
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 04 January 2018
Sec. Microbial Physiology and Metabolism
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02617
 Dong Jun1
Dong Jun1 Zoran Minic1
Zoran Minic1 Supriya V. Bhat1
Supriya V. Bhat1 Elizabeth M. Vanderlinde1,2Chris K. Yost1
Elizabeth M. Vanderlinde1,2Chris K. Yost1 Mohan Babu1
Mohan Babu1 Tanya E. S. Dahms1*
Tanya E. S. Dahms1*Post-translational modification expands the functionality of the proteome beyond genetic encoding, impacting many cellular processes. Cleavage of the carboxyl terminus is one of the many different ways proteins can be modified for functionality. Gel-electrophoresis and mass spectrometric-based techniques were used to identify proteins impacted by deficiency of a C-terminal protease, CtpA, in Rhizobium leguminosarum bv. viciae 3841. Predicted CtpA substrates from 2D silver stained gels were predominantly outer membrane and transport proteins. Proteins with altered abundance in the wild type and ctpA (RL4692) mutant, separated by 2D difference gel electrophoresis, were selected for analysis by mass spectrometry. Of those identified, 9 were the periplasmic solute-binding components of ABC transporters, 5 were amino acid metabolic enzymes, 2 were proteins involved in sulfur metabolism, and 1 each was related to carbon metabolism, protein folding and signal transduction. Alterations to ABC-binding-cassette transporters, nutrient uptake efficiency and to amino acid metabolism indicated an impact on amino acid metabolism and transport for the ctpA mutant, which was validated by measured amino acid levels.
Post-translational modification plays a key role in many cellular processes such as the cell cycle (David, 2012), signaling (Mowen and David, 2014), protein–protein interactions (Nogueira-Ferreira et al., 2013) and many others. To date, over 200 types of post-translational modifications have been identified, dramatically increasing the complexity of the proteome (Chandramouli and Qian, 2009). The most common modifications include phosphorylation, acetylation, glycosylation, amidation, hydroxylation, and methylation (Han and Martinage, 1993). Proteolytic post-translational modification is irreversible, ubiquitous and often activates or inactivates proteins by generating shorter protein chains with altered function.
The carboxyl terminal protease A (CtpA) of R. leguminosarum bv. viciae 3841 is part of a novel group of serine proteases involved in the maturation of other proteins (Gilbert et al., 2007; Rawlings, 2013). The crystal structure of CtpA purified from Scenedesmus obliquus shows three domains, including a PDZ domain capable of recognizing short amino acid motifs at the C-termini of target proteins (Liao et al., 2000; Lee and Zheng, 2010). The serine/lysine (Ser/Lys) catalytic dyad catalyzing the hydrolysis reaction of CtpA is distinct from other known serine proteases (Liao et al., 2000; Ekici et al., 2008). Using partially purified Ctp from spinach, Taguchi et al. (1993) showed a preference for P1 residues that are small and uncharged. Escherichia coli Tsp will proteolytically process a non-substrate when the WVAAA sequence is added to the C-terminus (Keiler et al., 1996) or when it replaces polar and charged (RSEYE) residues (Parsell et al., 1990). The cleavage site specificity of Ctp is broad, with Ala, Ser, Val, and to a lesser extent, Ile, Leu, Lys, or Arg, preferred at the P1 position (Keiler and Sauer, 1995), and these same residues plus Met, Tyr, or Trp at the P1′ position (Keiler et al., 1995).
CtpA is important for the photosynthetic system of higher plants and algae, helping to rapidly turnover the D1 protein at the reaction center of photosystem II (Bowyer et al., 1992). CtpA in E. coli cleaves the penicillin binding protein 3 (PBP3) (Hara et al., 1989, 1991) and in Borrelia burgdorferi it processes outer-membrane-associated proteins P13, BB0323, and OspC (Östberg et al., 2004) that form ion channels (Bárcena-Uribarri et al., 2014), help cells persist in vivo (Kariu et al., 2013), or assist in host invasion (Tilly et al., 2006), respectively.
To fully understand the cellular functions impacted by a protease, its substrates must be identified, along with the associated processing events that ultimately define its function. C-terminal proteolysis shortens protein chains to produce neo-C-termini, making it difficult to predict the protein’s functional sequence based on the genetic sequence (Tholen et al., 2013). To compound the problem, the widely diverse proteins found in cells have a dynamic range of expression levels, which includes the proteases and their target protein substrates (Vogel and Marcotte, 2012). Techniques developed to determine proteolytic function and substrate identity can be divided into gel electrophoresis- or LC-MS/MS (liquid chromatography-mass spectrometry/mass spectrometry)-based techniques (Otto et al., 2014). One- or two-dimensional (2D) electrophoresis has been successfully used to identify protease substrates (Agard and Wells, 2009) based on shifts from higher to lower molecular weight, reduction in spot intensity, or the appearance or disappearance of protein spots. Substrate spots can be excised and identified by MS, but the results are limited by the complexity of protein mixture and reproducibility (Chandramouli and Qian, 2009). The use of two-dimensional difference gel electrophoresis (2D DIGE) to separate fluorescent dye labeled protein samples allows determination of protein abundance by direct comparison of the dyes used to label treated and control samples (Bredemeyer et al., 2004). A variation of this approach, in which protease substrates of interest migrate off the diagonal on the second post-proteolysis separation, provides some improvement in substrate detection (Shao et al., 2007). In addition, 2D electrophoresis in combination with LC-MS/MS, allows a highly dynamic range in sensitivity, greatly improving throughput and proteome coverage (Chandramouli and Qian, 2009).
Altered substrate processing can impact many cellular processes, including cell envelope integrity. The R. leguminosarum ctpA null mutant 3845 has a compromised cell envelope (Gilbert et al., 2007), and is incapable of developing fully mature biofilms, consistent with its altered surface ultrastructure, greater roughness and stronger adhesion to hydrophilic surfaces (Jun et al., 2011). There was no change in the structure of the peptidoglycan peptide bridge for the ctpA mutant, so either PBPs are not substrates of CtpA or there is functional redundancy in the R. leguminosarum genome (Jun et al., unpublished). Here we use 2D electrophoresis and MS-based methods to identify proteins impacted by or potential substrates of CtpA and the influence of the ctpA mutation on cellular function.
All chemicals were purchased from Sigma (Canada) unless otherwise stated. R. leguminosarum bv. viciae 3841 and the ctpA mutant strain 3845 were grown at 30°C in Vincent’s minimal medium (VMM) (Vincent, 1970) or in tryptone-yeast (TY) medium (Beringer, 1974) supplemented as required with 100 μg/mL neomycin and 500 μg/mL streptomycin.
Starter cultures of R. leguminosarum were used to inoculate TY broth medium cultured to mid-exponential phase (OD600 = 0.6), cells harvested (20 min, 8000 × g), the pellets washed (5×, 10 mM Tris HCl, pH 8.5, 10 mM magnesium acetate), and sonicated (10 pulses 8×) in cell lysis buffer (30 mM Tris, pH 8.5, 7 M Urea, 2 M Thiourea, 4% [3-cholamidopropyldimethyl ammonio]-1-propane sulfonate (CHAPS) w/v, 1mM PMSF). Following centrifugation, the supernatant was precipitated and washed (80% acetone) and purified with the ReadyPrep 2D purification kit (Bio-Rad) to eliminate streaks. Concentrations of proteins dissolved in cell lysis buffer were determined by Bradford assay using the Coomassie Plus Reagent (Thermo Scientific) and absorbance at 595 nm (SynergyTM HT Multi-Mode Microplate Reader, BioTek).
Protein was dissolved in 2D rehydration buffer (8 M urea, 2 M thiourea, 4% DTT w/v, 2% CHAPS w/v, 0.1% Bio-lyte 3/10 ampholytes v/v with trace bromophenol blue, 300 μl final volume), loaded onto 17 cm immobilized pH gradient strips with pH ranges of 3–10 and 4–7 (Bio-Rad) covered with mineral oil and separated (14 h, 50 V). Narrow-ranged IPG strips were used for optimal resolution for isoelectric focussing (PROTEAN® i12TM IEF System, Bio-Rad) in rapid mode (30,000 Vh, 10,000 V), reduced from 43,000 Vh to eliminate vertical streaking and improve separation. After the first dimension, strips were incubated (15 min) sequentially in sodium dodecyl sulfate (SDS) equilibration buffer A (6 M urea, 2% SDS w/v, 0.375 M Tris, pH8.8, 20% glycerol w/v, 2% w/v dithiothreitol (DTT)) with slow shaking, followed by the same buffer with 2.5% iodoacetamide (w/v) replacing DTT. The strips were placed on top of the SDS – polyacrylamide gel electrophoresis (PAGE) gels and sealed with 0.5% agarose (w/v) for the second dimension at 4°C using 10–20% gradient acrylamide gels (Jule Biotechnologies) and proteins visualized using silver stain (sensitized in 0.02% Na2S2O3, chilled with 1% AgNO3) and developed (2% Na2CO3, 0.04% formaldehyde in millipore water).
Proteins from culture were extracted as described above and labeled with dyes according to the manufacturer’s instructions (Lumiprobe) using pH 8.5 for the rehydration buffer for efficient labeling. Protein (50 μg) from wild type and ctpA mutant strains were treated with the dyes [400 pmol/μl in dimethylformamide (DMF), Lumiprobe] cyanine 3 and cyanine 5 NHS ester (Cy3 and Cy5), respectively, the protein internal standard treated with Cy2, each incubated (30 min, 4°C, dark), reactions halted with lysine (1 μl, 10 mM), mixed by vortex and incubated (10 min, 4°C, dark). Two samples of each, labeled with different cyanine dyes, and internal standard were mixed and combined with 2× sample buffer [8 M urea, 2% CHAPS, 50 mM DTT, 0.2% Bio-lyte 3/10 ampholytes (v/v), trace bromophenol blue] and dissolved in 2D rehydration buffer (8 M urea, 2 M thiourea, 4% DTT w/v, 2% CHAPS w/v, 0.1% Bio-lyte 3/10 ampholytes v/v and trace of bromophenol blue) to a final volume of 300 μl. Samples were loaded onto 17 cm immobilized pH gradient strips with pH ranges of 3–6, 5–8, and 7–10 (Bio-Rad Laboratories), covered with mineral oil and separated (14 h, 50 V) followed by SDS – PAGE in the second dimension as outlined above, but without staining.
2D gels were imaged with a TyphoonTM Imager and processed using DeCyder Differential Analysis Software v6.5 (Amersham Pharmacia Biotech). Protein spots were detected (differential in-gel analysis), manually checked to exclude artifacts, aligned and analyzed (biological variation analysis). Spot intensities were normalized to the internal standard. For each spot, average abundance with standard deviation of each was compared and statistically analyzed using a student’s t-test.
Prior to spot-picking, 2D gels were stained with colloidal Coomassie Blue G250 according to the method described by Dyballa and Metzger (2009). Gel plugs were manually excised and washed (3×, 30% acetonitrile in 100 mM NH4HCO3 pH 8.5), dehydrated (15 min, 100% acetonitrile), dried (15 min, 35°C) in a Savant SpeedVac Concentrator (Thermo Electrition Corporation), trypsin solution (10 μl; 13 ng/μl in 50 mM NH4HCO3 pH 8.5, 5% acetonitrile) added, incubated on ice (30 min), covered with 50 mM NH4HCO3 (pH 8.5) containing 5% acetonitrile and digested overnight (37°C). Supernatants from digested samples were desalted (C18-ZipTips, Millipore, Bedford, MA, United States) and used for LC-MS/MS analysis.
Samples were analyzed by nanoLC coupled to the Orbitrap Elite mass spectrometer (MS, Thermo Fisher Scientific) following chromatographic separation of peptides on a Proxeon EASY nLC 1000 (Proxeon, Mississauga, ON, Canada) nano high-performance liquid chromatograph (HPLC). Samples directly injected onto a nano column (C18 column, 10 cm × 75 μm ID, 3 μm, 100 Å) with a eluent (water/acetonitrile/0.1% formic acid, 100 min, 0.30 μl/min) were separated with an acetonitrile gradient: 1–3% (2 min), 3–24% (3–74 min), 24–100% (75–90 min) and a final wash at 100% (91–100 min). Eluted peptides were delivered to the MS using positive electrospray ionization (250°C, 2.1 kV). Full-scan MS spectra (m/z 350–2000) were acquired in the Orbitrap at 60 000 (m/z 400) resolution using automatic gain control settings (1e6 for full FTMS scans and 5e4 for MS/MS scans). Peptides were fragmented with collision-induced dissociation (CID) in the linear ion trap when ion intensity was >1500 counts. The 15 most intense ions were isolated for ion trap CID with charge states ≥ 2 and sequentially isolated for fragmentation using normalized collision energy (35%), activation Q (0.250) and activation (10 ms). Ions selected for MS/MS were dynamically excluded for 30 s. The Orbitrap Elite MS was operated with Thermo XCalibur software.
RAW MS files were converted into mzXML files for database searching using SEQUEST-PVM v.27 (rev. 9) under standard workflow and a non-redundant rhizobial protein sequence FASTA file from the PATRIC database (Wattam et al., 2014). Search parameters allowed for post-translational modification of methionine by oxidation, and modification of cysteine by carbamidomethylation using precursor mass tolerances of 10 ppm and a fragment mass tolerance 0.6 Da. All peptide matches were filtered by XCorr, mass accuracy (<10 ppm): XCorr >1.5 for +2, +3, and +4 charged precursor ions. A stringent false-discovery rate (FDR) of 1% (or p < 0.01) was used to filter candidate peptides.
Wild type and ctpA mutant strains grown on VMM and TY agar plates for approximately 3–4 days were overlaid with 0.6% agar containing 30 mg/ml type VI Torula yeast RNA (Sigma) and incubated 1 day before the addition of 1 N HCl to precipitate undigested RNA and view the release of periplasmic RNase.
Following centrifugation, the supernatant containing cell-free TY media (1 mL) was collected from lag (8.5 h wt, 24 h ctpA), log (19.5 h wt, 34.5 h ctpA), early (23 h wt, 47 h ctpA) and late stationary (49 h wt, 68 h ctpA) phase cultures, frozen (-80°C), lyophilized, dissolved in 200 μL phenylisothiocyanate (PTIC)/water/ethanol/triethylamine solution (1:1:7:1, v/v/v/v), incubated (30 min) and stored frozen (-80°C). Frozen samples were lyophilized, mixed with methanol (200 μL), centrifuged (3 min, 10 000 × g) and the supernatants collected for HPLC (Agilent 1100 series) injection. Fourteen amino acids at physiologically relevant concentrations (mM) served as standards. PITC-amino acids (2 μL) were injected onto a reversed phase column (Viva C18, 5 μm, 250 × 4.6 mm (Cat# 9514575, Restek Corporation) with an inline filter and separated by HPLC, equipped with an autosampler and variable wavelength detector, using the method adapted from Dimova (2003). PITC derivatized amino acids were separated in 12 mM sodium phosphate buffer pH 5.5 (A) and methanol (B) at a flow rate of 0.5 mL/min by gradient elution: 65% A from 0 to 5 min; 65–40% A from 5 to 30 min, 40–5% A from 30 to 35 min, 5–90% A from 35 to 40 min, and 90–65% from 40 to 47 min. The relative amount of each PITC derivatized amino acid detected at 254 nm was calculated from peak area.
Approximately 350 spots were visualized on 2D gel images using a pI range of 3–10 in the first dimension, for which 20 were unique to the wild type and 6 unique to the ctpA mutant (Supplementary Figure S1). 2D separation with a pI range of 4–7 gave rise to 550 spots, with 16 unique to wild type and 18 unique to the ctpA mutant (Supplementary Figure S2 and Supplementary Table S1), indicating potential CtpA substrates or those indirectly influenced by the ctpA mutation. Based on electrophoretic patterns, proteins could be assigned to either potential CtpA substrates, or proteins that have higher or lower abundance in the ctpA mutant, and each was identified using the published genome of R. leguminosarum (Young et al., 2006).
Based on the processing sites studied in vitro (Keiler et al., 1995, 1996) and the published genome of R. leguminosarum (Young et al., 2006), the C-termini of putative cell envelope proteins were examined to identify amino acid residues preferred by C-terminal proteases in the P1, P2, and P3 positions for proteins having differential expression in the wild type and mutant. Proteins with appropriate pI and mass values for the wild type (CtpA processing) and mutant (no processing) from all the silver stained 2D gels having predicted processing sites are shown in Supplementary Table S2.
The more sensitive 2D DiGE electrophoresis showed that the majority of proteins had pIs between 4 and 8. Figure 1 shows representative DiGE images of four biological replicates for the wild type and ctpA mutant of R. leguminosarum. Approximately 1200 proteins could be visualized from 2D gels in the pI range 5–8, of which 651 appeared in all four replicates and 301 spots had significantly increased or decreased intensity in gels of the ctpA mutant strain. More than 800 proteins were detected using a pI range of 3 to 6, of which 332 protein spots appeared in all four replicates and 170 had higher or lower intensity in the ctpA mutant strain. In the pI range of 7–10 (data not shown), 500 proteins were detected in the 2D gels of which 142 protein spots appeared in all four replicates and 3 spots had significantly higher or lower intensity in the ctpA mutant strain. Twenty spots, showing significantly increased or reduced abundance in the ctpA mutant strain (p < 0.001), were selected for further analysis (Table 1).

FIGURE 1. Representative 2D-DIGE expression maps of Rhizobium leguminosarum bv. viciae 3841 (wt) and 3845 (ctpA mutant) labeled with Cy3 (green) and Cy5 (red), respectively. First dimension in (A) is pH 3-6 and in (B) is pH 5–8.
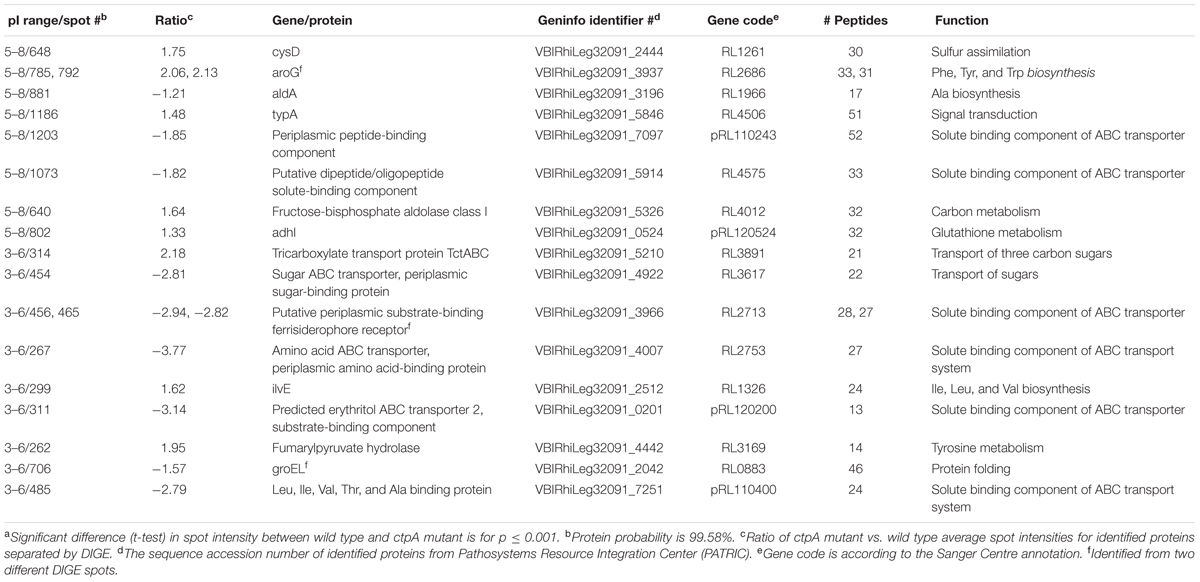
TABLE 1. Major protein components of the ctpA mutant from selected 2D-DIGE spots identified by mass spectrometrya.
Proteins from the selected 20 spots were further identified by LC-MS/MS and those constituting the majority of each are listed in Table 1. Some proteins could be linked to certain metabolic pathways using the KEGG database (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017). The Clusters of Orthologous Groups of proteins database was used to classify selected proteins of R. leguminosarum bv. viciae 3841 into six categories according to their function (Figure 2).
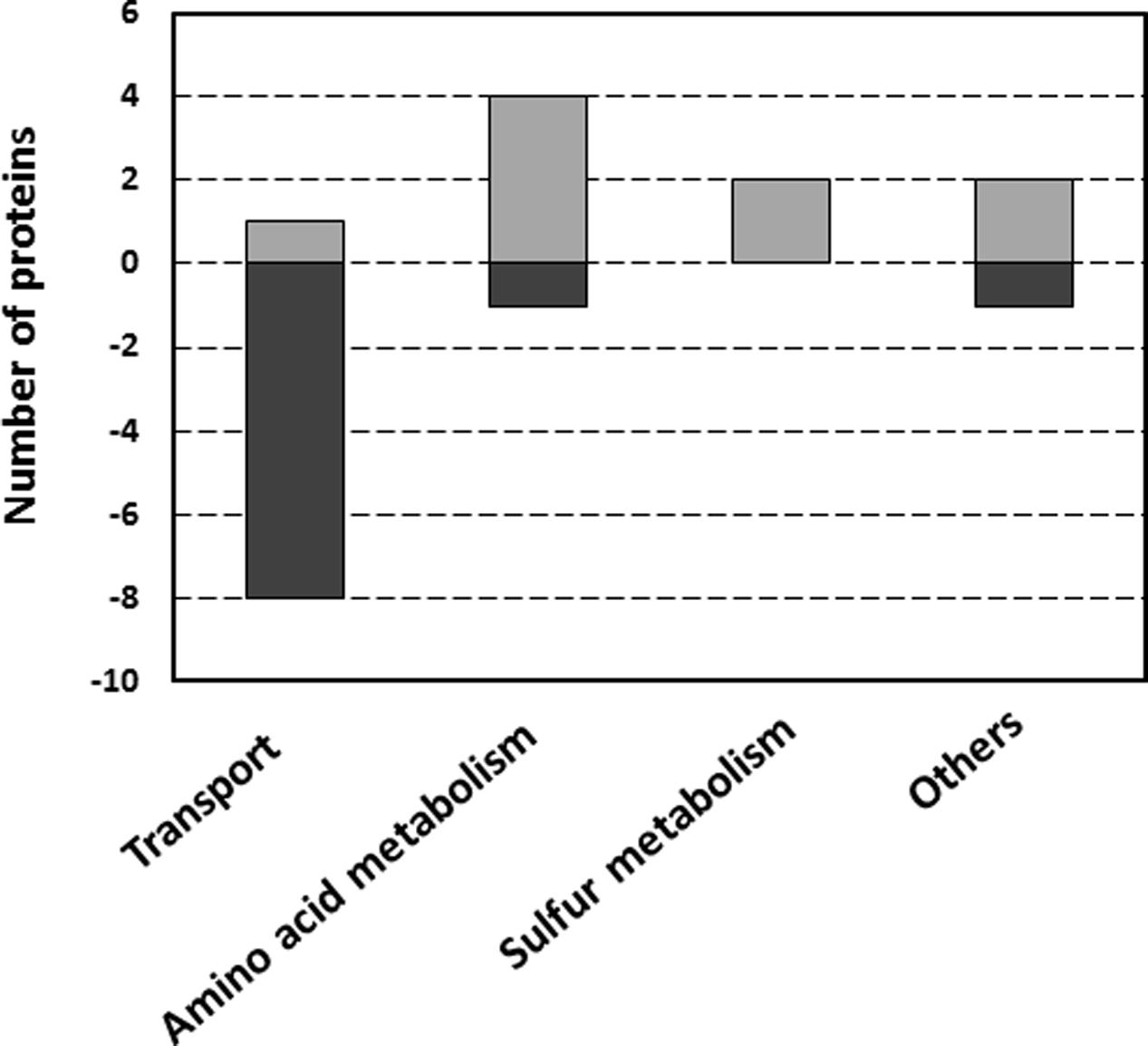
FIGURE 2. Functional classification of proteins with differential abundance in wild type and ctpA mutant R. leguminosarum, according to their biological function. The category “Others” includes one protein in each of the following categories: energy metabolism, protein folding and signal transduction. Assignments were made according to the Clusters of Orthologous Groups of proteins database.
The RNase release assay was used to test if the reduction of transporter proteins in the ctpA mutant cell envelope is the result of a destabilized outer membrane. Zones of clearing, indicating leakage of periplasmic RNase I into the agar, were observed surrounding the ctpA mutant colonies, but not wild type (data not shown).
To determine whether amino acid profiles were altered in the ctpA mutant, amino acid consumption was measured in lag, log, early and late stationary growth phases (esp and lsp, respectively) by HPLC. Fourteen amino acid standards relevant to this study were chromatographically separated, but high levels of Ile, Leu, and Trp, even in dilute samples (data not shown), precluded adequate resolution and further analysis. As expected, samples without PITC treatment lacked signal at 254 nm, serving as a negative control (Supplementary Figure S3). The relative amounts of PITC-amino acids obtained during the four growth phases for the wild-type and ctpA mutant are shown in Figure 3. In the mutant, there was a significant (p < 0.05) accumulation of Ala, Arg, Val, and Tyr during the late stationary phase, with Arg also accumulating at log and early stationary phase. There was a significant (p < 0.05) reduction in Glu and Thr during lag and log phases, Gly and Pro at log and early stationary phase, Asp and His during early stationary phase, and Met at early and late stationary phases in the mutant compared to wild type.
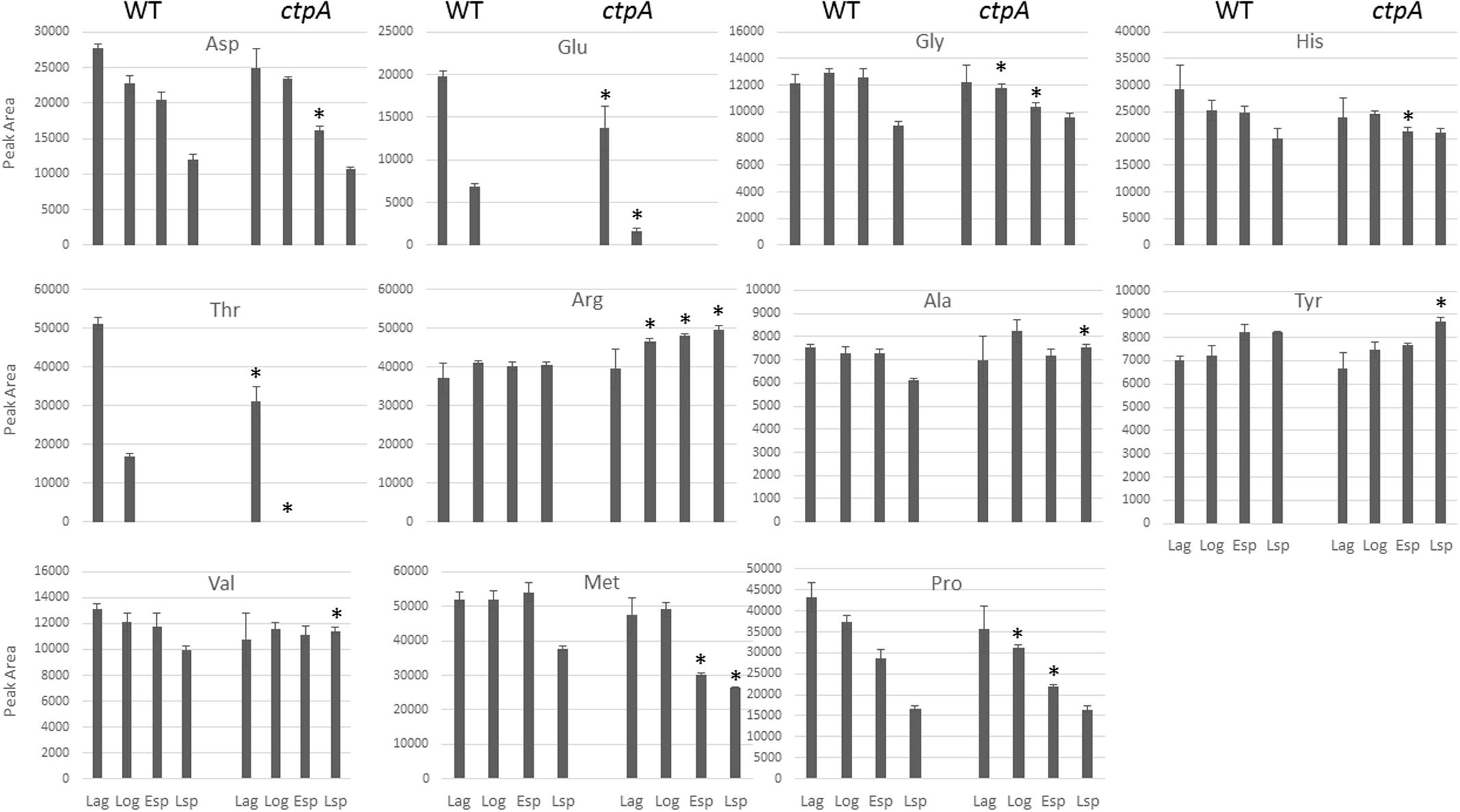
FIGURE 3. Histograms of peak areas for 11 amino acids from culture at lag, log, early and late stationary phases for the wild type (WT) and ctpA mutant. Columns with no bars represent signal not detected. Asterisks represent statistically significant differences relative to wild-type (p < 0.05) from a two-tailed student’s t-test.
In microbes, proteases have been linked to protein turnover, sporulation, conidial discharge, germination, nutrition and regulation of gene expression (Rao et al., 1998). Proteases are all capable of hydrolyzing the amide bond of peptide and protein substrates despite their varied mechanisms. Proteins expressed as precursors with a cleavable carboxyl-terminal extension (Bhargava and Spremulli, 2005) can be cleaved by CtpA during post-translational modification (Hara et al., 1991). The ctpA gene was identified in the genome of R. leguminosarum bv. viciae 3841 (Gilbert et al., 2007), with the CtpA protein sequence analysis showing highest similarity between members of the Rhizobiaceae order, such as Sinorhizobium meliloti, Agrobacterium tumefaciens (85% identity), and Mesorhizobium loti (75% identity). The R. leguminosarum CtpA has sequences homologous to the peptide binding motif and a catalytic dyad of a Ser protease, but its substrates are unknown. Searching for CtpA substrates is challenging but crucial for identifying the role of CtpA in the biological process of R. leguminosarum bv. viciae 3841, and understanding the downstream effect of CtpA in cellular physiology.
Carboxyl terminal protease is hypothesized to be transported into the periplasmic space through the inner membrane following cytoplasmic biosynthesis. Hara et al. (1991) and then Silber et al. (1992) demonstrated the localization of Prc in the cytoplasmic membrane and periplasm of E. coli. More recently, however, Hoge et al. (2011) detected only periplasmic CtpA in Pseudomonas aeruginosa which was not observable without the introduction of an expression vector harboring the ctpA gene, speculating that localization to the cytosol and inner membrane was a consequence of artificial CtpA overexpression.
Carboxyl terminal protease is not a highly specific protease, likely acting on a number of substrates, making it more difficult to identify its exact repertoire. Based on 2D electrophoretic patterns, proteins could be assigned to either potential CtpA substrates or proteins with higher or lower abundance in the ctpA mutant (Supplementary Table S2). While these proteins are potential targets of CtpA, actual processing is expected to be limited to those protein precursors having non-polar carboxyl termini. The majority of the proteins predicted in this manner were putative outer membrane proteins and transporter components. A detached outer membrane of the ctpA mutant, viewed by TEM (Jun et al., unpublished) and confirmed by the RNase assay, is consistent with previous data (Gilbert et al., 2007) and implies a loss of lipoproteins. Such proteins are widely distributed in Gram-negative bacteria and act as structural proteins to affix the outer or inner membrane to the peptidoglycan layer (Cascales et al., 2002). Indeed, the lipoproteins OspC and BB0323 are processed at the C-termini by CtpA in B. burgdorferi (Östberg et al., 2004; Kumru et al., 2011). The C-terminus of integral outer membrane porin P13 and BBA01 are also cleaved by CtpA in B. burgdorferi (Noppa et al., 2001; Pinne et al., 2006). So lipoproteins and porins are suspected targets for CtpA in R. leguminosarum bv. viciae 3841, but were not identified in this study, possibly since such a small proportion of proteins was characterized.
2D separation conditions were optimized using silver stain which is incompatible with mass spectrometry, so proteins were isolated by DIGE using optimal separation conditions with small pI ranges for identification by LC-MS/MS (Table 1). Identified proteins were distinct from those predicted (Supplementary Table S2), likely based on different pI ranges and the analysis of a relatively small number of spots, but both revealed a number of transport-related proteins. The majority of proteins identified by MS were related to transport and amino acid metabolism (Figures 2, 4), supported by validation studies showing altered levels of amino acids from the culture media of the wild type and mutant at lag, log, early and late stationary phases (Figure 3).
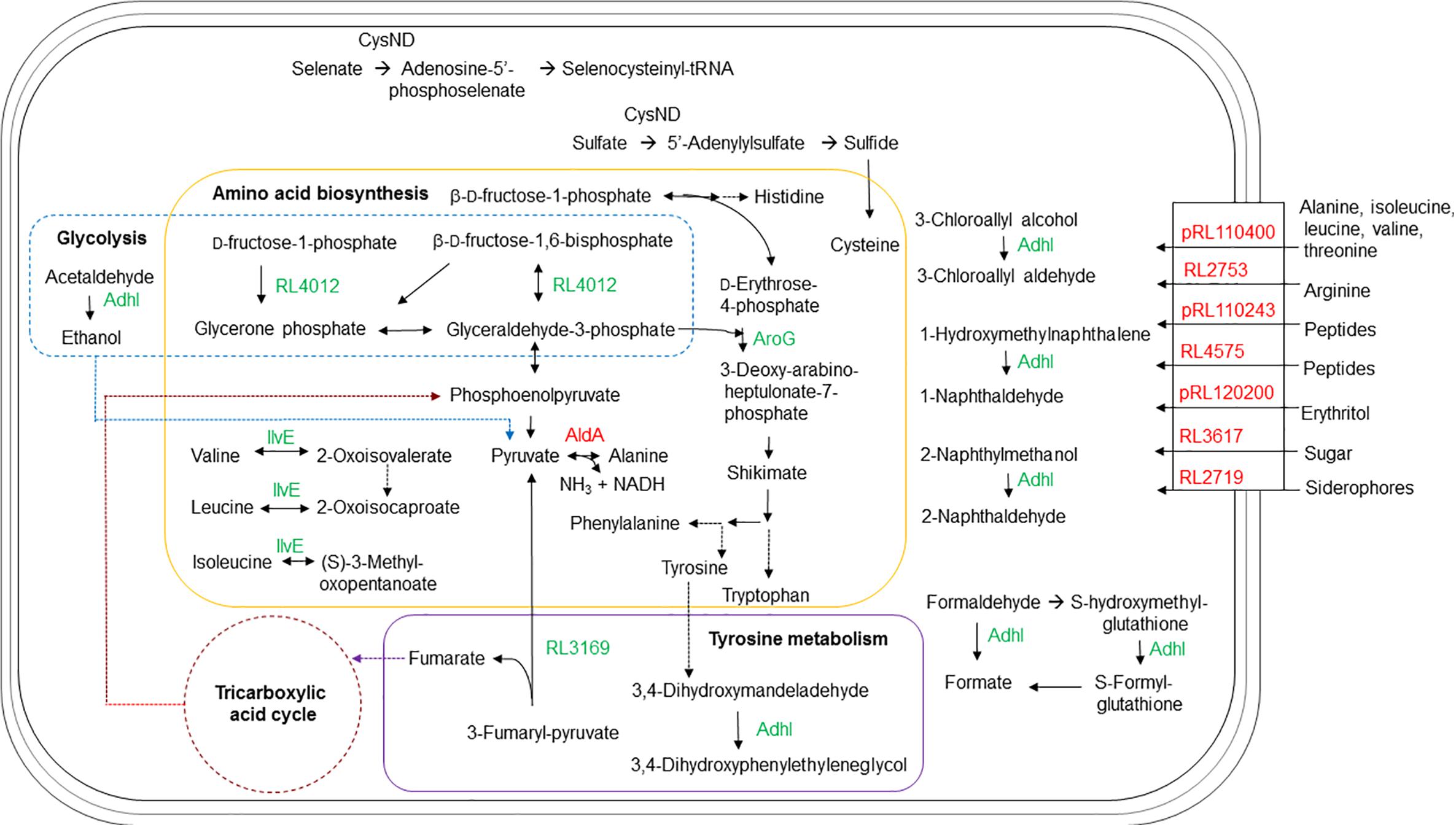
FIGURE 4. Schematic representation of the metabolic pathways and transport in R. leguminosarum bv. viciae affected by the ctpA mutation. In green are proteins with more abundance in the ctpA mutant and in red proteins have lesser abundance in the ctpA mutant. Solid arrows indicate a single step, while dashed arrows indicate multiple steps.
The putative solute-binding component of an ABC transporter encoded by RL3617 shares 98% similarity with its homolog ChvE in A. tumefaciens, a multiple sugar-binding periplasmic receptor (Wood et al., 2001) of the sugar ABC transporter (Kemner et al., 1997). The chvE mutant is slower growing, like the ctpA mutant (Gilbert et al., 2007), with a delayed chemotactic response to sugars (Shimoda et al., 1993). EryG encoded by pRL120200 is a periplasmic-binding protein for the erythritol ABC transporter (Yost et al., 2006) and its reduced abundance could impact the net transport of erythritol.
The gene products of RL4575 and pRL110243 are predicted to participate in nickel/peptide transport across the R. leguminosarum cell envelope. pRL110243 is the homolog of OppB in E. coli and Salmonella typhimurium, a hydrophobic integral membrane protein responsible for the transport of peptides across the cytoplasmic membrane (Pearce et al., 1992). RL4575 encodes a protein predicted as a putative solute-binding component of an ABC transporter containing a nickel/dipeptide/oligopeptide binding domain. Peptide uptake can play a major role in the nutrition for the organism, and a reduced abundance of these transporters may explain slower growth for the ctpA mutant (Gilbert et al., 2007).
The gene pRL110400 is predicted to encode the Ala-, Ile-, Leu-, Val-, and Thr-binding component of an ABC transporter in R. leguminosarum. The lower abundance of the pRL110400 gene product in the ctpA mutant is validated by the accumulation of Ala and Val at late stationary phase in the mutant’s growth media, but with no accumulation of Thr as might be expected (Figure 3). The periplasmic amino acid-binding protein encoded by RL2753 shares identity with two amino acid-binding proteins in E. coli, ArtJ (39%) and HisJ (41%), the former being the periplasmic binding component of the L-Arg ABC transport system (Wissenbach et al., 1995). The lower abundance of RL2753 is expected to impair amino acid uptake in the ctpA mutant, consistent with accumulation of Arg in the culture media of the ctpA mutant (Figure 3).
Many of the identified proteins are functionally connected (Supplementary Figure S4), with many involved in the ABC-binding-cassette transport system. Bacterial ABC transporters are involved in many biological processes, including multidrug resistance, protein secretion, quorum sensing, and in this case nutrient uptake (Higgins, 2001; Taga et al., 2001; Chang, 2003; Holland et al., 2005). In Gram-negative bacteria, ABC transporters consist of at least a periplasmic binding protein which binds solutes, a membrane-bound transport protein which interacts with the periplasmic protein and an ATP-binding protein which provides the energy required for transport (Higgins, 2001). In all cases, it was the periplasmic solute binding protein of ABC transporters that was impacted in the ctpA mutant (Table 1). A low abundance of periplasmic binding proteins in the ctpA mutant would affect solute uptake efficiency and response to chemotactic stimuli, further explaining its slow growth rate (Gilbert et al., 2007). The ctpA mutation broadly impacts the ABC transport system, which we attribute to a compromised outer membrane (Jun et al., unpublished).
Other than RL1966 (Table 1 and Figure 2), proteins involved in amino acid metabolism are found in higher abundance in the mutant, possibly to compensate for reduced nutrient uptake by impaired transport. Alanine dehydrogenase (AldA) encoded by RL1966 is the principle enzyme of de novo alanine biosynthesis, catalyzing the reversible conversion of pyruvate, ammonium and NADH to Ala (Lodwig et al., 2004). The lower abundance of AldA in the ctpA mutant indicates either less pyruvate or Ala in the cell, consistent with Ala accumulation at late stationary phase in the media (Figure 3). IlvE encoded by RL1326 is a branched-chain amino acid aminotransferase responsible for the last step of Ile, Leu, and Val biosynthesis, along with the first step in their degradation (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017), in accordance with altered Val levels. The gene aroG (RL2686) encodes the feedback regulated enzyme 3-deoxy-D-arabino-heptulosonate synthase (DAHP), part of the shikimate pathway that catalyzes the first step in the biosynthesis of Tyr, Phe, and Trp. Expression of AroG in Solanum lycopersicum and Arabidopsis plants increases levels of shikimate pathway metabolites, Phe, Tyr, and Trp, along with altered levels of Asn, Gln, Gly, Ile, N-acetyl-Glu and Thr (Tzin et al., 2012, 2013), consistent with altered amino acids levels in the ctpA mutant (Figure 3). RL3169 encoding a predicted protein shares 40% identity with a putative fumarylacetoacetate hydrolase YcgM in E. coli, which is involved in tyrosine degradation (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017). The higher abundance of Tyr in the mutant at late stationary phase is consistent with the slightly greater increase in RL2686 than RL3169 in the mutant (Table 1).
CysD, encoded by RL1261, corresponds to the putative sulfate adenylyltransferase subunit 2 which helps assimilate sulfur. Sulfur is an essential element incorporated into many molecules including the amino acids Cys and Met, as evidenced by reduced Met levels in the stationary phases (Figure 3). Putative alcohol dehydrogenase AdhI encoded by pRL120524 shares 58% identity with S-(hydroxymethyl) glutathione dehydrogenase frmA in E. coli (Gutheil et al., 1992). AdhI is involved in multiple metabolic pathways, including carbon metabolism, catabolism of aromatic compounds, fatty acids, and sugars (glycolysis), methane metabolism, tyrosine metabolism, chloroalkane and chloroalkene degradation (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017). RL4012 encodes fructose-bisphosphate aldolase, involved in several reactions of carbon metabolism, glycolysis, the pentose phosphate pathway, amino acid biosynthesis, methane metabolism, fructose and mannose metabolism. Higher levels of pRL120524 and RL4012 may compensate for reduced carbon and amino acid transport.
GroEL encoded by RL0883, detected in two gel spots (Table 1), is responsible for proper protein folding, is induced under stress conditions (Goulhen et al., 1998; Kusmierczyk and Martin, 2000; Klančnik et al., 2006) and plays an important role in the export of certain proteins (Kusukawa et al., 1989). Unlike E. coli, R. leguminosarum strain A3 has three genes encoding GroEL homologs (Rodríguez-Quiñones et al., 2005). Interestingly, GroEL is found to be down regulated in Bradyrhizobium japonicum under acidic conditions (Puranamaneewiwat et al., 2006), consistent with the ctpA mutant (Table 1) and suggesting it likely plays distinct roles in rhizobia.
TypA (tyrosine phosphorylated protein A) encoded by RL4506, a predicted protein in R. leguminosarum bv. viciae 3841, shares 56% identity with the GTP-binding protein TypA/BipA in E. coli K12. Disruption of E. coli typA alters protein expression and modification during exponential growth and carbon starvation (Freestone et al., 1998). TypA is involved in temperature-dependent regulation of E. coli cell surface polysaccharides (Rowe et al., 2000) and the survival of S. meliloti 1021 under stressful conditions (Kiss et al., 2004). Higher levels in the mutant may reflect carbon deficiency as a result of reduced carbon transport.
In summary, we show an impact to the ABC-binding-cassette transport system in the ctpA mutant and thus nutrient uptake efficiency that is consistent with its slow growth rate and a compromised outer membrane. The mutant appears to adapt by increasing a number of metabolic enzymes that would be capable of compensating inadequate nutrient transport. The predicted impact on amino acid metabolism and transport was validated by altered amino acid levels for the ctpA mutant.
DJ designed and completed most of the experiments, with some assistance from EV and CY, analyzed the data and prepared a first draft of the manuscript. ZM and SB completed the amino acid analysis. MB oversaw the proteomics data and provided data input into data interpretation. TD helped design all experiments, edited and polished the manuscript with editorial input from EV, SB, ZM, CY, and MB.
This work was supported by the Natural Sciences and Engineering Research Discovery Grants to TD (228206-2011), CY (288281-2017) and MB (20234-2012), and a Canada Foundation for Innovation Leaders Opportunity Fund to TD and CY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Hiroyuki Aoki for training and assistance with the 2D gels and pertinent discussions throughout this work. We thank Drs. Peter Leavitt and Tzu-Chiao Chao for giving us access to their 2D gel apparatus, and Dr. Vikram Misra and Noreen Rapin (College of Veterinary Medicine, University of Saskatcewan) for training on and access to their Typhoon imager and software.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02617/full#supplementary-material
Agard, N. J., and Wells, J. A. (2009). Methods for the proteomic identification of protease substrates. Curr. Opin. Chem. Biol. 13, 503–509. doi: 10.1016/j.cbpa.2009.07.026
Bárcena-Uribarri, I., Thein, M., Barbot, M., Sans-Serramitjana, E., Bonde, M., Mentele, R., et al. (2014). Study of the protein complex, pore diameter, and pore-forming activity of the Borrelia burgdorferi P13 porin. J. Biol. Chem. 289, 18614–18624. doi: 10.1074/jbc.M113.539528
Beringer, J. E. (1974). R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84, 188–198. doi: 10.1099/00221287-84-1-188
Bhargava, K., and Spremulli, L. L. (2005). Role of the N- and C-terminal extensions on the activity of mammalian mitochondrial translational initiation factor 3. Nucleic Acids Res. 33, 7011–7018. doi: 10.1093/nar/gki1007
Bowyer, J. R., Packer, J. C., McCormack, B. A., Whitelegge, J. P., Robinson, C., and Taylor, M. A. (1992). Carboxyl-terminal processing of the D1 protein and photoactivation of water-splitting in photosystem II. Partial purification and characterization of the processing enzyme from scenedesmus obliquus and Pisum sativum. J. Biol. Chem. 267, 5424–5433.
Bredemeyer, A. J., Lewis, R. M., Malone, J. P., Davis, A. E., Gross, J., Townsend, R. R., et al. (2004). A proteomic approach for the discovery of protease substrates. Proc. Natl. Acad. Sci. U.S.A. 101, 11785–11790. doi: 10.1073/pnas.0402353101
Cascales, E., Bernadac, A., Gavioli, M., Lazzaroni, J. C., and Lloubes, R. (2002). Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J. Bacteriol. 184, 754–759. doi: 10.1128/JB.184.3.754-759.2002
Chandramouli, K., and Qian, P.-Y. (2009). Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genomics Proteomics 2009:239204. doi: 10.4061/2009/239204
Chang, G. (2003). Multidrug resistance ABC transporters. FEBS Lett. 555, 102–105. doi: 10.1016/S0014-5793(03)01085-8
David, R. (2012). Post-translational modification: keeping cell cycle progression in check. Nat. Rev. Mol. Cell. Biol. 13:341. doi: 10.1038/nrm3363
Dimova, N. (2003). RP-HPLC Analysis of Amino acids with UV Detection. C. R. Acad. Bulg. Sci. 56, 75–78.
Dyballa, N., and Metzger, S. (2009). Fast and sensitive colloidal coomassie G-250 staining for proteins in polyacrylamide gels. J. Vis. Exp. 30:1431. doi: 10.3791/1431
Ekici, O. D., Paetze, M., and Dalbey, R. E. (2008). Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 17, 2023–2037. doi: 10.1110/ps.035436.108
Freestone, P., Trinei, M., Clarke, S. C., Nystrom, T., and Norris, V. (1998). Tyrosine phosphorylation in Escherichia coli. J. Mol. Biol. 279, 1045–1051. doi: 10.1006/jmbi.1998.1836
Gilbert, K. B., Vanderlinde, E. M., and Yost, C. K. (2007). Mutagenesis of the carboxy terminal protease CtpA decreases desiccation tolerance in Rhizobium leguminosarum. FEMS Microbiol. Lett. 272, 65–74. doi: 10.1111/j.1574-6968.2007.00735.x
Goulhen, F., Hafezi, A., Uitto, V.-J., Hinode, D., Nakamura, R., Grenier, D., et al. (1998). Subcellular localization and cytotoxic activity of the GroEL-like protein isolated from Actinobacillus actinomycetemcomitans. Infect. Immun. 66, 5307–5313.
Gutheil, W. G., Holmquist, B., and Vallee, B. L. (1992). Purification, characterization, and partial sequence of the glutathione-dependent formaldehyde dehydrogenase from Escherichia coli: a class III alcohol dehydrogenase. Biochemistry 31, 475–481. doi: 10.1021/bi00117a025
Han, K. K., and Martinage, A. (1993). Post-translational chemical modifications of proteins–III. current developments in analytical procedures of identification and quantitation of post-translational chemically modified amino acid(s) and its derivatives. Int. J. Biochem. 25, 957–970. doi: 10.1016/0020-711X(93)90108-Q
Hara, H., Nishimura, Y., Kato, J., Suzuki, H., Nagasawa, H., Suzuki, A., et al. (1989). Genetic analyses of processing involving C-terminal cleavage in penicillin-binding protein 3 of Escherichia coli. J. Bacteriol. 171, 5882–5889. doi: 10.1128/jb.171.11.5882-5889.1989
Hara, H., Yamamoto, Y., Higashitani, A., Suzuki, H., and Nishimura, Y. (1991). Cloning, mapping, and characterization of the Escherichia coli prc gene, which is involved in C-terminal processing of penicillin-binding protein 3. J. Bacteriol. 173, 4799–4813. doi: 10.1128/jb.173.15.4799-4813.1991
Higgins, C. F. (2001). ABC transporters: physiology, structure and mechanism–an overview. Res. Microbiol. 152, 205–210. doi: 10.1016/S0923-2508(01)01193-7
Hoge, R., Laschinski, M., Jaeger, K. E., Wilhelm, S., and Rosenau, F. (2011). The subcellular localization of a C-terminal processing protease in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 316, 23–30. doi: 10.1111/j.1574-6968.2010.02181.x
Holland, I. B., Schmitt, L., and Young, J. (2005). Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol. Membr. Biol. 22, 29–39. doi: 10.1080/09687860500042013
Jun, D., Signo, K. S. L., Vanderlinde, E. M., Yost, C. K., and Dahms, T. E. S. (2011). Atomic force microscopy of a ctpA mutant in Rhizobium leguminosarum reveals surface defects linking CtpA function to biofilm formation. Microbiology 157, 3049–3058. doi: 10.1099/mic.0.051045-0
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kariu, T., Yang, X., Marks, C. B., Zhang, X., and Pal, U. (2013). Proteolysis of BB0323 results in two polypeptides that impact physiologic and infectious phenotypes in Borrelia burgdorferi. Mol. Microbiol. 88, 510–522. doi: 10.1111/mmi.12202
Keiler, K. C., and Sauer, R. T. (1995). Identification of active site residues of the Tsp protease. J. Biol. Chem. 270, 28864–28868. doi: 10.1074/jbc.270.48.28864
Keiler, K. C., Silber, K. R., Downard, K. M., Papayannopoulos, I. A., Biemann, K., and Sauer, R. T. (1995). C-terminal specific protein degradation: activity and substrate specificity of the tsp protease. Protein Sci. 4, 1507–1515. doi: 10.1002/pro.5560040808
Keiler, K. C., Waller, P. R., and Sauer, R. T. (1996). Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271, 990–993. doi: 10.1126/science.271.5251.990
Kemner, J. M., Liang, X., and Nester, E. W. (1997). The Agrobacterium tumefaciens virulence gene chvE is part of a putative ABC-type sugar transport operon. J. Bacteriol. 179, 2452–2458. doi: 10.1128/jb.179.7.2452-2458.1997
Kiss, E., Huguet, T., Poinsot, V., and Batut, J. (2004). The typA gene is required for stress adaptation as well as for symbiosis of Sinorhizobium meliloti 1021 with certain Medicago truncatula lines. Mol. Plant Microbe Interact. 17, 235–244. doi: 10.1094/MPMI.2004.17.3.235
Klančnik, A., Botteldoorn, N., Herman, L., and Možina, S. S. (2006). Survival and stress induced expression of groEL and rpoD of Campylobacter jejuni from different growth phases. Int. J. Food Microbiol. 112, 200–207. doi: 10.1016/j.ijfoodmicro.2006.03.015
Kumru, O. S., Bunikis, I., Sorokina, I., Bergstrom, S., and Zuckert, W. R. (2011). Specificity and role of the Borrelia burgdorferi CtpA protease in outer membrane protein processing. J. Bacteriol. 193, 5759–5765. doi: 10.1128/JB.05622-11
Kusmierczyk, A. R., and Martin, J. (2000). High salt-induced conversion of Escherichia coli GroEL into a fully functional thermophilic chaperonin. J. Biol. Chem. 275, 33504–33511. doi: 10.1074/jbc.M006256200
Kusukawa, N., Yura, T., Ueguchi, C., Akiyama, Y., and Ito, K. (1989). Effects of mutations in heat-shock genes groES and groEL on protein export in Escherichia coli. EMBO J. 8, 3517–3521.
Lee, H. J., and Zheng, J. J. (2010). PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 8:8. doi: 10.1186/1478-811X-8-8
Liao, D. I., Qian, J., Chisholm, D. A., Jordan, D. B., and Diner, B. A. (2000). Crystal structures of the photosystem II D1 C-terminal processing protease. Nat. Struct. Biol. 7, 749–753. doi: 10.1038/78973
Lodwig, E., Kumar, S., Allaway, D., Bourdes, A., Prell, J., Priefer, U., et al. (2004). Regulation of L-alanine dehydrogenase in Rhizobium leguminosarum bv. viciae and its role in pea nodules. J. Bacteriol. 186, 842–849. doi: 10.1128/JB.186.3.842-849.2004
Mowen, K. A., and David, M. (2014). Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 15, 512–520. doi: 10.1038/ni.2873
Nogueira-Ferreira, R., Vitorino, R., Ferreira-Pinto, M. J., Ferreira, R., and Henriques-Coelho, T. (2013). Exploring the role of post-translational modifications on protein–protein interactions with survivin. Arch. Biochem. Biophys. 538, 64–70. doi: 10.1016/j.abb.2013.07.027
Noppa, L., Östberg, Y., Lavrinovicha, M., and Bergström, S. (2001). P13, an integral membrane protein of Borrelia burgdorferi, is C-terminally processed and contains surface-exposed domains. Infect. Immun. 69, 3323–3334. doi: 10.1128/IAI.69.5.3323-3334.2001
Östberg, Y., Carroll, J. A., Pinne, M., Krum, J. G., Rosa, P., and Bergstrom, S. (2004). Pleiotropic effects of inactivating a carboxyl-terminal protease, CtpA, in Borrelia burgdorferi. J. Bacteriol. 186, 2074–2084. doi: 10.1128/JB.186.7.2074-2084.2004
Otto, A., Becher, D., and Schmidt, F. (2014). Quantitative proteomics in the field of microbiology. Proteomics 14, 547–565. doi: 10.1002/pmic.201300403
Parsell, D. A., Silber, K. R., and Sauer, R. T. (1990). Carboxy-terminal determinants of intracellular protein degradation. Genes Dev. 4, 277–286. doi: 10.1101/gad.4.2.277
Pearce, S. R., Mimmack, M. L., Gallagher, M. P., Gileadi, U., Hyde, S. C., and Higgins, C. F. (1992). Membrane topology of the integral membrane components, OppB and OppC, of the oligopeptide permease of Salmonella typhimurium. Mol. Microbiol. 6, 47–57. doi: 10.1111/j.1365-2958.1992.tb00836.x
Pinne, M., Denker, K., Nilsson, E., Benz, R., and Bergström, S. (2006). The BBA01 protein, a member of paralog family 48 from Borrelia burgdorferi, is potentially interchangeable with the channel-forming protein P13. J. Bacteriol. 188, 4207–4217. doi: 10.1128/JB.00302-06
Puranamaneewiwat, N., Tajima, S., and Niamsup, H. (2006). Proteomic analysis of Bradyrhizobium japonicum USDA110 in acidic condition. Chiang Mai J. Sci. 33, 335–345.
Rao, M. B., Tanksale, A. M., Ghatge, M. S., and Deshpande, V. V. (1998). Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 62, 597–635.
Rawlings, N. D. (2013). “Protease families, evolution and mechanism of action,” in Proteases: Structure and Function, eds K. Brix and W. Stöcker (New York, NY: Springer), 1–36.
Rodríguez-Quiñones, F., Maguire, M., Wallington, E. J., Gould, P. S., Yerko, V., Downie, J. A., et al. (2005). Two of the three groEL homologues in Rhizobium leguminosarum are dispensable for normal growth. Arch. Microbiol. 183, 253–265. doi: 10.1007/s00203-005-0768-7
Rowe, S., Hodson, N., Griffiths, G., and Roberts, I. S. (2000). Regulation of the Escherichia coli K5 capsule gene cluster: evidence for the roles of H-NS, BipA, and integration host factor in regulation of group 2 capsule gene clusters in pathogenic E. coli. J. Bacteriol. 182, 2741–2745. doi: 10.1128/JB.182.10.2741-2745.2000
Shao, W., Yeretssian, G., Doiron, K., Hussain, S. N., and Saleh, M. (2007). The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 282, 36321–36329. doi: 10.1074/jbc.M708182200
Shimoda, N., Toyoda-Yamamoto, A., Aoki, S., and Machida, Y. (1993). Genetic evidence for an interaction between the VirA sensor protein and the ChvE sugar-binding protein of agrobacterium. J. Biol. Chem. 268, 26552–26558.
Silber, K. R., Keiler, K. C., and Sauer, R. T. (1992). Tsp: a tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc. Natl. Acad. Sci. U.S.A. 89, 295–299. doi: 10.1073/pnas.89.1.295
Taga, M. E., Semmelhack, J. L., and Bassler, B. L. (2001). The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol. 42, 777–793. doi: 10.1046/j.1365-2958.2001.02669.x
Taguchi, F., Yamamoto, Y., Inagaki, N., and Satoh, K. (1993). Recognition signal for the C-terminal processing protease of D1 precursor protein in the photosystem II reaction center. an analysis using synthetic oligopeptides. FEBS Lett. 326, 227–231. doi: 10.1016/0014-5793(93)81796-3
Tholen, S., Koczorowska, M. M., Lai, Z. W., Dengjel, J., and Schilling, O. (2013). “Limited and degradative proteolysis in the context of posttranslational regulatory networks: current technical and conceptional advances,” in Proteases: Structure and Function, eds K. Brix and W. Stöcker (New York, NY: Springer), 175–216.
Tilly, K., Krum, J. G., Bestor, A., Jewett, M. W., Grimm, D., Bueschel, D., et al. (2006). Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect. Immun. 74, 3554–3564. doi: 10.1128/IAI.01950-05
Tzin, V., Malitsky, S., Zvi, M. M. B., Bedair, M., Sumner, L., Aharoni, A., et al. (2012). Expression of a bacterial feedback-insensitive 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase of the shikimate pathway in Arabidopsis elucidates potential metabolic bottlenecks between primary and secondary metabolism. New Phytol. 194, 430–439. doi: 10.1111/j.1469-8137.2012.04052.x
Tzin, V., Rogachev, I., Meir, S., Moyal Ben Zvi, M., Masci, T., Vainstein, A., et al. (2013). Tomato fruits expressing a bacterial feedback-insensitive 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase of the shikimate pathway possess enhanced levels of multiple specialized metabolites and upgraded aroma. J. Exp. Bot. 64, 4441–4452. doi: 10.1093/jxb/ert250
Vincent, J. M. (1970). A Manual for the Practical Study of Root-Nodule Bacteria. Oxford: Blackwell Scientific.
Vogel, C., and Marcotte, E. M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232. doi: 10.1038/nrg3185
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucl. Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wissenbach, U., Six, S., Bongaerts, J., Ternes, D., Steinwachs, S., and Unden, G. (1995). A third periplasmic transport system for l-arginine in Escherichia coli: molecular characterization of the artPIQMJ genes, arginine binding and transport. Mol. Microbiol. 17, 675–686. doi: 10.1111/j.1365-2958.1995.mmi_17040675.x
Wood, D. W., Setubal, J. C., Kaul, R., Monks, D. E., Kitajima, J. P., Okura, V. K., et al. (2001). The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294, 2317–2323. doi: 10.1126/science.1066804
Yost, C. K., Rath, A. M., Noel, T. C., and Hynes, M. F. (2006). Characterization of genes involved in erythritol catabolism in Rhizobium leguminosarum bv. viciae. Microbiology 152, 2061–2074. doi: 10.1099/mic.0.28938-0
Keywords: ABC transporters, amino acid metabolism, C-terminal protease, proteomics, Rhizobium leguminosarum
Citation: Jun D, Minic Z, Bhat SV, Vanderlinde EM, Yost CK, Babu M and Dahms TES (2018) Metabolic Adaptation of a C-Terminal Protease A-Deficient Rhizobium leguminosarum in Response to Loss of Nutrient Transport. Front. Microbiol. 8:2617. doi: 10.3389/fmicb.2017.02617
Received: 11 October 2017; Accepted: 15 December 2017;
Published: 04 January 2018.
Edited by:
Satoshi Tsuneda, Waseda University, JapanReviewed by:
Michael F. Minnick, University of Montana, United StatesCopyright © 2018 Jun, Minic, Bhat, Vanderlinde, Yost, Babu and Dahms. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanya E. S. Dahms, dGFueWEuZGFobXNAdXJlZ2luYS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.