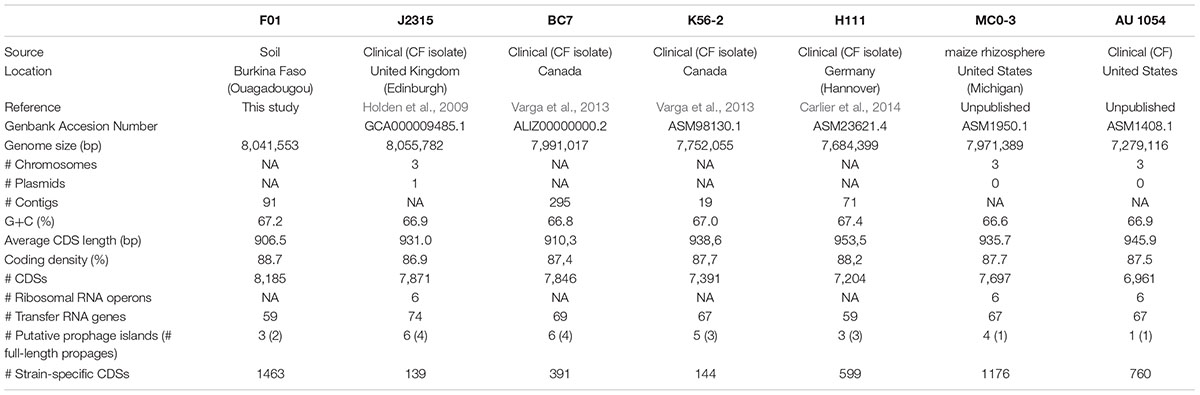
TABLE 1. General genomic features of the studied B. cenocepacia strains.
Genome Sequencing and Assembly
The draft genome of the strain F01 was performed by the DTAMB/Biofidal structure at the University of Lyon (France) using a Roche 454 GS Junior sequencer (454 Life Sciences, Branford, CT, United States) combined with an Illumina MiSeq approach (Illumina, San Diego, CA, United States). The 454 run led to 143,478 reads with an average read length of 597 bp. The Illumina MiSeq PE 2 × 300 led to 4,946,130 paired-end reads with a mean length of about 195 bp after trimming, a final coverage of × 73 being generated (Table 1). Reads were de novo assembled using the SPAdes version 3.10.1 (Bankevich et al., 2012). The contigs were reordered according to the genome sequence of the reference strain J2315 using the Mauve Contig Mover (Rissman et al., 2009) of the MAUVE software (Darling et al., 2004). The contigs that could not be replaced according to the J2315 genome sequence were placed at the end of the alignment.
The nucleotide sequences of the strain F01 was deposited into the European Nucleotide Archive2 with the accession number ERS2025400 (the study accession is PRJEB23651).
Genome Annotation and Comparative Genomics
Coding sequences (CDSs) predictions, as well as automatic and manual sequence annotations, were performed using the MicroScope platform pipeline at Genoscope (Vallenet et al., 2013). CDSs were predicted using AMIGene software (Bocs et al., 2003). Automatic functional annotation of the predicted CDSs was performed using the tools integrated in the MicroScope platform (Vallenet et al., 2009). Gene predicted to be involved in functions of interest were manually investigated by using the “genome browser” tool of the platform. Genomic islands and regions of genomic plasticity (RGPs) of each genome were identified using the “RGP finder” tool included in the MicroScope platform by searching for synteny breaks and HGT (horizontally transferred genes) features (Vernikos and Parkhill, 2006; Waack et al., 2006). Antibiotic resistance genes (ARGs), including intrinsic, mutation-driven and acquired resistance, were searched using the CARD database version 1.1.2 (Comprehensive Antibiotic Resistance Database; McArthur et al., 2013) included in the MicroScope platform. Prophage sequences were identified using the PHASTER tool (Arndt et al., 2016) and manually confirmed by eliminating false positive “incomplete prophages” (e.g., transposons). In addition, clustered regularly interspaced short palindromic repeats (CRISPRs) were investigated using the CRISPR Finder tool (Grissa et al., 2007).
Phylogenetic Analysis
The evolutionary relationships among the seven studied B. cenocepacia strains were determined from a concatenated alignment of the orthologous protein sequences of the core genome of these seven strains. Orthologous proteins were identified from bidirectional best-hit BLASTP searches of each strain proteome against J2315’s proteome with an e-value parameter threshold of 10e-5. Customized computer scripts were then used to extract the best reciprocal hits from all the strains and to align these protein sequences with Clustal omega (Sievers et al., 2011). The alignments were then filtered using Gblocks version 0.91 b (Talavera and Castresana, 2007) with default options and concatenated. A final alignment of 4,555 concatenated proteins (1,514,675 amino acids) was used for the phylogenetic analyses. A phylogenetic tree was reconstructed with the maximum-likelihood method by implementation in RAxML V7.9.5 (Stamatakis, 2006) with 1,000 bootstraps replicates. The phylogenetic tree root was confirmed to be positioned between the two genomovar IIIA and IIIB by reiterating the same protocol with 29 additional Burkholderia spp. genomes (data not shown).
Multilocus sequence typing (MLST) of the seven studied strains was performed using seven standard housekeeping genes (atpD, gltB, gyrB, lepA, phaC, trpB, and recA) according to the protocol and primers specified in a public database of MLST sequence data3 (Spilker et al., 2009). The sequences were obtained from the sequenced genomes.
Nitrate Respiration Test
Nitrate anaerobic respiration was evaluated using the API 20NE kit (bioMérieux, Marcy-l’Étoile, France), according to the manufacturer’s recommendations.
Antibiotic Resistance Test
Antibiotic resistances of the strain F01, as well as the six previously sequenced B. cenocepacia strains included in our study were determined by the agar diffusion method using a set of 16 antibiotic disks (Bio-Rad, Marnes-la-Coquett, France). The antibiotics tested corresponded essentially to the officially recommended drugs for use in human medicine against infections caused by non-fermenting gram negative bacilli, in particularly against B. cepacia: Ticarcillin (TIC, 75 μg), Ticarcillin + clavulanic acid (TCC, 75 + 10 μg), Piperacillin + tazobactam (PPT, 75 + 10 μg), Ceftazidime (CAZ, 30 μg), Cefepime (FEP, 30 μg), Cefpirome (CPO, 30 μg), Chloramphenicol (CHL, 30 μg), Imipenem (IPM, 10 μg), Meropenem (MEM, 10 μg), Ofloxacin (OFX, 5 μg), Levofloxacin (LVX, 5 μg), Ciprofloxacin (CIP, 5 μg), Pefloxacin (PEF, 5 μg), Minocycline (MNO, 30 μg), Colistin (CST, 50 μg), Trimethoprim + sulfamethoxazole (SXT, 23.75 + 1.25 μg). After 24 h at 35°C, the strains were classified as susceptible, intermediate or resistant according to French national guidelines4. P. aeruginosa CIP 76110 (ATCC 27853) was used as control.
Virulence Assays
Virulence was determined using the Dictyostelium discoïdeum social amoeba, as previously described (Froquet et al., 2009). Strains of P. aeruginosa PT5 and Klebsiella pneumoniae KpGe were used as negative and positive control for each assay. The axenic D. discoïdeum strain AX2 (kindly provided by Anne Vianney, CIRI, University Lyon1) was used for virulence assays. Overnight bacterial culture was diluted in Luria–Bertani (LB) medium to reach an OD600 nm of 1.5. Then, 1 mL of each bacterial suspension was plated on SM Agar (Formedium, Hunstanton, United Kingdom) medium. Plates were dried for 1 h to obtain a dry bacterial layer. Meanwhile, cells of D. discoïdeum were washed two times in PAS buffer (Page’s Amoeba Saline buffer; 2.5 mM KH2PO4, 4 mM MgSO4, 0.5 mM CaCl2, 2.5 mM, NA2HPO4, 0.05 mM (NH4)2FeII(SO4)2) by centrifugation at 1000 × g for 10 min. Suspension of amoebae was then adjusted to 2 × 106 cells per ml. This suspension was diluted in series until a final density of about 7800 cells per ml. Five microliters of each dilution were spotted on bacterial lawns. Plates were incubated at 22.5°C during 5 days. After incubation, appearance of phagocytic plaques was evaluated according three categories (Adamek et al., 2011): (i) non-virulent (less than 400 amoebae for lysis plaque formation), (ii) low-virulent (400–2500 amoebae for lysis plaque formation) and (iii) virulent (more than 2500 amoebae). Data shown are the means of two independent experiments, each performed in triplicate
Results
Core Genome Phylogeny and Multilocus Sequence Typing (MLST)
Sequencing of the recA gene of the strain F01 showed the same sequence as the B. cenocepacia stains J2315. Using reciprocal BLASTP, protein-coding genes having a 1:1 orthologous relationship to each other were identified across the seven studied B. cenocepacia. A total of 4,555 CDSs were identified which could be considered as the core genome for those strains. A final alignment of 1,514,675 amino acids was used for the phylogenetic tree reconstruction by maximum-likelihood method (Figure 1). Interestingly this phylogenetic tree is almost fully resolved, with bootstrap values ≥ 99%. Both strains belonging to genomovar IIIB (strains MC0-3 and AU 1054), as defined from the recA sequence, were clearly separated from strains belonging to genomovar IIIA. Moreover, the three strains belonging to the epidemic ET12 lineage (strains J2315, BC7 and K56-2), as defined by multilocus enzyme electrophoresis, grouped together. Interestingly, our new environmental F01 strain was closer to the ET12 lineage than the clinical strain H111.
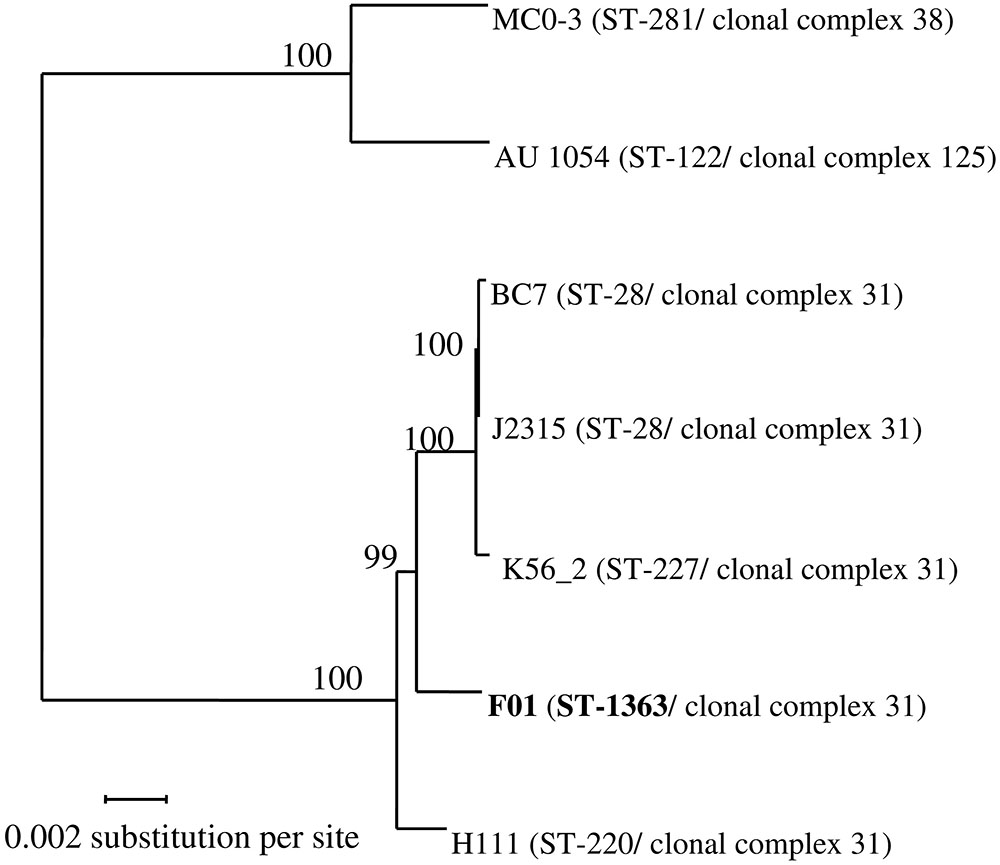
FIGURE 1. Phylogenetic tree from maximum-likelihood analysis of the core-genome alignments of seven Burkholderia cenocepacia strains. In total, 4,555 orthologous proteins were concatenated in an alignment of 1,514,675 amino acids. Bootstraps are indicated in each node. ST, Sequence Type (unique combination of 7 alleles; see Materials and Methods). Two strains belong to the same clonal complex if they share at least four identical alleles.
To further investigate these evolutionary relationships, a MLST analysis was performed (Figure 1). In this study, all genomovar IIIA strains belong to the same clonal complex (i.e., ≥4 alleles out of 7 are identical), while the two strains of genomovar IIIB belong to two different clonal complexes. In addition, all strains showed a different sequence type (ST), except the strains J2315 and BC7, which belongs to the ST-28. The ST of the F01 strain is new (ST-1363) because of a new allele for the atpD gene (atpD-437).
General Features of the F01 Genome and Comparative Genomics
The draft genome of F01 consisted of 8,041,553 bp organized in 91 contigs of size greater than 200 bp (Table 1). Therefore, F01 has a genome size close to the other B. cenocepacia strains, i.e., between 7,991,017 bp (for BC7) and 8,055,782 (for J2315). Moreover, the average GC content of the F01 genome was 67.2%, that is consistent with the average GC content of the other B. cenocepacia (66.9% ± 0.3; Table 1). The sequencing methods used in our study (Roche 454 and Illumina MiSeq) did not allow performing the complete genome assembly, because reads were too short to overcome some repeat regions. Thus, the number of chromosomes and presence of plasmids could not be determined in F01, as well as in the other draft genomes available (Table 1). Nevertheless, the “Genome Browser interface” available in the Microscope platform5) allowed to visualize a global synteny in F01 for the three chromosomes as well as for the plasmid of J2315 (data not shown). This synteny was also conserved in the other B. cenocepacia strains studied except for the plasmid that was not detected in the strains H111, MC0-3 and AU 1054.
In total, 8,185 predicted protein-CDSs were identified in the F01 genome that is noticeably higher than the average number of CDSs in the other B. cenocepacia strains (7495 bp ± 370). Another difference was the number of tRNAs, which was lower in F01 and H111, compared to the other strains (59 vs 67–74), although this last observation is questionable because of the sequencing technique used which may underestimate the number of repeated genes. Concerning the general genomic features, the most difference was the number of partial or full-length prophages detected (Table 1). Indeed, the ET12 lineage strains showed a higher number of (partial or full-length) prophages, with 5–6 prophages detected against 1–4 for the other B. cenocepacia strains. More generally, the number of full-length prophages (i.e., potentially functional prophages) seemed associated with the phylogenetic relationships, with 2–4 full-length prophages in genomovar IIIA and only one in genomovar IIIB. However, in genomovar IIIA, the environmental F01 strain presented the lowest number of full-length prophages (Table 1). In addition, no CRISPR system were detected in the seven B. cenocepacia genomes studied.
Comparative genomics by reciprocal Blast was then performed. Beside the core genome of 4,555 CDS that were used for phylogenetic analysis (Figure 1), particular attention was given to variable genes. First of all, we observed that the number of strain-specific genes varied between strains (Table 1). The low number of strain-specific genes observed in the ET12 lineage (from 139 to 391 genes) was probably due to the close phylogenetic relationships between the three strains J2315, BC7 and K56-2 (Figure 1). However, the highest number of strain-specific genes in the two environmental strains F01 and MC0-3 (1463 and 1176 genes, respectively) could not be phylogenetically explained, when compared to the clinical strains H111 and AU 1054 (599 and 760 genes, respectively), and could be associated with their environmental life style. Annotation of strain-specific genes mainly corresponded to proteins of unknown function (65.0% ± 13.3, compared to 16.7% for the core genome). Despite this lack of annotation, and beside the genes associated with phages, transposition or expression regulation, we focused on two gene categories. On the one hand, several genes specific to F01 were described as involved in xenobiotic degradation, in particular monoxygenases, dioxygenases, cyclases or cytochromes encoding genes, potentially responsible for the degradation of RDX (i.e., Hexahydro-1,3,5-trinitro-1,3,5-triazine) and chlorophenolic compounds (e.g., F01_v2_460557, F01_v2_460615 to F01_v2_460617). On the other hand, analysis of putative metabolic functions revealed more putative genes involved in nitrate respiration, compared to the other B. cenocepacia strains. However, although nitrate respiration was phenotypically confirmed by API gallery in F01, this feature was also found among some of our studied B. cenocepacia strains (e.g., J2315 and MC0-3).
Variable genes among our seven strains studied were also analyzed by two additional approaches. On one hand, a genomic comparison was performed between the two environmental strains (F01 and MC0-3) and the five other clinical strains. On the other hand, a comparison limited to the genomovar IIIA was performed between F01, H111 and the ET12 lineage strains (J2315, BC7 and K56-2). (i) The first comparison showed only eight genes present in all the clinical strains of B. cenocepacia and absent in the two environmental strains. Among these eight genes, the only functionally characterized gene (BCAM2106 in J2315) encodes a non-heme chloroperoxidase, associated with response to oxidative stress. Conversely, 23 genes specific to environmental strains and absent in clinical strains were also highlighted. These genes corresponded to a prophage partially conserved between the two environmental strains and absent in clinical strains (Table 2). (ii) The second comparison revealed 745 genes specific to the ET12 lineage strains and absent in the other genomovar IIIA strains. Most of these genes (62.1%) encoded putative proteins of unknown function, while best described genes corresponded to transposons, expression regulation genes and the four prophages specific to the ET12 lineage (Table 2).
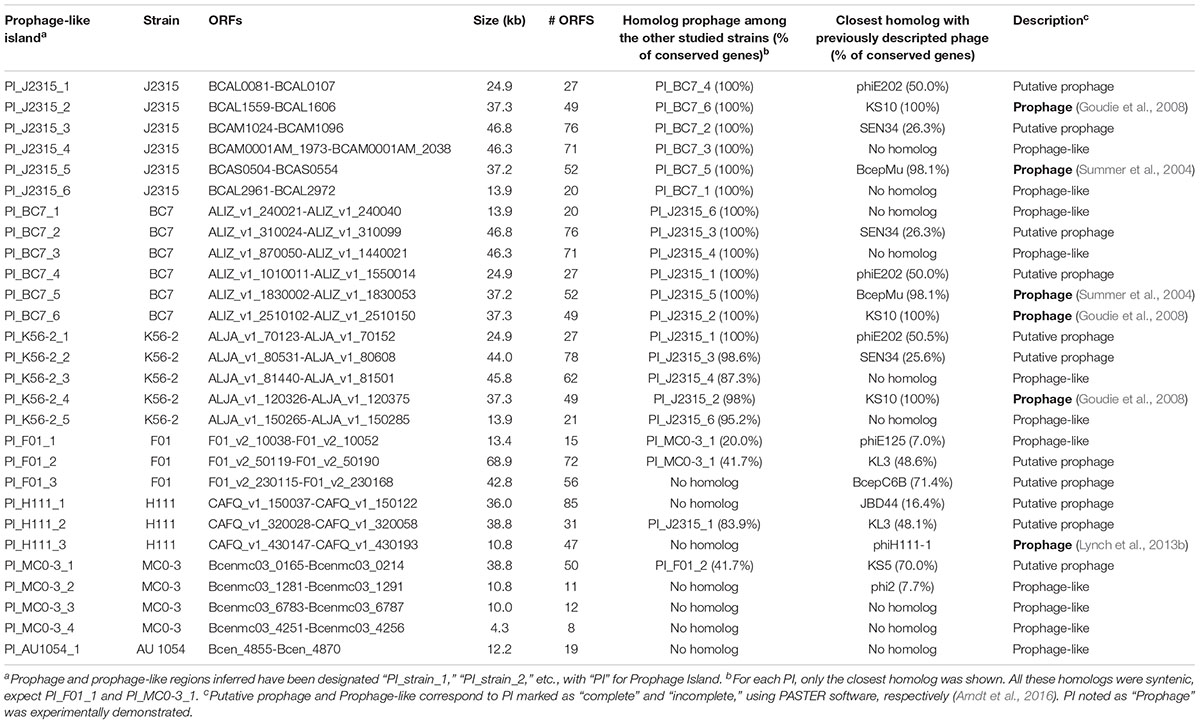
TABLE 2. Putative prophage islands (PI) identified in the studied B. cenocepacia strains.
Resistances to Antibiotics
Resistance phenotypes across 16 antibiotics and combinations were investigated using the disk diffusion method (Table 3). Five studied B. cenocepacia strains (J2315, K56-2, F01, H111 and MC0-3) showed a similar number of antibiotic resistances, with resistance to about half of the tested antibiotics. Indeed, all these strains were resistant to ticarcillin (alone or in combination with clavulanic acid), cefpirome, colistin, and ofloxacin. These same strains were susceptible to ceftazidime (a third generation cephalosporin), chloramphenicol, and minocycline (Table 3). Interestingly, no significant differences were found between the antibiotic resistance profiles of clinical (J2315, K56-2, and H111) and environmental (F01 and MC0-3) strains. The BC7 strain was the most susceptible of the strains tested, particularly against fluoroquinolones with susceptibility to ciprofloxacin and pefloxacin. The AU1054 strain was the most resistant of the B. cenocepacia strains studied, with a resistance to 13 out of the 16 antibiotics tested. Notably, AU1054 is resistant to ceftazidime, chloramphenicol, and the combination of trimethoprim and sulfamethoxazole (Table 3).
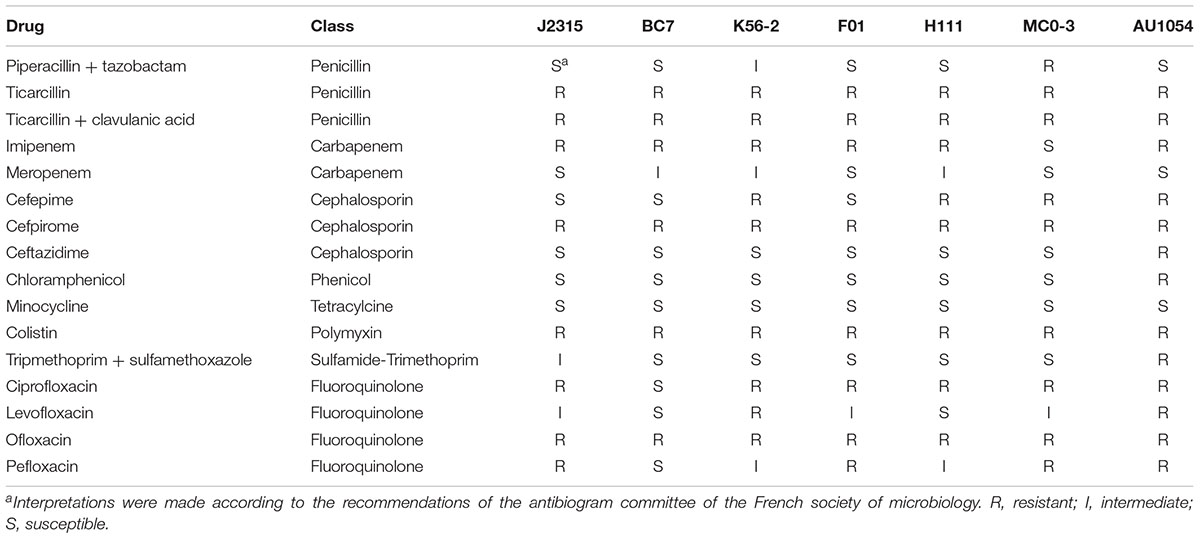
TABLE 3. Antibiotic resistances of seven studied B. cenocepacia strains.
In addition to the phenotypic assays, the comprehensive antibiotic resistance database (CARD), including resistance genes and mutations conferring antibiotic resistance, was used to predict the resistome of the strains studied (Supplementary Table S1). The resolution of the CARD database ontology is often at the antibiotic class level, which does not allow a direct comparison with resistance phenotypes. In addition, it is well known that ARGs must be expressed to provide effective resistance. Indeed, our genomic resistome analysis suggested potential resistance to all classes of antibiotics, even for strains revealing a susceptible phenotype (Supplementary Table S1). Differences observed between resistomes concern only genes encoding efflux pumps, notably the resistance nodulation cell-division (RND) pumps, as well as a specific allele of the katA gene involved in the resistance to isoniazid. Indeed, while all genomes contain both katA and katB genes, potentially conferring resistance to isoniazid, only katB was found in the F01 genome (Supplementary Table S1).
Because many RND efflux pumps involved in antibiotic resistance have already been described in B. cenocepacia, the genes encoding these pumps were exhaustively researched among the strains studied (Supplementary Table S3). 16 CDSs annotated as probably encoding 15 RND efflux pumps (HAE-1 and HME families) were found in J2315 (including a heterotrimeric RND pump). A role in antibiotic resistance was demonstrated for at least five of them (Bazzini et al., 2011b). In all the studied B. cenocepacia, only 13 of these 16 CDSs were highlighted, including the five efflux pumps involved in antibiotic resistance (Supplementary Table S3). The RND-12 pump, involved in copper efflux (HME family), is absent in H111, while the RND-15 pump, whose function is unknown, is absent in the two strains of genomovar IIIB (MC0-3 and AU1054), as confirmed by synteny study (Supplementary Table S3). Interestingly, genes potentially encoding the same RND pumps (HAE-1 and HME families) were found in the three strains of the ET12 lineage as well as in the environmental F01 strain (Supplementary Table S3). In H111, additional genes encoding for two other HAE-1 efflux pumps, and potentially involved in antibiotic resistance, were found. This strain therefore has genes encoding 16 potential RND pumps. In the two genomovar IIIB strains (MC0-3 and AU1054), additional genes encoding for three other efflux pumps were found, including a HAE-1 efflux pump homologous (orthologous) to one of the additional H111 pumps. Moreover, one of the genes encoding efflux pump is a pseudogene (Bcen_3622, RND-9). MC0-3 therefore has genes encoding 17 potential RND pumps.
Virulence
The virulence phenotype of the seven strains of interest was determined by using the D. discoïdeum model (Froquet et al., 2009). This assay measures the lowest number of amoeba cells required for effective bacterial predation. The higher the number of amoebae required for bacterial killing, the higher is the ability of bacteria to resist predation. Our results revealed that all B. cenocepacia strains can be considered as virulent, compared to the P. aeruginosa PT5 positive control (Figure 2). Indeed, these strains were able to resist predation of the maximum amount of amoebae tested, excepted for AU1054 which displayed a slightly less virulent phenotype compared to the other B. cenocepacia strains. This assay did not seem to display any significant difference of virulence between clinical and environmental strains (Figure 2).
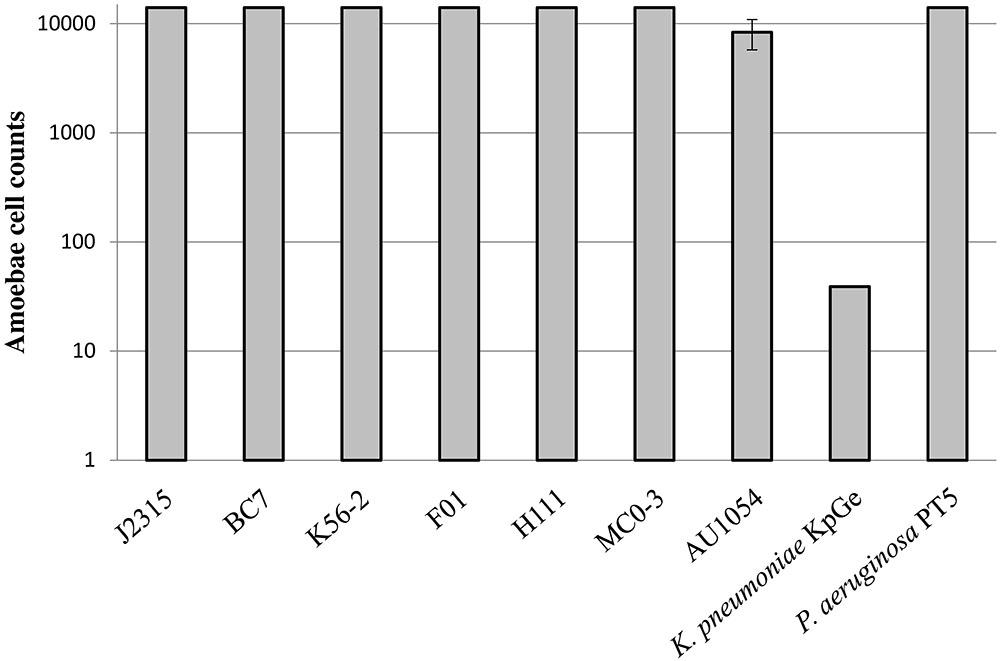
FIGURE 2. Virulence of B. cenocepacia strains determined using the social amoeba Dictyostelium discoïdeum. Strains of Pseudomonas aeruginosa PT5 and Klebsiella pneumoniae KpGe were used as positive and negative control, respectively. The axenic D. discoïdeum strain AX2 was used for virulence assays. Dilutions of amoebae were spotted on bacterial lawn plates and incubated during 5 days (n = 6).
Additionally, our analysis of the literature currently available on B. cenocepacia virulence genes (Aubert et al., 2008; Drevinek and Mahenthiralingam, 2010) highlighted that almost all virulence genes identified in members of the epidemiologic ET12 lineage are also present in the environmental strain F01, including genes encoding the cable pilus, previously described as ET12 lineage-specific (McClean and Callaghan, 2009) (Supplementary Table S2). Only the genes katA and wbcE (annotated as glycosyl transferase, and involved in lipopolysaccharide synthesis) were absent in the F01 genome. Finally, genes encoding proteins involved in QS, synthesis of cable pilus, iron acquisition or rhamnolipid synthesis were absent in the genomovar IIIB strains, as well as in the H111 strain (Supplementary Table S2).
Discussion
B. cenocepacia, especially strains from the ET12 lineage, are among the most prevalent Bcc members that colonize CF patients (Isles et al., 1984; Speert et al., 2002; Mahenthiralingam et al., 2005; Drevinek and Mahenthiralingam, 2010). However, despite that many virulence factors were described, the epidemiological success of some lineages is still misunderstood (Holden et al., 2009; McClean and Callaghan, 2009; Bazzini et al., 2011a; Gonyar et al., 2015). The B. cenocepacia species is characterized by a large variety of ecological niches, from patient-associated to environmental niches. Although some studies have shown that clinical strains of B. cenocepacia have a higher virulence than environmental strains (Pirone et al., 2008; Bevivino et al., 2012), the distinction between clinical and environmental strains based on phenotypic or genotypic criteria remains difficult (Bevivino et al., 2002; Peeters et al., 2017). Indeed, to our knowledge, strains belonging to the ET12 lineage had not yet been isolated from a non-clinical environment (Baldwin et al., 2007). Thus, this is the first time that an environmental strain phylogenetically related to the ET12 lineage, is described. The environmental F01 strain belongs to the same clonal complex and possess, to date, the phylogenetically closest genome to ET12 genomes, by comparison to other available B. cenocepacia genomes (Figure 1). In this context, this study provides a new evolutionary aspect to understanding the emergence of the ET12 lineage, and more generally, the epidemic risk represented by opportunistic pathogens.
The presence of genes involved in xenobiotic degradation and nitrate respiration suggested that F01 is well adapted to agricultural soil. Historically, F01 was isolated in 2008 from a soil characterized by an absence of amendment for at least 10 years (Youenou et al., 2016). A recent anthropogenic origin of F01 could therefore be excluded. Moreover, it is important to notice that another strain with a similar recA sequence (also similar to J2315 recA sequence) was isolated 3 years later from the same site (data not shown). Although we cannot affirm that this isolate is a direct descendant to F01, this strain displayed specific traits in common with F01 (i.e., same virulence phenotype, ability to metabolize nitrate, same pattern of antibiotic resistances) (data not shown). Taken together, these two isolates show that strains adapted to agricultural soils can be phylogenetically related to the ET12 epidemic lineage.
This observation raises the question of the specific pathogenic properties of strains involved in clinical infections, particularly when these strains have been described as opportunistic pathogens. So, among phylogenically related strains why some displayed an efficient success in clinical situations and not the others? Beyond the capacity for infection, are there specific properties of opportunistic pathogenic strains that explain their epidemiological success? In an attempt to answer these questions, we conducted in this present work a comparative study between ET12 strains and our new environmental strain of B. cenocepacia, focusing on two parameters that characterize clinical strains: antibiotic resistances and virulence.
Surprisingly, no significant differences in antibiotic resistance were found between ET12 strains and F01, as well as between clinical and environmental strains of genomovar IIIB (Table 3). As previously observed, the numerous RND pumps in B. cenocepacia are likely to represent the main genetic determinants responsible for the MDR phenotype frequently described in this bacterial species (Holden et al., 2009; Perrin et al., 2010; Bazzini et al., 2011b; Buroni et al., 2014). Interestingly, although this cannot be correlated with the phenotypes observed, some variation in the number of RND efflux pump genes between the B. cenocepacia strains were observed, while the other ARGs are almost all conserved in this bacterial species (Supplementary Table S3). Although the genes coding for RND pumps are generally found as conserved in a given bacterial species, we already identified horizontal gene transfers coding for such efflux pumps in environmental strains of Stenotrophomonas maltophilia, potentially associated with an MDR phenotype (Youenou et al., 2015; Bodilis et al., 2016). Because, both organic (e.g., PAHs; Hearn et al., 2003) and inorganic (e.g., metals; Nies, 1995) contaminants, as well as root exudates (Eda et al., 2011), were previously described as inducers and/or substrates of RND efflux pumps, a role of these environmental factors in the selection and dissemination of efflux-mediated resistances is probable (Martinez et al., 2009). Without necessarily directly inducing an increase in antibiotic resistance in these environmental opportunistic pathogenic strains, such factors could at least promote future clinical adaptability by selecting strains with multiple efflux pumps. Indeed, the emergence of multidrug-resistant bacteria is often linked to mutations that overexpress efflux pumps (Li et al., 2015).
As surprising as the results of antibiotic resistance phenotypes, all strains of B. cenocepacia, including F01, were virulent, according to our amoeba assay (Figure 2). In genomovar IIIB, we observed a probable non-significant slight decrease in virulence for the clinical strain (AU1054) compared to the environmental strain (MC0-3). The phenotypic virulence assay we used was validated by several studies on strains of B. cenocepacia or other opportunistic pathogenic species (Aubert et al., 2008; Froquet et al., 2009; Steinert, 2011). Moreover, the conservation of the virulence phenotype that we observed for all the B. cenocepacia tested is in agreement with the presence in their genome of most of the genes encoding the virulence factors already described in J2315 (Supplementary Table S2). Additional tests on other environmental strains of Burkholderia spp. showed virulence traits in all of the B. cenocepacia tested, while other Burkholderia spp. (e. g., B. dolosa and B. multivorans) were not virulent (data not shown). Although this should be deeply investigated, the virulence phenotype of Burkholderia spp. appears to be species-specific and not related to the clinical origin of the strains. This observation is in connection with other studies showing a significant number of opportunistic pathogenic bacteria in the rhizosphere. This environment could select bacterial virulence properties during interaction with plant roots or during the intense microbial competition present in the rhizosphere (Berg et al., 2005; Mendes et al., 2013). Recently, a study has even shown that a rhizospheric B. cenocepacia strain enhances its virulence during mice infection (Bragonzi et al., 2017). Consequently, although several Bcc strains have developed beneficial interactions with their plant hosts, the difficulties of distinguishing between pathogenic and harmless strains prevents the use of Bcc members as biocontrol agents (e.g., Vial et al., 2011).
Beyond the virulence, we also considered the epidemiological properties of ET12 strains, notably the genomic signatures specific to these clinical strains. Thus, Cable A is one of the few virulence factors previously described as specific to ET12 strains (Graindorge et al., 2010). However, we also identified the gene encoding this virulence factor in the genome of the environmental strain F01 (Supplementary Table S2), excluding an epidemiological signature associated with this gene. Similarly, the plasmid initially described in J2315 is found in all ET12 strains and in the environmental strain F01. An analysis of the databases revealed that this plasmid is also present in B. cenocepacia HI2424 (genomovar IIIB) and other Burkholderia species (B. thailandensis, B. gladioli, and B. glumae) (data not shown). Therefore, the occurrence of this plasmid in Burkholderia spp. does not appear to be related to particular species, environment, or virulence. Finally, the occurrence of prophages in the genome of ET12 strains appears to be the most interesting epidemiological signature (Table 2).
The most important differences, especially in genomovar IIIA, were therefore related to the number of prophages identified in the genomes (Table 1). The ET12 lineage strains displayed a noticeable greater number of (partial or full-length) prophages, especially compared to the phylogenetically related environmental F01 strain (i.e., 5–6 and 3 prophages, respectively). However, it is difficult to determine whether these prophages are directly involved into the ecological adaptation or whether they represent only habitat or lifestyle markers. On the one hand, some studies noticed the presence of genes encoding virulence factors into prophages (i.e., Sitkiewicz et al., 2006; Tobe et al., 2006). Several prophages have already been characterized in B. cenocepacia (Table 2; Summer et al., 2004; Goudie et al., 2008; Lynch et al., 2013b), without the presence of genes encoding new virulence factors detected (Summer et al., 2007). However, given the number of prophages not or only slightly characterized in B. cenocepacia (Table 2), a direct role of these prophages in the virulence and/or in particular epidemiological properties of the ET12 strains cannot be excluded. On the other hand, it can also be suggested that the substantial presence of prophages in the genome of ET12 strains is not the cause but the consequence of the epidemiological success of these strains. Thus, according to the “kill-the-winner” ecological model, dominant strains in an ecological niche are more likely to be targeted by phage attacks (Rodriguez-Valera et al., 2009). The ET12 strains, due to their epidemiological success, should indeed be at least transiently dominant in their ecological niche. The substantial presence of prophages in their genome would result from the numerous phage attacks that occurred. According to this model, the environmental strain F01 has probably never been so dominant in its telluric environment, which would be in accord with the lowest number of prophages in its genome. Finally, a third hypothesis could explain this variation in the number of prophages. A recent ecological model, “the piggyback-the-winner model,” predicts that phages integrate as prophages and undergo the lysogenic replication cycle instead of the lytic cycle when bacterial density and activity increase, especially in internal mucosal surfaces, such as human lungs (Silveira and Rohwer, 2016).
The dramatic decline in the effectiveness of antibiotics has generated renewed interest in phage therapy (Abedon et al., 2017). The use of bacteriophages as antibacterial therapeutics is especially important for targeting those pathogens for which antibiotic treatments are limited, e.g., in B. cenocepacia (Lynch et al., 2013a; Roszniowski et al., 2017). In this context, it is interesting to note that the absence of a self-defense CRISPR system makes these therapeutic approaches even more promising. Finally, the particular occurrence of prophage in the epidemiological ET12 strains reinforces the need to better understand the interactions between B. cenocepacia and bacteriophages, particularly in a clinical context.
Author Contributions
JB performed the bioinformatics analysis presented in the work. ED performed the virulence assays. EB performed the antibiotic resistance tests. All authors contributed to the design and interpretation of the results, as well as to writing the article, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
J2315, BC7, K56-2, H111, MCO-3, and AU1054 strains were kindly provided by Dr. John Varga and Dr. Joanna Goldberg, Emory University. The LABGeM (CEA/IG/Genoscope and CNRS UMR8030) and the France Génomique National infrastructure (funded as part of Investissement d’Avenir program managed by Agence Nationale pour la Recherche, contract ANR-10-INBS-09) are acknowledged for support within the MicroScope annotation platform.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00383/full#supplementary-material
TABLE S1 | Genomic resistome in seven studied B. cenocepacia strains.
TABLE S2 | Virulence genes in seven studied B. cenocepacia strains.
TABLE S3 | RND efflux systems in seven studied B. cenocepacia strains.
Footnotes
- ^http://www.ncbi.nlm.nih.gov/
- ^http://www.ebi.ac.uk/ena
- ^http://pubmlst.org/bcc/
- ^http://www.sfm-microbiologie.org/
- ^https://www.genoscope.cns.fr/agc/microscope
References
Aaron, S. D., Ferris, W., Henry, D. A., Speert, D. P., and MacDonald, N. E. (2000). Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am. J. Respir. Crit. Care Med. 161, 1206–1212. doi: 10.1164/ajrccm.161.4.9907147
Abedon, S. T., García, P., Mullany, P., and Aminov, R. (2017). Editorial: phage therapy: past, present and future. Front. Microbiol. 8:981. doi: 10.3389/fmicb.2017.00981
Adamek, M., Overhage, J., Bathe, S., Winter, J., Fischer, R., and Schwartz, T. (2011). Genotyping of environmental and clinical Stenotrophomonas maltophilia isolates and their pathogenic potential. PLoS One 6:e27615. doi: 10.1371/journal.pone.0027615
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aubert, D. F., Flannagan, R. S., and Valvano, M. A. (2008). A novel sensor kinase-response regulator hybrid controls biofilm formation and type VI secretion system activity in Burkholderia cenocepacia. Infect. Immun. 76, 1979–1991. doi: 10.1128/IAI.01338-07
Baldwin, A., Mahenthiralingam, E., Drevinek, P., Vandamme, P., Govan, J. R., Waine, D. J., et al. (2007). Environmental Burkholderia cepacia complex isolates in human infections. Emerg. Infect. Dis. 13, 458–461. doi: 10.3201/eid1303.060403
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bazzini, S., Udine, C., and Riccardi, G. (2011a). Molecular approaches to pathogenesis study of Burkholderia cenocepacia, an important cystic fibrosis opportunistic bacterium. Appl. Microbiol. Biotechnol. 92, 887–895. doi: 10.1007/s00253-011-3616-5
Bazzini, S., Udine, C., Sass, A., Pasca, M. R., Longo, F., Emiliani, G., et al. (2011b). Deciphering the role of RND efflux transporters in Burkholderia cenocepacia. PLoS One 6:e18902. doi: 10.1371/journal.pone.0018902
Berg, G., Eberl, L., and Hartmann, A. (2005). The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 7, 1673–1685. doi: 10.1111/j.1462-2920.2005.00891.x
Bevivino, A., Dalmastri, C., Tabacchioni, S., Chiarini, L., Belli, M. L., Piana, S., et al. (2002). Burkholderia cepacia complex bacteria from clinical and environmental sources in Italy: genomovar status and distribution of traits related to virulence and transmissibility. J. Clin. Microbiol. 40, 846–851. doi: 10.1128/JCM.40.3.846-851.2002
Bevivino, A., Pirone, L., Pilkington, R., Cifani, N., Dalmastri, C., Callaghan, M., et al. (2012). Interaction of environmental Burkholderia cenocepacia strains with cystic fibrosis and non-cystic fibrosis bronchial epithelial cells in vitro. Microbiology 158, 1325–1333. doi: 10.1099/mic.0.056986-0
Biddick, R., Spilker, T., Martin, A., and LiPuma, J. J. (2003). Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiol. Lett. 228, 57–62. doi: 10.1016/S0378-1097(03)00724-9
Bocs, S., Cruveiller, S., Vallenet, D., Nuel, G., and Médigue, C. (2003). AMIGene: annotation of MIcrobial genes. Nucleic Acids Res. 31, 3723–3726. doi: 10.1093/nar/gkg590
Bodilis, J., Youenou, B., Briolay, J., Brothier, E., Favre-Bonté, S., and Nazaret, S. (2016). Draft genome sequences of Stenotrophomonas maltophilia strains Sm32COP, Sm41DVV, Sm46PAILV, SmF3, SmF22, SmSOFb1, and SmCVFa1, isolated from different manures in France. Genome Announc. 4:e00841-16. doi: 10.1128/genomeA.00841-16
Bragonzi, A., Paroni, M., Pirone, L., Coladarci, I., Ascenzioni, F., and Bevivino, A. (2017). Environmental Burkholderia cenocepacia strain enhances fitness by serial passages during long-term chronic airways infection in mice. Int. J. Mol. Sci. 18:2417. doi: 10.3390/ijms18112417
Buroni, S., Matthijs, N., Spadaro, F., Van Acker, H., Scoffone, V. C., Pasca, M. R., et al. (2014). Differential roles of RND efflux pumps in antimicrobial drug resistance of sessile and planktonic Burkholderia cenocepacia cells. Antimicrob. Agents Chemother. 58, 7424–7429. doi: 10.1128/AAC.03800-14
Carlier, A., Agnoli, K., Pessi, G., Suppiger, A., Jenul, C., Schmid, N., et al. (2014). Genome sequence of Burkholderia cenocepacia H111, a cystic fibrosis airway isolate. Genome Announc. 2:e00298-14. doi: 10.1128/genomeA.00298-14
Chen, J. S., Witzmann, K. A., Spilker, T., Fink, R. J., and LiPuma, J. J. (2001). Endemicity and inter-city spread of Burkholderia cepacia genomovar III in cystic fibrosis. J. Pediatr. 139, 643–649. doi: 10.1067/mpd.2001.118430
Coenye, T. (2010). Social interactions in the Burkholderia cepacia complex: biofilms and quorum sensing. Fut. Microbiol. 5, 1087–1099. doi: 10.2217/fmb.10.68
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Smet, B., Mayo, M., Peeters, C., Zlosnik, J. E. A., Spilker, T., Hird, T. J., et al. (2015). Burkholderia stagnalis sp nov and Burkholderia territorii sp nov., two novel Burkholderia cepacia complex species from environmental and human sources. Int. J. Syst. Evol. Microbiol. 65, 2265–2271. doi: 10.1099/ijs.0.000251
Drevinek, P., and Mahenthiralingam, E. (2010). Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 16, 821–830. doi: 10.1111/j.1469-0691.2010.03237.x
Eda, S., Mitsui, H., and Minamisawa, K. (2011). Involvement of the SmeAB multidrug efflux pump in resistance to plant antimicrobials and contribution to nodulation competitiveness in Sinorhizobium meliloti. Appl. Environ. Microbiol. 77, 2855–2862. doi: 10.1128/AEM.02858-10
Foweraker, J. (2009). Recent advances in the microbiology of respiratory tract infection in cystic fibrosis. Br. Med. Bull. 89, 93–110. doi: 10.1093/bmb/ldn050
Froquet, R., Lelong, E., Marchetti, A., and Cosson, P. (2009). Dictyostelium discoideum: a model host to measure bacterial virulence. Nat. Protoc. 4, 25–30. doi: 10.1038/nprot.2008.212
Gibson, R. L., Burns, J. L., and Ramsey, B. W. (2003). Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168, 918–951. doi: 10.1164/rccm.200304-505SO
Gonyar, L. A., Fankhauser, S. C., and Goldberg, J. B. (2015). Single amino acid substitution in homogentisate 1,2-dioxygenase is responsible for pigmentation in a subset of Burkholderia cepacia complex isolates. Environ. Microbiol. Rep. 7, 180–187. doi: 10.1111/1758-2229.12217
Goudie, A. D., Lynch, K. H., Seed, K. D., Stothard, P., Shrivastava, S., Wishart, D. S., et al. (2008). Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genomics 9:615. doi: 10.1186/1471-2164-9-615
Govan, J., Brown, P., Maddison, J., Doherty, C., Nelson, J., Dodd, M., et al. (1993). Evidence for transmission of Pseudomonas cepacia by social contact in cystic-fibrosis. Lancet 342, 15–19. doi: 10.1016/0140-6736(93)91881-L
Graindorge, A., Menard, A., Neto, M., Bouvet, C., Miollan, R., Gaillard, S., et al. (2010). Epidemiology and molecular characterization of a clone of Burkholderia cenocepacia responsible for nosocomial pulmonary tract infections in a French intensive care unit. Diagn. Microbiol. Infect. Dis. 66, 29–40. doi: 10.1016/j.diagmicrobio.2009.06.008
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hearn, E. M., Dennis, J. J., Gray, M. R., and Foght, J. M. (2003). Identification and characterization of the emhABC efflux system for polycyclic aromatic hydrocarbons in Pseudomonas fluorescens cLP6a. J. Bacteriol. 185, 6233–6240. doi: 10.1128/JB.185.21.6233-6240.2003
Holden, M. T. G., Seth-Smith, H. M. B., Crossman, L. C., Sebaihia, M., Bentley, S. D., Cerdeno-Tarraga, A. M., et al. (2009). The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 191, 261–277. doi: 10.1128/JB.01230-08
Isles, A., Maclusky, I., Corey, M., Gold, R., Prober, C., Fleming, P., et al. (1984). Pseudomonas cepacia infection in cystic-fibrosis - an emerging problem. J. Pediatr. 104, 206–210. doi: 10.1016/S0022-3476(84)80993-2
Leitao, J. H., Sousa, S. A., Ferreira, A. S., Ramos, C. G., Silva, I. N., and Moreira, L. M. (2010). Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Appl. Microbiol. Biotechnol. 87, 31–40. doi: 10.1007/s00253-010-2528-0
Li, X.-Z., Plesiat, P., and Nikaido, H. (2015). The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi: 10.1128/CMR.00117-14
Lynch, K. H., Abdu, A. H., Schobert, M., and Dennis, J. J. (2013a). Genomic characterization of JG068, a novel virulent podovirus active against Burkholderia cenocepacia. BMC Genomics 14:574. doi: 10.1186/1471-2164-14-574
Lynch, K. H., Liang, Y., Eberl, L., Wishart, D. S., and Dennis, J. J. (2013b). Identification and characterization of ϕH111-1. Bacteriophage 3:e26649.
Mahenthiralingam, E., Baldwin, A., and Vandamme, P. (2002). Burkholderia cepacia complex infection in patients with cystic fibrosis. J. Med. Microbiol. 51, 533–538. doi: 10.1099/0022-1317-51-7-533
Mahenthiralingam, E., Bischof, J., Byrne, S. K., Radomski, C., Davies, J. E., Av-Gay, Y., et al. (2000). DNA-Based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 38, 3165–3173.
Mahenthiralingam, E., Urban, T. A., and Goldberg, J. B. (2005). The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3, 144–156. doi: 10.1038/nrmicro1085
Martinez, J. L., Sanchez, M. B., Martinez-Solano, L., Hernandez, A., Garmendia, L., Fajardo, A., et al. (2009). Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 33, 430–449. doi: 10.1111/j.1574-6976.2008.00157.x
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
McClean, S., and Callaghan, M. (2009). Burkholderia cepacia complex: epithelial cell-pathogen confrontations and potential for therapeutic intervention. J. Med. Microbiol. 58(Pt 1), 1–12. doi: 10.1099/jmm.0.47788-0
McKeon, S., McClean, S., and Callaghan, M. (2010). Macrophage responses to CF pathogens: JNK MAP kinase signaling by Burkholderia cepacia complex lipopolysaccharide. FEMS Immunol. Med. Microbiol. 60, 36–43. doi: 10.1111/j.1574-695X.2010.00712.x
Mendes, R., Garbeva, P., and Raaijmakers, J. M. (2013). The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663. doi: 10.1111/1574-6976.12028
Mullen, T., Markey, K., Murphy, P., McClean, S., and Callaghan, M. (2007). Role of lipase in Burkholderia cepacia complex (Bcc) invasion of lung epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 26, 869–877. doi: 10.1007/s10096-007-0385-2
Nies, D. (1995). The cobalt, zinc, and cadmium efflux system czcabc from alcaligenes-eutrophus functions as a cation-proton antiporter in Escherichia coli. J. Bacteriol. 177, 2707–2712. doi: 10.1128/jb.177.10.2707-2712.1995
Peeters, C., Cooper, V. S., Hatcher, P. J., Verheyde, B., Carlier, A., and Vandamme, P. (2017). Comparative genomics of Burkholderia multivorans, a ubiquitous pathogen with a highly conserved genomic structure. PLoS One 12:e0176191. doi: 10.1371/journal.pone.0176191
Perrin, E., Fondi, M., Papaleo, M. C., Maida, I., Buroni, S., Pasca, M. R., et al. (2010). Exploring the HME and HAE1 efflux systems in the genus Burkholderia. BMC Evol. Biol. 10:164. doi: 10.1186/1471-2148-10-164
Pirone, L., Bragonzi, A., Farcomeni, A., Paroni, M., Auriche, C., Conese, M., et al. (2008). Burkholderia cenocepacia strains isolated from cystic fibrosis patients are apparently more invasive and more virulent than rhizosphere strains. Environ. Microbiol. 10, 2773–2784. doi: 10.1111/j.1462-2920.2008.01697.x
Ramette, A., LiPuma, J. J., and Tiedje, J. M. (2005). Species abundance and diversity of Burkholderia cepacia complex in the environment. Appl. Environ. Microbiol. 71, 1193–1201. doi: 10.1128/AEM.71.3.1193-1201.2005
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Rodriguez-Valera, F., Martin-Cuadrado, A.-B., Rodriguez-Brito, B., Pašić, L., Thingstad, T. F., Rohwer, F., et al. (2009). Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 7, 828–836. doi: 10.1038/nrmicro2235
Roszniowski, B., Latka, A., Maciejewska, B., Vandenheuvel, D., Olszak, T., Briers, Y., et al. (2017). The temperate Burkholderia phage AP3 of the Peduovirinae shows efficient antimicrobial activity against B. cenocepacia of the IIIA lineage. Appl. Microbiol. Biotechnol. 101, 1203–1216. doi: 10.1007/s00253-016-7924-7
Schwager, S., Agnoli, K., Koethe, M., Feldmann, F., Givskov, M., Carlier, A., et al. (2013). Identification of Burkholderia cenocepacia strain H111 virulence factors using nonmammalian infection hosts. Infect. Immun. 81, 143–153. doi: 10.1128/IAI.00768-12
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Silveira, C. B., and Rohwer, F. L. (2016). Piggyback-the-Winner in host-associated microbial communities. NPJ Biofilms Microbiomes 2:16010. doi: 10.1038/npjbiofilms.2016.10
Sitkiewicz, I., Nagiec, M. J., Sumby, P., Butler, S. D., Cywes-Bentley, C., and Musser, J. M. (2006). Emergence of a bacterial clone with enhanced virulence by acquisition of a phage encoding a secreted phospholipase A2. Proc. Natl. Acad. Sci. U.S.A. 103, 16009–16014. doi: 10.1073/pnas.0607669103
Speert, D. P., Henry, D., Vandamme, P., Corey, M., and Mahenthiralingam, E. (2002). Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis. Can. Emerg. Infect. Dis. 8, 181–187. doi: 10.3201/eid0802.010163
Spilker, T., Baldwin, A., Bumford, A., Dowson, C. G., Mahenthiralingam, E., and LiPuma, J. J. (2009). Expanded multilocus sequence typing for Burkholderia species. J. Clin. Microbiol. 47, 2607–2610. doi: 10.1128/JCM.00770-09
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Steinert, M. (2011). Pathogen-host interactions in Dictyostelium, Legionella, Mycobacterium and other pathogens. Semin. Cell Dev. Biol. 22, 70–76. doi: 10.1016/j.semcdb.2010.11.003
Summer, E. J., Gill, J. J., Upton, C., Gonzalez, C. F., and Young, R. (2007). Role of phages in the pathogenesis of Burkholderia, or “Where are the toxin genes in Burkholderia phages?”. Curr. Opin. Microbiol. 10, 410–417. doi: 10.1016/j.mib.2007.05.016
Summer, E. J., Gonzalez, C. F., Carlisle, T., Mebane, L. M., Cass, A. M., Savva, C. G., et al. (2004). Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 340, 49–65. doi: 10.1016/j.jmb.2004.04.053
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tobe, T., Beatson, S. A., Taniguchi, H., Abe, H., Bailey, C. M., Fivian, A., et al. (2006). An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U.S.A. 103, 14941–14946. doi: 10.1073/pnas.0604891103
Tomich, M., Herfst, C. A., Golden, J. W., and Mohr, C. D. (2002). Role of flagella in host cell invasion by Burkholderia cepacia. Infect. Immun. 70, 1799–1806. doi: 10.1128/IAI.70.4.1799-1806.2002
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013). MicroScope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647. doi: 10.1093/bib/bbx113
Vallenet, D., Engelen, S., Mornico, D., Cruveiller, S., Fleury, L., Lajus, A., et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database 2009:bap021. doi: 10.1093/database/bap021
Vandamme, P., Holmes, B., Coenye, T., Goris, J., Mahenthiralingam, E., LiPuma, J. J., et al. (2003). Burkholderia cenocepacia sp. nov. - a new twist to an old story. Res. Microbiol. 154, 91–96. doi: 10.1016/S0923-2508(03)00026-3
Varga, J. J., Losada, L., Zelazny, A. M., Kim, M., McCorrison, J., Brinkac, L., et al. (2013). Draft genome sequences of Burkholderia cenocepacia ET12 lineage strains K56-2 and BC7. Genome Announc. 1:e00841-13. doi: 10.1128/genomeA.00841-13
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. doi: 10.1093/bioinformatics/btl369
Vial, L., Chapalain, A., Groleau, M.-C., and Deziel, E. (2011). The various lifestyles of the Burkholderia cepacia complex species: a tribute to adaptation. Environ. Microbiol. 13, 1–12. doi: 10.1111/j.1462-2920.2010.02343.x
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Youenou, B., Favre-Bonte, S., Bodilis, J., Brothier, E., Dubost, A., Muller, D., et al. (2015). Comparative genomics of environmental and clinical Stenotrophomonas maltophilia strains with different antibiotic resistance profiles. Genome Biol. Evol. 7, 2484–2505. doi: 10.1093/gbe/evv161
Youenou, B., Hien, E., Deredjian, A., Brothier, E., Favre-Bonté, S., and Nazaret, S. (2016). Impact of untreated urban waste on the prevalence and antibiotic resistance profiles of human opportunistic pathogens in agricultural soils from Burkina Faso. Environ. Sci. Pollut. Res. 23, 25299–25311. doi: 10.1007/s11356-016-7699-5
Keywords: Burkholderia cenocepacia, Burkina Faso, virulence factors, RND pumps, prophages, antibiotic resistance
Citation: Bodilis J, Denet E, Brothier E, Graindorge A, Favre-Bonté S and Nazaret S (2018) Comparative Genomics of Environmental and Clinical Burkholderia cenocepacia Strains Closely Related to the Highly Transmissible Epidemic ET12 Lineage. Front. Microbiol. 9:383. doi: 10.3389/fmicb.2018.00383
Received: 19 December 2017; Accepted: 20 February 2018;
Published: 06 March 2018.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, Hong KongReviewed by:
Silvia Buroni, University of Pavia, ItalyElena Perrin, Università degli Studi di Firenze, Italy
Copyright © 2018 Bodilis, Denet, Brothier, Graindorge, Favre-Bonté and Nazaret. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josselin Bodilis, josselin.bodilis@univ-rouen.fr
†Present address: Arnault Graindorge, Université Montpellier, Montpellier, France