 Jill S. McClary
Jill S. McClary Alexandria B. Boehm
Alexandria B. Boehm- Civil and Environmental Engineering, Stanford University, Stanford, CA, United States
The transcriptional response of Staphylococcus aureus strain Newman to sunlight exposure was investigated under both oxic and anoxic conditions using RNA sequencing to gain insight into potential mechanisms of inactivation. S. aureus is a pathogenic bacterium detected at recreational beaches which can cause gastrointestinal illness and skin infections, and is of increasing public health concern. To investigate the S. aureus photostress response in oligotrophic seawater, S. aureus cultures were suspended in seawater and exposed to full spectrum simulated sunlight. Experiments were performed under oxic or anoxic conditions to gain insight into the effects of oxygen-mediated and non-oxygen-mediated inactivation mechanisms. Transcript abundance was measured after 6 h of sunlight exposure using RNA sequencing and was compared to transcript abundance in paired dark control experiments. Culturable S. aureus decayed following biphasic inactivation kinetics with initial decay rate constants of 0.1 and 0.03 m2 kJ−1 in oxic and anoxic conditions, respectively. RNA sequencing revealed that 71 genes had different transcript abundance in the oxic sunlit experiments compared to dark controls, and 18 genes had different transcript abundance in the anoxic sunlit experiments compared to dark controls. The majority of genes showed reduced transcript abundance in the sunlit experiments under both conditions. Three genes (ebpS, NWMN_0867, and NWMN_1608) were found to have the same transcriptional response to sunlight between both oxic and anoxic conditions. In the oxic condition, transcripts associated with porphyrin metabolism, nitrate metabolism, and membrane transport functions were increased in abundance during sunlight exposure. Results suggest that S. aureus responds differently to oxygen-dependent and oxygen-independent photostress, and that endogenous photosensitizers play an important role during oxygen-dependent indirect photoinactivation.
Introduction
In the United States, pollution of recreational waters led to 23,481 beach closures in 2011 (National Resources Defense Council, 2012), and contact with polluted recreational waters can cause gastrointestinal illness, respiratory infections, and skin ailments (Cabelli et al., 1982; Haile et al., 1999; Colford et al., 2007). To prevent excess exposure to microbial pollution, recreational waters are traditionally monitored by the detection of culturable fecal indicator bacteria (FIB), such as Escherichia coli and enterococci, which requires processing times of ~18–24 h (US EPA, 2012). However, FIB concentrations are known to fluctuate on short timescales due to factors such as sunlight exposure and tides (Boehm et al., 2009; Russell et al., 2013; Corsi et al., 2016), calling into question the utility of FIB measurements that require long processing times. To address this issue, rapid detection methods and water quality modeling techniques have begun to be applied in recreational water quality monitoring (Wade et al., 2008; Thoe et al., 2015; He et al., 2016; Tryland et al., 2016). However, an incomplete understanding of the mechanisms leading to bacterial decay in coastal environments limits our ability to include these factors in water quality models and points to a need for improved understanding of these mechanisms.
Photoinactivation, or inactivation due to sunlight exposure, is an important process that modulates bacterial concentrations in environmental waters (Sassoubre et al., 2015) and can occur by both direct and indirect mechanisms. Direct photoinactivation involves the absorption of photons by vital cell components, like nucleic acids, which leads to cellular damage (Sinha and Häder, 2002). In contrast, during indirect photoinactivation, photons are absorbed by sensitizers (either endogenous or exogenous to the cell) which become excited and subsequently damage vital cell components either directly or through generation of reactive oxygen species (ROS) (Curtis et al., 1992). Several studies have identified ROS as one of the most important factors influencing photoinactivation of both bacteria and viruses in natural waters (Kohn and Nelson, 2007; Sassoubre et al., 2012; Maraccini et al., 2016b). However, the relative importance of direct and indirect photoinactivation mechanisms in environmental systems remains poorly understood. In engineered systems, advanced oxidation unit processes, which combine UV treatment with ROS or ROS precursors, are being increasingly considered for use in wastewater reuse treatment trains (Sun et al., 2016). The combination of ROS precursors and light exposure is also the basis of photodynamic therapy, which can be used for localized treatment of bacterial infections (Sabbahi et al., 2008). Due to the importance of photoinactivation in a range of contexts, a better understanding of direct and indirect photoinactivation mechanisms is needed.
Investigation into the transcriptional response of bacteria to sunlight stress can provide insights into photoinactivation mechanisms. Microarrays and RNA sequencing (RNA-seq) have been used to investigate the effects of sunlight exposure on gene expression in FIB, including Enterococcus faecalis (Sassoubre et al., 2014) and E. coli (Berney et al., 2006; Al-Jassim et al., 2017). A range of cellular processes are triggered by sunlight exposure, including DNA repair, oxidative stress response, virulence, and SOS response (Berney et al., 2006; Sassoubre et al., 2014; Al-Jassim et al., 2017). Evidence to date suggests that different species transcribe different genes in response to sunlight exposure. For example, following sunlight exposure, genes coding for superoxide dismutase, a highly conserved enzyme involved in oxidative stress response, were identified as upregulated in E. faecalis (Sassoubre et al., 2012, 2014) but downregulated in E. coli (Berney et al., 2006; Al-Jassim et al., 2017). This information allows us to gain insight into cells' ability to repair or respond to sunlight exposure and advances our understanding of bacterial fate in sunlight-exposed waters.
One bacterial pathogen of concern in recreational waters is Staphylococcus aureus, which is commonly detected in recreational beach water and sand (Charoenca and Fujioka, 1993; Goodwin et al., 2012; Levin-Edens et al., 2012; Hower et al., 2013) and can cause gastrointestinal, respiratory, and skin infections. Epidemiological studies have identified associations between recreational water contact and various skin ailments (Wade et al., 2008; Yau et al., 2009; Sinigalliano et al., 2010). Some studies have further identified relationships between staphylococci concentrations in beach water and skin ailments (Prüss, 1998), and between S. aureus skin infections and recreational water contact (Charoenca and Fujioka, 1995), indicating that recreational beaches may be a reservoir for pathogenic S. aureus in the environment. Recently, concern regarding particular strains of antibiotic resistant S. aureus that are able to spread within the community has grown. Compared to healthcare-associated strains, community-associated S. aureus have also been shown to be more virulent in mouse models, partially due to their ability to resist ROS-mediated killing by neutrophils (Voyich et al., 2005).
The present study investigates the transcriptional response of S. aureus suspended in clear seawater to sunlight exposure in order to gain insight into photoinactivation mechanisms and bacterial stress response. Experiments were performed under both oxic and anoxic conditions in order to differentiate between photostress responses associated with oxygen-mediated and non-oxygen-medated photoinactivation mechanisms. To our knowledge, this is the first study to evaluate genome-wide transcriptional response of a pathogenic bacterium under both oxygen-dependent and oxygen-independent photostress conditions.
Materials and Methods
Photoinactivation Experiments
Staphylococcus aureus photoinactivation under oxic and anoxic conditions was evaluated using an experimental design identical to a previously published study (McClary et al., 2017). In brief, S. aureus subsp. aureus str. Newman (ATCC 25904) was grown in chemostat cultures filled with 20 mL 25% Brain Heart Infusion (BHI) broth (Fluka Analytical, Steinheim, Germany). S. aureus was grown in chemostats in order to improve reproducibility between experimental replicates (Maraccini et al., 2015). After reaching a stable growth rate, bacteria were washed twice and resuspended in ~1 L sterile simulated seawater for a concentration of ~107 CFU/mL. The composition of simulated seawater was derived from Parker et al. (2013) and consisted of 424 mM sodium chloride, 0.87 mM sodium bromide, 29.2 mM sodium sulfate, 0.27 mM sodium carbonate, 1.83 mM sodium bicarbonate, 10.5 mM potassium chloride, 54.8 mM magnesium chloride, and 10.7 mM calcium chloride. The initial concentration of ~107 CFU/mL of S. aureus was chosen to allow for sufficient masses of mRNA to be extracted for sequencing. For experiments performed under anoxic conditions, the bacteria-seawater suspension was divided into two black PVC pipe reactors (described previously McClary et al., 2017), one experimental and one control. Reactors were sealed by fixing quartz glass plates to the top of the reactors with silicone sealant and were then sparged with nitrogen through rubber septa to remove oxygen from the water column and headspace. After sparging for ~30 min, reactors were held in the dark at 15°C with constant stirring for 12 h to acclimate to a cool, oligotrophic environment. For experiments performed under oxic conditions, reactors were set up identically but with quartz glass plates secured loosely with tape and without nitrogen sparging.
After 12 h of incubation at 15°C, the experimental reactor (oxic or anoxic) was placed in a 15°C recirculating water bath in a solar simulator (Atlas Suntest XLS+; Chicago, IL) equipped with a 1.1 kW xenon arc lamp and a glass filter to generate full spectrum sunlight (see Maraccini et al., 2015 for solar simulator light spectra). Reactors were exposed to 6 h of full spectrum sunlight. Six hours of sunlight exposure was chosen based on previous data showing significant changes in gene expression at this exposure duration (McClary et al., 2017). The control reactor was kept in the dark at 15°C during the photoinactivation experiments. Both reactors were constantly stirred, and samples were taken from the reactors as described below. For experiments performed under anoxic conditions, an equal volume of nitrogen was injected into the reactors during sampling events to keep the reactors anoxic and at constant pressure. Triplicate experiments were performed in both oxic and anoxic conditions to generate three biological replicates for each condition.
Culturability
To track S. aureus photoinactivation during experiments, 0.5-mL samples were taken from the experimental reactor every hour and from the control reactor every 3 h to determine culturability. Samples were diluted as necessary and appropriate dilutions were spread plated in duplicate on Brain Heart Infusion agar (BD Difco, Sparks, MD). After incubation at 37°C for 18–24 h, colonies were counted and sample concentrations were calculated in CFU/mL. Only dilutions resulting in countable colonies on duplicate plates were used to calculate sample concentrations. Inactivation rate constants were determined by non-linear regression using a biphasic first-order inactivation model:
where ln(C/C0) is the natural log-transformed relative concentration, f is the subpopulation fraction, k1 and k2 are the inactivation rate constants for the first and second phases, respectively, and FUVA+UVB is fluence in kJ/m2. Fluence was calculated as has been done previously based on wavelengths in the UVA & UVB spectra (280–400 nm) (Maraccini et al., 2016a; McClary et al., 2017). Rate constants were also determined using log-linear and shoulder log-linear decay models (Geeraerd et al., 2005), but the biphasic model resulted in the best fit as determined by minimizing residual standard error and so was used for all subsequent analysis.
RNA Stabilization, Extraction, and rRNA Removal
At the end of each experiment (i.e., after 6 h of sunlight exposure), 200-mL samples were taken from both the experimental and control reactors for RNA extraction. Samples were immediately centrifuged for 10 min at 10,000 × g, and bacterial pellets were treated with RNAProtect Bacterial Reagent (Qiagen, Hilden, Germany). After 5 min of incubation at room temperature, samples were centrifuged again and the supernatant discarded. Stabilized bacterial pellets were stored at −80°C until RNA extraction.
RNA extractions were performed as described previously (McClary et al., 2017). In brief, stored bacterial pellets were resuspended in 0.2 mg/mL lysostaphin (Sigma-Aldrich, St. Louis, MO) and incubated at 37°C to lyse cells. Further lysis was performed by addition of a 100:1 vol:vol solution of Buffer RLT (Qiagen) and β-mercaptoethanol (Sigma-Aldrich), followed by bead beating in Lysis Matrix B tubes with a FastPrep-24 cell homogenizer (MP Biomedicals, Solon, OH). After brief centrifugation, lysate was transferred to new tubes, 470 μL ethanol was added to each sample, and RNA was extracted using the RNeasy Mini Kit (Qiagen), following the manufacturer's instructions. After elution in 60 μL of RNase-free water warmed to 60°C, extracts were DNase-digested using the RNase-free DNase Set (Qiagen), following the manufacturer's instructions. Samples were then cleaned up using the RNeasy Mini Kit, with final elution in 40 μL of RNase-free water warmed to 60°C. DNase digestion was confirmed by a qPCR assay targeting the rexA gene of S. aureus as described previously (McClary et al., 2017). Primer and probe sequences for the qPCR assay are provided in Table 1. For each set of extractions, an extraction blank was processed in parallel to verify lack of contamination from protocol reagents.
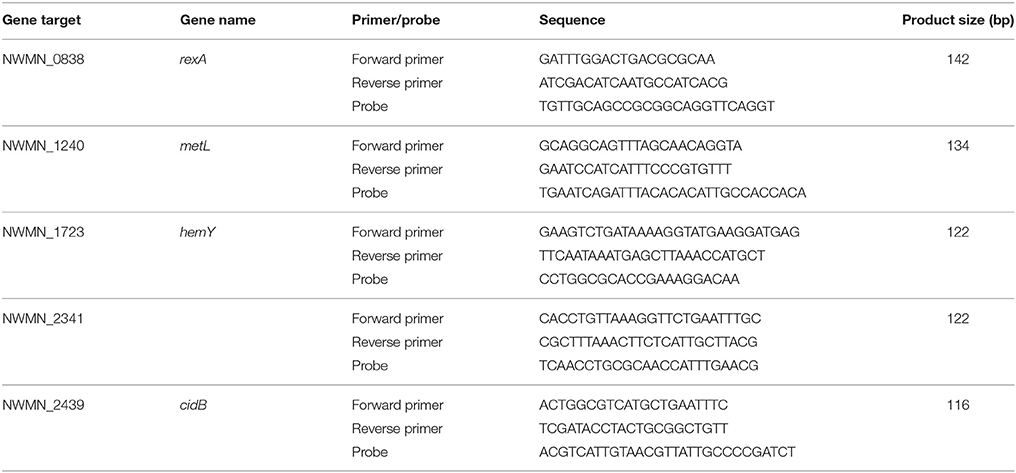
Table 1. Summary of primer and probe sequences used for RTqPCR reactions.
Total RNA samples were precipitated by adding 0.1 volume 3 M sodium acetate, 2.5 μL of 2 mg/mL glycogen, and 2.5 volumes 100% ethanol. The mixture was left overnight at −20°C before recovering precipitated RNA by centrifuging at 12,000 × g for 30 min at 4°C. RNA pellets were then washed twice in 1 mL ice cold 70% ethanol and recollected by centrifuging at 12,000 × g for 10 min at 4°C. After two ethanol washes, the RNA pellet was dissolved in 25 μL TE buffer. RNA precipitates were then depleted of rRNA using the MICROBExpress mRNA Enrichment Kit (Life Technologies, Carlsbad, CA), following the manufacturer's instructions. Five microliters from each extraction blank was also pooled and carried through the precipitation and rRNA-removal procedures as a negative control. Total RNA extracts, RNA precipitates, and rRNA-depleted samples were quantified on a Qubit v2.0 fluorometer or Nanodrop 1000, and RNA quality was confirmed on an Agilent 2100 Bioanalyzer at the Stanford Protein and Nucleic Acid Facility.
Library Preparation and Sequencing
Indexed sequencing libraries were prepared from rRNA-depleted samples, including the negative control, using the ScriptSeq v2 RNA-Seq Library Preparation Kit and ScriptSeq Index PCR Primers (Epicentre, Madison, WI) following the manufacturer's instructions. PCR amplification of the indexed libraries was performed for 14 cycles. An additional positive control sample was also included, consisting of a 101 nt RNA sequence coding for a portion of the grpE gene of Methanobacterium sp. MB1, obtained from Integrated DNA Technologies (San Diego, CA). This sequence was chosen as a control as it would not be expected to occur in any of the experimental samples. Library preparation of the positive control followed the manufacturer's instructions for Severely Fragmented RNA. Amplified indexed libraries were quantified on an Agilent 2100 Bioanalyzer at the Stanford Functional Genomics Facility.
A total of 14 indexed libraries were generated, with each index corresponding to an individual sample (Table 2). The 12 oxic & anoxic sample libraries were combined in equimolar ratios to generate a pooled library. The positive control was added to the pooled library at a 10-fold lower molar ratio. The average volume of the oxic & anoxic sample libraries that were pooled was calculated, and this volume of negative control was also added to the pooled library. The pooled library was then sequenced on an Illumina MiSeq machine at the Stanford Functional Genomics Facility, generating 75 bp paired-end reads.
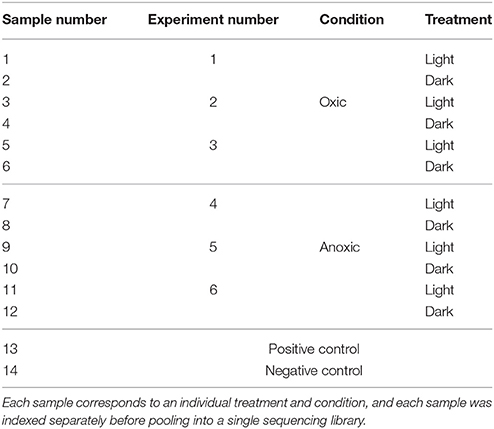
Table 2. Description of samples included for RNA sequencing.
Sequencing Data Analysis
Raw sequencing data was demultiplexed and quality scored by Illumina MiSeq software to generate fastq files for forward and reverse reads of each indexed sample library. Initial read quality was assessed in FastQC version 0.11.4. Adapter trimming and quality filtering was performed for paired-end reads using Trimmomatic version 0.36 with provided adapter Fasta files for TruSeq3, removing low quality bases from the beginning and end of reads, and dropping reads shorter than 75% of the amplicon length or with quality scores <30 (Bolger et al., 2014). Following quality filtering, RNA-seq reads were aligned to the S. aureus genome using STAR version 2.5.3a with default settings (Dobin et al., 2013), and count matrices were generated from the alignment output using the Bioconductor GenomicAlignments package (Gentleman et al., 2004; Lawrence et al., 2013). The S. aureus genome and gene annotation information used for alignment and read counting, respectively, were obtained from Ensembl (taxid: 426430). Separate count matrices were generated for oxic and anoxic experiments, and each count matrix was filtered to remove genes with low or no counts (i.e., counts ≤1 across all samples) and to remove counts mapped to rRNA genes. Data from the count matrices were then analyzed using DESeq2 (Love et al., 2014). First, the regularized-logarithm (rlog) transformation was applied to the count matrices and used to calculate Euclidean distances between samples. Visualization of the sample-to-sample distances using a distance matrix revealed that samples from one experiment (Experiment #4, Samples 7 & 8, Table 2) were outliers (Supplementary Figure 1), and so this experiment was dropped from further analysis. Next, non-transformed count matrices were used to determine differential expression between light and dark conditions using DESeq2. DESeq2 is capable of evaluating differential expression on as few as two biological replicates (Love et al., 2014; Sekulovic and Fortier, 2015), making this method most appropriate for use in this study. Genes with a false discovery rate (FDR) <25% were considered significantly differentially expressed. After identifying differentially expressed genes, gene functions were explored using the KEGG pathways database. All sequencing data analysis was performed in Linux and R version 3.4.1. RNA-seq data are deposited in the NCBI sequence read archive (SRA) under accession number SRP125691.
Reverse transcription qPCR (RTqPCR) confirmation of RNA-seq results was performed for four selected genes: metL, hemY, cidB, and NWMN_2341 (Table 1). These genes were selected based on (1) their observed expression changes from RNA-seq data analysis, and (2) the ability to develop efficient qPCR assays for these genes. Differential expression between light and dark samples by RTqPCR was based on calculating a relative expression ratio (R) using the Pfaffl method (Pfaffl, 2001) with rexA as the reference gene. rexA was used as a reference because we previously developed an RTqPCR assay for this gene (McClary et al., 2017) and the RNA sequencing data analysis demonstrated that rexA was not significantly differentially expressed. Significant differential expression was determined if R was ≥ 2 or ≤ 0.5 and if R ± standard error (SE) did not include 1. Further details on RTqPCR assays are provided in the Supplementary Material.
Results
Staphylococcus aureus Photoinactivation Kinetics in Oxic and Anoxic Conditions
Inactivation of S. aureus was observed during sunlight exposure under both oxic and anoxic conditions, as shown in Figure 1, and was discussed in our previous publication (McClary et al., 2017). Inactivation kinetics are biphasic under both conditions, displaying relatively fast inactivation followed by a period of slow or no inactivation. Non-linear regression was used to fit the observed data to biphasic first-order inactivation curves, and inactivation rate constants are presented in Table 3. The first-order rate constant during the initial phase of inactivation was larger in the oxic compared to anoxic condition (k1 ± SE = 0.1 ± 0.01 m2 kJ−1 in oxic conditions vs. 0.03 ± 0.002 m2 kJ−1 in anoxic; Z-test, P < 0.05). These rate constants are in agreement with those presented in our previous work (McClary et al., 2017). The first-order rate constants during the second phase of inactivation (k2) were 0.01 ± 0.005 m2 kJ−1 and −0.005 ±0.007 m2 kJ−1 in oxic and anoxic conditions, respectively. S. aureus continued to slowly decay following the initial rapid decay in oxic conditions. In anoxic conditions, k2 is not different from 0. No inactivation was observed in dark controls, suggesting that all observed inactivation was due to sunlight exposure. After 6 h of sunlight exposure [i.e., fluence (FUVA+UVB) between 427 and 687 kJ/m2], the concentration of cultivatable cells was at or below the limit of detection (i.e., ≤ 20 CFU/mL) in oxic experiments and was ~700 CFU/mL for anoxic sunlight experiments. After 6 h of dark incubation, the concentration of cultivatable cells in the control oxic and anoxic experiments remained steady at ~107 CFU/mL. These samples were used to investigate gene expression changes in sunlight-exposed experiments vs. dark controls.
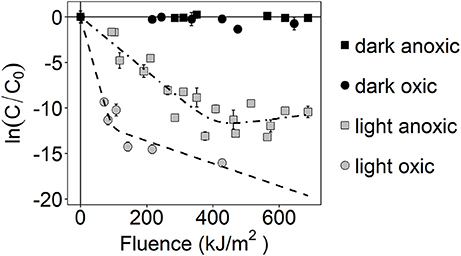
Figure 1. Photoinactivation kinetics of S. aureus, as measured by loss of culturability. ln(C/C0) is the natural-log transformed relative concentration. Error bars = ± standard deviation of technical replicates. Dashed lines are modeled biphasic inactivation curves.
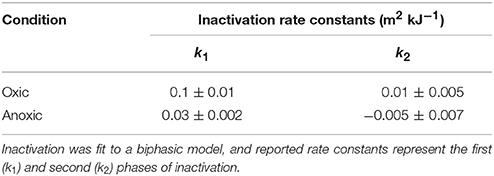
Table 3. Modeled inactivation rate constants of S. aureus under sunlight exposure.
Differential Gene Expression Due to Sunlight Exposure in Oxic and Anoxic Conditions
RNA sequencing was used to investigate changes in S. aureus gene expression as a result of sunlight exposure under oxic and anoxic conditions. A summary of sample-specific data generated by RNA sequencing is presented in Table 4. Sequencing resulted in ~21 million total reads, with an average of ~900,000 read pairs per sample. Quality filtering removed between 7 and 39% of read pairs per sample, and the resulting filtered reads aligned to the S. aureus genome at rates of at least 92%. As described in the Materials and Methods, based on Euclidean sample-to-sample distances generated from rlog-transformed count matrices, samples from one experiment (Experiment #4) clustered far from all other samples; samples from this experiment were subsequently removed from further gene expression analyses.
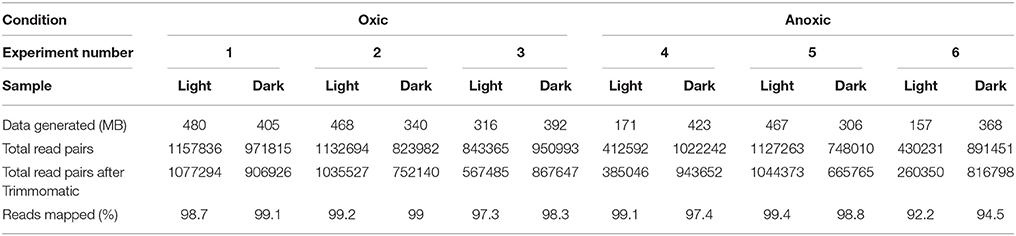
Table 4. Summary of sample-specific data generated by RNA sequencing.
To determine the effects of sunlight exposure on gene expression, differential expression analysis was carried out comparing sunlight-exposed samples from a single experimental condition (oxic or anoxic) to corresponding controls prepared identically and kept in the dark. Using this framework, a total of 71 differentially expressed genes were identified from oxic experiments (Table 5) and 18 from anoxic experiments (Table 6). Of these, three genes were differentially expressed under sunlight exposure in both the oxic and anoxic conditions: NWMN_1608 was increased in expression, while ebpS and NWMN_0867 were decreased in expression. Under both conditions, most differentially expressed genes showed reduced expression under sunlight-exposed conditions compared to the dark control; nine genes and two genes were significantly increased in abundance in sunlit oxic and anoxic conditions, respectively. Of the total number of differentially expressed genes, the proportions of genes showing increased expression under oxic and anoxic conditions are similar.
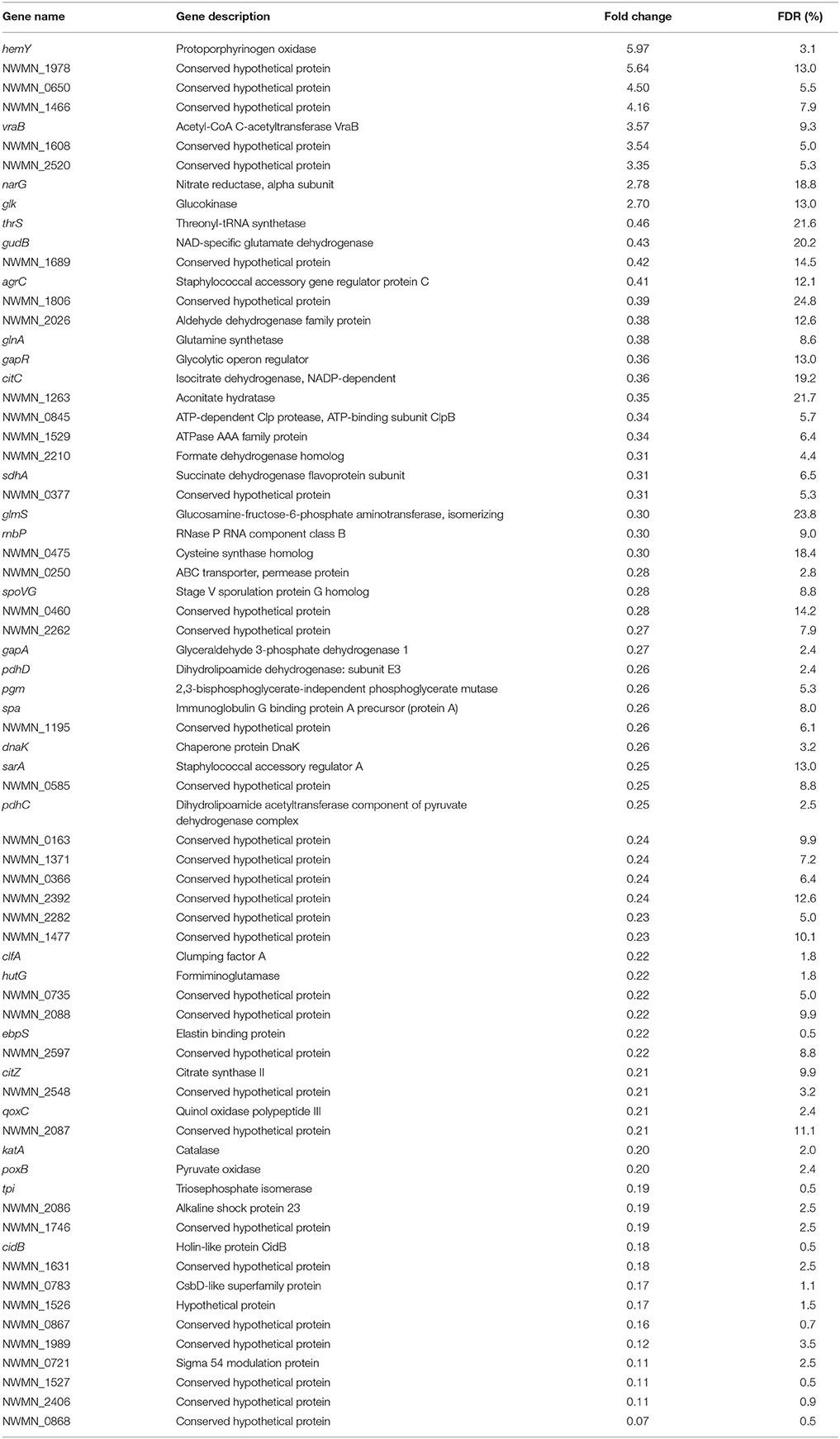
Table 5. List of significantly differentially expressed genes from oxic experiments.
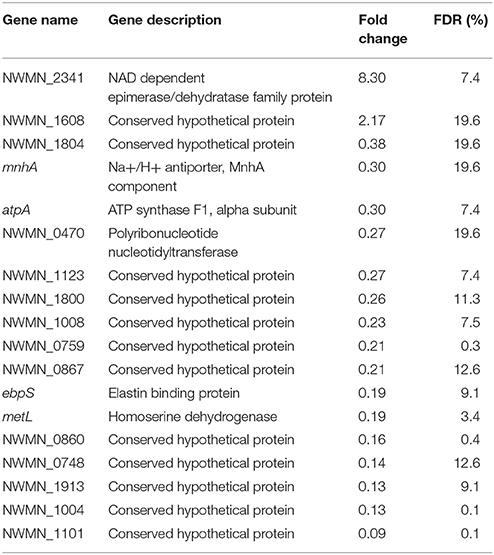
Table 6. List of significantly differentially expressed genes from anoxic experiments.
Functional Classification of Differentially Expressed Genes
The genome of S. aureus subsp. aureus str. Newman contains genes encoding 2,624 proteins, of which 1,051 are classified as hypothetical meaning that their function is unknown or unconfirmed. In the oxic condition, 30 differentially expressed genes (42% of 71) were assigned to functional pathways whereas for the anoxic condition, three differentially expressed genes (17% of 18) were assigned (Figure 2). Functional pathways with decreased expression due to sunlight exposure in both the oxic and anoxic conditions involved metabolism, environmental information processing, genetic information processing, cellular processes, and human disease. Expression of other genes involved in metabolism and environmental information processing were also induced by sunlight exposure in the oxic condition. Neither of the genes induced by sunlight exposure in the anoxic condition was assigned to functional pathways in KEGG.
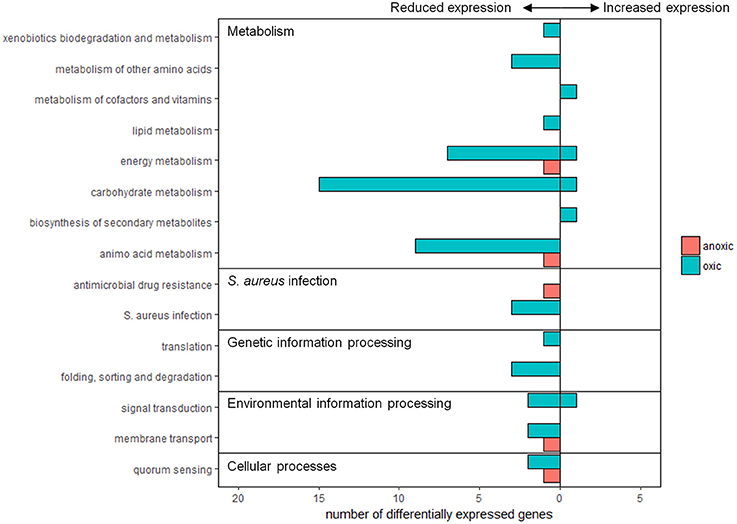
Figure 2. Summary of differentially expressed genes assigned to functional groups according to KEGG pathways. Pink bars represent expression from anoxic experiments, and blue bars represent expression from oxic experiments. Values to the left and right of the y-axis indicate genes with reduced or increased expression, respectively.
Differential Gene Expression Not Categorized to Functional Pathways
Differentially expressed genes not assigned to pathways include genes with no annotated function or with predicted functions not yet linked to specific S. aureus cell reactions or networks. In the oxic condition, 39 genes were differentially expressed but not assigned to KEGG functional pathways. Five hypothetical proteins showed increased expression; the remaining 34 differentially expressed genes not assigned to functional pathways in the oxic condition were decreased in expression following sunlight exposure. These included a glycolytic operon regulator (gapR), a subunit of Clp protease (NWMN_0845), an ATPase family protein (NWMN_1529), a component of RNase P (rnbP), an ABC transporter (NWMN_0250), a sporulation protein (spoVG), staphylococcal accessory regulator A (sarA), elastin binding protein (ebpS), an alkaline shock protein (NWMN_2086), holin-like protein CidB (cidB), a CsbD-like superfamily protein (NWMN_0783), sigma 54 modulation protein (NWMN_0721), and 22 hypothetical proteins.
In the anoxic condition, 15 genes were differentially expressed and not assigned to functional pathways. These included an epimerase/dehydratase family protein (NWMN_2341), a Na+/H+ antiporter (mnhA), a polyribonucleotide nuleotidyltransferase (NWMN_0470), and 12 hypothetical proteins. Of these, NWMN_2341 and a conserved hypothetical protein (NWMN_1608) were increased in expression; the expression of the remaining 13 genes was decreased following sunlight exposure.
Confirmation of Gene Expression with RTqPCR
Expression changes in the same samples analyzed by RNA sequencing were also measured using RTqPCR assays targeting four different genes: cidB, hemY, metL, and NWMN_2341. Fold changes of these genes detected by RTqPCR and RNA sequencing are shown for the oxic and anoxic cases in Figures 3, 4, respectively. As RTqPCR and RNA-seq use different methods to normalize the “baseline” expression level in samples, we opted not to compare the specific fold change values but rather to compare whether statistical analysis of each method concluded an increase, decrease, or no change in expression of the gene of interest. With this treatment of the data, RTqPCR and RNA-seq results were in agreement in most cases: 2/4 genes are in agreement in the oxic condition and 3/4 genes are in agreement in the anoxic condition. Exceptions were for metL in the anoxic samples, and cidB and hemY in the oxic samples. RNA sequencing detected significant decreases in expression for metL in the anoxic condition and cidB in the oxic condition, whereas RTqPCR did not detect any significant expression changes. Similarly, RNA sequencing detected a significant increase in expression of hemY in the oxic condition, while the fold change generated by RTqPCR was not significant. Others have also found that RTqPCR results do not always agree with RNA-seq or microarray results, usually in cases where significance is detected by one method but not by the other (Song et al., 2016; Al-Jassim et al., 2017).
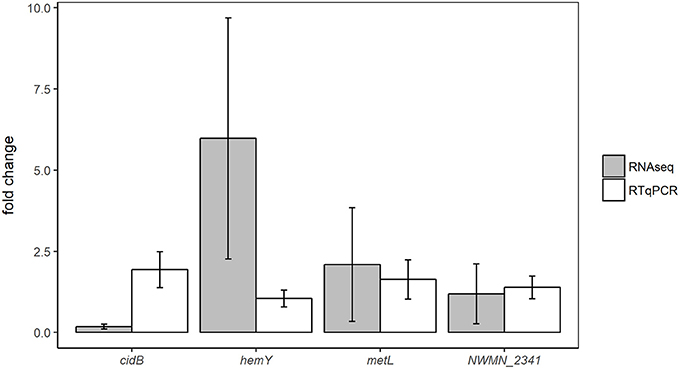
Figure 3. Comparison of differential expression results from oxic experiments using RNA-seq or RTqPCR. Error bars = ± SE.
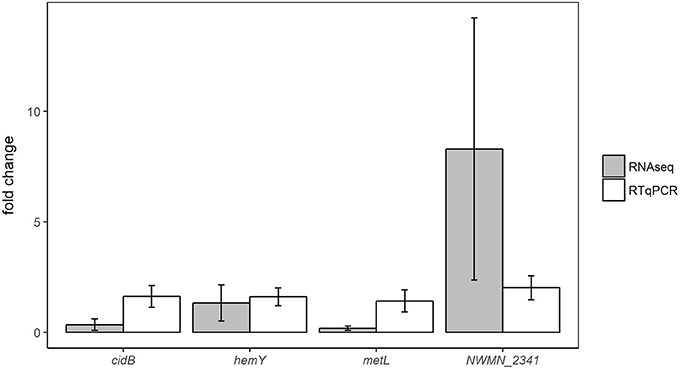
Figure 4. Comparison of differential expression results from anoxic experiments using RNA-seq or RTqPCR. Error bars = ± SE.
Discussion
To better understand the ways in which S. aureus responds to oxygen-mediated and non-oxygen-mediated photoinactivation, we used RNA sequencing to identify gene expression changes between oxic and anoxic sunlit reactors and their corresponding dark controls. After 6 h of sunlight exposure, concentrations of cultivatable S. aureus were reduced by more than four orders of magnitude in both oxic and anoxic conditions, and were reduced to levels at or below the limit of detection in the sunlit oxic treatment. Despite significant reduction in cultivatable cell concentration after 6 h of sunlight exposure, our previous work showed only slight reduction in the intact cell concentration during the same exposure period, as measured by fluorescence microscopy (McClary et al., 2017). The combination of intact cell membranes and detectable mRNA concentrations in these samples suggests the possibility that S. aureus entered a viable but non-culturable (VBNC) state under the sunlight stress condition, and these metrics have been used in previous studies to conclude the presence of VBNC cells (Liu et al., 2009; Chaisowwong et al., 2012; Pasquaroli et al., 2013). Additionally, samples collected after 6 h, which were analyzed by RNA sequencing, were collected during the second phase of the observed biphasic inactivation. This second phase of inactivation is often assumed to represent a resistant subpopulation of the bacterial community, a shift to a resistant phenotype, and/or a shift to a VBNC state, which could be triggered by environmental stresses (Brouwer et al., 2016). While the existence of a VBNC state is generally accepted within the scientific community, there remains uncertainty regarding what specific metrics must be used to define this state and differentiate from other non-growing states (Hammes et al., 2011; Ramamurthy et al., 2014; Pinto et al., 2015). Future work to characterize the transition of S. aureus into a VBNC state during sunlight exposure should include attempts at resuscitation of non-culturable cells.
To identify gene expression changes associated with oxic and anoxic photostress conditions, we used RNA sequencing and differential expression analysis with DESeq2 to compare mRNA transcript abundances between sunlight-exposed samples and control samples under either oxic or anoxic conditions, separately. To identify significant differential expression, we chose to consider genes identified by the DESeq2 program with FDR < 25%. Significant expression thresholds based on FDR are highly variable among previous microarray and RNA-seq studies, often ranging between 5 and 30%, while other studies base results on nominal p-values without correction for multiple hypothesis testing (Graham et al., 2005; Bore et al., 2007; Stasiewicz et al., 2011; Dhanjal et al., 2014; Sassoubre et al., 2014). We opted to consider significance based on FDR due to the importance of multiple hypothesis testing in detecting gene expression changes across the full genome, and we chose to set a somewhat liberal threshold at FDR < 25% based on our goals in this study to identify and explore overall transcriptional response to photostress conditions.
Overall, we identified 71 and 18 genes which were significantly differentially expressed after 6 h of sunlight exposure in oxic and anoxic conditions, respectively. This is comparable to the number of differentially expressed genes identified in E. faecalis during sunlight exposure using microarrays (Sassoubre et al., 2014), but is a smaller amount of genes than those identified in E. coli during sunlight exposure using RNA sequencing (Al-Jassim et al., 2017). Of the genes identified as differentially expressed, most showed significantly decreased expression in sunlight exposed reactors compared to their dark controls: 87 and 89% in oxic and anoxic conditions, respectively. Due to the fact that experiments were performed in oligotrophic conditions, it is possible that S. aureus in the sunlit experiments were forced to shut down transcription of cell functions not immediately necessary for combating the damaging effects of sunlight. In contrast, while control dark reactors were similarly oligotrophic, S. aureus in these reactors were exposed only to starvation stress and therefore were able to maintain a higher level of transcription in contrast to the sunlight-exposed cells. Additionally, sunlight exposure may lead to the direct mutation and degradation of mRNA transcripts in the sunlight-exposed samples. While the effects of UVA+UVB exposure on DNA have been more comprehensively investigated (Sinha and Häder, 2002; Rastogi et al., 2010), UVA+UVB can lead to degradation of RNA molecules through similar mechanisms (Swenson and Setlow, 1964; Qiao and Wigginton, 2016). It is therefore possible that mRNA transcripts were able to persist longer in the dark control reactors than in the sunlight-exposed reactors, and this differential persistence could also have an effect on the overall decreased gene expression detected in sunlight-exposed reactors. Another factor that may have influenced the overall changes in gene expression is a transition to a viable but non-culturable state. As mentioned previously, samples collected following sunlight exposure exhibited substantially reduced culturable cell numbers compared to those in dark controls. However, our previous work demonstrated that S. aureus cells remain intact in these samples (McClary et al., 2017), suggesting that cells remain viable but may be transitioning to a non-culturable state in the sunlight-exposed system. The difference between non-culturable cells in the sunlight-exposed samples and largely culturable cells in the dark control samples could control some of the transcriptome changes observed.
Due to the significant losses in S. aureus culturability observed after 6 h of sunlight exposure, genes identified with increased expression in the sunlight-exposed reactors relative to dark controls are hypothesized to be of great importance to the S. aureus photostress response. For the oxic case, genes with increased expression included hemY, vraB, narG, glk, and five conserved hypothetical proteins. The gene hemY, which was expressed in the oxic sunlight-exposed experiments ~6-fold more than in the dark controls, codes for a protoporphyrinogen oxidase and is involved in porphyrin metabolism. Porphyrins are well-known photosensitizers, and the use of synthetic or naturally occurring porphyrins for the enhancement of photoinactivation in applications like photodynamic therapy has been studied for many years (Jori and Brown, 2004; Ferro et al., 2007; Khlebtsov et al., 2013; Nakonieczna et al., 2016). Specifically, hemY catalyzes the oxidation of protoporphyrinogen (or coproporphyrinogen), yielding protoporphyrin (or coproporphyrin) and hydrogen peroxide. Despite the fact that this reaction yields potentially damaging hydrogen peroxide as well as the photosensitizer protoporphyrin, the enhancement of protoporphyrinogen oxidase activity would be required to metabolize and subsequently reduce the overall levels of endogenous porphyrins. A previous study in mice found that the use of a protoporphyrinogen oxidase inhibitor led to the buildup of endogenous porphyrin molecules and subsequently enhanced the effects of photodynamic therapy (Fingar et al., 1997). Additionally, in Bacillus subtilis, a Gram-positive bacterium with very similar hemY structure to that of S. aureus (Lobo et al., 2015), hemY mutants were found to accumulate endogenous coproporphyrin (Hansson and Hederstedt, 1994). In contrast, a recent study found that activation of hemY led to increased photosensitization in S. aureus (Surdel et al., 2017). Interestingly, of the four S. aureus strains tested in that study, activation of hemY in S. aureus Newman led to the least significant reduction in cell viability following light exposure (Surdel et al., 2017). We therefore hypothesize that oxygen-mediated indirect photoinactivation mechanisms in S. aureus are strongly dependent on levels of endogenous photosensitizers within the cells, and that the metabolism of photosensitizing porphyrins is potentially a more efficient stress response method under starvation conditions than the expression of antioxidant enzymes. This hypothesis should be explored in future work using mutants for specific genes in the porphyrin metabolism pathway, such as hemY, or by quantifying and identifying intracellular porphyrins (Nitzan and Kauffman, 1999; Fyrestam et al., 2015).
In addition to the increased expression of hemY, S. aureus also increased expression of vraB, narG, and glk following exposure to sunlight in oxic conditions. vraB codes for an acetyl-CoA acetyltransferase and is involved in the TCA cycle. Expression of vraB in S. aureus was previously found to be induced by other stresses, including treatment with the antibacterial compound berberine chloride (Wang et al., 2008) and exposure to Cr(VI) (Zhang et al., 2014), suggesting expression of vraB could be important for general S. aureus stress response. narG codes for the alpha subunit of nitrate reductase, a membrane-bound oxidoreductase enzyme. While narG is typically only regulated during anaerobic metabolism (Richardson et al., 2001), nitrate can also serve as an important precursor to reactive oxygen species like hydroxyl radical (Brezonik and Fulkerson-Brekken, 1998). S. aureus may therefore increase expression of narG in order to manage the potentially damaging effects of nitrate to the cell. S. aureus also increased expression of glk, coding for glucokinase, following sunlight exposure in oxic conditions. Glucokinase is involved in a range of metabolic functions, including metabolism of galactose and sucrose, as well as the biosynthesis of streptomycin. While overall more metabolism genes were observed to be decreased in expression following sunlight exposure, the increased expression of glk suggests that S. aureus remains metabolically active. Future work to identify S. aureus metabolism of specific substrates following sunlight exposure is warranted.
In the sunlit anoxic treatments, fewer genes were identified as differentially expressed. This could be because bacteria in anoxic experiments had been exposed to less overall stress due to the fact that oxygen-mediated photostress was not present in these systems. S. aureus in the anoxic experiments also decayed more slowly and better tracked the cell numbers in the dark controls, further pointing to the anoxic treatment being less stressful than the oxic. However, despite the fact that fewer differentially expressed genes were identified, we would like to stress the fact that, by using true biological replicates and carefully considered metrics of significant expression, the genes identified as differentially expressed are likely those that show the greatest expression changes and are most consistently differentially expressed in the anoxic photostress condition.
In the anoxic condition, two genes were identified as significantly increased in expression: NWMN_2341, coding for a NAD dependent epimerase/dehydratase family protein, and NWMN_1608, coding for a conserved hypothetical protein identified as a probable membrane transporter according to the UniProt database. NWMN_1608 is also the only gene identified as significantly increased in expression during sunlight exposure in both the oxic and anoxic conditions, suggesting its importance for the S. aureus photostress response. The increased expression of a probable membrane transporter could indicate that S. aureus are responding to membrane damage, or that the cells are attempting to increase the removal of toxic species from inside the cell. Cell membrane damage due to sunlight exposure could occur in an anoxic environment due to non-ROS radicals generated from endogenous cell components or direct UV damage of intermembrane proteins (Oppezzo et al., 2001; Kalisvaart, 2004). Our previous work suggests that sunlight exposure in anoxic conditions does lead to increased membrane damage in S. aureus (McClary et al., 2017). Additionally, previous work on the photostress response of E. coli confirmed the importance of efflux pumps in protecting E. coli from critical damage (Al-Jassim et al., 2017).
In conclusion, we have investigated gene expression changes associated with oxic and anoxic photostress in S. aureus in clear oligotrophic seawater. Results suggest that the photostress responses associated with oxygen-mediated and non-oxygen-mediated photoinactivation mechanisms are different from each other. Additionally, the increased expression of hemY in the oxic photostress condition suggests the importance of porphyrin metabolism for combating oxygen-mediated photoinactivation. While further work is needed to confirm that the gene expression changes described here correspond to protein level changes as well, this study helps to identify genes of importance for responding to different types of photostress. In particular, future work should focus on improving our understanding of types and concentrations of endogenous photosensitizers present in bacterial pathogens and fecal indicators, as these appear to play an important role in photoinactivation.
Author Contributions
AB and JM conceived and designed the study; JM wrote the manuscript, conducted experiments, and analyzed the data; AB and JM edited the manuscript; AB supervised the project; AB and JM read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Science Foundation (NSF) grants CBET-1334359. JM was supported by a NSF Graduate Research Fellowship (DGE-114747).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00249/full#supplementary-material
References
Al-Jassim, N., Mantilla-Calderon, D., Wang, T., and Hong, P.-Y. (2017). Inactivation of a virulent wastewater Escherichia coli and non-virulent commensal Escherichia coli DSM1103 strains and their gene expression upon solar irradiation. Environ. Sci. Technol. 51, 3649–3659. doi: 10.1021/acs.est.6b05377
Berney, M., Weilenmann, H.-U., and Egli, T. (2006). Gene expression of Escherichia coli in continuous culture during adaptation to artificial sunlight. Environ. Microbiol. 8, 1635–1647. doi: 10.1111/j.1462-2920.2006.01057.x
Boehm, A. B., Yamahara, K. M., Love, D. C., Peterson, B. M., McNeill, K., and Nelson, K. L. (2009). Covariation and photoinactivation of traditional and novel indicator organisms and human viruses at a sewage-impacted marine beach. Environ. Sci. Technol. 43, 8046–8052. doi: 10.1021/es9015124
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bore, E., Langsrud, S., Langsrud, Ø., Rode, T. M., and Holck, A. (2007). Acid-shock responses in Staphylococcus aureus investigated by global gene expression analysis. Microbiology 153, 2289–2303. doi: 10.1099/mic.0.2007/005942-0
Brezonik, P. L., and Fulkerson-Brekken, J. (1998). Nitrate-induced photolysis in natural waters: controls on concentrations of hydroxyl radical photo-intermediates by natural scavenging agents. Environ. Sci. Technol. 32, 3004–3010. doi: 10.1021/es9802908
Brouwer, A. F., Eisenberg, M. C., Remais, J. V., Collender, P. A., Meza, R., and Eisenberg, J. N. (2016). Modeling biphasic environmental decay of pathogens and implications for risk analysis. Environ. Sci. Technol. 51, 2186–2196. doi: 10.1021/acs.est.6b04030
Cabelli, V. J., Dufour, A. P., McCabe, L. J., and Levin, M. A. (1982). Swimming-associated gastroenteritis and water quality. Am. J. Epidemiol. 115, 606–616. doi: 10.1093/oxfordjournals.aje.a113342
Chaisowwong, W., Kusumoto, A., Hashimoto, M., Harada, T., Maklon, K., and Kawamoto, K. (2012). Physiological characterization of Campylobacter jejuni under cold stresses conditions: its potential for public threat. J. Vet. Med. Sci. 74, 43–50. doi: 10.1292/jvms.11-0305
Charoenca, N., and Fujioka, R. S. (1993). Assessment of Staphylococcus bacteria in Hawaii's marine recreational waters. Water Sci. Technol. 27, 283–289.
Charoenca, N., and Fujioka, R. S. (1995). Association of staphylococcal skin infections and swimming. Water Sci. Technol. 31, 11–17.
Colford, J. M., Wade, T. J., Schiff, K. C., Wright, C. C., Griffith, J. F., Sandhu, S. K., et al. (2007). Water quality indicators and the risk of illness at beaches with nonpoint sources of fecal contamination. Epidemiology 18, 27–35. doi: 10.1097/01.ede.0000249425.32990.b9
Corsi, S. R., Borchardt, M. A., Carvin, R. B., Burch, T. R., Spencer, S. K., Lutz, M. A., et al. (2016). Human and Bovine Viruses and bacteria at three great lakes beaches: environmental variable associations and health risk. Environ. Sci. Technol. 50, 987–995. doi: 10.1021/acs.est.5b04372
Curtis, T. P., Mara, D. D., and Silva, S. A. (1992). Influence of pH, oxygen, and humic substances on ability of sunlight to damage fecal coliforms in waste stabilization pond water. Appl. Environ. Microbiol. 58, 1335–1343.
Dhanjal, S., Singh, A. K., and Cameotra, S. S. (2014). Global gene expression analysis of bacterial stress response to elevated concentrations of toxic metalloids—selenium and arsenic. Geomicrobiol. J. 31, 480–492. doi: 10.1080/01490451.2013.822613
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Ferro, S., Ricchelli, F., Monti, D., Mancini, G., and Jori, G. (2007). Efficient photoinactivation of methicillin-resistant Staphylococcus aureus by a novel porphyrin incorporated into a poly-cationic liposome. Int. J. Biochem. Cell Biol. 39, 1026–1034. doi: 10.1016/j.biocel.2007.02.001
Fingar, V. H., Wieman, T. J., McMahon, K. S., Haydon, P. S., Halling, B. P., Yuhas, D. A., et al. (1997). Photodynamic therapy using a protoporphyrinogen oxidase inhibitor. Cancer Res. 57, 4551–4556.
Fyrestam, J., Bjurshammar, N., Paulsson, E., Johannsen, A., and Östman, C. (2015). Determination of porphyrins in oral bacteria by liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Bioanal. Chem. 407, 7013–7023. doi: 10.1007/s00216-015-8864-2
Geeraerd, A. H., Valdramidis, V. P., and Van Impe, J. F. (2005). GInaFiT, a freeware tool to assess non-log-linear microbial survivor curves. Int. J. Food Microbiol. 102, 95–105. doi: 10.1016/j.ijfoodmicro.2004.11.038
Gentleman, R., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Goodwin, K. D., McNay, M., Cao, Y., Ebentier, D., Madison, M., and Griffith, J. F. (2012). A multi-beach study of Staphylococcus aureus, MRSA, and enterococci in seawater and beach sand. Water Res. 46, 4195–4207. doi: 10.1016/j.watres.2012.04.001
Graham, M. R., Virtaneva, K., Porcella, S. F., Barry, W. T., Gowen, B. B., Johnson, C. R., et al. (2005). Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am. J. Pathol. 166, 455–465. doi: 10.1016/S0002-9440(10)62268-7
Haile, R. W., Witte, J. S., Gold, M., Cressey, R., McGee, C., Millikan, R. C., et al. (1999). The health effects of swimming in ocean water contaminated by storm drain runoff. Epidemiology 10, 355–363. doi: 10.1097/00001648-199907000-00004
Hammes, F., Berney, M., and Egli, T. (2011). Cultivation-independent assessment of bacterial viability. Adv. Biochem. Eng. Biotechnol. 124, 123–150. doi: 10.1007/10_2010_95
Hansson, M., and Hederstedt, L. (1994). Bacillus subtilis HemY is a peripheral membrane protein essential for protoheme IX synthesis which can oxidize coproporphyrinogen III and protoporphyrinogen IX. J. Bacteriol. 176, 5962–5970. doi: 10.1128/jb.176.19.5962-5970.1994
He, C., Post, Y., Dony, J., Edge, T., Patel, M., and Rochfort, Q. (2016). A physical descriptive model for predicting bacteria level variation at a dynamic beach. J. Water Health 14, 617–629. doi: 10.2166/wh.2016.206
Hower, S., Phillips, M. C., Brodsky, M., Dameron, A., Tamargo, M. A., Salazar, N. C., et al. (2013). Clonally related methicillin-resistant Staphylococcus aureus isolated from short-finned pilot whales (Globicephala macrorhynchus), human volunteers, and a bayfront cetacean rehabilitation facility. Microb. Ecol. 65, 1024–1038. doi: 10.1007/s00248-013-0178-3
Jori, G., and Brown, S. B. (2004). Photosensitized inactivation of microorganisms. Photochem. Photobiol. Sci. 3, 403–405. doi: 10.1039/b311904c
Kalisvaart, B. F. (2004). Re-use of wastewater: preventing the recovery of pathogens by using medium-pressure UV lamp technology. Water Sci. Technol. 50, 337–344.
Khlebtsov, B. N., Tuchina, E. S., Khanadeev, V. A., Panfilova, E. V., Petrov, P. O., Tuchin, V. V., et al. (2013). Enhanced photoinactivation of Staphylococcus aureus with nanocomposites containing plasmonic particles and hematoporphyrin. J. Biophotonics 6, 338–351. doi: 10.1002/jbio.201200079
Kohn, T., and Nelson, K. L. (2007). Sunlight-mediated inactivation of MS2 coliphage via exogenous singlet oxygen produced by sensitizers in natural waters. Environ. Sci. Technol. 41, 192–197. doi: 10.1021/es061716i
Lawrence, M., Huber, W., Pagès, H., Aboyoun, P., Carlson, M., Gentleman, R., et al. (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9:e1003118. doi: 10.1371/journal.pcbi.1003118
Levin-Edens, E., Soge, O. O., No, D., Stiffarm, A., Meschke, J. S., and Roberts, M. C. (2012). Methicillin-resistant Staphylococcus aureus from Northwest marine and freshwater recreational beaches. FEMS Microbiol. Ecol. 79, 412–420. doi: 10.1111/j.1574-6941.2011.01229.x
Liu, Y., Wang, C., Tyrrell, G., Hrudey, S. E., and Li, X. F. (2009). Induction of Escherichia coli O157:H7 into the viable but non-culturable state by chloraminated water and river water, and subsequent resuscitation. Environ. Microbiol. Rep. 1, 155–161. doi: 10.1111/j.1758-2229.2009.00024.x
Lobo, S. A., Scott, A., Videira, M. A., Winpenny, D., Gardner, M., Palmer, M. J., et al. (2015). Staphylococcus aureus haem biosynthesis: characterisation of the enzymes involved in final steps of the pathway. Mol. Microbiol. 97, 472–487. doi: 10.1111/mmi.13041
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–34. doi: 10.1186/s13059-014-0550-8
Maraccini, P. A., Mattioli, M. C. M., Sassoubre, L. M., Cao, Y., Griffith, J. F., Ervin, J. S., et al. (2016a). Solar inactivation of enterococci and Escherichia coli in natural waters: effect of water absorbance and depth. Environ. Sci. Technol. 50, 5068–5076. doi: 10.1021/acs.est.6b00505
Maraccini, P. A., Wang, D., McClary, J. S., and Boehm, A. B. (2015). Growth-dependent photoinactivation kinetics of Enterococcus faecalis. J. Appl. Microbiol. 118, 1226–1237. doi: 10.1111/jam.12773
Maraccini, P. A., Wenk, J., and Boehm, A. B. (2016b). Photoinactivation of eight health-relevant bacterial species: determining the importance of the exogenous indirect mechanism. Environ. Sci. Technol. 50, 5050–5059. doi: 10.1021/acs.est.6b00074
McClary, J. S., Sassoubre, L. M., and Boehm, A. B. (2017). Staphylococcus aureus strain newman photoinactivation and cellular response to sunlight exposure. Appl. Environ. Microbiol. 83, 1–14. doi: 10.1128/AEM.01052-17
Nakonieczna, J., Kossakowska-Zwierucho, M., Filipiak, M., Hewelt-Belka, W., Grinholc, M., and Bielawski, K. P. (2016). Photoinactivation of Staphylococcus aureus using protoporphyrin IX: the role of haem-regulated transporter HrtA. Appl. Microbiol. Biotechnol. 100, 1393–1405. doi: 10.1007/s00253-015-7145-5
National Resources Defense Council (2012). Testing the Waters: A Guide to Water Quality at Vacation Beaches. New York, NY.
Nitzan, Y., and Kauffman, M. (1999). Endogenous porphyrin production in bacteria by δ-aminolaevulinic acid and subsequent bacterial photoeradication. Lasers Med. Sci. 14, 269–277. doi: 10.1007/s101030050094
Oppezzo, O. J., and Pizarro, R. A. (2001). Sublethal effects of ultraviolet A radiation on Enterobacter cloacae. J. Photochem. Photobiol. B Biol. 62, 158–165. doi: 10.1016/S1011-1344(01)00180-4
Parker, K. M., Pignatello, J. J., and Mitch, W. A. (2013). Influence of ionic strength on triplet-state natural organic matter loss by energy transfer and electron transfer pathways. Environ. Sci. Technol. 47, 10987–10994. doi: 10.1021/es401900j
Pasquaroli, S., Zandri, G., Vignaroli, C., Vuotto, C., Donelli, G., and Biavasco, F. (2013). Antibiotic pressure can induce the viable but non-culturable state in Staphylococcus aureus growing in biofilms. J. Antimicrob. Chemother. 68, 1812–1817. doi: 10.1093/jac/dkt086
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 2002–2007. doi: 10.1093/nar/29.9.e45
Pinto, D., Santos, M. A., and Chambel, L. (2015). Thirty years of viable but nonculturable state research: unsolved molecular mechanisms. Crit. Rev. Microbiol. 41, 61–76. doi: 10.3109/1040841X.2013.794127
Prüss, A. (1998). Review of epidemiological studies on health effects from exposure to recreational water. Int. J. Epidemiol. 27, 1–9. doi: 10.1093/ije/27.1.1
Qiao, Z., and Wigginton, K. R. (2016). Direct and indirect photochemical reactions in viral RNA measured with RT-qPCR and mass spectrometry. Environ. Sci. Technol. 50, 13371–13379. doi: 10.1021/acs.est.6b04281
Ramamurthy, T., Ghosh, A., Pazhani, G. P., and Shinoda, S. (2014). Current perspectives on Viable but Non-Culturable (VBNC) pathogenic bacteria. Front. Public Heal. 2:103. doi: 10.3389/fpubh.2014.00103
Rastogi, R. P., Richa Kumar, A., Tyagi, M. B., and Sinha, R. P. (2010). Molecular mechanisms of ultraviolet radiation-induced dna damage and repair. J. Nucleic Acids 2010, 1–32. doi: 10.4061/2010/592980
Richardson, D. J., Berks, B. C., Russell, D. A., Spiro, S., and Taylor, C. J. (2001). Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cell. Mol. Life Sci. 58, 165–178. doi: 10.1007/PL00000845
Russell, T. L., Sassoubre, L. M., Wang, D., and Boehm, A. B. (2013). A coupled modeling and molecular biology approach to microbial source tracking at cowell beach, santa cruz, CA, United States. Environ. Sci. Technol. 47, 10231–10239. doi: 10.1021/es402303w
Sabbahi, S., Alouini, Z., Jemli, M., and Boudabbous, A. (2008). The role of reactive oxygen species in Staphylococcus aureus photoinactivation by methylene blue. Water Sci. Technol. 58, 1047–1054. doi: 10.2166/wst.2008.471
Sassoubre, L. M., Nelson, K. L., and Boehm, A. B. (2012). Mechanisms for photoinactivation of Enterococcus faecalis in seawater. Appl. Environ. Microbiol. 78, 7776–7785. doi: 10.1128/AEM.02375-12
Sassoubre, L. M., Ramsey, M. M., Gilmore, M. S., and Boehm, A. B. (2014). Transcriptional response of Enterococcus faecalis to sunlight. J. Photochem. Photobiol. B Biol. 130, 349–356. doi: 10.1016/j.jphotobiol.2013.12.013
Sassoubre, L. M., Yamahara, K. M., and Boehm, A. B. (2015). Temporal stability of the microbial community in sewage-polluted seawater exposed to natural sunlight cycles and marine microbiota. Appl. Environ. Microbiol. 81, 2107–2116. doi: 10.1128/AEM.03950-14
Sekulovic, O., and Fortier, L. C. (2015). Global transcriptional response of Clostridium difficile carrying the ϕCD38-2 prophage. Appl. Environ. Microbiol. 81, 1364–1374. doi: 10.1128/AEM.03656-14
Sinha, R. P., and Häder, D.-P. (2002). UV-induced DNA damage and repair: a review. Photochem. Photobiol. Sci. 1, 225–236. doi: 10.1039/b201230h
Sinigalliano, C. D., Fleisher, J. M., Gidley, M. L., Solo-Gabriele, H. M., Shibata, T., Plano, L. R., et al. (2010). Traditional and molecular analyses for fecal indicator bacteria in non-point source subtropical recreational marine waters. Water Res. 44, 3763–3772. doi: 10.1016/j.watres.2010.04.026
Song, Y., Rundberget, J. T., Evenseth, L. M., Xie, L., Gomes, T., Høgåsen, T., et al. (2016). Whole-organism transcriptomic analysis provides mechanistic insight into the acute toxicity of emamectin benzoate in Daphnia magna. Environ. Sci. Technol. 50, 11994–12003. doi: 10.1021/acs.est.6b03456
Stasiewicz, M. J., Wiedmann, M., and Bergholz, T. M. (2011). The transcriptional response of Listeria monocytogenes during adaptation to growth on lactate and diacetate includes synergistic changes that increase fermentative Acetoin production. Appl. Environ. Microbiol. 77, 5294–5306. doi: 10.1128/AEM.02976-10
Sun, P., Tyree, C., and Huang, C.-H. (2016). Inactivation of E. coli, Bacteriophage MS2 and Bacillus Spores under UV/H2O2 and UV/Peroxydisulfate Advanced Disinfection Conditions. Environ. Sci. Technol. 50, 4448–4458. doi: 10.1021/acs.est.5b06097
Surdel, M. C., Horvath, D. J., Lojek, L. J., Fullen, A. R., Simpson, J., Dutter, B. F., et al. (2017). Antibacterial photosensitization through activation of coproporphyrinogen oxidase. Proc. Natl. Acad. Sci. U.S.A. 114, E6652–E6659. doi: 10.1073/pnas.1700469114
Swenson, P. A., and Setlow, R. B. (1964). β-Galactosidase: inactivation of its messenger RNA by ultraviolet irradiation. Science 146, 791–795. doi: 10.1126/science.146.3645.791
Thoe, W., Gold, M., Griesbach, A., Grimmer, M., Taggart, M. L., and Boehm, A. B. (2015). Sunny with a chance of gastroenteritis: predicting swimmer risk at California beaches. Environ. Sci. Technol. 49, 423–431. doi: 10.1021/es504701j
Tryland, I., Braathen, H., Wennberg, A. C., Eregno, F., and Beschorner, A.-L. (2016). Monitoring of β -D-Galactosidase activity as a surrogate parameter for rapid detection of sewage contamination in urban recreational water. Water 8:65. doi: 10.3390/w8020065
Voyich, J. M., Braughton, K. R., Sturdevant, D. E., Whitney, A. R., Saïd-Salim, B., Porcella, S. F., et al. (2005). Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175, 3907–3919. doi: 10.4049/jimmunol.175.6.3907
Wade, T. J., Calderon, R. L., Brenner, K. P., Sams, E., Beach, M., Haugland, R., et al. (2008). High sensitivity of children to swimming-associated gastrointestinal illness. Epidemiology 19, 375–383. doi: 10.1097/EDE.0b013e318169cc87
Wang, D., Yu, L., Xiang, H., Fan, J., He, L., Guo, N., et al. (2008). Global transcriptional profiles of Staphylococcus aureus treated with berberine chloride. FEMS Microbiol. Lett. 279, 217–225. doi: 10.1111/j.1574-6968.2007.01031.x
Yau, V., Wade, T. J., de Wilde, C. K., and Colford, J. M. Jr. (2009). Skin-related symptoms following exposure to recreational water: a systematic review and meta-analysis. Water Qual. Exp. Heal. 1, 79–103. doi: 10.1007/s12403-009-0012-9
Keywords: Staphylococcus, sunlight, photoinactivation, transcription, RNA, sequencing
Citation: McClary JS and Boehm AB (2018) Transcriptional Response of Staphylococcus aureus to Sunlight in Oxic and Anoxic Conditions. Front. Microbiol. 9:249. doi: 10.3389/fmicb.2018.00249
Received: 28 November 2017; Accepted: 31 January 2018;
Published: 23 February 2018.
Edited by:
Peiying Hong, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Sebastien P. Faucher, McGill University, CanadaBin Cao, Nanyang Technological University, Singapore
Copyright © 2018 McClary and Boehm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandria B. Boehm, YWJvZWhtQHN0YW5mb3JkLmVkdQ==