 Vitaly Smelov1,2,3‡
Vitaly Smelov1,2,3‡ Alison Vrbanac4,†‡
Alison Vrbanac4,†‡ Eleanne F. van Ess5‡
Eleanne F. van Ess5‡ Marlies P. Noz5Raymond Wan4Carina Eklund2Tyler Morgan4Lydia A. Shrier6
Marlies P. Noz5Raymond Wan4Carina Eklund2Tyler Morgan4Lydia A. Shrier6 Blake Sanders4Joakim Dillner2
Blake Sanders4Joakim Dillner2 Henry J. C. de Vries7,8,9
Henry J. C. de Vries7,8,9 Servaas A. Morre5,10
Servaas A. Morre5,10 Deborah Dean4,11,12,13*
Deborah Dean4,11,12,13*- 1International Agency for Research on Cancer, World Health Organization, Lyon, France
- 2Karolinska Institute, Stockholm, Sweden
- 3North-Western State Medical University named after I.I. Mechnikov, St. Petersburg, Russia
- 4UCSF Benioff Children’s Hospital Oakland Research Institute, Oakland, CA, United States
- 5Laboratory of Immunogenetics, Department of Medical Microbiology and Infection Control, VU University Medical Center, Amsterdam, Netherlands
- 6Department of Pediatrics, Boston Children’s Hospital, Boston, MA, United States
- 7Center for Infection and Immunology Amsterdam, Academic Medical Center, Amsterdam, Netherlands
- 8Department of Dermatology, Academic Medical Center, Amsterdam, Netherlands
- 9STI Outpatient Clinic, Public Health Service of Amsterdam (GGD Amsterdam), Amsterdam, Netherlands
- 10Institute of Public Health Genomics, Department of Genetics and Cell Biology, Research Institute GROW (School for Oncology and Developmental Biology), Faculty of Health, Medicine and Life Sciences, University of Maastricht, Maastricht, Netherlands
- 11Department of Bioengineering, University of California, Berkeley, Berkeley, CA, United States
- 12Department of Bioengineering, University of California, San Francisco, San Francisco, CA, United States
- 13Departments of Medicine and Pediatrics, University of California, San Francisco, San Francisco, CA, United States
Chlamydia trachomatis (Ct) is the leading cause of bacterial sexually transmitted diseases worldwide. The Ct Multi Locus Sequence Typing (MLST) scheme is effective in differentiating strain types (ST), deciphering transmission patterns and treatment failure, and identifying recombinant strains. Here, we analyzed 323 reference and clinical samples, including 58 samples from Russia, an area that has not previously been represented in Ct typing schemes, to expand our knowledge of the global diversification of Ct STs. The 323 samples resolved into 84 unique STs, a 3.23 higher typing resolution compared to the gold standard single locus ompA genotyping. Our MLST scheme showed a high discriminatory index, D, of 0.98 (95% CI 0.97–0.99) confirming the validity of this method for typing. Phylogenetic analyses revealed distinct branches for the phenotypic diseases of lymphogranuloma venereum, urethritis and cervicitis, and a sub-branch for ocular trachoma. Consistent with these findings, single nucleotide polymorphisms were identified that significantly correlated with each phenotype. While the overall number of unique STs per region was comparable across geographies, the number of STs was greater for Russia with a significantly higher ST/sample ratio of 0.45 (95% CI: 0.35–0.53) compared to Europe or the Americas (p < 0.009), which may reflect a higher level of sexual mixing with the introduction of STs from other regions and/or reassortment of alleles. Four STs were found to be significantly associated with a particular geographic region. ST23 [p = 0.032 (95% CI: 1–23)], ST34 [p = 0.019 (95% CI: 1.1–25)]; and ST19 [p = 0.001 (95% CI: 1.7–34.7)] were significantly associated with Netherlands compared to Russia or the Americas, while ST 30 [p = 0.031 (95% CI: 1.1–17.8)] was significantly associated with the Americas. ST19 was significantly associated with Netherlands and Russia compared with the Americans [p = 0.001 (95% CI: 1.7–34.7) and p = 0.006 (95% CI: 1.5–34.6), respectively]. Additionally, recombinant strains were ubiquitous in the data set [106 (32.8%)], although Europe had a significantly higher number than Russia or the Americas (p < 0.04), the majority of which were from Amsterdam [43 (87.8%) of 49)]. The higher number of recombinants in Europe indicates selective pressure and/or adaptive diversification that will require additional studies to elucidate.
Introduction
The modern pathogenic Chlamydiaceae family has a rich evolutionary history, diverging from environmental Chlamydiales approximately seven million years ago (Horn et al., 2004). The human Chlamydiaceae spp. Chlamydia trachomatis (Ct) has infected human populations causing sexually transmitted diseases (STD) and the chronic ocular disease known as trachoma since the 27th century BC (Perkins and Hill, 2013). Trachoma was initially described in China and in the Eber’s Papyrus of Egypt, and subsequently spread to Europe during the Crusades (Perkins and Hill, 2013). While improvements in hygiene and sanitation have eliminated trachoma from many global populations, the disease is still endemic in many developing countries of Africa, Central and South America, the South Pacific and Asia in addition to aboriginal populations in Australia (Perkins and Hill, 2013). Presently, Ct is the leading cause of preventable blindness and bacterial sexually transmitted infections (STIs) worldwide (Rowley et al., 2012) with estimates of over 250 million trachoma cases and 110 million annual STI cases, according to the World Health Organization (WHO Sexually transmitted infections [STIs], 2015).
Ct has evolved to include 19 serological variants (serovars) based on antibody typing of the major outer membrane protein (MOMP) with over 60 ompA genotypes (Dean and Millman, 1997; Batteiger et al., 2014; Isaksson et al., 2016; Peuchant et al., 2016; Schillinger et al., 2016), the gold standard typing technique for all Chlamydiaceae spp. The serovars are designated A through K, Ba, Da, Ia, Ja, and L1-3, and L2a while the ompA genotypes or strains are denoted by the same or by a number or letter after the conventional serovar name (e.g., D1; Ga) for new genotypes (Batteiger et al., 2014). These strains are responsible for ocular, urogenital and rectal infections. Ocular infections include trachoma, a chronic ocular disease, and ophthalmia neonatorum (Darville, 2005), an infection acquired during passage through a Ct infected birth canal (WHO Sexually transmitted infections [STIs], 2015). Urogenital strains cause not only ocular infections, which usually present as unilateral conjunctivitis (Dean et al., 2008), but also can ascend from the endocervix to cause sequelae such as pelvic inflammatory disease, infertility and ectopic pregnancy (Mårdh, 2004; Blas et al., 2007; Baud et al., 2008). Rectal infections can progress to proctitis and inguinal syndrome (Sethi et al., 2009). While the later is caused primarily by the lymphogranuloma venereum strains (LGV) L1-3, L2a, L2b, and L2c, the former can be caused by most Ct strains, although strains B, Ba, and C are rarely detected in the urethra, endocervix or rectum (Danby et al., 2016; Labiran et al., 2016). Strain A is the only strain that is confined to the ocular mucosa (Dean, 2010).
Classification of Ct strains was conventionally performed by serotyping and more recently by ompA genotyping (Dean et al., 1992) since the organism is rarely cultured, a requirement for serotyping. Although ompA genotyping can be informative, the gene represents a mere 0.1% of the genome and is subject to immune selective pressure and recombination (Joseph et al., 2011, 2012). Finer, more holistic typing schemes are necessary to track recombination events, differentiate new and persistent infections (Götz et al., 2013), reinfection, transmission patterns and elucidate potential biomarkers. Three Multi Locus Sequence Typing (MLST) schemes have been developed for Ct (Klint et al., 2007; Pannekoek et al., 2008; Dean et al., 2009), of which only two meet the MLST criteria of using strictly housekeeping genes (Pannekoek et al., 2008; Dean et al., 2009). Our MLST scheme employs seven highly conserved housekeeping genes and successfully resolves reference and clinical Ct samples into LGV, trachoma, non-prevalent non-invasive urogenital, and prevalent non-invasive urogenital clonal complexes representing the respective diseases in addition to revealing evidence for recombination (Dean et al., 2009; Batteiger et al., 2014).
Partial and whole genome sequencing (WGS) have added considerably to our knowledge of the diversity of Ct and evidence for recombination. We initially bioinformatically identified recombination within ompA (Millman et al., 2001) and then among partial genome sequences of trachoma and sexually transmitted strains involving ompA and polymorphic membrane proteins (pmp) (Gomes et al., 2004, 2006, 2007). In the first publication of comparative WGS of Ct, we identified three major clades that were similar to the MLST disease-related clonal complexes with a subclade encompassing the trachoma strains within the non-prevalent non-invasive urogenital clade (Joseph et al., 2011). Subsequent WGS by our and other groups have substantiated these phylogenetic groupings as well as the recombinogenic nature of Ct (Harris et al., 2012; Joseph et al., 2012; Seth-Smith et al., 2013; Hadfield et al., 2017).
As whole genome sequencing remains cost-prohibitive for large sample sets and beyond the reach of most research laboratories, in this work, 323 Ct reference and clinical samples from 15 countries and 5 continents were analyzed by MLST to provide a more comprehensive analysis of the global diversification of Ct strain types. We included 60 new clinical samples from Amsterdam, Netherlands, eight from Boston, MA, United States, and 58 from St. Petersburg, Russia, a region that had not previously been evaluated by MLST.
Materials and Methods
Study Populations and Ethics
Information on populations and ethics for samples collected previously are included in the publications by Dean et al. (2009) and Batteiger et al. (2014). Russian women were enrolled in the original studies following verbal informed consent after approval by the Local Institutional Review Board at DO Ott Institute of Obstetrics and Gynecology and the Russian Academy of Medical Sciences (RAMS), St. Petersburg, Russia (Shipitsyna et al., 2007). The studies were additionally approved by the Department of Clinical Investigations and Intellectual Property, St. Petersburg Medical Academy of Postgraduate Studies (North-Western State Medical University named after I.I. Mechnikov since 2011), under the Federal Agency of Public Health and Social Development of Roszdrav, in accordance with the Declaration of Helsinki. For the 60 Dutch samples, each was obtained as previously described (Dean et al., 2009) according to the Declaration of Helsinki. The eight Boston samples were sent to Dr. Dean as de-identified samples with no trace to patient name and were not considered human subjects according to IRB at Children’s Hospital Oakland Research Institute and NIH guidelines.
For the Russian samples, endocervical swabs were obtained consecutively from January 2006 to January 2008 in two university clinics in St. Petersburg, Russia, as described elsewhere (Smelov et al., 2009). To minimize the potential for low response from the enrolled women, samples were collected without obtaining personal information. After removal of any mucopus with a cotton swab, a Dacron swab was inserted into the endocervix, rotated and placed in an empty 5 mL vials. Specimens were kept at 4°C (39°F) for up to 4 days before they were shipped to the laboratory where they were stored at 4°C (39°F). Within 1–3 days (Shipitsyna et al., 2007) all samples were tested for the presence of Ct by commercial NAAT assays (Shalepo et al., 2006; Smelov et al., 2009). Additionally, either a conventional PCR (Lytech, Moscow, Russia) or a real-time PCR (Central Research Institute of Epidemiology, Moscow, Russia) were used. The results were confirmed in Amsterdam, Netherlands by the commercial real-time PCR (TaqMan, Applied Biosystems, United States) (Morré et al., 1999; Smelov et al., 2009) and CE-IVD certificated Presto CT-NG Assays (Goffin Molecular Technologies, Beek, Netherlands) (de Waaij et al., 2015).
Ct Reference and Clinical Samples
A total of 323 reference and clinical samples were analyzed that included 58 endocervical samples from St. Petersburg, 60 additional endocervical samples from Amsterdam and eight endocervical samples from Boston, Massachusetts; 265 were already in the MLST database1 including the 20 reference strains: A/SA-1, A/HAR13, B/TW5/OT, Ba/Apache2, C/TW3/OT, D/UW3/Cx, Da/TW-448, E/Bour, F/IC-Cal3, G/UW57/Cx, H/UW4/Cx, I/UW12/Ur, Ia/UW202, J/UW36/Cx, Ja/UW92, K/UW36/Cx, L1/440, L2/434, L2a/UW396, L3/404.
ompA Genotyping and MLST Analysis
Genomic DNA was purified from clinical isolates and ompA genotyped as described previously (Dean et al., 2009; Batteiger et al., 2014). Only the samples from St. Petersburg were provided as swabs; DNA was extracted and purified for these samples using our protocol as described in Joseph et al. (2014). MLST analysis examined seven housekeeping genes: glyA, mdhC, pdhA, yhbG, pykF, lysS, and leuS, with primers as described (Dean et al., 2009) (Supplementary Table 1). All seven housekeeping genes were amplified and sequenced as described (Batteiger et al., 2014). A consensus sequence was created from the forward and reverse sequence. The genes for each of the St. Petersburg, Amsterdam and Boston samples were each concatenated and queried against the 265 samples in the MLST database in addition to including these samples in the database. Allelic numbers and STs were assigned based on this query as described previously (Batteiger et al., 2014).
Phylogenetic Analysis and Strain Clustering
Using the concatenated sequences, dataset strain clustering and Single Nucleotide Polymorphism (SNP) analyses were performed as described (Batteiger et al., 2014). Briefly, this included visualizing clusters of related STs and non-related STs using eBURST2 (Feil et al., 2004). Founder STs were identified by the highest number of single locus variants (SLV) branching from that particular ST (i.e., the clonal ancestor that diversifies into other STs). Clonal complexes generated by eBURST were defined as a group of STs separated by one SLV.
Phylogenetic trees were created by Maximum likelihood using the Symmetric+GI model, which provided the best fit for the data, in the R package phangorn (Schliep, 2010) to analyze the nucleotide sequence variation between the seven MLST loci for each ST. Tree nodes were verified with 1,000 bootstrap replicates. Alternative evolutionary pathways, such as horizontal gene transfer, were analyzed with SplitsTree3 using the splits decomposition method as described (Dean et al., 2009; Tamura et al., 2013). In addition, the sequence for each of the seven MLST loci for a sample were compared across the dataset and to the ompA genotype of the same sample to determine evidence for putative recombination.
Statistical Tests
Fisher’s exact test was performed in R4 to test for significant region-specific ompA and ST clustering; a p-value of <0.05 was considered significant. Confidence intervals were determined based on the method of Clopper-Pearson (Borkowf, 2006). Simpson’s Diversity Index, D, was calculated for the MLST data as described (Simpson, 1949; Hunter and Gaston, 1988). A D-value of ≥0.95 was considered ideal for molecular typing techniques (van Belkum et al., 2007). The ompA genotypes were excluded from the analysis. The Benjamin-Hochberg FDR method (Benjamini and Hochberg, 1995) was used to correct p-values for multiple comparisons.
Samples were classified as putative recombinants when the sequences of the seven gene sequences that comprise the ST or any of the seven genes were non-concordant with each other or with the ompA genotype of the same sample.
PROC FREQ in SAS was used to identify SNPs associated with disease phenotype and Haplotype as described previously (Dean et al., 2009). Levene’s test evaluated the variance across the dataset of the 323 samples. The Classification Index was used to determine significance of each SNP with a disease phenotype where a p-value of <0.05 was considered significant.
DnaSP v5.10 (Librado and Rozas, 2009) was used to calculate nucleotide (nt) and haplotype (hd) to determine the genetic diversity and differentiation for regional subgroups on the concatenated sequences of the seven MLST genes. DnaSP considers the frequency of variants (STs) present in a population and also genetic distances that separate these variants from each other. Genetic population differentiation between regional subgroups was assessed using the pairwise fixation index (Fst) in Arlequin v3.5 (Excoffier and Lischer, 2010) with significance testing by permutation.
Results
Characteristics and Geographic Distribution of Alleles
The characteristics of the alleles for each gene locus are shown in Supplementary Table 2. The number of alleles varied by gene locus, ranging from seven to 18, as did the number of polymorphic sites. We determined the allele frequencies by geographic region for the 78 alleles (Table 1). Thirty-two (41%) alleles were observed once. The highest number of unique alleles for a geographic region was in Western Europe at 16 alleles but the highest frequency was 43.3% for Russia, which was not statistically significant.
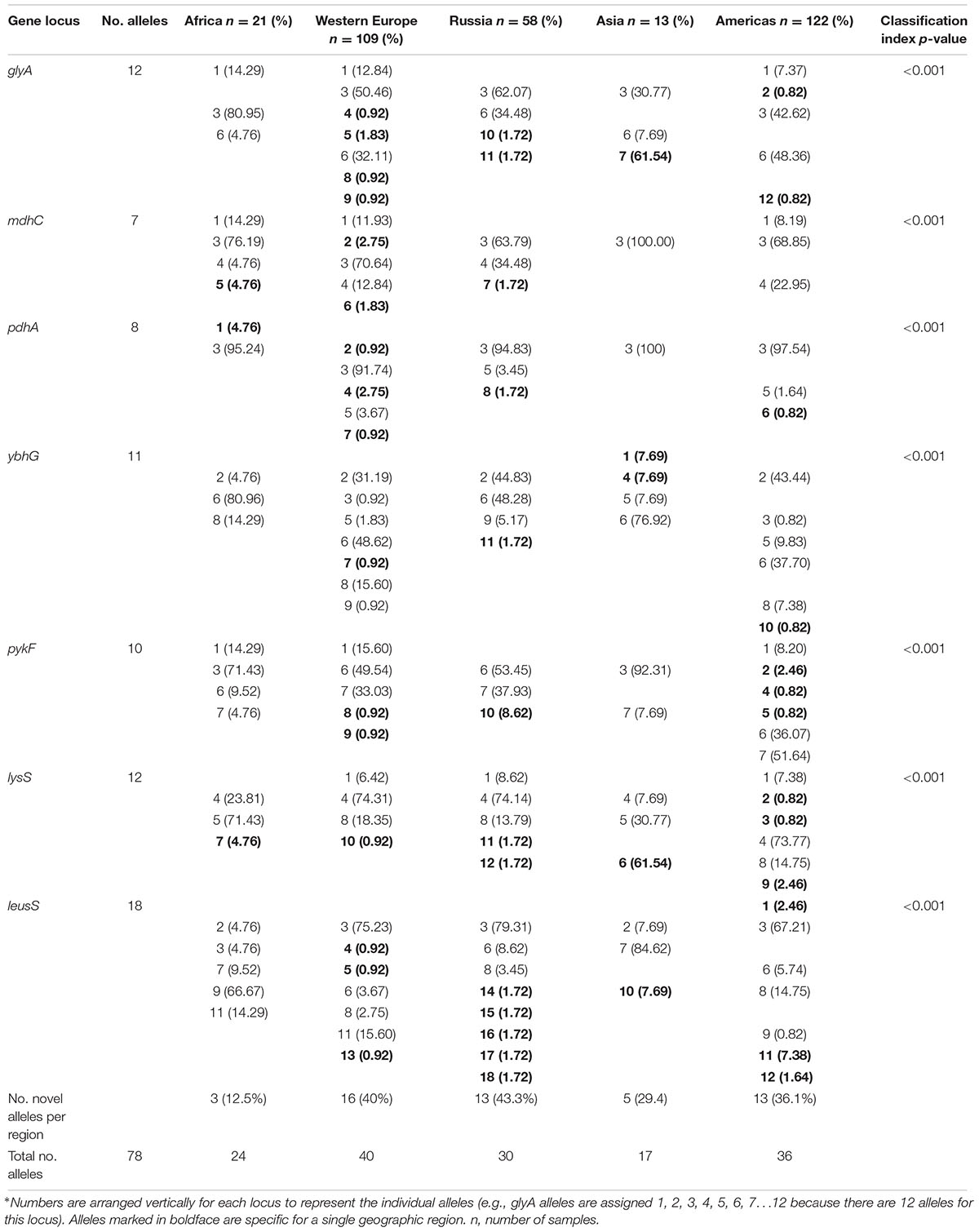
TABLE 1. Allele frequencies by geographic region by locus.
ST and ompA Distributions
For the 323 samples, 84 unique STs were identified (Supplementary Table 3). STs novel to the dataset were numbered consecutively in order of identification. Of those 84, 57 (67.9%) were singletons, with a relatively even distribution by geographic region (excluding Asia and Africa where the sample sizes were very small) with a higher percentage of singletons in Europe that was not significantly different (Table 2; p = 0.08). Table 2 also shows that the percentage of unique STs per region was highly similar. However, the ST/sample ratio was significantly greater for Russian than for European and American samples (p < 0.009). There were also significant differences in the distribution of STs (Table 3). Dutch females (n = 79) were significantly more likely to be infected with ST23 (p = 0.032; 95% CI: 1–23) and ST34 (p = 0.02; 95% CI: 1.1–25) compared to Russian females (n = 58) and with ST19 (p = 0.001; 95% CI: 1.7–34.7) compared to American females (n = 108). Supplementary Table 4 shows the distribution of STs by geographic region.
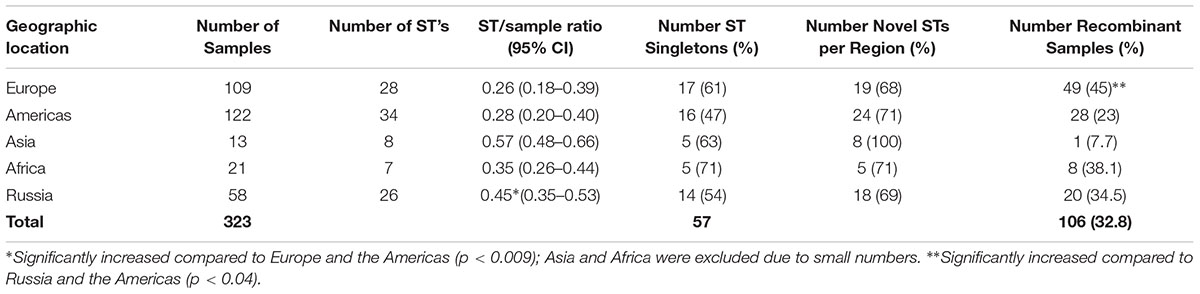
TABLE 2. Strain type (ST) diversity and recombinants by geographic region.
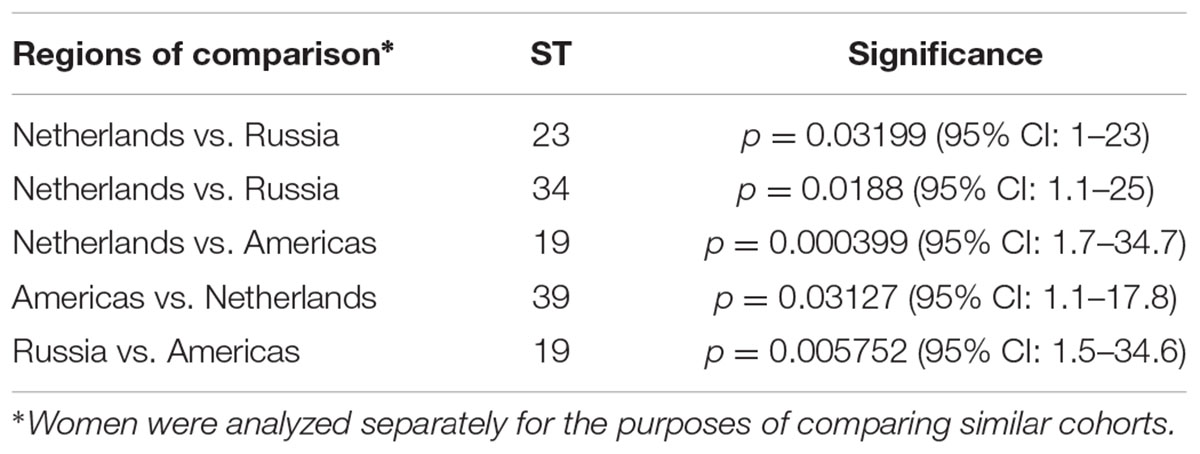
TABLE 3. Differences in ST distribution by geographic region.
There were 26 ompA genotypes observed in the dataset resulting in a 3.23 lower resolution than for STs. Excluding the STDs samples from South Africa, all samples available from Asia and Africa were from trachoma patients; 1 (7.1%) of 14 was a urogenital Da ompA genotype (ST 37) in Asia while 1 (5.9%) of 17 was a urogenital E genotype (ST 39) in Africa.
ompA genotype E (n = 72) was the most prevalent and associated with 18 STs with an ST to sample ratio of 0.25 (Supplementary Table 3). The distribution for the remaining ompA genotypes in descending order was G (38; 16 STs; ratio: 0.42); D (33; 16 STs; ratio: 0.48); F (30; 5 STs; ratio: 0.17); Ia (17; 4 STs; ratio: 0.25); K (15; 7 STs; ratio: 0.47); J (13; 9 STs; ratio: 0.69); B, H and I at 12 samples each (7, 6, and 3 STs, respectively; ratios: 0.58, 0.50, and.25, respectively); C (11: 5 STs; ratio: 0.45); A (9; 4 STs; ratio: 0.44); D2 and Ja at 4 samples each (1 and 3 STs, respectively; ratios: 0.25 and 0.75, respectively); Ba, D1, E6, F4 and Ia4 at 2 samples each (1 ST for each; ratio: 0.50 for each); and Da (1; 1 ST; ratio: 0.10).
The ompA distribution of urogenital strains varied across Europe, the Americas, and Russia. In Europe, ompA genotype D was significantly more prevalent than in other geographic regions (p = 0.046) and more frequent than the globally prevalent genotype E. In Russia, ompA genotype G was significantly more prevalent than in all other regions (p = 0.001). In the Americas, ompA genotype Ia was significantly more prevalent than the other regions (p = 0.001). In comparing female STD cohorts, Russian women were significantly more likely to be infected with E and G (p = 0.001 and 0.026, respectively) while Dutch women were significantly more likely to be infected with D and I (p = 0.026 and 0.002, respectively).
Simpson’s Discriminatory Index was 0.98 (95% CI 0.97–0.99) for MLST and 0.70 (95% CI 0.67–0.73) for ompA genotyping.
Based on DnaSP, nucleotide diversity was lower in Russia (Pi = 0.00216) compared to Netherlands (Pi = 0.00321) and North America (Pi = 0.00294) (Table 4). However, Netherlands and North American datasets contained isolates from men and LGV samples. The phylogenetically disparate LGV samples appeared to contribute substantially to nucleotide diversity; nucleotide diversity dropped from 0.00250 to 0.00196 in Dutch women when the four LGV isolates were removed. When comparing non-LGV isolates from women in these regions, Russian women had the highest nucleotide diversity (Pi = 0.00216), followed by North American women (Pi = 0.00210), and Dutch women (Pi = 0.00196).
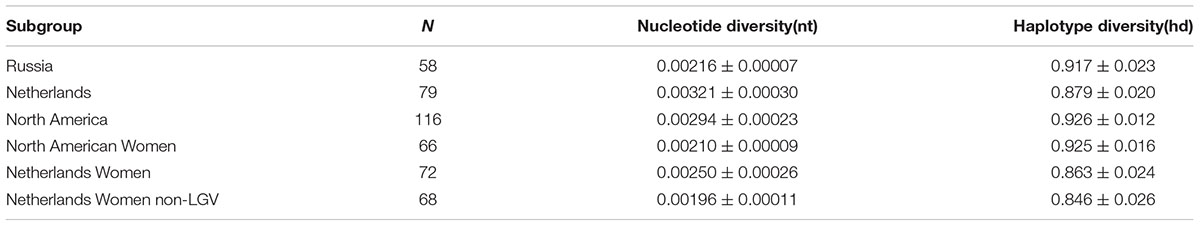
TABLE 4. Nucleotide and haplotype diversity.
Assessing population differentiation between regional subgroups by Fst revealed significant differences between most regional subgroup pairwise comparisons (Table 5). African and Asian subgroups exhibited high Fst values in all comparisons, indicating those regions were distinct from others in the dataset. This is not surprising given the small sample size and the fact that these samples are from two distinct trachoma populations. Though Western Europe was significantly different from all subgroups, the Russia and Americas subgroups were not significantly different by Fst. However, this could be attributed to the higher LGV representation in the Western European samples (15.5%) in comparison to the American samples (7.3%), and Russian samples (0%). Concordantly, when LGV samples were removed from the Western European dataset, Western Europe was no longer significantly different from Russia (Table 5). When evaluating only women without LGV from the Netherlands, Russia, and North America, the Russian women were not significantly different from the American and Netherlands women (Supplementary Table 8).
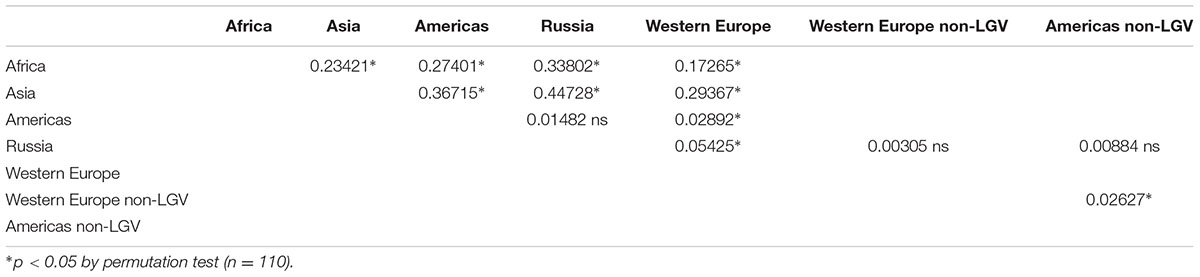
TABLE 5. Pairwise population differentiation (Fst) for regional subgroups.
Phylogenetic Relationships and Evidence for Recombination
The phylogenetic relationships were initially evaluated by eBURST, which revealed clonal clusters (CC) similar to what we reported previously (Figure 1) (Dean et al., 2009; Batteiger et al., 2014) but with the addition of an LGV cluster. These included CC-A encompassing trachoma STs, CC-B with non-invasive, non-prevalent urogenital STs, CC-C with non-invasive prevalent STs and CC-D that included LGV STs. The predicted founders were ST19, ST23, ST34, and ST39 (Supplementary Figure 1 and Table 5). The 57 singleton STs are denoted by a gray circle alone or within a colored circle, representing a specific geographic region, but were not associated with any specific region.
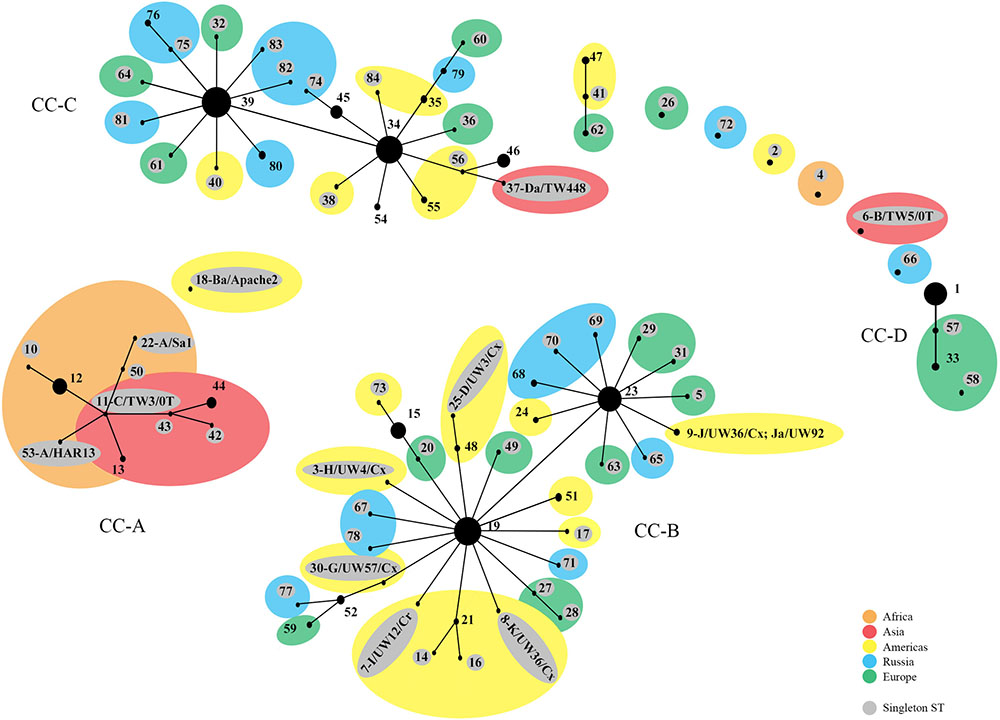
FIGURE 1. eBURST phylogeny for Chlamydia trachomatis geographic samples by ST. The numbers represent the Sequence Type (ST). STs that are linked differ at single multi-locus sequences (i.e., allelic number) and represent clonal groups. STs in the same clonal group are likely descended from the same recent ancestor, dependent on the region. Four distinct clonal clusters (CC) that correlate with disease phenotypes are shown: A, trachoma samples; B, non-prevalent urogenital samples; C, prevalent urogenital samples; and D, LGV samples. Large colored circles indicate what region multiple STs in a sub-group originated from. STs shaded gray are singleton STs. The area of the circle represents the number of samples of each ST. Orange, Africa; red, Asia; yellow, Americas; blue, Russia; green, Western Europe.
The tree (Figure 2) revealed ST branches similar to the eBURST clusters; both had branches or clusters for the disease phenotypic groups of LGV, non-invasive prevalent urogenital, and non-invasive non-prevalent urogenital STs. However, the trachoma STs formed a subgroup of the non-invasive non-prevalent urogenital branch. In addition, within disease groups, STs branched from central nodes by geographic region. These nodes contained, in general, large numbers of STs from diverse locations. For example, the founders ST19, ST23, ST34, and ST39 located at nodes in the tree contain STs from Europe, Russia, and the Americas. The amino acid tree showed a similar phylogeny (Supplementary Figure 1).
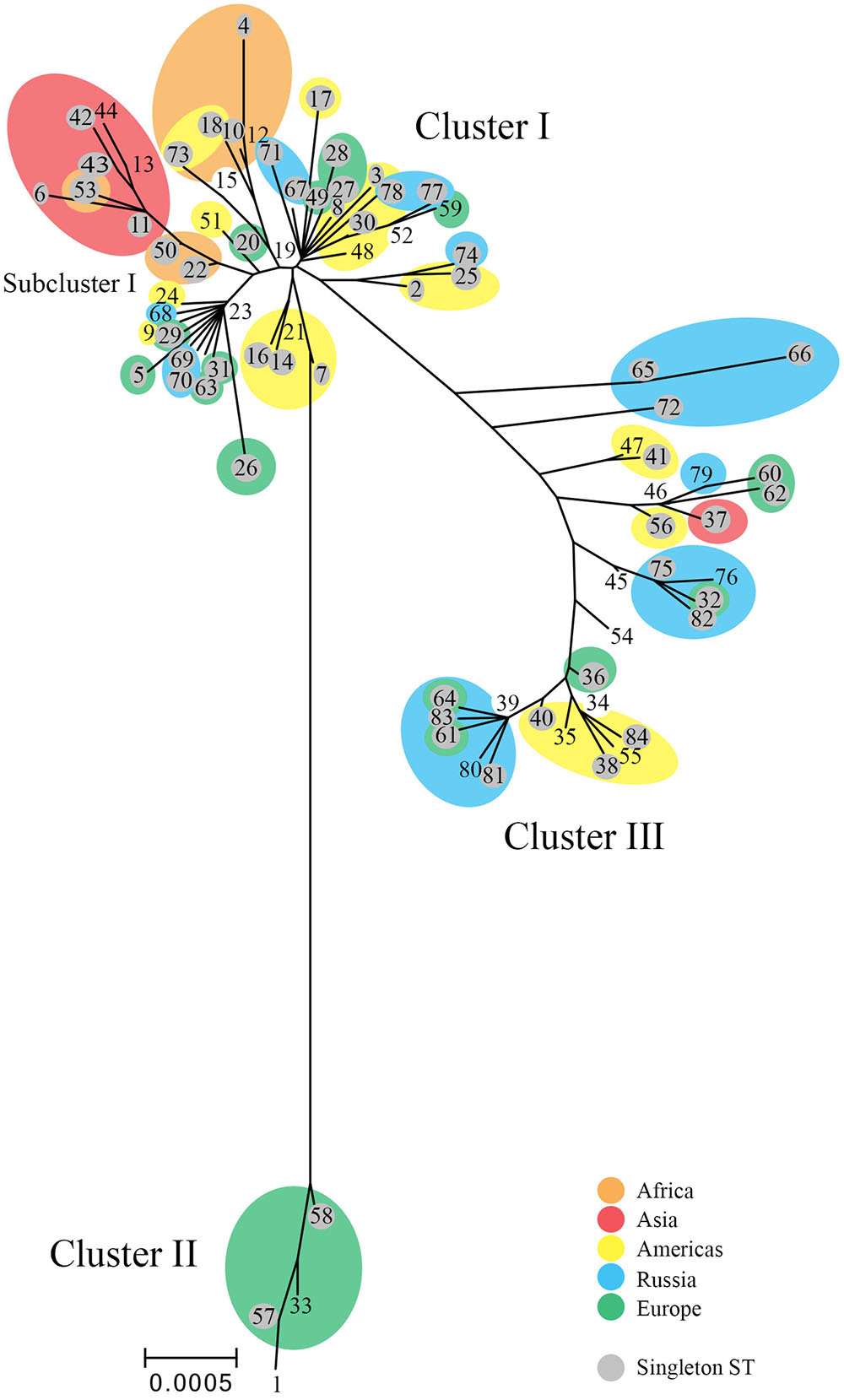
FIGURE 2. Phylogenetic tree for C. trachomatis nucleotide sequences by ST. The numbers represent the ST. The tree was constructed using the matrix of pairwise differences between the 84 ST concatenated nucleotide sequences for the seven MLST loci using the maximum composite likelihood method for estimating genetic distances. Only bootstrap values (1,000 replicates) of >70% are shown at branch points in the tree. STs (shaded gray) are singletons. Orange, Africa; red, Asia; yellow, Americas; blue, Russia; green, Western Europe. Scale bar indicates number of substitutions per site.
The Splitstree decomposition tree revealed evidence for a network structure consistent with homologous recombination (Figure 3). This was demonstrated by the interconnecting networks specifically among the ST founders ST19, ST23, ST34, and ST39 and other STs on the network in addition to the canonical evolutionary pathway shown in the tree (Figure 2). Supplementary Figure 2 shows the amino acid tree.
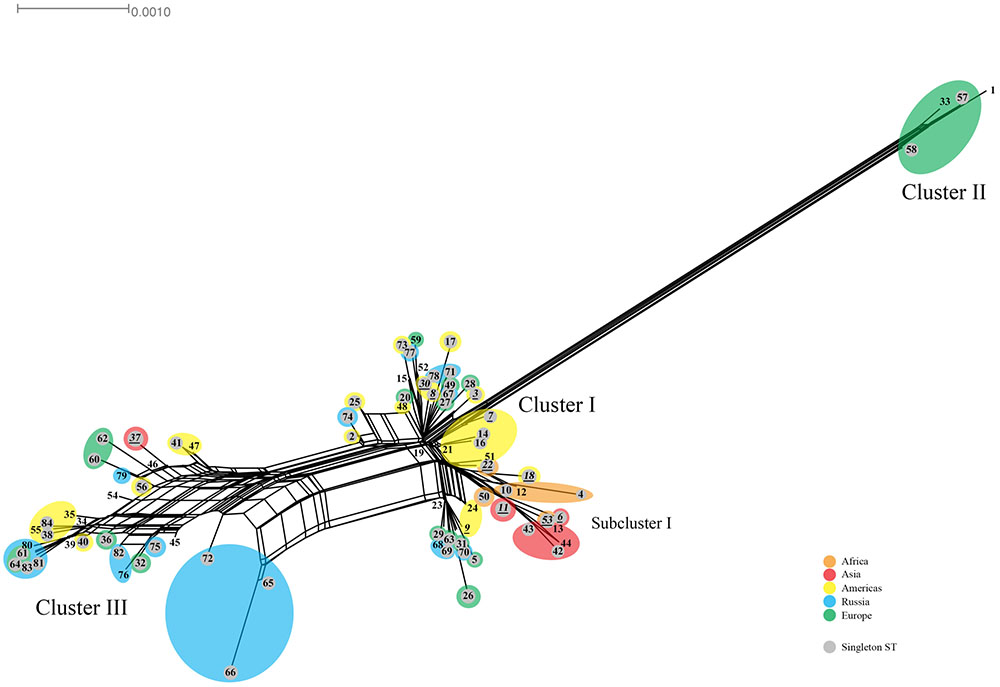
FIGURE 3. Splitstree for C. trachomatis nucleotide sequences by ST. The numbers represent the ST. The tree was obtained by using the seven loci for the 84 ST concatenated nucleotide sequences. STs (shaded gray) are singletons. Orange, Africa; red, Asia; yellow, Americas; blue, Russia; green, Western Europe. Scale bar indicates number of substitutions per site.
The Splitstree data are consistent with findings in the MLST and ompA genotyping data. Samples were classified as recombinant when the sequences of the seven genes that determine the ST were non-concordant with each other or with the seven gene sequences of the Ct genotype associated with the ompA genotype (Table 6), denoted in bold in Supplementary Table 3. There was no evidence of any recombination among the seven ST genes for any sample, although this would conceivably be possible.
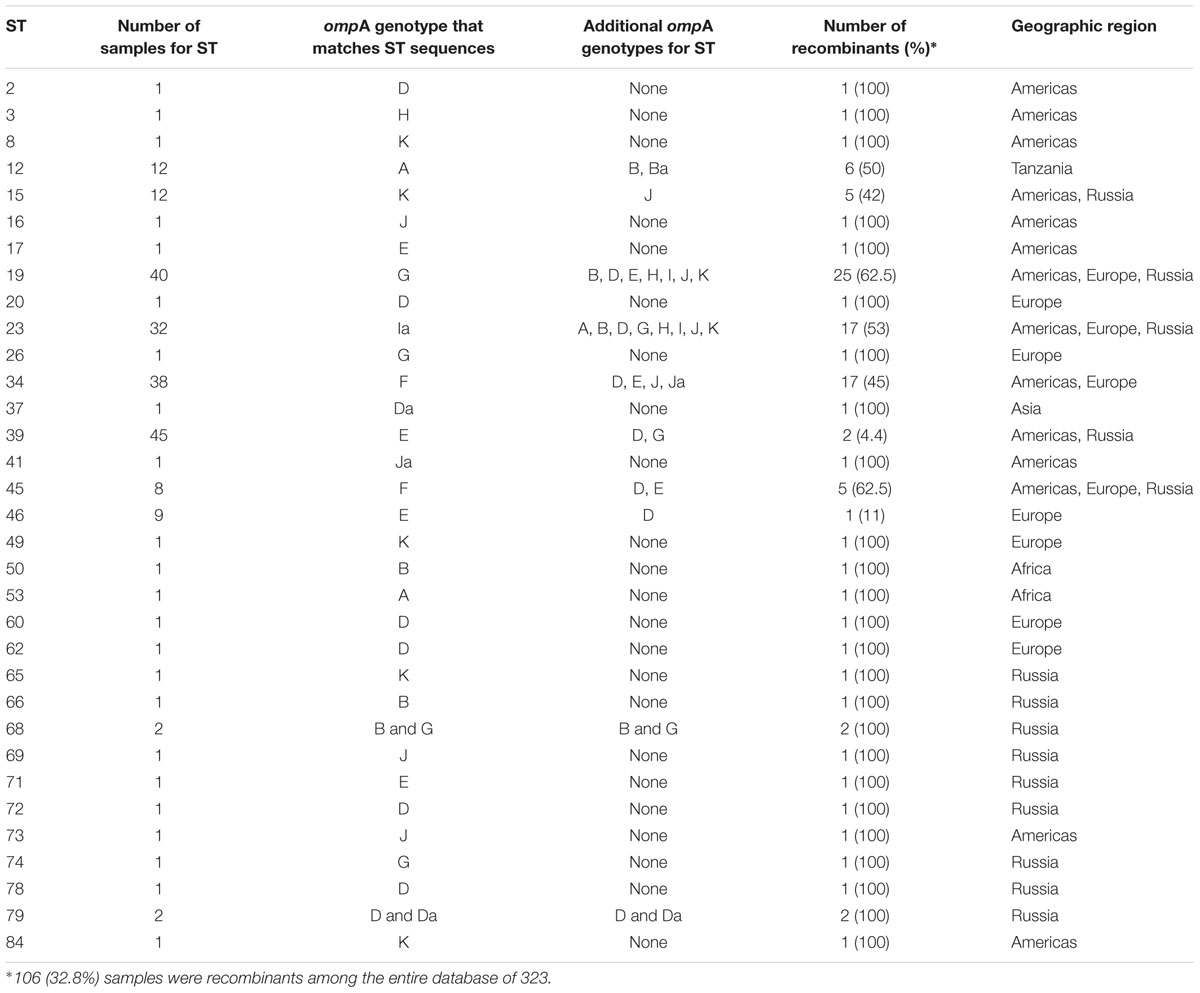
TABLE 6. Chlamydia trachomatis recombinant samples in the dataset.
A total of 106 (32.8%) samples were considered putative recombinants. Excluding Asia and Africa where the sample sizes were small, Europe had a significantly higher number of recombinants than Russia and the Americas (p < 0.04) (Table 2), where, of the 109 samples, 49 were recombinant with 43 (87.8%) from the Netherlands. While ompA genotypes were generally consistent across samples of the same ST, many cases of recombinant strains were observed. For example, ST19 (n = 40) was primarily associated with ompA genotype G (37.5%) but eight different ompA genotypes (B, D, E, G, H, I, J, K) were also associated with this ST. The majority of these samples were from St. Petersburg and Amsterdam (Supplementary Table 3). ST23 (n = 32) was also associated with eight ompA genotypes (B, D, G, H, I, Ia, J, K) but the most frequent was Ia (47%). In contrast, the most geographically prevalent ST was ST39 (n = 45) where 95% were associated with ompA genotypes E. There were also 24 singletons that were recombinants (Table 6).
Table 7 shows the ompA genotype, ST and allelic SNPs, if present, associated with each of the eight Boston samples added to the dataset. Under the column denoted ST sequence homology are the Ct ompA genotype sequences to which the sequences of the seven MLST genes are identical. For example, sample J/259b has four SNPs that are different from the sequences of the seven genes for reference and clinical J strains in the database. These SNPs are identical to the sequences of strains G and K, which suggests that this sample is a recombinant between J and G or K strains. Another example is sample D/256b; the ompA genotype is D but the MLST sequences of the seven genes are identical to reference and clinical F strains in the database. Therefore, it is a recombinant between D and F. Five (62.5%) of the eight Boston samples were putative recombinants. Supplementary Table 5 shows similar data for the 60 new samples from Amsterdam. For the Amsterdam samples, 43 (54.4%) of 79 samples were recombinants. Likewise, Supplementary Table 6 represents the data from St. Petersburg where 20 (34.5%) of 58 samples were recombinants.

TABLE 7. Evidence for recombination among the Boston, MA, United States C. trachomatis samples.
Disease Phenotypes Are Associated with Specific Single Nucleotide Polymorphisms (SNPs)
Table 8 shows the SNPs that are specific for each Haplotype and disease phenotype group. These groups include invasive urogenital disease caused by LGV strains (Haplotype 1), non-invasive urogenital disease group that includes the most prevalent worldwide strains including D, Da, E, and F (Haplotype 2), and trachoma caused by ocular A, B, Ba, and C strains (Haplotype 3). The non-invasive group includes clinical Ja strains that are recombinants of E and F strains. There are 14 SNPs that are specific for the LGV disease group, and each SNP independently identifies this group and Haplotype. The same is true for the non-invasive disease group. However, the two SNPs for the trachoma group together are specific for this group and Haplotype 3. Based on the Classification Index, the SNPs were not uniformly distributed.

TABLE 8. Single nucleotide polymorphisms (SNP) correlate with haplotype and disease phenotype.
Discussion
The evolution of straining-typing techniques for bacteria have progressed from identifying variations in gel electrophoresis patterns and melt curve analyses to sequencing single pathogen-specific genes and MLST. While typing based on WGS would be ideal, this remains out of reach given the current expense and lack, in general, of sufficient DNA from clinical samples. However, it should be mentioned that we and others have been developing techniques to enrich DNA recovery directly from urogenital and ocular patient sample types with some success (Seth-Smith et al., 2013; Joseph et al., 2014; Hadfield et al., 2017).
OmpA genotyping remains widely used among Chlamydia investigators for molecular epidemiologic and comparative studies of strains between STD and trachoma populations. However, ompA encodes for the MOMP, which is under immune selection. MLST offers a more robust typing scheme by employing 6–8 housekeeping genes as relatively immutable signatures for strain typing (Maiden et al., 1998; Dean et al., 2009) and has become an important tool for studying both the epidemiology and evolution of human pathogens (Urwin and Maiden, 2003), including Ct.
In this study, we included 58 samples from Russia and eight from Boston, regions that have not previously been represented in Ct typing schemes. The 323 reference and clinical samples resolved into 84 STs, representing a 3.23 higher typing resolution over ompA genotyping consistent with previous studies (Dean et al., 2009; Gravningen et al., 2012; Batteiger et al., 2014; Herrmann et al., 2015). The high discriminatory index D of 0.98 (95% CI 0.97–0.99) and narrow CI for our MLST scheme confirms the validity of this typing method.
We noted an overall high rate of novel STs (67.9%), which may be expected because entire populations were not sampled and the numbers are small for some areas such as Asia and Africa. For example, there were 109 samples from Europe, representing six different countries, and 19 (68%) of the 28 STs were novel. Our findings are similar to other studies in Europe where the rates for novel STs were 62% among high school students in Norway (Gravningen et al., 2012), 65% among Tunisian sex workers (Gharsallah et al., 2016) and 62% among young adults in Amsterdam (Versteeg et al., 2015). Additional novel STs are likely to be identified as new regions undergo MLST. Indeed in Russia, of the 26 STs that contained Russian samples, 18 (69%) were novel.
A significantly higher ST to sample ratio of 0.45 was identified for Russian compared to European and American samples (p < 0.009). This was not explained by the number of STs unique to a region as the percentages were relatively uniform across the geographies (Table 2). However, Russian women had the highest nucleotide diversity (Pi = 0.00216), followed by North American women (Pi = 0.00210), and Dutch women (Pi = 0.00196), excluding LGV strains that are present only in the female Dutch population and would therefore skew the data. There are no males in the Russian dataset and therefore only women were evaluated here for the three locals. However, when assessing population differentiation for women from the Netherlands, Russia, and North America, excluding those with LGV, there were no significant differences (Supplementary Table 8). A larger sample size for each group would likely provide better resolution of the data. The increased sample ratio for Russia may reflect sexual mixing among the Russian STD cohort with the introduction of STs from other regions or reassortment of existing alleles. Of the 30 alleles present in Russia, 23 (76.7%) were found in other geographic regions, supporting the geographic influx of STs. Reassortment of alleles that generate new STs is also possible given the higher frequency of novel alleles in Russia at 43.3%. It has been shown that recombinational replacements are the major contributors to clonal diversity in contrast to point mutations among bacteria (Spratt et al., 2001). In support of our hypothesis, Hadfield et al. (2017) has shown that Ct evolves within genomic ‘ecotypes’ but also outside of these niches via recombination, consistent with prior genomic studies (Harris et al., 2012; Joseph et al., 2012). However, without a larger sample size from St. Petersburg, the relative overall diversity of STs will remain unknown, and we can only speculate as to the degree of reassortment based on the alleles comprising the STs that represent only the currently sampled cohort of women in Russia.
Excluding singleton STs, STs 23 and 34 were significantly associated with female STD patients in Amsterdam. We had previously noted that ompA genotyping is valuable as a separate adjunctive typing method along with MLST as it allows comparison with strains typed only by ompA and also can provide evidence for transmission and treatment efficacy as well as putative recombination (Batteiger et al., 2014). But ompA should not be included as one of the genes in the MLST scheme because it is under immune selection (Maiden et al., 1998; Dean et al., 2009). We performed ompA genotyping on all samples in the database and, for ST23, the samples comprised Ia, B, D, G, H, I, J, and K genotypes while for ST34 they included genotypes F, D, D2, E, J, and Ja (Supplementary Table 3). The high number of ompA genotypes for these two STs suggests a high rate of recombination. For example, the sequences of the seven MLST genes matched those of reference strain Ia/UW202 for 47% of the ST23 samples where the ompA genotype was also Ia. However, the remaining samples that also matched the seven MLST Ia/UW202 sequences had B, D, G, H, I, J, and K ompA genotypes, indicating a mismatch between the MLST and ompA sequences, providing evidence for recombination (Supplementary Table 3). Similarly, STs such as 15, 19, and 34 had numerous samples, some of which were putative recombinants. These findings are supported by partial and WGSs where ompA has been shown to be involved in frequent exchange and is considered a hotspot for recombination (Gomes et al., 2007; Harris et al., 2012; Joseph et al., 2012). For example, phylogenetic analyses indicate clustering of ompA Ja genotypes, which are uncommon, with highly prevalent ompA, D, E, and F genotypes (similar to ST34) where hotspots of recombination were noted in ompA and pmpEFGH genomic regions (Gomes et al., 2007; Joseph et al., 2012). Clusters of ompA D genotypes with less prevalent strains G, Ia, and J, similar to our strains in ST23, have also been noted (Harris et al., 2012). In a recent study, ompA Ba and C trachoma strains isolated from Australian Aborigines were found to cluster with urogenital D, Da, E, and F strains with hotspots also involving ompA and pmpEFGH (Andersson et al., 2016). Of course, with WGS, many additional genes have been found to be involved in recombination (Harris et al., 2012; Joseph et al., 2012; Hadfield et al., 2017).
To confirm putative recombinants, we had described that the sequences of the seven MLST genes were individually aligned to the respective gene for all samples in the database and compared these results to the ompA genotype of the same sample. For example, Table 7 shows the five putative recombinants among the eight Boston samples. Sample D/256b had an ompA genotype of D but the seven MLST sequences were an exact match to the seven sequences of F. In some cases, one or more of the seven genes matched two different strains as was the case for samples J/253b and J/259b. Other examples of recombinants are shown in Supplementary Tables 6 and 7 for the Dutch and Russian samples, respectively. In addition, Splitstree analysis was performed and revealed ancillary evidence for a network structure consistent with homologous recombination (Figure 2). While these data confirm the recombinant nature of the samples, it is likely that there are other genetic regions that have undergone recombination and, therefore, it is not possible to determine the extent of genetic exchange unless WGS is performed (Joseph et al., 2011, 2012; Harris et al., 2012; Hadfield et al., 2017).
Overall, there were 106 (32.8%) putative recombinants (Table 2 and Supplementary Table 3), which is similar to our previous studies and those of other investigators (Dean et al., 2009; Gravningen et al., 2012; Batteiger et al., 2014). Each geographic region contained recombinants, although Europe had a significantly higher number than Russia and the Americas (p < 0.04) (Table 2). This result was skewed by the higher rate of recombinants among the Amsterdam population, which would be expected given that the samples came from individuals at high risk for STDs where sexual mixing and import of strains from tourists could increase the chances for multiple Ct strain infections and opportunities for recombination. Indeed, rates of Ct mixed infections as high as 6 to 16% have been reported among men who have sex with men and heterosexual populations, respectively, in Europe, including the Netherlands (Quint et al., 2011; Rodriguez-Dominguez et al., 2015).
The most geographically prevalent ST was 39 with 45 samples, 95% of which had an ompA genotype of E; there were only two recombinants in this ST: one from Boston with a D ompA genotype and one from St. Petersburg with a G genotype. E genotypes are known to be the most globally prevalent (Lysén et al., 2004; Millman et al., 2004; Spaargaren et al., 2004; Lee et al., 2006; Gharsallah et al., 2016) and the least recombinogenic based on whole genome sequencing (Joseph et al., 2011, 2012). In our dataset, there were 72 E genotypes with an ST to sample ratio of 0.25; only the F genotype, the 4th most prevalent genotype with 30 samples, had a lower ratio at 0.17. The lower ratios indicate greater fitness as these strains are prevalent worldwide and have fewer allelic variants that resolve into fewer STs. This is borne out by the fact that only eight (11%) of the 72 E genotypes and 0 of the 30 F genotypes were recombinants (Supplementary Table 3). A recent genome study that included 149 E strains supports our conclusions (Hadfield et al., 2017). Genotype D was also highly prevalent but had a much higher ratio at 0.48, and 29 (88%) of 33 samples were recombinants.
We previously found that phylogeny based on MLST resolved the STs along disease phenotype demarcations. As samples have been added to the database, the phenotype resolution has increased to include the LGV phenotype, denoted as clonal cluster-D (CC-D; Figure 1). Similarly, the tree shows the three main clusters with the trachoma STs as a Subcluster of Cluster I, which resembles those of other reports (Herrmann et al., 2015).
To determine whether the phenotypic groups could be more finely discriminated, we analyzed the database for SNPs that independently or together would identify a phenotype. As in our previous studies (Dean et al., 2009; Batteiger et al., 2014), specific SNPs correlated with LGV, non-invasive urogenital disease and trachoma. Haplotype 2, which included the non-invasive urogenital disease group required exclusion of strains that were recombinants, specifically D genotypes.
Author Contributions
Conceived and designed experiments: DD and VS. Wrote the paper: VS, AV, and DD. Assisted with editing the paper: EFvE, MPN, LAS, HJCdV, and SAM. Performed experiments: EFvE, MPN, TM, and BS. Analyzed data: VS, AV, EFvE, MPN, TM, BS, RW, and DD. Contributed reagents/materials/analysis tools: VS, LAS, HJCdV, JD, CE, SAM, and DD.
Funding
This work was funded in part by National Institutes of Health grants R01 AI 098843 to DD and R03 TW 007754 to DD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Payel Sen, Neda Dastgheyb, and Priyanka Manandhar for excellent technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02195/full#supplementary-material
Footnotes
- ^https://pubmlst.org/chlamydiales/
- ^http://eburst.mlst.net
- ^www.splitstree.org
- ^http://www.r-project.org
References
Andersson, P., Harris, S. R., Seth Smith, H. M., Hadfield, J., O’Neill, C., Cutcliffe, L. T., et al. (2016). Chlamydia trachomatis from Australian Aboriginal people with trachoma are polyphyletic composed of multiple distinctive lineages. Nat. Comm. 7:10688. doi: 10.1038/ncomms10688
Batteiger, B. E., Wan, R., Williams, J. A., He, L., Ma, A., Fortenberry, J. D., et al. (2014). Novel Chlamydia trachomatis strains in heterosexual sex partners, Indianapolis, Indiana, USA. Emerg. Infect. Dis. 20, 1841–1847. doi: 10.3201/2011.140604
Baud, D., Regan, L., and Greub, G. (2008). Emerging role of Chlamydia and Chlamydia-like organisms in adverse pregnancy outcomes. Curr. Opin. Infect. Dis. 21, 70–76. doi: 10.1097/QCO.0b013e3282f3e6a5
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi: 10.2307/2346101
Blas, M. M., Canchihuaman, F. A., Alva, I. E., and Hawes, S. E. (2007). Pregnancy outcomes in women infected with Chlamydia trachomatis: a population-based cohort study in Washington State. Sex. Transm. Infect. 83, 314–318. doi: 10.1136/sti.2006.022665
Borkowf, C. B. (2006). Constructing binomial confidence intervals with near nominal coverage by adding a single imaginary failure or success. Stat. Med. 25, 3679–3695. doi: 10.1002/sim.2469
Danby, C. S., Cosentino, L. A., Rabe, L. K., Priest, C. L., Damare, K. C., Macio, I. S., et al. (2016). Patterns of extragenital Chlamydia and Gonorrhea in women and men who have sex with men reporting a history of receptive anal intercourse. Sex. Transm. Dis. 43, 105–109. doi: 10.1097/OLQ.0000000000000384
Darville, T. (2005). Chlamydia trachomatis infections in neonates and young children. Semin. Pediatr. Infect. Dis. 16, 235–244. doi: 10.1053/j.spid.2005.06.004
Dean, D. (2010). “Pathogenesis of chlamydial ocular infections,” in Duane’s Foundations of Clinical Ophthalmology, eds W. Tasman and E. A. Jaeger (Philadelphia, PA: Lippincott Williams & Wilkins), 678–702. “Chapter 77.
Dean, D., Bruno, W. J., Wan, R., Gomes, J. P., Devignot, S., Mehari, T., et al. (2009). Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg. Infect. Dis. 15, 1385–1394. doi: 10.3201/eid1509.090272
Dean, D., Kandel, R. P., Adhikari, H. K., and Hessel, T. (2008). Multiple Chlamydiaceae species in trachoma: implications for disease pathogenesis and control. PLOS Med. 5:e14. doi: 10.1371/journal.pmed.0050014
Dean, D., and Millman, K. (1997). Molecular and mutation trends analyses of omp1 alleles for serovar E of Chlamydia trachomatis. Implications for the immunopathogenesis of disease. J. Clin. Invest. 99, 475–483. doi: 10.1172/JCI119182
Dean, D., Schachter, J., Dawson, C. R., and Stephens, R. S. (1992). Comparison of the major outer membrane protein variant sequence regions of B/Ba isolates: a molecular epidemiologic approach to Chlamydia trachomatis infections. J. Infect. Dis. 166, 383–392. doi: 10.1093/infdis/166.2.383
de Waaij, D. J., Dubbink, J. H., Peters, R. P., Ouburg, S., and Morré, S. A. (2015). Comparison of GMT presto assay and Roche cobas 4800 CT/NG assay for detection of Chlamydia trachomatis and Neisseria gonorrhoeae in dry swabs. J. Microbiol. Methods 118, 70–74. doi: 10.1016/j.mimet.2015.08.020
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Gharsallah, H., Bom, R. J. M., Bruisten, S. M., Himschoot, M., Frikha-Gargouri, O., and Hammami, A. (2016). Identification of a dominant Chlamydia trachomatis strain in patients attending sexual transmitted infection clinic and female sex workers in Tunisia using a high resolution typing method. Infect. Genet. Evol. 44, 444–449. doi: 10.1016/j.meegid.2016.08.002
Gomes, J. P., Bruno, W. J., Borrego, M. J., and Dean, D. (2004). Recombination in the genome of Chlamydia trachomatis involving the polymorphic membrane protein C relative to ompA and evidence for horizontal gene transfer. J. Bacteriol. 186, 4295–4306. doi: 10.1128/JB.186.13.4295-4306.2004
Gomes, J. P., Bruno, W. J., Borrego, M. J., Nunes, A., Florindo, C., and Dean, D. (2006). Polymorphisms in the nine polymorphic membrane proteins of Chlamydia trachomatis across all serovars: evidence for Da recombination and correlation with tissue tropism. J. Bacteriol. 188, 275–286. doi: 10.1128/JB.188.1.275-286.2006
Gomes, J. P., Bruno, W. J., Nunes, A., Santos, N., Florindo, C., Borrego, M. J., et al. (2007). Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 17, 50–60. doi: 10.1101/gr.5674706
Götz, H. M., Bom, R. J. M., Wolfers, M. E. G., Fennema, J., van den Broek, I. V. F., Speksnijder, A. G. C. L., et al. (2013). Use of Chlamydia trachomatis high-resolution typing: an extended case study to distinguish recurrent or persistent infection from new infection. Sex. Transm. Infect. 90, 155–160. doi: 10.1136/sextrans-2013-051218
Gravningen, K., Christerson, L., Furberg, A. S., Simonsen, G. S., Ödman, K., Ståhlsten, A., et al. (2012). Multilocus sequence typing of genital Chlamydia trachomatis in Norway reveals multiple new sequence types and a large genetic diversity. PLOS ONE 7:e34452. doi: 10.1371/journal.pone.0034452
Hadfield, J., Harris, S. R., Seth-Smith, H. M. B., Parmar, S., Andersson, P., Giffard, P. M., et al. (2017). Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing. Genome Res. 7, 1220–1229. doi: 10.1101/gr.212647.116
Harris, S. R., Clarke, I. N., Seth-Smith, H. M., Solomon, A. W., Cutcliffe, L. T., Marsh, P., et al. (2012). Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 44, 413–419, S1. doi: 10.1038/ng.2214
Herrmann, B., Isaksson, J., Ryberg, M., Tångrot, J., Saleh, I., Versteeg, B., et al. (2015). Global multilocus sequence type analysis of Chlamydia trachomatis strains from 16 countries. J. Clin. Microbiol. 53, 2172–2179. doi: 10.1128/JCM.00249-15
Horn, M., Collingro, A., Schmitz-Esser, S., Beier, C. L., Purkhold, U., Fartmann, B., et al. (2004). Illuminating the evolutionary history of chlamydiae. Science 304, 728–730. doi: 10.1126/science.1096330
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466.
Isaksson, J., Gallo Vaulet, L., Christerson, L., Ruettger, A., Sachse, K., Entrocassi, C., et al. (2016). Comparison of multilocus sequence typing and multilocus typing microarray of Chlamydia trachomatis strains from Argentina and Chile. J. Microbiol. Methods 127, 214–218. doi: 10.1016/j.mimet.2016.06.005
Joseph, S. J., Didelot, X., Gandhi, K., Dean, D., and Read, T. D. (2011). Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol. Direct 6:28. doi: 10.1186/1745-6150-6-28
Joseph, S. J., Didelot, X., Rothschild, J., de Vries, H. J. C., Morré, S. A., Read, T. D., et al. (2012). Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol. Biol. Evol. 29, 3933–3946. doi: 10.1093/molbev/mss198
Joseph, S. J., Li, B., Ghonasgi, T., Qin, Z. C., Dean, D., and Read, T. D. (2014). Direct amplification, sequencing and profiling of Chlamydia trachomatis strains in single and mixed infection clinical samples. PLOS ONE 9:e99290. doi: 10.1371/journal.pone.0109670
Klint, M., Fuxelius, H. H., Goldkuhl, R. R., Skarin, H., Rutemark, C., Andersson, S. G. E., et al. (2007). High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J. Clin. Microbiol. 45, 1410–1414. doi: 10.1128/JCM.02301-06
Labiran, C., Marsh, P., Zhou, J., Bannister, A., Clarke, I. N., Goubet, S., et al. (2016). Highly diverse MLVA-ompA genotypes of rectal Chlamydia trachomatis among men who have sex with men in Brighton, UK and evidence for an HIV-related sexual network. Sex. Transm. Infect. 92, 299–304. doi: 10.1136/sextrans-2015-052261
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lee, G., Park, J., Kim, B., Kim, S.-A., Yoo, C.-K., and Seong, W. K. (2006). OmpA genotyping of Chlamydia trachomatis from Korean female sex workers. J. Infect. 52, 451–454. doi: 10.1016/j.jinf.2005.08.019
Lysén, M., Osterlund, A., Rubin, C.-J., Persson, T., Persson, I., and Herrmann, B. (2004). Characterization of ompA genotypes by sequence analysis of DNA from all detected cases of Chlamydia trachomatis infections during 1 year of contact tracing in a Swedish County. J. Clin. Microbiol. 42, 1641–1647. doi: 10.1128/JCM.42.4.1641-1647.2004
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Mårdh, P.-A. (2004). Tubal factor infertility, with special regard to chlamydial salpingitis. Curr. Opin. Infect. Dis. 17, 49–52. doi: 10.1097/00001432-200402000-00010
Millman, K., Black, C. M., Johnson, R. E., Stamm, W. E., Jones, R. B., Hook, E. W., et al. (2004). Population-based genetic and evolutionary analysis of Chlamydia trachomatis urogenital strain variation in the United States. J. Bacteriol. 186, 2457–2465. doi: 10.1128/JB.186.8.2457-2465.2004
Millman, K., Tavere, S., and Dean, D. (2001). Recombination in the ompA gene but not the omcB gene of Chlamydia contribute to serovar specific differences in tissue tropism, immune surveillance and persistence of the organism. J. Bacteriol. 183, 5997–6008. doi: 10.1128/JB.183.20.5997-6008.2001
Morré, S. A., Van Valkengoed, I. G., Moes, R. M., Boeke, A. J., Meijer, C. J., and Van den Brule, A. J. (1999). Determination of Chlamydia trachomatis prevalence in an asymptomatic screening population: performances of the LCx and COBAS Amplicor tests with urine specimens. J. Clin. Microbiol. 37, 3092–3096.
Pannekoek, Y., Morelli, G., Kusecek, B., Morré, S. A., Ossewaarde, J. M., Langerak, A. A., et al. (2008). Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 8:42. doi: 10.1186/1471-2180-8-42
Perkins, E., and Hill, D. (eds) (2013). Scientific Foundations of Ophthalmology. Oxford: Butterworth-Heinemann.
Peuchant, O., Touati, A., Sperandio, C., Hénin, N., Laurier-Nadalié, C., Bébéar, C., et al. (2016). Changing pattern of Chlamydia trachomatis strains in Lymphogranuloma Venereum outbreak, France, 2010-2015. Emerg. Infect. Dis. 22, 1945–1947. doi: 10.3201/eid2211.160247
Quint, K. D., Bom, R. J., Quint, W. G. V., Bruisten, S. M., van der Loeff, M. F. S., Morré, S. A., et al. (2011). Anal infections with concomitant Chlamydia trachomatis genotypes among men who have sex with men in Amsterdam, the Netherlands. BMC Infect. Dis. 11:63. doi: 10.1186/1471-2334-11-63
Rodriguez-Dominguez, M., Gonzalez-Alba, J. M., Puerta, T., Menendez, B., Sanchez-Diaz, A. M., Canton, R., et al. (2015). High prevalence of co-infections by invasive and non-invasive Chlamydia trachomatis genotypes during the Lymphogranuloma Venereum outbreak in Spain. PLOS ONE 10:e0126145. doi: 10.1371/journal.pone.0126145
Rowley, J., Toskin, I., Ndowa, F., World Health Organization, and Reproductive Health and Research. (2012). Global Incidence and Prevalence of Selected Curable Sexually Transmitted Infections, 2008. Geneva: World Health Organization.
Schliep, K. P. (2010). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Schillinger, J. A., Katz, B. P., Markowitz, L. E., Braslins, P. G., Shrier, L. A., Madico, G., et al. (2016). Genotype-specific concordance of Chlamydia trachomatis genital infection within heterosexual partnerships. Sex. Transm. Dis. 43, 741–749. doi: 10.1097/OLQ.0000000000000525
Sethi, G., Allason-Jones, E., Richens, J., Annan, N. T., Hawkins, D., Ekbote, A., et al. (2009). Lymphogranuloma venereum presenting as genital ulceration and inguinal syndrome in men who have sex with men in London, UK. Sex. Transm. Infect. 85, 165–170. doi: 10.1136/sti.2008.034348
Seth-Smith, H. M., Harris, S. R., Skilton, R. J., Radebe, F. M., Golparian, D., Shipitsyna, E., et al. (2013). Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 5, 855–866. doi: 10.1101/gr.150037.112
Shalepo, K., Savicheva, A., Shipitsyna, E., Unemo, M., and Domeika, M. (2006). Diagnosis of Chlamydia trachomatis in Russia–in-house PCR assays may be effective but overall optimization and quality assurance are urgently needed. APMIS Acta Pathol. Microbiol. Immunol. Scand. 114, 500–507. doi: 10.1111/j.1600-0463.2006.apm_443.x
Shipitsyna, E., Shalepo, K., Savicheva, A., Unemo, M., and Domeika, M. (2007). Pooling samples: the key to sensitive, specific and cost-effective genetic diagnosis of Chlamydia trachomatis in low-resource countries. Acta Derm. Venereol. 87, 140–143. doi: 10.2340/00015555-0196
Smelov, V., Quint, K. D., Pleijster, J., Savelkoul, P. H. M., Shalepo, K., Shipitsyna, E., et al. (2009). Chlamydia trachomatis serovar distributions in Russian men and women: a comparison with Dutch serovar distributions. Drugs Today 45, Suppl B:33–38.
Spaargaren, J., Verhaest, I., Mooij, S., Smit, C., Fennema, H. S. A., Coutinho, R. A., et al. (2004). Analysis of Chlamydia trachomatis serovar distribution changes in the Netherlands (1986-2002). Sex. Transm. Infect. 80, 151–152. doi: 10.1136/sti.2003.006395
Spratt, B. G., Hanage, W. P., and Feil, E. J. (2001). The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr. Opin. Microbiol. 4, 602–606. doi: 10.1016/S1369-5274(00)00257-5
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Urwin, R., and Maiden, M. C. J. (2003). Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11, 479–487. doi: 10.1016/j.tim.2003.08.006
van Belkum, A., Tassios, P. T., Dijkshoorn, L., Haeggman, S., Cookson, B., Fry, N. K., et al. (2007). Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect. 13(Suppl. 3), 1–46. doi: 10.1111/j.1469-0691.2007.01786.x
Versteeg, B., Himschoot, M., van den Broek, I. V. F., Bom, R. J. M., Speksnijder, A. G. C. L., van der Loeff, M. F., et al. (2015). Urogenital Chlamydia trachomatis strain types, defined by high-resolution multilocus sequence typing, in relation to ethnicity and urogenital symptoms among a young screening population in Amsterdam, The Netherlands. Sex. Transm. Infect. 91, 415–422. doi: 10.1136/sextrans-2014051790
WHO Sexually transmitted infections [STIs] (2015). Available at: http://www.who.int/mediacentre/factsheets/fs110/en/ [Accessed June 5, 2015].
Keywords: Chlamydia trachomatis, MLST, recombination, global diversification, allele mixing, reassortment
Citation: Smelov V, Vrbanac A, van Ess EF, Noz MP, Wan R, Eklund C, Morgan T, Shrier LA, Sanders B, Dillner J, de Vries HJC, Morre SA and Dean D (2017) Chlamydia trachomatis Strain Types Have Diversified Regionally and Globally with Evidence for Recombination across Geographic Divides. Front. Microbiol. 8:2195. doi: 10.3389/fmicb.2017.02195
Received: 27 July 2017; Accepted: 25 October 2017;
Published: 13 November 2017.
Edited by:
Jorge Blanco, Universidade de Santiago de Compostela, SpainReviewed by:
Wilhelmina May Huston, University of Technology Sydney, AustraliaFernando Gonzalez-Candelas, Universitat de València, Spain
Carolyn Black, National Center for Emerging and Zoonotic Infectious Diseases (CDC), United States
Copyright © 2017 Smelov, Vrbanac, van Ess, Noz, Wan, Eklund, Morgan, Shrier, Sanders, Dillner, de Vries, Morre and Dean. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah Dean, ddean@chori.org
†Present address: Alison Vrbanac, Center for Immunity, Infection and Inflammation, University of California, San Diego, San Diego, CA, United States
‡These authors have contributed equally to this work.