 Katja Kriebel
Katja Kriebel Cathleen Hieke1
Cathleen Hieke1 Masanobu Nakata
Masanobu Nakata Bernd Kreikemeyer
Bernd Kreikemeyer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 30 January 2018
Sec. Infectious Agents and Disease
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.00053
This article is part of the Research TopicBacterial Colonization: Bystander Microbiota or the Initial Step of Chronic Disease?View all 15 articles
A wide range of bacterial species are harbored in the oral cavity, with the resulting complex network of interactions between the microbiome and host contributing to physiological as well as pathological conditions at both local and systemic levels. Bacterial communities inhabit the oral cavity as primary niches in a symbiotic manner and form dental biofilm in a stepwise process. However, excessive formation of biofilm in combination with a corresponding deregulated immune response leads to intra-oral diseases, such as dental caries, gingivitis, and periodontitis. Moreover, oral commensal bacteria, which are classified as so-called “pathobionts” according to a now widely accepted terminology, were recently shown to be present in extra-oral lesions with distinct bacterial species found to be involved in the onset of various pathophysiological conditions, including cancer, atherosclerosis, chronic infective endocarditis, and rheumatoid arthritis. The present review focuses on oral pathobionts as commensal and healthy members of oral biofilms that can turn into initiators of disease. We will shed light on the processes involved in dental biofilm formation and also provide an overview of the interactions of P. gingivalis, as one of the most prominent oral pathobionts, with host cells, including epithelial cells, phagocytes, and dental stem cells present in dental tissues. Notably, a previously unknown interaction of P. gingivalis bacteria with human stem cells that has impact on human immune response is discussed. In addition to this very specific interaction, the present review summarizes current knowledge regarding the immunomodulatory effect of P. gingivalis and other oral pathobionts, members of the oral microbiome, that pave the way for systemic and chronic diseases, thereby showing a link between periodontitis and rheumatoid arthritis.
The oral cavity is a unique habitat that allows for colonization of a wide variety of commensal microbial species, as it supplies a diversified nutrient influx as well as high humidity and variable oxygen concentrations. Furthermore, the existence of soft (gingiva) and non-shedding hard (teeth) tissues provides microorganisms with potential surfaces for adherence and subsequent interaction with various host cells. Colonization of the oral cavity in healthy individuals is based on balanced bacteria-host and interbacterial interactions. The continuous existence of dental plaques in gingival tissues and interactions of pathobionts with host cells cause inflammation, leading to periodontitis (PD). There is also increasing evidence suggesting an association of chronic PD with other types of systemic inflammatory diseases, such as atherosclerosis, infective endocarditis, diabetes, adverse pregnancy outcome, respiratory diseases, and rheumatoid arthritis (RA) (Li et al., 2000; Pihlstrom et al., 2005; Kim and Amar, 2006; Gaffen et al., 2014; Hajishengallis, 2015). This raises the question whether the periodontal microbiota is bystander or responsible for the initial step of chronic diseases. In the present review, the pathogenic mechanism of PD is introduced from the perspective of host bacteria/interbacterial interactions and host immune responses. Moreover, interactions of the pathobiont Porphyromonas gingivalis with host cells, as well as a possible link between the pathobiont and RA are discussed (Figure 1).
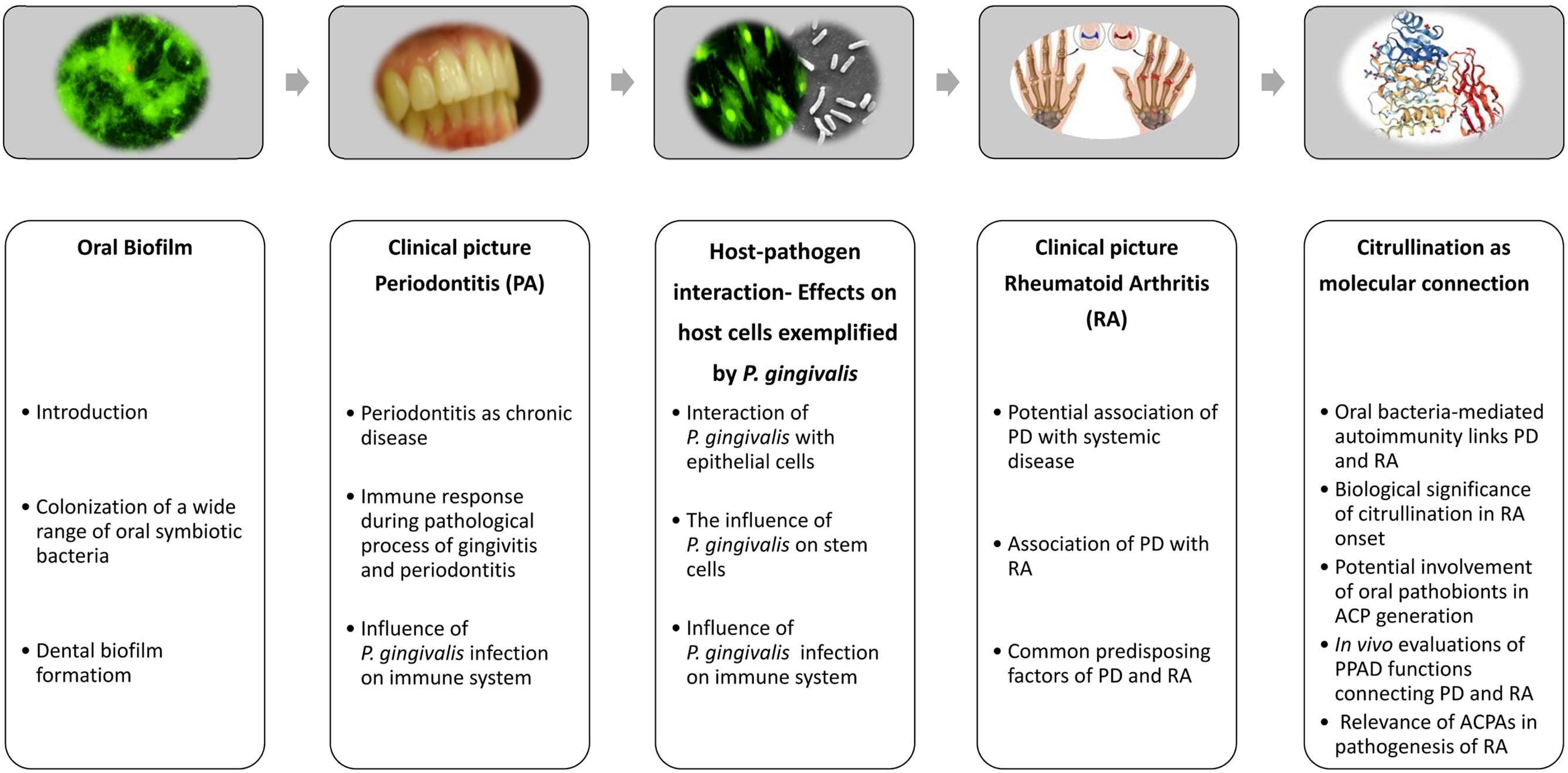
FIGURE 1. Brief overview of current concepts regarding onset of periodontitis and rheumatic arthritis, and deduced causal relationships between both diseases. After establishing a subgingival biofilm, oral pathobionts, including Porphyromonas gingivalis, induce periodontitis as a chronic disease, which is attributable to host-pathobiont interactions and deleterious host immune responses in periodontal tissues. Dysregulated citrullination caused by the pathobiont Porphyromonas gingivalis has been suspected to be a causative factor for onset of rheumatic arthritis. Parts of this figure were taken from freely available web resources: https://www.chirurgie-portal.de/innere-medizin/rheuma.html; www.rcsb.org/pdb/ngl/ngl.do?pdbid=5AK7 (Rosenstein and Hildebrand, 2015; Montgomery et al., 2016).
Initial bacterial host colonization occurs at birth, with Staphylococcus epidermidis and Streptococcus species detected within hours after birth (Nelson-Filho et al., 2013). The oral pioneer species Streptococcus salivarius, which has been detected within 8 h after birth (Rotimi and Duerden, 1981), represents the majority of oral bacteria with up to 98% of examined subjects showing its possession at the first tooth eruption (Cortelli et al., 2008). Dental structures and alterations in nutrition allow for further colonization of other bacterial species. Finally, the matured oral microbiome consists of hundreds of bacterial species, contributing to a complex community (Aas et al., 2005; Dewhirst et al., 2010). Gram-positive facultative anaerobic bacteria, such as the Streptococcus and Actinomyces genera, are predominant in healthy individuals, in whom a proper equilibrium between the oral microbiome and host immune responses is maintained with no signs of inflammation observed in the periodontium (Li et al., 2004; Jiao et al., 2014).
After tooth surfaces are cleaned, their immersion in the fluid environment of the oral cavity causes surface adsorption of a thin acquired pellicle, which is mainly composed of saliva glycoproteins, such as proline-rich proteins, α-amylase, statherin, mucins, and agglutinin (Heller et al., 2017). Coating of those solid surfaces with a pellicle leads to changes in surface charge and free energy, thus promoting bacterial adhesion (Weerkamp et al., 1988). Bacteria attach to tooth surfaces in a diverse manner, ranging from specific interactions between pellicle components and bacterial surface molecules to charge-mediated weak interactions (Nesbitt et al., 1992; Jenkinson, 1994; Oli et al., 2006; Kolenbrander et al., 2010). The predominant initial colonizers of teeth are Gram-positive facultative anaerobic cocci and rods, including Streptococcus and Actinomyces species. These initial colonizers provide a foundation for further development of dental biofilm. Streptococcus recognizes components in the pellicle, such as a specific interaction between a pilus protein of S. sanguinis and salivary α-amylase (Okahashi et al., 2011). Actinomyces binds to proline-rich proteins and statherin, a phosphate-containing protein (Li et al., 2001). Once the initial colonizers attach to the surface, a biofilm mass develops through continued growth and subsequent adsorption of other bacterial species via coaggregation.
The surface molecules of these early colonizers allow for coaggregation of Gram-negative bacteria possessing a lower level of adherence to the pellicle, including members of the genera Veillonella and Fusobacterium. Bacteria belonging to the genus Fusobacterium, such as Fusobacterium nucleatum, are able to coaggregate with both initial and late colonizers, thus are called bridge species and known to promote successful development of dental biofilm. For bridging neighboring bacteria, F. nucleatum utilizes surface molecules such as RadD, an arginine-inhibitable adhesin, and the fusobacterial apoptosis protein Fap2 (Kaplan et al., 2010). Habitat analysis of the oral microbiome has suggested that the genus Corynebacterium is strikingly specific for supragingival and subgingival plaque, with Corynebacterium matruchotii dominant among six species deposited in the Human Oral Microbiome Database (Dewhirst et al., 2010). Since this genus has been found in only trace amounts in saliva and other specimens from different anatomical sites, it is considered to have a specific role in dental biofilm formation. In fact, in a study that utilized combinational labeling and spectral imaging FISH (CLASI-FISH), Mark Welch et al. (2016) observed a complex microbial consortium, termed a hedgehog structure, mainly consisting of nine taxa arranged in an organized spatial framework, including Corynebacterium, Streptococcus, Porphyromonas, Haemophilus/Aggregatibacter, Neisseriaceae, Fusobacterium, Leptotrichia, Capnocytophaga, and Actinomyces. This plaque hedgehog represents a radially organized structure, of which the main framework is primarily composed of Corynebacterium with a multi-taxon filament-rich annulus and peripheral corncob structures. In the corn-cob structures, Corynebacterium filaments are surrounded primarily by Streptococcus, though Porphyromonas and Haemophilus/Aggregatibacter are also in close contact with streptococcal cells, while the filament-rich annulus is mainly composed of Fusobacterium, Leptotrichia, and Capnocytophaga. Thus, Corynebacterium organisms are considered to be bridge-species in regard to biofilm formation. Bridge-species further coaggregate late colonizers that have effects on PD pathogenesis.
Each anatomical site in the oral cavity possesses a distinct composition of biofilm members that affects the local environment by intrinsic metabolism. Stratification and selective interaction between distinct bacterial species in dental biofilms are conducted by mutually antagonistic and cooperative interactions, which are attributable to environmental/metabolite gradients and quorum sensing (Brown and Whiteley, 2007; Ramsey et al., 2011; Zhu and Kreth, 2012; Wessel et al., 2014). Tooth-related plaque biofilm can be generally classified based on location into supragingival, formed above the gingival margin, and subgingival, formed below the gingival margin. When a pathological dental pocket becomes formed between a tooth surface and gingiva during the course of PD onset, an anaerobic condition is built up. Moreover, major sources of nutrition for subgingival plaque bacteria are provided via inflammatory periodontal tissues and gingival cervical fluid originating in blood, since permeation of saliva components is more or less limited. Consequentially, subgingival plaque in the pocket is dominated by anaerobic and motile bacteria as compared with supragingival plaque, as noted in detail below. Interactions of obligate anaerobic bacteria, such as P. gingivalis, with host cells have been implicated in the pathogenesis of PD.
Establishment and maturation of periodontal dental biofilms are characterized by co-aggregation of opportunistic microorganisms caused by diverse factors, including poor oral hygiene. Infection of periodontal host cells as well as expression of virulence factors can provoke a local inflammatory response. Initial periodontal tissue inflammation is termed gingivitis and its pathology can be resolved by removal of dental biofilms (Figure 2). On the other hand, continuous existence of stable plaques, including accumulation of opportunistic bacterial species, supports long-lasting inflammation. A shift in the periodontal microbiome that accompanies an increase in Gram-negative anaerobic species is now accepted as an indicator of periodontal disease (Yano-Higuchi et al., 2000; Klein and Goncalves, 2003; Yang et al., 2004; Berezow and Darveau, 2011; He et al., 2015). Such a shift in composition affects host immune responses, and leads to dysbiosis between the oral microbiota and the host (Hajishengallis and Lamont, 2012). Therefore, following establishment of gingivitis, PD develops as a chronic inflammatory condition.
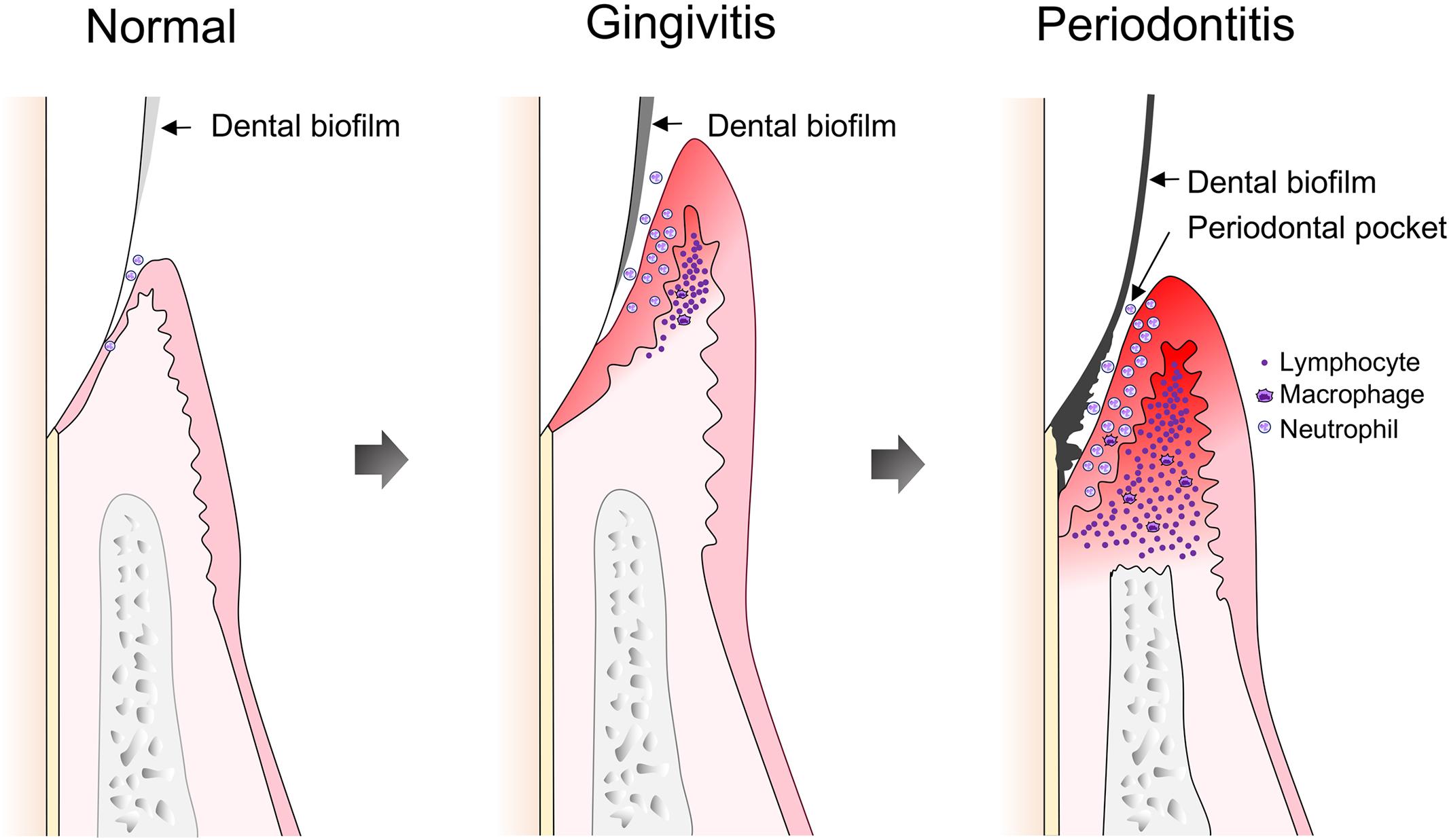
FIGURE 2. Development of gingivitis and periodontitis. Following dental plaque accumulation, neutrophils dominate the host immune response, accompanied by progression of an early or stable gingivitis lesion, along with increased infiltration of macrophages and T cells. The gingivitis lesion develops into a periodontitis lesion, which is characterized by formation of a pathogenic periodontal pocket and destruction of periodontal tissues. Infiltrated lymphocytes are dominated by B and plasma cells.
Periodontitis is characterized by irreversible and progressive degradation of periodontal tissues. With continuous inflammation, proliferation of epithelial cells connecting tooth surfaces and gingival tissues causes detachment of the cell layer, and subsequent formation of a pathogenic dental pocket between teeth and gingival tissues (Figure 2). The resulting micro-environment is characterized by reduced oxygen concentration or even anoxic areas. Mettraux et al. (1984) quantified oxygen concentrations in the periodontal pockets of patients with untreated PD and found them to range from 0.7 to 3.5%. On the other hand, the progression and severity of PD are strongly dependent on the quality and quantity of microorganisms harbored in periodontal plaque, as well as individual risk factors, e.g., age, genetic predisposition, systemic disorders, and lifestyle aspects, such as dental hygiene, nutrition, and smoking (Pihlstrom et al., 2005; van Dyke and Sheilesh, 2005; Hajishengallis and Lamont, 2012; Heaton and Dietrich, 2012; AlJehani, 2014).
Approximately 90% of microorganisms isolated from periodontal pockets are strictly anaerobic (Slots, 1977; Uematsu and Hoshino, 1992) and certain sets of bacteria have been frequently detected at elevated levels in periodontal lesions as compared with healthy tissues. Socransky et al. (1998) analyzed distribution of approximately 40 species in subgingival plaque using a DNA–DNA hybridization technique. Findings from DNA cluster analysis indicated that typical co-colonization of specific oral species, among which a cluster with the nomenclature “red complex” composed of the Gram-negative anaerobic species Tannerella forsythia, P. gingivalis, and Treponema denticola, is associated with increased pocket depth and bleeding upon clinical pocket probing, while the other four clusters examined were not shown to be associated with clinical parameters indicating periodontal disease. This pattern of oral colonization was also confirmed to exist in supragingival plaque samples (Haffajee et al., 2008). Aas et al. (2005) also identified the three bacterial species of the red complex as highly associated with disease status, which confirmed the colonization model by Socransky et al. (1998), and those findings were later supported by other studies (Dewhirst et al., 2010; Zarco et al., 2012; Wade, 2013; Duran-Pinedo and Frias-Lopez, 2015). Presently, the association of particular bacterial species within an intricate microbial community with periodontal health status is widely accepted.
Accumulation of opportunistic bacteria in periodontal plaques and their deleterious effects on host tissues via specific virulence factors provoke host immune responses. In response to microbial challenge, a massive cytokine response occurs, which triggers activation and recruitment of polymorphonuclear leukocytes (PMNs) in periodontal pockets (Figure 2). Their activation and oxidative burst contribute to periodontal homeostasis damage and subsequent degradation of periodontal tissues (Waddington et al., 2000; Kantarci et al., 2003; Graves, 2008). As compared to healthy individuals, the number of PMNs is increased in both periodontal pockets and the bloodstream of patients with chronic PD (Lakschevitz et al., 2013; Kolte et al., 2014), thus sustaining inflammation.
The concept of chronic PD as an immunological disease, which was proposed more than 40 years ago (Seymour et al., 1979), implies that a primary etiologic factor is bacterial infection that elicits a specific immune response by the host, triggering gingival inflammation and progression to chronic PD. Over the past 20 years, a number of studies have investigated and defined immune system components contributing to its pathogenesis. For example, it has been shown that both innate and adaptive immune systems are involved in PD onset, in which the roles of T- and B-lymphocytes are likely to be equally crucial (Gonzales, 2015). However, in regard to polarization of T-helper (Th) cell response, it remains elusive whether PD pathogenesis is driven by Th1, Th2, or Th17, or what role is adopted by regulatory T cells (Tregs) (Carvalho-Filho et al., 2016).
A plausible model for the pathological process of PD has been suggested, based on histopathological examinations of PD tissue sections. A pathological condition develops in sequential order and PD progression is subdivided into various stages, starting with initial lesion formation during the first 4 days after plaque accumulation. PMNs, i.e., neutrophils, dominate the host immune response, accompanied by the activation of complement component C3 via an alternative pathway. Subsequent production of the anaphylatoxins C3a and C5a leads to activation of mast cells with release of vasoactive substances that facilitate vascular permeability and development of edema. Moreover, mast cells release TNF-α, which up-regulates the expression of adhesion molecules on endothelial cells, allowing for increased PMN infiltration (Ohlrich et al., 2009). After approximately 4–7 days of plaque accumulation, the initial lesion progresses to an early or stable gingivitis lesion with increasing infiltration of macrophages and lymphocytes (Figure 2). Lymphocytes are predominantly T cells with a CD4-positive to CD8-positive ratio as high as 2:1, an activated phenotype that is at this point negative for the IL-2 receptor CD25. Since absence of CD25 indicates that T cells have proliferated elsewhere, characteristics of the early lesion indicate a delayed-type hypersensitivity reaction (DTH). The pathology can be stable for a certain period with equilibrium maintained between the immune system and microbiota, and inflammation confined to the gingiva. In cases when plaque is mechanically removed, the lesion will reversibly recover at this stage. However, if plaque accumulation is allowed to continue, and attachment between the gingiva epithelium and tooth surface is progressively lost, the stable lesion advances to an established or progressive PD lesion, characterized by a predominant response of B cells and plasma cells, high levels of IL-1 and IL-6, and periodontal tissue destruction, including alveolar bone loss (Ohlrich et al., 2009). The final stage, an advanced lesion, is also characterized by a dominance of B and plasma cells, while inflammatory status is exacerbated. Fibroblasts stimulated by IL-1β, TNF-α, and prostaglandin E2 secrete matrix metalloproteases (MMPs) that not only advance the lesion, but also accelerate bone loss (Figure 2). Palliative treatment of PD and complete removal of bacterial plaque improves the course of periodontitis and leads to arrest of the irreversible destruction of periodontal tissues.
Recent studies have shown the critical role of Th17 in maintenance of oral tissues. Individuals with a genetic defect in Th17 differentiation are susceptible to oral fungal infections (Liu et al., 2011; Moutsopoulos et al., 2015) and excess Th17 response in gingiva promotes inflammation, leading to deterioration related to periodontitis pathology (Eskan et al., 2012; Moutsopoulos et al., 2014). Dutzan et al. (2017) showed that the population of gingival IL-17-producing CD4+ T cells increases with age. Interestingly, Th17 responses are not dependent on colonization of commensal bacteria, which is totally different from those in the mucosa of other anatomical sites. Moreover, accumulation of gingival Th17 cells is dependent on physiological mechanical damage caused by mastication and subsequent induction of IL-6-mediated signals. Thus, mastication, a normal function of the oral cavity, shapes gingival immune homeostasis.
Even though the above sequence of events leading to chronic PD is feasible, it does not explain why the pathophysiological condition of an early lesion remains stable or even resolves in some individuals, while it progresses to B cell-driven progressive stages in others. The transition from a T cell- to B cell-rich lesion has been suggested to correlate with the transition from a Th1- to Th2-dominated response. Indeed, the pathological condition of chronic PD represents a pathology dominated by Th2 (Kinane and Bartold, 2007). Future research will be needed to investigate the involvement of Th17 and Treg cells, as well as the impact of environmental and genetic factors on susceptibility to chronic PD, and the underlying mechanisms for onset of PD-related systemic diseases.
The periodontal pathogen P. gingivalis infects gingival epithelial cells in the oral cavity, and its in vitro adherence to and internalization of epithelial cells have been well investigated. Dogan et al. (2000) observed invasion of primary epithelial cells by P. gingivalis, though it has been noted that the quantity of adherence and invasion of P. gingivalis are dependent on which human cell types and bacterial strains are investigated (Deshpande et al., 1998). For example, adherence rates of strain A7436 isolated from refractory PD to KB oral epithelial and human umbilical vein endothelial cells were found to be 1.1% and 0.5%, respectively (Deshpande et al., 1998). When comparing diverse P. gingivalis strains, the adherence rates vary, such as 0.5% for type strain W50 and 10.5% for strain 33277 to KB cells (Duncan et al., 1993; Dorn et al., 2000), while adhesion capacity also varies between cell types due to divergent interactions between P. gingivalis and the intrinsic cell surface. Furthermore, Saito et al. (2008) reported that strain ATCC 33 invaded Ca9-22 gingival epithelial cells as well as human aorta endothelial cells (HAEC) at higher rates as compared to strain W83, which might be explained by the highly fimbriated phenotype of strain ATCC33. Addition of Fusobacterium nucleatum strain TDC100 to that culture system increased the number of invaded bacteria for both strains (Saito et al., 2008). Pinnock et al. (2014) examined survival and bacterial release of P. gingivalis in a 3-D organotypic oral mucosal model, which was shown to mimic in vivo conditions. They found an increase of intracellular survival and bacterial release during incubation, as compared to monolayer experiments (Pinnock et al., 2014). These reports demonstrate that the interaction of epithelial cells with bacteria is dependent on a wide range of factors, while the high complexity of the oral cavity is not well represented by a monolayer cell culture system.
In addition to the composition of cells grown in various cell culture systems, host cell response itself is crucial for host signaling cascades. Toll-like receptors (TLRs) are pattern recognition receptors of epithelial cells as well as immune cells to recognize microbial molecules (Sugawara et al., 2006), which are involved in both intracellular and extracellular signaling pathways, culminating in activation of innate immune responses (Muzio and Mantovani, 2000). TLR2 and TLR4 respond to various bacterial factors, including lipoteichoic acid, lipopeptides, and lipopolysaccharide (LPS), and changes in their expression in gingival tissue during chronic PD have been demonstrated (Promsudthi et al., 2014). As an immunomodulation factor, P. gingivalis has effects on miRNA expression in host cells. miRNAs are single-stranded non-coding RNAs involved in regulatory processes, such as mRNA degradation and translational repression, and modulation of their expression results in dysregulation of proliferation and host cell immune responses (O’Connell et al., 2007; Aberdam et al., 2008). Gingival human oral keratinocytes incubated with heat-inactivated P. gingivalis exhibited up-regulation of miRNA-105, which is complementary to TLR2 mRNA (Benakanakere et al., 2009). Moreover, infection of primary gingival epithelial cells with viable P. gingivalis organisms was shown to significantly alter the expression of 14 miRNAs involved in regulation of apoptosis and cytokine secretion (Moffatt and Lamont, 2011). Overall, the oral cavity represents a complex network of numerous bacterial and/or host interactions, which can be disturbed by P. gingivalis via its utilization of epithelial cells to support its own survival.
The interactions of oral pathogens with differentiated cells, such as epithelial and bone cells, as well as stem cells and fibroblasts have been investigated. Stem cells can be isolated from various adult tissues, including bone marrow and gingiva (Barry and Murphy, 2004; Sonoyama et al., 2006; Tavian et al., 2006; Zhang et al., 2009; Jin et al., 2013). The source of human dental stem cells (hDSCs) is located in oral tissues and those exhibit the main characteristics of mesenchymal stem cells. hDSCs can be isolated from dental pulp and exfoliated deciduous teeth, as well as apical papilla, periodontal ligament, and dental follicle specimens (Gronthos et al., 2000; Miura et al., 2003; Seo et al., 2004; Jo et al., 2007; Sonoyama et al., 2008). The prominent presence of hDSCs in oral tissues provokes intriguing questions regarding whether P. gingivalis is able to interact with stem cells in tissues and, if so, what subsequent effects should be expected. The effect of the outer membrane component LPS of P. gingivalis on stem cells in regard to cell proliferation, viability, differentiating capacity, and immunomodulatory characteristics has been evaluated (Mysak et al., 2014; Chatzivasileiou et al., 2015; How et al., 2016), though the interaction of viable bacteria with stem cells remains poorly defined. Kriebel et al. (2013) demonstrated that stem cells and oral bacteria can be co-cultured under anaerobic conditions. In their system, oral microorganisms were less able to adhere to or internalize into human bone marrow stem cells (hBMSCs) in relation to gingival epithelial cells (Kriebel et al., 2013). Thereafter, additional studies revealed that human dental follicle stem cells (hDFSCs) elicit a reduced pro-inflammatory response following bacterial infection, as compared to differentiated cells (Biedermann et al., 2014). Furthermore, Kriebel et al. (2013) and Biedermann et al. (2014) showed that stem cell functions were influenced by oral bacteria in vitro, while Hieke et al. (2016) found that infection with viable bacteria induced distinct reactions by stem cells that were different from reactions to a single administration of LPS (Hieke et al., 2016). Thus, infected stem cells showed a reduced capacity for migration, though that finding is inconsistent with another study that demonstrated increased migration following stimulation with LPS (Chatzivasileiou et al., 2013). As compared with the analyses with LPS stimulation alone, data obtained in experiments with viable bacteria remain controversial. Additional studies are required to evaluate the reaction of stem cells to bacterial infection in human tissues.
Fibroblasts, the most predominant cell type in periodontal tissue, play important roles in tissue regeneration and PD-associated inflammation. They express TLRs, including TLR2 and 4 (Mahanonda et al., 2007), thus are considered to be involved in immune reactions to oral bacteria. The effects of P. gingivalis LPS on fibroblasts have been examined in regard to cell viability, immune response, and tissue repair, as well as the effects of cell signaling on those factors (Souza et al., 2010; Morandini et al., 2013; Sun et al., 2016). Furthermore, interactions of P. gingivalis with gingival fibroblasts have also been investigated, with the effects of immune modulating bacterial factors, the capsule, and gingipains, together with LPS, noted (O’Brien-Simpson et al., 2009; Brunner et al., 2010; Scheres and Crielaard, 2013). P. gingivalis can adhere to and invade human fibroblasts (Pathirana et al., 2008; Irshad et al., 2012; Zhang et al., 2014), and such bacterial infection induces secretion of pro-inflammatory mediators, including IL-6 and IL-8, via TLR-dependent and -independent pathways (Liu et al., 2014; Palm et al., 2015). In addition, P. gingivalis infection induces caspase-independent apoptosis as well as regulation of the inflammasome activation (Desta and Graves, 2007; Kuo et al., 2016). Such diverse responses of the infected fibroblasts affect biological functions and differentiation of other cell types, implicating their regulatory role in progression of periodontitis and consequent chronic inflammation (Zhang et al., 2014; Tzach-Nahman et al., 2017).
The dental pocket is constantly exposed to oral microorganisms and innate immunity components permanently interact with bacteria. During inflammation, various cell types, including neutrophils and macrophages, migrate to the site of infection, with the former the first line of defense against invading microorganisms. In addition to phagocytosis, exocytosis of granules, release of reactive oxygen species (ROS), and induction of neutrophil extracellular traps (NETs) serve as anti-microbial factors (Segal, 2005). Interestingly, P. gingivalis is able to modify neutrophil activity in a manner that promotes neutrophil survival. It has also been shown that prolongation of neutrophil survival caused by P. gingivalis results in accumulation of neutrophils in adult patients with PD (Gamonal et al., 2003), while later it was reported that neutrophils isolated from the blood of chronic PD patients were highly reactive to stimulation by P. gingivalis LPS, with increased release of the pro-inflammatory cytokine IL-8 (Restaino et al., 2007). Furthermore, neutrophils from patients with localized aggressive PD produce higher levels of ROS against P. gingivalis, as compared with those from healthy donors, and release of ROS results in secretion of pro-inflammatory cytokines, which can be advantageous to counteract the increased burden of P. gingivalis (Damgaard et al., 2016). As a strategy for escape from the host immune system, it has been shown that P. gingivalis has an ability to invade epithelial cells. Moreover, triggering of an immune response can be beneficial for colonizing deeper tissues of the host (Li et al., 2008). The working group of Hajishengallis noted that direct interaction of P. gingivalis with PMNs resulted in modulation of the neutrophil killing function via MyD88, an adaptor protein of TLR2 and TLR4 receptors (Hajishengallis et al., 2008; Maekawa et al., 2014).
Macrophage functions are also modulated by P. gingivalis. Macrophage migration-inhibitory factor (MIF) is involved in killing of bacteria by recruitment and activation of macrophages. Li et al. (2013) demonstrated that P. gingivalis is able to reduce the expression of MIF mRNA in deep-pocket tissues (Li et al., 2013). Furthermore, in vitro experiments demonstrated that treatment of macrophages with the lysine-specific gingipain Kgp impaired its migration to apoptotic neutrophils and reduced the anti-inflammatory effect of apoptotic cells, resulting a rapid inflammatory response, leading the authors to suggest that P. gingivalis promotes chronic inflammation by a gingipain-mediated defect in apoptotic cell clearance and resolution of tissue restoration (Castro et al., 2017).
Following infection of epithelial cells and fibroblasts, P. gingivalis can also indirectly modulate immune cell functions. In vitro experiments showed that P. gingivalis infection of oral epithelial cells inhibits neutrophil migration (Madianos et al., 1997). Also, exposure of live P. gingivalis strain W83 to fibroblasts from periodontal ligaments in vitro induced a reduction in expression of macrophage colony-stimulating factor (Scheres et al., 2009). Those findings demonstrated that immune cell functions are also indirectly influenced by P. gingivalis infection, with bacterial secreted factors potentially a part of this complex system.
Overviews regarding the interactions of various other oral pathogenic bacteria with eukaryotic animal cells and cells from human sources have been presented (Feng and Weinberg, 2006; Kebschull and Papapanou, 2011). In general, P. gingivalis modifies antimicrobial host response and causes an imbalance in immune responses, leading to prolongation of inflammatory status and continuous damage against periodontal tissues. General bacterial burden, variability of colonizing species, oral hygiene, and other individual risk factors have further impact on host immune response and the subsequent outcome of periodontal disease.
Specific oral pathobionts influence related systemic diseases, such as atherosclerosis, infective endocarditis, diabetes, adverse pregnancy outcome, respiratory diseases, and RA, with various hypotheses based on epidemiological and experimental data presented. First, these systemic diseases and PD share common confounding factors, including lifestyle and/or genetic predisposition, indicating the importance of common host backgrounds. In addition, oral dysbiosis may cause autoimmunity via immune response against oral microbiota and subsequent molecular mimicry/autoantibody generation, as reported in cases of RA, in which T cell subsets are shaped toward pro-inflammatory cytokine-producing cells that drive autoimmunity development (Hooper et al., 2012). Since improved prognosis of patients with systemic diseases, such as coronary heart disease and RA, has been demonstrated following treatment for PD, a periodontal immune response and/or plaque bacteria provide a link for the mutual relationship between PD and those diseases (Montebugnoli et al., 2005; Al-Katma et al., 2007; Ortiz et al., 2009). In the following sections, possible relevance to the etiologies of PD and RA is discussed.
A reciprocal relationship between PD and RA has been reported (de Pablo et al., 2009; Koziel et al., 2014), and is also implied by the fact that early Assyrians 2,500 years prior treated rheumatism by tooth extraction. Even though no reliable records regarding the effect of that treatment exist, the concept of a mutual relationship of chronic joint disease with PD has been noted. However, the exact molecular and cellular mechanisms linking PD and RA are only slowly being unraveled. Based on a review of recent literature, reports supporting the various scenarios mentioned above are introduced here.
The first factors linking RA and PD include lifestyle and genetic predisposition as common confounders. Indeed, smoking and aging have been identified as risk factors for both of those diseases (Eriksson et al., 2016). As for a common genetic predisposition, data presented thus far remain inconclusive. While the strongest association for RA has been found among alleles of the HLA-DRB1∗04 and ∗01 haplotype groups, which carry a shared epitope, there is no significant association between HLA class II antigens and PD (Stein et al., 2008). Findings of an epidemiological study indicated that the hypomethylated status of a single CpG in the IL-6 promoter region plays a role in pathogenesis of RA and PD (Ishida et al., 2012). In an investigation of cases with aggressive PD, a large candidate-gene association study found no definite evidence for a genetic link of PD with RA, while their results suggested that IRF5 and PRDM1 are shared susceptibility factors, both of which are involved in interferon-β signaling, and also associated with systemic lupus erythematosus and inflammatory bowel disease (Schaefer et al., 2014).
The second scenario, which has gained enormous attention, is oral dysbiosis as a prerequisite for pathogenic autoimmunity that leads to the onset of RA. Notably, the presence of P. gingivalis in PD lesions has been indicated as a link between both chronic inflammatory diseases (Rosenstein et al., 2004; Mikuls et al., 2009, 2014; Bartold et al., 2010). It was initially speculated that bacterial cell and/or toxin/metabolic byproducts can enter the systemic circulation from a clinically asymptomatic localized lesion containing pathogenic bacteria and spread to discrete anatomical sites, thereby initiating disease (Kumar, 2017). However, neither oral live bacteria nor their toxin/metabolic byproducts have been detected in the focus of the rheumatoid joint, though P. gingivalis DNA has been noted in synovial fluid (Reichert et al., 2013). On the other hand, immunological sequelae associated with oral pathobiont infection have gained considerable attention. These are formations of antibodies against citrullinated peptide antigens (ACPA) that precede the development of RA (Schellekens et al., 1998) and a molecular mimicry of bacterial proteins against host proteins, both of which raise autoantibodies.
Development of RA is attributable to production of ACPAs, the presence of which serves as a potent diagnostic marker for RA (Schellekens et al., 2000). Formation of ACPAs prior to RA onset has been the focus of intense research for the past two decades. While citrullination itself is a physiological process, the formation of antibodies against citrullinated peptide antigens is highly specific for RA (Schellekens et al., 1998). However, this specificity remains enigmatic, and whether these antibodies play an active role in the disease process or simply reflect an ongoing immune response has not been fully elucidated. On the other hand, the finding that citrullinated proteins accumulate in the joint provides a basis for interpreting the pathological condition in patients with RA. The pathology can be characterized as dysregulated citrullination, followed by release of neo-epitopes that breach immunological tolerance and trigger autoantibody formation.
Citrullination is mediated by peptidylarginine deaminase (PAD) enzymes. In humans, there are five different isotypes, PAD1-4, and PAD6, which exhibit a roughly 50–55% sequence similarity, and show distinct distributions in cells and tissues (Vossenaar et al., 2003; Zhang et al., 2004; Bicker and Thompson, 2013). Citrullination is the post-translational hydrolytic conversion of peptidyl-arginine into peptidyl-citrulline via deamination, a process that renders a reduction in the net positive charges of a given protein, thereby leading to increased hydrophobicity, protein unfolding, and altered intra- and inter-molecular interactions (Darrah et al., 2013). Physiologically, citrullination impacts gene regulation, terminal differentiation, and apoptosis, thus dysregulated citrullination is associated with numerous disorders, including autoimmune and neurodegenerative diseases (Witalison et al., 2015). PAD activity under a physiological condition is regulated by calcium concentration and a reducing environment (Arita et al., 2004). While full PAD activity in vitro requires millimolar amounts of calcium ion, intracellular nanomolar concentrations are likely to limit aberrant citrullination. Likewise, the oxidizing nature of the extracellular environment may provide protection from aberrant extracellular citrullination by PADs that may leak from activated or dying cells (Darrah et al., 2013). Of note, the citrullinome in RA is comprised of cytoplasmic and extracellular proteins, suggesting that both compartments are prone to dysregulated PAD activity. The major cell type for intracellular protein citrullination in the RA joint is represented by neutrophils, which are also the major source for soluble PAD2 and PAD4 released into synovial fluid (Romero et al., 2013; Spengler et al., 2015; Konig and Andrade, 2016). An important question then is what triggers hyper-citrullination in neutrophils? Among the various stimuli that trigger neutrophil activation and death, pore forming and membranolytic pathways that involve perforin and the complement membrane attack complex have been shown to induce intracellular calcium fluxes, a transient rise in intracellular calcium concentration and subsequent intracellular hyper-citrullination (Romero et al., 2013). Interestingly, the ability to provoke calcium influx-induced hyper-citrullination in neutrophils is definitely possessed by pore-forming immune mechanisms of the host, though that is also shared by bacterial calcium ionophores and pore-forming toxins (Konig et al., 2016). Due to limited conditions required for enzymatic activity, robust extracellular citrullination can only be maintained by a constant release of soluble PADs from dying cells and the presence of autoantibodies against PADs. Neutrophil NETosis and necrosis, as well as autophagy also contribute to extracellular hyper-citrullination via release of transiently active PAD enzymes (Spengler et al., 2015). Recently, it was shown that the presence of PAD3/PAD4 cross-reactive autoantibodies, which cause a decrease in calcium concentration required for catalysis, is associated with most erosive disease courses (Darrah et al., 2013; Navarro-Millán et al., 2016). As a consequence of hyper-citrullination, generation of neo-epitopes induced by changes in protein antigenicity might raise autoantibodies. Autoantibodies existing in synovial fluid opsonize target antigens and trigger a complement cascade, thus maintaining the vicious cycles of auto-inflammation and hyper-citrullination.
Bacterial toxins and enzymes have been reported to induce citrullination of host proteins and release of PADs. The pore-forming leukotoxin produced by Gram-negative Aggregatibacter actinomycetemcomitans kills human neutrophils and induces hyper-citrullination in neutrophils, thus contributing to dysregulated citrullination (Konig et al., 2016). Likewise, as a putative link between PD and RA, the pathobiont P. gingivalis expresses a prokaryotic PAD (PPAD), which is thus far unique among microorganisms. In contrast to human PADs, PPAD does not require calcium ion for its activity of citrullination of C-terminal arginine residues (Rodriguez et al., 2009; McGraw et al., 1999). Furthermore, while PADs are unable to catalyze free L-citrulline, PPAD can citrullinate both free and peptide-bound arginine (Abdullah et al., 2013). A cellular PPAD with an approximate size of 75–85 kDa and a secreted PPAD sized 47 kDa have been described (Konig et al., 2014). PPAD is extracellularly secreted or located in the outer membrane of P. gingivalis together with virulence factors, arginine-specific gingipains RgpA and RgpB, which cleave the carboxyl group of arginine residue in their own target proteins. The cleaved products exposing arginine residue at the carboxyl terminus are prone to rapid citrullination by PPAD (Wegner et al., 2010; Maresz et al., 2013).
Prokaryotic PAD also citrullinates human proteins, such as fibrin, vimentin, epidermal growth factor (EGF), fibrinogen, and α-enolase (McGraw et al., 1999; Mangat et al., 2010; Wegner et al., 2010; Montgomery et al., 2016). Therefore, host proteins modified by PPAD may function as antigens that induce generation of ACPAs (Vossenaar and van Venrooij, 2004; Moscarello et al., 2007; Nesse et al., 2012). As a consequence, citrullination of EGF results in defects in cell-cycle modulation. Since EGF activates cell proliferation, migration, repair, and regeneration of gingival epithelial cells, its citrullination hampers regeneration of damaged tissue. As a result, modification of human proteins mediated by PPAD likely induces a biological shift in the local environment (Pyrc et al., 2013). Also, PPAD may be important for interactions of P. gingivalis with eukaryotic cells, including neutrophils, macrophages, and epithelial cells (Quirke et al., 2014). Additionally, it has been shown that monocytes and macrophages exposed to viable P. gingivalis had increased extracellular citrullination levels, while the endogenous PAD level is not affected (Marchant et al., 2013). Bielecka et al. (2014) also showed that PPAD citrullinates the C-terminal arginine residue of the chemoattractant complement factor C5a, resulting in decreased chemotaxis of human neutrophils and release of pro-inflammatory cytokines from immune cells (Bielecka et al., 2014). However, the extent to which PPAD enzymatic activity affects the functions of oral host cells, including immune cells, remains elusive. As a consequence of citrullination of bacterial proteins, auto-citrullination of PPAD and antibodies against gingipains can be detected in both PD patients and healthy individuals. On the other hand, antibodies against citrullinated PPAD are specific to RA, suggesting that citrullinated PPAD is a member of early induced proteins that contribute to ACPA generation.
In the context of mechanisms that induce antibody cross-reactivity, P. gingivalis can citrullinate its own α-enolase, which shares an 82% sequence homology with human α-enolase, a finding that provides a convincing argument in terms of epitope spreading and molecular mimicry (Lundberg et al., 2008). Therefore, PD patients colonized with P. gingivalis might produce antibodies against citrullinated bacterial proteins homologous to human proteins and this molecular mimicry of the antigen may elicit an immune response to human tissues. Similarly, PPAD citrullination of host proteins allows for neo-epitope formation that triggers autoimmune responses.
Due to diverse factors that influence PD and RA, animal models have been utilized to detect a causal relationship between those diseases. Kinloch et al. (2011) demonstrated that citrullinated enolase induces experimental arthritis, and showed that enolase citrullinated by human PAD or P. gingivalis PPAD induces autoantibody production in DR4-IE-transgenic mice (Kinloch et al., 2011). Utilizing a collagen-induced arthritis (CIA) mouse model, Maresz et al. (2013) demonstrated that the ability of P. gingivalis strain W83 to augment CIA was dependent on PPAD activity. Moreover, infection with the wild-type strain, but not its PPAD-null mutant, induced elevated levels of autoantibodies to collagen type II (Maresz et al., 2013). These in vivo results emphasize the importance of PPAD as a potential virulence factor of P. gingivalis and a key component connecting PD and RA.
Recent findings have demonstrated that osteoclasts express PAD enzymes at all stages of their development, while detectable citrullinated proteins have also been detected on their cell surface (Harre et al., 2012; Krishnamurthy et al., 2016). ACPAs can therefore bind to osteoclast precursors and induce expression of IL-8, which acts as an autocrine growth factor and drives differentiation into mature bone-resorbing osteoclasts (Kopesky et al., 2014). While those findings account for bone-resorption mechanisms to some extent, they are clearly not sufficient to induce chronic synovial inflammation (Catrina et al., 2017). Indeed, animal models have shown that a single ACPA administration does not induce arthritis. However, if mild synovial inflammation already exists, severe joint disease reminiscent of human RA has been found to develop in the presence of ACPAs (Sohn et al., 2015). These findings suggest that ACPAs are important but not alone sufficient for inducing chronic inflammation, though they apparently play an active role in the disease processes leading to RA.
Oral pathobionts are constituents of a complex ecosystem and provide a mutually trophic metabolism together in a state of equilibrium with host factors and the immune system. Dental biofilms in each anatomical site are characterized by a distinct composition of bacterial species. A continuous existence of periodontal biofilm exacerbates host inflammatory response and drives a shift in the periodontal microbiome, which leads to onset of PD. As a red complex member, P. gingivalis affects the functions of various host cells and manipulates antimicrobial host response, thereby posing dysbiosis and prolongation of an inflammatory status along with periodontal tissue damage. Furthermore, recent findings indicate that particular periodontal pathobionts, such as P. gingivalis, have an exacerbating role for generation of ACPAs, which has been confirmed by epidemiological data showing an interrelationship between PD and RA. ACPAs activate immune response, including complement activation, and thus facilitate local hyper-citrullination, while they also activate osteoclasts to absorb bone and provide the basis for RA development. Imbalances in the oral microbiome shape the pro-inflammatory axis of the cytokine network, which offers a broad framework to comprehend the pathogeneses of autoimmunity and chronic inflammatory diseases (Hooper et al., 2012). However, the exact mechanisms by which oral pathobionts have effects on both the cytokine network and autoimmunity remain obscure. Further research is needed to evaluate the involvement of genetic/environmental susceptibility factors, citrullination, and cytokine networks in the reciprocal relationship of PD and RA. As shown in this review, it is currently not possible to finally conclude if oral pathobionts like P. gingivalis can simply be classified as bystander microbiota or if they are disease initiators, with the consequence that perhaps early prophylactic treatment could prevent systemic and chronic diseases, such as atherosclerosis, infective endocarditis, diabetes, adverse pregnancy outcome, respiratory diseases, and RA.
All authors conceived the concept for this review article and participated in writing the manuscript. Each equally contributed to reading, editing, and reviewing the manuscript.
Research performed in the laboratory of BM-H was supported by intramural funding (FORUN 889023). The work of MN was financed by JSPS KAKENHI (grant number 15KK0306).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Marcus Frank and the whole team of the Electron Microscopic Centre (EMZ) at the University Medicine Rostock for their support.
Aas, J. A., Paster, B. J., Stokes, L. N., Olsen, I., and Dewhirst, F. E. (2005). Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 43, 5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005
Abdullah, S. N., Farmer, E. A., Spargo, L., Logan, R., and Gully, N. (2013). Porphyromonas gingivalis peptidylarginine deiminase substrate specificity. Anaerobe 23, 102–108. doi: 10.1016/j.anaerobe.2013.07.001
Aberdam, D., Candi, E., Knight, R. A., and Melino, G. (2008). miRNAs, ‘stemness’ and skin. Trends Biochem. Sci. 33, 583–591. doi: 10.1016/j.tibs.2008.09.002
AlJehani, Y. A. (2014). Risk factors of periodontal disease: review of the literature. Int. J. Dent. 2014:182513. doi: 10.1155/2014/182513
Al-Katma, M. K., Bissada, N. F., Bordeaux, J. M., Sue, J., and Askari, A. D. (2007). Control of periodontal infection reduces the severity of active rheumatoid arthritis. J. Clin. Rheumatol. 13, 134–137. doi: 10.1097/RHU.0b013e3180690616
Arita, K., Hashimoto, H., Shimizu, T., Nakashima, K., Yamada, M., and Sato, M. (2004). Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 11, 777–783. doi: 10.1038/nsmb799
Barry, F. P., and Murphy, J. (2004). Mesenchymal stem cells: clinical applications and biological characterization. Int. J. Biochem. Cell Biol. 36, 568–584. doi: 10.1016/j.biocel.2003.11.001
Bartold, P. M., Marino, V., Cantley, M., and Haynes, D. R. (2010). Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 37, 405–411. doi: 10.1111/j.1600-051X.2010.01552.x
Benakanakere, M. R., Li, Q., Eskan, M. A., Singh, A. V., Zhao, J., Galicia, J. C., et al. (2009). Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J. Biol. Chem. 284, 23107–23115. doi: 10.1074/jbc.M109.013862
Berezow, A. B., and Darveau, R. P. (2011). Microbial shift and periodontitis. Periodontol. 2000 55, 36–47. doi: 10.1111/j.1600-0757.2010.00350.x
Bicker, K. L., and Thompson, P. R. (2013). The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 99, 155–163. doi: 10.1002/bip.22127
Biedermann, A., Kriebel, K., Kreikemeyer, B., and Lang, H. (2014). Interactions of anaerobic bacteria with dental stem cells: an in vitro study. PLOS ONE 9:e110616. doi: 10.1371/journal.pone.0110616
Bielecka, E., Scavenius, C., Kantyka, T., Jusko, M., Mizgalska, D., Szmigielski, B., et al. (2014). Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J. Biol. Chem. 289, 32481–32487. doi: 10.1074/jbc.C114.617142
Brown, S. A., and Whiteley, M. (2007). A novel exclusion mechanism for carbon resource partitioning in Aggregatibacter actinomycetemcomitans. J. Bacteriol. 189, 6407–6414. doi: 10.1128/JB.00554-07
Brunner, J., Scheres, N., El Idrissi, N. B., Deng, D. M., Laine, M. L., van Winkelhoff, A. J., et al. (2010). The capsule of Porphyromonas gingivalis reduces the immune response of human gingival fibroblasts. BMC Microbiol. 10:5. doi: 10.1186/1471-2180-10-5
Carvalho-Filho, P. C., Gomes-Filho, I. S., Meyer, R., Olczak, T., Xavier, M. T., and Trindade, S. C. (2016). Role of Porphyromonas gingivalis HmuY in immunopathogenesis of chronic periodontitis. Mediators Inflamm. 2016:7465852. doi: 10.1155/2016/7465852
Castro, S. A., Collighan, R., Lambert, P. A., Dias, I. H. K., Chauhan, P., Bland, C. E., et al. (2017). Porphyromonas gingivalis gingipains cause defective macrophage migration towards apoptotic cells and inhibit phagocytosis of primary apoptotic neutrophils. Cell Death Dis. 8:e2644. doi: 10.1038/cddis.2016.481
Catrina, A. I., Svensson, C. I., Malmstrom, V., Schett, G., and Klareskog, L. (2017). Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 13, 79–86. doi: 10.1038/nrrheum.2016.200
Chatzivasileiou, K., Kriebel, K., Steinhoff, G., Kreikemeyer, B., and Lang, H. (2015). Do oral bacteria alter the regenerative potential of stem cells? A concise review. J. Cell Mol. Med. 19, 2067–2074. doi: 10.1111/jcmm.12613
Chatzivasileiou, K., Lux, C. A., Steinhoff, G., and Lang, H. (2013). Dental follicle progenitor cells responses to Porphyromonas gingivalis LPS. J. Cell Mol. Med. 17, 766–773. doi: 10.1111/jcmm.12058
Cortelli, J. R., Aquino, D. R., Cortelli, S. C., Franco, G. C. N., Fernandes, C. B., Roman-Torres, C. V. G., et al. (2008). Detection of periodontal pathogens in oral mucous membranes of edentulous individuals. J. Periodontol. 79, 1962–1965. doi: 10.1902/jop.2008.080092
Damgaard, C., Kantarci, A., Holmstrup, P., Hasturk, H., Nielsen, C. H., and van Dyke, T. E. (2016). Porphyromonas gingivalis-induced production of reactive oxygen species, tumor necrosis factor-α, interleukin-6, CXCL8 and CCL2 by neutrophils from localized aggressive periodontitis and healthy donors: modulating actions of red blood cells and resolvin E1. J. Periodontal Res. 52, 246–254. doi: 10.1111/jre.12388
Darrah, E., Giles, J. T., Ols, M. L., Bull, H. G., Andrade, F., and Rosen, A. (2013). Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci. Transl. Med. 5:186ra165. doi: 10.1126/scitranslmed.3005370
de Pablo, P., Chapple, I. L., Buckley, C. D., and Dietrich, T. (2009). Periodontitis in systemic rheumatic diseases. Nat. Rev. Rheumatol. 5, 218–224. doi: 10.1038/nrrheum.2009.28
Deshpande, R., Khan, M., and Genco, C. (1998). Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect. Immun. 66, 5337–5343.
Desta, T., and Graves, D. T. (2007). Fibroblast apoptosis induced by Porphyromonas gingivalis is stimulated by a gingipain and caspase-independent pathway that involves apoptosis-inducing factor. Cell. Microbiol. 9, 2667–2675. doi: 10.1111/j.1462-5822.2007.00987.x
Dewhirst, F. E., Chen, T., Izard, J., Paster, B. J., Tanner, A. C. R., Yu, W. H., et al. (2010). The human oral microbiome. J. Bacteriol 192, 5002–5017. doi: 10.1128/JB.00542-10
Dogan, S., Gunzer, F., Guenay, H., Hillmann, G., and Geurtsen, W. (2000). Infection of primary human gingival fibroblasts by Porphyromonas gingivalis and Prevotella intermedia. Clin. Oral Investig. 4, 35–41. doi: 10.1007/s007840050111
Dorn, B., Burks, J., Seifert, K., and Progulske-Fox, A. (2000). Invasion of endothelial and epithelial cells by strains of Porphyromonas gingivalis. FEMS Microbiol. Lett. 187, 139–144. doi: 10.1111/j.1574-6968.2000.tb09150.x
Duncan, M. J., Nakao, S., Skobe, Z., and Xie, H. (1993). Interactions of Porphyromonas gingivalis with epithelial cells. Infect. Immun. 61, 2260–2265.
Duran-Pinedo, A. E., and Frias-Lopez, J. (2015). Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes Infect. 17, 505–516. doi: 10.1016/j.micinf.2015.03.014
Dutzan, N., Abusleme, L., Bridgeman, H., Greenwell-Wild, T., Zangerle-Murray, T., Fife, M. E., et al. (2017). On-going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity 46, 133–147. doi: 10.1016/j.immuni.2016.12.010
Eriksson, K., Nise, L., Kats, A., Luttropp, E., Catrina, A. I., Askling, J., et al. (2016). Prevalence of periodontitis in patients with established rheumatoid arthritis: a swedish population based case-control study. PLOS ONE 11:e0155956. doi: 10.1371/journal.pone.0155956
Eskan, M. A., Jotwani, R., Abe, T., Chmelar, J., Lim, J. H., Liang, S., et al. (2012). The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 13, 465–473. doi: 10.1038/ni.2260
Feng, Z., and Weinberg, A. (2006). Role of bacteria in health and disease of periodontal tissues. Periodontol. 2000 40, 50–76. doi: 10.1111/j.1600-0757.2005.00148.x
Gaffen, S. L., Herzberg, M. C., Taubman, M. A., and van Dyke, T. E. (2014). Recent advances in host defense mechanisms/therapies against oral infectious diseases and consequences for systemic disease. Adv. Dent. Res. 26, 30–37. doi: 10.1177/0022034514525778
Gamonal, J., Sanz, M., O’Connor, A., Acevedo, A., Suarez, I., Sanz, A., et al. (2003). Delayed neutrophil apoptosis in chronic periodontitis patients. J. Clin. Periodontol. 30, 616–623. doi: 10.1034/j.1600-051X.2003.00350.x
Gonzales, J. R. (2015). T- and B-cell subsets in periodontitis. Periodontol. 2000 69, 181–200. doi: 10.1111/prd.12090
Graves, D. (2008). Cytokines that promote periodontal tissue destruction. J. Periodontol. 79, 1585–1591. doi: 10.1902/jop.2008.080183
Gronthos, S., Mankani, M., Brahim, J., Robey, P., and Shi, S. (2000). Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 13625–13630. doi: 10.1073/pnas.240309797
Haffajee, A. D., Socransky, S. S., Patel, M. R., and Song, X. (2008). Microbial complexes in supragingival plaque. Oral Microbiol. Immunol. 23, 196–205. doi: 10.1111/j.1399-302X.2007.00411.x
Hajishengallis, G. (2015). Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44. doi: 10.1038/nri3785
Hajishengallis, G., and Lamont, R. J. (2012). Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 27, 409–419. doi: 10.1111/j.2041-1014.2012.00663.x
Hajishengallis, G., Wang, M., Bagby, G. J., and Nelson, S. (2008). Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J. Immunol. 181, 4141–4149. doi: 10.4049/jimmunol.181.6.4141
Harre, U., Georgess, D., Bang, H., Bozec, A., Axmann, R., Ossipova, E., et al. (2012). Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Invest. 122, 1791–1802. doi: 10.1172/JCI60975
He, J., Li, Y., Cao, Y., Xue, J., and Zhou, X. (2015). The oral microbiome diversity and its relation to human diseases. Folia Microbiol. 60, 69–80. doi: 10.1007/s12223-014-0342-2
Heaton, B., and Dietrich, T. (2012). Causal theory and the etiology of periodontal diseases. Periodontol. 2000 58, 26–36. doi: 10.1111/j.1600-0757.2011.00414.x
Heller, D., Helmerhorst, E. J., and Oppenheim, F. G. (2017). Saliva and serum protein exchange at the tooth enamel surface. J. Dent. Res. 96, 437–443. doi: 10.1177/0022034516680771
Hieke, C., Kriebel, K., Engelmann, R., Müller-Hilke, B., Lang, H., and Kreikemeyer, B. (2016). Human dental stem cells suppress PMN activity after infection with the periodontopathogens Prevotella intermedia and Tannerella forsythia. Sci. Rep. 6:39096. doi: 10.1038/srep39096
Hooper, L. V., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
How, K. Y., Song, K. P., and Chan, K. G. (2016). Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7:53. doi: 10.3389/fmicb.2016.00053
Irshad, M., van der Reijden, W. A., Crielaard, W., and Laine, M. L. (2012). In vitro invasion and survival of Porphyromonas gingivalis in gingival fibroblasts; role of the capsule. Arch. Immunol. Ther. Exp. 60, 469–476. doi: 10.1007/s00005-012-0196-8
Ishida, K., Kobayashi, T., Ito, S., Komatsu, Y., Yokoyama, T., Okada, M., et al. (2012). Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J. Periodontol. 83, 917–925. doi: 10.1902/jop.2011.110356
Jenkinson, H. F. (1994). Cell surface protein receptors in oral streptococci. FEMS Microbiol. Lett. 121, 133–140. doi: 10.1111/j.1574-6968.1994.tb07089.x
Jiao, Y., Hasegawa, M., and Inohara, N. (2014). The role of oral pathobionts in dysbiosis during periodontitis development. J. Dent. Res. 93, 539–546. doi: 10.1177/0022034514528212
Jin, H. J., Bae, Y. K., Kim, M., Kwon, S. J., Jeon, H. B., Choi, S. J., et al. (2013). Comparative analysis of human mesenchymal stem cells from bone marrow, adipose tissue, and umbilical cord blood as sources of cell therapy. Int. J. Mol. Sci. 14, 17986–18001. doi: 10.3390/ijms140917986
Jo, Y., Lee, H., Kook, S., Choung, H., Park, J., Chung, J., et al. (2007). Isolation and characterization of postnatal stem cells from human dental tissues. Tissue Eng. 13, 767–773. doi: 10.1089/ten.2006.0192
Kantarci, A., Oyaizu, K., and van Dyke, T. E. (2003). Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J. Periodontol. 74, 66–75. doi: 10.1902/jop.2003.74.1.66
Kaplan, C. W., Ma, X., Paranjpe, A., Jewett, A., Lux, R., Kinder-Haake, S., et al. (2010). Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 78, 4773–4778. doi: 10.1128/IAI.00567-10
Kebschull, M., and Papapanou, P. (2011). Periodontal microbial complexes associated with specific cell and tissue responses. J. Clin. Periodontol. 38, 17–27. doi: 10.1111/j.1600-051X.2010.01668.x
Kim, J., and Amar, S. (2006). Periodontal disease and systemic conditions: a bidirectional relationship. Odontology 94, 10–21. doi: 10.1007/s10266-006-0060-6
Kinane, D. F., and Bartold, P. M. (2007). Clinical relevance of the host responses of periodontitis. Periodontol. 2000 43, 278–293. doi: 10.1111/j.1600-0757.2006.00169.x
Kinloch, A. J., Alzabin, S., Brintnell, W., Wilson, E., Barra, L., Wegner, N., et al. (2011). Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian α-enolase and arthritis in DR4-IE-transgenic mice. Arthritis Rheum. 63, 3818–3823. doi: 10.1002/art.30639
Klein, M. I., and Goncalves, R. B. (2003). Detection of Tannerella forsythensis (Bacteroides forsythus) and Porphyromonas gingivalis by polymerase chain reaction in subjects with different periodontal status. J. Periodontol. 74, 798–802. doi: 10.1902/jop.2003.74.6.798
Kolenbrander, P. E., Palmer, R. J. Jr., Periasamy, S., and Jakubovics, N. S. (2010). Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8, 471–480. doi: 10.1038/nrmicro2381
Kolte, R. A., Kolte, A. P., and Deshpande, N. M. (2014). Assessment and comparison of anemia of chronic disease in healthy subjects and chronic periodontitis patients: a clinical and hematological study. J. Indian Soc. Periodontol. 18, 183–186. doi: 10.4103/0972-124X.131321
Konig, M. F., Abusleme, L., Reinholdt, J., Palmer, R. J., Teles, R. P., Sampson, K., et al. (2016). Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 8, 369ra176. doi: 10.1126/scitranslmed.aaj1921
Konig, M. F., and Andrade, F. (2016). A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front. Immunol. 7:461. doi: 10.3389/fimmu.2016.00461
Konig, M. F., Paracha, A. S., Moni, M., Bingham, C. O., and Andrade, F. (2014). Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann. Rheum. Dis. 74, 2054–2061. doi: 10.1136/annrheumdis-2014-205385
Kopesky, P., Tiedemann, K., Alkekhia, D., Zechner, C., Millard, B., Schoeberl, B., et al. (2014). Autocrine signaling is a key regulatory element during osteoclastogenesis. Biol. Open 3, 767–776. doi: 10.1242/bio.20148128
Koziel, J., Mydel, P., and Potempa, J. (2014). The link between periodontal disease and rheumatoid arthritis: an updated review. Curr. Rheumatol. Rep. 16:408. doi: 10.1007/s11926-014-0408-9
Kriebel, K., Biedermann, A., Kreikemeyer, B., and Lang, H. (2013). Anaerobic co-culture of mesenchymal stem cells and anaerobic pathogens-A new in vitro model system. PLOS ONE 8:e78226. doi: 10.1371/journal.pone.0078226
Krishnamurthy, A., Joshua, V., Haj Hensvold, A., Jin, T., Sun, M., Vivar, N., et al. (2016). Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 75, 721–729. doi: 10.1136/annrheumdis-2015-208093
Kumar, P. S. (2017). From focal sepsis to periodontal medicine: a century of exploring the role of the oral microbiome in systemic disease. J. Physiol. 595, 465–476. doi: 10.1113/JP272427
Kuo, H. C., Chang, L. C., Chen, T. C., Lee, K. C., Lee, K. F., Chen, C. N., et al. (2016). Sterol regulatory element-binding protein-1c regulates inflammasome activation in gingival fibroblasts infected with high-glucose-treated Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 6:195. doi: 10.3389/fcimb.2016.00195
Lakschevitz, F. S., Aboodi, G. M., and Glogauer, M. (2013). Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PLOS ONE 8:e68983. doi: 10.1371/journal.pone.0068983
Li, J., Helmerhorst, E. J., Leone, C. W., Troxler, R. F., Yaskell, T., Haffajee, A. D., et al. (2004). Identification of early microbial colonizers in human dental biofilm. J. Appl. Microbiol. 97, 1311–1318. doi: 10.1111/j.1365-2672.2004.02420.x
Li, L., Michel, R., Cohen, J., Decarlo, A., and Kozarov, E. (2008). Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. 8:26. doi: 10.1186/1471-2180-8-26
Li, T., Khah, M. K., Slavnic, S., Johansson, I., and Strömberg, N. (2001). Different type 1 fimbrial genes and tropisms of commensal and potentially pathogenic Actinomyces spp. with different salivary acidic proline-rich protein and statherin ligand specificities. Infect. Immun. 69, 7224–7233. doi: 10.1128/IAI.69.12.7224-7233.2001
Li, X., Kolltveit, K. M., Tronstad, L., and Olsen, I. (2000). Systemic diseases caused by oral infection. Clin. Microbiol. Rev. 13, 547–558. doi: 10.1128/CMR.13.4.547-558.2000
Li, X., Lan, H. Y., Huang, X. R., Zhang, C., and Jin, L. J. (2013). Expression profile of macrophage migration-inhibitory factor in human gingiva and reconstituted human gingival epithelia stimulated by Porphyromonas gingivalis lipopolysaccharide. J. Periodontal Res. 48, 527–532. doi: 10.1111/jre.12035
Liu, J., Wang, Y., and Ouyang, X. (2014). Beyond toll-like receptors: Porphyromonas gingivalis induces IL-6, IL-8, and VCAM-1 expression through NOD-mediated NF-κB and ERK signaling pathways in periodontal fibroblasts. Inflammation 37, 522–533. doi: 10.1007/s10753-013-9766-0
Liu, L., Okada, S., Kong, X. F., Kreins, A. Y., Cypowyj, S., Abhyankar, A., et al. (2011). Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208, 1635–1648. doi: 10.1084/jem.20110958
Lundberg, K., Kinloch, A., Fisher, B. A., Wegner, N., Wait, R., Charles, P., et al. (2008). Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 58, 3009–3019. doi: 10.1002/art.23936
Madianos, P. N., Papapanou, P. N., and Sandros, J. (1997). Porphyromonas gingivalis infection of oral epithelium inhibits neutrophil transepithelial migration. Infect. Immun. 65, 3983–3990.
Maekawa, T., Krauss, J. L., Abe, T., Jotwani, R., Triantafilou, M., Triantafilou, K., et al. (2014). Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 15, 768–778. doi: 10.1016/j.chom.2014.05.012
Mahanonda, R., Sa-Ard-Iam, N., Montreekachon, P., Pimkhaokham, A., Yongvanichit, K., Fukuda, M. M., et al. (2007). IL-8 and IDO expression by human gingival fibroblasts via TLRs. J. Immunol. 178, 1151–1157. doi: 10.4049/jimmunol.178.2.1151
Mangat, P., Wegner, N., Venables, P. J., and Potempa, J. (2010). Bacterial and human peptidylarginine deiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 12:209. doi: 10.1186/ar3000
Marchant, C., Smith, M. D., Proudman, S., Haynes, D. R., and Bartold, P. M. (2013). Effect of Porphyromonas gingivalis on citrullination of proteins by macrophages in vitro. J. Periodontol. 84, 1272–1280. doi: 10.1902/jop.2012.120103
Maresz, K. J., Hellvard, A., Sroka, A., Adamowicz, K., Bielecka, E., Koziel, J., et al. (2013). Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLOS Pathog. 9:e1003627. doi: 10.1371/journal.ppat.1003627
Mark Welch, J. L., Rossetti, B. J., Rieken, C. W., Dewhirst, F. E., and Borisy, G. G. (2016). Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. U.S.A. 113, E791–E800. doi: 10.1073/pnas.1522149113
McGraw, W. T., Potempa, J., Farley, D., and Travis, J. (1999). Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect. Immun. 67, 3248–3256.
Mettraux, G. R., Gusberti, F. A., and Graf, H. (1984). Oxygen tension (pO2) in untreated human periodontal pockets. J. Periodontol. 55, 516–521. doi: 10.1902/jop.1984.55.9.516
Mikuls, T. R., Payne, J. B., Reinhardt, R. A., Thiele, G. M., Maziarz, E., Cannella, A. C., et al. (2009). Antibody responses to Porphyromonas gingivalis in subjects with rheumatoid arthritis and periodontitis. Int. Immunopharmacol. 9, 38–42. doi: 10.1016/j.intimp.2008.09.008
Mikuls, T. R., Payne, J. B., Yu, F., Thiele, G. M., Reynolds, R. J., Cannon, G. W., et al. (2014). Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 66, 1090–1100. doi: 10.1002/art.38348
Miura, M., Gronthos, S., Zhao, M., Lu, B., Fisher, L. W., Robey, P. G., et al. (2003). SHED: stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. U.S.A. 100, 5807–5812. doi: 10.1073/pnas.0937635100
Moffatt, C. E., and Lamont, R. J. (2011). Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect. Immun. 79, 2632–2637. doi: 10.1128/IAI.00082-11
Montebugnoli, L., Servidio, D., Miaton, R. A., Prati, C., Tricoci, P., Melloni, C., et al. (2005). Periodontal health improves systemic inflammatory and haemostatic status in subjects with coronary heart disease. J. Clin. Periodontol. 32, 188–192. doi: 10.1111/j.1600-051X.2005.00641.x
Montgomery, A. B., Kopec, J., Shrestha, L., Thezenas, M. L., Burgess-Brown, N. A., Fischer, R., et al. (2016). Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: implications for autoimmunity in rheumatoid arthritis. Ann. Rheum. Dis. 75, 1255–1261. doi: 10.1136/annrheumdis-2015-207656
Morandini, A. C., Chaves Souza, P. P., Ramos-Junior, E. S., Brozoski, D. T., Sipert, C. R., Souza Costa, C. A., et al. (2013). Toll-like receptor 2 knockdown modulates interleukin (IL)-6 and IL-8 but not stromal derived factor-1 (SDF-1/CXCL12) in human periodontal ligament and gingival fibroblasts. J. Periodontol. 84, 535–544. doi: 10.1902/jop.2012.120177
Moscarello, M. A., Mastronardi, F. G., and Wood, D. D. (2007). The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem. Res. 32, 251–256. doi: 10.1007/s11064-006-9144-5
Moutsopoulos, N. M., Konkel, J., Sarmadi, M., Eskan, M. A., Wild, T., Dutzan, N., et al. (2014). Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 6:229ra40. doi: 10.1126/scitranslmed.3007696
Moutsopoulos, N. M., Lionakis, M. S., and Hajishengallis, G. (2015). Inborn errors in immunity: unique natural models to dissect oral immunity. J. Dent. Res. 94, 753–758. doi: 10.1177/0022034515583533
Muzio, M., and Mantovani, A. (2000). Toll-like receptors. Microbes Infect. 2, 251–255. doi: 10.1016/S1286-4579(00)00303-8
Mysak, J., Podzimek, S., Sommerova, P., Lyuya-Mi, Y., Bartova, J., Janatova, T., et al. (2014). Porphyromonas gingivalis: major periodontopathic pathogen overview. J. Immunol. Res. 2014:476068. doi: 10.1155/2014/476068
Navarro-Millán, I., Darrah, E., Westfall, A. O., Mikuls, T. R., Reynolds, R. J., Danila, M. I., et al. (2016). Association of anti-peptidyl arginine deiminase antibodies with radiographic severity of rheumatoid arthritis in African Americans. Arthritis Res. Ther. 18:241. doi: 10.1186/s13075-016-1126-7
Nelson-Filho, P., Borba, I. G., Mesquita, K. S., Silva, R. A., Queiroz, A. M., and Silva, L. A. (2013). Dynamics of microbial colonization of the oral cavity in newborns. Braz. Dent. J. 24, 415–419. doi: 10.1590/0103-6440201302266
Nesbitt, W. E., Beem, J. E., Leung, K. P., and Clark, W. B. (1992). Isolation and characterization of Actinomyces viscosus mutants defective in binding salivary proline-rich proteins. Infect. Immun. 60, 1095–1100.
Nesse, W., Westra, J., van der Wal, J. E., Abbas, F., Nicholas, A. P., Vissink, A., et al. (2012). The periodontium of periodontitis patients contains citrullinated proteins which may play a role in ACPA (anti-citrullinated protein antibody) formation. J. Clin. Periodontol. 39, 599–607. doi: 10.1111/j.1600-051X.2012.01885.x
O’Brien-Simpson, N. M., Pathirana, R. D., Walker, G. D., and Reynolds, E. C. (2009). Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect. Immun. 77, 1246–1261. doi: 10.1128/IAI.01038-08
O’Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G., and Baltimore, D. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U.S.A. 104, 1604–1609. doi: 10.1073/pnas.0610731104
Ohlrich, E. J., Cullinan, M. P., and Seymour, G. J. (2009). The immunopathogenesis of periodontal disease. Aust. Dent. J. 54, S2–S10. doi: 10.1111/j.1834-7819.2009.01139.x
Okahashi, N., Nakata, M., Terao, Y., Isoda, R., Sakurai, A., Sumitomo, T., et al. (2011). Pili of oral Streptococcus sanguinis bind to salivary amylase and promote the biofilm formation. Microb. Pathog. 50, 148–154. doi: 10.1016/j.micpath.2011.01.005
Oli, M. W., McArthur, W. P., and Brady, L. J. (2006). A whole cell BIAcore assay to evaluate P1-mediated adherence of Streptococcus mutans to human salivary agglutinin and inhibition by specific antibodies. J. Microbiol. Methods. 65, 503–511. doi: 10.1016/j.mimet.2005.09.011
Ortiz, P., Bissada, N. F., Palomo, L., Han, Y. W., Al-Zahrani, M. S., Panneerselvam, A., et al. (2009). Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J. Periodontol. 80, 535–540. doi: 10.1902/jop.2009.080447
Palm, E., Demirel, I., Bengtsson, T., and Khalaf, H. (2015). The role of toll-like and protease-activated receptors in the expression of cytokines by gingival fibroblasts stimulated with the periodontal pathogen Porphyromonas gingivalis. Cytokine 76, 424–432. doi: 10.1016/j.cyto.2015.08.263
Pathirana, R. D., O’Brien-Simpson, N. M., Visvanathan, K., Hamilton, J. A., and Reynolds, E. C. (2008). The role of the RgpA-Kgp proteinase-adhesin complexes in the adherence of Porphyromonas gingivalis to fibroblasts. Microbiology 154, 2904–2911. doi: 10.1099/mic.0.2008/019943-0
Pihlstrom, B. L., Michalowicz, B. S., and Johnson, N. W. (2005). Periodontal diseases. Lancet 366, 1809–1820. doi: 10.1016/S0140-6736(05)67728-8
Pinnock, A., Murdoch, C., Moharamzadeh, K., Whawell, S., and Douglas, C. W. (2014). Characterisation and optimisation of organotypic oral mucosal models to study Porphyromonas gingivalis invasion. Microbes Infect. 16, 310–319. doi: 10.1016/j.micinf.2014.01.004
Promsudthi, A., Poomsawat, S., and Limsricharoen, W. (2014). The role of Toll-like receptor 2 and 4 in gingival tissues of chronic periodontitis subjects with type 2 diabetes. J. Periodontal Res. 49, 346–354. doi: 10.1111/jre.12112
Pyrc, K., Milewska, A., Kantyka, T., Sroka, A., Maresz, K., Kozieł, J., et al. (2013). Inactivation of epidermal growth factor by Porphyromonas gingivalis as a potential mechanism for periodontal tissue damage. Infect. Immun. 81, 55–64. doi: 10.1128/IAI.00830-12
Quirke, A. M., Lugli, E. B., Wegner, N., Hamilton, B. C., Charles, P., Chowdhury, M., et al. (2014). Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 73, 263–269. doi: 10.1136/annrheumdis-2012-202726
Ramsey, M. M., Rumbaugh, K. P., and Whiteley, M. (2011). Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLOS Pathog. 7:e1002012. doi: 10.1371/journal.ppat.1002012
Reichert, S., Haffner, M., Keyßer, G., Schäfer, C., Stein, J. M., Schaller, H.-G., et al. (2013). Detection of oral bacterial DNA in synovial fluid. J. Clin. Periodontol. 40, 591–598. doi: 10.1111/jcpe.12102
Restaino, C. G., Chaparro, A., Valenzuela, M., Vernal, R., Silva, A., Puente, J., et al. (2007). Stimulatory response of neutrophils from periodontitis patients with periodontal pathogens. Oral Dis. 13, 474–481. doi: 10.1111/j.1601-0825.2006.01323.x
Rodriguez, B., Stitt, B. L., and Ash, D. E. (2009). Expression of peptidylarginine deiminase from Porphyromonas gingivalis in Escherichia coli: enzyme purification and characterization. Arch. Biochem. Biophys. 488, 14–22. doi: 10.1016/j.abb.2009.06.010
Romero, V., Fert-Bober, J., Nigrovic, P. A., Darrah, E., Haque, U. J., Lee, D. M., et al. (2013). Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci. Transl. Med. 5:209ra150. doi: 10.1126/scitranslmed.3006869
Rosenstein, E. D., Greenwald, R. A., Kushner, L. J., and Weissmann, G. (2004). Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 28, 311–318. doi: 10.1007/s10753-004-6641-z
Rose A. S., and Hildebrand, P. W. (2015). NGL Viewer: a web application for molecular visualization. Nucl. Acids Res. 43, W576–W579. doi: 10.1093/nar/gkv402
Rotimi, V. O., and Duerden, B. I. (1981). The development of the bacterial flora in normal neonates. J. Med. Microbiol. 14, 51–62. doi: 10.1099/00222615-14-1-51
Saito, A., Inagaki, S., Kimizuka, R., Okuda, K., Hosaka, Y., Nakagawa, T., et al. (2008). Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol. Med. Microbiol. 54, 349–355. doi: 10.1111/j.1574-695X.2008.00481.x
Schaefer, A. S., Jochens, A., Dommisch, H., Graetz, C., Jockel-Schneider, Y., Harks, I., et al. (2014). A large candidate-gene association study suggests genetic variants at IRF5 and PRDM1 to be associated with aggressive periodontitis. J. Clin. Periodontol. 41, 1122–1131. doi: 10.1111/jcpe.12314
Schellekens, G. A., De Jong, B. A., van den Hoogen, F. H., van de Putte, L. B., and van Venrooij, W. J. (1998). Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 101, 273–281. doi: 10.1172/JCI1316
Schellekens, G. A., Visser, H., de Jong, B. A., van den Hoogen, F. H., Hazes, J. M., Breedveld, F. C., et al. (2000). The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 43, 155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3
Scheres, N., and Crielaard, W. (2013). Gingival fibroblast responsiveness is differentially affected by Porphyromonas gingivalis: implications for the pathogenesis of periodontitis. Mol. Oral Microbiol. 28, 204–218. doi: 10.1111/omi.12016
Scheres, N., Laine, M. L., de Vries, T. J., Everts, V., and van Winkelhoff, A. J. (2009). Gingival and periodontal ligament fibroblasts differ in their inflammatory response to viable Porphyromonas gingivalis. J. Periodontal Res. 45, 262–270. doi: 10.1111/j.1600-0765.2009.01229.x
Segal, A. W. (2005). How neutrophils kill microbes. Annu. Rev. Immunol. 23, 197–223. doi: 10.1146/annurev.immunol.23.021704.115653
Seo, B., Miura, M., Gronthos, S., Bartold, P. M., Batouli, S., Brahim, J., et al. (2004). Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 364, 149–155. doi: 10.1016/S0140-6736(04)16627-0
Seymour, G. J., Powell, R. N., and Davies, W. I. (1979). The immunopathogenesis of progressive chronic inflammatory periodontal disease. J. Oral Pathol. 8, 249–265. doi: 10.1111/j.1600-0714.1979.tb01826.x
Slots, J. (1977). The predominant cultivable microflora of advanced periodontitis. Scand. J. Dent. Res. 85, 114–121. doi: 10.1111/j.1600-0722.1977.tb00541.x
Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., and Kent, R. L. Jr. (1998). Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x
Sohn, D. H., Rhodes, C., Onuma, K., Zhao, X., Sharpe, O., Gazitt, T., et al. (2015). Local Joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol. 67, 2877–2887. doi: 10.1002/art.39283
Sonoyama, W., Liu, Y., Fang, D., Yamaza, T., Seo, B., Zhang, C., et al. (2006). Mesenchymal stem cell-mediated functional tooth regeneration in swine. PLOS ONE 1:e79. doi: 10.1371/journal.pone.0000079
Sonoyama, W., Liu, Y., Yamaza, T., Tuan, R. S., Wang, S., Shi, S., et al. (2008). Characterization of the apical papilla and its residing stem cells from human immature permanent teeth: a pilot study. J. Endod. 34, 166–171. doi: 10.1016/j.joen.2007.11.021
Souza, P. P., Palmqvist, P., Lundgren, I., Lie, A., Costa-Neto, C. M., Lundberg, P., et al. (2010). Stimulation of IL-6 cytokines in fibroblasts by toll-like receptors 2. J. Dent. Res. 89, 802–807. doi: 10.1177/0022034510366898
Spengler, J., Lugonja, B., Ytterberg, A. J., Zubarev, R. A., Creese, A. J., Pearson, M. J., et al. (2015). Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 67, 3135–3145. doi: 10.1002/art.39313
Stein, J. M., Machulla, H. K., Smeets, R., Lampert, F., and Reichert, S. (2008). Human leukocyte antigen polymorphism in chronic and aggressive periodontitis among Caucasians: a meta-analysis. J. Clin. Periodontol. 35, 183–192. doi: 10.1111/j.1600-051X.2007.01189.x
Sugawara, Y., Uehara, A., Fujimoto, Y., Kusumoto, S., Fukase, K., Shibata, K., et al. (2006). Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J. Dent. Res. 85, 524–529. doi: 10.1177/154405910608500609
Sun, J., Nemoto, E., Hong, G., and Sasaki, K. (2016). Modulation of stromal cell-derived factor 1 alpha (SDF-1α) and its receptor CXCR4 in Porphyromonas gingivalis-induced periodontal inflammation. BMC Oral Health 17:26. doi: 10.1186/s12903-016-0250-8
Tavian, M., Zheng, B., Oberlin, E., Crisan, M., Sun, B., Huard, J., et al. (2006). The vascular wall as a source of stem cells. Ann. N. Y. Acad. Sci. 1044, 41–50. doi: 10.1196/annals.1349.006
Tzach-Nahman, R., Nashef, R., Fleissig, O., Palmon, A., Shapira, L., Wilensky, A., et al. (2017). Oral fibroblasts modulate the macrophage response to bacterial challenge. Sci. Rep. 7:11516. doi: 10.1038/s41598-017-11771-3
Uematsu, H., and Hoshino, E. (1992). Predominant obligate anaerobes in human periodontal pockets. J. Periodontal Res. 27, 15–19. doi: 10.1111/j.1600-0765.1992.tb02080.x
van Dyke, T. E., and Sheilesh, D. (2005). Risk factors for periodontitis. J. Int. Acad. Periodontol. 7, 3–7.
Vossenaar, E. R., and van Venrooij, W. J. (2004). Citrullinated proteins: sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res. Ther. 6, 107–111. doi: 10.1186/ar1184
Vossenaar, E. R., Zendman, A. J., Van Venrooij, W. J., and Pruijn, G. J. (2003). PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–1118. doi: 10.1002/bies.10357
Waddington, R. J., Moseley, R., and Embery, G. (2000). Reactive oxygen species: a potential role in the pathogenesis of periodontal diseases. Oral Dis. 6, 138–151. doi: 10.1111/j.1601-0825.2000.tb00325.x
Wade, W. G. (2013). The oral microbiome in health and disease. Pharmacol. Res. 69, 137–143. doi: 10.1016/j.phrs.2012.11.006
Weerkamp, A. H., Uyen, H. M., and Busscher, H. J. (1988). Effect of zeta potential and surface energy on bacterial adhesion to uncoated and saliva-coated human enamel and dentin. J. Dent. Res. 67, 1483–1487. doi: 10.1177/00220345880670120801
Wegner, N., Wait, R., Sroka, A., Eick, S., Nguyen, K. A., Lundberg, K., et al. (2010). Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 62, 2662–2672. doi: 10.1002/art.27552
Wessel, A. K., Arshad, T. A., Fitzpatrick, M., Connell, J. L., Bonnecaze, R. T., Shear, J. B., et al. (2014). Oxygen limitation within a bacterial aggregate. mBio 5:e00992. doi: 10.1128/mBio.00992-14
Witalison, E. E., Thompson, P. R., and Hofseth, L. J. (2015). Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr. Drug Targets 16, 700–710. doi: 10.2174/1389450116666150202160954
Yang, H. W., Huang, Y. F., and Chou, M. Y. (2004). Occurrence of Porphyromonas gingivalis and Tannerella forsythensis in periodontally diseased and healthy subjects. J. Periodontol. 75, 1077–1083. doi: 10.1902/jop.2004.75.8.1077
Yano-Higuchi, K., Takamatsu, N., He, T., Umeda, M., and Ishikawa, I. (2000). Prevalence of Bacteroides forsythus, Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans in subgingival microflora of Japanese patients with adult and rapidly progressive periodontitis. J. Clin. Periodontol. 27, 597–602. doi: 10.1034/j.1600-051x.2000.027008597.x
Zarco, M. F., Vess, T. J., and Ginsburg, G. S. (2012). The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Dis. 18, 109–120. doi: 10.1111/j.1601-0825.2011.01851.x
Zhang, J., Dai, J., Zhao, E., Lin, Y., Zeng, L., Chen, J., et al. (2004). cDNA cloning, gene organization and expression analysis of human peptidylarginine deiminase type VI. Acta Biochim. Pol. 51, 1051–1058.
Zhang, Q., Shi, S., Liu, Y., Uyanne, J., Shi, Y., and Le, A. (2009). Mesenchymal stem cells derived from human gingiva are capable of immunomodulatory functions and ameliorate inflammation-related tissue destruction in experimental colitis. J. Immunol. 183, 7787–7798. doi: 10.4049/jimmunol.0902318
Zhang, W., Ju, J., Rigney, T., and Tribble, G. (2014). Porphyromonas gingivalis infection increases osteoclastic bone resorption and osteoblastic bone formation in a periodontitis mouse model. BMC Oral Health 14:89. doi: 10.1186/1472-6831-14-89
Keywords: oral biofilm, periodontitis, rheumatic arthritis, P. gingivalis, peptidylarginine deiminase
Citation: Kriebel K, Hieke C, Müller-Hilke B, Nakata M and Kreikemeyer B (2018) Oral Biofilms from Symbiotic to Pathogenic Interactions and Associated Disease –Connection of Periodontitis and Rheumatic Arthritis by Peptidylarginine Deiminase. Front. Microbiol. 9:53. doi: 10.3389/fmicb.2018.00053
Received: 30 November 2017; Accepted: 10 January 2018;
Published: 30 January 2018.
Edited by:
Mattias Collin, Lund University, SwedenReviewed by:
Ana Carolina Morandini, University of the Pacific, United StatesCopyright © 2018 Kriebel, Hieke, Müller-Hilke, Nakata and Kreikemeyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernd Kreikemeyer, YmVybmQua3JlaWtlbWV5ZXJAbWVkLnVuaS1yb3N0b2NrLmRl Masanobu Nakata, bmFrYXRhQGRlbnQub3Nha2EtdS5hYy5qcA==
†Present address: Katja Kriebel, Institute of Microbiology, University of Rostock, Rostock, Germany
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.