 Jordan G. Kueneman
Jordan G. Kueneman Sophie Weiss3
Sophie Weiss3 Valerie J. McKenzie
Valerie J. McKenzie- 1Department of Ecology and Evolutionary Biology, University of Colorado Boulder, Boulder, CO, United States
- 2Smithsonian Tropical Research Institute, Panama City, Panama
- 3Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, United States
Global amphibian decline linked to fungal pathogens has galvanized research on applied amphibian conservation. Skin-associated bacterial communities of amphibians have been shown to mediate fungal skin infections and the development of probiotic treatments with antifungal bacteria has become an emergent area of research. While exploring the role of protective bacteria has been a primary focus for amphibian conservation, we aim to expand and study the other microbes present in amphibian skin communities including fungi and other micro-eukaryotes. Here, we characterize skin-associated bacteria and micro-eukaryotic diversity found across life stages of Cascades frog (Rana cascadae) and their associated aquatic environments using culture independent 16S and 18S rRNA marker-gene sequencing. Individuals of various life stages of Cascades frogs were sampled from a population located in the Trinity Alps in Northern California during an epidemic of the chytrid fungus, Batrachochytrium dendrobatidis. We filtered the bacterial sequences against a published database of bacteria known to inhibit B. dendrobatidis in co-culture to estimate the proportion of the skin bacterial community that is likely to provide defense against B. dendrobatidis. Tadpoles had a significantly higher proportion of B. dendrobatidis-inhibitory bacterial sequence matches relative to subadult and adult Cascades frogs. We applied a network analysis to examine patterns of correlation between bacterial taxa and B. dendrobatidis, as well as micro-eukaryotic taxa and B. dendrobatidis. Combined with the published database of bacteria known to inhibit B. dendrobatidis, we used the network analysis to identify bacteria that negatively correlated with B. dendrobatidis and thus could be good probiotic candidates in the Cascades frog system.
Introduction
Microbial symbionts of vertebrate hosts facilitate many biological processes, such as nutrient acquisition, host behavior, training of the adaptive immune system and protection against environmental pathogens (Bravo et al., 2011; Ezenwa et al., 2012; Tuddenham and Sears, 2015). Thus, microbial associations can contribute to host phenotypes and may influence survival (Fierer et al., 2012). The amphibian skin microbiome has become an active area of research due to the protective role that some bacteria provide against Batrachochytrium dendrobatidis, an emerging fungal skin pathogen (Harris et al., 2009; Kueneman et al., 2016). This aquatic pathogen, now the agent of a global pandemic, invades the keratinized epidermis of amphibians resulting in cell disruption and osmotic imbalances that can lead to death (Voyles et al., 2009). The use of 16S rRNA marker gene sequencing to characterize bacterial communities on amphibians has enabled many researchers to investigate the factors that shape those communities (McKenzie et al., 2012; Fitzpatrick and Allison, 2014; Jani and Briggs, 2014; Kueneman et al., 2014, 2015; Loudon et al., 2014; Walke et al., 2014). However, most studies have overlooked the fungal and micro-eukaryotic diversity that also occurs on amphibian skin and have yet to explore their function. Here, we use a parallel approach (18S rRNA marker gene sequencing) to explore micro-eukaryotic diversity alongside bacterial diversity on the skin of endangered Cascades frogs (Rana cascadae).
Bacterial-derived skin defenses have been experimentally shown to mitigate B. dendrobatidis infection (Harris et al., 2009; Jani and Briggs, 2014; Kueneman et al., 2016), however, interactions of other fungal taxa and micro-eukaryotes with B. dendrobatidis remain underexplored. Currently, it is unknown whether specific micro-eukaryotes may help mitigate infection of B. dendrobatidis. In plant systems, numerous experiments have found that some fungi are capable of excluding or reducing the effects of fungal pathogens. Examples include various leaf and root rots found in legumes, cacao, cotton, Dutch Elm, and fruit trees (Cotty and Bayman, 1993; Del Rio et al., 2002; Kabaluk et al., 2010). It is possible that symbiotic fungi on amphibians may be able to competitively exclude fungal pathogens through direct and indirect competition, however, this hypothesis remains to be tested on the amphibian skin.
In the absence of direct experimentation, network analyses provide one way to explore patterns of correlations within complex communities, identify microbes that negatively correlate with a pathogen, and infer possible antagonistic interactions (Rebollar et al., 2016a). While a negative correlation with a pathogen in a network does not confirm an antagonistic interaction, it is suggestive and should be tested independently. Microbes that are negatively correlated can help direct research to test specific interactions mechanistically and assess antibiosis that occurs with competing microbial taxa (Bletz et al., 2013). Previous field studies and lab experiments have demonstrated that individuals of species which have a higher proportion of their skin bacterial community composed of B. dendrobatidis-inhibitory taxa have lower abundance of B. dendrobatidis on their skin (Lam et al., 2010; Kueneman et al., 2016; Rebollar et al., 2016b). Thus, B. dendrobatidis-inhibitory bacteria negatively interacting with B. dendrobatidis within these networks are of special interest for conservation.
Cascades frogs are found in the Cascade and Olympic Mountains in the Pacific Northwest region of the United States. They have declined precipitously as a result of trout reintroduction and, more recently, B. dendrobatidis (Piovia-Scott et al., 2011). In Northern California, the Cascades frog population at Section Line Lake experienced a decline in juvenile frogs (>99%) attributed to B. dendrobatidis between 2009 and 2012 (Piovia-Scott et al., 2015). We had the opportunity to non-destructively sample the skin microbes from individuals in 2011, thus capturing a time point during an active B. dendrobatidis epidemic. Studying an epidemic in progress provides a unique perspective for examining the role of amphibian skin microbial communities in defense against pathogens, specifically by identifying microbes that have strong patterns of negative correlation with B. dendrobatidis. As Cascades frogs are imperiled, protective microbes can potentially offer new tools to augment the conservation of this species (Garcia et al., 2006; Piovia-Scott et al., 2015).
Here, we present a dataset that includes both the micro-eukaryotes and bacteria collected from the skin of wild R. cascadae. Our previous work contributed to a bacterial database of more than 1,200 isolates from amphibians around the globe that have demonstrated inhibitory action against B. dendrobatidis when tested in co-culture assays (Woodhams et al., 2015). No such database exists for the micro-eukaryotes, as of yet. We matched the bacterial sequence reads from the Cascades frogs against the B. dendrobatidis-inhibitory database to estimate the bacterial members of the skin community that are B. dendrobatidis-inhibitory. The aims of this study are threefold: (1) to characterize skin-associated micro-eukaryotes, including fungal skin communities among life-history stages of R. cascadae, (2) to examine correlational patterns among diverse microbes (bacteria and fungi), with a particular focus on interactions with the B. dendrobatidis pathogen, and (3) to identify candidate anti-B. dendrobatidis microbial taxa by leveraging the B. dendrobatidis-inhibitory sequence database.
Materials and Methods
Amphibian and Environmental Sampling
Individuals of Rana cascadae were caught and sampled in the Trinity Alps of Northern California, August of 2011. All individuals were captured using a dip net, handled with new nitrile gloves and sampled on the same day. In order to remove any transient microbes from the environment, each individual was rinsed twice with 50 mL of sterile water. Each amphibian was then sampled using a sterile cotton-tipped swab. Swabbing consisted of brushing over the entire ventral surface and limbs of the amphibian for 30 s. Tadpoles were swabbed uniformly over entire body for 30 s (McKenzie et al., 2012). All sampling was done non-destructively and individuals were released back into the lake. Lake water samples were collected by moving a swab through the water for 30 s at a depth of 40 cm. Sediment samples were collected via embedding the swab into the sediment for 30 s. Swabbing amphibians and the environment for the same amount of time (30 s) was part of the effort to standardize the sampling across sample types. Each swab was stored in its original sterile container and stored on ice for transfer to a 20°C freezer for storage until DNA extraction. Micro-eukaryotic communities from R. cascadae at Section Line, an alpine lake in the Trinity Alps of California, included tadpoles (N = 4), subadults (N = 6), adults (N = 11), sediment (N = 3), and lake water (N = 2). Permits and authorization were granted by California Fish and Game and the University of Colorado Institutional Animal Care and Use Committee.
DNA Extraction and Sample Processing
DNA extraction was completed utilizing the MoBio Power Soil extraction kit (MoBio Laboratories, Carlsbad, CA, United States). We used the standard MoBio protocol with minor adjustments including incubating samples in 65°C for 10 min after the addition of C1, vortexing the PowerBead tubes horizontally for 2 min, and allowing solution C6 to sit on the filter for 5 min before the final elution (Fierer et al., 2008; Lauber et al., 2008). Extraction controls were included. The PCR recipe was comprised of: 12 μL PCR water, 10 μL 5 Prime Master Mix, 1.0 μL of the forward and reverse primers at 10 uM concentrations, 1.0 μL MgCl2, and 1.0 μL genomic DNA. For bacteria, PCR primers (515f/806r) were used to target the V4 region of the 16S rRNA gene and amplify region 533–786 in the Escherichia coli strain 83972 sequence (greengenes accession no. prokMSA_id:470367). The reverse PCR primer contained a 12-base error correcting Golay barcode developed in Caporaso et al. (2010). For micro-eukaryotes, primers 1391f/EukB were used (Amaral-Zettler et al., 2009). The PCR profiles included an initial denaturation step of 94°C for 3 min, followed by 35 cycles of 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s, and final extension at 72°C for 10 min. The PCR was performed in triplicate and combined after amplification. Extraction controls were processed and showed no amplification. Amplicons were quantified using the Quant-IT Picogreen dsDNA reagent and were pooled into one sample per plate by combining equal concentrations of each amplicon. These pools of DNA were cleaned using the MoBio UltraClean PCR clean-up DNA purification kit. Following cleanup, samples were again quantified using PicoGreen reagent with equal concentrations and pooled together one final time before sequencing. A NanoDrop spectrophotometer was used to determine the purity and DNA concentration of this pool. Finally, prepared DNAs and control samples were sequenced using an Illumina HiSeq 2000 instrument at the BioFrontiers Institute Next-Generation Genomics Facility at the University of Colorado Boulder, Boulder, CO, United States.
Sequence Filtering and Processing
18S amplicons were sequenced on one Illumina MiSeq run at the University of Colorado Boulder, Boulder, CO, United States, yielding 150 base pair reads. All analyses were performed using QIIME v1.9.0 (Caporaso et al., 2010), unless otherwise stated. Sequences were filtered and assigned to samples using default settings and Cutadapt v1.2 (MARTIN, M) was used to trim primers from the combined forward and reverse sequences. Low abundance OTUs (i.e., less than 0.00005 proportional abundance) were removed (Bokulich et al., 2013) according to the subsampling open reference protocol (Rideout et al., 2014) using eukaryotic reference library Silva111 (Yilmaz et al., 2013). The no-template sequencing control was clear of unintended DNA amplification. Sequences were assigned to amphibian and environmental samples collected from the research location of Section Line Lake. Total micro-eukaryotic sequences were less abundant on tadpoles limiting the utility of rarefaction. Thus, to visualize the full extent of diversity within each lifestage, we use a proportional taxonomic abundance table (Figures 1A,B). However, we acknowledge that comparison across lifestages in Figures 1A,B is invalid, due to differences in library size. To give the viewer an idea of original library size used to make the proportions, average library size is indicated in the Figure 1 legend. To confirm our approach, and compare micro-eukaryotic communities (18S) combined across all lifestages, we used a rarefaction depth of 330 micro-eukaryotic sequences per sample (Supplementary Table 1 and Supplementary Figures A,C). Rarefaction reduced samples sizes in the micro-eukaryotic dataset to tadpoles (N = 2), subadults (N = 6), adults (N = 9), sediment (N = 3), and lake water (N = 2). Rarefied data was used in part due to the >10x library size difference between tadpoles and other life stages (Weiss et al., 2017). Data was not rarefied in the correlation (Network) or differential abundance analyses (ANCOM) to maximize statistical power. Bacterial community (16S) sequence filtering and processing was done on a rarefied dataset of 19,900 sequences per sample. Bacterial samples tadpoles (N = 4), subadults (N = 8), adults (N = 12), sediment (N = 3), and lake water (N = 2) (Figure 3).
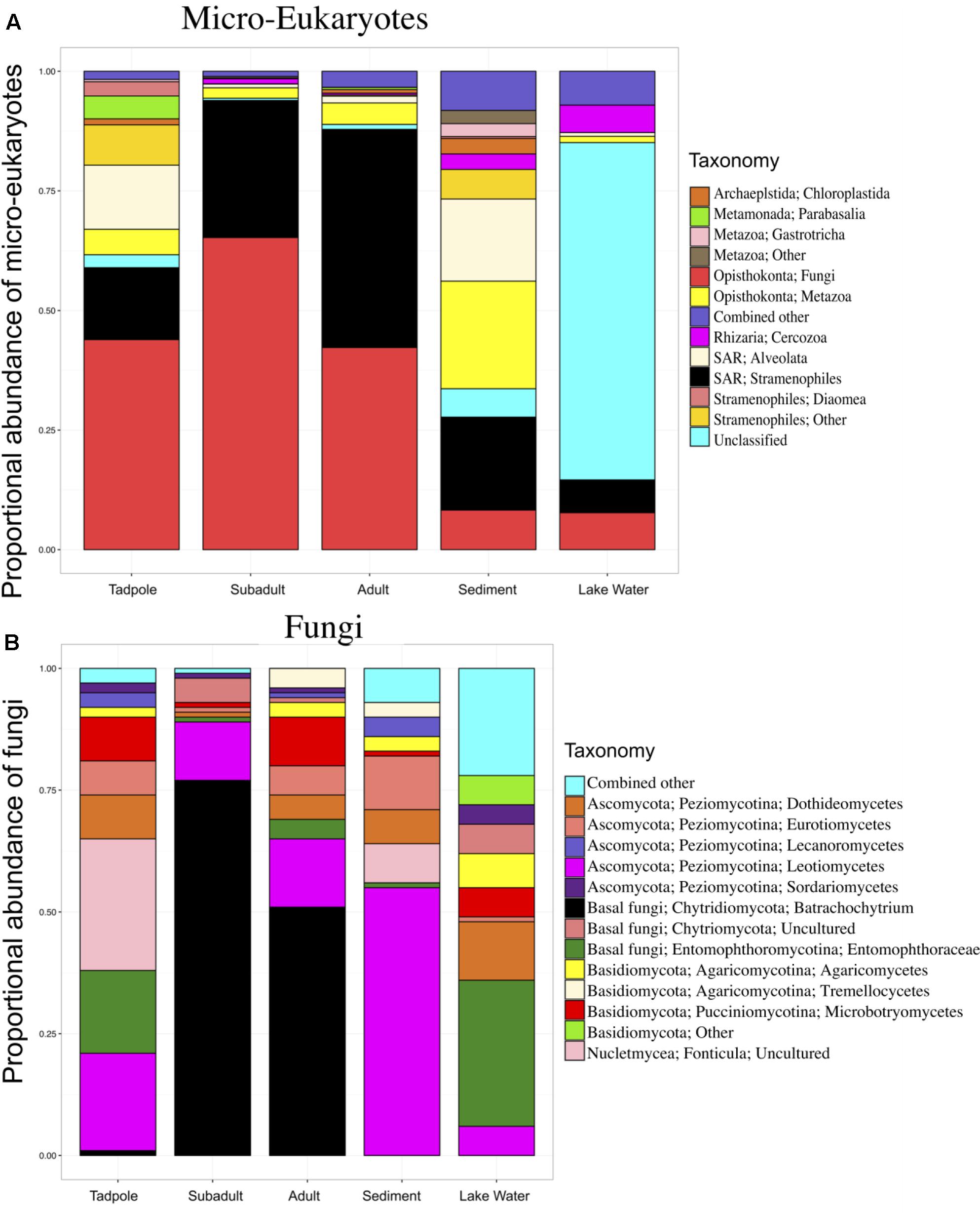
FIGURE 1. Skin micro-eukaryotic taxa on Rana cascadae from Section Line at each life stage, collected on the same day. The proportional abundance of (A) micro-eukaryote and (B) Fungal OTU sequences per major taxon across lifestages: tadpoles (N = 4), subadults (N = 6), adults (N = 11), sediment (N = 3), and lake water (N = 2). Data for both figures is based on the proportional abundance of each microbial taxon per individual. OTUs with lower than 0.5% total abundance were grouped into the category combined other. Average number of sequences per sample type; tadpoles (N = 213), subadults (N = 2,661), adults (N = 3,490), sediment (N = 125,818) and lake water (N = 58,062).
Network Analysis
For the network analyses 16S and 18S datasets were merged and sediment and water samples were removed, resulting in 20 amphibian samples remaining with library sizes greater than 37,800. SparCC (Sparse Correlations for Compositional data) relies on sample proportions; therefore, analysis was done on the raw proportions without rarefying or other normalization. Then, OTUs not present in at least half the samples, and having total sum across samples less than 10, were removed prior to network analysis. This is because correlation detection performance degrades significantly with increased number of zero counts (Weiss et al., 2017). For SparCC in particular, performance degradation may be due to pseudo count addition. In this analysis ‘edges’ are positive and negative correlations with B. dendrobatidis. In the network, ‘edges’ with correlation values of 0.35 or higher were included. For SparCC (Friedman and Alm, 2012), correlation values are just as precise as p-value thresholds and computationally much faster (Weiss et al., 2017). In this study, we used default SparCC parameters. Since, we were mostly interested in B. dendrobatidis-inhibitory OTUs, only predicted ‘edges’ with B. dendrobatidis are shown in the network (Figure 2). A complete list of both positive and negative bacterial and micro-eukaryotic correlations with B. dendrobatidis can be found on the data repository DRYAD.
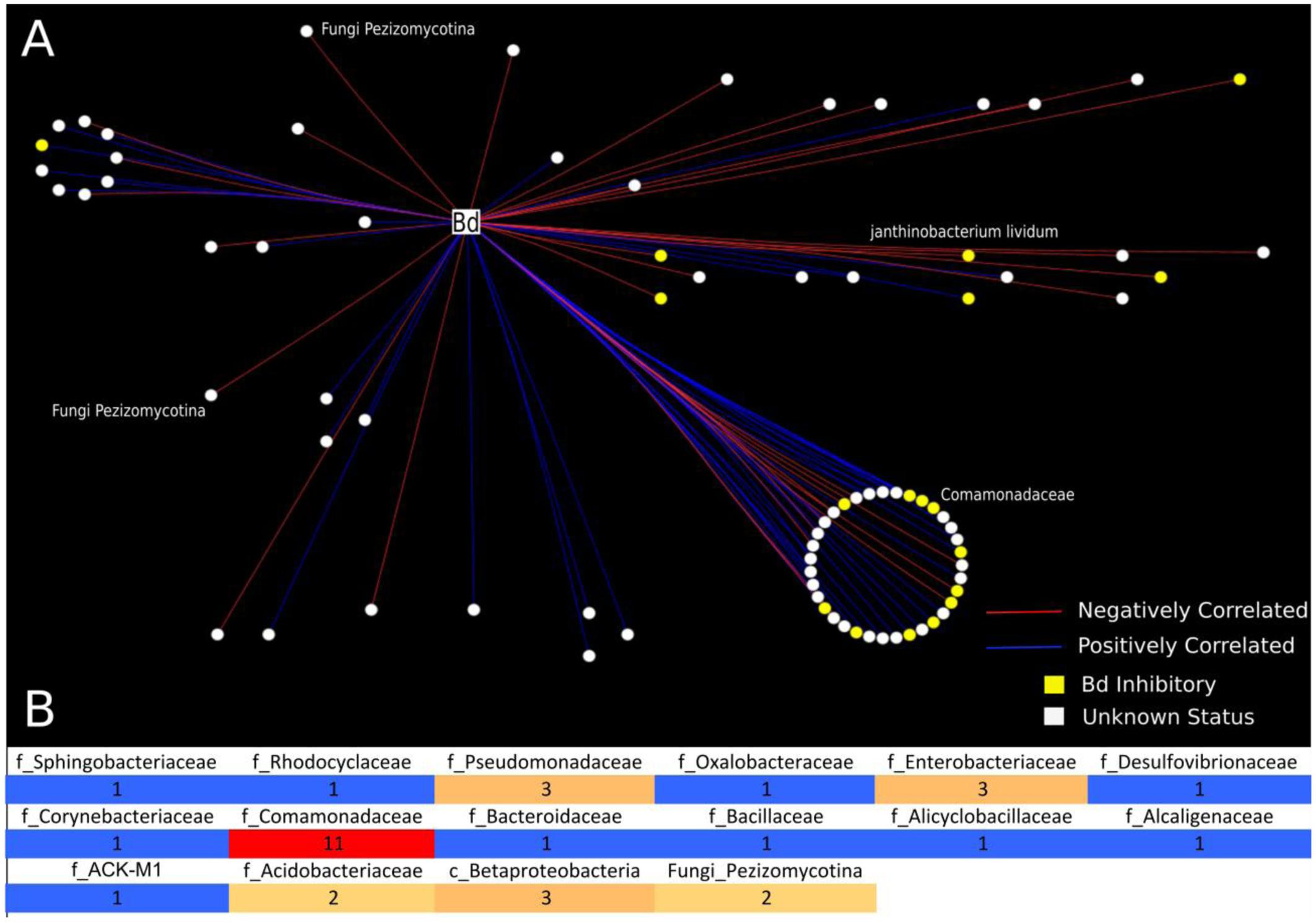
FIGURE 2. Analysis of bacterial and fungal OTUs that correlate with B. dendrobatidis found on Cascades frogs at Section Line. This analysis included 21 frog individuals for which we had successful sequencing yields for both 16S and 18S: tadpoles (N = 4), subadults (N = 6), adults (N = 11). (A) Network analysis is depicting only significantly correlating bacterial and fungal OTUs with B. dendrobatidis. All circular nodes represent OTUs (either bacteria or fungi) with significant correlation with pathogen B. dendrobatidis in the network. Red lines indicate negative correlation between two OTUs. Blue lines indicate positive correlation between two OTUs. Yellow nodes = (B. dendrobatidis-inhibitory), White nodes (Unknown Status). The large circle represents bacterial OTUs of the family Comomonadaceae; the smaller circle shows taxa clustering at the order level (Burkholderiales). Two fungal OTUs (Pezizomycotina) were found to negatively correlate with pathogen B. dendrobatidis. (B) Heatmap is depicting the number of negatively correlating OTUs between bacterial and fungal OTUs and fungal taxon B. dendrobatidis. All positively correlated interactions are excluded from the heatmap. The numbers in the heatmap indicate the frequency of negative correlations between bacterial and fungal groups calculated as the sum of negative OTU correlations per bacterial taxon and B. dendrobatidis, which are also depicted as red lines in A. Taxonomic levels are described as f = Family, c = Class, and Fungi = subdivision.
Batrachochytrium dendrobatidis-Inhibitory Bacterial Isolates Database
Culturing of bacterial isolates from diverse amphibians across the globe, including R. cascadae, have been collated into a B. dendrobatidis-inhibitory database including ∼1,200 isolates that have been tested and shown to inhibit B. dendrobatidis in co-culture (Roth et al., 2013; Woodhams et al., 2015). The bacterial isolates within this database were Sanger sequenced (16S rRNA gene, 1,500 bp), then trimmed to 100 bp and used to pick OTUs with Greengenes reference database (August 2013 version). To explore the overlap between the B. dendrobatidis-inhibitory database and the naturally occurring microbial community of R. cascadae, we trimmed inhibitory OTUs based on sequences trimmed to the first 100 bp beyond primer 515f (GTGCCAGCMGCCGCGGTAA) to match the Illumina reads (819 isolates clustering to 304 OTUs). Then, we use expanded the dataset to include expected inhibitory OTUs within 0.1 Jukes-Cantor distances on the Greengenes phylogenetic tree (7,459 OTUs). This approach resulted in 7,459 unique expected inhibitory OTUs, forming the basis of the B. dendrobatidis-inhibitory database used in this study (available on Dryad). Using this strategy introduces some uncertainty, discussed below, however, it also provides a more comprehensive list of potential B. dendrobatidis-inhibitory taxa OTUs.
Results
Total micro-eukaryotic sequences per sample ranged from 53 to 157,156, with a median observation level of 1,666. We observed 500 unique OTUs before rarefaction. Rarefication to a sequencing depth of 330 sequences per sample reduced the total number of unique OTUs to 255. Eukaryotic taxa found on amphibians included Fungi, Stramenopiles, and Metazoa. Analysis of Composition of Microbes (ANCOM) (Mandal et al., 2015) revealed six OTUs with significant differential abundance across life stages. Importantly, B. dendrobatidis was significantly lower on tadpoles and found highest on subadults. Both subadult and adult skin communities appeared to be dominated by pathogen B. dendrobatidis (Figure 1B). The five other taxa significantly differing across sample types were found to be more abundant in lake water (Supplementary Table 2). Qualitatively, fungi are the most abundant micro-eukaryotic taxa found on all life stages of R. cascadae and fungi shared between all amphibian life stages and sediment included Leotiomycetes and Eurotiomycetes. OTUs in the group Alveolata were found on both tadpoles and in the sediment (Figure 1A). Inter-sample variation for the proportional abundance of eukaryotic taxa across sample types was explored. Here, we see the proportional abundance of B. dendrobatidis is highest on all six subadults, compared with eight out of eleven adults revealing high proportional abundance of B. dendrobatidis (Supplementary Figure B). The combined patterns for proportion abundance using the full dataset are supported by the proportion abundance patterns using the rarefied dataset (Supplementary Figure C).
A list of the most abundant micro-eukaryotic OTUs found on R. cascadae, whether or not they were known to be pathogenic, and a brief description of where else they have been observed, is found in Supplementary Table 1. Surprisingly, we found nine OTUs of fungi, six OTUs of Stramenopiles, and five OTUs of Metazoa represented the majority of the micro-eukaryotic community. Of these 20 OTUs seven are believed to be pathogenic, and eight are believed to not have any negative consequences for the host. We did not find additional information for the remaining four OTUs.
The unequal and relatively small sample size of this dataset limits the strength of statistical measurements of diversity as well as identification of biologically relevant taxa. In particular, only a few tadpoles were found at this location and 18S sequencing yielded low library sizes for micro-eukaryotes. In contrast, 16S sequencing for bacteria yielded sufficient library sizes for comparison to other samples. The low library sizes for micro-eukaryotes warranted the multiple normalization methods described in the methods section. The normalization methods chosen were used to maximize information from the data while maintaining statistical integrity. We found Shannon diversity of micro-eukaryotes and fungi was higher in the sediment compared to amphibian samples. We also report no difference in Shannon diversity across the amphibian lifestages. These results are limited by the sample-sizes across groups and consequently the results and discussion of alpha diversity are otherwise excluded from the main text. A brief methods, results, and discussion of alpha diversity metrics are found in the Supplementary Figure A.
Using network analysis, we discovered that only two fungal OTUs, both Pezizomycota, were negatively correlated with B. dendrobatidis (Figures 2A,B). We observed that the majority of negative correlations occurred between bacterial OTUs that have not yet been challenged in co-culture (unknown status) and that the majority of both positively and negatively B. dendrobatidis-correlated bacterial taxa are in the class Burkholderiales and family Comomonadaceae (Figure 2A). Additional groups that negatively correlate with B. dendrobatidis are shown in a heat map (Figure 2B). Higher taxonomic resolution for both positive and negative correlation ‘edges’ can be found with supportive data materials on DRYAD. Specifically, taxa that negatively correlate with B. dendrobatidis and also match expected B. dendrobatidis-inhibitory database included Comamonadaceae (3) – OTU’s 270402, 536916, and 823696; Enterobacteriaceae (2) – OTUs 537871 and 783638; Pseudomonadaceae (1) – OTU 279948, and Bacillus (1) – OTU 321618. Janthinobacterium lividum (OTU 351280), a common antifungal bacterial isolate, was also found to negatively correlate with B. dendrobatidis (Figure 2A). Taxa listed here warrant increased attention for future conservation applications.
The proportion of B. dendrobatidis-inhibitory bacteria was highest on tadpoles compared to subadults and adults (ANOVA, DF = 2, F = 6.14; p = 0.0079, Figure 3). The composition of expected B. dendrobatidis-inhibitory bacteria is shown in Figure 3B. Tadpole OTUs matching to B. dendrobatidis-inhibitory taxa were primarily in the family Pseudomonadaceae. Subadult OTUs primarily matched to members in the family Comamonadaceae, and more specifically an OTU in the genus Ramlibacter. Adult OTUs matched similarly to those of subadults, but also matched to low abundance inhibitory OTUs. Qualitatively, Enterobacteriaceae and Bacillus OTUs were comparatively very low in their sequence abundance, and Pseudomonadaceae, Comamonadaceae, and Janthinobacterium lividum were detected across all lifestages. A group significance (Kruskal-Wallis) test revealed that several OTUs matching to Pseudomonas as well as J. lividum were more abundant on tadpoles.
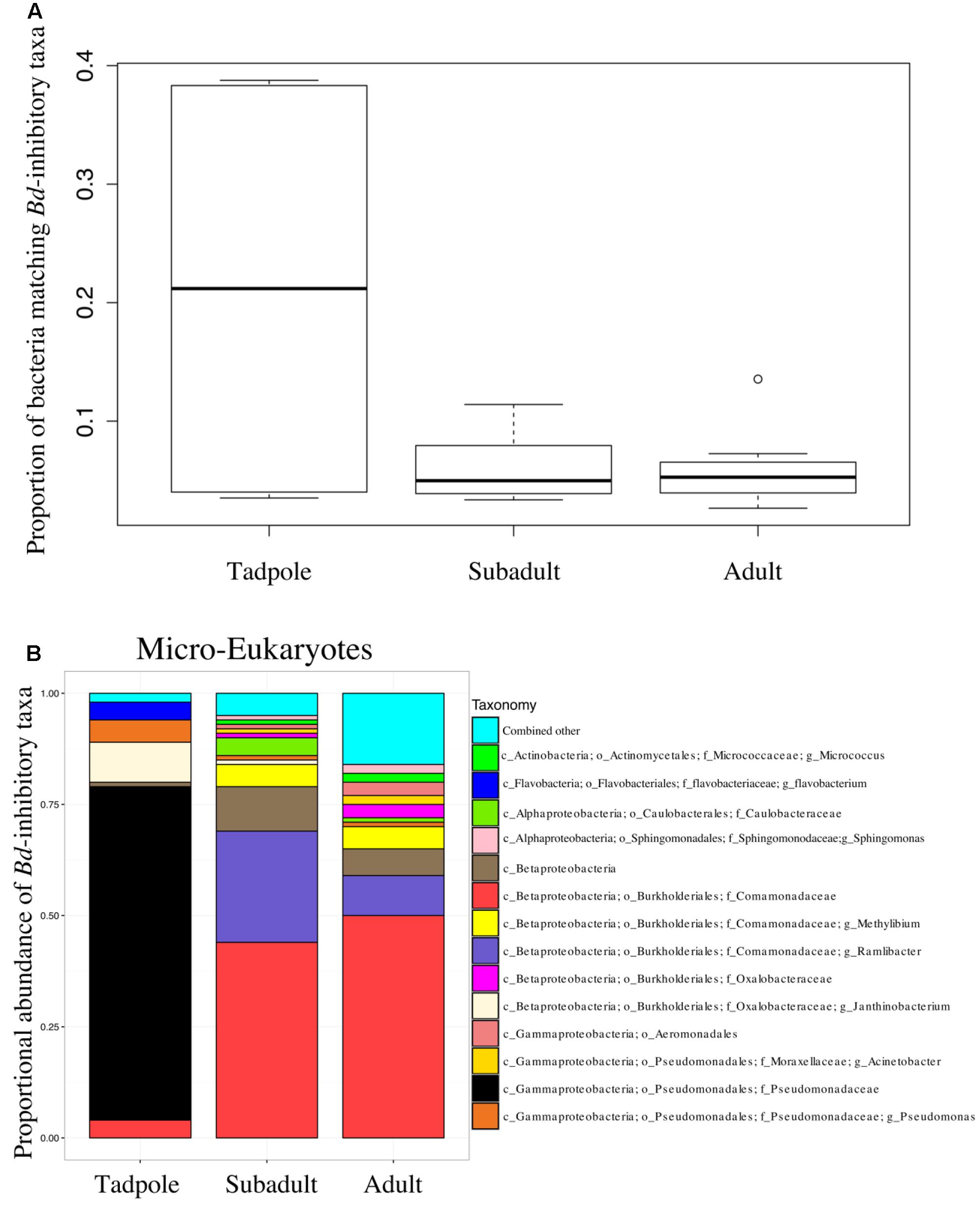
FIGURE 3. (A) The proportion of sequences that match the B. dendrobatidis-inhibitory database found on individuals. There is a higher proportion of the skin community that is B. dendrobatidis-inhibitory on tadpoles compared with subadults and adults; ANOVA, DF = 2, F = 6.143; p = 0.00793. Calculation for A and B are conducted on a rarefied dataset of 19,900 sequences per sample; Lifestages: tadpoles (N = 4), subadults (N = 6), adults (N = 11), sediment (N = 3), and lake water (N = 2). (B) B. dendrobatidis-inhibitory skin bacteria on Rana cascadae from Section Line at each life stage, collected on the same day. OTUs with lower than 1% total abundance are grouped into the category combined other.
Discussion
One previous study has also used 18S rRNA marker gene sequencing to examine the micro-eukaryotic skin community of amphibian boreal toad (Anaxyrus boreas) (Kueneman et al., 2015), thus allowing for some comparison. In R. cascadae we detect a higher proportion of B. dendrobatidis-inhibitory taxa on tadpoles compared with post-metamorphic individuals (Figure 3). This same pattern was found for Anaxyrus boreas (Kueneman et al., 2015). The dominant taxonomy of B. dendrobatidis-inhibitory bacteria found on tadpoles of both R. cascadae and A. boreas are not equivalent. This suggests that each species maintains its own unique protective assemblages (Pseudomonadales for R. Cascadae and Burkholderiales in A. boreas), which may have differential ability to inhibit diverse fungal groups. Comparing other micro-eukaryotes of R. cascadae to A. boreas qualitatively, we find that the class Stramenopiles is shared across life stages. Taxa in class Alveolata are present among tadpoles in the Cascades frog and the boreal toad, but are uniquely absent in post-metamorphic stages of Cascades frogs. Thus, we observe important similarities and differences in the diversity of micro-eukaryotes across amphibian life stages of two different amphibian families (Ranidae and Bufonidae).
Interactions between naturally occurring communities of microbial taxa on amphibian skin can be difficult to infer. However, we show that network analysis can be a useful tool to predict interactions between diverse taxa on the skin of R. cascadae. To date, the sequence database for B. dendrobatidis-inhibitory bacterial taxa (Woodhams et al., 2015) includes 37 bacterial isolates from R. cascadae (Roth et al., 2013), but only five isolates were found from the individuals sampled at Section Line in this dataset. Additionally, OTUs that match these five inhibitory isolates are relatively rare in our dataset of negatively correlating bacterial taxa with B. dendrobatidis. Thus, we utilize a B. dendrobatidis-inhibitory dataset, described in the methods section, which enables us to consider taxa that are closely related to B. dendrobatidis-inhibitory taxa isolated from other amphibian species. We report possible negative interactions that may occur between bacterial and micro-eukaryote taxa and B. dendrobatidis (Figure 2). Specific taxa, identified as negatively correlating with B. dendrobatidis, should be tested experimentally in the lab to confirm activity against B. dendrobatidis. After additional confirmation, these bacterial taxa can be referenced by the B. dendrobatidis-inhibitory database and better inform risk assessment of individuals, life stages, or populations of R cascadae, as well as other amphibian species. Only one bacterial taxon, Janthinobacterium lividum, identified here as negatively correlating with B. dendrobatidis, and matching the B. dendrobatidis-inhibitory database, has been tested as a probiotic on amphibians (Harris et al., 2009; Becker et al., 2011; Walke et al., 2015; Kueneman et al., 2016). However, its activity against B. dendrobatidis on various amphibian species offered mixed results, and its efficacy as a probiotic for R. cascadae remains to be explored in vivo. Intriguingly, J. lividum was found most abundantly on tadpoles. Additionally, isolates of the fungal group Pezizomycota (shown to negatively correlate with B. dendrobatidis) and more specifically Pezizomycetidae, should also be tested for their bioactivity against B. dendrobatidis. Taken together, taxa that negatively correlate with B. dendrobatidis and also match the B. dendrobatidis-inhibitory database may serve to direct additional studies aimed at conservation of R. cascadae and should be tested in vivo.
While the biology of many of the micro-eukaryotic taxa identified in this study remain unknown, several taxa identified, beyond B. dendrobatidis, have been previously shown to be antagonistic/pathogenic to free living organisms and could possibly influence the ecology of the frog skin microbiome (Supplementary Table 1). For example, Entomophthora culicis and Leptolegnia caudata are known parasites and pathogens of insects, respectively (Kerwin, 1982). Additionally, Aphanomyces invadans and Karlodinium micrum were found to be pathogens of fish and their eggs and Saprolegnia species are pathogens of both amphibians and fish, including their eggs (Bisht et al., 1996). Surprisingly, Leptolegnia caudata and Aphanomyces invadans were found to positively correlate with B. dendrobatidis. It is unknown at this point if the detection of these taxa signifies secondary infections or if they may have facilitated B. dendrobatidis infections. The ecology and consequence of the aforementioned taxa should be considered in future studies of R. cascadae health. Additionally, network analysis tools, while having reasonable false positive rates on OTUs with <50% zeroes, need improvement. Many tools have poor detection of ecological relationships other than mutualism and commensalism. SparCC, while attempting to account for compositions, is also one of the strongest network analysis techniques for detecting competitive mutual exclusion (Weiss et al., 2017). Detection of multispecies relationships is also difficult. In this study, increased library size would have provided more insight. Despite these weaknesses, correlation techniques have proven to be useful here and in a variety of additional studies.
In this study, we acknowledge the limitations to the predictive power of the B. dendrobatidis-inhibitory database. This database lacks sufficient information on bacterial isolates that do not exhibit bioactivity against B. dendrobatidis. This is because isolates cultured from amphibians were tested in co-culture with B. dendrobatidis, and then Sanger sequenced. Isolates that did not show inhibition against B. dendrobatidis are often not sequenced and thus not included in the database. Consequently, it is not possible to fully consider microbial interactions not captured by the database. To address the limitation that many B. dendrobatidis inhibitory microbes are also not present in the database we utilize an expanded list of OTUs to capture additional bacterial taxa that may be biologically important. Additional limitations to the predictive power of the B. dendrobatidis-inhibitory dataset exist: For example, (1) most isolates are cultured from adults, and thus the database may be biased toward microbial taxa that colonize the skin of post-metamorphic individuals (Woodhams et al., 2015). (2) In captivity, bacterial taxa may interact differently with B. dendrobatidis than they do in their natural environments. (3) Bacterial taxa that are inhibitory to B. dendrobatidis growth under certain temperature regimes are known to behave differently at other temperature regimes (Woodhams et al., 2014). (4) B. dendrobatidis-inhibitory taxa can act differently in the context of other microbes in the environment, or at certain densities (Woodhams et al., 2014). (5) Bacterial strains of the same species also behave differently with respect to B. dendrobatidis. (6) There is intrinsic bias for amplification of taxa based on the primers used. Specifically, pairs of primers for both bacteria and micro-eukaryotes have limitations for equal sequencing across all groups (Tedersoo et al., 2015; Tedersoo and Lindahl, 2016). (7) Lastly, many of the naturally occurring microbial OTUs are absent from the B. dendrobatidis-inhibitory database which limit the strength of its predictive power. The limitations listed here are universal to the state of extracting meaningful microbial interactions from microbial marker-gene surveys, when there is limited system specific knowledge. Indeed, while metagenomic tools may introduce error, they can also help to direct future studies to address specific knowledge gaps.
The role of skin microbial communities in protecting individuals from B. dendrobatidis is an area open to discovery that may help inform the defenses of R. cascadae against B. dendrobatidis and provide tools for enhanced conservation efforts of R. cascadae in the future. Important to the inferences made from this study, a particularly lethal strain of pathogen B. dendrobatidis has been isolated from Section Line Lake and tested in an experimental setting against R. cascadae reared from eggs. Individuals reared from eggs collected from Section Line survived longer and were healthier than other local populations tested against the Section Line B. dendrobatidis isolate (Piovia-Scott et al., 2015). In addition, individuals from Section Line survived longer against an extraneous B. dendrobatidis isolate, compared to other local populations tested (Piovia-Scott et al., 2015). The evidence shows that R. cascadae at Section Line interact with an aggressive strain of B. dendrobatidis and thus it is particularly important to understand their defenses against the pathogen. Unfortunately, subadults retained very high infection intensities (Figure 1B), and high mortality for this age group is observed in population demographics (Piovia-Scott et al., 2015). This dwindling age group is responsible for the continued decline of this remnant population of the Cascades frog. The function of skin microbial associations on amphibians has yet to be fully explored. This contribution begins to address knowledge gaps regarding the diversity and role of micro-eukaryotes in the health of amphibians, in identifying naturally occurring microbial interactions that may protect R. cascadae from fungal disease epidemics, and in direct conservation research for the most promising protective bacterial groups.
Author Contributions
JK and VM conceived the study, JK collected the data, JK and SW analyzed the data, JK, VM, and SW wrote the paper.
Funding
This project was supported by: an NSF grant (DEB: 1146284 to VM), a grant from the John S. Templeton Foundation to VM, an EBIO summer grant to JK, and JK was supported by an NSF GRFP and a grant from the Simons Foundation (429440, WTW).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jonah Piovia-Scott provided expert field knowledge and Laura Wegener-Parfrey provided the preprocessing steps of the micro-eukaryotic data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02350/full#supplementary-material
References
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W., and Huse, S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hyper variable regions of small-subunit ribosomal RNA genes. PLOS ONE 4:e6372. doi: 10.1371/journal.pone.0006372
Bravo, J. A., Forsythe, P., Chew, M. V., Escaravage, E., Savignac, H. M., Dinan, T. G., et al. (2011). Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. U.S.A. 108, 16050–16055. doi: 10.1073/pnas.1102999108
Becker, M. H., Harris, R. N., Minbiole, K. P., Schwantes, C. R., Rollins-Smith, L. A., Reinert, L. K., et al. (2011). Towards a better understanding of the use of probiotics for preventing chytridiomycosis in Panamanian golden frogs. Ecohealth 8, 501–506. doi: 10.1007/s10393-012-0743-0
Bisht, G. S., Joshi, C.H., and Khulbe, R. D. (1996). Watermolds: potential biological control agents of malaria vector Anopheles culicifacies. Curr. Sci. 70, 393–395.
Bletz, M. C., Loudon, A. H., Becker, M. H., Bell, S. C., Woodhams, D. C., Minbiole, K. P., et al. (2013). Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16, 807–820. doi: 10.1111/ele.12099
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cotty, P. J., and Bayman, P. (1993). Competitive exclusion of a toxigenic strain of Aspergillus flavus by an atoxigenic strain. Phytopathology 83, 1283–1287. doi: 10.1094/Phyto-83-1283
Del Rio, L. E., Martinson, C. A., and Yang, X. B. (2002). Biological control of Sclerotinia stem rot of soybean with Sporidesmium sclerotivorum. Plant Dis. 86, 999–1004. doi: 10.1094/PDIS.2002.86.9.999
Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science 338, 198–199. doi: 10.1126/science.1227412
Fierer, N., Ferrenberg, S., Flores, G. E., González, A., Kueneman, J., Legg, T., et al. (2012). From animalcules to an ecosystem: application of ecological concepts to the human microbiome. Annu. Rev. Ecol. Evol. Syst. 43, 137–155. doi: 10.1146/annurev-ecolsys-110411-160307
Fierer, N., Hamady, M., Lauber, C. L., and Knight, R. (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 17994–17999. doi: 10.1073/pnas.0807920105
Fitzpatrick, B. M., and Allison, A. L. (2014). Similarity and differentiation between bacteria associated with skin of salamanders (Plethodon jordani) and free-living assemblages. FEMS Microbiol. Ecol. 88, 482–494. doi: 10.1111/1574-6941.12314
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLOS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Garcia, T. S., Romansic, J. M., and Blaustein, A. R. (2006). Survival of three species of anuran metamorphs exposed to UV-B radiation and the pathogenic fungus Batrachochytrium dendrobatidis. Dis. Aquat. Organ. 72:163. doi: 10.3354/dao072163
Harris, R. N., Brucker, R. M., Walke, J. B., Becker, M. H., Schwantes, C. R., Flaherty, D., et al. (2009). Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. doi: 10.1038/ismej.2009.27
Jani, A. J., and Briggs, C. J. (2014). The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl. Acad. Sci. U.S.A. 111, E5049–E5058. doi: 10.1073/pnas.1412752111
Kabaluk, J. T., Svircev, A. M., Goettel, M. S., and Woo, S. G. (2010). The Use and Regulation of Microbial Pesticides in Representative Jurisdictions Worldwide. St. Paul, MN: International Organization for Biological Control.
Kerwin, J. L. (1982). Chemical control of the germination of asexual spores of Entomophthora culicis, a fungus parasitic on dipterans. Microbiology 128, 2179–2186. doi: 10.1099/00221287-128-9-2179
Kueneman, J. G., Parfrey, L. W., Woodhams, D. C., Archer, H. M., Knight, R., McKenzie, V. J., et al. (2014). The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250. doi: 10.1111/mec.12510
Kueneman, J. G., Woodhams, D. C., Harris, R., Archer, H. M., Knight, R., and McKenzie, V. J. (2016). Probiotic treatment restores protection against lethal fungal infection lost during amphibian captivity. Proc. R. Soc. 283, 1839–1553. doi: 10.1098/rspb.2016.1553
Kueneman, J. G., Woodhams, D. C., Van Treuren, W., Archer, H. M., Knight, R., and McKenzie, V. J. (2015). Inhibitory bacteria reduce fungi on early life stages of endangered Colorado boreal toads (Anaxyrus boreas). ISME J. 10, 934–944 doi: 10.1038/ismej.2015.168
Lam, B. A., Walke, J. B., Vredenburg, V. T., and Harris, R. N. (2010). Proportion of individuals with anti-Batrachochytrium dendrobatidis skin bacteria is associated with population persistence in the frog Rana muscosa. Biol. Conserv. 143, 529–531. doi: 10.1016/j.biocon.2009.11.015
Lauber, C. L., Strickland, M. S., Bradford, M. A., and Fierer, N. (2008). The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 40, 2407–2415. doi: 10.1016/j.soilbio.2008.05.021
Loudon, A. H., Woodhams, D. C., Parfrey, L. W., Archer, H., Knight, R., McKenzie, V., et al. (2014). Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J. 8, 830–840. doi: 10.1038/ismej.2013.200
Mandal, S., Van Treuren, W., White, R. A., Eggesbø, M., Knight, R., and Peddada, S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26:27663.
McKenzie, V. J., Bowers, R. M., Fierer, N., Knight, R., and Lauber, C. L. (2012). Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 6, 588–596. doi: 10.1038/ismej.2011.129
Piovia-Scott, J., Pope, K. L., Lawler, S. P., Cole, E. M., and Foley, J. E. (2011). Factors related to the distribution and prevalence of the fungal pathogen Batrachochytrium dendrobatidis in Rana cascadae and other amphibians in the Klamath Mountains. Biol. Conserv. 144, 2913–2921. doi: 10.1016/j.biocon.2011.08.008
Piovia-Scott, J., Pope, K., Worth, S. J., Rosenblum, E. B., Poorten, T., Refsnider, J., et al. (2015). Correlates of virulence in a frog-killing fungal pathogen: evidence from a California amphibian decline. ISME J. 9, 1570–1578. doi: 10.1038/ismej.2014.241
Rebollar, E. A., Antwis, R. E., Becker, M. H., Belden, L. K., Bletz, M. C., Brucker, R. M., et al. (2016a). Using “omics” and integrated multi-omics approaches to guide probiotic selection to mitigate chytridiomycosis and other emerging infectious diseases. Front. Microbiol. 7:68. doi: 10.3389/fmicb.2016.00068
Rebollar, E. A., Hughey, M. C., Medina, D., Harris, R. N., Ibáñez, R., and Belden, L., K. (2016b). Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J. 10, 1682–1695. doi: 10.1038/ismej.2015.234
Rideout, J. R., He, Y., Navas-Molina, J. A., Walters, W. A., Ursell, L. K., Gibbons, S. M., et al. (2014). Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2:545. doi: 10.7717/peerj.545
Roth, T., Foley, J., Worth, J., Piovia-Scott, J., Pope, K., and Lawler, S. (2013). Bacterial flora on Cascades frogs in the Klamath mountains of California. Comp. Immunol. Microbiol. Infect. Dis. 36, 591–598. doi: 10.1016/j.cimid.2013.07.002
Tedersoo, L., Anslan, S., Bahram, M., Põlme, S., Riit, T., Liiv, I., et al. (2015). Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10, 1–43. doi: 10.3897/mycokeys.10.4852
Tedersoo, L., and Lindahl, B. (2016). Fungal identification biases in microbiome projects. Environ. Microbiol. Rep. 8, 774–779. doi: 10.1111/1758-2229.12438
Tuddenham, S., and Sears, C. L. (2015). The intestinal microbiome and health. Curr. Opin. Infect. Dis. 28, 464–470. doi: 10.1097/QCO.0000000000000196
Voyles, J., Young, S., Berger, L., Campbell, C., Voyles, W. F., and Dinudom, A., et al. (2009). Pathogenesis of chytridiomycosis, a cause of catastrophic amphibian declines. Science 326, 582–585. doi: 10.1126/science.1176765
Walke, J. B., Becker, M. H., Loftus, S. C., House, L. L., Cormier, G., Jensen, R. V., et al. (2014). Amphibian skin may select for rare environmental microbes. ISME J. 8, 2207–2217. doi: 10.1038/ismej.2014.77
Walke, J. B., Becker, M. H., Loftus, S. C., House, L. L., Teotonio, T. L., Minbiole, K. P. C., et al. (2015). Community structure and function of amphibian skin microbes: an experiment with bullfrogs exposed to a Chytrid fungus. PLOS ONE 10:e0139848. doi: 10.1371/journal.pone.0139848
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Woodhams, D. C., Alford, R. A., Antwis, R. E., Archer, H., Becker, M. H., Belden, L. K., et al. (2015). Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96, 595–595. doi: 10.1890/14-1837.1
Woodhams, D. C., Brandt, H., Baumgartner, S., Kielgast, J., Küpfer, E., Tobler, U., et al. (2014). Interacting symbionts and immunity in the amphibian skin mucosome predict disease risk and probiotic effectiveness. PLOS ONE 9:e96375. doi: 10.1371/journal.pone.0096375
Keywords: microbiome, micro-eukaryote, network, skin, Rana cascadae, Batrachochytrium dendrobatidis, Cascades frog
Citation: Kueneman JG, Weiss S and McKenzie VJ (2017) Composition of Micro-eukaryotes on the Skin of the Cascades Frog (Rana cascadae) and Patterns of Correlation between Skin Microbes and Batrachochytrium dendrobatidis. Front. Microbiol. 8:2350. doi: 10.3389/fmicb.2017.02350
Received: 29 June 2017; Accepted: 15 November 2017;
Published: 08 December 2017.
Edited by:
Ana E. Escalante, National Autonomous University of Mexico, MexicoReviewed by:
Corinne Lee Richards-Zawacki, University of Pittsburgh, United StatesGerman Bonilla Rosso, University of Lausanne, Switzerland
Copyright © 2017 Kueneman, Weiss and McKenzie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerie J. McKenzie, dmFsZXJpZS5tY2tlbnppZUBjb2xvcmFkby5lZHU=