 Taha Soliman1,2,3*
Taha Soliman1,2,3* James D. Reimer2,4
James D. Reimer2,4 Sung-Yin Yang1,5
Sung-Yin Yang1,5 Alejandro Villar-Briones6Michael C. Roy6
Alejandro Villar-Briones6Michael C. Roy6 Holger Jenke-Kodama1*
Holger Jenke-Kodama1*- 1Microbiology and Biochemistry of Secondary Metabolites Unit, Okinawa Institute of Science and Technology Graduate University (OIST), Kunigami, Japan
- 2Molecular Invertebrate Systematics and Ecology Laboratory, Graduate School of Engineering and Science, University of the Ryukyus, Nishihara, Japan
- 3Genetics and Genetic Engineering Research Group, National Institute of Oceanography and Fisheries (NIOF), Cairo, Egypt
- 4Tropical Biosphere Research Center, University of the Ryukyus, Nishihara, Japan
- 5Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
- 6Imaging and Instrumental Analysis Section, Okinawa Institute of Science and Technology Graduate University (OIST), Kunigami, Japan
Microbial community diversity and chemodiversity were investigated in marine sediments adjacent to the Okinawan “Kaichu-Doro” Causeway, which was constructed 46 years ago to connect a group of four islands (Henza-jima, Miyagi-jima, Ikei-jima, Hamahiga-jima) to the Okinawan main island. This causeway was not built on pilings, but by land reclamation; hence, it now acts as a long, thin peninsula. The construction of this causeway was previously shown to have influenced the surrounding marine ecosystem, causing ecosystem fragmentation and loss of water circulation. In this study, we collected sediment cores (n = 10) from five paired sites in 1 m water depths. Each pair of sites consisted of one site each on the immediate north and south sides of the causeway. Originally the members of each pair were much closer to each other (<150 m) than to other pairs, but now the members of each pair are isolated by the causeway. Each core was 60–80 cm long and was divided into 15-cm layers. We examined the vertical diversity of microbial communities and chemical compounds to determine the correlation between chemodiversity and microbial communities among marine sediment cores and layers. Principal coordinate analyses (PCoA) of detected compounds and of bacterial and archaeal operational taxonomic units (OTUs) revealed that the north and south sides of the causeway are relatively isolated, with each side having unique microbial OTUs. Additionally, some bacterial families (e.g., Acidaminobacteraceae, Rhizobiaceae, and Xanthomonadaceae) were found only on the south side of Kaichu-Doro. Interestingly, we found that the relative abundance of OTUs for some microbial families increased from top to bottom, but this was reversed in some other families. We conclude that the causeway has altered microbial community composition and metabolite profiles in marine sediments.
Introduction
Anthropogenic impacts such as pollution, large-scale coastal construction, and overexploitation of marine resources have affected many marine ecosystems (Islam and Tanaka, 2004), including those of the shallow waters in the Ryukyu Archipelago in southern Japan (Reimer et al., 2015). Okinawa-jima, the main island, located in the central Ryukyu Archipelago, is situated in a marine area with high biodiversity that is under threat from anthropogenic impacts (Roberts et al., 2002). The causeway “Kaichu-Doro” is one example of large-scale coastal development on Okinawa. This causeway was built about 46 years ago (1971–1974) to connect four smaller islands (Henza-jima I., Miyagi-jima I., Ikei-jima I., Hamahiga-jima I.) to Okinawa, effectively cutting a tidal mud flat into two separate areas, a northern side in Kin Bay and a southern side in the Pacific Ocean (Reimer et al., 2015, Figure 1).
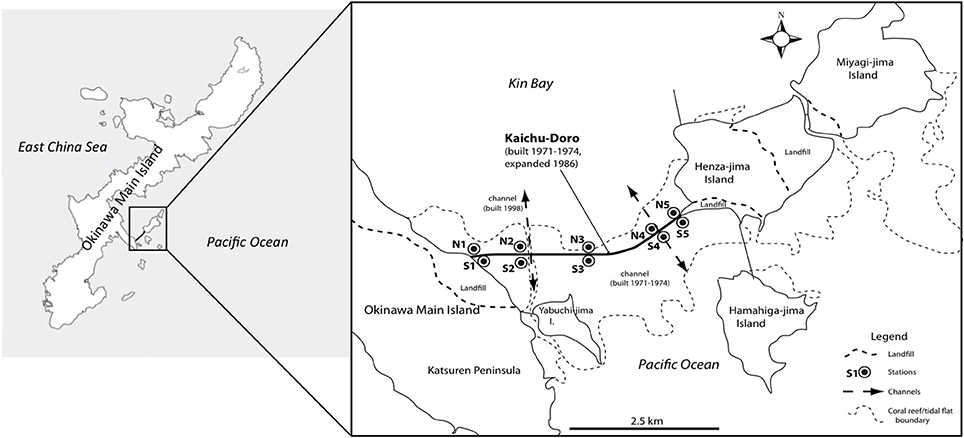
Figure 1. Sampling sites at the causeway, Kaichu-Doro, Okinawa Main Island, Japan (adapted from Reimer et al., 2015).
Microbial communities and their metabolites are critical to the health of marine ecosystems (Das et al., 2006). Previous studies have reported the importance of marine microbes in mangrove ecosystems in photosynthesis (Vethanayagam, 1991), nitrogen fixation (Toledo et al., 1995), methanogenesis (Mohanraju and Natarajan, 1992), and the production of antibiotics and enzymes (Kathiresan and Bingham, 2001). In other ecosystems, such as coral reefs, microbial communities in marine sediment are a source of phosphorus and nitrogen, and corals support bacterial activity by producing mucus, which contains proteins and polysaccharides (Wild et al., 2004).
In general, marine sediment contains numerous microbes (bacteria, archaea, fungi, and other eukaryotes) and it has been challenging to estimate their abundance and distribution in sediments. However, recently, next generation sequencing (NGS) has allowed acquisition of DNA sequences from various sediments with increasing accuracy (Quail et al., 2012), and studies on microbial diversity within marine sediments have accelerated (e.g., Acosta-Gonzalez and Marques, 2016; Fan and Xing, 2016; Galand et al., 2016; Hu et al., 2016; Stagars et al., 2016; Walsh et al., 2016).
The term chemodiversity is generally used to describe the diversity of chemical compounds in a specified context. Chemodiversity studies are often restricted to a certain group of organisms, such as Aspergillus species (Frisvad and Larsen, 2015) or to a specific set of plant secondary metabolites (Moore et al., 2014). However, chemodiversity really embraces all metabolites present in complex environments, like soil or water. For instance, recent studies have analyzed the influence of soils on wines produced from two proximate vineyards in the Bourgogne area of France (Roullier-Gall et al., 2014) and climate-dependent differences in the distribution of dissolved organic matter in lakes (Kellerman et al., 2014). Here, we define chemodiversity as the aggregate of all detectable chemical compounds within marine sediment cores, without focusing on particular producers or subsets of natural products.
The aim of this study was to follow-up on a previous investigation by Reimer et al. (2015), which showed differences in taxonomic diversity and abundance on either side of the Kaichu-Doro causeway. In this study, we specifically investigated compositional differences of microbial communities (bacteria, archaea, fungi and other eukaryotes) and quantitative chemodiversity across a vertical gradient within 1 m sediment cores taken from various sites along the causeway. We then examined the correlation between chemical compounds and microbial communities among marine sediment layers. The results of the present study will be useful not only to furnish baseline marine microbe diversity data for the Kaichu-Doro causeway, but can also serve as a model for examining long-term impacts of coastal construction on marine sediment microbial communities.
Materials and Methods
Cores
Marine sediment cores were collected as described in Reimer et al. (2015) from the north (N1–N5) and south (S1–S5) sides or “coasts” of Kaichu-Doro (Figure 1). Cores were approximately 60–80 cm in depth (Figure 1), and were transported and stored at −80°C at the University of the Ryukyus, Okinawa, Japan. Core sediments were separated into vertical layers ~15 cm in length (total n = 47) and sediment dating was based on sedimentation rates (Reimer et al., 2015), and designated N1A (north core 1 layer A), N1B (north core 1 layer B), etc. (A–F from top to bottom). Additional information about cores, such as sampling dates, time, characteristics of sediments, and a full description of the study area were detailed in Reimer et al. (2015).
DNA Extraction and PCR
Sediments of each layer were mixed well. Triplicate samples of total genomic DNA were extracted from 0.5 g of each layer of core sediment using a MO BIO PowerSoil® DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) following the manufacturer's protocol. Polymerase chain reaction (PCR) amplifications were conducted using 16S rRNA gene primers [341F, 5′-CCTACGGGNGGCWGCAG-3′; 805R, 5′-GACTACHVGGGTATCTAATCC-3′ (Herlemann et al., 2011)] for the V3-V4 region of bacteria, 16S rRNA gene primers [340F, 5′-CCCTAYGGGGYGCASCAG-3′ (Gantner et al., 2011); 915R, 5′-GTGCTCCCCCGCCAATTCCT-3′ (Stahl and Amann, 1991)] for archaea, internal transcribed spacer (ITS) region primers [ITS3, 5′-GCATCGATGAAGAACGCAGC-3′; ITS4, 5′-TCCTCCGCTTATTGATATGC-3′ (White et al., 1990)] for fungi, and 18S rRNA primers [1380F, 5′-CCCTGCCHTTTGTACACAC-3′; 1510R, 5′-CCTTCYGCAGGTTCACCTAC-3′ (Amaral-Zettler et al., 2009)] for other eukaryotes. PCR amplifications were conducted in 20 μL total volume containing 4 μL (10 ng/μL) of microbial template genomic DNA, 0.6 μL (10 mM) of each forward and reverse primer, 4.8 μL of PCR-grade water, and 10 μL of 2 × KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Boston, MA, USA). PCR conditions were 95°C for 5 min (initial denaturing), 30 cycles of 20 s at 98°C, 20 s at optimum annealing temperatures, and 30 s at 72°C, and a final extension at 72°C for 5 min. Amplicons were confirmed and sized using gel electrophoresis (1.2% and 1 × TAE buffer). PCR products were cleaned up using AMPure XP beads (Agencourt ®AMPure ®XP kit, Beckman Coulter, USA), according to the Illumina MiSeq protocol for amplicon preparation. Amplicon products were sequenced using the Illumina MiSeq platform at the DNA Sequencing Section of the Okinawa Institute of Science and Technology (OIST) Graduate University (Onna, Okinawa, Japan).
Methanol Extraction of Samples
One gram of sediment sample (wet weight) was transferred to a 15-mL Falcon tube and twice extracted with 5 mL methanol. The extraction was carried out by vortexing (30 s), and sonication (10 min), followed by centrifugation (10 min, 9,000 g, 10°C). Pooled methanol extracts were transferred to a new Falcon tube (15 mL) and dried in a vacuum concentrator (40°C, TOMY Speed Vac). One mL of methanol was added to the resulting dried extract, vortexed (1 min), and sonicated (10 min). Finally, the suspension was transferred to an Eppendorf tube (2 mL) and centrifuged (10 min, 14,000 g, 10°C) to give a clear methanol solution. The clear methanol solution was decanted in an Eppendorf tube (2 mL) and dried in the vacuum concentrator (40°C). The resulting extract was finally dissolved in methanol (200 mL) and centrifuged (10 min, 14,000 g, 10°C) to give a clean methanol extract. The methanol extract was either analyzed immediately or stored at −30°C.
Liquid Chromatography-Mass Spectrometry (LC-MS)
A Thermo Scientific Mass Spectrometer (Q Exactive Plus, Bremen, Germany) was used for mass spectrometry (MS) data collection. The mass spectrometer was equipped with an ultra-high-pressure liquid chromatography (UHPLC) (Dionex Ultimate 3000, Thermo Scientific), an auto-sampler (HTC PAL, CTC Analytics, Zwingen, Switzerland), and a heated electrospray ionization source-II (H-ESI). Two tandem spectra were generated for each targeted compound in positive ion mode. H-ESI probe conditions were set as follows: sheath gas flows 35, auxiliary gas flow 10, sweep gas flow 0, spray voltage 3.5 kV, capillary temperature at 290°C, S-lens RF level at 70.0, and auxiliary gas heater at 350°C. Full MS spectra were acquired at 70,000 resolution AGC target 3e6, maximum IT 100 ms, and full mass range m/z 250–1,200 Da. Data-dependent MS2 spectrum was configured to select top ions from the MS1 spectrum, with 35,000 resolution, AGC target 1e5, maximum IT 50 ms, loop count 5, optimized collision energy (NCE 35), and isolation window (4.0 m/z).
Clean samples were separated on an Ascentis Express C18 column (150 × 2.1 mm, 2.7 μm, Supelco). An 18-min step-gradient was used for compound separation (10% B for 0.0-2.0 min, 10 to 25% B for 2.0–2.5 min, 25 to 100% B for 2.5–10.0 min, hold 100% B until 14.0 min, equilibration 10% B from 14.1 to 18.0 min, where solvent A was aqueous-acetonitrile 95:5 and solvent B was aqueous-acetonitrile 10:90. Both solvents contained 0.1% formic acid. A flow rate of 300 μL/min was used and the column temperature was maintained at 40°C). A 15 μL (20 μL loop) sample was injected using the auto-sampler and triplicate runs were carried out for each sample.
Data Analyses
The Illumina MiSeq (V3 kit) produced 2 × 300 bp paired-end sequences, and quality control of raw fastq data files was performed using FastQC v0.11.4 (Andrews, 2010). High-throughput sequence raw data were imported into CLC Genomics Workbench version 9.0.1 with plugins CLC Microbial Genomics Module version 1.3.1 (QIAGEN, Aarhus A/S, http://www.clcbio.com) according to quality scores from Illumina pipeline 1.8, in order to achieve the highest quality sequences for clustering. Sequence data were trimmed using 0.05 as a limit for quality scores with 2 as the maximum number of ambiguities. The optional merge paired reads method was run with default settings (mismatch cost = 1; minimum score = 40; gap cost = 4 and maximum unaligned end mismatch = 5). Sequence reads were clustered and chimeric sequences detected using an identity of 97% as the Operational Taxonomic Unit (OTU) threshold. Reference OTU data used in the present study were downloaded from the Greengenes database (DeSantis et al., 2006) for 16S rRNA (bacteria and archaea), the Unite database (Koljalg et al., 2013) for ITS (fungi), and Silva (Quast et al., 2013) for 18S rRNA (eukaryotes). Relative abundances of the most abundant OTUs among all four-sediment layers for all 10 cores were estimated at the family level for archaea, bacteria, eukaryotes, and fungi. OTU relative abundances are presented in separate figures for high and low abundance values of each microbial community based on the number of sequences per OTU. High-throughput sequencing data from this study were deposited in the Sequence Read Archive (SRA) of GenBank under Accession number SRP077849. LC-MS raw data were processed and analyzed using Mzmine version 2.02 (Pluskal et al., 2010), XCMS R package (Smith et al., 2006) and MetaboAnalyst version 3.0 (Xia et al., 2015). Our strategy for analyzing the quantitative chemodiversity of sediment cores was to perform an initial general screening of methanol extracts by mass spectrometry (LC-MS) without focusing on specific metabolites or compound groups and without attempting to identify the compounds. The initial screening resulted in thousands of peaks, as would be expected for an environment as complex as marine sediment. In order to reduce the complexity of analyses, we selected the most significant MS signals based on p-values (p ≤ 0.01) and Relative Standard Deviations (RSD ≤ 10%) among layers of sampled cores. This procedure resulted in a selection of 195 peaks (see Table S1).
Overall, Good's estimator of coverage [(1–(singletons/individuals)) × 100] and Shannon's entropy were estimated for all microbial communities in the present study. A Permutational Multivariate Analysis of Variance (PERMANOVA) test was conducted based on Bray-Curtis (Beals, 1984) and Weighted UniFrac (Lozupone et al., 2011) distances among cores, and separately among the top layer (A). PERMANOVA parameters were measured using 100,000 permutations using CLC Genomics Workbench version 9.0.1 and the data grouped based on layers (AN, BN, CN, DN, EN, AS, BS, CS, DS, ES, FS), cores (N1–N5 and S1–S5) and locations (north and south). Principal Coordinates Analysis (PCoA) was used to characterize all layers and the top layer (A) among cores based on Bray-Curtis for both community structure and chemodiversity. Additionally, alpha diversity (rarefaction curves) was calculated based on Chao 1 measurements. PCoA and Alpha diversity were conducted for microbial communities using CLC Microbial Genomics Module version 1.3.1. Partial Least Squares Discriminant Analysis (PLS) estimated the weighted sum of absolute regression coefficients among the microbial communities from the top layers (A) of the cores. The heatmap visualization was estimated based on Euclidean matrix distances, and Ward's clustering algorithm was utilized for chemodiversity features and OTU abundances as identified by t-test analysis (p < 0.05) among all layers. Seawater environmental data obtained by Reimer et al. (2015) were interpreted and discussed with microbial community top layers (A).
Results
Numbers of Gene Sequences and Coverage
Numbers of predicted operational taxonomic units (OTUs) defined by nucleotide sequence identity higher than 95% were estimated for all communities (bacteria = 6,039; archaea = 517; fungi = 580; other eukaryotes = 764). Predicted OTUs, reads in OTUs, filtered reads, Good's coverage, and Shannon entropy values of each layer are summarized in Table 1. All layers showed a Good's sequencing coverage of at least 95%. Characteristic changes in OTU-based diversity, as measured by the number of different OTUs and Shannon entropy could not be observed for most of the cores. Only in cores N5 and S2 did the diversity of all communities steadily increase from the top to the bottom layers. In general, the diversity of fungal and other eukaryotes were much more constant among layers compared to bacteria and archaea.
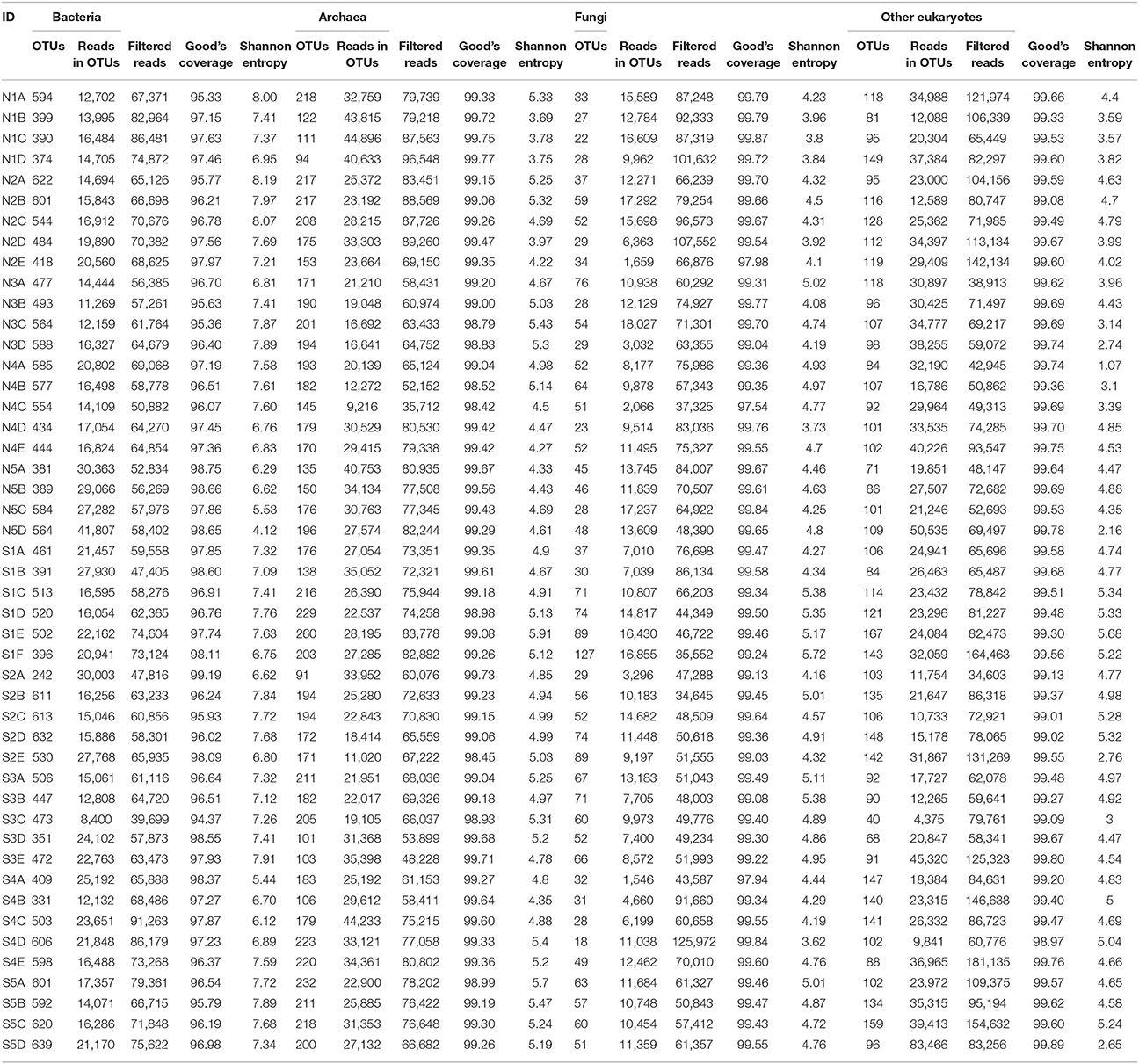
Table 1. Summary of reads, operational taxonomic unit (OTU) numbers, and overall parameters of microbial communities from sites at Kaichu-Doro, Okinawa, Japan.
Bacterial and Archaeal Communities
In bacterial communities, the relative abundance of OTUs of the class Deltaproteobacteria (phylum Proteobacteria) clearly decreased from top to bottom layers in cores N2, N3, S1, S2, and S3, but this trend was reversed in core N4 (Figures 2A,C). Likewise, the Atribacteria (formerly called candidate phylum OP8) and SHA-20 showed the same patterns of distribution among layers (Figures 2A,C). However, other phyla such as Firmicutes, Actinobacteria, Chloroflexi, Cyanobacteria, Planctomycetes, and Bacteroidetes showed fluctuations in OTU values among all layers from both the north and south sides of the causeway (Figures 2C,D). In cores S4 and N4, the photosynthetic Chloroflexi showed an unusual distribution as their proportions increased in deeper layers. The family Bacillaceae was abundant in cores N5 (49%) and S4 (38%) and the order Bacillales (unclassified family) was high in the same two cores (N5 and S4) compared to other cores (Figures 2C,D). In addition, the family Helicobacteraceae (Proteobacteria) made up the highest percentages of OTUs in cores N2, N4, and S1 (Figures 2B,D). In addition, the family Moraxellaceae (Proteobacteria) showed high relative abundance in layers N5C-D of core N5 (Figure 2A). Unique OTUs of bacterial phyla Bacteroidetes (order: Bacteroidales), Proteobacteria (order: Rhizobiales; families: Rhizobiaceae, Rhodobacteraceae, Pseudomonadaceae, Xanthomonadaceae; genera: Rhodobacter, Paracoccus, Thiothrix, Dechloromonas), and Verrucomicrobia (genus: Luteolibacter) were present only in cores from the south side of Kaichu-Doro (Table 2).
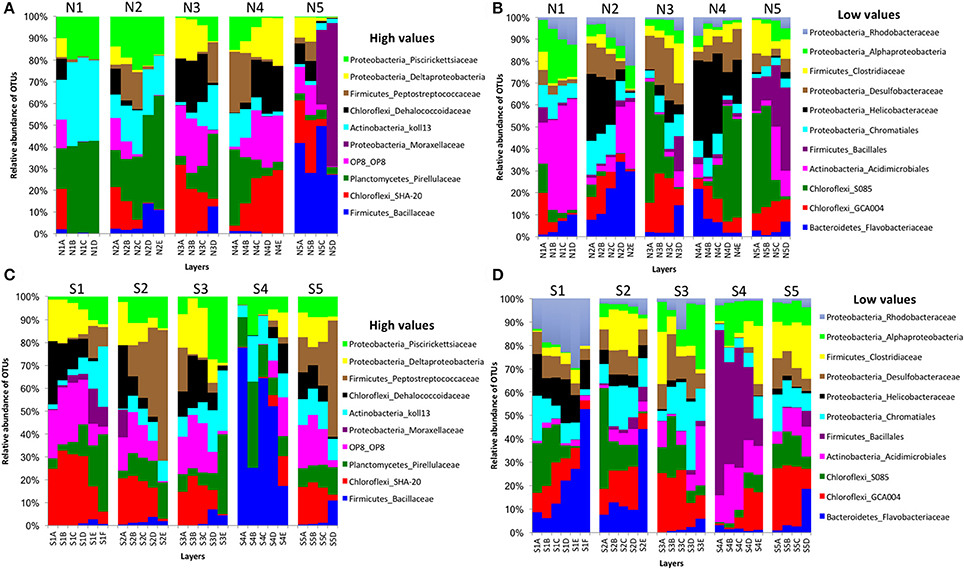
Figure 2. Relative abundances of bacterial OTUs between north (N) and south (S) sites at Kaichu-Doro among layers (A–F). High (A) and low (B) OTU abundance on the north; high (C) and low (D) OTU abundances values on the south.
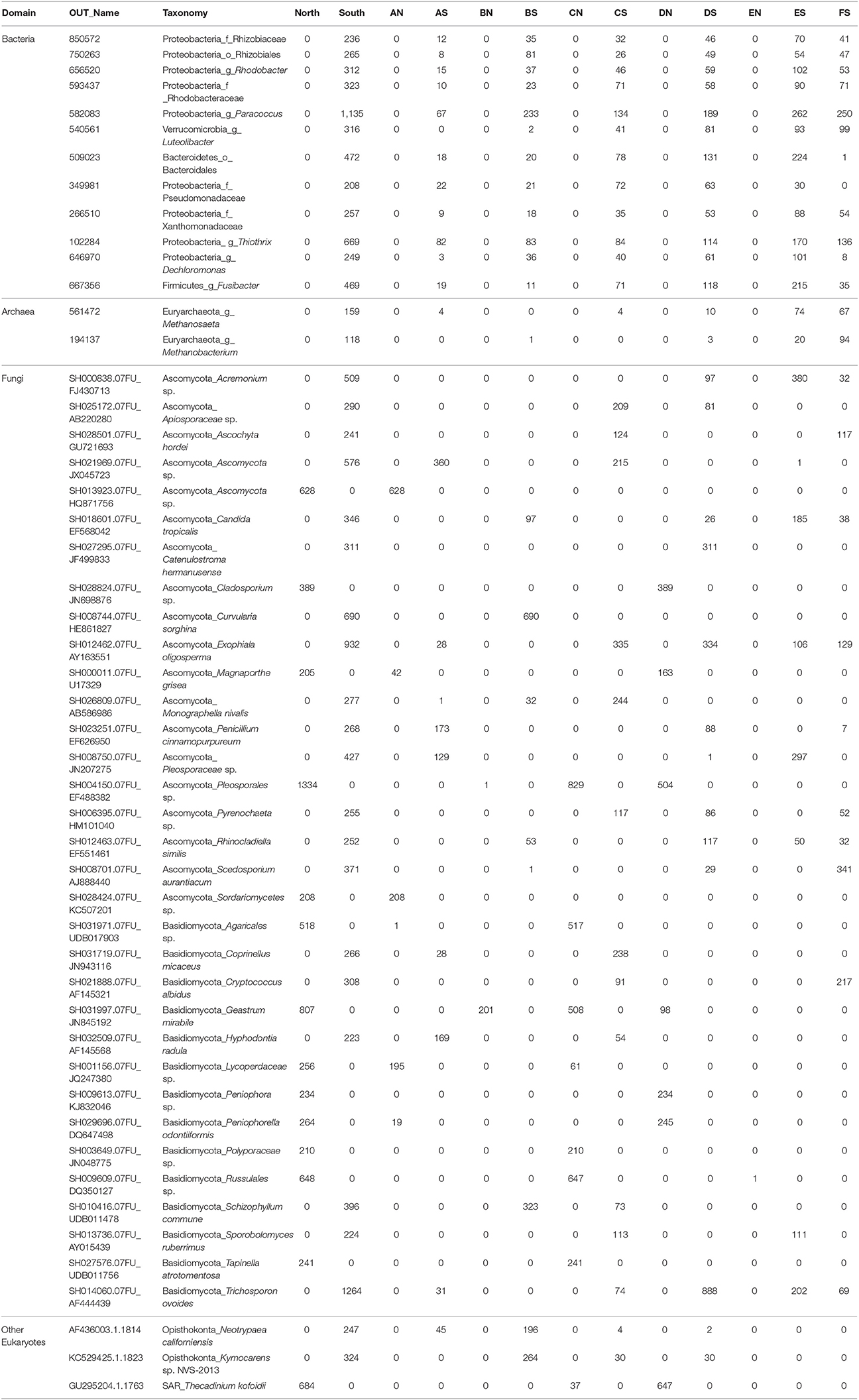
Table 2. Unique operational taxonomic units (OTUs) of 47 marine sediment layers of microbial communities among layers between the north and south sides of Kaichu-Doro, Okinawa, Japan.
For archaeal communities, we obtained 1,285,885 archaea-specific reads out of a total of 3,394,675 filtered sequence reads. The diversity of archaeal OTUs was lower than the diversities found for other organism groups. The dominant phyla were Crenarchaeota, Euryarchaeota, and Thaumarchaeota (Table 1 and Figure 3). OTUs in the family Crenarchaeaceae were most abundant in cores N1, N2 and S3-S5, and the family Nitrososphaeraceae was abundant only in core S4 in the top layers (Figure 3D). Group pMC1 of the phylum Euryarchaeota had the highest relative abundance in layers C-E of cores S3 and S4 (Figure 3D). OTUs of the phylum Euryarchaeota varied from top to bottom for cores from both sides (from 9 to 22%; Figures 3A–D). Unique OTUs of the genera Methanosaeta and Methanobacterium were only seen on the south side of Kaichu-Doro (159 and 118 sequences, respectively; Table 2).
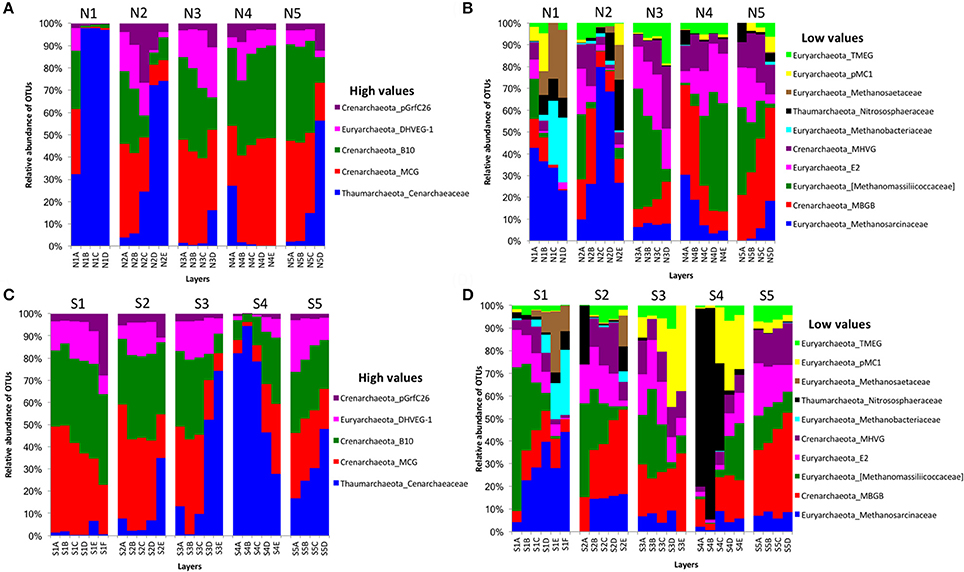
Figure 3. Relative abundances of archaeal OTUs between north (N) and south (S) side sites at Kaichu-Doro among layers (A–F). High (A) and low (B) OTU abundance on the north; high (C) and low (D) OTU abundances values on the south.
Fungal and Other Eukaryotic Communities
We obtained 498,660 fungal reads and 580 total predicted OTUs from a total of 3,112,892 filtered sequence reads targeting the ITS-3 and ITS-4 regions (Table 1). Fungal OTU abundances indicated variations among layers from cores on both sides of Kaichu-Doro (Figures 4A–D). Incertae sedis were abundant among all layers of all cores (Figures 4A,C). A relatively high abundance of OTUs of the family Exobasidiaceae was present in N1C and N3C with small abundances in other cores (Figures 4B,D). We estimated 13 and 20 unique OTUs among fungal taxa from the north and south sides of Kaichu-Doro, respectively (Table 2).
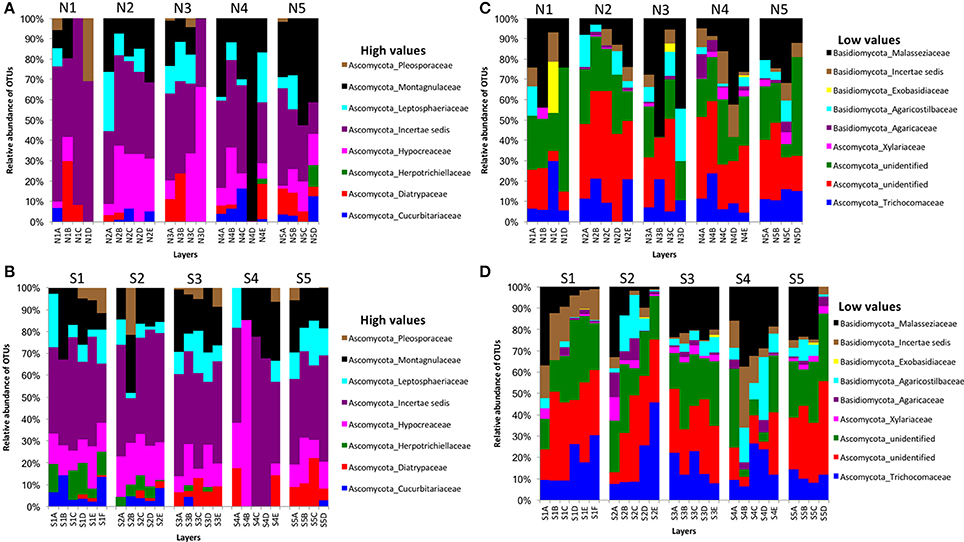
Figure 4. Relative abundances of fungal OTUs between north (N) and south (S) side sites at Kaichu-Doro among layers (A–F). High (A) and low (B) OTU abundance on the north; high (C) and low (D) OTU abundances values on the south.
Partial sequencing of the eukaryotic 18S rRNA gene produced 764 total predicted OTUs, comprising 1,278,706 specific reads from a total of 4,009,753 filtered reads (Table 1). The relative abundance of eukaryote OTUs, such as Animalia, Bacillariophyceae, Bacillariophytina, Gastropoda, and Crustacea increased from top to bottom on the north side of Kaichu-Doro, and this pattern was reversed for Agaricomycotina, Craniata, Novel Apicomplexa Class 2, and Thoracosphaeraceae. Gastropoda and Crustacea were abundant in cores N3B-D and S2 (among all layers), (Figures 5A,C). In addition, the relative OTU abundance of Pseudoperkinsidae was high in cores S1, layers C-F from the south side, and in Core N1 layer D (Figures 4B,D). Neotrypaea californiensis (ghost shrimp, 247 sequences) and Kymocarens sp. NVS-2013 (flatworm, 324 sequences) were unique to the south side of Kaichu-Doro, and Thecadinium kofoidii (dinoflagellate, 684 sequences) was unique to the north side (Table 2).
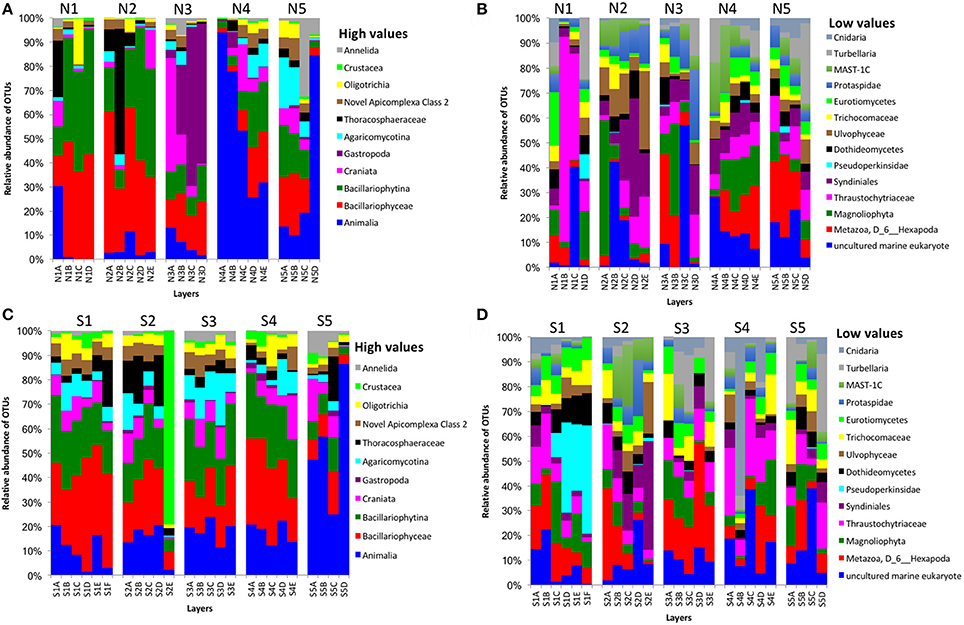
Figure 5. Relative abundances of other eukaryotes OTUs between north (N) and south (S) side sites at Kaichu-Doro among layers (A–F). High (A) and low (B) OTU abundance on the north; high (C) and low (D) OTU abundances values on the south.
Statistical Analyses of Community Structure and Chemodiversity
To test statistically whether there were differences in structures of microbial communities with regard to location, we performed permutational multivariate analyses of variance (PERMANOVA) for each type of community among layers and cores, and compared community composition of north and south sites as well. Differences in overall layers of all communities were insignificant (p values; bacteria = 0.8612, archaea = 0.5020, fungi = 0.3577 and other eukaryotes = 0.2945), but were significant in chemodiversity analyses (p < 0.002). This means that there was no clear vertical trend for changes in community composition. In contrast, the PERMANOVA test of composition of community structure and chemodiversity among all cores showed significant differences (p < 0.0001). However, the total comparison between north and south showed significant differences for other eukaryotic communities and chemodiversity (p < 0.002), but not for bacteria (0.0887), for archaea (0.3423), or for fungi (0.1315). Additionally, PERMANOVA was used to test only layer A among all north and south sites. It revealed insignificant differences (p-values; bacteria = 0.8174, archaea = 0.8095, fungi = 0.2698, other eukaryotes = 0.8968).
Sea Water Environment and Microbial Communities in the Top Layer
From the PCoA result of seawater environmental data (Reimer et al., 2015), sites N1, N2, N3, S3, and S5 had similar features. S4 and N4 were similar, and S1, S2, and N5 were distinct from other groups (Figure 6A). However, chemodiversity (Figure 6B) showed different patterns, with north and south sites separated, except for S4, which resembled north sites.
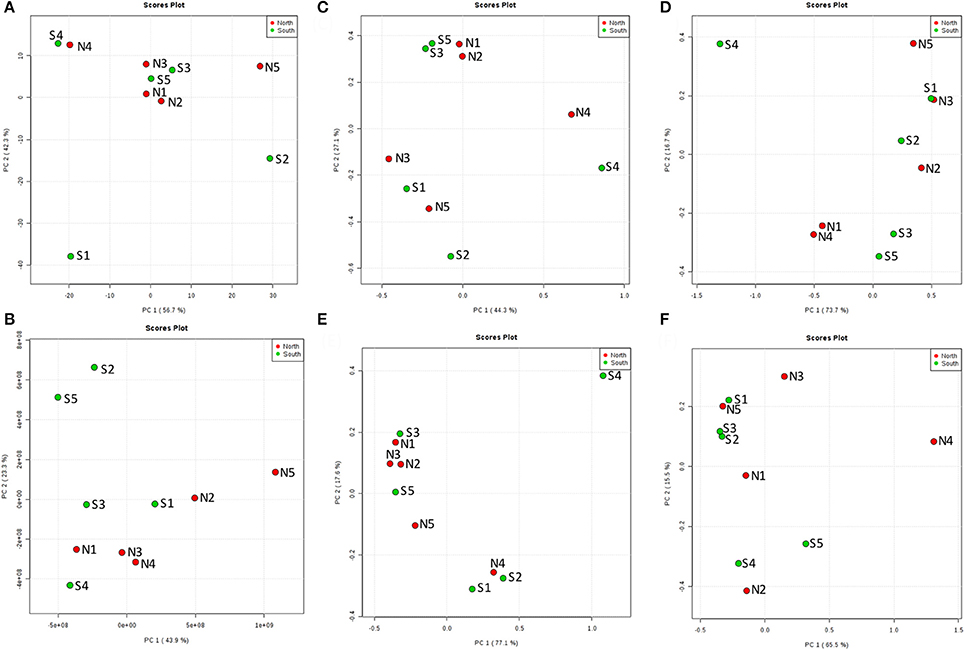
Figure 6. Principal Coordinates Analysis (PCoA) of the top layer (A) among 10 cores for north and south sides of the Kaichu-Doro causeway. (A) Environmental factors; (B) chemodiversity (LC-MS); (C) bacteria; (D) archaea; (E) fungi; (F) other eukaryotes.
Similar patterns could be found throughout all layers. Although the number of compounds and microbial communities per layer did not show clear differences within or among cores, Principle Coordinate Analysis (PCoA) of compounds indicated two relatively discrete clusters corresponding to the north and south sides of the causeway among all layers (Figure S1A). Similarly, PCoA showed the same general clustering pattern for the OTUs of overall bacterial and archaeal communities (Figures S1B,C). In contrast, PCoA revealed no pattern of clearly clustered groups among fungal and other eukaryote OTUs (Figures S1D,E). While targeting high abundance OTUs and metabolites, differences of south and north were more pronounced (Figures 7A–E). Metabolite classes from north and south were very distinct (Figure 7A) throughout layers and sites. The distribution of high-abundance bacterial OTUs was also bimodal (Figure 7). In addition, it spread into five clusters: (1) S3, S4 and S5; (2) N1C, D, N2D, E, and N3D; (3) N2 to N5, with S1F and S2E from the deepest layers of south cores; (4) N1A, B, and S4A, B; (5) S1, S2 and S3B. For other dominant OTUs in archaea (Figure 7C), fungi (Figure 7D) and eukaryotes (Figure 7E), there were no clear groupings for sites or layers. Alpha rarefaction curves were constructed for each core sample showing numbers of OTUs (Chao 1), based on minimum numbers of reads (Figure S2). Core N5 showed low numbers of OTUs of archaea and other eukaryotes, but core S4 showed the highest number of OTUs in both communities (Figures S2B,D). However, core S5 presented the lowest number of OTUs of bacteria, archaea, and fungi (Figures S2A–C).
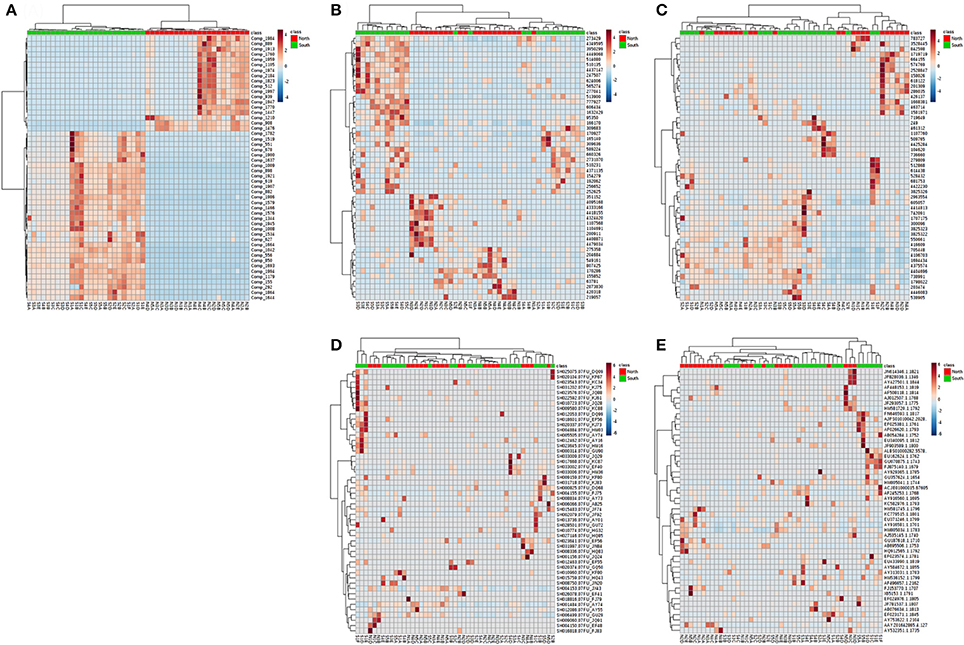
Figure 7. Heatmap for the hierarchical clustering (using Euclidean matrix distances, and Ward's clustering algorithm) for chemodiversity features and OTU abundances among all layers of the Kaichu-Doro causeway. (A) Metabolites; (B) bacteria; (C) archaea; (D) fungi; (E) other eukaryotes.
PLS-DA showed there were certain microbial groups that had strong correlations with the north or south side (Figures 8A–D). For bacteria, two classes from Chloroflexi, Dehalococcoidaceae and GCA004, were mainly found in the south, while of three Firmicutes classes, the Peptostreptococcacea was more common in the north, whereas the Bacillales and Clostridiaceae were found more in the south. For archaea, there were only two Crenarchaeota, predominately found in the north. Trichocomaceae and one unidentified class of fungi from Ascomycota and class Basidiomycota from Malasseziaceae were identified more frequently in the north (Figures 8A–D).
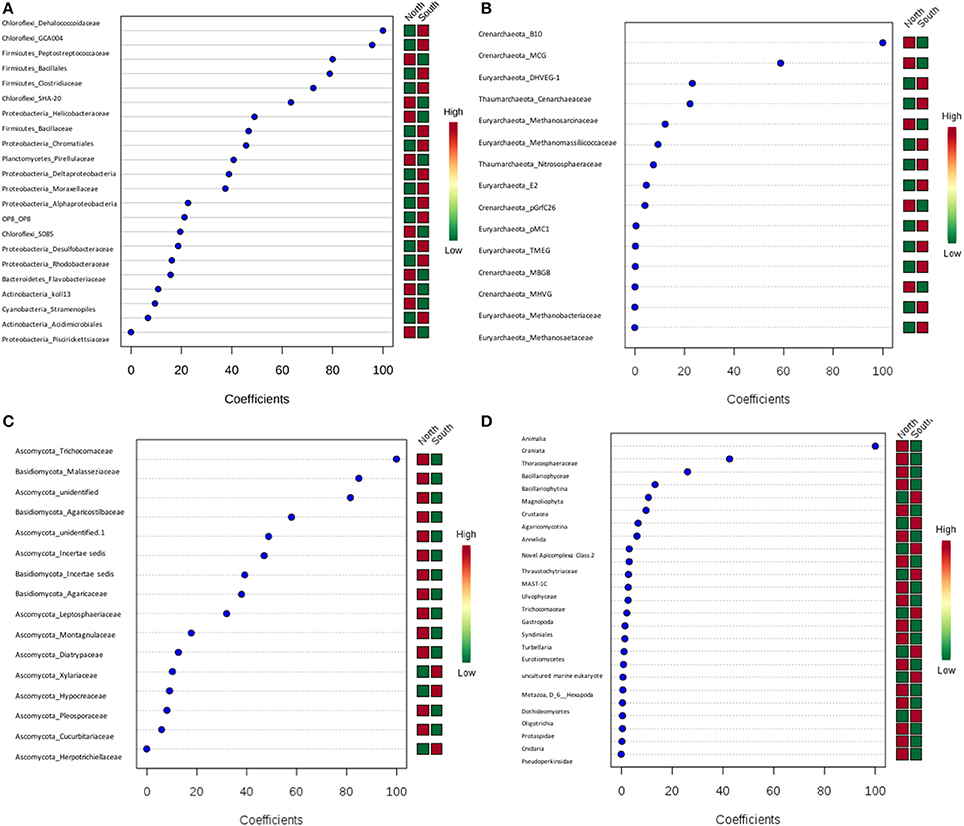
Figure 8. Partial least squares Discriminant Analysis (PLS-DA) based on weighted sum of absolute regression coefficients. Colored boxes on the right indicate the relative abundance of corresponding OTUs in north and south sides. (A) Bacteria; (B) archaea; (C) fungi; (D) other eukaryotes.
Discussion
Microbial communities have previously been investigated from shallow to deep ocean waters, as well as from marine sediments using next-generation sequencing (NGS) (DeLong et al., 2006; Brown et al., 2009; Hu et al., 2016; Walsh et al., 2016). Recently, high-throughput sequencing technology was used to examine microbial communities in deep-sea sediments and to investigate sediment-hosted CO2 in the Okinawa Trough (Inagaki et al., 2006; Zhang J. et al., 2015). However, despite such applications of this technology, many ecosystems throughout the world remain to be investigated. In the present study, we examined the vertical distribution of microbial communities from marine sediment cores. Another novelty of the current study was the analyses of chemodiversity along the causeway to investigate possible correlations between microbial communities and compounds detectable in sediment cores.
Our study indicates that some groups of organisms may have relatively definite depth profiles based on the vertical composition of microbial communities in the cores, with either an increase in abundance from top to bottom, or vice-versa. Such patterns were particularly common in bacterial communities (Figures 2A–D). Previously, most studies have reported on variations of microbial community composition in the water column and gyres, with various results by depth (Brown et al., 2009; Treusch et al., 2009; Ghiglione et al., 2012; Fan and Xing, 2016; Walsh et al., 2016). However, the present study focused on the diversity of microbial communities among marine sediment layers on a small geographic scale in shallow water. Therefore, we expected that our results would diverge from those of previous studies due to habitat differences and specific characteristics of sediment and water. Microbial composition is affected by many environmental factors, including temperature, dissolved oxygen, nutrients, and pH (Baril et al., 2012; Teixiera and Merquior, 2014; Mandic-Mulec et al., 2015). In addition, some other environmental factors have also been identified as important determinants of sediment microbial composition, such as sedimentological characteristics and organic matter content (Dang et al., 2008, 2009, 2013).
Overall, among bacterial communities, the phyla Actinobacteria (21%), Chloroflexi (23%), Firmicutes (25%), and Proteobacteria (18%) were dominant among all samples (n = 47) in the present study. Some bacterial families showed a specific pattern within some cores. For instance, the family Bacillaceae and order Bacillales (unclassified family) of the phylum Firmicutes were dominant among all layers of cores N5 and S4 (Figures 2A,C). The reason for the high abundance of Bacillaceae and Moraxellaceae in N5 could be due to a shrimp farm nearby, since these forms commonly dominate shrimp pond water (Vanderzant et al., 1971). The family Bacillaceae is widely distributed in soil, marine sediments, air, food, and other environments (Mandic-Mulec et al., 2015; Krishnamoorthy et al., 2017). It is important in degradation of soil organic matter and in phosphorus solubilization, and it is able to form resistance endospores (Mandic-Mulec et al., 2015). Some studies of soils have reported that members of the family Bacillaceae interact and colonize organic matter (Siala et al., 1974; Toljander et al., 2006; Mandic-Mulec et al., 2015; Krishnamoorthy et al., 2017). The family Moraxellaceae is found in naturally saline environments and is adapted in low temperatures, but little is known about its ecological role, such as its role in the degradation of organic compounds (Wery et al., 2003; Teixiera and Merquior, 2014). The Eubacteria is known for transforming toxic halogenated ethenes (e.g., tetrachloroethene) to non-toxic ethene (Maymo-Gatell et al., 1997). The high abundance of the Chloroflexi family Dehalococcoidaceae found along this causeway indicates that it might be contaminated by industrial organic pollutants, such as halogenated pollutants, as reported by previous studies in another marine environment (e.g., Wang et al., 2016; Zinder, 2016). Sites S1, S2, and N3 have a high abundance of Dehalococcoidaceae in the top layer, which may be a response to wastewater input, or garbage (Reimer et al., 2015). Interestingly, photosynthetic Chloroflexi was distributed in deep layers of cores N4 and S4. Kurladze and Ivanova (2008) reported that photosynthetic Chloroflexi are capable of chemoheterotrophic growth under aerobic conditions and they can switch from anoxic photosynthesis to aerobic respiration. Additionally, we assume that Chloroflexi were detected in the deep layers of the cores and therefore, photosynthetic activity can be excluded as they are much too far away from the photic zone. Chloroflexi sometimes show diel migration in intertidal microbial mats. However, such diel migration is limited to the mm range. One can speculate that the Chloroflexi of cores N4 and S4 permanently live without photosynthetic activity showing a chemoorganotrophic lifestyle, which may be a result of niche differentiation.
The archaeal family Cenarchaeaceae was more abundant in bottom layers than top layers of cores N1-2, N5, and S2-3 and showed a reversed distribution in core S4. In addition, it was the most abundant family in core N1. The families Cenarchaeaceae and Nitrososphaeraceae belong to phylum Thaumarchaeota, and they are found in marine sediments of marginal seas of the western Pacific Ocean (e.g., Dang et al., 2009, 2010, 2013), including the East China Sea (e.g., Dang et al., 2008). The Thaumarchaeota are chemolithoautotrophic or mixotrophic ammonia oxidizers, playing very important roles in nitrification and inorganic carbon fixation among marine Cenarchaeaceae (Dang and Chen, 2017). The Methanosarcinaceae was distributed among all ten cores and in all four layers of sediment, and was the most abundant group in three cores (N1, N2, and S1). Additionally, the Methanobacteriaceae and Methanosaetaceae were most abundant in cores N1 and S1. Previous studies have demonstrated that Methanobacteriaceae and Methanosarcinaceae are predominant in wastewater treatment sludge, and can adapt to high carbon concentrations (Fei et al., 2015; Kuroda et al., 2015; Fan and Xing, 2016; Hu et al., 2016).
In fungal communities of the present study, Ascomycota were abundant and more frequent than Basidiomycota among all cores. The frequencies of Ascomycota and Basidiomycota shows the similar relative abundance in different climatic origin of marine sediments such as Arctic marine sediments (Zhang T. et al., 2015; Zhang et al., 2016). However, furthermore investigations are needed to address the ecological and biological functions of these fungi, and their adaptations in different environments.
The marine environment may influence some microbial communities in the top layer of the sediment (Figure 6A). However, ocean currents in this area complicate the composition of both macro- and micro-marine biota. The causeway faces a bay to the south with two nearby islands, and it opens to a large bay in north (Figure 1) with tidal currents flowing through channels at sites 2 and 4. The biota surveyed by Reimer et al. (2015) showed variable species diversity and richness along the causeway, which may also influence the composition of microbiota at different sites. Nevertheless, the results of our chemodiversity survey indicate that effects of this causeway have been major, and even with construction of the two channels, differences between the north and south sides are conspicuous (Figure S1, Figure 7) despite their original close proximity. There were more unique OTUs on the south than the north (Table 2). In a previous study, Reimer et al. (2015) reported that while both sides were similar in regard to many marine taxa, there were clear differences in fish communities between the north and south sides, commensurate with our results. Environmental factors may explain some microbial communities at sites N1, N2, S3, and S5 (Figure 6C), archaean communities at sites S3 and S5 (Figure 6D), and fungal communities at sites N1, N2, N3, S3 and S5 (Figure 6E). Eukaryote and other microbial communities (Figure 6F) seem uninfluenced by environmental factors. Despite the channels under the causeway that were built at sites 2 and 4 to mitigate the effect of the causeway, there are few similarities in the environmental factors, chemodiversity, and microbial communities, except at site 4 (Figure 6B).
As a follow-up to Reimer et al. (2015), this study provides crucial baseline information, not only for future changes, but also valuable data for pollution monitoring and control. Based upon environmental data, sites N1, N2, N3, N4, S3, and S4 have been considered less polluted (Reimer et al., 2015), but our data from microbial communities implies there may be pollution. Microbial community composition may have originally been very diverse due to different marine environmental factors; however, chemodiversity results imply that this causeway has caused the south and north sides to differentiate. Finally, Serratia marcescens, an opportunistic human pathogen and agent of coral diseases, as previously reported at site S1 (Reimer et al., 2015), and some species of Bacillus (Bacillaceae), Moraxella (Moraxellaceae), and Clostridium (Clostridiaceae), all found in our study, are known to be human and mammalial pathogens. As fishing and recreational activities are popular in the waters around Kaichu-Doro, pollution management is needed to improve water quality and to prevent potential disease outbreaks. Additionally, our results show that even 40+ years after causeway construction the north and south communities show lasting effects of such coastal development.
Author Contributions
TS: design the experimental work, DNA extraction, PCR, LC-MS etc., data analysis, and wrote and review the paper; JR: design the experimental work, sample collection, data analysis, and review the paper; S-YY: data analysis and review the paper; AV-B: experimental work, data analysis (LC-MS), and review the paper; MR: experimental work, data analysis (LC-MS), and review the paper; HJ-K: design the experimental work, sample collection, provide kits, machine and chemicals, data analysis, and review the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by an internal grant from Okinawa Institute of Science and Technology, Graduate University (OIST) to HJ-K. The authors thank Dr. Thanai Paxton (Waters Corporation, Japan) for his general guidance in compound analyses. We thank the OIST DNA Sequencing and Instrumental Analysis sections for preparing and sequencing the libraries, and for sample preparation for LC-MS analysis. The authors are grateful to Dr. Steven D. Aird and the reviewers for their valuable comments that improved the paper. The initial study of Kaichu-Doro was supported by grants to JR from Risona Bank's Okumura Corporation Trust Fund for Construction, Environment and Technology, and from the Nissei Foundation's Research Grant for Environmental Issues. Additional funding to JR came from the Rising Star Program and the International Research Hub Project for Climate Change and Coral Reef/Island Dynamics, both at the University of the Ryukyus.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02451/full#supplementary-material
Table S1. Raw data for 195 peaks of LC-MS data.
Figure S1. Principal Component Analyses (PCA) of metabolite profiling and overall microbial communities (including 47 samples of various layers). (A) compounds; (B) bacteria; (C) archaea; (D) fungi; (E) other eukaryotes.
Figure S2. Alpha rarefaction curves representing the number of OTUs (Chao 1) based on the minimum number of reads in each domain of the top layer among 10 cores from the north and the south. (A) bacteria; (B) archaea; (C) fungi; (D) other eukaryotes.
References
Acosta-Gonzalez, A., and Marques, S. (2016). Bacterial diversity in oil-polluted marine coastal sediments. Curr. Opin. Biotechnol. 38, 24–32. doi: 10.1016/j.copbio.2015.12.010
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W., and Huse, S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE 4:e6372. doi: 10.1371/journal.pone.0006372
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Baril, E., Coroller, L., Couvert, O., El Jabri, M., Leguerinel, I., Postollec, F., et al. (2012). Sporulation boundaries and spore formation kinetics of Bacillus spp. as a function of temperature, pH and a(w). Food Microbiol. 32, 79–86. doi: 10.1016/j.fm.2012.04.011
Beals, E. W. (1984). Bray-Curtis ordination: an effective strategy for analysis of multivariate ecological data. Adv. Ecol. Res. 14, 1–55. doi: 10.1016/S0065-2504(08)60168-3
Brown, M. V., Philip, G. K., Bunge, J. A., Smith, M. C., Bissett, A., Lauro, F. M., et al. (2009). Microbial community structure in the North Pacific ocean. ISME J. 3, 1374–1386. doi: 10.1038/ismej.2009.86
Dang, H., and Chen, C. A. (2017). Ecological energetic perspectives on responses of nitrogen-transforming chemolithoautotrophic microbiota to changes in the marine environment. Front. Microbiol. 8:1246. doi: 10.3389/fmicb.2017.01246
Dang, H., Li, J., Zhang, X., Li, T., Tian, F., and Jin, W. (2009). Diversity and spatial distribution of amoA-encoding archaea in the deep-sea sediments of the tropical West Pacific Continental Margin. J. Appl. Microbiol. 106, 1482–1493. doi: 10.1111/j.1365-2672.2008.04109.x
Dang, H., Luan, X. W., Chen, R., Zhang, X., Guo, L., and Klotz, M. G. (2010). Diversity, abundance and distribution of amoA-encoding archaea in deep-sea methane seep sediments of the Okhotsk Sea. FEMS Microbiol. Ecol. 72, 370–385. doi: 10.1111/j.1574-6941.2010.00870.x
Dang, H., Zhang, X., Sun, J., Li, T., Zhang, Z., and Yang, G. (2008). Diversity and spatial distribution of sediment ammonia-oxidizing crenarchaeota in response to estuarine and environmental gradients in the Changjiang Estuary and East China Sea. Microbiology 154, 2084–2095. doi: 10.1099/mic.0.2007/013581-0
Dang, H., Zhou, H., Yang, J., Ge, H., Jiao, N., Luan, X., et al. (2013). Thaumarchaeotal signature gene distribution in sediments of the northern South China Sea: an indicator of the metabolic intersection of the marine carbon, nitrogen, and phosphorus cycles? Appl. Environ. Microbiol. 79, 2137–2147. doi: 10.1128/AEM.03204-12
Das, S., Lyla, P. S., and Khan, S. A. (2006). Marine microbial diversity and ecology: importance and future perspectives. Curr. Sci. 90, 1325–1335.
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N. U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean's interior. Science 311, 496–503. doi: 10.1126/science.1120250
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Fan, X., and Xing, P. (2016). The vertical distribution of sediment archaeal community in the “black bloom” disturbing Zhushan Bay of Lake Taihu. Archaea 2016:8232135. doi: 10.1155/2016/8232135
Fei, X., Zekkos, D., and Raskin, L. (2015). Archaeal community structure in leachate and solid waste is correlated to methane generation and volume reduction during biodegradation of municipal solid waste. Waste Manag. 36, 184–190. doi: 10.1016/j.wasman.2014.10.027
Frisvad, J. C., and Larsen, T. O. (2015). Chemodiversity in the genus Aspergillus. Appl. Microbiol. Biotechnol. 99, 7859–7877. doi: 10.1007/s00253-015-6839-z
Galand, E. P., Lucas, S., Fagervold, S. K., Peru, E., Pruski, A. M., Vetion, G., et al. (2016). Disturbance increases microbial community diversity and production in marine sediments. Front. Microbiol. 7:1950. doi: 10.3389/fmicb.2016.01950
Gantner, S., Andersson, A. F., Alonso-Saez, L., and Bertilsson, S. (2011). Novel primers for 16S rRNA-based archaeal community analyses in environmental samples. J. Microbiol. Methods 84, 12–18. doi: 10.1016/j.mimet.2010.10.001
Ghiglione, J. F., Galand, P. E., Pommier, T., Pedros-Alio, C., Maas, E. W., Bakker, K., et al. (2012). Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 109, 17633–17638. doi: 10.1073/pnas.1208160109
Herlemann, D. P. R., Labrenz, M., Jurgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579. doi: 10.1038/ismej.2011.41
Hu, A., Wang, H., Li, J., Liu, J., Chen, N., and Yu, C. P. (2016). Archaeal community in a human-disturbed watershed in southeast China: diversity, distribution, and responses to environmental changes. Appl. Microbiol. Biotechnol. 100, 4685–4698. doi: 10.1007/s00253-016-7318-x
Inagaki, F., Kuypers, M. M., Tsunogai, U., Ishibashi, J., Nakamura, K., Treude, T., et al. (2006). Microbial community in a sediment-hosted CO2 lake of the southern Okinawa Trough hydrothermal system. Proc. Natl. Acad. Sci. U.S.A. 103, 14164–14169. doi: 10.1073/pnas.0606083103
Islam, M. S., and Tanaka, M. (2004). Impacts of pollution on coastal and marine ecosystems including coastal and marine fisheries and approach for management: a review and synthesis. Mar. Pollut. Bull. 48, 624–649. doi: 10.1016/j.marpolbul.2003.12.004
Kathiresan, K., and Bingham, B. L. (2001). Biology of mangroves and mangrove ecosystems. Adv. Mar. Biol. 40, 81–251. doi: 10.1016/S0065-2881(01)40003-4
Kellerman, A. M., Dittmar, T., Kothawala, D. N., and Tranvik, L. J. (2014). Chemodiversity of dissolved organic matter in lakes driven by climate and hydrology. Nat. Commun. 5:3804. doi: 10.1038/ncomms4804
Koljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Krishnamoorthy, S., Annaselvi, M., Murthy, A. R., and Iyer, N. R. (2017). Nutritional behavior, morphogenesis cycle and sediment consolidation capabilities of the calcareous bacteria derived from coastal marine sediments. Geomicrobiol. J. 34, 795–803. doi: 10.1080/01490451.2016.1271840
Kurladze, G. V., and Ivanova, L. B. (2008). Cell Differentiation Research Developments. New York, NY: Nova Publishers.
Kuroda, K., Hatamoto, M., Nakahara, N., Abe, K., Takahashi, M., Araki, N., et al. (2015). Community composition of known and uncultured archaeal lineages in anaerobic or anoxic wastewater treatment sludge. Microb. Ecol. 69, 586–596. doi: 10.1007/s00248-014-0525-z
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. doi: 10.1038/ismej.2010.133
Mandic-Mulec, I., Stefanic, P., and van Elsas, J. D. (2015). Ecology of Bacillaceae. Microbiol. Spectr. 3:TBS-0017-2013. doi: 10.1128/microbiolspec.TBS-0017-2013
Maymo-Gatell, X., Chien, Y., Gossett, J. M., and Zinder, S. H. (1997). Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276, 1568–1571. doi: 10.1126/science.276.5318.1568
Mohanraju, R., and Natarajan, R. (1992). Methanogenic bacteria in mangrove sediments. Hydrobiologia 247, 187–193. doi: 10.1007/BF00008218
Moore, B. D., Andrew, R. L., Kulheim, C., and Foley, W. J. (2014). Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytol. 201, 733–750. doi: 10.1111/nph.12526
Pluskal, T., Castillo, S., Villar-Briones, A., and Oresic, M. (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11:395. doi: 10.1186/1471-2105-11-395
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Reimer, J. D., Yang, S. Y., White, K. N., Asami, R., Fujita, K., Hongo, C., et al. (2015). Effects of causeway construction on environment and biota of subtropical tidal flats in Okinawa, Japan. Mar. Pollut. Bull. 94, 153–167. doi: 10.1016/j.marpolbul.2015.02.037
Roberts, C. M., McClean, C. J., Veron, J. E. N., Hawkins, J. P., Allen, G. R., McAllister, D. E., et al. (2002). Marine biodiversity hotspots and conservation priorities for tropical reefs. Science 295, 1280–1284. doi: 10.1126/science.1067728
Roullier-Gall, C., Lucio, M., Noret, L., Schmitt-Kopplin, P., and Gougeon, R. (2014). How subtle is the “Terroir” effect? chemistry-related signatures of two “Climats de Bourgogne”. PLoS ONE 9:e97615. doi: 10.1371/journal.pone.0097615
Siala, A., Hill, I. R., and Gray, T. R. G. (1974). Populations of spore-forming bacteria in an acid forest soil, with special reference to Bacillus-Subtilis. J. Gen. Microbiol. 81, 183–190. doi: 10.1099/00221287-81-1-183
Smith, C. A., Want, E. J., O'Maille, G., Abagyan, R., and Siuzdak, G. (2006). XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787. doi: 10.1021/ac051437y
Stagars, M. H., Ruff, S. E., Amann, R., and Knittel, K. (2016). High diversity of anaerobic alkane-degrading microbial communities in marine seep sediments based on (1-methylalkyl) succinate synthase genes. Front. Microbiol. 6:1511. doi: 10.3389/fmicb.2015.01511
Stahl, D. A., and Amann, R. (1991). “Development and application of nucleic acid probes,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: John Wiley), 205–248.
Teixiera, L. M., and Merquior, V. L. C. (2014). “The family Moraxellaceae,” in The Prokarytoes—Gammaproteobacteria 4th Edn., eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer-Verlag), 443–476.
Toledo, G., Bashan, Y., and Soeldner, A. (1995). Cyanobacteria and black mangroves in northwestern Mexico - colonization, and diurnal and seasonal nitrogen-fixation on aerial roots. Can. J. Microbiol. 41, 999–1011. doi: 10.1139/m95-139
Toljander, J. F., Artursson, V., Paul, L. R., Jansson, J. K., and Finlay, R. D. (2006). Attachment of different soil bacteria to arbuscular mycorrhizal fungal extraradical hyphae is determined by hyphal vitality and fungal species. FEMS Microbiol. Lett. 254, 34–40. doi: 10.1111/j.1574-6968.2005.00003.x
Treusch, A. H., Vergin, K. L., Finlay, L. A., Donatz, M. G., Burton, R. M., Carlson, C. A., et al. (2009). Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J. 3, 1148–1163. doi: 10.1038/ismej.2009.60
Vanderzant, C., Nickelson, R., and Judkins, P. W. (1971). Microbial flora of pond-reared brown shrimp (Penaeus aztecus). Appl. Microbiol. 21, 916–921.
Vethanayagam, R. R. (1991). Purple photosynthetic bacteria from a tropical mangrove environment. Mar. Biol. 110, 161–163. doi: 10.1007/BF01313103
Walsh, E. A., Kirkpatrick, J. B., Rutherford, S. D., Smith, D. C., Sogin, M., and D'Hondt, S. (2016). Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 10, 979–989. doi: 10.1038/ismej.2015.175
Wang, K., Ye, X., Zhang, H., Chen, H., Zhang, D., and Liu, L. (2016). Regional variations in the diversity and predicted metabolic potential of benthic prokaryotes in coastal northern Zhejiang, East China Sea. Sci. Rep. 6:38709. doi: 10.1038/srep38709
Wery, N., Gerike, U., Sharman, A., Chaudhuri, J. B., Hough, D. W., and Danson, M. J. (2003). Use of a packed-column bioreactor for isolation of diverse protease-producing bacteria from antarctic soil. Appl. Environ. Microbiol. 69, 1457–1464. doi: 10.1128/AEM.69.3.1457-1464.20030
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Ampli cation and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in Pcr Protocols: A Guide To Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York, NY: Academic Press), 315–322.
Wild, C., Huettel, M., Klueter, A., Kremb, S. G., Rasheed, M. Y. M., and Jorgensen, B. B. (2004). Coral mucus functions as an energy carrier and particle trap in the reef ecosystem. Nature 428, 66–70. doi: 10.1038/nature02344
Xia, J., Sinelnikov, I. V., Han, B., and Wishart, D. S. (2015). MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257. doi: 10.1093/nar/gkv380
Zhang, J., Sun, Q. L., Zeng, Z. G., Chen, S., and Sun, L. (2015). Microbial diversity in the deep-sea sediments of Iheya North and Iheya Ridge, Okinawa Trough. Microbiol. Res. 177, 43–52. doi: 10.1016/j.micres.2015.05.006
Zhang, T., Wang, N. F., Zhang, Y. Q., Liu, H. Y., and Yu, L. Y. (2015). Diversity and distribution of fungal communities in the marine sediments of Kongsfjorden, Svalbard (High Arctic). Sci. Rep. 5:14524. doi: 10.1038/srep14524
Zhang, T., Wang, N. F., Zhang, Y. Q., Liu, H. Y., and Yu, L. Y. (2016). Diversity and distribution of aquatic fungal communities in the Ny-Alesund region, svalbard (High Arctic): aquatic fungi in the Arctic. Microb. Ecol. 71, 543–554. doi: 10.1007/s00248-015-0689-1
Keywords: bacteria, archaea, eukaryotes, metagenomics, NGS, chemodiversity
Citation: Soliman T, Reimer JD, Yang S-Y, Villar-Briones A, Roy MC and Jenke-Kodama H (2017) Diversity of Microbial Communities and Quantitative Chemodiversity in Layers of Marine Sediment Cores from a Causeway (Kaichu-Doro) in Okinawa Island, Japan. Front. Microbiol. 8:2451. doi: 10.3389/fmicb.2017.02451
Received: 20 July 2017; Accepted: 27 November 2017;
Published: 11 December 2017.
Edited by:
Hongyue Dang, Xiamen University, ChinaReviewed by:
Paris Sepulveda Lavin, University of Antofagasta, ChileWim Vyverman, Ghent University, Belgium
Copyright © 2017 Soliman, Reimer, Yang, Villar-Briones, Roy and Jenke-Kodama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taha Soliman, dGFoYXNvbGltYW4yMDAwQHlhaG9vLmNvbQ==
Holger Jenke-Kodama, aG9sZ2VyLmplbmtlLmtvZGFtYUBnbWFpbC5jb20=