 Marina Sánchez-Hidalgo
Marina Sánchez-Hidalgo Ignacio González
Ignacio González Cristian Díaz-Muñoz
Cristian Díaz-Muñoz Germán Martínez
Germán Martínez Olga Genilloud
Olga Genilloud
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 13 March 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00369
This article is part of the Research TopicMicrobial Taxonomy, Phylogeny and BiodiversityView all 28 articles
Actinomycetes have been extensively exploited as one of the most prolific secondary metabolite-producer sources and continue to be in the focus of interest in the constant search of novel bioactive compounds. The availability of less expensive next generation genome sequencing techniques has not only confirmed the extraordinary richness and broad distribution of silent natural product biosynthetic gene clusters among these bacterial genomes, but also has allowed the incorporation of genomics in bacterial taxonomy and systematics. As part of our efforts to isolate novel strains from unique environments, we explored lichen-associated microbial communities as unique assemblages to be studied as potential sources of novel bioactive natural products with application in biotechnology and drug discovery. In this work, we have studied the whole genome sequences of two new Amycolatopsis strains (CA-126428 and CA-128772) isolated from tropical lichens, and performed a comparative genomic analysis with 41 publicly available Amycolatopsis genomes. This work has not only permitted to infer and discuss their taxonomic position on the basis of the different phylogenetic approaches used, but has also allowed to assess the richness and uniqueness of the biosynthetic pathways associated to primary and secondary metabolism, and to provide a first insight on the potential role of these bacteria in the lichen-associated microbial community.
The class Actinobacteria was defined for Gram-positive bacteria with high genomic G+C content (over 55%), among which are included major families of actinomycetes that produce almost 75% of all known secondary metabolites, many of them of high relevance for human health and biotechnology industry (Barka et al., 2016). These compounds have been shown to include a wide range of industrial and medical applications, as drugs (i.e., antifungals, antibacterials, antitumorals, or immunosuppresors), herbicides and plant growth promoting agents among others (Genilloud et al., 2011; Sharma et al., 2014; Genilloud, 2017a). Within the actinomycetes, the genus Streptomyces is the most prolific and most studied producer of secondary metabolites, but members of the families Pseudonocardiaceae and Micromonosporaceae have also shown to produce a broad diversity of bioactive molecules. Among Pseudonocardiaceae, members of the genus Amycolatopsis produce several relevant secondary metabolites, such as balhimycin, vancomycin, avoparcin, ristomycin, chelocardin, chloroeremomycin, ECO-0501 and rifamycin (Chen et al., 2016; Kumari et al., 2016). More recently, other antibiotics have been described from some Amycolatopsis strains such as macrotermycins A–D (Beemelmanns et al., 2017), pargamicins B–D (Hashizume et al., 2017) and rifamorpholines A–E (Xiao et al., 2017). In addition, the importance of Amycolatopsis strains in industrial processes such as bioremediation (heavy metal immobilization, herbicide and polymers biodegradation) and bioconversion (wuxistatin and vanillin production) (Dávila Costa and Amoroso, 2014), has been clearly demonstrated.
Members of the genus Amycolatopsis were originally misidentified as Streptomyces, then as Nocardia to be finally recognized as belonging to a new genus with species lacking mycolic acid in their cell wall (Lechevalier et al., 1986). A total of 70 Amycolatopsis species have been recognized so far (http://www.bacterio.net/amycolatopsis.html) and isolated from a broad diversity of environments ranging from soils, plants, and ocean sediments to clinical sources. Most species of Amycolatopsis belong to two major subclades: A. methanolica (AMS) and A. orientalis (AOS) (Tang et al., 2016). Several AOS species have been shown to synthesize antibiotics, while AMS strains show a biotechnological potential for the overproduction of aromatic amino acids and bioremediation (Tang et al., 2016).
Recent advances in the field of next generation sequencing (NGS), have allowed an exponential increase of bacterial genome sequences available in public databases, being the genomes of Streptomyces spp. the most intensively studied (Loman and Pallen, 2015; Chen et al., 2016; Kumari et al., 2016). In the case of Amycolatopsis, as of June 2017, 54 sequencing genome projects had been assembled (www.ncbi.nlm.nih.gov/assembly) from which 30 belong to type strains. The 9 genome projects that have been completely sequenced have shown that Amycolatopsis strains contain comparatively large genomes (from 5 to 10 Mb) in the form of a circular chromosome. The analysis of these genomes with improved algorithms has revealed that Amycolatopsis strains harbor many more cryptic biosynthetic gene clusters (BGCs) than previously estimated. Eleven BGCs from Amycolatopsis have been characterized and compiled in the MIBiG (Minimum Information about a Biosynthetic Gene cluster) Repository (Table 1) (http://mibig.secondarymetabolites.org, Medema et al., 2015). Moreover, this increased availability of new genome sequences in public databases is allowing a deeper characterization of microbial biosynthetic potential using genomic data as well as a new standardization of new taxonomic approaches based on full genome sequence information (Colston et al., 2014).
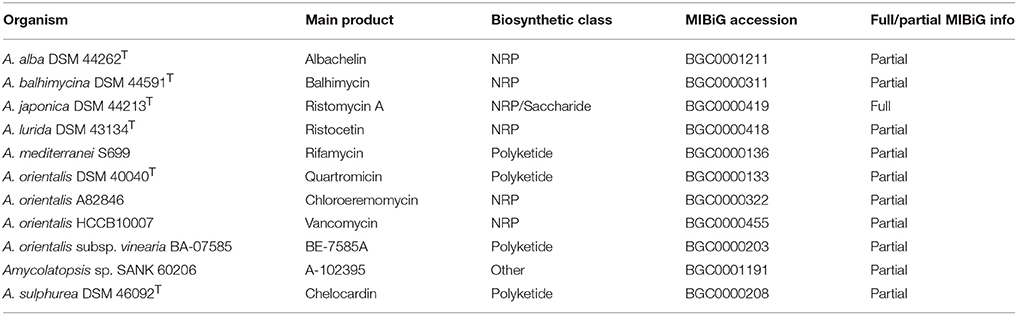
Table 1. Biosynthetic gene clusters from Amycolatopsis strains described in the MIBiG database.
The emergence of antimicrobial resistance against frequently used antibiotics has brought to light the urgent need of novel antibacterial compounds, and the necessity to look for alternative isolation sources and new drug discovery strategies to identify novel chemical classes of compounds (Adamek et al., 2017). As part of our integrated antibiotic discovery programs, we selected two bioactive Amycolatopsis strains (CA-128772 and CA-126428) previously isolated in our laboratory from lichens collected respectively in tropical areas from Hawaii and Reunion islands (González et al., 2005). Lichens are symbiotic associations of a fungal mycobiont, one or more algal or cyanobacterial photobionts and a diverse community of associated microbes, and represent important sources of natural products, mostly produced by the mycobiont. The Alphaproteobacteria are the predominant lichen-associated bacteria, but Actinobacteria, Firmicutes, Betaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria have been also identified (Cardinale et al., 2006; Aschenbrenner et al., 2016). These lichen-associated bacterial communities have been suggested to play important roles in the symbiosis, being of special interest the populations of the orders Burkholderiales and Actinomycetales, well known as prolific secondary metabolite producers (Calcott et al., 2017). Recent studies have confirmed that lichen-associated bacteria produce new bioactive substances, especially among Streptomyces species (González et al., 2005; Cardinale et al., 2006; Parrot et al., 2015; Calcott et al., 2017; Liu et al., 2017). However, few actinomycetes belonging to the genus Amycolatopsis have been isolated so far from these sources (González et al., 2005; Liu et al., 2017), what triggered our interest to study their biosynthetic potential and suggest a role in this unique environment.
In this work, we have established the taxonomic position and mined the draft genomes of two new strains of Amycolatopsis, CA-126428 and CA-128772. The study has permitted to perform a comparative analysis with all publicly available complete or draft Amycolatopsis genomes, with a specific focus on the richness and diversity of their BGCs.
Genomic DNAs from strains CA-126428 and CA-128772 were extracted and purified as previously described (Kieser et al., 2000) from strains grown in ATCC-2 liquid medium [0.5% yeast extract (Difco, Franklin Lakes, NJ, USA), 0.3% beef extract (Difco), 0.5% peptone (Difco), 0.1% dextrose (Difco), 0.2% starch from potato (Panreac, Barcelona, Spain), 0.1% CaCO3 (E. Merck, Darmstadt, Germany), and 0.5% NZ amine E (Sigma, St Louis, MO, USA)].
Genomes of strains CA-126428 and CA-128772 were sequenced de novo by Macrogen (Seoul, Korea; http://www.macrogen.com/) and Service XS (Leiden, the Netherlands; http://www.servicexs.com), respectively, using the Illumina HiSeq 2500 platform. Paired-end libraries were created using the NEBNext Ultra DNA library prep kit (New England Biolabs). The quality and yield after sample preparation was measured with the Fragment Analyzer (AATI), and the size of the resulting product was consistent with the expected size of 500–700 bp. Clustering and DNA sequencing was performed according to manufacturer's protocol. A concentration of 15.0 pM DNA was used. Image analysis, base calling and quality check was performed with the Illumina data analysis pipeline RTA v1.18.64 and Bcl2fastq v2.17. A dataset of at least 1.3 Gb per sample was delivered. Prior to assembly, the reads were trimmed for adapter sequences and filtered for sequence quality. Presumed adapter sequences were removed from the read when the bases matched a sequence in the adapter sequence set (TruSeq adapters) with 2 or less mismatches and an alignment score of at least 12. Bases with phred scores below Q22 were removed from the reads. Glimmer v3.2 (Aggarwal and Ramaswamy, 2002) was used for annotation.
Since the sequences of the 16S rRNA genes were incomplete in the genomes, PCR primers FD1 and RP2 were used to amplify the nearly full-length 16S rRNA genes of the strains CA-126428 and CA-128772 (Weisburg et al., 1991). PCR products were sequenced by Secugen (Madrid, Spain; http://www.secugen.es/) with primers FD1, RP2, 1100R, and 926F (Lane, 1991). Partial sequences were assembled and edited using the Assembler contig editor component of Bionumerics 5.10 analysis software (Applied Maths NV, Sint-Martens-Latem, Belgium).
The identification of the closest match sequences was performed at the EzBiocloud server (http://www.ezbiocloud.net/identify) (Yoon et al., 2017).
Strains CA-128772 and CA-126428 belong to MEDINA's microbial collection and were previously isolated from arboricolous lichens collected in humid tropical forests from Hawaii and dry tropical forests from Reunion islands, respectively, as previously described (González et al., 2005). In brief, each lichen sample (300–500 mg) was washed twice with sterile water and homogenized in 30 ml of sterile water using a blender. Serial dilutions were plated on selective actinomycete isolation media (González et al., 2005). Individual colonies were isolated and grown at 28°C on YME agar medium (0.4% yeast extract, 1% malt extract, 0.4% glucose and 0.2% Bacto-agar).
The complete 16S rRNA sequences of 70 Amycolatopsis type strains were downloaded from the List of Prokaryotic Names with Standing in Nomenclature (LPSN) database (http://www.bacterio.net/amycolatopsis.html) (Supplementary Table 1). The sequences ranged from 1,351 to 1,530 bp. A set of publicly available 41 complete and draft genomes from Amycolatopsis strains and the 16S rRNA gene sequences of Amycolatopsis non-type strains were downloaded from NCBI (https://www.ncbi.nlm.nih.gov) (Supplementary Table 1).
The 16S rRNA sequences were aligned with the MEGA 7.0.26 package (http://www.megasoftware.net) (Kumar et al., 2016) using Clustawl. The pairwise deletion Neighbor-Joining method of the MEGA 7.0.26 package (http://www.megasoftware.net) (Kumar et al., 2016), corrected with Jukes-Cantor algorithms and a bootstrap of 1,000 replicates, was used to construct a phylogenetic tree based on the complete 16S rRNA sequences. Micromonospora chalcea DMS 43026T (Foulerton, 1905) 16S rRNA gene was used as outgroup. Genomic distances were calculated with the Kimura-2 parameter model included also in the MEGA software.
Four single-copy housekeeping genes present in all the genomes were used for MLSA analysis: atpD (ATP synthase F1, beta subunit), dnaK (Hsp70 chaperone), recA (recombinase A) and rpoB (RNA polymerase beta subunit) (Sentausa and Fournier, 2013; Glaeser and Kämpfer, 2015). The sequences of each of these genes were extracted from the Amycolatopsis genomes using the Multi-tBlastN algorithm (https://blast.ncbi.nlm.nih.gov) and were subsequently concatenated with Geneious 9.1.8 software (Biomatters, www.geneious.com, (Kearse et al., 2012), generating a sequence of approximately 7.8 Kb. A phylogenetic tree was constructed using the MEGA 7.0.26. pairwise deletion Neighbor-Joining and Jukes-Cantor algorithms methods and a bootstrap of 1,000 replicates (Kumar et al., 2016). Micromonospora chalcea DMS 43026T (Foulerton, 1905) concatenated housekeeping genes were used as outgroup. Genomic distances were calculated with the Kimura-2 parameter model included also in the MEGA software.
The genomes were functionally characterized in the KEGG Database (http://www.kegg.jp) (Kanehisa et al., 2016a). Ortholog K numbers were assigned by the BlastKOALA sequence similarity tool (http://www.kegg.jp/blastkoala/) (Kanehisa et al., 2016b). KEGG mapper tool (http://www.kegg.jp/kegg/tool/map_pathway.html) was used to reconstruct and compare the metabolic pathways.
The presence of BGCs in the genomes was analyzed using antiSMASH 4.0, (http://antismash.secondarymetabolites.org) (Blin et al., 2017) including Clusterfinder with a minimum probability of 60%.
Genome comparisons were performed on both complete and draft genomes. The contigs of draft genomes were concatenated with Geneious 9.1.8 software (Biomatters, www.geneious.com), (Kearse et al., 2012) to facilitate analysis.
Gene synteny analysis was assessed with progressiveMauve (http://darlinglab.org/mauve/), (Darling et al., 2010). To compare and visualize genomic regions from Mauve results we used the R-package genoPlotR (http://genoplotr.r-forge.r-project.org/) (Guy et al., 2011).
The genome sequence similarity between Amycolatopsis strains was evaluated with the Genome-to-Genome Distance Calculator (GGDC) 2.1 online software (http://ggdc.dsmz.de), (Auch et al., 2010; Meier-Kolthoff et al., 2013, 2014).
The Average Nucleotide Identity (ANI) and the orthology of genome sequences were calculated with OrthoANI, (http://www.ezbiocloud.net/sw/oat), (Lee et al., 2016).
The genomes of strains CA-126428 and CA-128772 were sequenced, assembled and annotated by external providers and their characteristics are summarized in Table 2. A total of 2 × 10,113,591 paired reads for CA-128772 and 2 × 6,420,119 paired reads for CA-126428 were generated with theoretical coverage >100 ×. All reads were assembled to a draft genome of 10.5 Mb for CA-126428 and 10.2 Mb for CA-128772, with a G+C content of 71.4 and 71.8%, respectively. These G+C content values are similar to those of some Amycolatopsis complete genomes, such as the strains of A. mediterranei U32 and S699 (Kumari et al., 2016). A total of 188 contigs and 9626 coding DNA sequences (CDSs) were obtained for CA-126428, while 121 contigs and 10986 CDSs were assembled for CA-128772. The inconsistency between the number of CDS and the length of the genomes can be explained by the N50 and the average length of the contigs. As it is shown in Table 2, the genome sequence from strain CA-128772 presents a higher N50 value (186,760) and a higher average contig length (83,989 bp) than strain CA-126428 (N50 = 139,227, average length = 55,808 bp) suggesting that the CA-126428 genome is more fragmented and probably more CDSs are missed.

Table 2. NGS statistic data of CA-126428 and CA-128772 draft genomes.
These Whole Genome Shotgun projects were deposited at DDBJ/ENA/GenBank under the accession numbers PPHF00000000 (strain CA-126428) and PPHG00000000 (strain CA-128772). The versions described in this paper are versions PPHF01000000 and PPHG01000000 (Supplementary Table 1).
The molecular identification of strains CA-128772 and CA-126428 was based on the comparison of their 16S rRNA gene sequences to reference type strains sequences using the EzBiocloud server. Both strains showed to contain only one 16S rRNA copy. The 16S rRNA gene sequences were deposited at GenBank under the accession numbers MG800320 (strain CA-126428) and MG799844 (strain CA-128772) (Supplementary Table 1). Strain CA-128772 showed the highest similarity values with A. pretoriensis DSM 44654T (99.79%), A. rifamycinica DSM 46095T (99.58%), A. lexingtoniensis DSM 44653T (99.58%) and A. tolypomycina DSM 44544T (99.50%). With respect to isolate CA-126428, the highest similarity values were obtained with A. mediterranei DSM 43304T (99.51%), A. kentuckyensis DSM 44652T (99.44%), A. rifamycinica DSM 46095T (99.44%) and A. pretoriensis DSM 44654T (99.37%).
To confirm the taxonomic assignment and the existing phylogenetic relationships between our isolates and other reference and non-type Amycolatopsis strains (Supplementary Table 1), a phylogenetic tree was built based on the complete 16S rRNA gene sequences, using Micromonospora chalcea DSM 43026T as outgroup (Figure 1). Amycolatopsis species group into three major subclades including the mesophilic or moderately thermophilic A. orientalis (AOS) subclade, the thermophilic A. methanolica (AMS) subclade, and the mesophilic A. taiwanensis (ATS) subclade (Figure 1) (Tang et al., 2016). The AOS subclade includes previously defined groups A-E and G (Everest and Meyers, 2009, 2011) and groups H, I, and J (Tang et al., 2016). The ATS and AMS subclades include the F group (Everest and Meyers, 2009, 2011), that was latter divided in F1 (ATS) and F2 (AMS) (Tang et al., 2016).
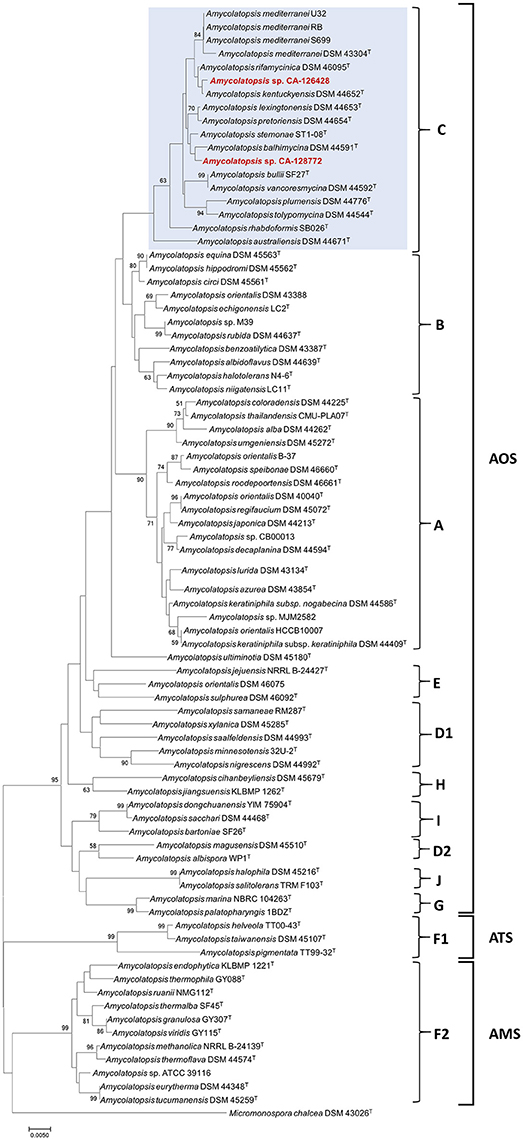
Figure 1. Phylogenetic NJ tree based on 16S rRNA gene sequences from 82 Amycolatopsis strains. The scale bar indicates 5 nucleotide substitutions per 1,000 nucleotides, and the node numbers are percentage bootstrap values based on 1,000 resampled datasets. Bootstrap values below 50% are not shown. The AOS, ATS and AMS subclades are indicated, as well as the groups A-J. The strains grouped with CA-126428 and CA-128772 isolates (group C) are blue-shaded.
The phylogenetic tree associates both strains CA-126428 and CA-128772 to the AOS subclade, containing 36 type species from which three have a full genome sequence, as well as three non-type strains of A. mediterranei (RB, S699 and U32), five non-type strains of A. orientalis (DSM 43388, DSM 46075, HCCB10007, CB00013, and B-37) and two strains of Amycolatopsis sp. (M39 and CB00013). Both CA-126428 and CA-128772 strains, together with the three non-type A. mediterranei strains, are included in a branch containing 13 type strains, identified as group C (Figure 1). Within the AMS subclade, another non-type genome-sequenced strain, Amycolatopsis sp. ATCCC 39116, was included (Figure 1). In general, the topology of the groups A, B, C, F1, F2, G, H, and I is conserved in our tree despite the additional species that were not previously analyzed (Figure 1). The only exceptions are the group D, that is divided in two groups (D1 and D2), and the strain A. ultiminotia DSM 45180T, that is no longer included in group E (Figure 1).
The closest genetic distances were determined for strain CA-128772 with A. pretoriensis DSM 44654T (0.003) and for strain CA-126428 with A. kentuckyensis DSM 44652T (0.001) (Supplementary Figure 1). The distances among the strains in group C ranged between 0.001 and 0.02, and no distance was observed among the non-type A. mediterranei strains. Strains A. helveola TT00-43T, A. taiwanensis DSM 45107T and A. pigmentata TT99-32T showed the highest evolutionary distances (0.07–0.09) with the rest of strains. Nonetheless, these values are very low and show that these species are very closely-related.
Two new 16S rRNA- and MLSA-based phylogenetic trees were constructed only including the 43 genome-sequenced strains (Figure 2). The MLSA tree was constructed from four-gene (atpD, dnaK, recA, and rpoB) concatenated nucleotide sequences. As shown in Figure 2, the three AOS, ATS and AMS subclades are present in both trees, but some differences are observed in the organization of the groups in AOS subclade. The same A, B and C groups are defined in both trees, whereas in the MLSA tree, group B includes strains from group E and group A includes strains from group D1. In the 16S rRNA-based tree strains from groups E, G, and J clustered together, as well as strains from D1 and I groups, whereas in the MLSA tree, strains from groups G and J are clustered.

Figure 2. Phylogenetic NJ trees based on 16S rRNA gene sequences (Left) and four concatenated sequences (atpD, dnaK, recA and rpoB) (MLSA, Right) from 43 genome-sequenced Amycolatopsis strains. The scale bars indicate the percentage of difference in the nucleotide sequences, and the node numbers are percentage bootstrap values based on 1,000 resampled datasets. Bootstrap values below 50% are not shown. The AOS, ATS and AMS subclades are indicated, as well as the groups A–J. The strains grouped with CA-126428 and CA-128772 isolates (group C) are blue-shaded.
The composition of group C, that contains strains CA-126428 and CA-128772, is conserved in both 16S rRNA and MLSA trees, except for the strain A. saalfeldensis DSM 44993T, which is not included in the group in the MLSA tree. However, the relative position of the strains differs in both trees. In the 16S rRNA tree strain CA-126428 is closely related to the strains A. rifamycinica DSM 46095T, A. kentuckyensis DSM 44652T and A. mediterranei, and strain CA-128772 is closely related to A. balhimycina DSM 44591T, A. lexingtoniensis DSM 44653T and A. pretoriensis DSM 44654T. In contrast, in the MLSA tree, both strains cluster together and are closely related to A. rifamycinica DSM 46095T and the A. mediterranei strains. Nevertheless none of these relationships were supported by the bootstrap values, suggesting that both strains could represent novel species within the genus. Bootstrap values of group C (100% in MLSA tree and >50% in 16S rRNA tree) support the significance of the MLSA-based phylogeny. The genomic distances based on both 16S rRNA and MLSA phylogenetic analyses are shown in Figure 3. The 16S rRNA sequence distances ranged from 0 to 0.096 (mean 0.039), while MLSA distances present a broader range between 0 and 0.582 (mean 0.071).
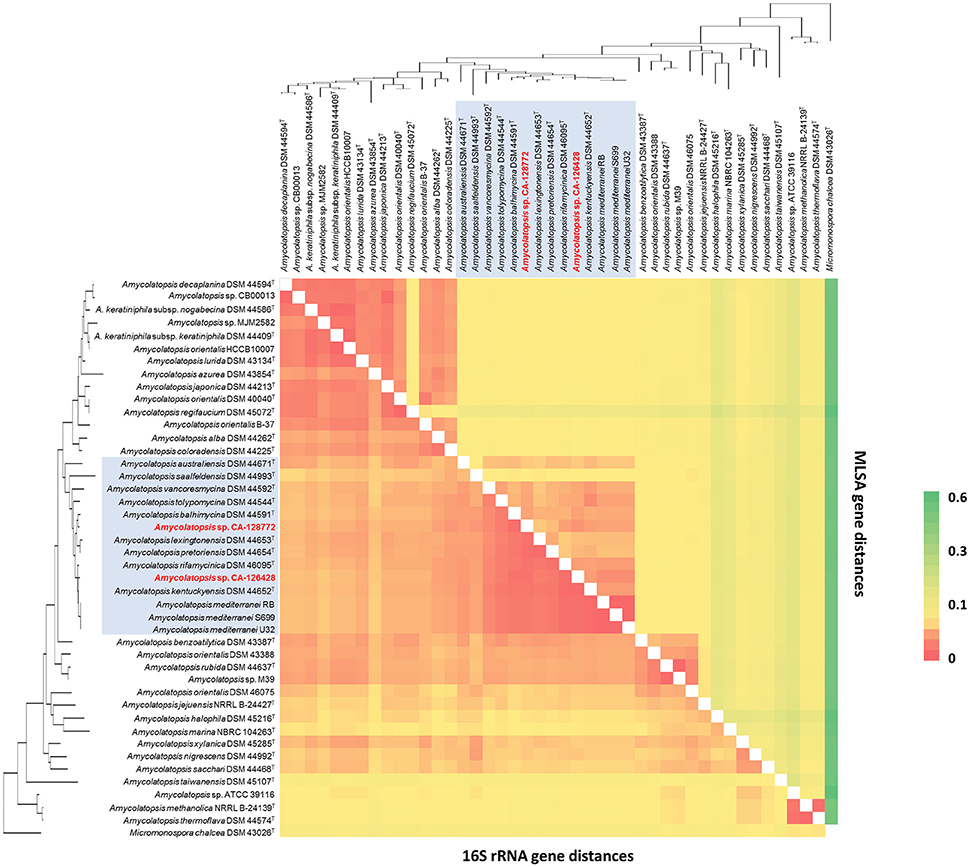
Figure 3. 16S rRNA (Bottom left) vs. MLSA (Top right) genomic distances heatmap using the Kimura 2-parameter model. Self-genome comparisons occur on a diagonal line stretching from the top left to the bottom right corners. The strains are ordered in the same way as the 16S phylogenetic tree in Figure 2, which has been placed upon both axes for orientation. The strains belonging to group C have been shaded in blue for clarification. The heatmap legend is shown on the right.
Previous whole genome sequence studies (Tang et al., 2016) have shown that some Amycolatopsis strains present multiple copies of 16S rRNA genes, which is not the case of strains CA-126428 and CA-128772. The 16S rRNA sequence identities for some inter-species pairs are higher than those of the corresponding intra-species pairs and this fact may influence the structure of Amycolatopsis 16S rRNA gene phylogenetic trees (Tang et al., 2016). The lack of resolution at the species level of 16S rRNA gene-based phylogenies can be overcome by MLSA methods (Glaeser and Kämpfer, 2015). The observed inconsistency between MLSA and 16S rRNA gene phylogenies reinforce the idea that MLSA allows a much more precise species delineation within bacteria (Stackebrandt et al., 2002; Konstantinidis and Tiedje, 2007; Thompson et al., 2013). However, given that not all housekeeping gene sequences are available for all species, some improvements are needed to make MLSA more generally applicable. The selection of an universal set of genes for all the prokaryotes, the standardization of the number and length of genes to be used or a low time consuming calculation method have been recently proposed (Glaeser and Kämpfer, 2015).
In the case of the strains of Amycolatopsis, it has been shown that a 315 bp variable fragment of the gyrB gene has a higher resolution power than the 16S rRNA gene (Everest and Meyers, 2009). This partial gyrB gene sequence, together with other housekeeping genes, has been proposed as a good candidate to be used for MLSA of this genus. Unfortunately, we were not able to use the gyrB sequence as a MLSA marker, since the sequences were partial or not present in some genomes.
The overall similarity of the Amycolatopsis genomes was analyzed using several approaches.
First, we performed an overall analysis using the Genome-to-Genome Distance Calculator (GGDC) (Auch et al., 2010; Meier-Kolthoff et al., 2013, 2014), that calculates digital (in silico) DNA-DNA hybridization (dDDH) from the intergenomic distances and quantifies the G+C content. No G+C content differences were found among A. mediterranei strains. The highest G+C content differences were found between A. halophila DSM 45216Tand the strains from group C (about 4% difference) (Supplementary Figure 2). In the case of strain CA-128772, the maximum and minimal G+C content differences were found with A. balhimycina DSM 44591T (1.07%), and with A. rifamycinica DSM 46095T (0.1%). In strain CA-126428, the maximum G+C content difference was also observed with A. balhimycina DSM 44591T (0.62%), whereas the minimum difference (0.15%) was with A. lexingtonensis DSM 44653T. According to Meier-Kolthoff et al. (2014), within-species differences in the G+C content are almost exclusively below 1%. The observed differences below 1% between different species are probably due to the incompleteness of most of the genomes analyzed and/or inaccuracies of the species descriptions (Meier-Kolthoff et al., 2014). dDDH analysis showed that, in general, the re-association values among Amycolatopsis strains were in the range of 20%. However, re-association values higher than 70% were observed between some of the genomes analyzed corresponding to non-type species, indicating that they may belong to the same species (Supplementary Figure 3). This is the case of A. orientalis B-37 and A. orientalis DSM 40040T with a 72.1% of re-association; A. orientalis HCCB10007 showed a 72.5% of re-association with A. keratiniphila subsp. keratiniphila DSM 44409T and a 71.9% with A. keratiniphila subsp. nogabecina DSM 44586T; A. japonica DSM 44213T showed a 72.7% of re-association with Amycolatopsis sp. MJM2582 and a 88.3% with Amycolatopsis sp. CB00013 suggesting both strains to belong to A. japonica; Amycolatopsis sp. M39 and A. rubida DSM 44637T had a 93.3% of re-association; A. keratiniphila subsp. nogabecina DSM 44586T and A. keratiniphila subsp. keratiniphila DSM 44409T showed a 90.1%; Amycolatopsis sp. CB00013 had a 72.6% of re-association with Amycolatopsis sp. MJM2582. The rest of the dDDH obtained were below 70%, indicating that these strains belong to different species.
The re-association values between strains of group C ranged from 23 to 54%, except for the three species strains of A. mediterranei (U32, S699, and RB), with a 100% of re-association. In the case of the two new strains of our study, strain CA-128772 showed the maximum re-association value (45.3%) with A. vancoresmycina DSM 44592T, and strain CA-126428 had the maximum value (54.4%) with the three A. mediterranei strains, well below the threshold defined for strains of the same species. These results suggest that both CA-128772 and CA-126428 isolates may represent new Amycolatopsis strains.
Another approach used to study the similarity of the Amycolatopsis genomes was to perform an analysis of the Average Nucleotide Identity (ANI) and the orthology (OrthoANI) (Lee et al., 2016). The comparison of CA-128772 and CA-126428 genomes with the rest of Amycolatopsis genomes showed that the maximum ANI and OrthoANI values were obtained with strains included in group C. However, none of them reach the ANI threshold range (95–96%) for species delineation (Table 3). Again, as previously observed with the dDDH values, strain CA-128772 showed the highest similarity (92.0 and 91.35%) with A. vancoresmycina DSM 44592T, while strain CA-126428 showed the highest similarity (93.97 and 93.03%) with A. mediterranei U32. These results suggest again that CA-128772 and CA-126428 isolates may represent new Amycolatopsis species.
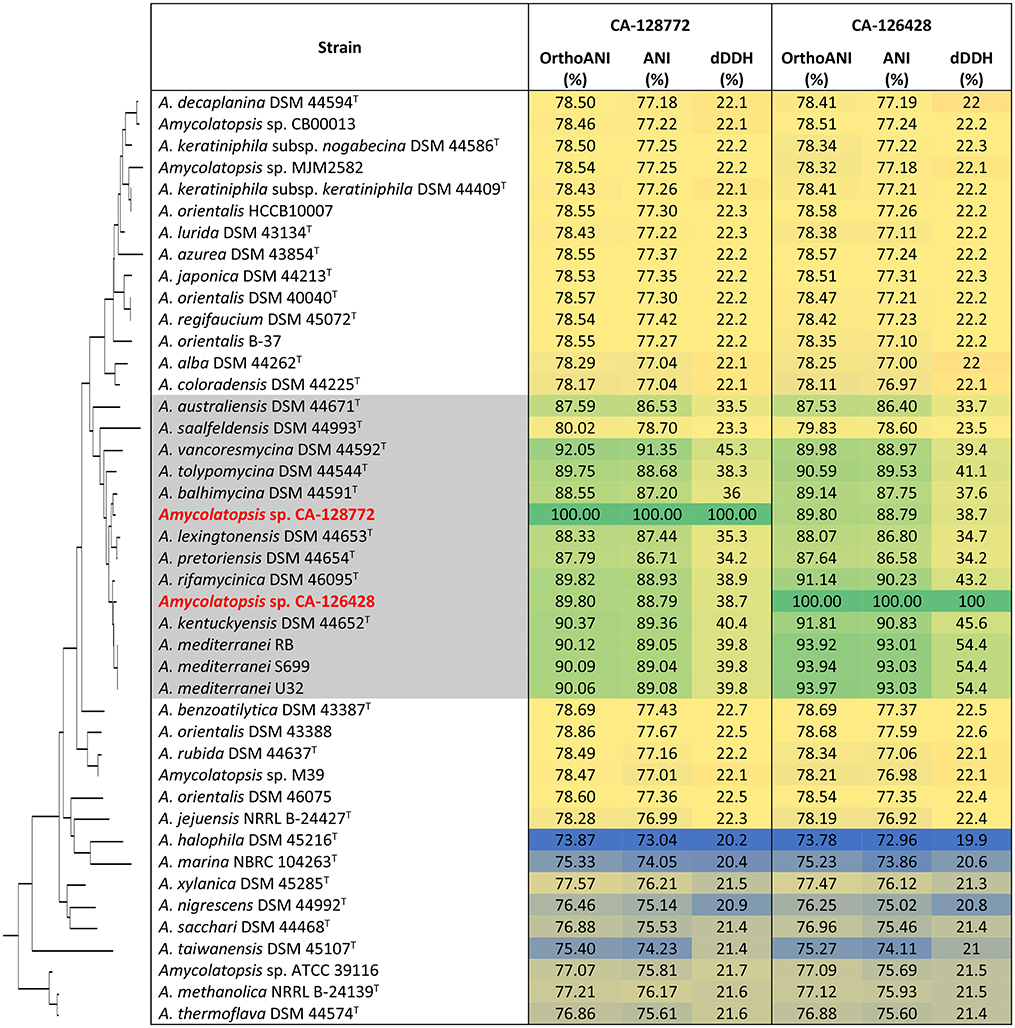
Table 3. OrthoANI and ANI calculations of CA-128772 and CA-126428 genomes against other Amycolatopsis genomes.
Several studies have confirmed that GGDC analysis yields higher correlations with classical DDH than ANI softwares (Auch et al., 2010; Meier-Kolthoff et al., 2013, 2014). Moreover, dDDH calculation is independent of genome length and is thus robust against the use of incomplete draft genomes (Auch et al., 2010).
To compare the relative organization of the genomes, we also performed a gene synteny progressiveMauve analysis (Darling et al., 2010) of the concatenated draft genomes of strains CA-126428 and CA-128772 with nine Amycolatopsis complete genomes (Figure 4). The complete genomes were linearized at position 1 of their sequences. Once again, the high similarity of the three A. mediterranei strains was clearly observed. As previously described (Tang et al., 2016), highly conserved core regions were detected in the left and right arms of all the genomes, except for A. lurida DSM 43134T, which has a different genome rearrangement, and strains CA-126428 and CA-128772. Since the latter are draft concatenated contigs and not complete genomes, the order of the regions is altered, but homologous regions can be observed in the alignment (Figure 4). Other central regions of the chromosomes are also conserved among the genomes, especially in the A. orientalis, A. mediterranei, A. japonica and A. keratiniphila strains.

Figure 4. Comparative analyses of CA-126428 and CA-128772 draft genomes with 9 complete Amycolatopsis genomes. Horizontal straight lines represent the genomes and vertical colored bars represent different conserved genes. The vertical bars are connected by corresponding colored thin lines. Black arrows represent the position of the secondary metabolite biosynthetic pathways predicted by antiSMASH.
Due to the lack of resolution at the species level of the 16S rRNA gene-based phylogenies, and the difficulty to design an universal set of primers for MLSA analysis, our results derived from whole-genome content comparisons arise, as previously reported for other strains (Colston et al., 2014), as the most valuable tool to discriminate between bacterial species. As more bacterial genomes become available, the use of whole genome sequences opens new opportunities for the characterization of bacterial species.
As saprophytic bacteria, actinomycetes have a well-coordinated carbohydrate and nitrogen catabolic systems that allow the adaptation and the efficient utilization of the resources in the nutrient-limited conditions of the environment. These bacteria can produce a large diversity of extracellular enzymes to digest complex polymeric substrates and import the resulting monomers and oligomers to be used as nutrients for catabolism and anabolism and biomass generation (Genilloud, 2017b).
With the aim to analyze the global primary metabolism of strains CA-126428 and CA-128772, their genomes were functionally characterized in the KEGG PATHWAY Database (http://www.genome.jp/kegg/pathway.html) using the BlastKOALA sequence similarity tool (http://www.kegg.jp/blastkoala/). The KEGG BlastKOALA tool mapped 31.8% of the CA-126428 predicted proteins (3063 proteins) to KEGG ortholog groups, while only 19% of the CA-128772 predicted proteins (1882) could be mapped. Supplementary Table 2 presents the distribution of pathways and the number of genes annotated with BlastKOALA, and shows that the content and distribution of different metabolic pathways are very similar between both strains. Although not enough data are available because of the incompleteness of the genomes, some observations on their primary metabolism have been achieved.
A wide variety of extracellular polysaccharide-degrading enzymes have been mapped in the genomes of both Amycolatopsis strains including amylases, endoglucanases, xylanases, chitinases, and beta-glucosidases. However, no ligninases, agarases, mannanases, or cellulases have been detected. As in the case of many actinomycetes, some multiple transport systems to uptake specific carbohydrates are present in both genomes: such as ABC permeases and specific phosphoenolpyruvate-dependent phosphotransferase systems (PTS). In most bacteria, the PTS system plays a major role in the carbon catabolite repression (CCR), a global control system that ensures the preferential use of the different carbon sources available. CCR is mediated by the glycolytic enzyme glucose kinase, for which an ortholog has been identified in both strains. Glucose kinase converts glucose to glucose-6-phosphate, a substrate of different enzymes and pathways (glucose phosphate isomerase, glucose-6-phosphate dehydrogenase, pentose phosphate pathway, and glucose-6-phosphatase) and also plays a key role mediating CCR for enzymes involved in primary and secondary metabolism and precursors for natural product synthesis (Kwakman and Postma, 1994).
Actinomycetes carbohydrate primary metabolism is complex and characterized by the presence of multiple isoenzymes involved in single catalytic steps of glycolysis, the pentose phosphate and TCA cycle pathways. Normally, the genes encoding those enzymes do not cluster in operons and are scattered in the chromosome, subjected to different regulation depending on the carbon sources or the development stage (Genilloud, 2017b). The most important glycolytic pathway is the Embden-Meyerhof (EM) pathway (Salas et al., 1984), which is used to generate ATP and provide precursors for secondary metabolism. The EM pathway is regulated at the level of the glycolytic enzyme phosphofructokinase (Pfk) (Genilloud, 2017b), an enzyme that has not been identified so far in the genomes of strains CA-126428 and CA-128772, suggesting a different type of regulation. The enzymes involved in glucose metabolism via glycolysis and the pentose phosphate pathway (PPP) (Salas et al., 1984), as well as those involved in the tricarboxylic acid cycle (TCA), essential in the supply of precursors to secondary metabolism, have been mapped as expected in both isolates CA-126428 and CA-128772. The Entner-Doudoroff (ED) pathway is unusual in actinomycetes (Gunnarsson et al., 2004), although some of them contain homologs of 6-phosphogluconate dehydratase gene, as is the case of the strain CA-128772. Whether this pathway is active or not on this strain should be further investigated.
Amycolatopsis, as other actinomycetes, require nitrogen to ensure biomass production and to synthesize a large diversity of secondary metabolites from amino acids that are used as key precursors. Since actinomycetes are frequently isolated from nitrogen-poor environments, these bacteria have developed a complex system to efficiently retrieve nitrogen. A large diversity of extracellular proteases and peptidases are produced, as well as amino acid and oligopeptide transport systems. Some orthologous genes of these systems are also present in the genomes of strains CA-126428 and CA-128772, such as serine protease PepD, leucyl aminopeptidase, methionyl aminopeptidase, D-aminopeptidase, carboxypeptidases, amino acid, and oligopeptide transport system permeases. Different routes are followed to catabolize the families of amino acids introduced by permeases upon action of the extracellular proteases. Strains CA-126428 and CA-128772 have orthologous of most of the genes involved in these reactions, although some differences have been found between them. For example, histidine can be processed via formyl glutamic acid in the strain CA-128772, but in the case of CA-126428, it seems that histidine is processed via ergothioneine. Proline catabolism involves a proline oxidase and a pyrroline-5-carboxylate dehydrogenase to form glutamate, but the proline oxidase activity has not been mapped so far in any of the genomes. Valine dehydrogenase is responsible of the deamination of valine, leucine, and isoleucine. However, this enzyme has only been detected in strain CA-128772. Another enzyme with the same function, a branched-chain amino acid aminotransferase, is present both in CA-126428 and CA-128772. The enzymes involved in the catabolism of lysine, which usually follows the cadaverine pathway to generate glutarate, are absent in both strains. Only enzymes lysine N6-hydroxylase and lysine 2,3-aminomutase, involved in the conversion of L-lysine to N6-hydroxylysin and L-β-lysine, respectively, have been located. Enzymes involved in alanine catabolism have not been found either.
The enzymes of the shikimate pathway have been mapped in the isolates CA-126428 and CA-128772. An important number of secondary metabolites are derived from intermediates of the shikimate pathway, which is responsible of the formation of chorismate, a precursor of tryptophan, phenylalanine and tyrosine, as well as prephenate, anthranilate and p-aminobenzoate.
Interestingly, both strains possess genes involved in the degradation of aromatic compounds such as toluene, benzoate, fluorobenzoate or xylene, a trait that could be very useful for their application in bioremediation processes. So far, the genus Amycolatopsis has not been deeply studied in the field of bioremediation, with the exception of Amycolatopsis tucumanensis DSM45259T, which is the only species of this genus found to be resistant to copper (Albarracin et al., 2010). Another Amycolatopsis strain, M3-1, is able to degrade the herbicide ZJ0273, and several Amycolatopsis strains possess the capacity to degrade polylactic acid (PLA) (Dávila Costa and Amoroso, 2014).
In addition, we identified in both strains genes involved in antimicrobial resistance to vancomycin, beta-lactam antibiotics and cationic antimicrobial peptides. The presence of vancomycin resistance genes has been applied to the discovery of glycopeptide-producing strains using different screening approaches (Thaker et al., 2013; Truman et al., 2014), as it was the case of the ristomycin A producer Amycolatopsis sp. MJM2582 (Truman et al., 2014). The analysis of these genes, together with other genome mining approaches, highlights the potential to produce secondary metabolites by strains CA-128772 and CA-126428.
The former analysis of the different primary metabolism pathways present in the genomes in study supports the ability of both Amycolatopsis strains to utilize a broad variety of sources to ensure the supply of basic nutrients. Particularly in lichens, symbiotic partners contributions allow to colonize extreme environments and to tolerate harsh conditions. Despite the lack of information about the potential role of these strains as part of the microbial community associated to the lichen, there is also accumulating evidence that the bacterial counterparts may also contribute given their metabolic capabilities, providing carbohydrates, nitrogen sources and secondary metabolites to the microbial consortia (Scherlach and Hertweck, 2017). Future research in the physiology and primary metabolism regulation of Amycolatopsis spp., as well as its influence on lichen symbiosis and secondary metabolism, will benefit from the new genomic approaches and genomic-scale metabolic analyses.
Primary and secondary metabolisms are deeply interconnected in a complex network of regulatory signals that sense the environment and ensure the survival and adaptation of the microbial community (Genilloud, 2017b). The capacity to produce multiple secondary metabolites depends on the use of the available precursors and building blocks provided by the primary metabolism. In contrast to what has been observed in primary metabolism, the genes related with the production of secondary metabolites are frequently clustered, and their expression is modulated by transcriptional regulators (Genilloud, 2017b).
We used the antiSMASH algorithm (Blin et al., 2017) to search for putative BGCs clusters in all the Amycolatopsis genomes described in Supplementary Table 1. The secondary metabolite classes examined cover all common secondary metabolites in actinomycetes (butyrolactone, ectoine, fatty acid, indole, NRPS, RiPP, saccharide, siderophore, PKS-I, PKS-II, PKS-III, and terpene) and are shown in Figure 5. As it might be expected, the number of predicted secondary metabolite biosynthetic gene clusters depends on the completeness of the genomes and the size of the contigs. Overall, the number of polyketide (PKS) and non-ribosomal peptide (NRPS) BGCs is similar among all the Amycolatopsis strains and correlates with the completeness of the genomes. In the case of A. kentuckyensis DSM 44652T and A. lexingtoniensis DSM 44653T, a great number of putative PKS-I and NRPS pathways were predicted; however, these are very fragmented genomes and thus the number of predicted ORFs and BGCs increases, especially in the case of repeating, multi-modular proteins such as PKS and NRPS (Klassen and Currie, 2012).
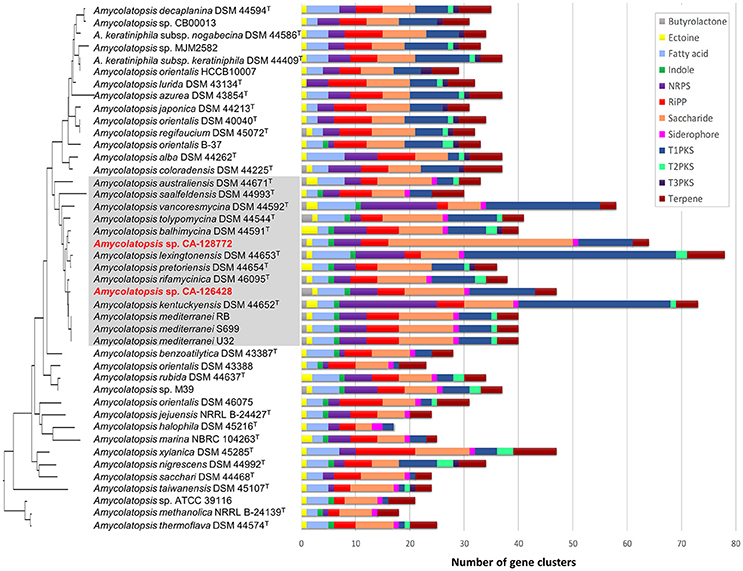
Figure 5. Most frequent secondary metabolite biosynthetic gene clusters predicted by antiSMASH in the genomes of Amycolatopsis strains. The strains are ordered in the same way as the 16S rRNA phylogenetic tree in Figure 2, which has been placed on the left axis for orientation. The strains of group C have been shaded in gray for clarification. The length of each horizontal bar corresponds with the number of BGCs, and the color code is indicated on the right.
The antiSMASH analysis of Amycolatopsis sequenced genomes detected many secondary metabolite BGCs when only a few metabolites have been reported to be produced by these strains, suggesting that even these well-studied species have the potential to produce new molecules (Chen et al., 2016). In the case of strains CA-126428 and CA-128772, antiSMASH predicted as many as 140 pathways for both strains given the high fragmentation level (Supplementary Table 3). In spite of the high number of predicted BGCs, only 70 clusters from CA-126428 and 55 clusters from CA-128772 show some degree of homology with known BGCs in MIBiG, and among them, only five clusters from each strain show equal or more than 50% of homology (Supplementary Table 3) suggesting the high new biosynthetic potential encoded in the genomes of the isolates CA-126428 and CA-128772.
Strain CA-128772 is richer than strain CA-126428 in saccharide pathways (Figure 5, Supplementary Table 3). A predominance of saccharide gene clusters among microbial genomes was previoulsy found in a global analysis of prokaryotic biosynthetic gene clusters (Cimermancic et al., 2014). Cell wall-associated saccharides play key roles in microbe-host and microbe-microbe interactions, while other diffusible saccharides have antibacterial activity (Cimermancic et al., 2014). The functions of many of the putative saccharide BGCs are still unknown since they are not closely related to any known gene cluster. In the case of strain CA-128772, approximately 40% of the predicted saccharide clusters do not show homology to any known cluster, in contrast to the 27% in strain CA-126428 (Supplementary Table 3).
We focused our analyses on the predicted BGC with more than 70% homology with known BGCs from the MIBiGC database (Figure 6) (Medema et al., 2015). Figure 6 shows the pathways predicted in each strain (yellow boxes), as well as the secondary metabolites that have been detected in culture fermentations (green or red boxes if the pathway has been or not predicted, respectively). Interestingly, strains CA-126428 and CA-128772 show only two and three BGCs, respectively, with more than 70% homology with known pathways (Figure 6). The rest of pathways (Figure 5, Supplementary Table 3) show low homology with known BGCs. As stated above, this fact reflects the biosynthetic potential of the strains, which may host novel BGCs encoding new secondary metabolites.
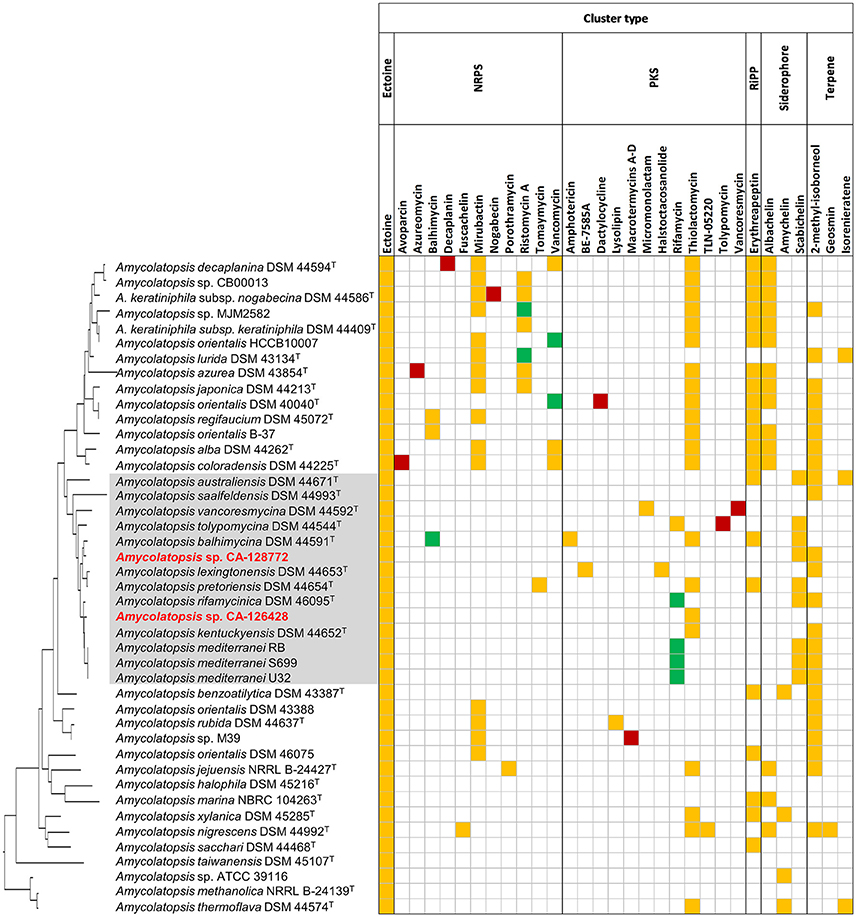
Figure 6. Biosynthetic gene clusters predicted in the genomes of Amycolatopsis with more than 70% homology with known BGCs. Yellow: predicted pathways whose encoded metabolites have not been detected in culture; green: predicted pathways whose encoded metabolites have been detected in culture; red: compounds detected in culture but not predicted by antiSMASH. The strains are ordered in the same way as the 16S rRNA phylogenetic tree in Figure 2, which has been placed on the left axis for orientation. The strains of group C have been shaded in gray for clarification.
All the strains, including CA-126428 and CA-128772, were predicted to produce ectoine (1,4,5,6-tetrahydro-2-methyl-4-pyrimidinecarboxylic acid), a compatible solute with a considerable biotechnological importance that acts as stress protectant and stabilizes macromolecules against severe environmental conditions (Hamedi et al., 2013). The production of ectoine has been widely described in salt-tolerant Streptomyces strains (Nett et al., 2009; Zhao et al., 2016), and the antiSMASH database (https://antismash-db.secondarymetabolites.org) shows the presence of the ectoine BGC in a high number of strains belonging to the Proteobacteria and Actinobacteria. This fact suggests the capacity of adaptation of the members of this genus to different environments, since the production of ectoine may act as a protectant against desiccation or drought periods. Nevertheless, we have no further evidences about the potential role of the production of ectoine by Amycolatopsis in the lichen-associated microbial community.
In addition, our strains were only predicted to produce three compounds. The strain CA-128772, was predicted to produce 2-methylisoborneol, a widespread odorous-terpenoid compound (Nett et al., 2009) also shown in another 23 strains. Surprisingly, the production of this compound was not identified in any of the strains from subclades ATS and AMS. This strain was also predicted to synthetize the siderophore schabichelin (Kodani et al., 2013) as well as 9 of the 14 strains of group C (Figure 6). Strain CA-126428, as well as another 21 strains, contained a cluster encoding thiolactomycin, a thiotetronate antibiotic first described in 1982 with a broad antibacterial activity against a wide spectrum of Gram-positive and Gram-negative bacteria (Yurkovich et al., 2017). Interestingly, the production of thiolactomycin was predicted in nearly all the strains from group A except for A. lurida DSM 43134T, whereas within group C only 4 of the 14 strains may produce this compound, as well as A. thermoflava DSM 44574T, a strain from the AMS subclade (Figure 6).
An additional group of 21 strains were predicted to produce the RiPP class III lantibiotic erythreapeptin (Völler et al., 2012), from which only strains A. australiensis DSM 44671T, A. balhimycina DSM 44591T and A. pretoriensis DSM 44654T belonged to group C. As in the case of thiolactomycin, the production of erythreapeptin was predicted in nearly all the strains conforming group A (Figure 6), with the exception of A. lurida DSM 43134T.
Interestingly, only five strains showed a BGC with more than 70% homology with the rifamycin pathway of the ansamycin class of antibiotics, and another five strains with the vancomycin glycopeptide pathway (Chen et al., 2016). Curiously, the strains predicted to contain PKS pathways associated to rifamycin belong exclusively to group C, while strains predicted to contain a vancomycin related pathways are only present in group A.
Other biosynthetic pathways have been predicted exclusively in specific groups. These are the cases of the clusters encoding the polyketides amphotericin (Caffrey et al., 2001), BE-7585A (Sasaki et al., 2010), halstoctacosanolide (Tohyama et al., 2006), micromonolactam (Skellam et al., 2013) and the non-ribosomal peptide tomaymycin (Li et al., 2009) that were also detected only in individual strains from group C (Figure 6). The production of the glycopeptide antibiotic ristomycin A (Spohn et al., 2014) was predicted only in 7 of 14 strains from group A. Other predicted clusters are involved in the production of several siderophores such as mirubactin (Giessen et al., 2012), predicted in 11 strains from group A and in another 4 strains conforming group B, abachelin (Kodani et al., 2015), predicted in 12 strains from group A and some strains from groups D1, G, and E, and amychelin (Seyedsayamdost et al., 2011), predicted in two strains of the AMS subclade and in two strains of another AOS subgroups.
The relative position of the BGC homologous to known pathways is shown in Figure 4 (black arrows). In strains CA-126428 and CA-128772, the observed BGC distribution is not the order in the genome given that they correspond to draft concatenated and not complete genomes. Our comparison shows that strains A. keraniphila subsp. nogabecina DSM 44586T and A. japonica DSM 44213T, and strains A. mediterranei, respectively, share similar BGCs organization, with most of the BGCs located in the non-core regions in agreement with Xu et al. (2014).
Despite the large number of BGCs detected in our strains in study, only four of them can be highly correlated to known families of compounds, with a high prevalence of BGCs showing low homology to any annotated cluster.
The sequencing and comparative analysis of Amycolatopsis CA-126428 and CA-128772 genomes has contributed to improve our understanding of the taxonomic diversity and metabolic potential of the species of this genus. Both MLSA and 16S rRNA phylogenies consistently show that strains CA-126428 and CA-128772 belong to the group C of the AOS subclade. The different relative position of the strains in the 16S rRNA and MLSA phylogenies, as well as the differences observed from the genomic comparisons, suggest that strains CA-126428 and CA-128772 may represent new species, requiring further investigations to explore the uniqueness and the role of these new strains in the lichen associated microbial community.
All the Amycolatopsis strains analyzed have shown a large genomic potential to produce different classes of specialized metabolites restricted to few groups of species. In addition, the results of our analysis support previous reports suggesting actinomycetes as a still untapped source of novel compounds and the relevance in modern natural products drug discovery of the application of genome-based mining approaches of these species to foster the discovery of new natural products encoded by cryptic or poorly expressed BGCs.
MS-H and OG designed the experiments; MS-H, IG, and CD-M performed the experiments; GM configured the bioinformatic software; and MS-H and OG wrote the manuscript.
This study was supported by Fundación MEDINA, Centro de Excelencia en Medicamentos Innovadores en Andalucía, Granada, Spain.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00369/full#supplementary-material
Adamek, M., Spohn, M., Stegmann, E., and Ziemert, N. (2017). “Mining bacterial genomes for secondary metabolite gene clusters,” in Antibiotics, Methods in Molecular Biology, ed P. Sass (New York, NY: Humana Press), 23–47.
Aggarwal, G., and Ramaswamy, R. (2002). Ab initio gene identification: prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14. doi: 10.1007/BF02703679
Albarracin, V. H., Alonso-Vega, P., Trujillo, M. E., Amoroso, M. J., and Abate, C. M. (2010). Amycolatopsis tucumanensis sp. nov., a copper-resistant actinobacterium isolated from polluted sediments. Int. J. Syst. Evol. Microbiol. 60, 397–401. doi: 10.1099/ijs.0.010587-0
Aschenbrenner, I. A., Cernava, T., Berg, G., and Grube, M. (2016). Understanding microbial multi-species symbioses. Front. Microbiol. 7:180. doi: 10.3389/fmicb.2016.00180
Auch, A. F., von Jan, M., Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Barka, E. A., Vatsa, P., Sanchez, L., Gaveau-vaillant, N., Jacquard, C., Klenk, H. P., et al. (2016). Taxonomy, physiology, and natural products of actinobacteria. Microbiol. Mol. Biol. Rev. 80, 1–43. doi: 10.1128/MMBR.00019-15
Beemelmanns, C., Ramadhar, T. R., Kim, K. H., Klassen, J. L., Cao, S., Wyche, T. P., et al. (2017). Macrotermycins A–D, glycosylated macrolactams from a termite-associated Amycolatopsis sp. M39. Org. Lett. 19, 1000–1003. doi: 10.1021/acs.orglett.6b03831
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 1–6. doi: 10.1093/nar/gkx319
Caffrey, P., Lynch, S., Flood, E., Finnan, S., and Oliynyk, M. (2001). Amphotericin biosynthesis in Streptomyces nodosus: deductions from analysis of polyketide synthase and late genes. Chem. Biol. 8, 713–723. doi: 10.1016/S1074-5521(01)00046-1
Calcott, M. J., Ackerley, D. F., Knight, A., Keyzers, R. A., and Owen, J. G. (2017). Secondary metabolism in the lichen symbiosis. Chem. Soc. Rev. doi: 10.1039/c7cs00431a. [Epub ahead of print].
Cardinale, M., Puglia, A. M., and Grube, M. (2006). Molecular analysis of lichen-associated bacterial communities. FEMS Microbiol. Ecol. 57, 484–495. doi: 10.1111/j.1574-6941.2006.00133.x
Chen, S., Wu, Q., Shen, Q., and Wang, H. (2016). Progress in understanding the genetic information and biosynthetic pathways behind Amycolatopsis antibiotics, with implications for the continued discovery of novel drugs. ChemBioChem. 17, 119–128. doi: 10.1002/cbic.201500542
Cimermancic, P., Medema, M. H., Claesen, J., Kurita, K., Wieland Brown, L. C., Mavrommatis, K., et al. (2014). Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421. doi: 10.1016/j.cell.2014.06.034
Colston, S. M., Fullmer, M. S., Beka, L., Lamy, B., Peter Gogarten, J., and Graf, J. (2014). Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test case. MBio 5, 1–13. doi: 10.1128/mBio.02136-14
Darling, A. E., Mau, B., and Perna, N. T. (2010). ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Dávila Costa, J. S., and Amoroso, M. J. (2014). Current biotechnological applications of the genus Amycolatopsis. World J. Microbiol. Biotechnol. 30, 1919–1926. doi: 10.1007/s11274-014-1622-3
Everest, G. J., and Meyers, P. R. (2009). The use of gyrB sequence analysis in the phylogeny of the genus Amycolatopsis. Antonie Van Leeuwenhoek 95, 1–11. doi: 10.1007/s10482-008-9280-9
Everest, G. J., and Meyers, P. R. (2011). Evaluation of the antibiotic biosynthetic potential of the genus Amycolatopsis and description of Amycolatopsis circi sp. nov., Amycolatopsis equina sp. nov. and Amycolatopsis hippodromi sp. nov. J. Appl. Microbiol. 111, 300–311. doi: 10.1111/j.1365-2672.2011.05058.x
Genilloud, O. (2017a). Actinomycetes: still a source of novel antibiotics. Nat. Prod. Rep. 34, 1203–1232. doi: 10.1039/C7NP00026J
Genilloud, O. (2017b). “Physiology of actinobacteria,” in Biology and Biotechnology of Actinobacteria, eds J. Wink, F. Mohammadipanah, and J. Hamedi (Cham: Springer), 151–180.
Genilloud, O., González, I., Salazar, O., Martín, J., Tormo, J. R., and Vicente, F. (2011). Current approaches to exploit actinomycetes as a source of novel natural products. J. Ind. Microbiol. Biotechnol. 38, 375–389. doi: 10.1007/s10295-010-0882-7
Giessen, T. W., Franke, K. B., Knappe, T. A., Kraas, F. I., Bosello, M., Xie, X., et al. (2012). Isolation, structure elucidation, and biosynthesis of an unusual hydroxamic acid ester-containing siderophore from Actinosynnema mirum. J. Nat. Prod. 75, 905–914. doi: 10.1021/np300046k
Glaeser, S. P., and Kämpfer, P. (2015). Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst. Appl. Microbiol. 38, 237–245. doi: 10.1016/j.syapm.2015.03.007
González, I., Ayuso-Sacido, A., Anderson, A., and Genilloud, O. (2005). Actinomycetes isolated from lichens: evaluation of their diversity and detection of biosynthetic gene sequences. FEMS Microbiol. Ecol. 54, 401–415. doi: 10.1016/j.femsec.2005.05.004
Gunnarsson, N., Mortensen, U. H., Sosio, M., and Nielsen, J. (2004). Identification of the Entner–Doudoroff pathway in an antibiotic-producing actinomycete species. Mol. Microbiol. 52, 895–902. doi: 10.1111/j.1365-2958.2004.04028.x
Guy, L., Kultima, J. R., Andersson, S. G. E., and Quackenbush, J. (2011). GenoPlotR: comparative gene and genome visualization in R. Bioinformatics 27, 2334–2335. doi: 10.1093/bioinformatics/btq413
Hamedi, J., Mohammadipanah, F., and Ventosa, A. (2013). Systematic and biotechnological aspects of halophilic and halotolerant actinomycetes. Extremophiles 17, 1–13. doi: 10.1007/s00792-012-0493-5
Hashizume, H., Sawa, R., Yamashita, K., Nishimura, Y., and Igarashi, M. (2017). Structure and antibacterial activities of new cyclic peptide antibiotics, pargamicins B, C and D, from Amycolatopsis sp. ML1-hF4. J. Antibiot. (Tokyo). 70, 699–704. doi: 10.1038/ja.2017.34
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016a). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kanehisa, M., Sato, Y., and Morishima, K. (2016b). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F., and Hopwood, D. A. (2000). “Preparation and analysis of genomic and plasmid DNA,” in Practical Streptomyces Genetics, Vol. 1, eds T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood (Norwich: John Innes Foundation), 161–210.
Klassen, J. L., and Currie, C. R. (2012). Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics 13:14. doi: 10.1186/1471-2164-13-14
Kodani, S., Bicz, J., Song, L., Deeth, R. J., Ohnishi-Kameyama, M., Yoshida, M., et al. (2013). Structure and biosynthesis of scabichelin, a novel tris-hydroxamate siderophore produced by the plant pathogen Streptomyces scabies 87.22. Org. Biomol. Chem. 11, 4686–4694. doi: 10.1039/c3ob40536b
Kodani, S., Komaki, H., Suzuki, M., Hemmi, H., and Ohnishi-Kameyama, M. (2015). Isolation and structure determination of new siderophore albachelin from Amycolatopsis alba. Biometals 28, 381–389. doi: 10.1007/s10534-015-9842-z
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kumari, R., Singh, P., and Lal, R. (2016). Genetics and genomics of the genus Amycolatopsis. Indian J. Microbiol. 56, 233–246. doi: 10.1007/s12088-016-0590-8
Kwakman, J. H., and Postma, P. W. (1994). Glucose kinase has a regulatory role in carbon catabolite repression in Streptomyces coelicolor. J. Bacteriol. 176, 2694–2698. doi: 10.1128/jb.176.9.2694-2698.1994
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: John Wiley & Sons), 115–175.
Lechevalier, M. P., Prauser, H., Labeda, D. P., and Ruan, J. S. (1986). Two new genera of nocardioform actinomycetes: Amycolata gen. nov. and Amycolatopsis gen. nov. Int. J. Syst. Bacteriol. 36, 29–37. doi: 10.1099/00207713-36-1-29
Lee, I., Kim, Y. O., Park, S. C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Li, W., Chou, S., Khullar, A., and Gerratana, B. (2009). Cloning and characterization of the biosynthetic gene cluster for tomaymycin, an SJG-136 monomeric analog. Appl. Environ. Microbiol. 75, 2958–2963. doi: 10.1128/AEM.02325-08
Liu, C., Jiang, Y., Wang, X., Chen, D., Chen, X., Wang, L., et al. (2017). Diversity, antimicrobial activity, and biosynthetic potential of cultivable actinomycetes associated with lichen symbiosis. Microb. Ecol., 74, 570–584. doi: 10.1007/s00248-017-0972-4
Loman, N. J., and Pallen, M. J. (2015). Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 13, 1–9. doi: 10.1038/nrmicro3565
Medema, M. H., Kottmann, R., Yilmaz, P., Cummings, M., Biggins, J. B., Blin, K., et al. (2015). Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 11, 625–631. doi: 10.1038/nchembio.1890
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Klenk, H. P., and Göker, M. (2014). Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64, 352–356. doi: 10.1099/ijs.0.056994-0
Nett, M., Ikeda, H., and Moore, B. S. (2009). Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384. doi: 10.1039/b817069j
Parrot, D., Antony-Babu, S., Intertaglia, L., Grube, M., Tomasi, S., and Suzuki, M. T. (2015). Littoral lichens as a novel source of potentially bioactive actinobacteria. Sci. Rep. 5:15839. doi: 10.1038/srep15839
Salas, J. A., Quirós, L. M., and Hardisson, C. (1984). Pathways of glucose catabolism during germination of Streptomyces spores. FEMS Microbiol. Lett. 22, 229–233. doi: 10.1111/j.1574-6968.1984.tb00732.x
Sasaki, E., Ogasawara, Y., and Liu, H. W. (2010). A biosynthetic pathway for BE-7585A, a 2-thiosugar-containing angucycline-type natural product. J. Am. Chem. Soc. 132, 7405–7417. doi: 10.1021/ja1014037
Scherlach, K., and Hertweck, C. (2017). Mediators of mutualistic microbe–microbe interactions. Nat. Prod. Rep. doi: 10.1039/C7NP00035A. [Epub ahead of print].
Sentausa, E., and Fournier, P. E. (2013). Advantages and limitations of genomics in prokaryotic taxonomy. Clin. Microbiol. Infect. 19, 790–795. doi: 10.1111/1469-0691.12181
Seyedsayamdost, M. R., Traxler, M. F., Zheng, S. L., Kolter, R., and Clardy, J. (2011). Structure and biosynthesis of amychelin, an unusual mixed-ligand siderophore from Amycolatopsis sp. AA4. J. Am. Chem. Soc. 133, 11434–11437. doi: 10.1021/ja203577e
Sharma, M., Dangi, P., and Choudhary, M. (2014). Actinomycetes: source, identification, and their applications. Int. J. Curr. Microbiol. App. Sci. 3, 801–832. Available online at: https://www.ijcmas.com/Archives-15.php
Skellam, E. J., Stewart, A. K., Strangman, W. K., and Wright, J. L. (2013). Identification of micromonolactam, a new polyene macrocyclic lactam from two marine Micromonospora strains using chemical and molecular methods: clarification of the biosynthetic pathway from a glutamate starter unit. J. Antibiot. (Tokyo). 66, 431–441. doi: 10.1038/ja.2013.34
Spohn, M., Kirchner, N., Kulik, A., Jochim, A., Wolf, F., Muenzer, P., et al. (2014). Overproduction of ristomycin A by activation of a silent gene cluster in Amycolatopsis japonicum MG417-CF17. Antimicrob. Agents Chemother. 58, 6185–6196. doi: 10.1128/AAC.03512-14
Stackebrandt, E., Frederiksen, W., Garrity, G. M., Grimont, P. A. D., Kampfer, P., Maiden, M. C. J., et al. (2002). Report of the ad hoc comittee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 52, 1043–1047. doi: 10.1099/ijs.0.02360-0.02360
Tang, B., Xie, F., Zhao, W., Wang, J., Dai, S., Zheng, H., et al. (2016). A systematic study of the whole genome sequence of Amycolatopsis methanolica strain 239T provides an insight into its physiological and taxonomic properties which correlate with its position in the genus. Synth. Syst. Biotechnol. 1, 169–186. doi: 10.1016/j.synbio.2016.05.001
Thaker, M. N., Wang, W., Spanogiannopoulos, P., Waglechner, N., King, A. M., Medina, R., et al. (2013). Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol. 31, 922–927. doi: 10.1038/nbt.2685
Thompson, C. C., Chimetto, L., Edwards, R. A., Swings, J., Stackebrandt, E., and Thompson, F. L. (2013). Microbial genomic taxonomy. BMC Genomics 14:913. doi: 10.1186/1471-2164-14-913
Tohyama, S., Kakinuma, K., and Eguchi, T. (2006). The complete biosynthetic gene cluster of the 28-membered polyketide macrolactones, halstoctacosanolides, from Streptomyces halstedii HC34. J. Antibiot. (Tokyo) 59, 44–52. doi: 10.1038/ja.2006.7
Truman, A. W., Kwun, M. J., Cheng, J., Yang, S. H., Suh, J. W., and Hong, H. J. (2014). Antibiotic resistance mechanisms inform discovery: identification and characterization of a novel Amycolatopsis strain producing ristocetin. Antimicrob. Agents Chemother. 58, 5687–5695. doi: 10.1128/AAC.03349-14
Völler, G. H., Krawczyk, J. M., Pesic, A., Krawczyk, B., Nachtigall, J., and Süssmuth, R. D. (2012). Characterization of new class III lantibiotics-erythreapeptin, avermipeptin and griseopeptin from Saccharopolyspora erythraea, Streptomyces avermitilis and Streptomyces griseus demonstrates stepwise N-terminal leader processing. ChemBioChem 13, 1174–1183. doi: 10.1002/cbic.201200118
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S Ribosomal DNA amplification for phylogenetic study. J. Biotechnol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Xiao, Y. S., Zhang, B., Zhang, M., Guo, Z. K., Deng, X. Z., Shi, J., et al. (2017). Rifamorpholines A–E, potential antibiotics from locust-associated actinobacteria Amycolatopsis sp. Hca4. Org. Biomol. Chem. 15, 3909–3916. doi: 10.1039/C7OB00614D
Xu, L., Huang, H., Wei, W., Zhong, Y., Tang, B., Yuan, H., et al. (2014). Complete genome sequence and comparative genomic analyses of the vancomycin-producing Amycolatopsis orientalis. BMC Genomics 15:363. doi: 10.1186/1471-2164-15-363
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yurkovich, M. E., Jenkins, R., Sun, Y., Tosin, M., and Leadlay, P. F. (2017). The polyketide backbone of thiolactomycin is assembled by an unusual iterative polyketide synthase. Chem. Commun. 53, 2182–2185. doi: 10.1039/C6CC09934C
Keywords: Amycolatopsis, secondary metabolites, phylogeny, whole genome sequence, biosynthetic gene clusters
Citation: Sánchez-Hidalgo M, González I, Díaz-Muñoz C, Martínez G and Genilloud O (2018) Comparative Genomics and Biosynthetic Potential Analysis of Two Lichen-Isolated Amycolatopsis Strains. Front. Microbiol. 9:369. doi: 10.3389/fmicb.2018.00369
Received: 09 October 2017; Accepted: 16 February 2018;
Published: 13 March 2018.
Edited by:
Jesus L. Romalde, Universidade de Santiago de Compostela, SpainReviewed by:
Martha E. Trujillo, Universidad de Salamanca, SpainCopyright © 2018 Sánchez-Hidalgo, González, Díaz-Muñoz, Martínez and Genilloud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga Genilloud, b2xnYS5nZW5pbGxvdWRAbWVkaW5hYW5kYWx1Y2lhLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.