
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 March 2018
Sec. Terrestrial Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00294
 Annelein Meisner1,2,3*
Annelein Meisner1,2,3* Samuel Jacquiod4
Samuel Jacquiod4 Basten L. Snoek5,6,7
Basten L. Snoek5,6,7 Freddy C. ten Hooven5
Freddy C. ten Hooven5 Wim H. van der Putten5,7
Wim H. van der Putten5,7It is increasingly acknowledged that climate change is influencing terrestrial ecosystems by increased drought and rainfall intensities. Soil microbes are key drivers of many processes in terrestrial systems and rely on water in soil pores to fulfill their life cycles and functions. However, little is known on how drought and rainfall fluctuations, which affect the composition and structure of microbial communities, persist once original moisture conditions have been restored. Here, we study how simulated short-term drying and re-wetting events shape the community composition of soil fungi and prokaryotes. In a mesocosm experiment, soil was exposed to an extreme drought, then re-wetted to optimal moisture (50% WHC, water holding capacity) or to saturation level (100% WHC). Composition, community structure and diversity of microbes were measured by sequencing ITS and 16S rRNA gene amplicons 3 weeks after original moisture content had been restored. Drying and extreme re-wetting decreased richness of microbial communities, but not evenness. Abundance changes were observed in only 8% of prokaryote OTUs, and 25% of fungal OTUs, whereas all other OTUs did not differ between drying and re-wetting treatments. Two specific legacy response groups (LRGs) were observed for both prokaryotes and fungi. OTUs belonging to the first LRG decreased in relative abundance in soil with a history of drought, whereas OTUs that increased in soil with a history of drought formed a second LRG. These microbial responses were spread among different phyla. Drought appeared to be more important for the microbial community composition than the following extreme re-wetting. 16S profiles were correlated with both inorganic N concentration and basal respiration and ITS profiles correlated with fungal biomass. We conclude that a drying and/or an extreme re-wetting history can persist in soil microbial communities via specific response groups composed of members with broad phylogenetic origins, with possible functional consequences on soil processes and plant species. As a large fraction of OTUs responding to drying and re-wetting belonged to the rare biosphere, our results suggest that low abundant microbial species are potentially important for ecosystem responses to extreme weather events.
Future climate is expected to include more variable drought and rainfall events (IPCC, 2012; Fischer and Knutti, 2016). These enhanced fluctuations will directly affect soil microbes that rely on water to fulfill their life cycles and activities (Vos et al., 2013). Fluctuations in soil moisture conditions influence the regulation of microbial activities such as respiration and growth (Manzoni et al., 2012; Meisner et al., 2015). For example, a pulse of respiration often occurs when water is added to dry soil (Birch, 1958; Kim et al., 2012). This is a general phenomenon that occurs in many regions in the world, such as arctic (Clein and Schimel, 1994; Meisner et al., 2017), temperate (Pulleman and Tietema, 1999; Meisner et al., 2013a), and arid regions (Miller et al., 2005; Chowdhury et al., 2011; Warren, 2014). The amount of water that is added during re-wetting determines the microbial response to the drying and re-wetting event (Orchard and Cook, 1983; Evans et al., 2014; Lado-Monserrat et al., 2014). Drying and re-wetting is considered a carbon loss from the microbial community (Schimel et al., 2007), as the carbon seems to be respired and not used for biomass incorporation via microbial growth (Blazewicz et al., 2014; Meisner et al., 2015). Whereas, the microbial growth, respiration rates and biomass are recovered 1 week after re-wetting (Lundquist et al., 1999; Meisner et al., 2013a), relatively little is known on how the composition of the microbial community recovers within weeks after re-wetting.
Drying and re-wetting can affect the composition of soil communities (Fierer et al., 2003; Barnard et al., 2013; Evans et al., 2014; Hartmann et al., 2017). Since terrestrial microbial communities are important regulators of many ecosystem services such as plant performance, bioremediation, and carbon cycling (Vidali, 2001; Schimel and Schaeffer, 2012; Philippot et al., 2013), soil moisture history may have important implications for these “higher-order” processes (Hawkes et al., 2017). For example, a past drought in the field can affect the bacterial composition when soil is exposed to drying and re-wetting events in the laboratory (Evans and Wallenstein, 2012). In addition, N mineralization and inorganic N availability is increased upon drying and re-wetting events (Meisner et al., 2013b). These so-called “legacy effects” provide a source of variation affecting microbes, but the extent and amplitude of their contribution to soil microbiome composition remains unclear.
Soil microbes have different strategies to cope with fluctuating moisture availability (Lennon et al., 2012). Bacteria have been shown to respond sensitively, tolerant or opportunistically to drying and re-wetting (Evans and Wallenstein, 2014). Bacterial sensitivity can be due to increased injuries in viable cells (Mackey and Derrick, 1984; Nocker et al., 2012), which may lead to a decrease in microbial activity (Kieft et al., 1987). Cell damage is difficult to repair during drought due to a decrease in microbial activity (Potts, 1994). As such, the fraction of “dormant-over-active” cells is likely to increase in drying soils (Manzoni et al., 2014) and more free niches will become available upon re-wetting. Microbes can tolerate drought when they produce protective molecules (Schimel et al., 2007), such as osmolytes (Warren, 2014). Opportunistic microbes can colonize free niches that become available upon re-wetting. Fast responding microbes (Placella et al., 2012) may affect the composition of the slower responders in the microbial community via priority effects (Fukami, 2015). Abundant microbes can take advantage of niches that become available after drought, however, sub-dominant and rare microbes may respond opportunistically as well (Aanderud et al., 2015; Lynch and Neufeld, 2015), so that growth rates of rare bacteria may not necessarily be different from those that are abundant (Kurm et al., 2017). The consequence of all these different response strategies is that extreme drought may imprint a legacy signature in the soil microbiome composition that can last for weeks after the end of the drought event.
Unlike bacteria, soil fungi are often less affected by drought due to their extended and exploratory hyphal structures (Bapiri et al., 2010; Yuste et al., 2011; de Vries et al., 2012a; Barnard et al., 2013). However, contrasting findings for the capacity of soil fungi to resist alterations in moisture conditions have been reported. The composition of fungal communities can differ between dry and wet conditions (Hawkes et al., 2011; Cregger et al., 2012; Acosta-Martínez et al., 2014; Barnes et al., 2018) and their biomass may increase, decrease, or remain unaffected by drought or irrigation (Scheu and Parkinson, 1994; Gordon et al., 2008; Haugwitz et al., 2014; Hartmann et al., 2017). Despite these contrasting findings, some fungal species are sensitive bioindicators of changes in soil moisture (Kaisermann et al., 2015). Therefore, fungal species may not only tolerate drought stress, but may respond sensitively or opportunistically (Crowther et al., 2014) just as bacteria.
Our aim was to test how extreme fluctuations in soil moisture content may result in a legacy in the composition of soil microbial communities. We identified legacy effects by applying high throughput molecular approaches. We tested the overall hypothesis that extreme drought and re-wetting events enact a legacy in soil via a changed composition of the microbial community after soil moisture conditions have been restored. More specifically, we predicted that: (1) an extreme drying and re-wetting event will decrease the number of abundant or sub-abundant OTUs, (2) fungal communities will also respond to changed moisture conditions by tolerant, opportunistic and sensitive response patterns, and (3) some rare microbes will increase in relative abundance. We further tested if the microbiome profiles were correlated to the previously measured soil processes, such as inorganic N concentration, N mineralization rates and respiration rates (see Meisner et al., 2013b for details on soil processes and nutrients). In order to test our hypotheses, we performed a mesocosm experiment and exposed soil to a 4 weeks drying treatment after which soils were either re-wetted to optimal (50% WHC, water holding capacity), or to saturated moisture conditions (100% WHC). The optimal soil moisture content is in between 50 and 70% WHC (Ilstedt et al., 2000; Setia et al., 2011). Soil was left to recover for 18 days, after which we extracted DNA and analyzed the composition of 16S rRNA gene and ITS amplicons.
At November 3, 2009, soil was collected from five locations in the nature reserve Millingerwaard, which is located in the Gelderse Poort region along the Rhine River delta in the Netherlands (N51° 52.224′ E5° 59.494′). In the week before sampling, there was in total 22 mm of rain spread among the different days and the average air temperature was 11.5°C1. Coarse fragments and plant material were removed by sieving through a 10-mm sieve, and the remaining soil was homogenized and placed in the greenhouse where later on the experiment was carried out (see Meisner et al., 2013b for details). The soil is considered a Sandy soil (Meisner et al., 2011).
We tested how a history of an extreme drying and/or a re-wetting affected the composition of the microbial community 18 days after the soil was re-wetted to original conditions or re-wetted to saturation. Thereto, 32 microcosms were filled with 7L of soil (equivalent to 6000 g of dry soil) and left to settle for 2 weeks before the experiments started in a greenhouse with a temperature of 21°C (±2°C) for 16 h (±2°C) and 16°C (±2°C) for 8 h. The mesocosms were exposed to one of the four treatments: constant moisture content (Moist or “C”), drought stress for 28 days with re-wetting to 50% WHC (drought or “D”), extreme re-wetting to 100% WHC (extreme re-wetting or “ER”), or a combination of the drying and extreme re-wetting (drought and extreme re-wetting or “DR”). Soil samples were collected 18 days after the drought stress had stopped, at day 46 of the experiment. Soil was sampled with a small auger by taking ca. 100 gram (based on dry soil) from 5 to 7 places in the 7L mesocosms. Then, this soil sample was homogenized, after which a subsample was taken, put on ice and stored in the -80°C freezer as soon as possible after sampling. The non-frozen soil samples were used for the following measurements: Inorganic Nitrogen, Arginine Ammonification, basal respiration, substrate induced respiration, pH, ergosterol as fungal biomass indicator and total microbial biomass. The data have been published in a previous study (see Meisner et al., 2013b for details).
To characterize the bacterial and fungal soil communities we used barcode sequencing. Soil samples previously stored at -80°C were defrosted and from 250 mg of dried soil, total DNA was extracted for each sample using the PowerSoil DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA, United States) according to the manufacturer’s instructions. The composition of the prokaryotic community was determined by targeting a fragment of the 16S rRNA gene with amplicon sequencing. The PCR mixture contained 10 μl of 5 Prime Hot mastermix (QuantaBio), 1.25 μl BSA, 11.75 μl molecular grade water, 0.5 μl of 10 μm of the 515F, 0.5 μl of 10 μm 806R primers for amplification (Bates et al., 2011) and 1 μl template. The PCR conditions were the following: 94°C for 5 min, followed by 35 cycles of 94°C for 45 s, 50°C for 60 s, and 72°C for 1.30 min, followed by a final elongation of 10 min at 72°C. The composition of the fungal community was determined by targeting the ITS region. The PCR master mix contained 17.1 μl molecular grade water, 1 μl 5 μM DNtPs, 1 μl 25 mM MgCl2, 2.5 μl 10× PCR reaction buffer with MgCl2 (Roche), 1.25 BSA, 0.5 μl of 10 μm ITS4 (Ihrmark et al., 2012) primer and 0.5 μl of 10 μm fITS9 primer (Ihrmark et al., 2012), 0.15 μl of Taq DNA Polymerase (Roche) and 1 μl of template. PCR conditions were composed of an initial denaturation at 95°C for 10 min, followed by 35 cycles of denaturation at 94°C for 45 s, annealing at 54°C for 60 s and elongation at 72°C for 1.3 min followed with a final elongation at 72°C for 10 min. Both 16S and ITS PCR products were purified using the Agencourt AMPure XP PCR Purification kit (Beckman Coulter, Inc., Danvers, MA, United States). A PCR product/AMPure bead ratio of 1:0.7 was used. Elution is done with 25 μl preheated (55°C) Milli-Q water with an incubation time of 5 min. All other steps were performed according to the manufacturer’s protocol. After purification both amplicons were sequenced using the Illumina MiSeq platform for 300 bp paired-end reads.
The raw paired-end Miseq data was processed into an annotated OTU table using the following pipeline. The RDP extension to PANDASeq (Masella et al., 2012) Assembler (Cole et al., 2014) was used to merge the raw reads using a minimum overlap of 150 bp and a minimum PHRED score of 25. Primer sequences were removed from the FASTQ files using Flexbar version 2.5 (Dodt et al., 2012). Sequences were converted to FASTA format and concatenated into one file. Sequence clustering was done using VSEARCH version 1.0.10 (Rognes et al., 2016) at 97% identity and using usearch_global with the default settings to map quality controlled reads against the OTU centroids. The strategy used for this was de-replication, sorting by abundance (at least 2 sequences) and clustering using the UCLUST smallmem algorithm (Edgar, 2010). Thereafter, chimeric sequences were detected and removed using the UCHIME algorithm (Edgar et al., 2011) implemented in VSEARCH. Lastly, taxonomic classification for each OTU was obtained by using the RDP Classifier version 2.10 using the bootstrap value of 80% and classification was done on full-length entries (Cole et al., 2014). The pipeline was made with Snakemake (Koster and Rahmann, 2012) as available at DOI: https://doi.org/10.5281/zenodo.597131 (de Hollander, 2016). This pipeline was also used for ITS with the following adjustments: (1) ITS2 regions where extracted using ITSx 1.0.11 (Bengtsson-Palme et al., 2013). (2) Using the UNITE database (Kõljalg et al., 2013) provided by RDP the sequences were classified. OTUs without affiliation to at least kingdom level were excluded from the downstream beta-diversity analysis as the carry little information.
Summary tables describing each sample are presented in Supplementary Tables S1, S2. The raw sequencing counts were used directly to estimate the sequencing depth completeness via rarefaction curves using the package vegan in R (Supplementary Figure S1). Although amplicon profiles didn’t achieved asymptotic approximation (Supplementary Figure S1), sequencing depth was sufficient to analyze the core majority of this soil microbial diversity with a satisfactory rarefaction threshold (Schöler et al., 2017). There were on average 23808 OTUs per sample for 16S (Supplementary Table S1) of which three were excluded from further analysis due to too few counts or too low richness. For ITS, there were on average 4106 OTUs per sample (Supplementary Table S2) of which one sample was excluded from further analysis due to too few counts. The R software (R Core Team, 2017) and PAST software were used (Hammer et al., 2001). Raw fastq files are available on the European Nucleotide Archive (ENA) and have accession number PRJEB23318.
Diversity indices were analyzed as described in previous work (Jacquiod et al., 2016; Nunes et al., 2016). The Shannon diversity index (H) was calculated as H = -∑ pi ln (pi), where pi is the proportional abundance of OTU i in the mesocosms. Shannon diversity indices (H, Gaussian glm model fit) and sample richness (R, Poisson glm model fit) were calculated on rarefied data at 50,000 counts per sample for 16S rRNA gene profiles and 12,000 counts per sample for ITS profiles in order to avoid biases that may come from uneven numbers. Univariate statistical analysis was done in R version 3.1.1. We used the multcomp package version 1.3.6 (Hothorn et al., 2008) for the ANOVA corrected with Tukey post hoc test.
The multivariate analysis was done with the raw and non-rarefied contingency tables using the R software version 3.0.2 with the functions vegdist, hclust, rda, Adonis in package vegan and function dudi.pca, bca, randtest, s.class, in package ade4. A log10 transformation was needed to improve normality of data. Principal Component Analysis (PCA) was performed after center-scaling normalization. A pattern search was applied to the original PCAs by grouping replicates together in order to perform a Between Group Analysis (BGA). The statistical significance of the selected grouping factor was tested with a Monte-Carlo simulation involving 10,000 permutations. Complementing PERMANOVA tests were performed on the Euclidean distance profiles using 10,000 permutations in order to assess differences across the four legacies. An ANOVA was performed with phyla as response variables and treatment as explanatory variable. Differences between the four treatments were identified using a post hoc Tukey–Kramer test and false discovery rate multiple correction test (functions glm, cld, gltht in package multcomp, FDR, P < 0.05). Redundancy analysis (RDA) were performed as described before (Nunes et al., 2016) using function rda in vegan package, after center-scaling normalization for both ITS and 16S rRNA gene amplicon profiles using the following explanatory variables: four moisture legacies as well as the soil measurements, inorganic nitrogen, arginine ammonification, basal respiration, substrate induced respiration, pH, ergosterol, total microbial biomass (see for details: Meisner et al., 2013b). A permutation test was performed using 999 permutations to test the robustness of the model for discrimination of 16S or ITS profiles.
We classified microbial OTUs into Legacy Response groups (LRGs), which are defined as groups of organisms with similar response to a change in their environment. Here, we apply this concept to define groups of OTUs that may respond differently to a legacy effect of drought. LRGs were extracted from microbiomes as described previously (Nunes et al., 2016; Jacquiod et al., 2018). Fine changes in the soil microbiomes at the OTU level were extracted using negative binomial distribution and generalized linear model (nbGLM) (Robinson et al., 2010; Schöler et al., 2017). Significance of OTU changes was inferred with a quasi-likelihood F-test (QLF) under post hoc false discovery rate multiple correction test with the package edgeR in R (FDR, p < 0.05) (Robinson et al., 2010). This method has been suggested recently as one of the most accurate ways to extract significantly responding OTUs by minimizing the risk of error (Thorsen et al., 2016). LRGs aggregating OTUs based on similar response patterns were identified using hierarchical clustering and heatmaps representation, followed by Monte-Carlo simulation for statistical validation. Enrichment of phylogenetic groups in LRGs was tested using a hypergeometric test. We asked for each phylogenetic level and each phylogenetic group if we found more of that specific group in the LRG than expected by chance depending on the size of the LRG and on the occurrence of that group in the total sample. We determined the proportion of OTUs in LRGs belonging to rare microbes. Thereto, we considered the OTUs that had a relative abundance below 0.01% as rare (Galand et al., 2009).
The legacy of drying and/or extreme re-wetting appeared as decreased OTU richness for both ITS and 16S-based profiles, but not the evenness (Figure 1). In both profiles, the lowest richness was observed when soils were exposed to drying and extreme re-wetting, while the highest richness was obtained in the moist control soil. The 16S rRNA gene profiles showed a gradual decrease in richness, in descending order with the following soil history: moist control, extreme re-wetting, drying, drying and extreme re-wetting.
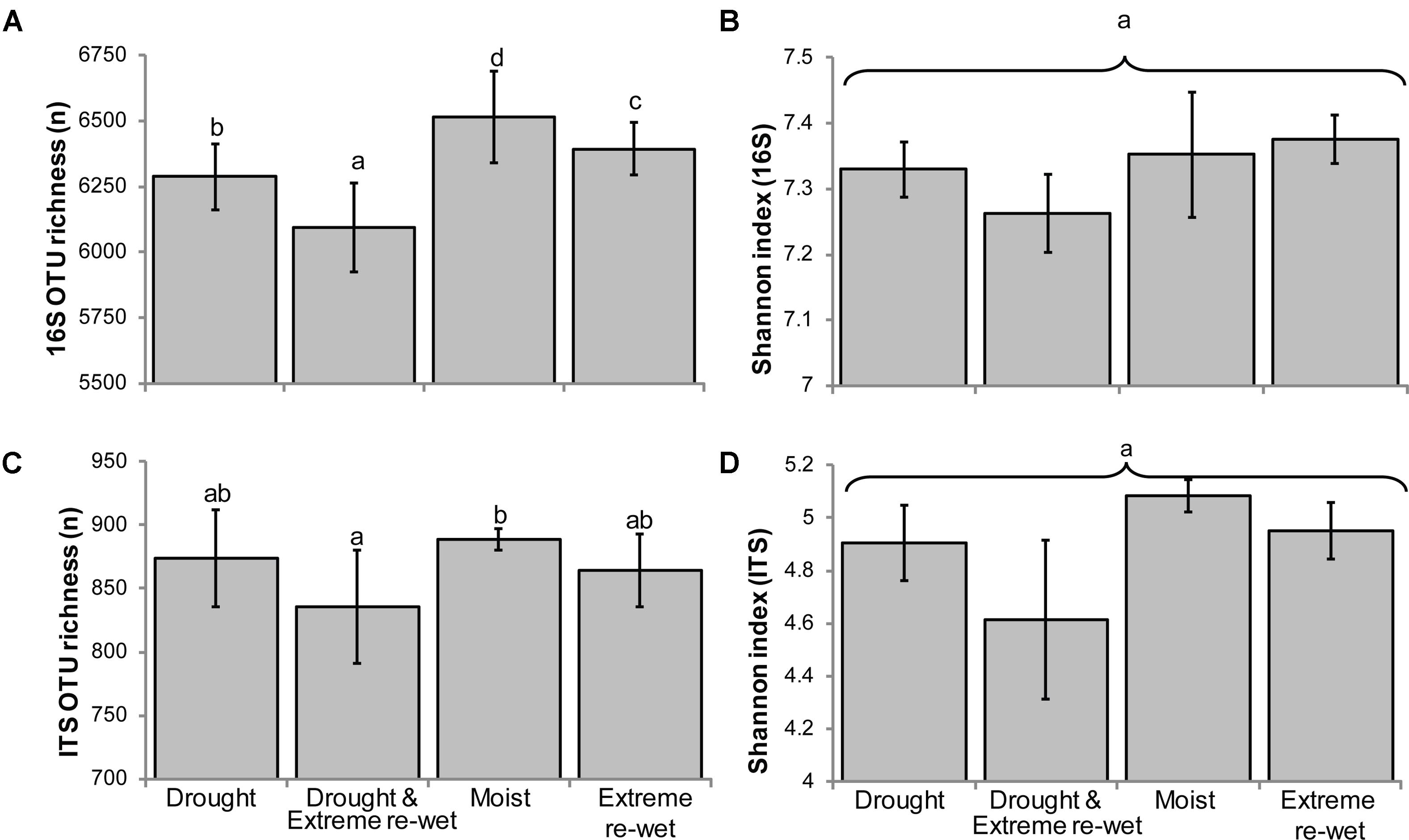
FIGURE 1. Richness and evenness of ITS and 16S rRNA gene amplicon profiles according to drying and re-wetting treatments (Average ± SEM). The chart displays the richness (A) and the Shannon indices (B) for 16S rRNA gene profiles and the richness (C) and Shannon indices (D) for ITS. Letters denote differences at P < 0.05 for a post hoc Tukey HSD correction test. ANOVA was run with the Poisson distribution for the richness.
The ITS and 16S profiles were separated in soil with a history of drying and/or extreme re-wetting (Figure 2). However, there was also variation within the different treatments as demonstrated by the spread in the replicates of the control and extreme re-wetting for 16S, and drying and extreme re-wetting for the ITS. Drought legacy explained 8.4% of the PERMANOVA variation in 16S profiles and 6.1% of the variation in ITS profiles, whereas the legacy of extreme re-wetting only explained 4.3% of the variation in ITS and none in 16S (Table 1). Redundancy analysis revealed that the microbiome profiles of 16S correlated with inorganic N content and soil respiration (Figure 3A). In addition, the microbiome profiles for ITS correlated with the amount of ergosterol (Figure 3B).
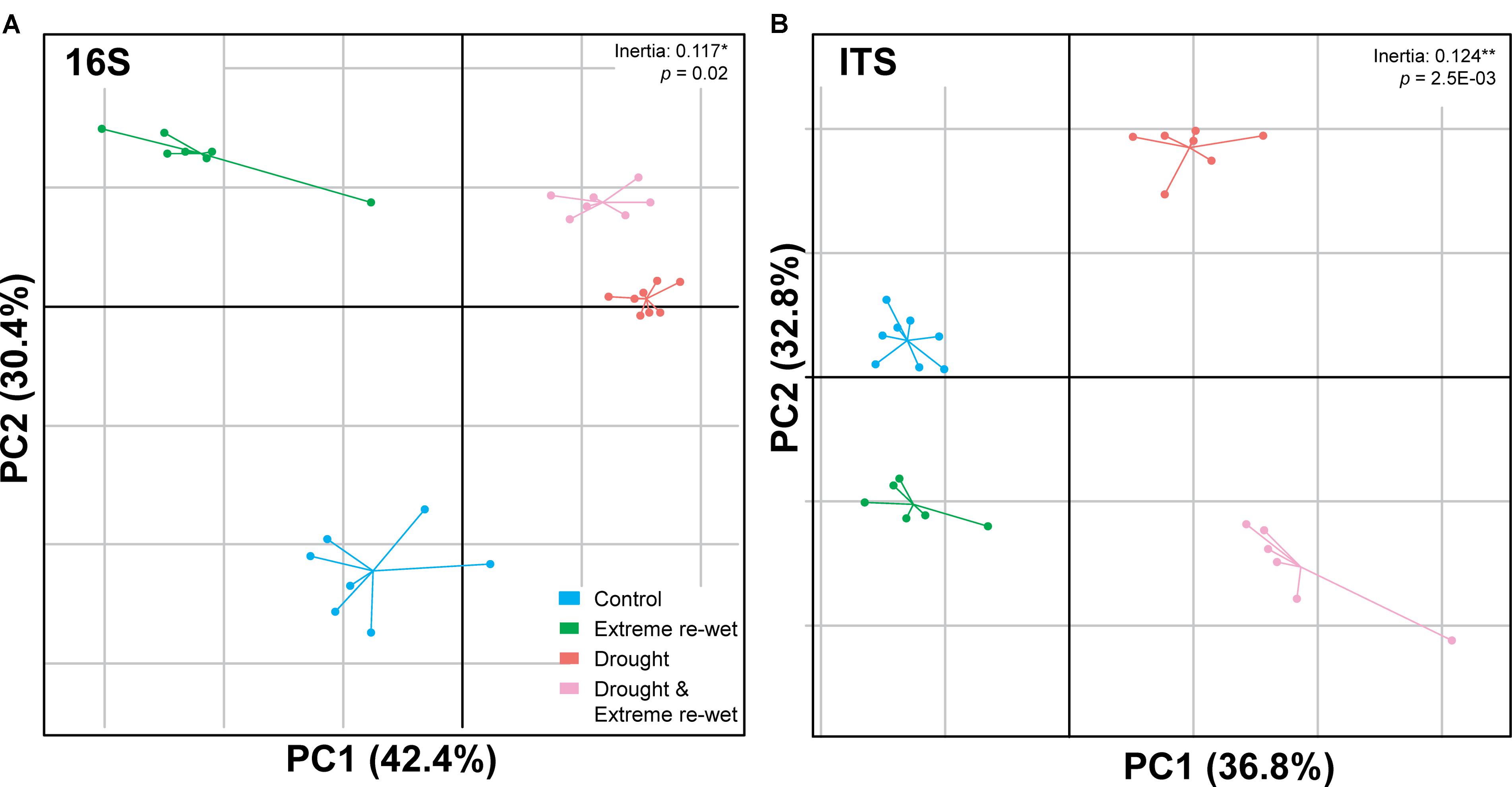
FIGURE 2. Between Group Analysis (BGA) of the soil microbial communities. The figure shows constrained principal component analysis (PCA) of the microbiome profiles after applying sample grouping, according to replicates for four different moisture treatments for 16S rRNA gene profiles (A) and ITS profiles (B). Non-random distribution of the BGA grouping was tested using a Monte-Carlo simulation with 10.000 permutations (ITS: p = 2.5E–03∗∗; 16S: p = 0.02∗).

TABLE 1. PERMANOVA on Euclidean distance matrix of the microbiomes.
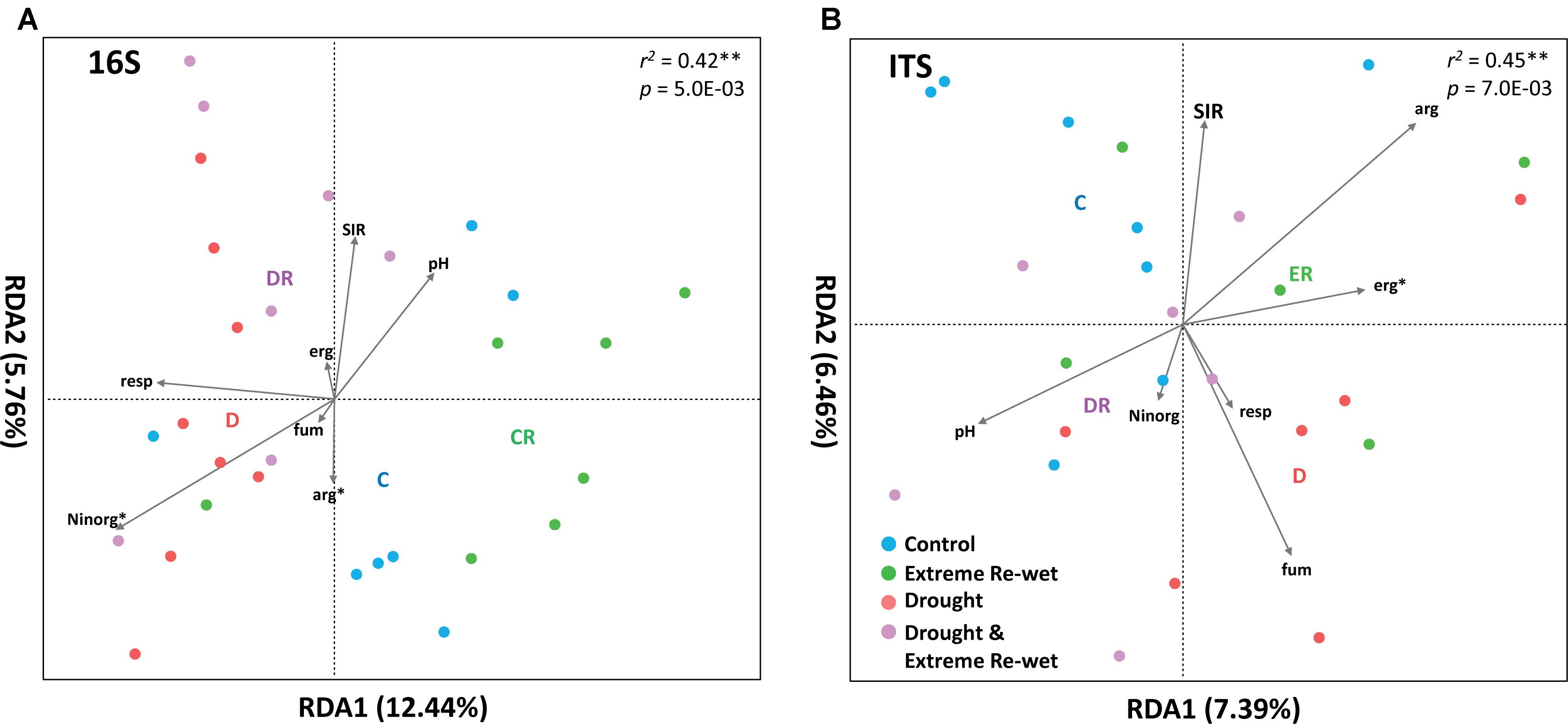
FIGURE 3. Redundancy analysis of prokaryotes (A) and fungi (B) with as explanatory variables: Moist, Extreme Re-wetting, Drought or Drought and Extreme re-wetting. In addition, we have used the following continues explanatory variables: Ninorg (N inorganic), resp (Basal Respiration), SIR (Substrate Induced respiration), erg (ergosterol content, which is a measure of fungal biomass), Fum (total biomass measured with fumigation extraction), arg (arginine ammonification). The R2 indicated the fit of the model and the P-value the significance of all axis (tested with a permutation test, n = 999).
Two LRGs were identified for 428 prokaryote OTUs, representing 5–8% of the total number of sequences, and 1.8% of the total number of OTUs in this study (428/23880). Prokaryote OTUs were either decreased in relative abundance in soil with a history of drought (LRG wet) or increased in relative abundance in soil with a history of drought (LRG dry; Figure 4; see heat maps in Supplementary Figure S2). OTUs belonging to the phylum Cyanobacteria, Chloroflexi, and Verrucomicrobia were enriched in LRG wet (Supplementary Table S3 and Figure 4) and OTUs belonging to the phylum Thaumarchaeota, and Proteobacteria were enriched in LRG dry (Supplementary Table S4 and Figure 4). OTUs belonging to Bacteroidetes were enriched in both LRG dry and LRG wet. This is mostly driven by the response of Sphingobacteriaceae and Flavobacteriaceae in LRG wet and by the response of Chitinophagaceae and Cytophagaceae in LRG dry. Responding OTUs were less rare than the total amount of rare OTUs in the community, which was 92% of the OTUs. For LRG wet, ca. 80% of the OTUs belonged to the rare biosphere in the non-drought soil and ca. 92% in the dried soil (Supplementary Figure S3A). For LRG dry, ca. 80% belonged to the rare biosphere in the non-drought soil and this decreased to ca. 70% in the dried soil (Supplementary Figure S3B). As such, some OTUs that were more abundant decreased in relative abundance when exposed to drought in LRG wet whereas rare OTUs become more abundant in LRG dry (Supplementary Figure S3). Although the initial drought had the most structuring effect on OTUs belonging to the two responding groups, the extreme re-wetting also affected some OTUs when further analyzing if there were LRG within soils with or without a history of drought (see Supplementary Figure S4).
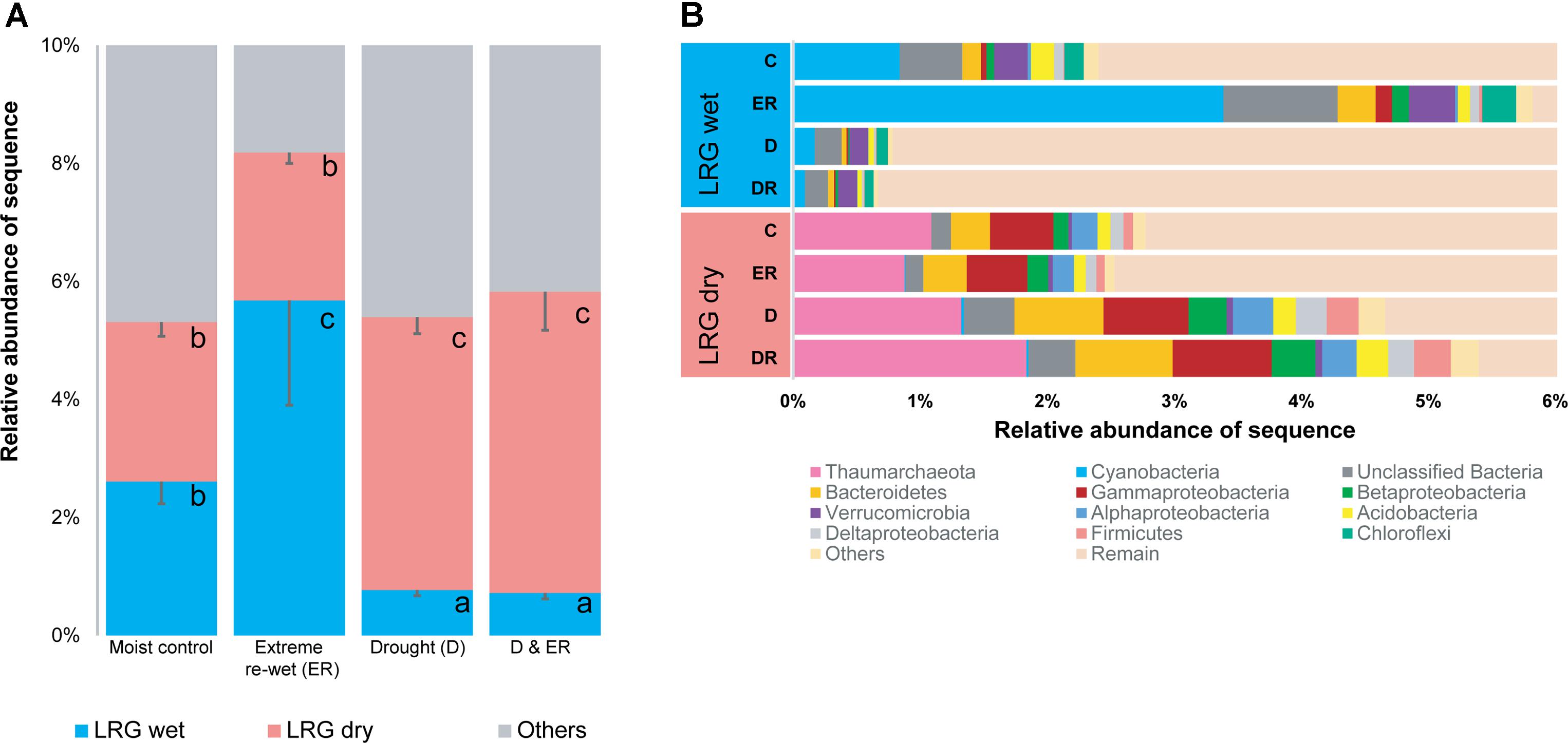
FIGURE 4. The relative abundance of the Legacy Response Groups (LRGs) for 16S amplicons (A) and the taxonomic affiliation of each group (B). Letters denote differences among the four treatments within one LRG at P < 0.05 (ANOVA, Tukey HSD post hoc test).
Two LRGs were also identified for 113 fungal OTUs, accounting for ∼20% of ITS sequences, and 12.3% of the total number of OTUs (Figure 5, see heat maps in Supplementary Figure S5). Fungal OTUs were either decreased in relative abundance in soils with a history of drought (LRG wet) or increased in relative abundance by drought legacy (LRG dry). OTU belonging to the Ascomycota were enriched in the LRG dry group (Supplementary Table S5) although there were also responding OTUs that belonged to the Ascomycota that decreased in relative abundance due to drought history. Sordariomycetes, Diaporthales, unclassified Sordariomycetes, Sordariales, Eurotiomycetes and Dothideomycetes were the most responsive Ascomycota within LRG dry as OTUs belonging to these orders were increased in relative abundance by drought legacy. Sordariomycetes and Leotiomycetes were orders that were decreased in relative abundance by drought legacy. There was no clear linkage between others OTUs and their phylogenetic origins. About half the OTUs can be considered rare in the moist control soils, and a higher number of these rare OTUs were observed in the soil exposed to drought for LRG wet (Supplementary Figure S6). However, these responding OTUs were less rare than the total amount of rare OTUs in the community, which was 77%. As previously seen for prokaryotes, the extreme re-wetting had an effect within the overruling legacy effect of drought (Supplementary Figure S7).
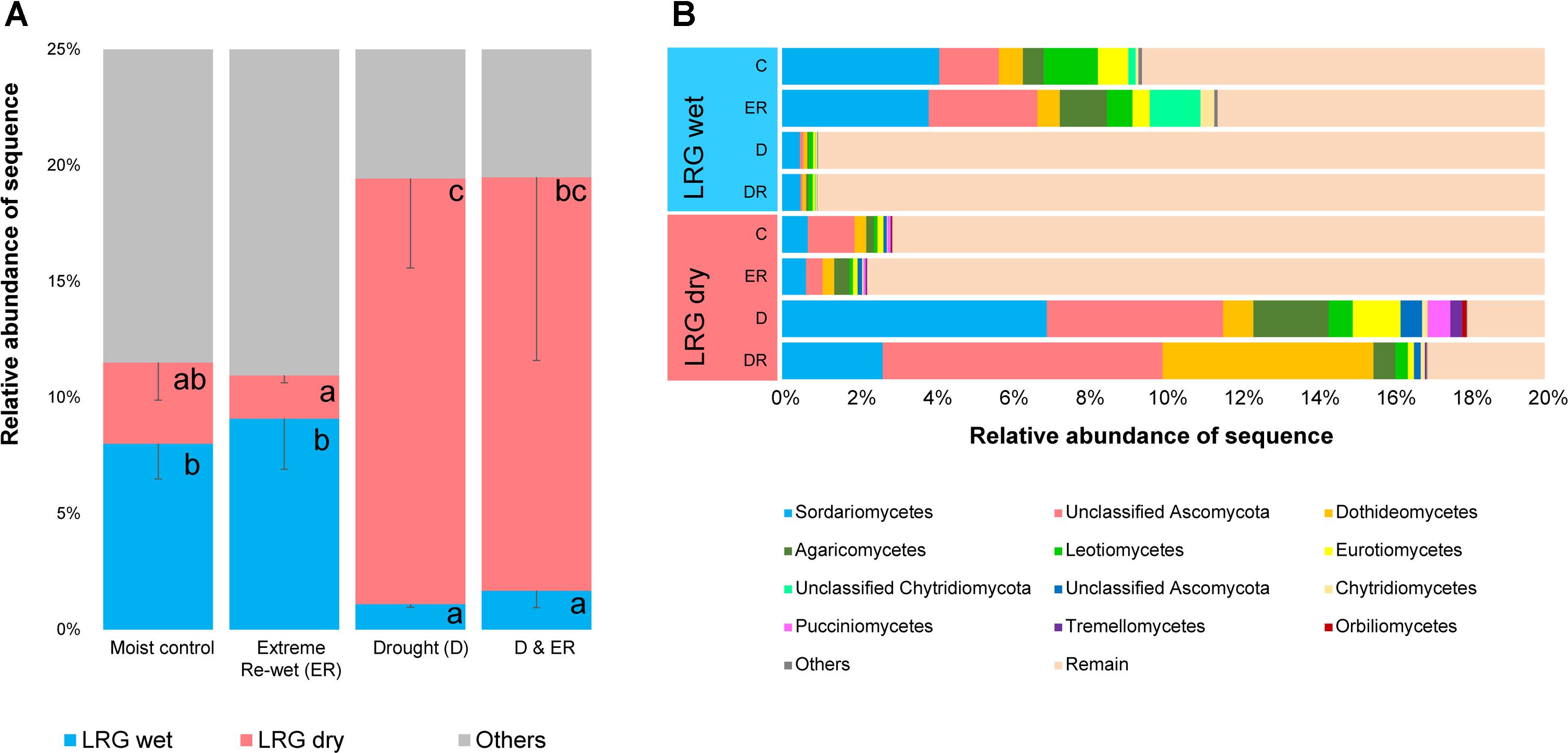
FIGURE 5. The relative abundance of the Legacy Response Groups for ITS amplicons (A) and the taxonomic affiliation of each group (B). Letters in the left panel denote differences among the four treatments within one LRG at P < 0.05 (ANOVA, Tukey HSD post hoc test).
Our results supported our overall hypothesis that extreme simulated drying and/or extreme re-wetting leave a legacy in the composition of the soil microbial community after abiotic conditions have been restored. We found that soils with a history of drying and/or extreme re-wetting differed in composition of prokaryotic and fungal communities. Fungi and prokaryotes responded not only sensitively, but also resistant and opportunistically to a history of drought, which has also been shown for soils exposed to multiple drying and re-wetting cycles (Evans and Wallenstein, 2014). OTUs were grouped in distinct LRGs, regardless of their phylogenetic origin, as the responses of bacteria and fungi were spread among different phyla, classes, and orders. For example, Bacteroides OTUs decreased and increased in relative abundance in soil with a history of drought, which is similar as observed in other studies that measured the response directly to drought (Evans and Wallenstein, 2014). In addition, Chloroflexi can decrease in relative abundance under drought conditions (our study), increase in relative abundance under aridity (Maestre et al., 2015), or has a mixed response to altered moisture conditions (Hartmann et al., 2017). This would suggest that adaptation to extreme moisture fluctuation in soils is likely to have evolved independently in different microbial phyla, leading to the parallel selection and development of similar responders in separated phyla. One possible explanation for responses being spread among phyla is that microbes with oligotrophic and copiotrophic strategies have been suggested to be widespread among phylogenetic groups (Ho et al., 2017). This suggests that resources becoming available for the microbial communities upon re-wetting can be used by a wide range of species with a copiotrophic strategy that belong to different phyla. The initial fast responding species may be important for the composition of slower responding species (Fukami, 2015). As such, the initial copiotrophic strategy may be important for the successional outcome of the microbial re-colonization of empty niches that became available after the drought event.
Our results partly support our first hypothesis that the history of drying and/or extreme re-wetting event decreases the relative abundance of sub-dominant taxa, as this was only the case for bacteria and fungi belonging to the first response group (LRG wet). Cyanobacteria seem to have responded sensitive to drought, because they decreased in soil with this legacy, which is in line with studies on Cyanobacteria in soil crusts (Williams et al., 2008; Hagemann et al., 2017). As Cyanobacteria might have a slow recovery upon re-wetting (Williams et al., 2008), they will most likely not have recovered 3 weeks after the drought period had stopped.
Certain bacterial groups increased in abundance in soils with a history of drought. There are three reasons why some OTUs increased in relative abundance in soil with a history of drought (LRG dry). First, OTUs belonging to certain Prokaryotes may have resisted the drought stress. For example, the most abundant group of Archaea, Thaumarchaeota, increased in relative abundance in soil with a history of drought. This group has been found in extreme environments (Stieglmeier et al., 2014) and can be highly abundant in dessert soil (Shi et al., 2016). Second, bacteria with an opportunistic strategy have likely belonged to the fast responders that increased their abundance after re-wetting (Placella et al., 2012). For example, OTUs that belonged to Alpha-, Beta- and Gammaproteobacteria increased in relative abundance in soil with a history of drought (Figure 3). These classes have been identified as typical copiotrophs with high growth rates (Philippot et al., 2010). Third, some groups of bacteria may have responded via microbial facilitation mechanisms. For example Cyanobacteria and Acidobacteria can produce extracellular polysaccharides that can create moist micro-niches that may benefit other bacteria during drought conditions (Kielak et al., 2016). As there were some OTUs from both groups present in soil that have experienced a drought legacy (Figure 3), these moist micro-niches may have helped other bacteria to survive the drought period.
Our results contrast previous reports showing that fungi are more resistant to drought than bacteria (Barnard et al., 2013; Meisner et al., 2013a). Instead, a short-term history of drought remained in the soil as a legacy effect in the composition of the fungal community. The fungi responded sensitive, tolerant or opportunistic to drying and re-wetting. Long-term drought treatments have been observed to affect the composition of fungi (Sayer et al., 2017). In addition, the composition of fungal community is shaped by rainfall amounts (Hawkes et al., 2011), and can differ between wet and dry seasons (Cregger et al., 2012; Acosta-Martínez et al., 2014). As such, the fungal communities differ from wet to drought conditions and remain present when soil has a short-term drought history. Responding fungal OTUs occurred in Ascomycota; in particular the Sordariomycetes and Dothideomycetes appeared to be major responders to drought history (Figure 4). Some fungi can likely respond to small moisture fluctuations, because they can use an opportunistic strategy thereby changing the composition of the fungal community (Kaisermann et al., 2015).
The history of extreme re-wetting could be identified for fungi and prokaryotes when soils with a history of drought were analyzed separately from the other soils (Supplementary Figures S4, S7). This suggests that drought has a major contribution to overall changes in the composition of the microbial community. As a consequence, a second dichotomy was identified between OTUs reacting to extreme re-wetting, leading to the identification of two additional sub-response groups. Gamma- and Betaproteobacteria were increasing or decreasing in relative abundance when re-wetting to maximum moisture. This is consistent with an earlier study where Proteobacteria have been identified as a group that clearly responded to re-wetting (Evans et al., 2014). The increase in relative abundance of Cyanobacteria and decrease in Firmicutes was clearly linked to the maximum water addition in soils without a history of drought. The same trends were observed for fungi, although the phylogenetic linkage was not as obvious as for bacteria since a wide diversity of lineages were always affected positively and negatively by the extreme re-wetting.
Our work supports the third hypothesis that some rare microbes will increase in relative abundance due to drought legacy. This supports the view that there are a high number of rare taxa in the microbial community, which is common in soil (Lynch and Neufeld, 2015). Members of the rare biosphere can be highly active and contribute to processes in soil even if they have low-ranked abundance (Pester et al., 2010; Pedrós-Alió, 2012). A large fraction of the rare bacteria can respond within days after re-wetting (Aanderud et al., 2015). In addition, the growth rates of low-abundant microbes are not necessarily different from abundant ones (Kurm et al., 2017). As such, the history of drought may potentially lead to altered composition of the microbial community through effects on rare members of the soil microbial community, without obvious consequences on the overall evenness indices.
Our results suggest that drying and re-wetting events can cause a legacy effect in the microbial communities due to a direct effect of moisture. The consequences of altered composition of microbial community due to a history of drought can modify the performance of plant species in communities when other abiotic conditions were kept constant (Meisner et al., 2013b). In addition, changes in soil microbial communities due to drying and/or extreme re-wetting influenced fitness of a rapidly reproducing plant species (Lau and Lennon, 2012). Moreover, changes in soil microbial communities due to drought legacies can affect plant-soil feedbacks (Kaisermann et al., 2017). As such, extreme weather effects on composition of the microbial community may also have the potential to influence the composition and functioning of plant species in terrestrial ecosystems.
Fluctuations between drying and re-wetting do not only affect the composition of soil microbial communities, but can also boost soil fertility by increasing nitrogen availability for plants (Birch, 1958; Jarvis et al., 2007; Meisner et al., 2013b). Previous work showed that drought legacy effects resulted into increased available nitrogen and soil respiration rates (Meisner et al., 2013b), which also seems to be correlated with composition of the prokaryotes (Figure 3). On the one hand, this indicates that the abiotic changes during drought affected nitrogen availability and soil functions and therewith the composition of the microbial community. On the other hand, one of the major changes during drought is a decrease in microbial activity (Meisner et al., 2017) and biomass (Kieft et al., 1987), which increases the number of niches available for microbes to recolonize upon re-wetting. Increased nitrogen availability has also been suggested to positively correlate with plant growth performance following a period of drought (Lebedjantzev, 1924; de Vries et al., 2012b). For example, pre-season drought effects may increase invasiveness of non-native plants that shift range as a consequence of climate warming (Meisner et al., 2013b). Drought can also affect the composition of plant species directly (Kardol et al., 2010). Fluctuations in drying and extreme re-wetting events and magnitudes have also been suggested to affect soil microbial communities via changes in plant composition (de Vries et al., 2012b; Evans et al., 2014). Future research is needed in order to reveal how drought legacies can affect the interactions between plants and soil microbes at different temporal, for example successional, stages of ecosystem development. Therefore, we conclude that effects of extreme drying and re-wetting may remain in the soil as a legacy effect in the composition of the microbial community. These legacy effects may explain why the composition of terrestrial plant communities change even once the extreme weather event ends (Meisner et al., 2013b).
Sequences are deposited on the European Nucleotide Archive (ENA) and have accession number PRJEB23318.
AM and WvdP designed the experiments. FtH performed the lab analysis. BS and SJ analyzed the data. AM wrote the first draft of the manuscript and all other authors were involved in revising the manuscript text. All authors approved the submitted version of the manuscript.
The research was supported by an international career grant from the Swedish Research Council (VR, Grant No. 330-2014-6430) and Marie Sklodowska Curie Actions (Cofund Project INCA 600398) to AM, and an ERC-Adv grant 26055290 (SPECIALS) to WvdP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Gerlinde de Deyn and Wietse de Boer for discussion about the design of the main experiment. This is NIOO publication 6490.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00294/full#supplementary-material
Aanderud, Z. T., Jones, S. E., Fierer, N., and Lennon, J. T. (2015). Resuscitation of the rare biosphere contributes to pulses of ecosystem activity. Front. Microbiol. 6:24. doi: 10.3389/fmicb.2015.00024
Acosta-Martínez, V., Cotton, J., Gardner, T., Moore-Kucera, J., Zak, J., Wester, D., et al. (2014). Predominant bacterial and fungal assemblages in agricultural soils during a record drought/heat wave and linkages to enzyme activities of biogeochemical cycling. Appl. Soil Ecol. 84, 69–82. doi: 10.1016/j.apsoil.2014.06.005
Bapiri, A., Bååth, E., and Rousk, J. (2010). Drying-rewetting cycles affect fungal and bacterial growth differently in an arable soil. Microb. Ecol. 60, 419–428. doi: 10.1007/s00248-010-9723-5
Barnard, R. L., Osborne, C. A., and Firestone, M. K. (2013). Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 7, 2229–2241. doi: 10.1038/ismej.2013.104
Barnes, C. J., Van Der Gast, C. J., Mcnamara, N. P., Rowe, R., and Bending, G. D. (2018). Extreme rainfall affects assembly of the root-associated fungal community. New Phytol. doi: 10.1111/nph.14990 [Epub ahead of print].
Bates, S. T., Berg-Lyons, D., Caporaso, J. G., Walters, W. A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917. doi: 10.1038/ismej.2010.171
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4, 914–919. doi: 10.1111/2041-210x.12073
Birch, H. F. (1958). The effect of soil drying on humus decomposition and nitrogen availability. Plant Soil 10, 9–31. doi: 10.1007/BF01343734
Blazewicz, S. J., Schwartz, E., and Firestone, M. K. (2014). Growth and death of bacteria and fungi underlie rainfall-induced carbon dioxide pulses from seasonally dried soil. Ecology 95, 1162–1172. doi: 10.1890/13-1031.1
Chowdhury, N., Yan, N., Islam, M. N., and Marschner, P. (2011). The extent of drying influences the flush of respiration after rewetting in non-saline and saline soils. Soil Biol. Biochem. 43, 2265–2272. doi: 10.1016/j.soilbio.2011.07.013
Clein, J. S., and Schimel, J. P. (1994). Reduction in microbial activity in Birch litter due to drying and rewetting events. Soil Biol. Biochem. 26, 403–406. doi: 10.1016/0038-0717(94)90290-9
Cole, J. R., Wang, Q., Fish, J. A., Chai, B. L., Mcgarrell, D. M., Sun, Y. N., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Cregger, M. A., Schadt, C. W., Mcdowell, N. G., Pockman, W. T., and Classen, A. T. (2012). Response of the soil microbial community to changes in precipitation in a semiarid ecosystem. Appl. Environ. Microbiol. 78, 8587–8594. doi: 10.1128/aem.02050-12
Crowther, T. W., Maynard, D. S., Crowther, T. R., Peccia, J., Smith, J. R., and Bradford, M. A. (2014). Untangling the fungal niche: the trait-based approach. Front. Microbiol. 5:579. doi: 10.3389/fmicb.2014.00579
de Hollander, M. (2016). GitLab Community Edition: Open Source Software to Collaborate on Code [Online]. Available at: https://gitlab.bioinf.nioo.knaw.nl/amplicon metagenomics/illumina_paired_end
de Vries, F. T., Liiri, M. E., Bjornlund, L., Bowker, M. A., Christensen, S., Setala, H. M., et al. (2012a). Land use alters the resistance and resilience of soil food webs to drought. Nat. Clim. Change 2, 276–280. doi: 10.1038/nclimate1368
de Vries, F. T., Liiri, M. E., Bjørnlund, L., Setälä, H. M., Christensen, S., and Bardgett, R. D. (2012b). Legacy effects of drought on plant growth and the soil food web. Oecologia 170, 821–833. doi: 10.1007/s00442-012-2331-y
Dodt, M., Roehr, J. T., Ahmed, R., and Dieterich, C. (2012). FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905. doi: 10.3390/biology1030895
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Evans, S. E., and Wallenstein, M. D. (2012). Soil microbial community response to drying and rewetting stress: does historical precipitation regime matter? Biogeochemistry 109, 101–116. doi: 10.1007/s10533-011-9638-3
Evans, S. E., and Wallenstein, M. D. (2014). Climate change alters ecological strategies of soil bacteria. Ecol. Lett. 17, 155–164. doi: 10.1111/ele.12206
Evans, S. E., Wallenstein, M. D., and Burke, I. C. (2014). Is bacterial moisture niche a good predictor of shifts in community composition under long-term drought? Ecology 95, 110–122. doi: 10.1890/13-0500.1
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Influence of drying-rewetting frequency on soil bacterial community structure. Microb. Ecol. 45, 63–71. doi: 10.1007/s00248-002-1007-2
Fischer, E. M., and Knutti, R. (2016). Observed heavy precipitation increase confirms theory and early models. Nat. Clim. Change 6, 986–991. doi: 10.1038/nclimate3110
Fukami, T. (2015). Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu. Rev. Ecol. Evol. Syst. 46, 1–23. doi: 10.1146/annurev-ecolsys-110411-160340
Galand, P. E., Casamayor, E. O., Kirchman, D. L., and Lovejoy, C. (2009). Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. U.S.A. 106, 22427–22432. doi: 10.1073/pnas.0908284106
Gordon, H., Haygarth, P. M., and Bardgett, R. D. (2008). Drying and rewetting effects on soil microbial community composition and nutrient leaching. Soil Biol. Biochem. 40, 302–311. doi: 10.1016/j.soilbio.2007.08.008
Hagemann, M., Henneberg, M., Felde, V. J., Berkowicz, S. M., Raanan, H., Pade, N., et al. (2017). Cyanobacterial populations in biological soil crusts of the northwest Negev Desert, Israel - effects of local conditions and disturbance. FEMS Microbiol. Ecol. doi: 10.1093/femsec/fiw228 [Epub ahead of print].
Hammer,Ø, Harper, D., and Ryan, P. D. (2001). Paleontological statistics software package for education and data analysis. Palaeontol. Electronica 4, 1–9.
Hartmann, M., Brunner, I., Hagedorn, F., Bardgett, R. D., Stierli, B., Herzog, C., et al. (2017). A decade of irrigation transforms the soil microbiome of a semi-arid pine forest. Mol. Ecol. 26, 1190–1206. doi: 10.1111/mec.13995
Haugwitz, M. S., Bergmark, L., Priemé, A., Christensen, S., Beier, C., and Michelsen, A. (2014). Soil microorganisms respond to five years of climate change manipulations and elevated atmospheric CO2 in a temperate heath ecosystem. Plant Soil 374, 211–222. doi: 10.1007/s11104-013-1855-1
Hawkes, C. V., Kivlin, S. N., Rocca, J. D., Huguet, V., Thomsen, M. A., and Suttle, K. B. (2011). Fungal community responses to precipitation. Glob. Chang. Biol. 17, 1637–1645. doi: 10.1111/j.1365-2486.2010.02327.x
Hawkes, C. V., Waring, B. G., Rocca, J. D., and Kivlin, S. N. (2017). Historical climate controls soil respiration responses to current soil moisture. Proc. Natl. Acad. Sci. U.S.A. 114, 6322–6327. doi: 10.1073/pnas.1620811114
Ho, A., Di Lonardo, D. P., and Bodelier, P. L. E. (2017). Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 93:fix006. doi: 10.1093/femsec/fix006
Hothorn, T., Bretz, F., and Westfall, P. (2008). Simultaneous inference in general parametric models. Biom. J. 50, 346–363. doi: 10.1002/bimj.200810425
Ihrmark, K., Bödeker, I. T. M., Cruz-Martinez, K., Friberg, H., Kubartova, A., Schenck, J., et al. (2012). New primers to amplify the fungal ITS2 region – evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 82, 666–677. doi: 10.1111/j.1574-6941.2012.01437.x
Ilstedt, U., Nordgren, A., and Malmer, A. (2000). Optimum soil water for soil respiration before and after amendment with glucose in humid tropical acrisols and a boreal mor layer. Soil Biol. Biochem. 32, 1591–1599. doi: 10.1016/S0038-0717(00)00073-0
IPCC (2012). Managing the Risks of Extreme Events and Disasters to Advance Climate Change Adaptation. A Special Report of Working Groups I and II of the Intergovernmental Panel on Climate Change, eds C. B. Field, V. Barros, T. F. Stocker, D. Qin, D. J. Dokken, K. L. Ebi, et al. (Cambridge: Cambridge University Press).
Jacquiod, S., Cyriaque, V., Riber, L., Al-Soud, W. A., Gillan, D. C., Wattiez, R., et al. (2018). Long-term industrial metal contamination unexpectedly shaped diversity and activity response of sediment microbiome. J. Hazard. Mater. 344, 299–307. doi: 10.1016/j.jhazmat.2017.09.046
Jacquiod, S., Stenbæk, J., Santos, S. S., Winding, A., Sørensen, S. J., and Priemé, A. (2016). Metagenomes provide valuable comparative information on soil microeukaryotes. Res. Microbiol. 167, 436–450. doi: 10.1016/j.resmic.2016.03.003
Jarvis, P., Rey, A., Petsikos, C., Wingate, L., Rayment, M., Pereira, J., et al. (2007). Drying and wetting of Mediterranean soils stimulates decomposition and carbon dioxide emission: the “Birch effect”. Tree Physiol. 27, 929–940. doi: 10.1093/treephys/27.7.929
Kaisermann, A., De Vries, F. T., Griffiths, R. I., and Bardgett, R. D. (2017). Legacy effects of drought on plant–soil feedbacks and plant–plant interactions. New Phytol. 215, 1413–1424. doi: 10.1111/nph.14661
Kaisermann, A., Maron, P. A., Beaumelle, L., and Lata, J. C. (2015). Fungal communities are more sensitive indicators to non-extreme soil moisture variations than bacterial communities. Appl. Soil Ecol. 86, 158–164. doi: 10.1016/j.apsoil.2014.10.009
Kardol, P., Cregger, M. A., Campany, C. E., and Classen, A. T. (2010). Soil ecosystem functioning under climate change: plant species and community effects. Ecology 91, 767–781. doi: 10.1890/09-0135.1
Kieft, T. L., Soroker, E., and Firestone, M. K. (1987). Microbial biomass response to a rapid increase in water potential when dry soil is wetted. Soil Biol. Biochem. 19, 119–126. doi: 10.1016/0038-0717(87)90070-8
Kielak, A. M., Barreto, C. C., Kowalchuk, G. A., Van Veen, J. A., and Kuramae, E. E. (2016). The Ecology of Acidobacteria: moving beyond genes and genomes. Front. Microbiol. 7:744. doi: 10.3389/fmicb.2016.00744
Kim, D. G., Vargas, R., Bond-Lamberty, B., and Turetsky, M. R. (2012). Effects of soil rewetting and thawing on soil gas fluxes: a review of current literature and suggestions for future research. Biogeosciences 9, 2459–2483. doi: 10.5194/bg-9-2459-2012
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Koster, J., and Rahmann, S. (2012). Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. doi: 10.1093/bioinformatics/bts480
Kurm, V., Van Der Putten, W. H., De Boer, W., Naus-Wiezer, S., and Hol, W. H. G. (2017). Low abundant soil bacteria can be metabolically versatile and fast growing. Ecology 98, 555–564. doi: 10.1002/ecy.1670
Lado-Monserrat, L., Lull, C., Bautista, I., Lidón, A., and Herrera, R. (2014). Soil moisture increment as a controlling variable of the “Birch effect”. Interactions with the pre-wetting soil moisture and litter addition. Plant Soil 379, 21–34. doi: 10.1007/s11104-014-2037-5
Lau, J. A., and Lennon, J. T. (2012). Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. U.S.A. 109, 14058–14062. doi: 10.1073/pnas.1202319109
Lebedjantzev, A. N. (1924). Drying of soil, as one of the natural factors in maintaining soil fertility. Soil Sci. 18, 419–447. doi: 10.1097/00010694-192412000-00001
Lennon, J. T., Aanderud, Z. T., Lehmkuhl, B. K., and Schoolmaster, D. R. (2012). Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93, 1867–1879. doi: 10.1890/11-1745.1
Lundquist, E. J., Scow, K. M., Jackson, L. E., Uesugi, S. L., and Johnson, C. R. (1999). Rapid response of soil microbial communities from conventional, low input, and organic farming systems to a wet/dry cycle. Soil Biol. Biochem. 31, 1661–1675. doi: 10.1016/s0038-0717(99)00080-2
Lynch, M. D. J., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Mackey, B. M., and Derrick, C. M. (1984). Conductance measurements of the lag phase of injured Salmonella-typhimurium. J. Appl. Bacteriol. 57, 299–308. doi: 10.1111/j.1365-2672.1984.tb01394.x
Maestre, F. T., Delgado-Baquerizo, M., Jeffries, T. C., Eldridge, D. J., Ochoa, V., Gozalo, B., et al. (2015). Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. U.S.A. 112, 15684–15689. doi: 10.1073/pnas.1516684112
Manzoni, S., Schaeffer, S. M., Katul, G., Porporato, A., and Schimel, J. P. (2014). A theoretical analysis of microbial eco-physiological and diffusion limitations to carbon cycling in drying soils. Soil Biol. Biochem. 73, 69–83. doi: 10.1016/j.soilbio.2014.02.008
Manzoni, S., Schimel, J. P., and Porporato, A. (2012). Responses of soil microbial communities to water-stress: results from a meta-analysis. Ecology 93, 930–938. doi: 10.1890/11-0026.1
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31
Meisner, A., Bååth, E., and Rousk, J. (2013a). Microbial growth responses upon rewetting soil dried for four days or one year. Soil Biol. Biochem. 66, 188–192. doi: 10.1016/j.soilbio.2013.07.014
Meisner, A., De Boer, W., Verhoeven, K. J. F., Boschker, H. T. S., and Van Der Putten, W. H. (2011). Comparison of nutrient acquisition in exotic plant species and congeneric natives. J. Ecol. 99, 1308–1315. doi: 10.1111/j.1365-2745.2011.01858.x
Meisner, A., De Deyn, G. B., De Boer, W., and Van Der Putten, W. H. (2013b). Soil biotic legacy effects of extreme weather events influence plant invasiveness. Proc. Natl. Acad. Sci. U.S.A. 110, 9835–9838. doi: 10.1073/pnas.1300922110
Meisner, A., Leizeaga, A., Rousk, J., and Bååth, E. (2017). Partial drying accelerates bacterial growth recovery to rewetting. Soil Biol. Biochem. 112, 269–276. doi: 10.1016/j.soilbio.2017.05.016
Meisner, A., Rousk, J., and Bååth, E. (2015). Prolonged drought changes the bacterial growth response to rewetting. Soil Biol. Biochem. 88, 314–322. doi: 10.1016/j.soilbio.2015.06.002
Miller, A. E., Schimel, J. P., Meixner, T., Sickman, J. O., and Melack, J. M. (2005). Episodic rewetting enhances carbon and nitrogen release from chaparral soils. Soil Biol. Biochem. 37, 2195–2204. doi: 10.1016/j.soilbio.2005.03.021
Nocker, A., Fernandez, P. S., Montijn, R., and Schuren, F. (2012). Effect of air drying on bacterial viability: a multi parameter viability assessment. J. Microbiol. Methods 90, 86–95. doi: 10.1016/j.mimet.2012.04.015
Nunes, I., Jacquiod, S., Brejnrod, A., Holm, P. E., Johansen, A., Brandt, K. K., et al. (2016). Coping with copper: legacy effect of copper on potential activity of soil bacteria following a century of exposure. FEMS Microbiol. Ecol. 92:fiw175. doi: 10.1093/femsec/fiw175
Orchard, V. A., and Cook, F. J. (1983). Relationship between soil respiration and soil-moisture. Soil Biol. Biochem. 15, 447–453. doi: 10.1016/0038-0717(83)90010-x
Pedrós-Alió, C. (2012). The rare bacterial biosphere. Annu. Rev. Mar. Sci. 4, 449–466. doi: 10.1146/annurev-marine-120710-100948
Pester, M., Bittner, N., Deevong, P., Wagner, M., and Loy, A. (2010). A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 4, 1591–1602. doi: 10.1038/ismej.2010.75
Philippot, L., Andersson, S. G. E., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529. doi: 10.1038/nrmicro2367
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and Van Der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Placella, S. A., Brodie, E. L., and Firestone, M. K. (2012). Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc. Natl. Acad. Sci. U.S.A. 109, 10931–10936. doi: 10.1073/pnas.1204306109
Pulleman, M., and Tietema, A. (1999). Microbial C and N transformations during drying and rewetting of coniferous forest floor material. Soil Biol. Biochem. 31, 275–285. doi: 10.1016/s0038-0717(98)00116-3
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Robinson, M. D., Mccarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Sayer, E. J., Oliver, A. E., Fridley, J. D., Askew, A. P., Mills, R. T. E., and Grime, J. P. (2017). Links between soil microbial communities and plant traits in a species-rich grassland under long-term climate change. Ecol. Evol. 7, 855–862. doi: 10.1002/ece3.2700
Scheu, S., and Parkinson, D. (1994). Changes in bacterial and fungal biomass-c, bacterial and fungal biovolume and ergosterol content after drying, remoistening and incubation of different layers of cool temperate forest soils. Soil Biol. Biochem. 26, 1515–1525. doi: 10.1016/0038-0717(94)90093-0
Schimel, J., Balser, T. C., and Wallenstein, M. (2007). Microbial stress-response physiology and its implications for ecosystem function. Ecology 88, 1386–1394. doi: 10.1890/06-0219
Schimel, J., and Schaeffer, S. (2012). Microbial control over carbon cycling in soil. Front. Microbiol. 3:348. doi: 10.3389/fmicb.2012.00348
Schöler, A., Jacquiod, S., Vestergaard, G., Schulz, S., and Schloter, M. (2017). Analysis of soil microbial communities based on amplicon sequencing of marker genes. Biol. Fertil. Soils 53, 485–489. doi: 10.1007/s00374-017-1205-1
Setia, R., Marschner, P., Baldock, J., Chittleborough, D., and Verma, V. (2011). Relationships between carbon dioxide emission and soil properties in salt-affected landscapes. Soil Biol. Biochem. 43, 667–674. doi: 10.1016/j.soilbio.2010.12.004
Shi, Y., Adams, J. M., Ni, Y., Yang, T., Jing, X., Chen, L., et al. (2016). The biogeography of soil archaeal communities on the eastern Tibetan Plateau. Sci. Rep. 6:38893. doi: 10.1038/srep38893
Stieglmeier, M., Alves, R. J. E., and Schleper, C. (2014). “The phylum Thaumarchaeota,” in The Prokaryotes: Other Major Lineages of Bacteria and The Archaea, eds E. Rosenberg, E. F. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 347–362.
Thorsen, J., Brejnrod, A., Mortensen, M., Rasmussen, M. A., Stokholm, J., Al-Soud, W. A., et al. (2016). Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome 4:62. doi: 10.1186/s40168-016-0208-8
Vidali, M. (2001). Bioremediation. An overview. Pure Appl. Chem. 73, 1163–1172. doi: 10.1351/pac200173071163
Vos, M., Wolf, A. B., Jennings, S. J., and Kowalchuk, G. A. (2013). Micro-scale determinants of bacterial diversity in soil. FEMS Microbiol. Rev. 37, 936–954. doi: 10.1111/1574-6976.12023
Warren, C. R. (2014). Response of osmolytes in soil to drying and rewetting. Soil Biol. Biochem. 70, 22–32. doi: 10.1016/j.soilbio.2013.12.008
Williams, W. J., Eldridge, D. J., and Alchin, B. M. (2008). Grazing and drought reduce cyanobacterial soil crusts in an Australian Acacia woodland. J. Arid Environ. 72, 1064–1075. doi: 10.1016/j.jaridenv.2007.11.017
Keywords: climate change, soil, Birch effect, re-wetting, bacteria, fungi, microbial communities
Citation: Meisner A, Jacquiod S, Snoek BL, ten Hooven FC and van der Putten WH (2018) Drought Legacy Effects on the Composition of Soil Fungal and Prokaryote Communities. Front. Microbiol. 9:294. doi: 10.3389/fmicb.2018.00294
Received: 06 November 2017; Accepted: 08 February 2018;
Published: 07 March 2018.
Edited by:
Martin Hartmann, Swiss Federal Institute for Forest, Snow and Landscape Research, SwitzerlandReviewed by:
Zachary B. Freedman, West Virginia University, United StatesCopyright © 2018 Meisner, Jacquiod, Snoek, ten Hooven and van der Putten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annelein Meisner, QW5uZWxlaW4uTWVpc25lckBiaW9sLmx1LnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.