 Paula Díaz
Paula Díaz Manuel Valenzuela Valderrama
Manuel Valenzuela Valderrama Jimena Bravo
Jimena Bravo Andrew F. G. Quest
Andrew F. G. Quest- 1Cellular Communication Laboratory, Instituto de Ciencias Biomédicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 2Advanced Center for Chronic Diseases, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 3Center for Molecular Studies of the Cell, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 4Instituto de Investigación e Innovación en Salud, Facultad de Ciencias de la Salud, Universidad Central de Chile, Santiago, Chile
Helicobacter pylori (H. pylori) infection is the major risk factor associated with the development of gastric cancer. The transition from normal mucosa to non-atrophic gastritis, triggered primarily by H. pylori infection, initiates precancerous lesions which may then progress to atrophic gastritis and intestinal metaplasia. Further progression to dysplasia and gastric cancer is generally believed to be attributable to processes that no longer require the presence of H. pylori. The responses that develop upon H. pylori infection are directly mediated through the action of bacterial virulence factors, which drive the initial events associated with transformation of infected gastric cells. Besides genetic and to date poorly defined environmental factors, alterations in gastric cell stress-adaptive mechanisms due to H. pylori appear to be crucial during chronic infection and gastric disease progression. Firstly, H. pylori infection promotes gastric cell death and reduced epithelial cell turnover in the majority of infected cells, resulting in primary tissue lesions associated with an initial inflammatory response. However, in the remaining gastric cell population, adaptive responses are induced that increase cell survival and proliferation, resulting in the acquisition of potentially malignant characteristics that may lead to precancerous gastric lesions. Thus, deregulation of these intrinsic survival-related responses to H. pylori infection emerge as potential culprits in promoting disease progression. This review will highlight the most relevant cellular adaptive mechanisms triggered upon H. pylori infection, including endoplasmic reticulum stress and the unfolded protein response, autophagy, oxidative stress, and inflammation, together with a subsequent discussion on how these factors may participate in the progression of a precancerous lesion. Finally, this review will shed light on how these mechanisms may be exploited as pharmacological targets, in the perspective of opening up new therapeutic alternatives for non-invasive risk control in gastric cancer.
Introduction
Worldwide, gastric cancer (GC) is the fifth most commonly diagnosed malignancy, and the third leading cause of cancer-related deaths per year (IARC, 2014; Lordick et al., 2014). Helicobacter pylori (H. pylori) is a Gram-negative, flagellated, microaerophilic bacterium that grows in close association with the lining of the stomach and the presence of this infection is identified as the major known risk factor associated with the development of GC (Suerbaum and Michetti, 2002). Epidemiological studies report that 2–3% of H. pylori-infected individuals eventually develop GC (Herrera and Parsonnet, 2009).
Generally, GC is viewed as the consequence of a multifactorial process, involving the host responses, bacterial virulence, diet, and other environmental factors (Valenzuela et al., 2015). The intestinal-type adenocarcinoma is the most frequent GC diagnosed (Hu et al., 2012). GC development is a multistep process initiated by the transition of normal mucosa to chronic superficial gastritis (non-atrophic gastritis), triggered primarily by H. pylori infection. Gastritis may progress to atrophic gastritis, then intestinal metaplasia, and finally to dysplasia and adenocarcinoma (Correa, 1992; Correa and Houghton, 2007). Prospective studies have shown that antibiotic-mediated eradication of H. pylori significantly reduces the incidence of precancerous lesions and thus highlights the role of H. pylori infection in early stages of gastric carcinogenesis (Mera et al., 2005). Indeed, removal of the bacterium with antibiotics can contribute to regression of atrophic gastritis, however this course of action is no longer effective once the disease has progressed to the stage of intestinal metaplasia (Massarrat et al., 2012). In accordance, eradication of H. pylori in patients with metaplasia and dysplasia does not reduce the risk of GC (Chen et al., 2016). These data suggest that the transition from atrophic gastritis to intestinal metaplasia is a crucial step in the progression towards GC and underscore the role of H. pylori in the initiation of the multistep cascade leading to precancerous lesions.
The inflammatory response that develops upon H. pylori infection is directly mediated through the action of a variety of bacterial virulence factors on host gastric epithelial cells (Peek and Crabtree, 2006). Pathogenicity of H. pylori is attributed to bacterial factors including, but not limited to, urease, vacuolating cytotoxin A (VacA), cag pathogenicity island, cytotoxin-associated gene A (CagA), peptidoglycan outer membrane proteins (e.g., BabA, SabA, OipA), and γ-glutamyl transpeptidase (GGT) (Polk and Peek, 2010; Valenzuela et al., 2013).
Besides genetic and environmental factors, alterations in gastric cell adaptive mechanisms due to H. pylori provoked stress appear to be crucial during chronic infection and gastric disorders. Initially, the H. pylori-induced effects observed during gastritis (and throughout the beginning of intestinal metaplasia) are an increase in apoptosis and a reduction in cell turnover in the majority of infected epithelial cells, thus resulting in atrophy, phenotypic changes, and the development of primary tissue lesions associated with the initial inflammatory response (Polk and Peek, 2010). However, in the remaining gastric cell population, adaptive responses are induced that increase cell survival and proliferation, and thus the acquisition of potentially malignant characteristics that allow the progression of gastric precancerous lesions, invasion, and metastasis. As summarized in Figure 1, deregulation of intrinsic survival-related responses to H. pylori infection may emerge as potential culprits in favoring disease progression.
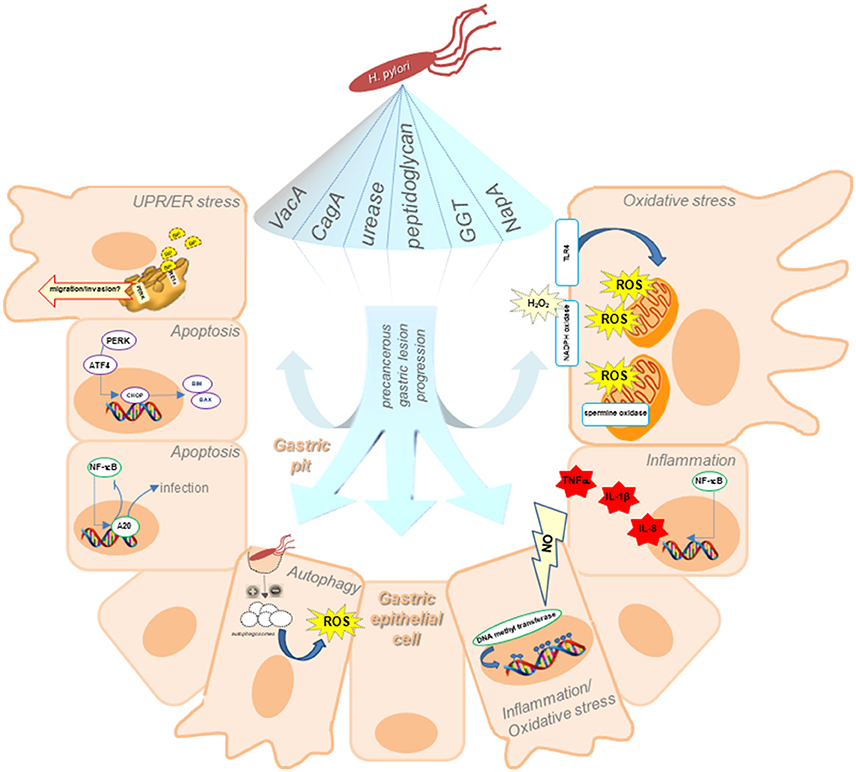
Figure 1. Schematic illustration of our current understanding of adaptive cellular mechanisms triggered upon H. pylori infection, including ER stress and the UPR, autophagy, oxidative stress, and inflammation, indicating how they may participate in precancerous lesion progression. Responses in host gastric epithelial cells located in the gastric pits triggered upon H. pylori infection are attributable to the action of bacterial virulence factors. ER stress associated with H. pylori infection, leads to an increase in BiP, suggesting that H. pylori-induction of ER stress is relevant in early stages of GC precancerous lesions. The ER stress sensor PERK may also facilitate tumor development by increasing the migratory and invasive potential of gastric cells. Unresolved ER stress results in apoptosis. ER-stress induced apoptosis is mediated by the transcription of CHOP, leading to expression of the pro-apoptotic proteins Bim and Bax. Moreover, H. pylori benefits from NF-κB activation and negatively regulates apoptosis via A20 deubiquitinylase activity, thereby promoting persistence of the infection. Inhibition (or activation) of autophagy, resulting in accumulation of autophagosomes within the cell at the beginning of the precancerous cascade are depicted as increasing ROS production leading to persistent oxidative stress, which in turn promotes the acquisition of characteristics, favoring invasion and metastasis. Long-term inflammation of the gastric mucosa generates significant amounts of nitric oxide (NO), which alters the transcriptional regulation in gastric cells by increasing DNA methyl transferase activity. The resulting hypermethylation of gene promoter sequences leads to epigenetic changes in gene expression. Additionally, NF-κB target genes include those representing polymorphisms associated with an increased risk for GC in patients, such as TNFα, IL-1β, and IL-8. Gastric cells produce ROS in response to H. pylori infection by inducing pro-oxidant activities, such as the host spermine oxidase, NADPH oxidase or generating ROS from mitochondria following activation of TLR4 signaling.
Endoplasmic Reticulum Stress and the Unfolded Protein Response are Associated with the Pathogenesis of H. pylori-Induced Gastric Tumourigenesis
Protein folding stress at the endoplasmic reticulum (ER) is involved in the pathogenesis of a variety of human diseases (Hetz, 2012). The accumulation of unfolded and/or misfolded proteins within the ER induces ER stress, which can be resolved by an adaptive mechanism termed the unfolded protein response (UPR) (Hetz, 2012).
Activation of ER stress was found to be significantly associated with H. pylori-positive GC (92%) (Baird et al., 2013). In intestinal metaplasia, levels of key modulators of the UPR activated by ER stress such as HSPA5 (encoding for binding immunoglobulin protein; BiP), C/EBP-homologous protein [CHOP; positively controlled by the protein kinase R (PKR)-like ER kinase (PERK) pathway], and XBP1 (downstream of inositol-requiring enzyme-1 alpha; IRE1-α), were reportedly higher in H. pylori-positive subjects compared to earlier precancerous stages (i.e., non-atrophic and atrophic gastritis) (Baird et al., 2013). This suggests that H. pylori-induction of ER stress may play a significant role in the early stages of precancerous lesion formation.
Above a certain threshold, unresolved ER stress results in apoptosis and evasion of apoptotic cell death is recognized as one of the hallmarks of cancer cells (Hanahan and Weinberg, 2011). Thus, ER stress-induced cell death in the mucosa after H. pylori infection may favor the progression from precancerous lesions into GC. ER-stress induced apoptotic cell death is mediated by the transcription of CHOP (Tabas and Ron, 2011). In line with this, it was reported that CHOP is transcriptionally upregulated following incubation of gastric cells with H. pylori bacterial virulence factor VacA (Akazawa et al., 2013). Moreover, silencing of the PERK gene (EIF2AK3) attenuated VacA-mediated phosphorylation of eukaryotic initiation factor-2 alpha (eIF2α), expression of BH3-only protein Bim and Bax, as well as cell death induced by VacA (Akazawa et al., 2013), indicating that ER stress may lead to apoptotic cell death during VacA-induced toxicity (Akazawa et al., 2013). In line with this hypothesis, it has been recently reported that H. pylori benefits from NF-κB activation and negatively regulates apoptotic cell death via a deubiquitinylase activity, thereby promoting persistence of the infection (Lim et al., 2017).
Furthermore, besides epithelial cells, dendritic cell alteration is also observed in the H. pylori infected gastric mucosa. Treatment of dendritic cells with purified H. pylori VacA was shown to induce translocation of cytoplasmic Bax and cytochrome c release from mitochondria and thus bring about apoptosis (Kim et al., 2015). Indeed, suppression of ER stress appears to result in a significant inhibition of the VacA-induced apoptotic cell death in dendritic cells, suggesting that ER stress is also critical for regulation of dendritic cell apoptosis in response to VacA stimulation (Kim et al., 2015).
The role of the ER stress sensor PERK in tumor development remains controversial (Ranganathan et al., 2008). In agreement with the classical role of PERK in the UPR, some studies suggest that PERK activation inhibits tumor cell proliferation leading to apoptosis (Akazawa et al., 2013; Huber et al., 2013; Martín-Pérez et al., 2014). However, other reports have shown that PERK activation facilitates tumor development by promoting tumor cell survival and enhancing angiogenesis (Atkins et al., 2013; Mujcic et al., 2013). Consistent with this latter role, PERK promotes tumourigenesis (Bobrovnikova-Marjon et al., 2010) and PERK-deficient cells display a reduced ability to form solid tumors in nude mice (Spiotto et al., 2010). More recently, a new role for PERK signaling in promoting cancer cell migration and invasion was proposed based on experiments evaluating the effects of moderate PERK activation on medulloblastoma cell migration and invasion (Jamison et al., 2015). Overall, these observations support the idea that, besides its classical role in UPR and apoptosis, following H. pylori infection, PERK expression also promotes human gastric cell migration and invasion and thus the acquisition of an aggressive phenotype.
H. pylori-Mediated Autophagy Favors the Progression of Gastric Cancer Precursor Lesions
The link between ER stress and autophagy is well established, and in the context of H. pylori, it has been demonstrated that the secreted antigen HP0175 can regulate PERK, which in turn activates the transcription of ATF4 and CHOP leading to the induction of autophagy in gastric epithelial cells (Halder et al., 2015).
Autophagy is a conserved mechanism by which the eukaryotic cell seeks to maintain cellular homeostasis; it is also induced as a survival response upon exposure to various stress stimuli, including deprivation of nutrients and infection by microbial pathogens (He and Klionsky, 2009). Briefly, cellular components destined for degradation are sequestered within the autophagosome that fuses with the lysosome (autolysosome), where the cargo is degraded by lysosomal enzymes and recycled back to the cell (Russell et al., 2014).
More recently, autophagy has also come to be recognized as an important intracellular defense mechanism that challenges pathogenic bacteria after their internalization by the host cell. Several types of pathogens have developed different strategies to either escape from lysosomal degradation, or control and modulate autophagy to their own benefit (Lerena et al., 2010). In the context of H. pylori infection, Terebiznik et al. (2009) reported that infection of gastric epithelial cells with H. pylori for short periods (6 h) induced autophagy in a manner that was independent of the virulence factors CagA and urease, but dependent on VacA and the latter factor alone was sufficient to induce this effect (Terebiznik et al., 2009). This suggested that autophagy acts as a mechanism by which infected cells limit toxin-induced cellular damage and thus favor cell survival. In a subsequent study, Raju et al. (2012) showed that prolonged exposure to VacA (24 h) disrupted the antiphagocytic pathway and defective autophagosomes were reported to accumulate within the cells (Raju et al., 2012). Moreover, these authors showed that p62, a selective substrate for autophagy-mediated degradation, increased in gastric biopsies of patients infected with the toxic s1m1 VacA strain in comparison with those infected with a nontoxic strain (VacA s2m2). Additionally, it was reported that the effect of H. pylori in the regulation of autophagy was linked to changes in the expression of autophagy-related genes in gastric cells and macrophages (Castaño-Rodríguez et al., 2015). In line with this, silencing by methylation of the microtubule-associated protein 1 light chain 3 variant 1 (MAP1LC3Av1), an important protein in the autophagy process, was observed in gastric tissue infected with H. pylori (Muhammad et al., 2017). The available literature suggests that H. pylori modulates autophagy in the host and points toward VacA as a possible mediator; however, the reported results provide contradictory mechanisms concerning how precisely VacA might regulate this process. In addition, although the effect of virulence factor CagA in autophagy was initially discarded by Terebiznik et al. (2009), this factor has recently been identified as a negative regulator of autophagy in gastric mucosa of patients and AGS cells infected with a non-functional VacA strain (VacA s1m2) (Li et al., 2017). Consequently, CagA might cause the inhibition of autophagy through the c-Met-PI3K/Akt-mTOR signaling pathway. Moreover, these authors also found higher expression of p62 in gastric biopsies of patients infected with CagA+ strains in comparison with those infected with CagA− strains (Li et al., 2017). Considering the latter observation, the differences found in the literature can be explained in part by the use of different cell lines and bacterial strains, however, the possibility that additional H. pylori virulence factors are likely to be involved in the regulation of autophagy in gastric cells merits further consideration. Thus, autophagy acts as a quality control system in early stages of cancer, and one may infer that the inhibition of this process at the beginning of the precancerous cascade should increase reactive oxygen species (ROS) production leading to persistent oxidative stress, which in turn causes selective pressure for the acquisition of characteristics, such as increased growth, invasion, and metastasis (Azad et al., 2009). This would, in the case of GC, favor the progression of precursor lesions. In summary, a strong connection between H. pylori infection and autophagy is emerging that may prove relevant to GC progression.
H. pylori-Associated Inflammation
Chronic inflammation contributes to the pathogenesis of several types of cancer (Fernandes et al., 2015) and is particularly relevant in the case of H. pylori-associated GC (Valenzuela et al., 2015). H. pylori infection is invariably associated with gastritis; however, only a minor proportion of individuals progress through the pre-neoplastic cascade (Peek and Blaser, 2002). Particularly, the interplay between host responses and bacterial virulence factors seems most relevant in determining the extent of inflammation and the clinical outcome (Chmiela et al., 2017), which in turn may be favored by a permissive environment.
Beyond representing an innate response to pathogens, long-term inflammation of the gastric mucosa generates significant amounts of the diffusible molecule nitric oxide (Nam et al., 2004), which contributes not only to the damage of nucleotide bases in host DNA, but also alters the transcriptional regulation in gastric cells by increasing DNA methyl transferase activity (Huang et al., 2012). In doing so, epigenetic changes involving the hypermethylation of DNA sequences, particularly in tumor suppressor genes (coding and non-coding) are observed in the gastric mucosa of infected individuals (Kaise et al., 2008; Valenzuela et al., 2015). Accordingly, the promoter region of the tumor suppressor E-Cadherin is frequently hypermethylated in adult patients infected with H. pylori (Perri et al., 2007). Also, the demethylating agent 5-azacytidine has been demonstrated to reduce GC incidence in the Mongolian gerbil model of infection (Niwa et al., 2013). Additionally, chronic exposition to inflammatory cytokines is associated with deregulation of the Hedgehog/GLI signaling pathway, which is known to be involved in the maintenance of gastric physiology (Wessler et al., 2017). In doing so, chief cells are reprogrammed toward a metaplastic phenotype, which together with the dysregulated differentiation of infiltrating gastric stem cells (e.g., bone marrow-derived cells), contributes to the so-called mosaicism in GC (Zhang et al., 2016; Leushacke et al., 2017; Mills and Goldenring, 2017).
T-cell responses are essential for the generation of the inflammatory response following H. pylori infection and gastric carcinogenesis as demonstrated in animal models of infection (Kandulski et al., 2010). Notably, H. pylori has developed mechanisms to persist in the gastric niche suppressing inflammation at sites of colonization (Every et al., 2011). Accordingly, some virulence factors have been shown to control the extent of inflammatory responses, not only by inhibiting T-cell activation, but also by reducing phagocytosis or by promoting evasion of toll-like receptor (TLR) recognition, as well as by inducing tolerogenic effects in dendritic cells (Mejías-Luque and Gerhard, 2017). Well documented in this context are the actions of bacterial virulence factors, such as the secreted vacuolating toxin VacA and GGT, as well as the oncogenic cytotoxin CagA. Present in more virulent and carcinogenic strains, CagA is injected via the type IV secretion system encoded in the bacterial pathogenicity island (CagPAI) (Miyaji et al., 2000; Cha et al., 2010; Mejías-Luque and Gerhard, 2017). GGT, on the other hand, is a soluble factor that provokes oxidative stress, loss of survivin and enhanced apoptosis in gastric cells (Valenzuela et al., 2013). In addition, H. pylori secreted peptidyl prolyl cis, trans-isomerase (HP0175) causes a gastric T-cell (Th17) response in patients with GC; leading to the production of IL-17 and IL-21, matrix degradation and the induction of pro-angiogenic pathways in response to HP0175 (Amedei et al., 2014).
The balance between these contradictory responses seems to be determined largely by intrinsic host-specific mechanisms. In agreement with this concept, knock-out animal models have revealed the importance of some host factors in modulating inflammation. For instance, TLR9 signaling has demonstrated anti-inflammatory effects by controlling exacerbated IL-17 production (Varga et al., 2016). Alternatively, the trefoil factor 1 suppresses H. pylori-induced inflammation by antagonizing NF-κB activation (Soutto et al., 2015). The NF-κB protein is a master regulator of the inflammatory transcriptional response to bacterial molecules, mainly in response to injected factors, such as peptidoglycan and CagA (Sokolova and Naumann, 2017). Importantly, certain NF-κB target genes such as TNFα, IL-1β, IL-2, and IL-8, present polymorphisms that are associated with increased risk for GC in patients (Valenzuela et al., 2015; Melchiades et al., 2017). However, it is important to consider the participation of other cell-signaling pathways in the production of inflammatory molecules. For instance, activation of the epidermal growth factor receptor (EGFR) is also associated with inflammation, DNA damage and gastric carcinogenesis (Sierra et al., 2017).
H. pylori and Oxidative/Nitrosative Stress
Oxidative stress in the gastric mucosa as a consequence of H. pylori infection is a crucial contributing factor to gastric carcinogenesis. Reportedly, infection was shown to correlate with increased damage due to oxidative stress of the gastric mucosa (Butcher et al., 2017). Although reversible upon bacterial eradication (Pignatelli et al., 2001), the consequences of oxidative stress are evident through observed changes in global lipid and protein expression (Baek et al., 2004) and an increase in damaged biomolecules, such as DNA (modification of bases, telomere shortening) (Obst et al., 2000; Lee et al., 2016). Additionally, anti-oxidant capacity is also reduced due to decreased levels of antioxidant molecules, such a glutathione (GSH) (Shirin et al., 2001) in the gastric mucosa of H. pylori-infected patients. The sources of reactive oxygen and nitrogen species are mainly neutrophils, macrophages and the gastric cells themselves (Valenzuela et al., 2015). Thus, in addition to the inflammatory cells, gastric cells produce ROS in response to H. pylori infection in different ways. For instance, by inducing pro-oxidant activities, such as the host spermine oxidase enzyme, which also promotes GC risk associated with H. pylori CagA cytotoxin activity (Chaturvedi et al., 2011), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Cha et al., 2010) or generating ROS from mitochondria following activation of TLR4 signaling (Yuan et al., 2013). Also, H. pylori-neutrophil-activating protein A (NapA) activity promotes host gastric cell production of reactive oxygen intermediates (Wang et al., 2006b). Furthermore, the secreted bacterial GGT has emerged as a relevant pathogenic factor associated with oxidative stress and inflammation (Gong et al., 2010). GGT activity is associated with oxidation of membrane lipids using GSH as a substrate in the presence of iron (Valenzuela et al., 2015). Nitric oxide generated by the activity of iNOS in macrophages, lymphocytes and gastric cells of the infected mucosa yields highly toxic peroxynitrite, which damages proteins and DNA by generating nitrotyrosine and DNA adducts, respectively (Sakaguchi et al., 1999; Cherdantseva et al., 2014; Valenzuela et al., 2015).
On the other hand, gastric cells can escape from the oxidative stress induced by H. pylori infection by producing scavenger molecules, such as metallothioneins, which have been shown to be crucial factors in protecting against H. pylori-induced gastric erosive lesions in an animal model (Mita et al., 2008). Other relevant antioxidant mechanisms include those involving global regulation of energy metabolism, such as the AMP-activated protein kinase (AMPK) (Zhao et al., 2015) or the cyto-protective activity of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Yanaka, 2011). Such a harmful environment may represent major challenges for infecting bacteria, comparable even to that of acid stress. However, H. pylori is a well-adapted bacteria, able to resist oxidative stress due to mechanisms that permit the successful colonization and persistence in the gastric niche (Wang et al., 2006a). For instance, isogenic mutants deficient in superoxide dismutase (Seyler et al., 2001), NADPH quinone reductase (Wang and Maier, 2004), thioredoxin (Windle et al., 2000), and catalase (KatA) (Harris et al., 2003) activities are sensitive to oxidative stress and are largely defective in host colonization (Seyler et al., 2001). Furthermore, thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stress (McGee et al., 2006). Interestingly, the bacteria itself reportedly also produces ROS (Ding et al., 2007).
Table 1 summarizes the adaptive mechanisms due to H. pylori infection that could promote the acquisition of potentially malignant characteristics and favor disease progression to GC.
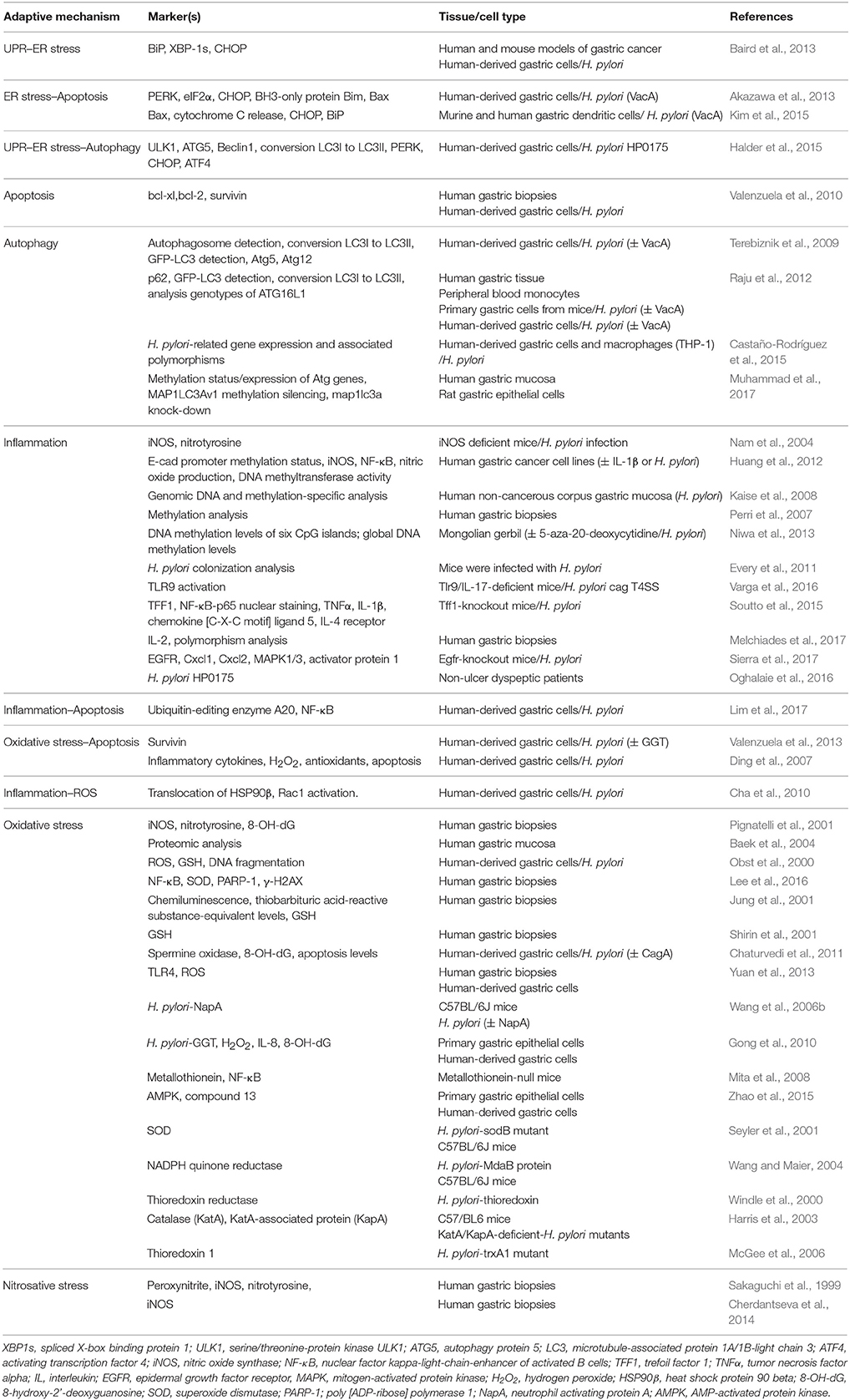
Table 1. Summary of literature references linking H. pylori infection to induction of the UPR, ER stress, apoptosis, autophagy, inflammation, and oxidative stress.
Discussion-Potential Therapeutic Strategies
Understanding the regulation of adaptive chronic mechanisms by H. pylori may open up therapeutic options to prevent the progression of GC precursor lesions.
In line with this, PERK has emerged as potential therapeutic target in cancer. The compound GSK2606414 (GlaxoSmithKline®) was the first reported small molecular inhibitor of PERK, which is highly specific (Axten et al., 2012). When administered orally, GSK2606414 inhibits tumor growth in a dose-dependent manner in mice with human pancreatic BxPC3 tumors (Axten et al., 2012). Moreover, a second PERK inhibitor, GSK2656157, was developed for preclinical studies exhibiting promising results in xenograft mouse models bearing human tumors (Atkins et al., 2013; Axten et al., 2013). More recently, a new role for PERK signaling in promoting cancer cell migration and invasion has been proposed. Medulloblastoma cell lines treated with GSK2606414 exhibited a significant reduction in the level of phospho-eIF2α and concomitant suppression of cell migration and invasion (Jamison et al., 2015). Overall, these observations support the idea that, besides its classical role in UPR and apoptosis, PERK also promotes an aggressive cellular phenotype by increasing the migratory and invasive potential. With this in mind, it is not surprising that there is considerable interest in defining whether PERK contributes to tumor progression and whether it represents a suitable therapeutic target in cancer (Pytel et al., 2016).
On the other hand, cancer cells are thought to use autophagy as a source of energy in the unfavorable metastatic environment, and a number of clinical trials are now revealing the promising role of chloroquine, an anti-malaria drug and autophagy inhibitor, that also suppresses tumor growth and metastasis (Kimura et al., 2013; Maes et al., 2014). In addition, a potential role for gastrin in the regulation of autophagy has been recently proposed, opening up new avenues for the treatment of GC by targeting autophagy in combination with conventional cytostatic drugs (Rao et al., 2017). Furthermore, the use of statins has been reported to enhance H. pylori eradication by antibiotics and to reduce H. pylori-mediated inflammation by promoting autophagy (Liao et al., 2016); however, the mechanisms underlying these effects remain unclear.
Studies using pharmacological approaches to modulate inflammation or knock-out animal models of infection lacking cytokine receptors have highlighted the crucial importance of pro-inflammatory responses in promoting H. pylori-associated gastric carcinogenesis (Waghray et al., 2010; Huang et al., 2013; Oshima et al., 2014). This may point towards the importance of protective natural dietary compounds and micronutrients in reducing inflammation and preventing carcinogenesis (Wang, 2014; Noto and Peek, 2015; Zitvogel et al., 2017). Thus, natural anti-inflammatory compounds may hold considerable promise in preventing H. pylori-associated gastric pathologies (Wang, 2014).
In summary, the mechanisms described here may be exploited as pharmacological targets, in the perspective of opening up new therapeutic alternatives for non-invasive risk control in GC. This possibility is particularly intriguing when dealing with GC, which represents the leading cause of cancer deaths in several countries (IARC, 2014). Many highly specific inhibitors to regulate and/or control these adaptive mechanisms are already available and being evaluated in clinical trials for other cancers.
Author Contributions
PD, MV, and JB drafted the manuscript with support from AQ. PD drafted summary figure and table. PD, MV, JB, and AQ critically revised the article. All authors read and approved the final article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported CONICYT-FONDAP 15130011, Fondecyt 1130250, 1170925 (AQ); Fondecyt 3170140 (PD); Fondecyt 1171615 (MV); and Fondecyt 3160384 (JB). Dr. Gareth Owen is gratefully acknowledged for careful revision of the final manuscript.
References
Akazawa, Y., Isomoto, H., Matsushima, K., Kanda, T., Minami, H., Yamaghchi, N., et al. (2013). Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE 8:e82322. doi: 10.1371/journal.pone.0082322
Amedei, A., Munari, F., Bella, C. D., Niccolai, E., Benagiano, M., Bencini, L., et al. (2014). Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern. Emerg. Med. 9, 303–309. doi: 10.1007/s11739-012-0867-9
Atkins, C., Liu, Q., Minthorn, E., Zhang, S. Y., Figueroa, D. J., Moss, K., et al. (2013). Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002. doi: 10.1158/0008-5472.CAN-12-3109
Axten, J. M., Medina, J. R., Feng, Y., Shu, A., Romeril, S. P., Grant, S. W., et al. (2012). Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. doi: 10.1021/jm300713s
Axten, J. M., Romeril, S. P., Shu, A., Ralph, J., Medina, J. R., Feng, Y., et al. (2013). Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 4, 964–968. doi: 10.1021/ml400228e
Azad, M. B., Chen, Y., and Gibson, S. B. (2009). Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid. Redox Signal. 11, 777–790. doi: 10.1089/ars.2008.2270
Baek, H. Y., Lim, J. W., Kim, H., Kim, J. M., Kim, J. S., Jung, H. C., et al. (2004). Oxidative-stress-related proteome changes in Helicobacter pylori-infected human gastric mucosa. Biochem. J. 379, 291–299. doi: 10.1042/bj20031208
Baird, M., Woon Ang, P., Clark, I., Bishop, D., Oshima, M., Cook, M. C., et al. (2013). The unfolded protein response is activated in Helicobacter-induced gastric carcinogenesis in a non-cell autonomous manner. Lab. Invest. 93, 112–122. doi: 10.1038/labinvest.2012.131
Bobrovnikova-Marjon, E., Grigoriadou, C., Pytel, D., Zhang, F., Ye, J., Koumenis, C., et al. (2010). PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29, 3881–3895. doi: 10.1038/onc.2010.153
Butcher, L. D., den Hartog, G., Ernst, P. B., and Crowe, S. E. (2017). Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 3, 316–322. doi: 10.1016/j.jcmgh.2017.02.002
Castaño-Rodríguez, N., Kaakoush, N. O., Goh, K. L., Fock, K. M., and Mitchell, H. M. (2015). Autophagy in Helicobacter pylori infection and related gastric cancer. Helicobacter 20, 353–369. doi: 10.1111/hel.12211
Cha, B., Lim, J. W., Kim, K. H., and Kim, H. (2010). HSP90β interacts with Rac1 to activate NADPH oxidase in Helicobacter pylori-infected gastric epithelial cells. Int. J. Biochem. Cell Biol. 42, 1455–1461. doi: 10.1016/j.biocel.2010.04.015
Chaturvedi, R., Asim, M., Romero-Gallo, J., Barry, D. P., Hoge, S., de Sablet, T., et al. (2011). Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 141, 1696–1708.e2. doi: 10.1053/j.gastro.2011.07.045
Chen, H. N., Wang, Z., Li, X., and Zhou, Z. G. (2016). Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: evidence from a meta-analysis. Gastric Cancer 19, 166–175. doi: 10.1007/s10120-015-0462-7
Cherdantseva, L. A., Potapova, O. V., Sharkova, T. V., Belyaeva, Y. Y., and Shkurupiy, V. A. (2014). Association of Helicobacter pylori and iNOS production by macrophages and lymphocytes in the gastric mucosa in chronic gastritis. J. Immunol. Res. 2014:762514. doi: 10.1155/2014/762514
Chmiela, M., Karwowska, Z., Gonciarz, W., Allushi, B., and Staczek, P. (2017). Host pathogen interactions in Helicobacter pylori related gastric cancer. World J. Gastroenterol. 23, 1521–1540. doi: 10.3748/wjg.v23.i9.1521
Correa, P. (1992). Human gastric carcinogenesis: a multistep and multifactorial process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52, 6735–6740.
Correa, P., and Houghton, J. (2007). Carcinogenesis of Helicobacter pylori. Gastroenterology 133, 659–672. doi: 10.1053/j.gastro.2007.06.026
Ding, S. Z., Minohara, Y., Fan, X. J., Wang, J., Reyes, V. E., Patel, J., et al. (2007). Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect. Immun. 75, 4030–4039. doi: 10.1128/IAI.00172-07
Every, A. L., Ng, G. Z., Skene, C. D., Harbour, S. N., Walduck, A. K., Mcguckin, M. A., et al. (2011). Localized suppression of inflammation at sites of Helicobacter pylori colonization. Infect. Immun. 79, 4186–4192. doi: 10.1128/IAI.05602-11
Fernandes, J. V., Cobucci, R. N., Jatobá, C. A., Fernandes, T. A., de Azevedo, J. W., and de Araújo, J. M. (2015). The role of the mediators of inflammation in cancer development. Pathol. Oncol. Res. 21, 527–534. doi: 10.1007/s12253-015-9913-z
Gong, M., Ling, S. S., Lui, S. Y., Yeoh, K. G., and Ho, B. (2010). Helicobacter pylori gamma-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology 139, 564–573. doi: 10.1053/j.gastro.2010.03.050
Halder, P., Datta, C., Kumar, R., Sharma, A. K., Basu, J., and Kundu, M. (2015). The secreted antigen, HP0175, of Helicobacter pylori links the unfolded protein response (UPR) to autophagy in gastric epithelial cells. Cell. Microbiol. 17, 714–729. doi: 10.1111/cmi.12396
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Harris, A. G., Wilson, J. E., Danon, S. J., Dixon, M. F., Donegan, K., and Hazell, S. L. (2003). Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 149, 665–672. doi: 10.1099/mic.0.26012-0
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi: 10.1146/annurev-genet-102808-114910
Herrera, V., and Parsonnet, J. (2009). Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 15, 971–976. doi: 10.1111/j.1469-0691.2009.03031.x
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hu, B., El Hajj, N., Sittler, S., Lammert, N., Barnes, R., and Meloni-Ehrig, A. (2012). Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 3, 251–261. doi: 10.3978/j.issn.2078-6891.2012.021
Huang, F. Y., Chan, A. O., Lo, R. C., Rashid, A., Wong, D. K., Cho, C. H., et al. (2013). Characterization of interleukin-1β in Helicobacter pylori-induced gastric inflammation and DNA methylation in interleukin-1 receptor type 1 knockout IL-1R1−/− mice. Eur. J. Cancer 49, 2760–2770. doi: 10.1016/j.ejca.2013.03.031
Huang, F. Y., Chan, A. O., Rashid, A., Wong, D. K., Cho, C. H., and Yuen, M. F. (2012). Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer 118, 4969–4980. doi: 10.1002/cncr.27519
Huber, A. L., Lebeau, J., Guillaumot, P., Pétrilli, V., Malek, M., Chilloux, J., et al. (2013). p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol. Cell 49, 1049–1059. doi: 10.1016/j.molcel.2013.01.009
Jamison, S., Lin, Y., and Lin, W. (2015). Pancreatic endoplasmic reticulum kinase activation promotes medulloblastoma cell migration and invasion through induction of vascular endothelial growth factor A. PLoS ONE 10:e0120252. doi: 10.1371/journal.pone.0120252
Jung, H. K., Lee, K. E., Chu, S. H., and Yi, S. Y. (2001). Reactive oxygen species activity, mucosal lipoperoxidation and glutathione in Helicobacter pylori-infected gastric mucosa. J. Gastroenterol. Hepatol. 16, 1336–1340. doi: 10.1046/j.1440-1746.2001.02647.x
Kaise, M., Yamasaki, T., Yonezawa, J., Miwa, J., Ohta, Y., and Tajiri, H. (2008). CpG island hypermethylation of tumor-suppressor genes in H. pylori-infected non-neoplastic gastric mucosa is linked with gastric cancer risk. Helicobacter 13, 35–41. doi: 10.1111/j.1523-5378.2008.00572.x
Kandulski, A., Malfertheiner, P., and Wex, T. (2010). Role of regulatory T-cells in H. pylori-induced gastritis and gastric cancer. Anticancer Res. 30, 1093–1103.
Kim, J. M., Kim, J. S., Kim, N., Ko, S. H., Jeon, J. I., and Kim, Y. J. (2015). Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells. J. Gastroenterol. Hepatol. 30, 99–108. doi: 10.1111/jgh.12663
Kimura, T., Takabatake, Y., Takahashi, A., and Isaka, Y. (2013). Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 73, 3–7. doi: 10.1158/0008-5472.CAN-12-2464
Lee, W. P., Hou, M. C., Lan, K. H., Li, C. P., Chao, Y., Lin, H. C., et al. (2016). Helicobacter pylori-induced chronic inflammation causes telomere shortening of gastric mucosa by promoting PARP-1-mediated non-homologous end joining of DNA. Arch. Biochem. Biophys. 606, 90–98. doi: 10.1016/j.abb.2016.07.014
Lerena, M. C., Vázquez, C. L., and Colombo, M. I. (2010). Bacterial pathogens and the autophagic response. Cell. Microbiol. 12, 10–18. doi: 10.1111/j.1462-5822.2009.01403.x
Leushacke, M., Tan, S. H., Wong, A., Swathi, Y., Hajamohideen, A., Tan, L. T., et al. (2017). Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat. Cell Biol. 19, 774–786. doi: 10.1038/ncb3541
Li, N., Tang, B., Jia, Y. P., Zhu, P., Zhuang, Y., Fang, Y., et al. (2017). Helicobacter pylori CagA protein negatively regulates autophagy and promotes inflammatory response via c-Met-PI3K/Akt-mTOR signaling pathway. Front. Cell. Infect. Microbiol. 7:417. doi: 10.3389/fcimb.2017.00417
Liao, W. C., Huang, M. Z., Wang, M. L., Lin, C. J., Lu, T. L., Lo, H. R., et al. (2016). Statin decreases Helicobacter pylori burden in macrophages by promoting autophagy. Front. Cell. Infect. Microbiol. 6:203. doi: 10.3389/fcimb.2016.00203
Lim, M. C. C., Maubach, G., Sokolova, O., Feige, M. H., Diezko, R., Buchbinder, J., et al. (2017). Pathogen-induced ubiquitin-editing enzyme A20 bifunctionally shuts off NF-κB and caspase-8-dependent apoptotic cell death. Cell Death Differ. 24, 1621–1631. doi: 10.1038/cdd.2017.89
Lordick, F., Allum, W., Carneiro, F., Mitry, E., Tabernero, J., Tan, P., et al. (2014). Unmet needs and challenges in gastric cancer: the way forward. Cancer Treat. Rev. 40, 692–700. doi: 10.1016/j.ctrv.2014.03.002
Maes, H., Kuchnio, A., Peric, A., Moens, S., Nys, K., De Bock, K., et al. (2014). Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26, 190–206. doi: 10.1016/j.ccr.2014.06.025
Martín-Pérez, R., Palacios, C., Yerbes, R., Cano-González, A., Iglesias-Serret, D., Gil, J., et al. (2014). Activated ERBB2/HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Res. 74, 1766–1777. doi: 10.1158/0008-5472.CAN-13-1747
Massarrat, S., Haj-Sheykholeslami, A., Mohamadkhani, A., Zendehdel, N., Rakhshani, N., Stolte, M., et al. (2012). Precancerous conditions after H. pylori eradication: a randomized double blind study in first degree relatives of gastric cancer patients. Arch. Iran. Med. 15, 664–669.
McGee, D. J., Kumar, S., Viator, R. J., Bolland, J. R., Ruiz, J., Spadafora, D., et al. (2006). Helicobacter pylori thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stresses. J. Biol. Chem. 281, 3290–3296. doi: 10.1074/jbc.M506139200
Mejías-Luque, R., and Gerhard, M. (2017). Immune evasion strategies and persistence of Helicobacter pylori. Curr. Top. Microbiol. Immunol. 400, 53–71. doi: 10.1007/978-3-319-50520-6_3
Melchiades, J. L., Zabaglia, L. M., Sallas, M. L., Orcini, W. A., Chen, E., Smith, M., et al. (2017). Polymorphisms and haplotypes of the interleukin 2 gene are associated with an increased risk of gastric cancer. The possible involvement of Helicobacter pylori. Cytokine 96, 203–207. doi: 10.1016/j.cyto.2017.04.020
Mera, R., Fontham, E. T., Bravo, L. E., Bravo, J. C., Piazuelo, M. B., Camargo, M. C., et al. (2005). Long term follow up of patients treated for Helicobacter pylori infection. Gut 54, 1536–1540. doi: 10.1136/gut.2005.072009
Mills, J. C., and Goldenring, J. R. (2017). Metaplasia in the stomach arises from gastric chief cells. Cell Mol. Gastroenterol. Hepatol. 4, 85–88. doi: 10.1016/j.jcmgh.2017.03.006
Mita, M., Satoh, M., Shimada, A., Okajima, M., Azuma, S., Suzuki, J. S., et al. (2008). Metallothionein is a crucial protective factor against Helicobacter pylori-induced gastric erosive lesions in a mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G877–884. doi: 10.1152/ajpgi.00251.2007
Miyaji, H., Azuma, T., Ito, S., Abe, Y., Gejyo, F., Hashimoto, N., et al. (2000). Helicobacter pylori infection occurs via close contact with infected individuals in early childhood. J. Gastroenterol. Hepatol. 15, 257–262. doi: 10.1046/j.1440-1746.2000.02070.x
Muhammad, J. S., Nanjo, S., Ando, T., Yamashita, S., Maekita, T., Ushijima, T., et al. (2017). Autophagy impairment by Helicobacter pylori-induced methylation silencing of MAP1LC3Av1 promotes gastric carcinogenesis. Int. J. Cancer 140, 2272–2283. doi: 10.1002/ijc.30657
Mujcic, H., Nagelkerke, A., Rouschop, K. M., Chung, S., Chaudary, N., Span, P. N., et al. (2013). Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 19, 6126–6137. doi: 10.1158/1078-0432.CCR-13-0526
Nam, K. T., Oh, S. Y., Ahn, B., Kim, Y. B., Jang, D. D., Yang, K. H., et al. (2004). Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 53, 1250–1255. doi: 10.1136/gut.2003.030684
Niwa, T., Toyoda, T., Tsukamoto, T., Mori, A., Tatematsu, M., and Ushijima, T. (2013). Prevention of Helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev. Res. (Phila) 6, 263–270. doi: 10.1158/1940-6207.CAPR-12-0369
Noto, J. M., and Peek, R. M. Jr. (2015). Micronutrients: a double-edged sword in microbial-induced gastric carcinogenesis. Trends Cancer 1, 136–144. doi: 10.1016/j.trecan.2015.07.002
Obst, B., Wagner, S., Sewing, K. F., and Beil, W. (2000). Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 21, 1111–1115. doi: 10.1093/carcin/21.6.1111
Oghalaie, A., Saberi, S., Esmaeili, M., Ebrahimzadeh, F., Barkhordari, F., Ghamarian, A., et al. (2016). Helicobacter pylori peptidyl prolyl isomerase expression is associated with the severity of gastritis. J. Gastrointest. Cancer 47, 375–380. doi: 10.1007/s12029-016-9849-x
Oshima, H., Ishikawa, T., Yoshida, G. J., Naoi, K., Maeda, Y., Naka, K., et al. (2014). TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 33, 3820–3829. doi: 10.1038/onc.2013.356
Peek, R. M. Jr., and Blaser, M. J. (2002). Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37. doi: 10.1038/nrc703
Peek, R. M. Jr., and Crabtree, J. E. (2006). Helicobacter infection and gastric neoplasia. J. Pathol. 208, 233–248. doi: 10.1002/path.1868
Perri, F., Cotugno, R., Piepoli, A., Merla, A., Quitadamo, M., Gentile, A., et al. (2007). Aberrant DNA methylation in non-neoplastic gastric mucosa of H. pylori infected patients and effect of eradication. Am. J. Gastroenterol. 102, 1361–1371. doi: 10.1111/j.1572-0241.2007.01284.x
Pignatelli, B., Bancel, B., Plummer, M., Toyokuni, S., Patricot, L. M., and Ohshima, H. (2001). Helicobacter pylori eradication attenuates oxidative stress in human gastric mucosa. Am. J. Gastroenterol. 96, 1758–1766. doi: 10.1111/j.1572-0241.2001.03869.x
Polk, D. B., and Peek, R. M. Jr. (2010). Helicobacter pylori: gastric cancer and beyond. Nat. Rev. Cancer 10, 403–414. doi: 10.1038/nrc2857
Pytel, D., Majsterek, I., and Diehl, J. A. (2016). Tumor progression and the different faces of the PERK kinase. Oncogene 35, 1207–1215. doi: 10.1038/onc.2015.178
Raju, D., Hussey, S., Ang, M., Terebiznik, M. R., Sibony, M., Galindo-Mata, E., et al. (2012). Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142, 1160–1171. doi: 10.1053/j.gastro.2012.01.043
Ranganathan, A. C., Ojha, S., Kourtidis, A., Conklin, D. S., and Aguirre-Ghiso, J. A. (2008). Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 68, 3260–3268. doi: 10.1158/0008-5472.CAN-07-6215
Rao, S. V., Solum, G., Niederdorfer, B., Nørsett, K. G., Bjørkøy, G., and Thommesen, L. (2017). Gastrin activates autophagy and increases migration and survival of gastric adenocarcinoma cells. BMC Cancer 17:68. doi: 10.1186/s12885-017-3055-5
Russell, R. C., Yuan, H. X., and Guan, K. L. (2014). Autophagy regulation by nutrient signaling. Cell Res. 24, 42–57. doi: 10.1038/cr.2013.166
Sakaguchi, A. A., Miura, S., Takeuchi, T., Hokari, R., Mizumori, M., Yoshida, H., et al. (1999). Increased expression of inducible nitric oxide synthase and peroxynitrite in Helicobacter pylori gastric ulcer. Free Radic. Biol. Med. 27, 781–789. doi: 10.1016/S0891-5849(99)00124-0
Seyler, R. W. Jr., Olson, J. W., and Maier, R. J. (2001). Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect. Immun. 69, 4034–4040. doi: 10.1128/IAI.69.6.4034-4040.2001
Shirin, H., Pinto, J. T., Liu, L. U., Merzianu, M., Sordillo, E. M., and Moss, S. F. (2001). Helicobacter pylori decreases gastric mucosal glutathione. Cancer Lett. 164, 127–133. doi: 10.1016/S0304-3835(01)00383-4
Sierra, J. C., Asim, M., Verriere, T. G., Piazuelo, M. B., Suarez, G., Romero-Gallo, J., et al. (2017). Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut. doi: 10.1136/gutjnl-2016-312888. [Epub ahead of print].
Sokolova, O., and Naumann, M. (2017). NF-κB signaling in gastric cancer. Toxins (Basel) 9:E119. doi: 10.3390/toxins9040119
Soutto, M., Chen, Z., Katsha, A. M., Romero-Gallo, J., Krishna, U. S., Piazuelo, M. B., et al. (2015). Trefoil factor 1 expression suppresses Helicobacter pylori-induced inflammation in gastric carcinogenesis. Cancer 121, 4348–4358. doi: 10.1002/cncr.29644
Spiotto, M. T., Banh, A., Papandreou, I., Cao, H., Galvez, M. G., Gurtner, G. C., et al. (2010). Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 70, 78–88. doi: 10.1158/0008-5472.CAN-09-2747
Suerbaum, S., and Michetti, P. (2002). Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186. doi: 10.1056/NEJMra020542
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. doi: 10.1038/ncb0311-184
Terebiznik, M. R., Raju, D., Vázquez, C. L., Torbricki, K., Kulkarni, R., Blanke, S. R., et al. (2009). Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5, 370–379. doi: 10.4161/auto.5.3.7663
Valenzuela, M. A., Canales, J., Corvalán, A. H., and Quest, A. F. (2015). Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 21, 12742–12756. doi: 10.3748/wjg.v21.i45.12742
Valenzuela, M., Bravo, D., Canales, J., Sanhueza, C., Díaz, N., Almarza, O., et al. (2013). Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J. Infect. Dis. 208, 1131–1141. doi: 10.1093/infdis/jit286
Valenzuela, M., Perez-Perez, G., Corvalan, A. H., Carrasco, G., Urra, H., Bravo, D., et al. (2010). Helicobacter pylori-induced loss of the inhibitor-of-apoptosis protein survivin is linked to gastritis and death of human gastric cells. J. Infect. Dis. 202, 1021–1030. doi: 10.1086/656143
Varga, M. G., Piazuelo, M. B., Romero-Gallo, J., Delgado, A. G., Suarez, G., Whitaker, M. E., et al. (2016). TLR9 activation suppresses inflammation in response to Helicobacter pylori infection. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G852–G858. doi: 10.1152/ajpgi.00175.2016
Waghray, M., Zavros, Y., Saqui-Salces, M., El-Zaatari, M., Alamelumangapuram, C. B., Todisco, A., et al. (2010). Interleukin-1β promotes gastric atrophy through suppression of Sonic Hedgehog. Gastroenterology 138, 562–572.e2. doi: 10.1053/j.gastro.2009.10.043
Wang, G., Alamuri, P., and Maier, R. J. (2006a). The diverse antioxidant systems of Helicobacter pylori. Mol. Microbiol. 61, 847–860. doi: 10.1111/j.1365-2958.2006.05302.x
Wang, G., Hong, Y., Olczak, A., Maier, S. E., and Maier, R. J. (2006b). Dual Roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect. Immun. 74, 6839–6846. doi: 10.1128/IAI.00991-06
Wang, G., and Maier, R. J. (2004). An NADPH quinone reductase of Helicobacter pylori plays an important role in oxidative stress resistance and host colonization. Infect. Immun. 72, 1391–1396. doi: 10.1128/IAI.72.3.1391-1396.2004
Wang, Y. C. (2014). Medicinal plant activity on Helicobacter pylori related diseases. World J. Gastroenterol. 20, 10368–10382. doi: 10.3748/wjg.v20.i30.10368
Wessler, S., Krisch, L. M., Elmer, D. P., and Aberger, F. (2017). From inflammation to gastric cancer-the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun. Signal. 15, 15. doi: 10.1186/s12964-017-0171-4
Windle, H. J., Fox, A., Ní Eidhin, D., and Kelleher, D. (2000). The thioredoxin system of Helicobacter pylori. J. Biol. Chem. 275, 5081–5089. doi: 10.1074/jbc.275.7.5081
Yanaka, A. (2011). Sulforaphane enhances protection and repair of gastric mucosa against oxidative stress in vitro, and demonstrates anti-inflammatory effects on Helicobacter pylori-infected gastric mucosae in mice and human subjects. Curr. Pharm. Des. 17, 1532–1540. doi: 10.2174/138161211796196945
Yuan, X., Zhou, Y., Wang, W., Li, J., Xie, G., Zhao, Y., et al. (2013). Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 4:e794. doi: 10.1038/cddis.2013.334
Zhang, S., Kim, W., Pham, T. T., Rogers, A. B., Houghton, J. M., and Moss, S. F. (2016). Native and bone marrow-derived cell mosaicism in gastric carcinoma in H. pylori-infected p27-deficient mice. Oncotarget 7, 69136–69148. doi: 10.18632/oncotarget.12049
Zhao, H., Zhu, H., Lin, Z., Lin, G., and Lv, G. (2015). Compound 13, an alpha1-selective small molecule activator of AMPK, inhibits Helicobacter pylori-induced oxidative stresses and gastric epithelial cell apoptosis. Biochem. Biophys. Res. Commun. 463, 510–517. doi: 10.1016/j.bbrc.2015.05.059
Keywords: Helicobacter pylori, precancerous lesion, gastric cancer, endoplasmic reticulum stress, autophagy, inflammation, oxidative stress
Citation: Díaz P, Valenzuela Valderrama M, Bravo J and Quest AFG (2018) Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 9:5. doi: 10.3389/fmicb.2018.00005
Received: 16 October 2017; Accepted: 04 January 2018;
Published: 22 January 2018.
Edited by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Nagendran Tharmalingam, Alpert Medical School, Brown University, United StatesMario M. D'Elios, University of Florence, Italy
Copyright © 2018 Díaz, Valenzuela Valderrama, Bravo and Quest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew F. G. Quest, YXF1ZXN0QG1lZC51Y2hpbGUuY2w=