 Xiaofei Ji1†
Xiaofei Ji1† Ying Wang2†
Ying Wang2† Jiaojiao Li1Qianyu Rong1Xingxing Chen1Ying Zhang1Xiaoning Liu2Boqing Li1*Huilin Zhao1*
Jiaojiao Li1Qianyu Rong1Xingxing Chen1Ying Zhang1Xiaoning Liu2Boqing Li1*Huilin Zhao1*- 1Department of Pathogenic Biology, School of Basic Medical Sciences, Binzhou Medical University, Yantai, China
- 2Central Laboratory, Huai'an First People's Hospital, Nanjing Medical University, Huai'an, China
Helicobacter pylori is a Gram-negative, microaerophilic bacterium associated with human gastric diseases. Further investigations on virulence genes are still required to clarify the pathogenic mechanism of H. pylori and the heterogeneous problem of infection. In order to develop an efficient and accurate method to study gene functions in H. pylori pathogenesis, an unmarked deletion method for both a single gene and a large fragment was established based on the FLP-FRT recombination system. Using this method, the gene hp0788, encoding an outer membrane protein (HofF), was deleted. Deletion of hp0788 did not affect growth or motility of H. pylori, but reduced the adherence of the bacteria to gastric epithelial cells. The apoptosis of GES-1 cells caused by H. pylori infection was also reduced by the defection of hp0788. These suggest that hp0788 takes part in the bacterium-host interaction and plays an important role in H. pylori infection. Furthermore, a large genomic fragment deletion from hp0541 to hp0547 in cag pathogenicity island was also successfully achieved using FLP-FRT method. The innovative application of the FLP-FRT recombination system in H. pylori to construct unmarked deletion would provide a helpful tool for further function research of putative pathogenic genes and contribute to the understanding of H. pylori pathogenesis.
Introduction
Helicobacter pylori is a main pathogen in our gastric mucosa causing gastrointestinal diseases. It was reported that more than half of the population has been infected by this bacterium, and about 20% of them developed severe diseases according to clinical statistics (Forman, 1998; Peek and Blaser, 2002; Kato et al., 2004; Parsonnet and Forman, 2004; Sugiyama, 2004). Extensive researches have been done to identify H. pylori virulence factors and characterize their roles in bacterial colonization and pathogenesis. The functions of vacuolating cytotoxin A (VacA), cytotoxin associated gene A (CagA) and many important adhesins have been studied and described in detail (Ferrero et al., 1992; Haas et al., 1993; Atherton et al., 1995; Blaser et al., 1995; Backert et al., 2000, 2011; Oleastro and Menard, 2013; de Bernard and Josenhans, 2014; Zhang et al., 2014). However, the exact pathogenic mechanism, especially the different outcomes of an infection by H. pylori, has not been elucidated (Shanks and El-Omar, 2009; Alzahrani et al., 2014; Floch et al., 2017). It has been suggested that comprehensive effect of infecting strain virulence, host genetics and environmental factors lead to the final outcome of an infection (Yamaoka, 2008; Posselt et al., 2013). However, the current understanding of this complicated process is limited.
The outer membrane proteins (OMPs) usually play important roles in bacterial adherence, which is the first step of the colonization. H. pylori has a large set of OMPs, and genes encoding these proteins occupy 4% genetic sequences of the genome (Alm et al., 2000). These numerous OMPs could be divided into five families: Hop and Hor proteins, Hof proteins, Hom proteins, iron-regulated OMPs, and efflux pump OMPs. Many of these OMPs have been deeply studied on their roles in the adhesion process (Alm et al., 2000; Oleastro and Menard, 2013).
In order to explore functional genes in H. pylori colonization in the gerbil stomach, Kavermann and co-workers constructed 960 mutants using a signature tagged mutagenesis (STM) approach. Finally 47 genes were identified as essential for gastric colonization through screening these STM mutants, and hp0788 was one of these genes. The protein encoded by hp0788 was named HofF, which is a member of the Hof family. A recent report about Helicobacter heilmannii, one of the most predominant Helicobacter species in feline stomach, showed that HofF can act as an adhesin participating in bacterial adherence. The deficiency of HofF in H. heilmannii resulted in lower level in binding to gastric epithelial cells and gastric colonization in the stomach (Cheng et al., 2016).
Gene knockout, an important method to identify and characterize functional genes, was achieved 20 years ago in H. pylori, and various virulence genes have been studied through this way (Ferrero et al., 1992; Haas et al., 1993; Kahrs et al., 1995; Bauerfeind et al., 1996; Copass et al., 1997; Yuan et al., 2003). Moreover, unmarked deletion, the more accurate operation for gene knockout, was also used successfully in H. pylori through a sucrose-based counterselection system by Copass et al. (1997) and a rpsL-mediated method by Dailidiene et al. (2006).
The FLP-FRT recombination system is a useful tool for genetic engineering. It was found in Saccharomyces cerevisiae and has been proved to be effective in diverse bacterial species (Cox, 1983; Hoang et al., 1998; Chiang and Mekalanos, 2000; Schweizer, 2003; Stephan et al., 2004; Tracy et al., 2008; Ishikawa and Hori, 2013; Wang et al., 2014). For gene deletion, two FRT (FLP recombinase recognition target) sites were introduced into two terminals of a target gene on the same orientation. The target DNA sequence between FRT sites was further excised under the action of the FLP recombinase. The FLP-FRT recombination system could be used to excise a single gene, a large DNA segment or multiple genes repeatedly, which is the advantage compared to other site-specific mutagenesis or deletion methods used in H. pylori (Leprince et al., 2012; Wang et al., 2014). To our knowledge, this genetic manipulation system has not yet been used in H. pylori.
In this study, the FLP-FRT recombination system was applied in H. pylori to generate unmarked deletions. hp0788, a gene encoding an outer membrane protein HofF, was deleted by this method. The function of this gene in H. pylori pathogenesis was further analyzed through the co-cultured system of GES-1 cells and the mutant. Furthermore, a large genomic fragment deletion in cag pathogenicity island (from hp0541 to hp0547, about 10 kbp) was obtained using this method.
Materials and Methods
Bacterial Strains, Cell Line, and Growth Conditions
H. pylori 26695 (ATCC700392) was used for construction of unmarked deletion in this study. H. pylori strains were grown on chocolate agar plates supplemented with 10% sheep's blood, and cultivated at 37°C under microaerobic conditions (85% N2, 10% CO2, 5% O2). Brain Heart Infusion broth supplemented with 10% fetal bovine serum (FBS) was used as liquid medium to test the growth of H. pylori with shaking at 100 rpm under microaerophilic conditions after inoculation. The motility of H. pylori was determined on soft agar (0.4%) as described by Worku and Belogolova (Worku et al., 2004; Belogolova et al., 2013). Escherichia coli strains were routinely cultured at 37°C in Luria-Bertani medium. Antibiotics were used at the following concentrations when needed: 15 mg/l kanamycin (Km) or 10 mg/l chloramphenicol (Cm) for H. pylori; 100 mg/l ampicillin (Ap) or 30 mg/l chloramphenicol (Cm) for E. coli. The human gastric epithelial cell line GES-1 (ATCC, Rockville, MD) was used to study the pathogenicity of H. pylori mutant strains. GES-1 cells were cultured with Dulbecco's Modified Eagle Medium (DMEM) containing 10% FBS. In co-cultured system of H. pylori and GES-1 cells, the value of multiplicity of infection (MOI) was 200:1. Bacterial strains and plasmids used in this study are listed in Table 1, and primers are listed in Table 2.
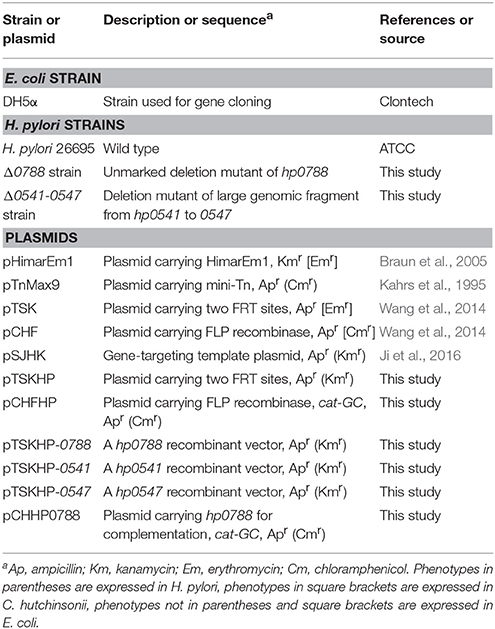
Table 1. Strains and plasmids used in this study.

Table 2. Primers used in this study.
Construction of Gene-Targeting Template Plasmid pTSKHP Carrying FRT Sites
Plasmid pTSK carrying erythromycin-resistant cassette flanked by two FRT sites was used as the backbone (Wang et al., 2014). As the erythromycin-resistance gene (ermF) on pTSK does not work effectively in H. pylori, a kanamycin-resistance gene (aphA) was used to replace ermF in pTSK. pTSK was digested with BssH II into two fragments, and the larger backbone fragment was recovered followed by treated with alkaline phosphatase. aphA was amplified from pHimarEm1 with primers BssHII-KmF and BssHII-KmR, digested with BssH II, and ligated with the backbone. The resulting plasmid, named pTSKHP, contains a kanamycin-resistant cassette flanked by two FRT sites with the same orientation (Figure 1A).
Figure 1. Illustration of the unmarked deletion of hp0788 by FLP-FRT method. (A) Schematic representations of plasmids pTSKHP and pCHFHP. (B) The process of unmarked deletion of hp0788 by FLP-FRT method. A gene-targeting cassette containing homologous arms (H1 and H2), the kanamycin-resistance gene and FRT sites was firstly transformed into H. pylori by electroporation. The genetic recombination at hp0788 locus was verified by PCR with diagnostic primers (test1/kt2 and kt3/test4). Plasmid pCHFHP was then transformed into the validated transformants to delete the kanamycin resistance gene between two FRT sites. Only one FRT site was left on the locus of hp0788 at last. Black filled boxes indicate the homologous arms; black arrows indicate the approximate locations and orientations of primers; open arrows indicate orientations and arrangements of genes; open boxes indicate residual fragment of gene hp0788. (C) Diagnostic PCR for verification of genetic recombination at hp0788 locus. Lane M, DNA molecular weight standard (DL10000, Takara); Lane 1 and 2, PCR products amplified using primers test1 and kt2 from WT and the mutant; Lane 3 and 4, PCR products amplified using primers kt3 and test4 from WT and the mutant. (D) PCR verification of the deletion of aphA gene at hp0788 locus. Lane M, DNA molecular weight standard (DL10000, Takara); Lane 1-5, PCR products from different tested mutants using primers test1 and test4.
Construction of the FLP Recombinase Expression Plasmid pCHFHP
The FLP expression plasmid pCHF (Wang et al., 2014) in Cytophaga hutchinsonii was used as the backbone to construct the FLP recombinase expression plasmid in H. pylori. The pHel1 fragment (Heuermann and Haas, 1995) containing a H. pylori-specific replicon was amplified by PCR using the primers pHel1-1 and pHel1-2, and inserted into pCHF after digested with Sac I and Sal I, to generate pCHF-p. The chloramphenicol-resistance gene (cat) on pCHF could not be expressed in H. pylori. Therefore, another chloramphenicol-resistance gene (catGC) from pTnMax9 was amplified with primers cm-F and cm-R to replace cat after digested with Sal I and Sph I. The yielding plasmid was named pCHFHP (Figure 1A).
Unmarked Deletion of hp0788
The process of unmarked deletion of hp0788 was illustrated in Figure 1B. Double-crossover recombination plasmid for hp0788 was constructed as follows. An 828 bp fragment spanning the first 203 bp of hp0788 and its upstream sequence was used as the upstream homologous arm (H1). It was amplified with primers 0788H1F and 0788H1R, digested with Sac I and Sal I, and then ligated into the corresponding sites of pTSKHP. Downstream homologous arm (H2) was a 1105 bp fragment containing the last 69 bp of hp0788 and its downstream sequence. The fragment was amplified with primers 0788H2F and 0788H2R. After digested with Kpn I and BamH I, it was also ligated into the corresponding sites of pTSKHP, yielding the plasmid pTSKHP-0788.
pTSKHP-0788 was transformed into H. pylori by electroporation as previously described (Ji et al., 2016). The transformants were selected with 15 mg/l of kanamycin and confirmed by PCR with diagnostic primers (test1/kt2 and kt3/test4). The verified transformant cells were used as recipient cells for transforming pCHFHP by electroporation. After incubation at 37°C on chloramphenicol selective plates for 7–10 days, the transformant colonies were inoculated into fresh medium without antibiotics for the elimination of pCHFHP.
Finally, diagnostic PCR with primers test1 and test4 was preformed to verify the excision of aphA gene in the final transformant cells. The cells were also streaked on serum plates containing kanamycin or chloramphenicol to confirm the loss of the exogenous resistance genes.
RT-PCR Analysis
Total RNA of wild-type H. pylori and hp0788 mutant was isolated using the RNApure Bacteria Kit (CWBIO, Beijing, China). Traces of DNA in extractive RNA were eliminated and first-strand cDNA was synthesized through HiFiScript cDNA Synthesis Kit (CWBIO, Beijing, China). The cDNA was used as the template to carry out PCR with primers listed in Table 2 (RT0786-1, RT0786-2, RT0787-1, RT0787-2, RT0788-1, RT0788-2, RT0789-1, RT0789-2, RT0790-1, and RT0790-2). In PCR control reactions, RNA without reverse transcription was used as the template to determine whether the RNA was free of genomic DNA.
Complementation of hp0788 Mutant
The replicative plasmid pCHFHP was used to complement hp0788 in the mutant. A fragment containing Xba I, digested from the plasmid pSJHK with Kpn I and Sph I, was inserted in the corresponding sites of pCHFHP to add the restriction enzyme site XbaI, generating pCHHPK. The fragment containing hp0788, 240 bp upstream of the start codon, and 28 bp downstream of the stop codon was amplified with primers C0788-1 and C0788-2. The amplicon was digested with Xba I and Kpn I and ligated into the corresponding sites of pCHHPK, yielding pCHHP0788. The plasmid pCHHP0788 was then electroporated into the hp0788 mutant and transformants were selected by chloramphenicol resistance.
Deletion of a Large Genomic Fragment in cag Pathogenicity Island
The genomic fragment from hp0541 to hp0547 in cag pathogenicity island was selected for gene targeting in this study. The schematic representation of the deletion of this region was shown in Figure 7A. First, one of the terminal genes of this genomic fragment, hp0541, was deleted with the same procedure as described above. Then, the other terminal gene hp0547, was also subjected to unmarked deletion. Finally, the transformants were confirmed by diagnostic PCR with primers T1 and T4.
Western Blot Analysis for CagA
H. pylori cells were harvested and washed with phosphate-buffered saline (PBS, pH 7.4). The pellets were subjected to ultrasonication. After centrifugation at 16,000 g for 5 min at 4°C, cell debris were removed and the cell lysates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins in the SDS-PAGE gel were transferred onto a 0.45 μm Immobilon-P PVDF membrane (Millipore, MA, USA). After blocked with skim milk, the membrane was incubated with monoclonal mouse antibody (anti-CagA) (Santa Cruz Biotechnology, CA, USA) and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Cowin Biotech, Beijing, China) successively. Finally, proteins on the membrane were detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore, MA, USA) according to the instructions.
Measuring the Adherence of H. pylori to GES-1 Cells
The GES-1 cells (1 × 105 cells/2 ml/well) were plated in 6-well plates in DMEM medium with 10% FBS at 37°C in a 5% CO2 incubator overnight. Cells were washed with PBS and co-cultured with H. pylori (2 × 107 colony forming units/ml) in 1 ml of DMEM medium for another 30 min incubation. Subsequently, cells were harvested with 1% trypsin-EDTA and collected by centrifugation. After washed three times with PBS to remove non-adherent bacteria, the remaining bound H. pylori on the cells were measured through plate counting and the urease test. The urease test was carried out as the procedure described by Ki et al. (2010).
Morphology Determination of GES-1 Cells in the Co-cultured System with H. pylori
GES-1 cells at a concentration of 1 × 105 cells/2 ml/well were plated into 6-well plates overnight in DMEM with 10% FBS. H. pylori cells at mid-exponential phase were collected and added into the GES-1 cells culture at an MOI of 200:1. At the time points of 0, 8, and 16 h in co-culture, morphologic characteristics of GES-1 cells were detected through both crystal violet staining and phalloidin labeling methods. Crystal violet staining was carried out as routine procedure. In phalloidin labeling, cells were firstly fixed by paraformaldehyde for half an hour followed by washed with PBS and labeled by fluor-labeled phalloidin (Invitrogen, Waltham, MA). The images of the stained or labeled cells were finally captured through inverted fluorescence microscope (Olympus, Tokyo, Japan).
Cell Apoptosis Determination
GES-1 cells were co-cultured with H. pylori as mentioned above. After 0, 8, or 16 h, GES-1 cells were immediately harvested to detect the cell apoptosis rate by Annexin V-FITC Apoptosis Detection Kit (KeyGEN BioTECH, Nanjing, China). According to the instructions, the assay was performed as follows. 1 × 105 GES-1 cells were firstly suspended with 500 μl Annexin binding buffer containing 5 μl FITC-conjugated Annexin V antibody and 5 μl propidium iodide. Then the mixture was incubated at room temperature for 15 min in the dark. Finally, the relative number of apoptotic cells in the mixture was determined using flow cytometry (Zhang et al., 2016).
Cell Viability Determination
Cell Counting Kit-8 (CCK-8) (KeyGEN BioTECH, Nanjing, China) was used to determine the cell viability according to the instructions. GES-1 cells were plated in 96-well plates with an initial density of 103 cells/well, and H. pylori cells were added at an MOI of 200:1. After co-incubation at 37°C for 0, 8, or 16 h, 10 μl of kit reagent was added for another 3 h incubation. The absorbance at 450 nm of the sample was measured by a microplate reader.
IL-8 Secretion Determination
GES-1 cells were infected with H. pylori strains as previously described. The supernatant of the culture was collected at 8 h, and IL-8 was measured by ELISA using an HS Human IL-8 kit (Neobioscience, Shenzhen, China), according to the manufacturer's instructions.
Statistical Analysis
Statistical analysis was carried out using t-test, and a P-value below 0.05 was considered as significant difference.
Results
Application of FLP-FRT System on Unmarked Deletion of hp0788 in H. pylori
In order to apply the FLP-FRT recombination system to achieve genetic manipulation in H. pylori, plasmids pTSKHP carrying FRT sites and pCHFHP expressing FLP recombinase were constructed. The essential gene involved in H. pylori colonization, hp0788 (Kavermann et al., 2003), was selected as a target to construct an unmarked deletion.
pTSKHP contains a kanamycin-resistance gene (aphA) flanked by two FRT sites. For gene targeting, homologous arms were inserted into the multiple cloning sites upstream and downstream of the resistance gene (aphA) on pTSKHP. After gene replacement by recombination, aphA flanked by two FRT sites would be inserted into the targeted site on the genome. Plasmid pCHFHP contains FLP recombinase coding gene (flp), which locates downstream of the chloramphenicol-resistance gene (catGC) directly and will be co-transcribed with catGC. The schematic diagrams of pTSKHP and pCHFHP are shown in Figure 1A.
As shown in Figure 1B, the hp0788 recombinant vector (pTSKHP-0788) was constructed according to the procedure and transformed into H. pylori cells by electroporation. Kanamycin-resistant colonies were selected and verified by PCR. As shown in Figure 1C, two expected bands spanning the neighboring genes and the kanamycin resistance gene were amplified in the mutant from both upstream and downstream, which were absent from the wild-type strain. This result confirmed that hp0788 had been replaced by the kanamycin resistance gene. Then, the kanamycin resistance gene was ejected with the transformation of the flp carrying plasmid pCHFHP. In order to verify the gene deletion between two FRT sites, diagnostic PCR with primers test1/test4 was performed and the results are shown in Figure 1D. A band with an expected size was amplified from each tested transformant, which indicated that the kanamycin resistance gene was lost. Sequencing analysis of the mutants confirmed the deletion of hp0788.
RT-PCR Analysis of the Deletion of hp0788
The arrangement of hp0788 and surrounding genes in the genome of H. pylori is shown in Figure 2A. To investigate whether the transcription of the surrounding genes is affected by the deletion of hp0788, RT-PCR was performed as previously described. In the wild type of H. pylori, amplicons corresponding to hp0786, hp0787, hp0788, hp0789, and hp0790 were all present, while the hp0788 amplicon was not obtained in the hp0788 mutant (Figure 2B). This result indicated that hp0788 was deleted. The existence of amplicons corresponding to hp0786, hp0787, hp0789, and hp0790 in the mutant suggests that the deletion of hp0788 do not affect the transcription of surrounding genes. All the amplicons were confirmed by sequencing analysis.
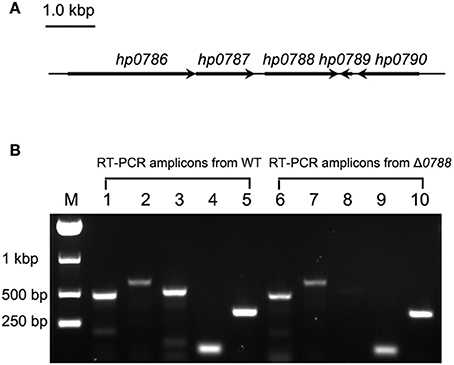
Figure 2. RT-PCR analysis of the transcription of hp0788 and the surrounding genes. (A) Illustration of hp0788 and the surrounding genes. (B) RT-PCR analysis of the transcription of hp0788 and the surrounding genes in the wild type H. pylori (WT) and hp0788 deleted mutant (Δ0788). PCR reactions were performed using primers RT0786-1, RT0786-2, RT0787-1, RT0787-2, RT0788-1, RT0788-2, RT0789-1, RT0789-2, RT0790-1, and RT0790-2. Lane M, Marker; Lane 1 to 5, PCR reactions performed with WT cDNA; Lane 6 to 10, PCR reactions performed with Δ0788 cDNA. The following primers were used: RT0786-1 and RT0786-2 for lane 1 and 6; RT0787-1 and RT0787-2 for lane 2 and 7; RT0788-1 and RT0788-2 for lane 3 and 8; RT0789-1 and RT0789-2 for lane 4 and 9; RT0790-1 and RT0790-2 for lane 5 and 10.
The Deletion of hp0788 Resulted in No Significant Change in Bacterial Growth and Motility, but Reduced the Adherence of H. pylori to Epithelial Cells
Kavermann et al. reported that hp0788 is an essential gene for H. pylori colonization in the gerbil stomach (Kavermann et al., 2003). However, the exact pathogenic role of this gene is not clear. Therefore, the characteristics of the hp0788-deleted mutant were identified in this paper.
First, bacterial morphology, growth curve, cell motility and adherence to GES-1 cells were determined to analyze the phenotypes of the hp0788-deleted mutant. The hp0788 deficient strain exhibited similar morphology and structure to the wild-type strain according to the observation under transmission electron microscopy (Figure S1). As shown in Figure 3A, the mutant had a similar growth rate with the wild type except that the final cell density of the mutant was a little higher. The results of the motility assay reveal that the deletion of hp0788 do not change the motility of the bacteria either (Figure 3B). However, the adherence of H. pylori to GES-1 cells was changed due to the deletion of hp0788 according to plate counting and the urease test. For the wild type, the mean number of bacteria per cell is 37.7, while the numerical value is 30.2 for the mutant (Figure 3C). The results of urease test also illustrated the reduction in adherence of H. pylori to GES-1 cells (Figure S2).
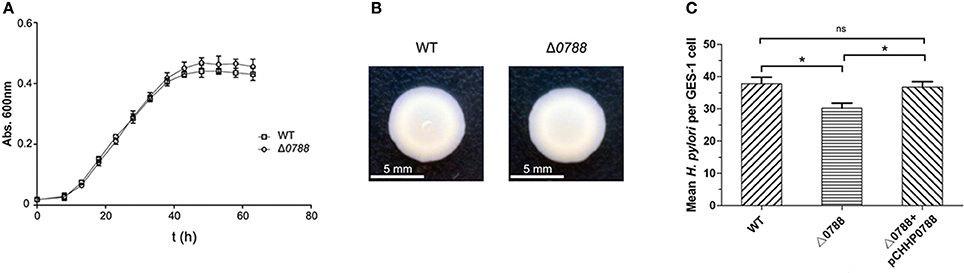
Figure 3. Phenotypic characteristics of the wild-type (WT) H. pylori, the mutant (Δ0788), and the complemented strain (Δ0788+ pCHHP0788). (A) Growth curves of H. pylori in Brain Heart Infusion broth supplemented with 10% fetal bovine serum. (B) The motility of H. pylori cells on soft agar. (C) The adherence of H. pylori to GES-1 cells. Statistically significant *p < 0.05; ns, not significant.
To verify that the reduction in adherence was caused by the deletion of hp0788, complementation of the mutant was carried out as described in section Materials and Methods. As shown in Figure 3C, the reduction in adherence of H. pylori to GES-1 cells was almost restored in complemented strain. These results imply that HofF acts as an adhesin in the infection process of H. pylori, which is consistent with the previous reports (Kavermann et al., 2003; Cheng et al., 2016).
Morphological Changes of H. pylori-Infected GES-1 Cells
GES-1 cells were co-cultured with H. pylori as previously described, and morphological changes of GES-1 cells were detected. Figure 4A shows the images of GES-1 cells stained with crystal violet (magnification, 100×). Without infected by H. pylori, GES-1 cells exhibited agglomerated growth at 8 and 16 h. The number of GES-1 cells at 16 h was higher than the number of cells at 8 h, revealing the proliferation of GES-1 cells. After co-cultured with H. pylori (both the wild type and the mutant), GES-1 cells was more dispersed. The numbers of GES-1 cells at 8 and 16 h were lower than the control, which indicated that the proliferation of GES-1 cells was inhibited. Figure 4B shows the images of GES-1 cells labeled by fluor-labeled phalloidin (magnification, 200×). Without infected by H. pylori, GES-1 cells were regular and intact. After co-cultured with H. pylori (both the wild type and the mutant), some cells turned irregular and even became round. No obvious differences were observed between cells cultured with the wild type and with the mutant.
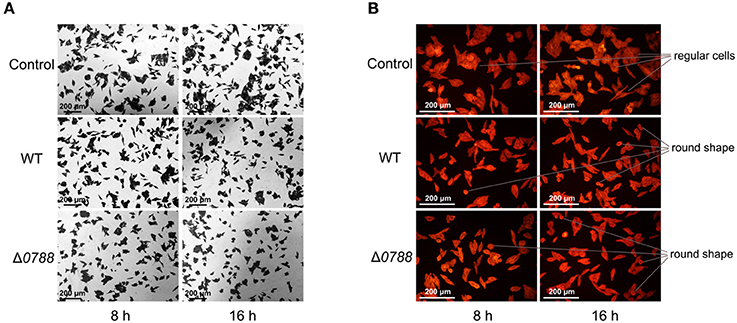
Figure 4. Microscopic observation of GES-1 cells in the co-incubation with H. pylori. (A) GES-1 cells were stained with crystal violet (magnification, 100×). (B) GES-1 cells were labeled with phalloidin (magnification, 100×). Control, non-infected GES-1 cells; WT, GES-1 cells were infected with the wild type of H. pylori; Δ0788, GES-1 cells were infected with hp0788 deleted mutant. Regular cells and round shape cells were pointed out.
The Deletion of hp0788 Decreased the Virulence of H. pylori to Cause Cell Apoptosis and Viability Decline
Cell apoptosis assay was performed as described in section Materials and Methods, and the relative number of apoptotic cells was determined using flow cytometry. Figure 5A shows the representative flowcharts, in which apoptosis cells occurred in the second and fourth quadrants. Apoptosis rates of GES-1 cells are calculated from the statistical analysis of apoptotic cells, which is shown in Figure 5B. Cell apoptosis rate of GES-1 cells was very low (about 4.00 ± 3.15%) without infection with H. pylori. After infected by the wild-type H. pylori, the apoptosis rate of GES-1 cells increased to 15.73 ± 7.84% after 8 h and reached 25.26 ± 5.81% after 16 h. For the co-incubation with the mutant, cell apoptosis rate increased to12.46 ± 12.3% and 21.13 ± 10.09% at 8 and 16 h, respectively. Compared with cells infected with the wild type strain, those cells infected with mutant strain showed a reduction in apoptosis. When the complemented strain was used to infect GES-1 cells, the apoptosis rate of GES-1 cells recovered to 15.29 ± 13.1% at 8 h and 25.57 ± 9.46% at 16 h, respectively. These results suggest that the deletion of hp0788 disrupts the action of H. pylori to the GES-1 cells in cell apoptosis.
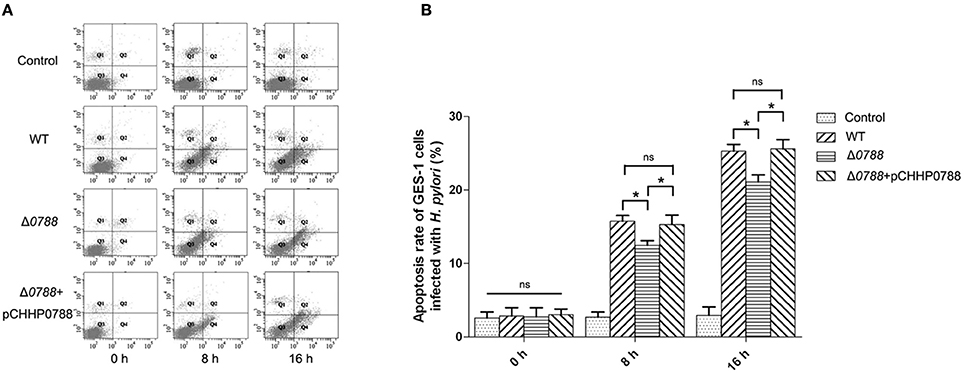
Figure 5. Apoptosis analysis of GES-1 cells infected with H. pylori. GES-1 cells were infected with the wild type (WT), Δ0788 or the complemented strain (Δ0788+ pCHHP0788) at an MOI of 200:1. The apoptosis rates of GES-1 cells at different time (0, 8, and 16 h) were detected using AnnexinV-FITC/PI double staining combined with flow cytometry. Control, non-infected GES-1 cells; WT, GES-1 cells infected with the wild type of H. pylori; Δ0788, GES-1 cells infected with hp0788 deleted mutant; Δ0788+ pCHHP0788, GES-1 cells infected with the complemented strain. (A) Representative flowcharts. Apoptotic cells occurred in the second and fourth quadrants. (B) Cell apoptosis rate of GES-1 cells. Apoptosis rates of GES-1 cells were calculated from the statistical analysis of apoptotic cells. Statistically significant *p < 0.05; ns, not significant.
The viabilities of GES-1 cells after infection with the wild-type H. pylori, the Δ0788 mutant and the complemented strain were also comparatively analyzed. As shown in Figure 6, cell viability of the control without being co-cultured with H. pylori increased with time. In the samples co-cultured with either the wild-type or the mutant strain, cell viability decreased significantly. When infected by the wild type, the value of cell viability decreased to 53.43 or 41.76% relative to the initial value after 8 or 16 h. But with infection by the mutant, the value of cell viability retained 66.07 and 49.84% after 8 and 16 h, respectively. When the complemented strain was used, the value of cell viability decreased again, to 52.49 and 42.68% after 8 and 16 h, which was basically of the same level with wild type strain. These results show that deletion of hp0788 decreases the effect of H. pylori on the viability of infected GES-1 cells. The tendency of cellular damage of infected GES-1 cells is consistent with the results of the cell apoptosis and viability assay.
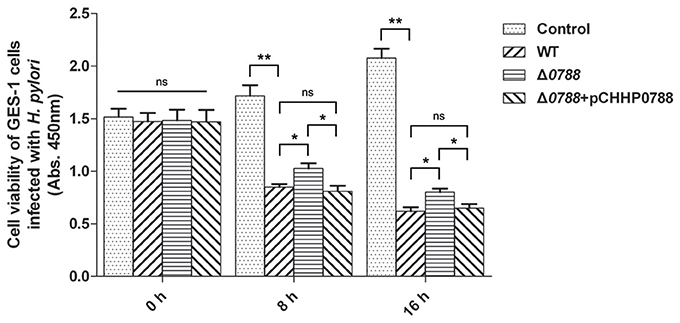
Figure 6. Cell viability of GES-1 cells infected with H. pylori. GES-1 cells were in the co-incubation with the wild type (WT), Δ0788 or the complemented strain (Δ0788+ pCHHP0788). Cell viability was determined by absorbance at 450 nm after using CCK-8. Control, non-infected GES-1 cells; WT, GES-1 cells infected with the wild type of H. pylori; Δ0788, GES-1 cells infected with hp0788 deleted mutant; Δ0788+ pCHHP0788, GES-1 cells infected with the complemented strain. Statistically significant **p < 0.01, *p < 0.05; ns, not significant.
Deletion of a Large Genomic Fragment in H. pylori Based on the FLP-FRT Recombination System
The cag pathogenicity island (cagPAI) is a very important region for the pathogenicity of H. pylori and the functions of genes within this region are still incompletely clear. So the genomic fragment from hp0541 to hp0547 (about 10 kbp) on cagPAI which contains cagA gene and several genes encoding the type IV secretion system was selected as the target region.
As shown in Figure 7A, the deletion of this region was started with the disruption of hp0541 using pTSKHP-0541, followed by transformation of pCHFHP to obtain the unmarked hp0541 mutant (Δ0541). Then, hp0547 was replaced by the resistant gene on pTSKHP flanked by two FRT sites in Δ0541 mutant. The fragment containing the resistant gene and the other genes in this region will be evicted by the transformation of pCHFHP. The new junction fragments amplified from resultant colonies are shown in Figure 7B. A band with expected size was present in the mutant, while no band was amplified in the wild-type strain, mainly because the undeleted fragment is too long to be amplified using regular PCR procedure. The DNA sequencing of the amplified fragment from the mutant confirmed the deletion. After cultured without antibiotics, the mutant cells showed sensitive to both kanamycin and chloramphenicol (Figure 7C), suggesting the exogenous antibiotic-resistance genes had been eliminated.
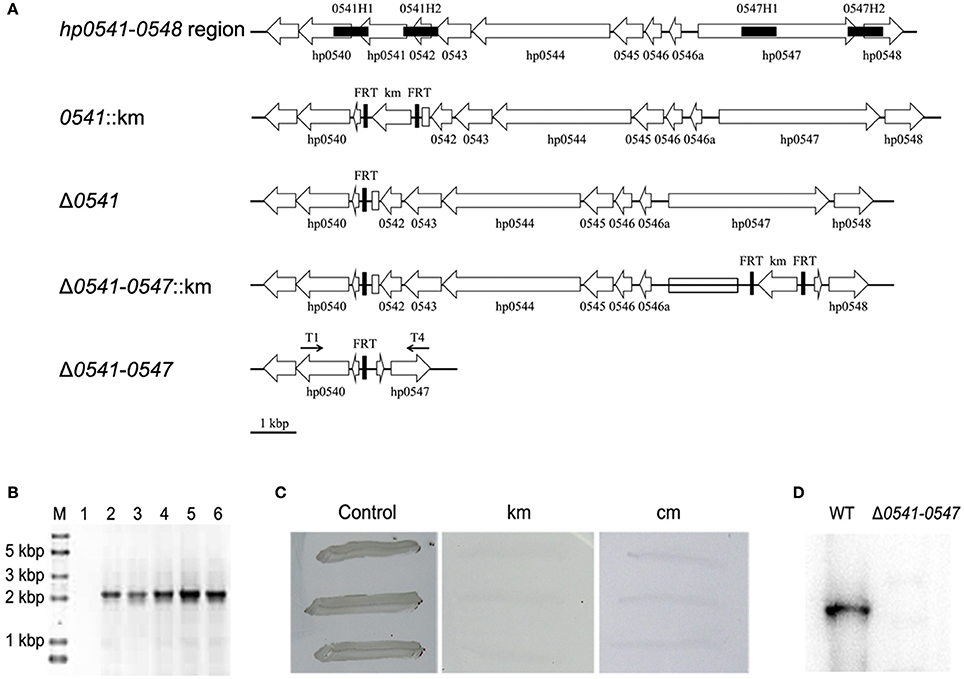
Figure 7. Deletion of a large genomic fragment (hp0541-0547) in H. pylori by the FLP-FRT recombination system. (A) Schematic representation of the deletion procedure. First, recombinant vector pTSKHP-0541 combined with the helper plasmid pCHFHP were used to obtain the hp0541-deletion mutant (Δ0541) leaving only one FRT in hp0541 locus. The replacement of hp0547 with a kanamycin-resistance gene and another two FRT sites was performed with recombinant vector pTSKHP-0547 in Δ0541. Finally, the helper plasmid pCHFHP was transformed into the mutant cells again to excise the genomic fragments between FRT sites, leaving only one FRT on this locus. The deletion of genetic fragments was verified by PCR with primers T1 and T4. Black filled boxes show homologous arms; black arrows indicate orientations and approximate locations of primers; open arrows show orientations and arrangements of genes; open boxes indicate residual fragments of genes hp0541 and hp0547. (B) Diagnostic PCR for verification of the deletion from hp0541 to hp0547. Lane M, DNA molecular weight standard (Trans2K Plus II DNA Marker, TransGen); Lane 1, PCR product from the wild type using primers T1 and T4; Lane 2-6, PCR products from different tested mutants using primers T1 and T4. (C) Antibiotic sensitivity test of the mutant cells. The mutants were streaked on fresh medium without antibiotics or containing kanamycin or chloramphenicol. (D) Western blot detection of CagA in the wild-type strain and the Δ0541-0547 mutant.
Western blot of CagA was performed to examine the deletion on protein level. The result showed that CagA was present in the wild type, but absent from the mutant strain (Figure 7D). This result also indicated that the large fragment from hp0541 to hp0547 had been excised. This mutant provides a useful material for further study on the pathogenic role of the cag pathogenicity island.
It is clear that CagA and cagPAI could induce secretion of interleukin 8 (IL-8) in the infected cells, such as AGS cells and GES-1 cells (Fischer et al., 2001; Odenbreit et al., 2002). To further confirm the deleted mutant, IL-8 production was measured, and the results are shown in Figure 8. Consistent with the results reported by other articles (Fischer et al., 2001; Odenbreit et al., 2002; Belogolova et al., 2013), IL-8 secretion in GES-1 cells was much lower following infection with the mutant (Δ0541-0547) compared with the wild type.
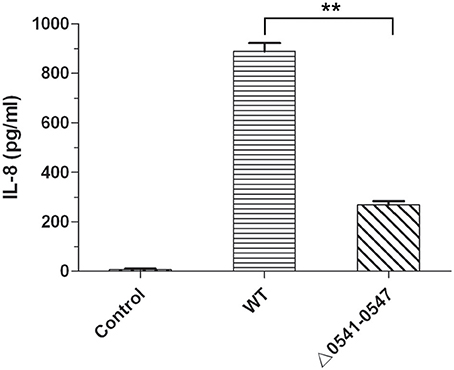
Figure 8. IL-8 production of GES-1 cells infected with H. pylori. IL-8 was measured by ELISA using an HS Human IL-8 kit. Control, non-infected GES-1 cells; WT, GES-1 cells infected with the wild type of H. pylori; Δ0541-0547, GES-1 cells infected with hp0541-0547 deleted mutant. Data represent means and standard deviations of at least three independent experiments. Statistically significant **p < 0.01.
Discussion
Although, many virulence genes have been deeply studied, the pathogenic mechanism of H. pylori is not entirely clear (Shanks and El-Omar, 2009; Alzahrani et al., 2014; Floch et al., 2017). H. pylori has a high mutation rate itself and frequent genetic exchange and recombination with other H. pylori strains, which results in extensive genetic diversity (Suerbaum et al., 1998; Bjorkholm et al., 2001). Very little is known about the impact of genetic variation of virulence genes on disease outcomes. Therefore, more putative pathogenic genes are needed for further study to better understand the detailed pathogenic mechanism and clarify what determines different outcomes of the infection by H. pylori.
Construction of gene-knockout mutants is an important method for exploring novel virulence factors in pathogens. Among methods of disrupting genes, such as transposon mutagenesis (Haas et al., 1993; Kahrs et al., 1995), gene-targeting by single or double crossover homologous recombination (Ferrero et al., 1992; Bauerfeind et al., 1996; Yuan et al., 2003), unmarked deletion is the most accurate method without introduction of exogenous genes or polar effect. Copass and co-workers reported unmarked mutagenesis in H. pylori in 1997 through a sacB-mediated method (Copass et al., 1997). However, this method was proved not work well by following studies, as it is difficult to find the appropriate sucrose concentration for killing sacB-containing H. pylori cells to select expected deletion (Dailidiene et al., 2006). Dailidiene and co-workers indicated rpsL is a better contraselectable marker to construct unmarked deletion in H. pylori. With this method, the efficiency of deletion selection was considerable. However, the yield of desired deletion was obviously depending on the genome location of the targeted locus (Dailidiene et al., 2006). In addition, the eviction step in these methods was based on intrinsic homologous recombination, which usually resulted in incomplete deletion mutants. In this study, FLP-FRT recombination system was successfully applied in H. pylori, and unmarked deletion of a single gene was obtained through a two-step transformation. Compared with the rpsL-mediated unmarked deletion, the ejection of resistance genes through the FLP-FRT system is site-specific and more efficient. Moreover, the necessary step of construction streptomycin-resistant mutant in rpsL-mediated method is no longer needed. Unmarked deletions of several genes besides hp0788 in H. pylori have also been achieved using this approach (unpublished data), which demonstrates the effectiveness of this method. Furthermore, the deletion of large DNA fragment was also achieved using this method, which has not been reported in H. pylori. This gene targeting method provides an efficient tool for further study of novel pathogenic genes in H. pylori.
The gene hp0788 encodes a Hof protein, which belongs to an important OMP family in H. pylori. Kavermann and co-workers first reported that hp0788 was essential for H. pylori colonization in the gerbil stomach (Kavermann et al., 2003). Recent report from Cheng and co-workers showed that the hofF gene is also essential for H. heilmannii colonization in the gastric mucosa (Cheng et al., 2016). The suspect pathway of HofF affecting Helicobacter colonization is that HofF can regulate MUC13 expression through IL-1β secretion, which further affects the bacterial colonization (Liu et al., 2014; Cheng et al., 2016). In this study, gene hp0788 in H. pylori was deleted unmarkedly through FLP-FRT recombination system. Deletion of hp0788 reduced the adherence of H. pylori to epithelial cells, which reconfirms the role of HofF as an adhesin in bacterial infection. In addition, HofF may take part in H. pylori pathogenesis. It was found that H. pylori strain lacking HofF had impaired abilities to reduce the viability and induce apoptosis in infected GES-1 cells. We speculate that HofF also acts as an effector protein playing roles in the cell-contact interaction between H. pylori and the host cells. To better understand the role of HofF in H. pylori infection, the changes in signal transmission caused by the deletion of HofF are being further studied in our lab, which may provide more insights into the mechanism of H. pylori pathogenesis.
Ethics Statement
This article does not contain any studies with human participants or animals performed by any of the authors. The genetic modification of H. pylori was used only for basic research, which was under supervision and approved by Medical Ethics Committee of Binzhou Medical University.
Author Contributions
XJ, YW, HZ, and BL: conceive and design the experiments; Perform the experiments: XJ, YW, HZ, JL, QR, and XC; XJ, YW, HZ: writing the paper; YZ, XL, and BL: Revising the paper; All authors approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81501718, 81471561, and 81702054) and Natural Science Foundation of Shandong Province (ZR2015PC021, ZR2014CP020, and ZR2017BC011).
Conflict of Interest Statement
The method of unmarked deletion in H. pylori through FLP-FRT recombination system was submitted in patent application (CN106086054A), and the patent application is under substantive examination presently.
Acknowledgments
We sincerely thank Prof. Lu (Shandong University, China) for providing plasmids (pHimarEm1, pTSK, and pCHF) and Dr. Yongtao Zhu (University of Wisconsin-Milwaukee, USA) for linguistic advice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02357/full#supplementary-material
References
Alm, R. A., Bina, J., Andrews, B. M., Doig, P., Hancock, R. E., and Trust, T. J. (2000). Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68, 4155–4168. doi: 10.1128/IAI.68.7.4155-4168.2000
Alzahrani, S., Lina, T. T., Gonzalez, J., Pinchuk, I. V., Beswick, E. J., and Reyes, V. E. (2014). Effect of Helicobacter pylori on gastric epithelial cells. World J. Gastroenterol. 20, 12767–12780. doi: 10.3748/wjg.v20.i36.12767
Atherton, J. C., Cao, P., Peek, R. M. Jr., Tummuru, M. K., Blaser, M. J., and Cover, T. L. (1995). Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 270, 17771–17777. doi: 10.1074/jbc.270.30.17771
Backert, S., Clyne, M., and Tegtmeyer, N. (2011). Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal. 9:28. doi: 10.1186/1478-811X-9-28
Backert, S., Ziska, E., Brinkmann, V., Zimny-Arndt, U., Fauconnier, A., Jungblut, P. R., et al. (2000). Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell. Microbiol. 2, 155–164. doi: 10.1046/j.1462-5822.2000.00043.x
Bauerfeind, P., Garner, R. M., and Mobley, L. T. (1996). Allelic exchange mutagenesis of nixA in Helicobacter pylori results in reduced nickel transport and urease activity. Infect. Immun. 64, 2877–2880.
Belogolova, E., Bauer, B., Pompaiah, M., Asakura, H., Brinkman, V., Ertl, C., et al. (2013). Helicobacter pylori outer membrane protein HopQ identified as a novel T4SS-associated virulence factor. Cell. Microbiol. 15, 1896–1912. doi: 10.1111/cmi.12158
Björkholm, B., Sjölund, M., Falk, P. G., Berg, O. G., Engstrand, L., and Andersson, D. I. (2001). Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 98, 14607–14612. doi: 10.1073/pnas.241517298
Blaser, M. J., Perez-Perez, G. I., Kleanthous, H., Cover, T. L., Peek, R. M., Chyou, P. H., et al. (1995). Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55, 2111–2115.
Braun, T. F., Khubbar, M. K., Saffarini, D. A., and McBride, M. J. (2005). Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J. Bacteriol. 187, 6943–6952. doi: 10.1128/JB.187.20.6943-6952.2005
Cheng, L., Mirko, R., Sara, L., Medea, P., Caroline, B., Eva, B., et al. (2016). The Helicobacter heilmannii hofE and hofF genes are essential for colonization of the gastric mucosa and play a role in IL-1β-induced gastric MUC13 expression. Helicobacter 21, 504–522. doi: 10.1111/hel.12307
Chiang, S. L., and Mekalanos, J. J. (2000). Construction of a Vibrio cholerae vaccine candidate using transposon delivery and FLP recombinase-mediated excision. Infect. Immun. 68, 6391–6397. doi: 10.1128/IAI.68.11.6391-6397.2000
Copass, M., Grandi, G., and Rappuoli, R. (1997). Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect. Immun. 65, 1949–1952.
Cox, M. M. (1983). The FLP protein of the yeast 2-microns plasmid: expression of a eukaryotic genetic recombination system in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 80, 4223–4227. doi: 10.1073/pnas.80.14.4223
Dailidiene, D., Dailide, G., Kersulyte, D., and Berg, D. E. (2006). Contraselectable streptomycin susceptibility determinant for genetic manipulation and analysis of Helicobacter pylori. Appl. Environ. Microbiol. 72, 5908–5914. doi: 10.1128/AEM.01135-06
de Bernard, M., and Josenhans, C. (2014). Pathogenesis of Helicobacter pylori infection. Helicobacter 19(Suppl. 1), 11–18. doi: 10.1111/hel.12160
Ferrero, R. L., Cussac, V., Courcoux, P., and Labigne, A. (1992). Construction of isogenic urease-negative mutants of Helicobacter pylori by allelic exchange. J. Bacteriol. 174, 4212–4217. doi: 10.1128/jb.174.13.4212-4217.1992
Fischer, W., Püls, J., Buhrdorf, R., Gebert, B., Odenbreit, S., and Haas, R. (2001). Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 42, 1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x
Floch, P., Mégraud, F., and Lehours, P. (2017). Helicobacter pylori strains and gastric MALT lymphoma. Toxins 9:E132. doi: 10.3390/toxins9040132
Forman, D. (1998). Helicobacter pylori infection and cancer. Br. Med. Bull. 54, 71–78. doi: 10.1093/oxfordjournals.bmb.a011682
Haas, R., Meyer, T. F., and van Putten, J. P. (1993). Aflagellated mutants of Helicobacter pylori generated by genetic transformation of naturally competent strains using transposon shuttle mutagenesis. Mol. Microbiol. 8, 753–760. doi: 10.1111/j.1365-2958.1993.tb01618.x
Heuermann, D., and Haas, R. (1995). Genetic organization of a small cryptic plasmid of Helicobacter pylori. Gene 165, 17–24. doi: 10.1016/0378-1119(95)00469-M
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/S0378-1119(98)00130-9
Ishikawa, M., and Hori, K. (2013). A new simple method for introducing an unmarked mutation into a large gene of non-competent Gram-negative bacteria by FLP/FRT recombination. BMC Microbiol. 13:86. doi: 10.1186/1471-2180-13-86
Ji, X., Zhao, H., Zhang, Y., Chen, X., Li, J., and Li, B. (2016). Construction of novel plasmid vectors for gene knockout in Helicobacter pylori. Curr. Microbiol. 73, 897–903. doi: 10.1007/s00284-016-1140-7
Kahrs, A. F., Odenbreit, S., Schmitt, W., Heuermann, D., Meyer, T. F., and Haas, R. (1995). An improved TnMax mini-transposon system suitable for sequencing, shuttle mutagenesis and gene fusions. Gene 167, 53–57. doi: 10.1016/0378-1119(95)00671-0
Kato, M., Asaka, M., Shimizu, Y., Nobuta, A., Takeda, H., and Sugiyama, T. (2004). Relationship between Helicobacter pylori infection and the prevalence, site and histological type of gastric cancer. Aliment. Pharmacol. Ther. 20(Suppl. 1), 85–89. doi: 10.1111/j.1365-2036.2004.01987.x
Kavermann, H., Burns, B. P., Angermuller, K., Odenbreit, S., Fischer, W., Melchers, K., et al. (2003). Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 197, 813–822. doi: 10.1084/jem.20021531
Ki, M. R., Ghim, S. Y., Hong, I. H., Park, J. K., Hong, K. S., Ji, A. R., et al. (2010). In vitro inhibition of Helicobacter pylori growth and of adherence of cagA-positive strains to gastric epithelial cells by Lactobacillus paraplantarum KNUC25 isolated from kimchi. J. Med. Food 13, 629–634. doi: 10.1089/jmf.2009.1265
Leprince, A., de Lorenzo, V., Völler, P., van Passel, M. W., and Martins dos Santos, V. A. (2012). Random and cyclical deletion of large DNA segments in the genome of Pseudomonas putida. Environ. Microbiol. 14, 1444–1453. doi: 10.1111/j.1462-2920.2012.02730.x
Liu, C., Smet, A., Blaecher, C., Flahou, B., Ducatelle, R., Linden, S., et al. (2014). Gastric de novo Muc13 expression and spasmolytic polypeptide-expressing metaplasia during Helicobacter heilmannii infection. Infect. Immun. 82, 3227–3239. doi: 10.1128/IAI.01867-14
Odenbreit, S., Kavermann, H., Püls, J., and Haas, R. (2002). CagA tyrosine phosphorylation and interleukin-8 induction by Helicobacter pylori are independent from alpAB, HopZ and bab group outer membrane proteins. Int. J. Med. Microbiol. 292, 257–266. doi: 10.1078/1438-4221-00205
Oleastro, M., and Ménard, A. (2013). The role of Helicobacter pylori outer membrane proteins in adherence and pathogenesis. Biology 2, 1110–1134. doi: 10.3390/biology2031110
Parsonnet, J., and Forman, D. (2004). Helicobacter pylori infection and gastric cancer–for want of more outcomes. JAMA 291, 244–245. doi: 10.1001/jama.291.2.244
Peek, R. M. Jr., and Blaser, M. J. (2002). Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37. doi: 10.1038/nrc703
Posselt, G., Backert, S., and Wessler, S. (2013). The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 11:77. doi: 10.1186/1478-811X-11-77
Schweizer, H. P. (2003). Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol. 5, 67–77. doi: 10.1159/000069976
Shanks, A. M., and El-Omar, E. M. (2009). Helicobacter pylori infection, host genetics and gastric cancer. J. Dig. Dis. 10, 157–164. doi: 10.1111/j.1751-2980.2009.00380.x
Stephan, J., Stemmer, V., and Niederweis, M. (2004). Consecutive gene deletions in Mycobacterium smegmatis using the yeast FLP recombinase. Gene 343, 181–190. doi: 10.1016/j.gene.2004.08.028
Suerbaum, S., Smith, J. M., Bapumia, K., Morelli, G., Smith, N. H., Kunstmann, E., et al. (1998). Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 95, 12619–12624. doi: 10.1073/pnas.95.21.12619
Sugiyama, T. (2004). Development of gastric cancer associated with Helicobacter pylori infection. Cancer Chemother. Pharmacol. 54(Suppl. 1), S12–S20. doi: 10.1007/s00280-004-0881-3
Tracy, E., Ye, F., Baker, B. D., and Munson, R. S. Jr. (2008). Construction of non-polar mutants in Haemophilus influenzae using FLP recombinase technology. BMC Mol. Biol. 9:101. doi: 10.1186/1471-2199-9-101
Wang, Y., Wang, Z., Cao, J., Guan, Z., and Lu, X. (2014). FLP-FRT-based method to obtain unmarked deletions of CHU_3237 (porU) and large genomic fragments of Cytophaga hutchinsonii. Appl. Environ. Microbiol. 80, 6037–6045. doi: 10.1128/AEM.01785-14
Worku, M. L., Karim, Q. N., Spencer, J., and Sidebotham, R. L. (2004). Chemotactic response of Helicobacter pylori to human plasma and bile. J. Med. Microbiol. 53(Pt 8), 807–811. doi: 10.1099/jmm.0.45636-0
Yamaoka, Y. (2008). Roles of the plasticity regions of Helicobacter pylori in gastroduodenal pathogenesis. J. Med. Microbiol. 57(Pt 5), 545–553. doi: 10.1099/jmm.0.2008/000570-0
Yuan, J. P., Li, T., Shi, X. D., Hu, B. Y., Yang, G. Z., Tong, S. Q., et al. (2003). Deletion of Helicobacter pylori vacuolating cytotoxin gene by introduction of directed mutagenesis. World J. Gastroenterol. 9, 2251–2257. doi: 10.3748/wjg.v9.i10.2251
Zhang, J., Qian, J., Zhang, X., and Zou, Q. (2014). Outer membrane inflammatory protein A, a new virulence factor involved in the pathogenesis of Helicobacter pylori. Mol. Biol. Rep. 41, 7807–7814. doi: 10.1007/s11033-014-3673-9
Keywords: Helicobacter pylori, unmarked deletion, FLP-FRT, hp0788, cell apoptosis
Citation: Ji X, Wang Y, Li J, Rong Q, Chen X, Zhang Y, Liu X, Li B and Zhao H (2017) Application of FLP-FRT System to Construct Unmarked Deletion in Helicobacter pylori and Functional Study of Gene hp0788 in Pathogenesis. Front. Microbiol. 8:2357. doi: 10.3389/fmicb.2017.02357
Received: 28 February 2017; Accepted: 15 November 2017;
Published: 29 November 2017.
Edited by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2017 Ji, Wang, Li, Rong, Chen, Zhang, Liu, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boqing Li, c2RsaWJvcWluZ0AxNjMuY29t
Huilin Zhao, emhhb2h1aWxpbjE5ODRAMTYzLmNvbQ==
†These authors have contributed equally to this work.