 Ya Zhang
Ya Zhang Masaaki Kitajima2
Masaaki Kitajima2 Wen-Tso Liu
Wen-Tso Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 October 2017
Sec. Microbiotechnology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.02036
This article is part of the Research Topic Microbial Safety in Water Resources View all 10 articles
The occurrence of pathogenic bacteria in drinking water distribution systems (DWDSs) is a major health concern, and our current understanding is mostly related to pathogenic species such as Legionella pneumophila and Mycobacterium avium but not to bacterial species closely related to them. In this study, genomic-based approaches were used to characterize pathogen-related species in relation to their abundance, diversity, potential pathogenicity, genetic exchange, and distribution across an urban drinking water system. Nine draft genomes recovered from 10 metagenomes were identified as Legionella (4 draft genomes), Mycobacterium (3 draft genomes), Parachlamydia (1 draft genome), and Leptospira (1 draft genome). The pathogenicity potential of these genomes was examined by the presence/absence of virulence machinery, including genes belonging to Type III, IV, and VII secretion systems and their effectors. Several virulence factors known to pathogenic species were detected with these retrieved draft genomes except the Leptospira-related genome. Identical clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins (CRISPR-Cas) genetic signatures were observed in two draft genomes recovered at different stages of the studied system, suggesting that the spacers in CRISPR-Cas could potentially be used as a biomarker in the monitoring of Legionella related strains at an evolutionary scale of several years across different drinking water production and distribution systems. Overall, metagenomics approach was an effective and complementary tool of culturing techniques to gain insights into the pathogenic characteristics and the CRISPR-Cas signatures of pathogen-related species in DWDSs.
Over 500 waterborne or water-based pathogens of potential concern in drinking water (e.g., Legionella pneumophila, Escherichia coli O157:H7, Mycobacterium avium, and Cryptosporidium parvum) have been included in the Candidate Contaminant List by the US Environmental Protection Agency (EPA; Ashbolt, 2015). The traditional approach to identify these pathogens is through cultivation and then biochemical/serological tests or 16S rRNA gene-based phylogeny analysis (Lye and Dufour, 1993; Edberg et al., 1996; Stelma et al., 2004). However, identifying pathogens at species level does not always translate into health risks as some strains of the same species are more pathogenic than others (Schmidt and Schaechter, 2012).
Alternatively, comparative genomic analysis has become an effective way to evaluate the pathogenicity potential. It is reported that pathogens infect host through a multi-step process from entering the host, adhering to host tissues, penetrating or evading host defenses, damaging host tissues, to exiting the host. As a result, various virulence factors (VFs) are required for pathogenic species during the infection process, which can be divided into several general groups based on the conservation of similar mechanisms, such as adhesins, invasins, toxins, protein secretion systems, and antibiotic resistance mechanisms (Finlay and Falkow, 1997; Wilson et al., 2002). Thus, the presence of a set of virulence machinery in a bacterial genome has been used to define pathogenic subpopulations (Chapman et al., 2006; Cazalet et al., 2008; Bouzid et al., 2013; Foley et al., 2013; Picardeau, 2017). The knowledge on virulence machinery and the functions of key VFs in the literature have facilitated the usage of virulence machinery to evaluate health risks associated with pathogens in drinking water distribution systems (DWDSs; Wu et al., 2008; Huang et al., 2014). Secretion systems are essential for the transportation of proteins (i.e., effectors) from the cytoplasm into host cells or host environments to enhance attachment to eukaryotic cells, scavenge resources in an environmental niche, and disrupt target cell functions (Green and Mecsas, 2016). Some secretion systems are dedicated for bacteria-host interaction, such as the type III secretion system (T3SS) in Chlamydia (Betts-Hampikian and Fields, 2010), the type IVB secretion system (T4BSS, Dot/Icm) in Lg. pneumophila (Voth et al., 2012), and the type VII secretion system (T7SS) in Mycobacterium (Costa et al., 2015). The deletion of these secretion systems could result in a substantial decrease in virulence (Costa et al., 2015). In addition, several other VFs have also been reported for pathogens including those facilitating attachment and invasion (e.g., cell wall, type IV pili) and endotoxins (i.e., lipopolysaccharides, LPS; Schroeder et al., 2010; Favrot et al., 2013; Tortora et al., 2013.
While the identification of pathogens of potential concern in DWDSs is an important task, recent studies have often detected pathogens simultaneously together with their closely related species, which are often present at higher abundance. These include, for example, Lg. pneumophila-related species such as Lg. dumoffii (Hsu et al., 1984), Lg. sainthelensis (Rodriguez-Martinez et al., 2015), and Lg. jordanis (Hsu et al., 1984; Kao et al., 2014), and M. avium-related species such as Mycobacterium gordonae (Falkinham et al., 2001; Lalande et al., 2001; Vaerewijck et al., 2005), Mycobacterium immunogenum (Gomez-Alvarez and Revetta, 2016a), and Mycobacterium chelonae (Gomez-Alvarez and Revetta, 2016b). Some of these species have been associated with illness and infections in clinical environments, including Lg. dumoffii (Yu et al., 2002), M. gordonae (Lalande et al., 2001), M. immunogenum (Wilson et al., 2001), and M. chelonae (Lowry et al., 1990). As pathogens and their closely related species often share ecological niches (predominantly in biofilms), genetic exchange through conjugation and transformation occurs between the two groups, sometimes involving VFs (Gimenez et al., 2011; Gomez-Valero et al., 2011). However, it is not clear whether they possess similar VFs as observed in pathogens.
Furthermore, in DWDSs, pathogens and their closely related species mostly reside within biofilms where protozoa predation and viral lysis occur more frequently than bulk water, and have developed mechanisms to resist predation by inhibiting phagosome acidification and lysosome fusion of protozoa (Hilbi et al., 2001; Tilney et al., 2001). Phage DNA can be integrated into bacterial genomes by horizontal gene transfer as prophages, which are major contributors to differences among individuals within a bacterial species (Bobay et al., 2014). To protect bacteria from phage lysis, encountered foreign DNA fragments can be integrated into a clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins (CRISPR-Cas) locus as spacers (Makarova et al., 2015). Through addition of spacers at one end of the CRISPR array and conservation of spacers at the other end (the leader distal end), the CRISPR-Cas system participates in a constant evolutionary battle between phages and bacteria (Deveau et al., 2010; Sun et al., 2016). This mechanism has been used as a vital tool for strain typing in epidemiology for the recognition of outbreaks and identification of infection sources (Horvath et al., 2008; Shariat and Dudley, 2014). Nevertheless, it is not clear how intracellular growth and phage integration might impact the genomic composition and virulence of pathogen-related species.
In this study, metagenomics analysis instead of cultivation based methods was carried out to investigate virulence machinery and genomic signatures as the result of phage integration in pathogens-related species in a drinking water production and distribution system. A groundwater-derived drinking water system studied previously (Ling et al., 2016; Zhang et al., 2017) was used as a model system. It consists of abstraction, softening, recarbonation, disinfection, filtration, and final distribution with a disinfectant residual (free chlorine). Samples of microbial biomass from 10 locations of the water production process and the distribution system were collected and community metagenomes sequenced (Zhang et al., 2017). Coupling digital droplet PCR (ddPCR) with metagenomics, draft genomes affiliated with known pathogen genera were recovered to reveal their abundance, diversity, potential pathogenicity, genetic exchange, and distribution across an urban drinking water system.
Microbial biomass samples from different stages of the treatment processes and different locations in the distribution system were collected from a groundwater-sourced drinking water system. Detailed description of the studied drinking water system can be found in a previous study (Zhang et al., 2017) and in Figure S1. Briefly, these samples were from raw water (RW), immediately before filtration and chlorination (BC), finished water (FW) prior to distribution, three taps (DS1-DS3), two retired water mains (PB1-PB2), 14 household water meters (WM, combined into one sample), and five premise plumbing pipe reactors (PR, combined into one sample). The three tap water sampling sites (DS1-3) were located approximately one mile apart from each other to represent different locations within the DWDS. For water-phase samples (including RW, BC, FW, and DS1-3), a 10-min flushing (the cold-water side) was carried out before each sampling event to minimize the influence of premise plumbing before installing point-of-use water purifiers (Toray Industries Inc. Japan). Approximately 2,000 L of water was filtered during each sampling event at each site over a time span of 48 hrs. Water purifiers were collected at the end of each sampling event and transported to the laboratory in cools (the Department of Civil and Environmental Engineering, University of Illinois at Urbana-Champaign). They were disassembled after arriving at the laboratory and cells were washed off from the multilayer hollow fiber membrane with phosphate-buffered saline (PBS) through sonication (Symphony™ Ultrasonic Cleaners, VWR). The obtained mixture was filtered through 0.22 μm membranes and the membranes with cells were stored at −80°C. To obtain a better representation of the average composition, water-phase sampling was repeated four times, in June, July, August, and September 2014, except the BC sample due to membrane blockage (Zhang et al., 2017).
For biofilm samples, PB1 was a 2.25-inch cast iron water main installed in 1968 and PB2 was a 1.5-inch cast iron water main installed prior to 1927. Each pipe was cut into two 12-inch long pieces on site with an effort to minimize contamination. Additionally, 14 water meters were obtained through the local drinking water plant. For the PR sample, five galvanized pipes of the plumbing system of a dormitory were obtained within the service area of the studied system, which were installed before World War II (size = 2 inch, OD = 2.375 inch, ID = 2.067 inch, length = 14 feet). Detailed description and handling of these samples could be found in our previous study (Zhang et al., 2017). The biofilm samples were swabbed off the surfaces, re-suspended in PBS, and collected by filtering through 0.22 μm membranes. All the membranes with cells were stored at −80°C. Genomic DNA (gDNA) was extracted using FastDNA® SPIN Kit for Soil (MP Biomedicals, Carlsbad, CA, USA) from these membranes with cells following manufacturer's protocol with an elution volume of 50 μl. The effect of different DNA extraction methods on the quantity and quality of DNA yields from drinking water biofilms had been evaluated and published in a previous study (Hwang et al., 2012).
ddPCR was used to quantify total Bacteria and Archaea 16S rRNA genes and pathogens of potential concern, including Mycobacterium spp., M. tuberculosis complex, Legionella spp., Lg. pneumophila, Pseudomonas aeruginosa, and Aeromonas hydrophila, in the combined samples submitted for metagenomic sequencing, except DS1 and DS3 due to not enough gDNA. TaqMan-based ddPCR assays using primer/probe sets specific to each target (Table S1) were performed with a QX200™ Droplet Digital™ PCR System using ddPCR™ Supermix for Probes (Bio-Rad, Pleasanton, CA, USA). In addition, three eukaryotic groups (amoebae), Naegleria fowleri, Acanthamoeba spp., and Balamuthia madrillaris, were tested with TaqMan-based real-time PCR assays using primer/probe sets specific to internal transcribed spacer (ITS)/18S rRNA gene of each target (Table S1). Real-time PCR was performed with a CFX96™ Real-Time PCR Detection System using SsoAdvanced™ Universal Probes Supermix (Bio-Rad, Pleasanton, CA, USA). Because of the large variations in the number of ITS/18S rRNA genes in different eukaryotic species, only cycle threshold (CT) values were reported. Positive control (standard plasmid DNA) and negative control (H2O) were included in every ddPCR and real-time PCR reaction to ensure the successful amplification and the absence of contamination, respectively.
16S rRNA gene amplicon analysis was carried out using a universal primer set targeting the V4-V5 hypervariable regions of both the Bacteria and Archaea domains (515F: 5′-GTGCCAGCMGCCGCGGTAA-3′ and 909R: 5′-CCCGTCAATTCMTTTRAGT-3′) using the Illumina Miseq platform with dual indexing strategy as described in a previous study (Zhang et al., 2017). DNA libraries for metagenomic sequencing were prepared by combining all the extracted gDNA from each sampling site due to the requirement of a relatively large amount of gDNA (>0.1 μg). The prepared library was paired-end sequenced on Illumina HiSeq2500 platforms (Illumina, Inc., San Diego, CA, USA) as described previously (Zhang et al., 2017).
The obtained paired-end 16S rRNA gene sequences were aligned with Mothur (Kozich et al., 2013). The resulting sequences were screened for chimeras by the UCHIME algorithm implemented in USEARCH 6.1 and processed using the de novo OTU picking workflow in QIIME as described previously (Zhang et al., 2017). EMIRGE was used to reconstruct nearly full-length SSU genes in metagenomes (Miller, 2013).
Draft genomes are presented as a set of sequence fragments or contigs, which are the most common form of genome assemblies obtained using metagenomics sequencing binning pipelines and account for two thirds of the bacterial genomes available in the GenBank database (Nagarajan et al., 2010; Edwards and Holt, 2013). Figure 1 illustrates the workflow of draft genome recovery used in this study. All the metagenomic datasets were trimmed using SolexaQA2 based on a cutoff of 20 by phred scores (Cox et al., 2010) and assembled using Megahit (Li et al., 2015). High-quality contigs (~2.0 × 108 bp for each metagenome) were obtained at this step, to which >85.0% of the raw reads could be mapped except the RW sample. The longest contig in each metagenome was >4.0 × 105 bp. More details of the assemblies could be found in our previous study (Zhang et al., 2017). The obtained contigs were binned based on metagenomics read coverage, tetranucleotide frequency, and the occurrence of unique marker genes by using both MaxBin 2.0 (Wu et al., 2016) and MetaBAT (Kang et al., 2015) to minimize the contamination of each bin. These two binning methods employed different clustering methods for the determination of different bins: MaxBin compares the distributions of distances between and within the same bins whereas MetaBAT clusters contigs iteratively by modified K-methods algorithm. Bins of pathogen-related species from the two binning tools were compared and assessed with CheckM (Parks et al., 2015) and ProDeGe (Tennessen et al., 2016), followed by manual curation. The curated bins with ≥90% completeness and ≥15-fold coverage were finalized as draft genomes. Details of each step in the pipeline had been reviewed and summarized by Sangwan et al. (2016) and a step-by-step tutorial of the workflow supplied with a sample dataset had been available by Edwards and Holt (2013). Percentages of reads mapped over the refined genome bins were estimated by Burrow-Wheeler Aligner-mem (Li and Durbin, 2009). The entire workflow was computed on a high-performance workstation (DELL precision T7600) equipped with 136 GB memory.
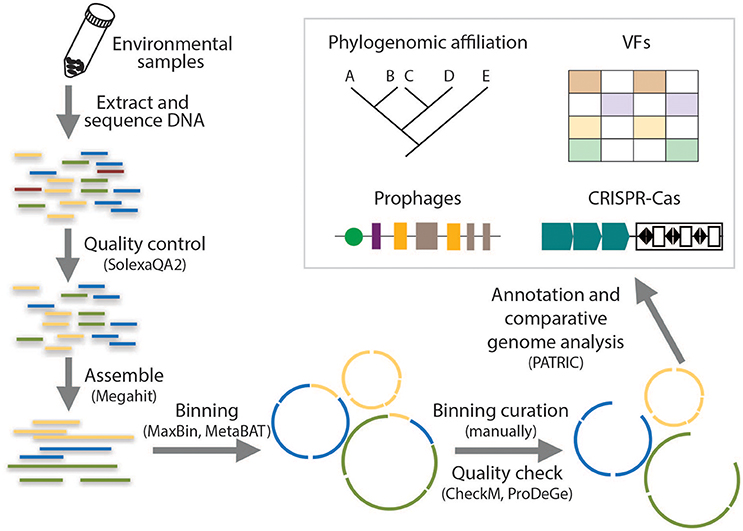
Figure 1. Flowing chart on the analysis of metagenomic data for the reconstruction of draft genomes and the identification of genetic signatures.
Draft genomes of pathogen-related species retrieved were uploaded into PATRIC for annotation and feature identification (Wattam et al., 2014). VFs of different pathogens were collected from the literature and the VF database (VFDB, http://www.mgc.ac.cn/VFs/; Chen et al., 2012). Reported virulence genes within Lg. pneumophila included: the type II secretion system (T2SS, Lsp) for growth at low temperatures (Soderberg et al., 2008); the T4ASS (Lvh, F-type, and P-type) associated with conjugal DNA transfer and potentially in virulence (Gomez-Valero et al., 2011); the T4BSS (Dot/Icm) translocating several hundred effector proteins to support intracellular growth (Burstein et al., 2016); T4BSS-type effectors such as ralF, lidA, sdhA, and lepAB genes (Newton et al., 2010); type IV pili (pilB,C,D) involving in the entry to host cells, biofilm development, formation, type II protein secretion, and horizontal gene transfer (Schroeder et al., 2010); LPS transport (Lpt) proteins; and mip (macrophage infectivity potentiator) gene associated with the ability of Lg. pneumophila to replicate in eukaryotic cells (Newton et al., 2010).
For M. tuberculosis, the reported VFs included: the T7SS, also known as the ESX pathway (ESX-1 to ESX-5) to secrete proteins across their complex cell envelope (Houben et al., 2014); early secretory antigenic target (ESAT6), esxA, H, and N; culture filtrate protein-10 kDa (CFP-10), esxB, G, and M (Li et al., 2005); pe/ppe genes unique to mycobacteria and abundant in pathogenic mycobacteria (Sampson, 2011); antigen 85 (ag85) complex and mycolic acid cyclopropane synthase (pcaA) required for the biosynthesis of major components of the cell envelope (Favrot et al., 2013); adhesin (hbhA); phospholipase C (plcC); and oxidative stress reducer (ahpC; Forrellad et al., 2013).
For leptospires, some potential VFs identified in the literature included: lipL32, mce, invA, atsE, mviN, rfb for attachment and invasion and asd, trpE, and sphH for amino acid biosynthesis (Ren et al., 2003; Ko et al., 2009; Fouts et al., 2016).
For Parachlamydia, known VFs included: negative regulator of the T3SS, SctW; protein kinase, Pkn5; translocated actin-recruiting phosphoprotein, tarp; inclusion membrane proteins IncA to IncG; translocator protein, CopB; modulation of host cell apoptosis, CADD; and Mip (Greub, 2009; Betts-Hampikian and Fields, 2010; Collingro et al., 2011; Croxatto et al., 2013). Furthermore, genes coding for nucleotide transporters that import host cell ATP in exchange for ADP (ntt) were part of the complex involving in bacteria-host interaction, but were generally not considered as VFs (Schmitz-Esser et al., 2004; Haferkamp et al., 2006).
PhyloPhlAn (Segata et al., 2013) was used to construct phylogenomic trees based on draft genomes and reference genomes. The constructed trees were visualized using iTOL (Letunic and Bork, 2016).
ARGs and CRISPR-Cas regions were screened with PATRIC. The identified CRISPR loci and ARGs were confirmed with CRISPRfinder (Grissa et al., 2007) and ResFinder (Zankari et al., 2012), respectively. Identified CRIPSR-Cas loci were classified into the current system consisting of two classes, five types, and 16 subtypes (type I-A to I-F and I-U, type II-A to II-C, type III-A to III-D, type IV, and type V) based on cas genes and additional signature genes (Makarova et al., 2015). Additionally, we investigated the possible targets (protospacers) of spacers in CRISPR-Cas arrays within the obtained draft genomes using CRISPRTarget to search against all the available databases (i.e., GenBank-Phage, GenBank-Environmental, RefSeq-Plasmid, RefSeq-Viral, and RefSeq-Bacteria), which was combined with the known features of each subtype that had been reported to be essential for target recognition, such as protospacer adjacent motifs (PAMs) and seed regions (Biswas et al., 2013). Extra weighting was given to known PAMs: 5′-GG-3′ for I-F (Mojica et al., 2009) at the 3′ region of protospacer and 5′-CCN-3′ for II-B (Fonfara et al., 2014) at the 5′ region of protospacer. Moreover, we also manually examined seed sequences (8-nt for Type I-F and 13-nt for Type II-B) within the match. PHAST was used to identify prophage sequences in these draft genomes (Zhou et al., 2011).
The nine draft genomes reconstructed in this study are deposited in GenBank under the BioProject PRJNA323575 with BioSamples SAMN07572181- SAMN07572189.
A combination of different molecular biological techniques, namely, 16S rRNA gene amplicon sequencing, metagenomics, and ddPCR/real-time PCR was employed to investigate the diversity and quantity of potential pathogens in the drinking water production and distribution system. Regarding prokaryotes, Figure 2 shows that in general, the distribution system samples contained the highest relative abundance of Mycobacterium spp. and Legionella spp. in comparison with samples from the treatment process. The highest level of Mycobacterium spp. was detected with the PR sample with a relative abundance of 1.3 × 10−1 and an absolute concentration of 3.3 × 104 copies/ng-gDNA by ddPCR (Table S2). The BC sample contained the highest level of Legionella spp.: a relative abundance of 4.7 × 10−3 based on 16S rRNA amplicon analysis and a concentration of 40.9 copies/ng-gDNA by ddPCR. Despite the occurrence of potential pathogens at the genus level, known pathogenic species, including M. tuberculosis complex, Lg. pneumophila, and A. hydrophila were not detected (Table S2). Additionally, sequences related to Candidatus Protochlamydia spp., Parachlamydia spp., and Leptospira spp. were also detected (Figure 2). Candidatus Protochlamydia spp. and Parachlamydia spp. were endosymbionts of amoeba and emerging agents of pneumonia (Greub, 2009). Notably, Candidatus Protochlamydia spp. were detected in all the distribution water phase samples.
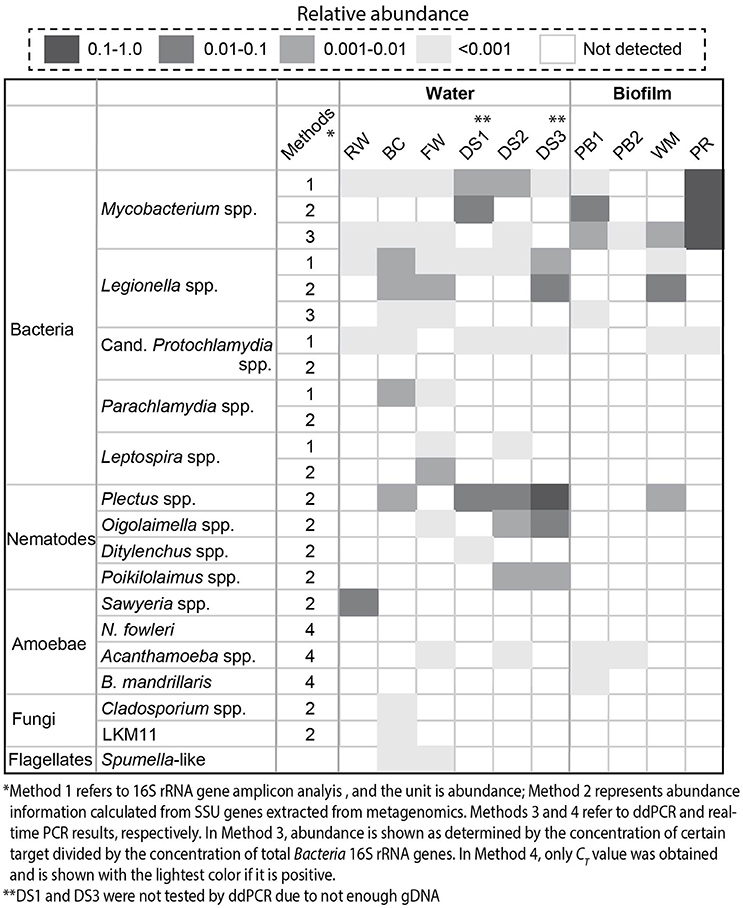
Figure 2. Detected potential pathogens and eukaryotes (nematodes, amoebae, fungi, and flagellates) by 16S rRNA gene amplicon analysis (1), SSU genes extracted from metagenomes (2), ddPCR (3), and real-time PCR (4). Here, relative abundance was reported. The concentration of specific pathogens determined with ddPCR was summarized in Table S2. We divided the samples into water and biofilm phases.
Meanwhile, we could identify various eukaryotes, such as nematodes, amoebae, and flagellates with metagenomics and real-time PCR that co-existed with these potential pathogens. Plectus spp. were the most abundant nematodes detected in the system and present in half of the samples. For amoebae, Acanthamoeba spp. were observed in FW, DS2, PB1, and PB2 while Sawyeria spp. were only found in RW.
Nine draft genomes closely related to known pathogens were successfully recovered from the metagenomes of BC, FW, DS1-3, and PR with ≥90% completeness and ≥15-fold coverage (Table 1). The phylogenomic tree in Figure 3 showed that four draft genomes were affiliated with Legionella (BC.3.64, FW.3.37, DS3.009, BC.3.72; Figure 3A), three with Mycobacterium (DS1.3.26, DS2.013, PR.002; Figure 3B), one with Leptospira (FW.030; Figure 3C), and one with Parachlamydia (BC.030; Figure 3D). In Figure 3A, different species of Legionella were observed to co-exist in the same niche, i.e., BC.3.64 and BC.3.72 in the BC sample. FW.3.37 was observed to be 99.7% similar to BC.3.64 in the average nucleotide identity (ANI) based on 400 marker genes. These three draft genomes probably represented new species of Legionella as they did not cluster together with any known species. A fourth draft genome, DS3.009, was affiliated with Lg. drozanskii. For Mycobacterium draft genomes, all three (DS1.3.26, DS2.013, PR.002) were closely related to M. gordonae. The Leptospira draft genome FW.030 was outside of the cluster containing mostly saprophytic species. Last, draft genome BC.030 fell between Pa. acanthamoebae and Candidatus Protochlamydia amoebophila. Collectively, five of the draft genomes retrieved were not closely related to any known isolated species, possibly due to the limitation of cultivation methods to recover microorganisms from drinking water systems so far.
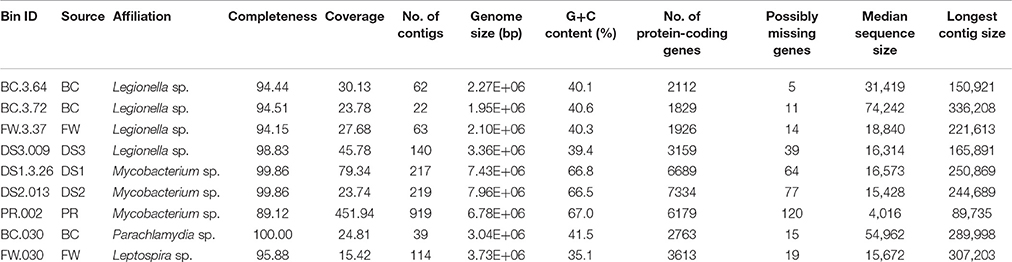
Table 1. General features of the recovered genomes of pathogen-related species.
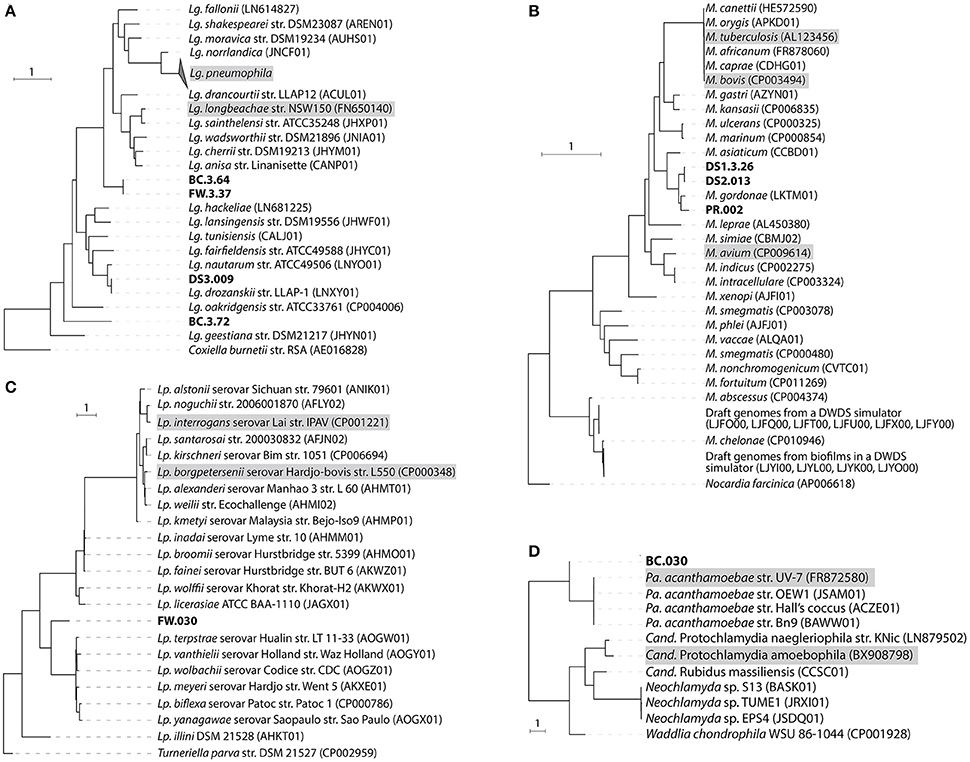
Figure 3. Phylogenomic tree of recovered draft genomes constructed based on up to 400 conserved protein sequences. (A) Legionella; (B) Mycobacterium; (C) Leptospira; (D) Parachlamydia. The nine draft genomes recovered from this study were bold. Known pathogenic species were shaded with gray. Scale bar, 1 expected substitutions per site.
Figure 4 (also see Table S3) indicated the presence and absence of VFs affiliated with secretion systems, effectors, attachment and invasion, endotoxins (e.g., lipopolysaccharides), and amino acid biosynthesis found in the recovered draft genomes and their related reference genomes. For Legionella in the secretion system category, the T2SS and T4BSS were the major pathogenesis systems observed in all draft genomes recovered. By contrast, the T4ASS, associated with conjugal DNA transfer, was detected in BC.3.64 and DS3.009 but absent in BC.3.72 and FW.3.37 possibly due to non-existence in these bacteria or the inability or poor efficiency to retrieve and assemble sequences pertaining to these hypervariable regions (Pop, 2009; Gomez-Valero et al., 2011). In the effectors category, T4BSS-assicated VFs including lidA, sdhA, and lepAB genes but not ralF were detected in three of the four draft genomes. In addition, all draft genomes contained LPS transport related genes, lptA and lptE. Last, the mip gene was observed in BC.3.64, FW.3.37, and DS3.009, but not BC.3.72.
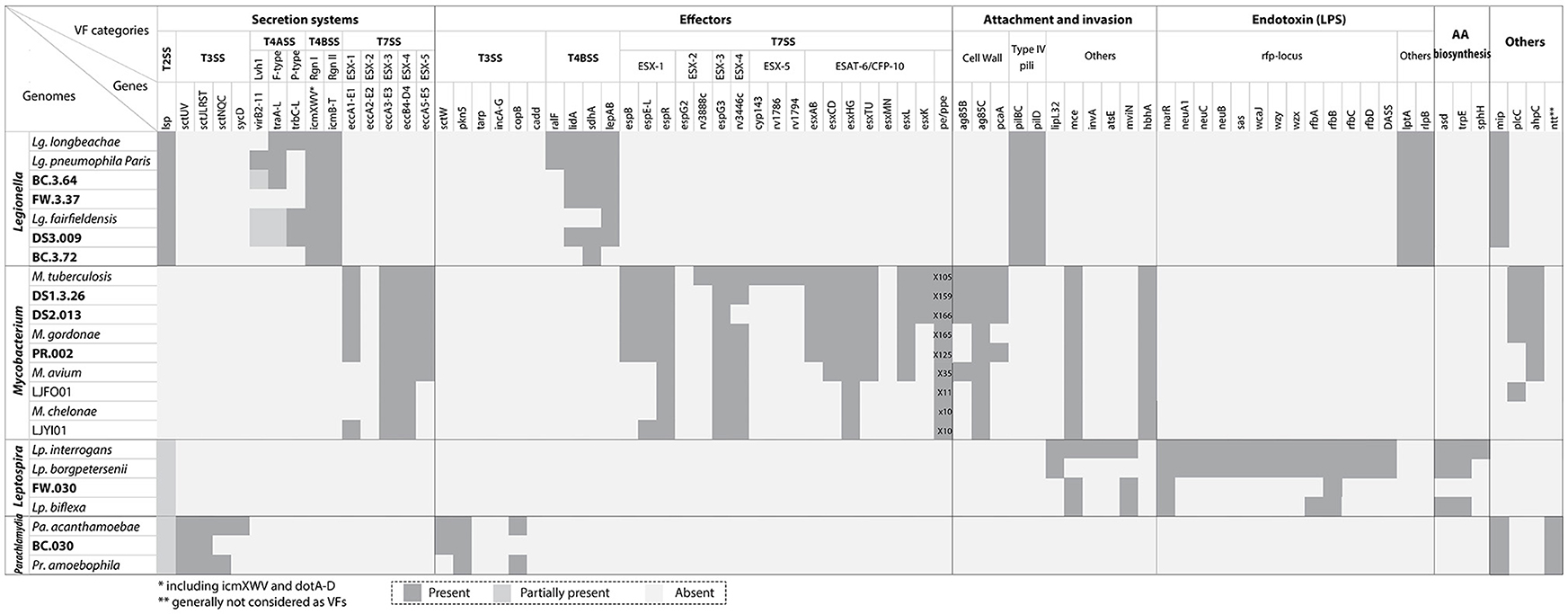
Figure 4. VFs identified with the draft genomes recovered in this study and related genomes from public databases. VFs were grouped based on general categories (secretion systems and associated effectors, attachment and invasion, endotoxin, amino acid biosynthesis, and others). The genomes were organized by their taxonomic affiliations. There were some shared VFs among different genera, including T2SS among Legionella, Leptospira, and Parachlamydia, the mip gene between Legionella and Parachlamydia, and the mce gene between Mycobacterium and Leptospira. The accession number of known genomes were listed in Table S3.
For Mycobacterium, ESX-1, ESX-3, and ESX-5 T7SSs were observed in all Mycobacterium draft genomes recovered. Effectors belonging to ESX-1 and ESX-3 could also be detected, including esxAB and TU, but not effectors belonging to ESX-5 (cyp143, rv1786, rv1794, and esxMN). For the pe/ppe multigene family, all the recovered draft genomes contained more than 100 such genes, which was comparable to those observed in pathogenic species. Other VFs detected included cell envelop biosynthesis, ag85 (except in PR.002) and pcaA; adhesin, hbhA; phospholipase C, plcC; and oxidative stress reducer, ahpC. For Leptospira, the known VFs were mainly associated with the attachment and invasion, endotoxin and amino acid biosynthesis categories, and among them four (i.e., mce1B, mviN, marR, and rfbD) were detected in FW.030. The T2SS was partially present in Leptospira spp., including FW.030, but the association of the T2SS with virulence had not been experimentally tested (Picardeau, 2017). For Parachlamydia, VFs were mainly observed in the T3SS and associated effector categories. Two VFs, the T2SS (partially) and mip in the “others” category were also observed. As Parachlamdia spp. and Candidatus Protochlamydia spp. were intracellular bacteria of amoebae like Legionella spp., they also possessed T2SSs and Mip systems. Five ntt genes were observed with BC.030, putatively belonging to three NTT isoforms (NTT1-3) as shown in Figure S2 (Haferkamp et al., 2006). Last, several ARGs related to the resistance of aminoglycoside (moderate level), beta-lactam, and chloramphenicol (antimicrobial peptides) could be detected in the Legionella draft genome DS3.009. All the Mycobacterium recovered draft genomes possessed the aac(2′)-Ic gene, which was universally distributed among all Mycobacterium spp. (Ainsa et al., 1997; Table S4).
CRISPR-Cas genetic signatures, which are defense systems used by prokaryotes against viruses and not associated with pathogenicity, could be an effective tool to discriminate and monitor sub-lineages of pathogen-related species across the studied drinking water production and distribution system. Figure 5 indicates the type of CRISPR-Cas systems identified in the draft genomes recovered and in several published Lg. pneumophila genomes. Among the three known subtypes of Lg. pneumophila (I-F, II-B, and I-C), this study detected type I-F with BC.3.64 and FW.3.37 based on cas gene clusters. The type I-F CRISPR-Cas observed in these two draft genomes was almost identical, i.e., 99% sequence similarity for cas1 gene and 100% sequence similarity for the remaining cas genes (Table S5). Together with the findings of phylogenomic classification and genome similarity (99.7%; Figure 3), BC.3.64 and FW.3.37 were very likely to belong to a closely-related population originated from the same ancestor traveling from upstream (BC) to downstream (FW) of the studied drinking water production and distribution system. There was not enough information to determine whether the strain was alive at the BC site or whether filtration and chlorination had inactivated the strain in FW. Their cas gene clusters shared relatively low protein sequence similarities (from <40–76%) with other type I-F CRISPR-Cas loci of Lg. pneumophila (Table S5). Last, a Type II-B CRISPR-Cas locus was detected with Leptospira draft genome FW.030 (Figure S3).
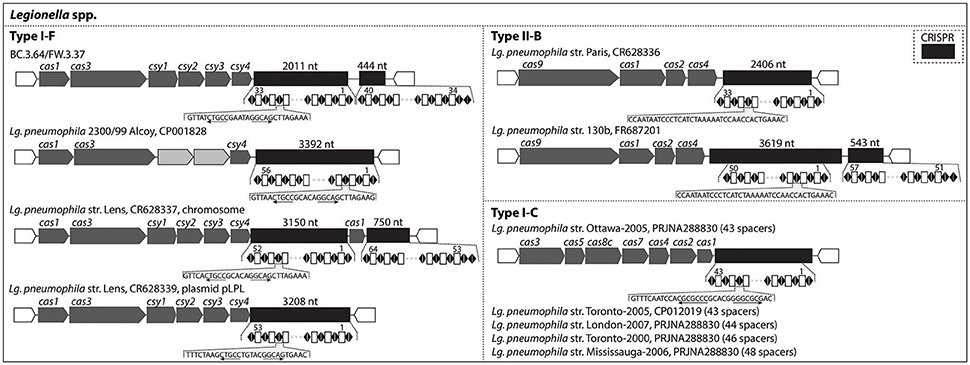
Figure 5. CRISPR-Cas loci identified in the draft genomes recovered in this study and known genomes of Legionella. They were organized according to the subtypes (Type I-F, II-B, and I-C) of CRISPR-Cas loci.
Table 2 shows the types of prophages found in the recovered draft genomes. Initially, 36 potential prophage sequences were identified using PHAST (Figure S4) and they were reduced to 16 by considering the presence of genes encoding integrases and/or cI-type repressors (Fan et al., 2014; Figure S5). The lengths of prophage regions varied from 9.5 to 40.1 kbp. Six were associated with Legionella draft genomes, seven with Mycobacterium draft genome, and one each with Parachlamydia and Leptospira. An intact prophage (37.1 kbp) was recovered from PR.002. Shared prophage structures were observed between BC.3.64 and FW.3.37 and between DS1.3.26 and DS2.013. In addition, DS2.013 contained as many as five prophage sequences, which was rare for Mycobacterium genomes. Last, a prophage region identified in FW.030 showed sequence similarities to Pandoravirus salinus which was the largest virus reported so far with genomes up to 2.5 Mb and restricted to Acanthamoeba as hosts (Philippe et al., 2013).
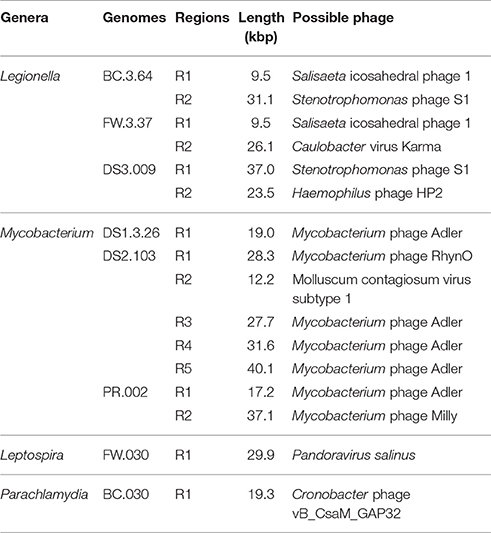
Table 2. Prophages identified in the retrieved draft genomes.
Virulence machinery characterized by genomic analysis has been used to define pathogenicity for many known pathogens, such as E. coli (Chapman et al., 2006), Salmonella (Foley et al., 2013), Cryptosporidium (Bouzid et al., 2013), Lg. pneumophila (Cazalet et al., 2008), and Leptospira (Picardeau, 2017). This approach is used here to evaluate the potential pathogenicity of those draft genomes of pathogen-related species recovered from an urban drinking water system. Legionella-related draft genomes found at two different locations of the water production process (i.e., BC.3.64 and FW.3.37) shared almost identical genomic sequences and possessed almost all known VFs to Lg. pneumophila and Lg. longbeachae. Another strain found during the water production process (i.e., BC.3.72) was clustered outside of known pathogenic Legionella clusters, and possessed fewer virulence genes than the other three recovered strains (i.e., BC.3.64, FW.3.37, and DS3.009). While the finding that most of the draft genomes encoded a high number of VFs may raises concerns on their pathogenicity, previous studies on closely related species/strains of pathogenic Aeromonas found no correlations between the presence/absence of VFs and extraintestinal infections (Havelaar et al., 1992; Lye et al., 2007). Thus, further studies combining microbiological (e.g., cultivation and animal models), genomic, and metabolic (e.g., transcriptomics and proteomics) methods should be carried out to understand the role of these VFs at the level of gene expression, protein function and regulation, and interaction with host immune system to confirm the virulence of these strains for immunocompromised individuals. This framework, once established, can be transferred into a novel pathogen surveillance program that enables virulence assessment of a broad range of heterotrophic bacteria found in potable water to possibly identify currently unknown pathogens.
All three Mycobacterium-related draft genomes recovered were closely related to M. gordonae, which is less virulent than M. tuberculosis, but contained a high number of genes (over 100) related to pe/ppe and T7SS. In comparison, genomes of M. immunogenum (LJFO01) and M. chelonae (LJYI01) isolated from a chloraminated DWDS simulator in previous studies (Gomez-Alvarez and Revetta, 2016a,b) lacked ESX-1 or ESX-5 and contained fewer pe/ppe genes. Due to the prevalence of M. gordonae in tap water and biofilms, particularly in groundwater-derived drinking water systems (Vaerewijck et al., 2005), special attention to this group would be necessary. Pathogenic Leptospira are the causative agent of leptospirosis, which is the most widespread zoonotic disease infecting both human and animals (Evangelista and Coburn, 2010). In this study, the Leptospira-related genome FW.030 obtained did not contain most of the VFs known for Lp. interrogans and thus was likely not pathogenic. Among Parachlamydiaceae, only few strains such as Pa. acanthamoebae and Candidatus Pr. naegleriophila have been considered as emerging pathogens, causing mainly respiratory infections, while many others including Neochlamydia hartmannellae and Pr. amoebophila might be environmental strains or endosymbionts (Corsaro and Greub, 2006; Lamoth et al., 2011). Therefore, the pathogenic potential of Parachlamydia-related genome BC.030 remains to be determined.
Due to the high genome plasticity of Legionella species, molecular typing by a single marker gene has been difficult. For instance, the mip gene is associated with the ability of Lg. pneumophila to replicate in eukaryotic cells, and has been extensively used as a biomarker to detect the presence/absence of Lg. pneumophila in a sample (Gomez-Valero et al., 2009). It was detected in three Legionella-related draft genomes constructed in this study: BC.3.64 and FW.3.37 were closely related to Lg. fallonii, and DS3.009 to Lg. drozanskii (Figure S6). However, the mip gene was limited in differentiating the Lg. pneumophila subspecies fraseri from other subspecies (Figure S4). Thus, the European Working Group for Legionella Infections (EWGLI) has suggested that a combination of several biomarkers, including flaA, pilE, asd, mip, mompS, proA, and neuA, should be used to effectively identify Lg. pneumophila (Fry et al., 2000; Gaia et al., 2005; Ratzow et al., 2007). However, phylogenetic incongruence (i.e., different lineages of the same strain indicated by different biomarkers) and limitations (i.e., the inability of some biomarkers to discriminate certain strains) in the discriminatory power of these multiple biomarkers could still occur because of differences in selection pressures associated with individual biomarkers.
Alternatively, spacers in CRISPR-Cas can be used as a biomarker in the monitoring of certain Legionella strains at an evolutionary scale of several years across drinking water production and distribution systems. The pattern of adding new spacers at one end of the CRISPR array and conserving spacers among common ancestors at the other end has been demonstrated with Legionella strains collected in Canada and Europe (CRISPR Type I-C and Type II-B; Ginevra et al., 2012; Lück et al., 2015; Rao et al., 2016). The longest time for these spacers to remain conserved among these strains and a Leptospirillum strain previously studied was reported to be 5 years or longer (Sun et al., 2016). As shown in Figure 5, Type I-F Cas loci were detected in the genomes of Lg. pneumophila str. 2300/99 Alcoy and str. Lens (both in the chromosome and plasmid). The two draft genomes recovered in our study, BC.3.64 and FW.3.37, also contained type I-F CRISPR-Cas loci, but the spacers were different from str. 2300/99 Alcoy and str. Lens. With 100% sequence similarity in CRISPR and high overall genomic similarity, these two genomes were likely derived from the same ancestor. Thus a specific CRISPR-Cas biomarker could be developed and used to monitor the distribution of this strain within the drinking water system studied. Furthermore, Types II-B and I-C were detected in a variety of Lg. pneumophila strains (Figure 5) and Type II-B was detected in 75.0% of the 400 Lg. pneumophila strains collected in a previous study (Ginevra et al., 2012). With more than 600 Legionella genomes available with NCBI's website and the diversity of CRISPR-Cas Types (I-C, I-F, and II-B) known among these strains, CRISPR-Cas spacers will be a promising biomarker for monitoring the distribution of Legionella at the strain level in samples taken from various drinking water systems, across different water bodies, and between patients over several years. However, cautions are needed when applying this method over a relatively large evolutionary scale as previous reports on Yersinia pestis, Streptococcus thermophiles, and Leptospirillum suggested that CRISPR loci could also evolve via internal deletion of spacers in the CRISPR array (Pourcel et al., 2005; Horvath et al., 2008; Sun et al., 2016).
The interaction between bacteria and viruses in drinking water systems or, more broadly, in oligotrophic environments is not well understood (Lehtola et al., 2004; Liu et al., 2015; Guidi et al., 2016). Table 3 shows only 26 out of the 119 identified CRISPR-Cas spacers matched to entries in databases including GenBank-Phage, GenBank-Environmental, RefSeq-Plasmid, RefSeq-Viral, and RefSeq-Bacteria. Among them, 13 spacers matched sequences in other Lg. pneumophila strains. Two commonly observed targets were a 30-kb unstable genetic element previously identified in Lg. pneumophila str. RC1 and a 60-kb plasmid in Lg. pneumophila str. Lens. Likely, these elements were originated from bacteriophages in environments and incorporated into Lg. pneumophila genomes as mobile genetic elements such as prophages and plasmids. When the DNA of Lg. pneumophila was damaged or under other stress conditions, prophages could be excised, replicated, and ultimately used to lyse the host and spread into the environment. Ecologically, it would be rational for other Lg. pneumophila strains to incorporate their fragments into CRISPR systems so that they had the ability to destroy them when being attacked (Rao et al., 2016).
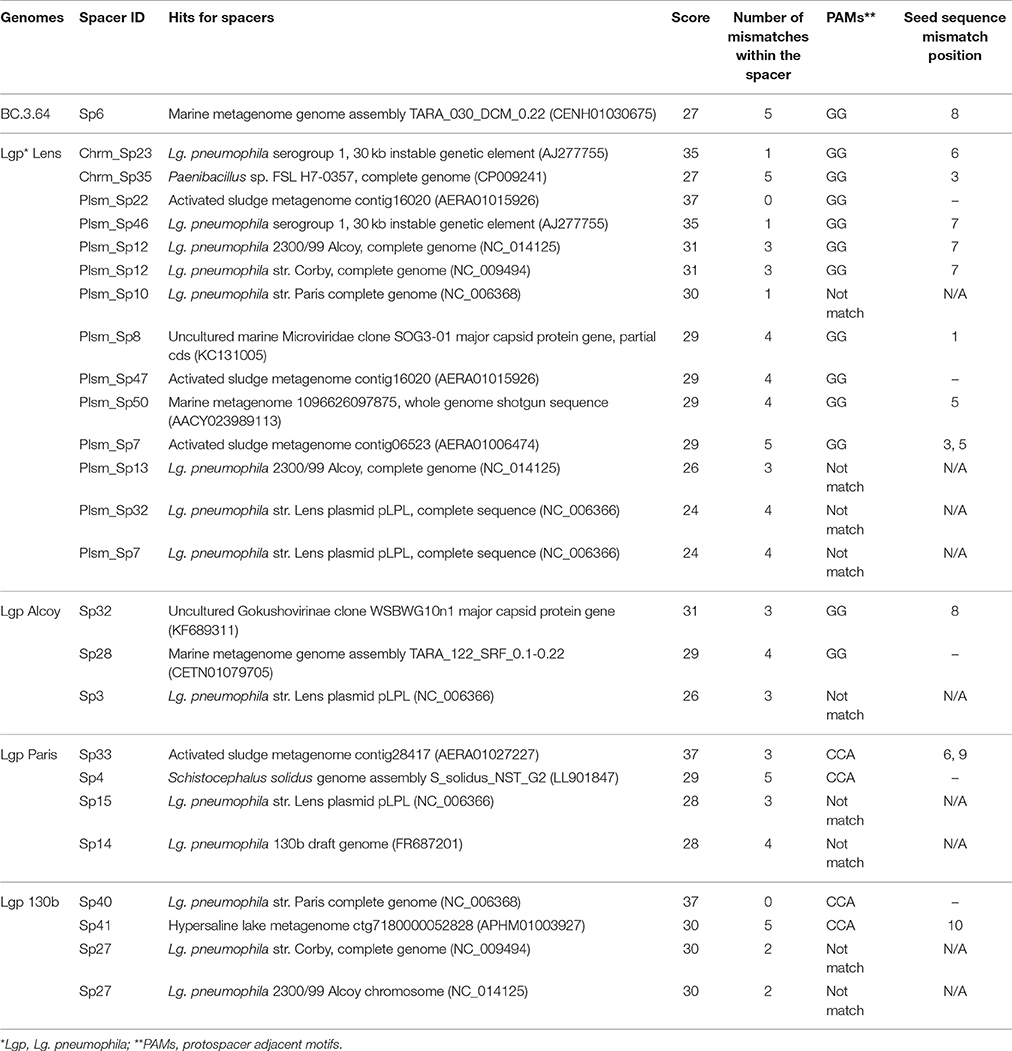
Table 3. Potential targets of CRISPR-Cas spacers in Legionella-related genomes.
We also observed near-perfect matches of four spacers in CRISPR-Cas to one activated sludge metagenome (AERA01; More et al., 2014). It has been reported that wastewater treatment plants (WWTPs) contained 10–1,000 times higher viral concentration than in natural aquatic environments, making WWTP an important reservoir and source of viruses (Edwards and Rohwer, 2005; Tamaki et al., 2012). In the studied drinking water production and distribution system, we estimated that the viral concentration was ~104 viruses/ml based on the bacterial cell counts published previously (Zhang et al., 2017) with the general rule that viral count is 10 times of the bacterial count (Maranger and Bird, 1995). Additionally, spacers detected in the BC.3.64 and FW.3.37 genomes recovered here and Lg. pneumophila 2300/99 Alcoy matched to contigs in marine metagenomes (AACY02; Venter et al., 2004). Although the matches are not perfect (except one) to organisms in WWTPs or marine environment, the evolving nature of spacers by mutations at CRISPR loci allows us to speculate that WWTPs and marine environments were possible sources of these spacers. Those Legionella strains could have come from water bodies under the influence of wastewater or seawater, such as flooded sewers or coastal groundwater.
This study observed that the prophage exhibiting high sequence similarity to Pandoravirus could co-exist with Acanthamoeba spp., Parachlamydia spp., Legionella spp., and Mycobacterium spp. in the FW sample. So far, free-living amoebae in drinking water systems are reported to be an ideal shelter to provide nutritional requirements for the growth of Legionella (Breiman et al., 1990; Dupuy et al., 2016), and are the only reported host of Pandoravirus (Philippe et al., 2013). Various giant viruses, including Mimivirus, Mamavirus, and Pandoravirus, have been detected in amoebae and were reported to be involved in lateral gene transfer between viruses and bacteria (La Scola et al., 2003, 2008; Philippe et al., 2013). While the detection of Parachlamydia in drinking water systems is rare (Thomas et al., 2008), previous studies have suggested that Chlamydiae were likely prevalent in aquatic environments (Barret et al., 2013; Lagkouvardos et al., 2014). These observations all support amoebae as the “hub” connecting viruses and intracellular bacteria, and facilitating the genetic exchange between pathogens and their closely related species (Gimenez et al., 2011; Gomez-Valero et al., 2011). Thus, developing control strategies to eukaryotic populations, e.g., filtration with 1 μm membranes, whose size is larger than bacteria but smaller than amoebae, could be an effective means to suppress the growth and spreading of pathogens in DWDSs (Wadowsky et al., 1988).
In summary, our study demonstrates that metagenomics analysis can be used to determine the presence of VFs in potential pathogens in drinking water production and distribution systems. Future studies combining microbiological, genomic, and metabolic methods at the level of gene expression, protein function and regulation, and bacteria-host interaction can help determine the relationship between the presence of these VFs and pathogenicity in immunocompromised individuals, especially for environmental strains recovered from drinking water systems. Furthermore, the development of genomics analysis can serve as a new platform for the detection, strain typing, and monitoring of pathogens, which can provide novel insights into the surveillance and control of waterborne or water-based pathogens. Characteristic regions in bacterial genomes, such as CRISPR-Cas studied here, can be used in combination with the traditional biomarkers to facilitate and simplify the subtyping of pathogens of potential concern and monitor the distribution of the same strains across different environmental niches.
YZ designed and carried out the experiments, analyzed the obtained data, and wrote the manuscript. MK and AW carried out the experiments to quantify the pathogens and participated in the manuscript writing process. WL designed and carried out the experiments, analyzed the obtained data, and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02036/full#supplementary-material
Ainsa, J. A., Perez, E., Pelicic, V., Berthet, F. X., Gicquel, B., and Martin, C. (1997). Aminoglycoside 2′-N-acetyltransferase genes are universally present in mycobacteria: characterization of the aac(2′)-lc gene from Mycobacterium tuberculosis and the aac(2′)-ld gene from Mycobacterium smegmatis. Mol. Microbiol. 24, 431–441. doi: 10.1046/j.1365-2958.1997.3471717.x
Ashbolt, N. J. (2015). Microbial contamination of drinking water and human health from community water systems. Curr. Environ. Health Rep. 2, 95–106. doi: 10.1007/s40572-014-0037-5
Barret, M., Egan, F., and O'Gara, F. (2013). Distribution and diversity of bacterial secretion systems across metagenomic datasets. Environ. Microbiol. Rep. 5, 117–126. doi: 10.1111/j.1758-2229.2012.00394.x
Betts-Hampikian, H. J., and Fields, K. A. (2010). The chlamydial type III secretion mechanism: revealing cracks in a tough nut. Front. Microbiol. 1:114. doi: 10.3389/fmicb.2010.00114
Biswas, A., Gagnon, J. N., Brouns, S. J. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827. doi: 10.4161/rna.24046
Bobay, L. M., Touchon, M., and Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. U.S.A. 111, 12127–12132. doi: 10.1073/pnas.1405336111
Bouzid, M., Hunter, P. R., Chalmers, R. M., and Tyler, K. M. (2013). Cryptosporidium pathogenicity and virulence. Clin. Microbiol. Rev. 26, 115–134. doi: 10.1128/CMR.00076-12
Breiman, R. F., Fields, B. S., Sanden, G. N., Volmer, L., Meier, A., and Spika, J. S. (1990). Association of shower use with legionnaires-disease - possible role of amebas. JAMA 263, 2924–2926. doi: 10.1001/jama.1990.03440210074036
Burstein, D., Amaro, F., Zusman, T., Lifshitz, Z., Cohen, O., Gilbert, J. A., et al. (2016). Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat. Genet. 48, 167–175. doi: 10.1038/ng.3481
Cazalet, C., Jarraud, S., Ghavi-Helm, Y., Kunst, F., Glaser, P., Etienne, J., et al. (2008). Multigenome analysis identifies a worldwide distributed epidemic Legionella pneumophila clone that emerged within a highly diverse species. Genome Res. 18, 431–441. doi: 10.1101/gr.7229808
Chapman, T. A., Wu, X. Y., Barchia, I., Bettelheim, K. A., Driesen, S., Trott, D., et al. (2006). Comparison of virulence gene profiles of Escherichia coli strains isolated from healthy and diarrheic swine. Appl. Environ. Microbiol. 72, 4782–4795. doi: 10.1128/AEM.02885-05
Chen, L. H., Xiong, Z. H., Sun, L. L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–D645. doi: 10.1093/nar/gkr989
Collingro, A., Tischler, P., Weinmaier, T., Penz, T., Heinz, E., Brunham, R. C., et al. (2011). Unity in variety-the pan-genome of the chlamydiae. Mol. Biol. Evol. 28, 3253–3270. doi: 10.1093/molbev/msr161
Corsaro, D., and Greub, G. (2006). Pathogenic potential of novel Chlamydiae and diagnostic approaches to infections due to these obligate intracellular bacteria. Clin. Microbiol. Rev. 19, 283–297. doi: 10.1128/CMR.19.2.283-297.2006
Costa, T. R. D., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Cox, M. P., Peterson, D. A., and Biggs, P. J. (2010). SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485
Croxatto, A., Murset, V., Chassot, B., and Greub, G. (2013). Early expression of the type III secretion system of Parachlamydia acanthamoebae during a replicative cycle within its natural host cell Acanthamoeba castellanii. Pathog. Dis. 69, 159–175. doi: 10.1111/2049-632X.12065
Deveau, H., Garneau, J. E., and Moineau, S. (2010). CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475–493. doi: 10.1146/annurev.micro.112408.134123
Dupuy, M., Binet, M., Bouteleux, C., Herbelin, P., Soreau, S., and Hechard, Y. (2016). Permissiveness of freshly isolated environmental strains of amoebae for growth of Legionella pneumophila. FEMS Microbiol. Lett. 363:fnw022. doi: 10.1093/femsle/fnw022
Edberg, S. C., Gallo, P., and Kontnick, C. (1996). Analysis of the virulence characteristics of bacteria isolated from bottled, water cooler, and tap water. Microb Ecol Health D 9, 67–77. doi: 10.3109/08910609609166445
Edwards, D. J., and Holt, K. E. (2013). Beginner's guide to comparative bacterial genome analysis using next-generation sequence data. Microb. Inform. Exp. 3:2. doi: 10.1186/2042-5783-3-2
Edwards, R. A., and Rohwer, F. (2005). Viral metagenomics. Nat. Rev. Microbiol. 3, 504–510. doi: 10.1038/nrmicro1163
Evangelista, K. V., and Coburn, J. (2010). Leptospira as an emerging pathogen: a review of its biology, pathogenesis and host immune responses. Future Microbiol. 5, 1413–1425. doi: 10.2217/fmb.10.102
Falkinham, J. O., Norton, C. D., and LeChevallier, M. W. (2001). Factors influencing numbers of Mycobacterium avium, Mycobacterium intracellulare, and other mycobacteria in drinking water distribution systems. Appl. Environ. Microbiol. 67, 1225–1231. doi: 10.1128/AEM.67.3.1225-1231.2001
Fan, X. Y., Xie, L. X., Li, W., and Xie, J. P. (2014). Prophage-like elements present in Mycobacterium genomes. BMC Genomics 15:243. doi: 10.1186/1471-2164-15-243
Favrot, L., Grzegorzewicz, A. E., Lajiness, D. H., Marvin, R. K., Boucau, J., Isailovic, D., et al. (2013). Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat.Commun. 4:2748. doi: 10.1038/ncomms3748
Finlay, B. B., and Falkow, S. (1997). Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 61, 136–169.
Foley, S. L., Johnson, T. J., Ricke, S. C., Nayak, R., and Danzeisen, J. (2013). Salmonella pathogenicity and host adaptation in chicken-associated serovars. Microbiol. Mol. Biol. Rev. 77, 582–607. doi: 10.1128/MMBR.00015-13
Fonfara, I., Le Rhun, A., Chylinski, K., Makarova, K. S., Lecrivain, A. L., Bzdrenga, J., et al. (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–2590. doi: 10.1093/nar/gkt1074
Forrellad, M. A., Klepp, L. I., Gioffre, A., Garcia, J. S. Y., Morbidoni, H. R., Santangelo, M. D., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Fouts, D. E., Matthias, M. A., Adhikarla, H., Adler, B., Amorim-Santos, L., Berg, D. E., et al. (2016). What makes a bacterial species pathogenic? comparative genomic analysis of the genus Leptospira. PLoS Negl. Trop. Dis. 10:e0004403. doi: 10.1371/journal.pntd.0004403
Fry, N. K., Bangsborg, J. M., Bernander, S., Etienne, J., Forsblom, B., Gaia, V., et al. (2000). Assessment of intercentre reproducibility and epidemiological concordance of Legionella pneumophila serogroup 1 genotyping by amplified fragment length polymorphism analysis. Eur. J. Clin. Microbiol. Infect. Dis. 19, 773–780. doi: 10.1007/s100960000359
Gaia, V., Fry, N. K., Afshar, B., Luck, P. C., Meugnier, H., Etienne, J., et al. (2005). Consensus sequence-based scheme for epidemiological typing of clinical and environmental isolates of Legionella pneumophila. J. Clin. Microbiol. 43, 2047–2052. doi: 10.1128/JCM.43.5.2047-2052.2005
Gimenez, G., Bertelli, C., Moliner, C., Robert, C., Raoult, D., Fournier, P. E., et al. (2011). Insight into cross-talk between intra-amoebal pathogens. BMC Genomics 12:542. doi: 10.1186/1471-2164-12-542
Ginevra, C., Jacotin, N., Diancourt, L., Guigon, G., Arquilliere, R., Meugnier, H., et al. (2012). Legionella pneumophila sequence Type 1/Paris pulsotype subtyping by spoligotyping. J. Clin. Microbiol. 50, 696–701. doi: 10.1128/JCM.06180-11
Gomez-Alvarez, V., and Revetta, R. P. (2016a). Draft genome sequences of six Mycobacterium immunogenum strains obtained from a chloraminated drinking water distribution system simulator. Genome Announc. 4:e01538–15. doi: 10.1128/genomeA.01538-15
Gomez-Alvarez, V., and Revetta, R. P. (2016b). Whole-genome sequences of four strains closely related to members of the Mycobacterium chelonae group, isolated from biofilms in a drinking water distribution system simulator. Genome Announc. 4:e01539–15. doi: 10.1128/genomeA.01539-15
Gomez-Valero, L., Rusniok, C., and Buchrieser, C. (2009). Legionella pneumophila: population genetics, phylogeny and genomics. Infect. Genet. Evol. 9, 727–739. doi: 10.1016/j.meegid.2009.05.004
Gomez-Valero, L., Rusniok, C., Jarraud, S., Vacherie, B., Rouy, Z., Barbe, V., et al. (2011). Extensive recombination events and horizontal gene transfer shaped the Legionella pneumophila genomes. BMC Genomics 12:536. doi: 10.1186/1471-2164-12-536
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4. doi: 10.1128/microbiolspec.VMBF-0012-2015
Greub, G. (2009). Parachlamydia acanthamoebae, an emerging agent of pneumonia. Clin. Microbiol. Infect. 15, 18–28. doi: 10.1111/j.1469-0691.2008.02633.x
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Guidi, L., Chaffron, S., Bittner, L., Eveillard, D., Larhlimi, A., Roux, S., et al. (2016). Plankton networks driving carbon export in the oligotrophic ocean. Nature 532, 465–470. doi: 10.1038/nature16942
Haferkamp, I., Schmitz-Esser, S., Wagner, M., Neigel, N., Horn, M., and Neuhaus, H. E. (2006). Tapping the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia amoebophila. Mol. Microbiol. 60, 1534–1545. doi: 10.1111/j.1365-2958.2006.05193.x
Havelaar, A. H., Schets, F. M., van Silfhout, A., Jansen, W. H., Wieten, G., and van der Kooij, D. (1992). Typing of Aeromonas strains from patients with diarrhoea and from drinking water. J. Appl. Bacteriol. 72, 435–444. doi: 10.1111/j.1365-2672.1992.tb01857.x
Hilbi, H., Segal, G., and Shuman, H. A. (2001). Icm/dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol. Microbiol. 42, 603–617. doi: 10.1046/j.1365-2958.2001.02645.x
Horvath, P., Romero, D. A., Coute-Monvoisin, A. C., Richards, M., Deveau, H., Moineau, S., et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190, 1401–1412. doi: 10.1128/JB.01415-07
Houben, E. N. G., Korotkov, K. V., and Bitter, W. (2014). Take five - Type VII secretion systems of Mycobacteria. Biochim. Biophys. Acta 1843, 1707–1716. doi: 10.1016/j.bbamcr.2013.11.003
Hsu, S. C., Martin, R., and Wentworth, B. B. (1984). Isolation of Legionella species from drinking water. Appl. Environ. Microbiol. 48, 830–832.
Huang, K. L., Zhang, X. X., Shi, P., Wu, B., and Ren, H. Q. (2014). A comprehensive insight into bacterial virulence in drinking water using 454 pyrosequencing and Illumina high-throughput sequencing. Ecotoxicol. Environ. Saf. 109, 15–21. doi: 10.1016/j.ecoenv.2014.07.029
Hwang, C. C., Ling, F. Q., Andersen, G. L., LeChevallier, M. W., and Liu, W. T. (2012). Evaluation of methods for the extraction of DNA from drinking water distribution system biofilms. Microbes Environ. 27, 9–18. doi: 10.1264/jsme2.ME11132
Kang, D. W. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Kao, P. M., Hsu, B. M., Hsu, T. K., Ji, W. T., Huang, P. H., Hsueh, C. J., et al. (2014). Application of TaqMan fluorescent probe-based quantitative real-time PCR assay for the environmental survey of Legionella spp. and Legionella pneumophila in drinking water reservoirs in Taiwan. Sci. Tot. Environ. 490, 416–421. doi: 10.1016/j.scitotenv.2014.04.103
Ko, A. I., Goarant, C., and Picardeau, M. (2009). Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat. Rev. Microbiol. 7, 736–747. doi: 10.1038/nrmicro2208
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
La Scola, B., Audic, S., Robert, C., Jungang, L., de Lamballerie, X., Drancourt, M., et al. (2003). A giant virus in amoebae. Science 299, 2033–2033. doi: 10.1126/science.1081867
La Scola, B., Desnues, C., Pagnier, I., Robert, C., Barrassi, L., Fournous, G., et al. (2008). The virophage as a unique parasite of the giant mimivirus. Nature 455, 100–104. doi: 10.1038/nature07218
Lagkouvardos, I., Weinmaier, T., Lauro, F. M., Cavicchioli, R., Rattei, T., and Horn, M. (2014). Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 8, 115–125. doi: 10.1038/ismej.2013.142
Lalande, V., Barbut, F., Varnerot, A., Febvre, M., Nesa, D., Wadel, S., et al. (2001). Pseudo-outbreak of Mycobacterium gordonae associated with water from refrigerated fountains. J. Hosp. Infect. 48, 76–79. doi: 10.1053/jhin.2000.0929
Lamoth, F., Jaton, K., Vaudaux, B., and Greub, G. (2011). Parachlamydia and Rhabdochlamydia: emerging agents of community-acquired respiratory infections in children. Clin. Infect. Dis. 53, 500–501. doi: 10.1093/cid/cir420
Lehtola, M. J., Miettinen, K. T., Keinanen, M. M., Kekki, T. K., Laine, O., Hirvonen, A., et al. (2004). Microbiology, chemistry and biofilm development in a pilot drinking water distribution system with copper and plastic pipes. Water Res. 38, 3769–3779. doi: 10.1016/j.watres.2004.06.024
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, D. H., Liu, C. M., Luo, R. B., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L. L., Bannantine, J. P., Zhang, Q., Amonsin, A., May, B. J., Alt, D., et al. (2005). The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 12344–12349. doi: 10.1073/pnas.0505662102
Ling, F. Q., Hwang, C. A., LeChevallier, M. W., Andersen, G. L., and Liu, W. T. (2016). Core-satellite populations and seasonality of water meter biofilms in a metropolitan drinking water distribution system. ISME J. 10, 582–595. doi: 10.1038/ismej.2015.136
Liu, H., Yuan, X. C., Xu, J., Harrison, P. J., He, L., and Yin, K. D. (2015). Effects of viruses on bacterial functions under contrasting nutritional conditions for four species of bacteria isolated from Hong Kong waters. Sci. Rep. 5:14217. doi: 10.1038/srep14217
Lowry, P. W., Becksague, C. M., Bland, L. A., Aguero, S. M., Arduino, M. J., Minuth, A. N., et al. (1990). Mycobacterium chelonae infection among patients receiving high-flux dialysis in a hemodialysis clinic in California. J. Infect. Dis. 161, 85–90. doi: 10.1093/infdis/161.1.85
Lück, C., Brzuszkiewicz, E., Rydzewski, K., Koshkolda, T., Sarnow, K., Essig, A., et al. (2015). Subtyping of the Legionella pneumophila “Ulm” outbreak strain using the CRISPR–Cas system. Int. J. Med. Microbiol. 305, 828–837. doi: 10.1016/j.ijmm.2015.08.001
Lye, D. J., and Dufour, A. P. (1993). Virulence characteristics of heterotrophic bacteria commonly isolated from potable water. Environ Toxic Water 8, 13–23. doi: 10.1002/tox.2530080103
Lye, D. J., Rodgers, M. R., Stelma, G., Vesper, S. J., and Hayes, S. L. (2007). Characterization of Aeromonas virulence using an immunocompromised mouse model. Curr. Microbiol. 54, 195–198. doi: 10.1007/s00284-006-0381-2
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Maranger, R., and Bird, D. F. (1995). Viral abundance in aquatic systems - a comparison between marine and fresh-waters. Mar. Ecol. Prog. Ser. 121, 217–226. doi: 10.3354/meps121217
Miller, C. S. (2013). Assembling full-length rRNA genes from short-read metagenomic sequence datasets using EMIRGE. Method Enzymol. 531, 333–352. doi: 10.1016/B978-0-12-407863-5.00017-4
Mojica, F. J. M., Diez-Villasenor, C., Garcia-Martinez, J., and Almendros, C. (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740. doi: 10.1099/mic.0.023960-0
More, R. P., Mitra, S., Raju, S. C., Kapley, A., and Purohit, H. J. (2014). Mining and assessment of catabolic pathways in the metagenome of a common effluent treatment plant to induce the degradative capacity of biomass. Bioresour. Technol. 153, 137–146. doi: 10.1016/j.biortech.2013.11.065
Nagarajan, N., Cook, C., Di Bonaventura, M., Ge, H., Richards, A., Bishop-Lilly, K. A., et al. (2010). Finishing genomes with limited resources: lessons from an ensemble of microbial genomes. BMC Genomics 11:242. doi: 10.1186/1471-2164-11-242
Newton, H. J., Ang, D. K. Y., van Driel, I. R., and Hartland, E. L. (2010). Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23, 274–298. doi: 10.1128/CMR.00052-09
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Philippe, N., Legendre, M., Doutre, G., Coute, Y., Poirot, O., Lescot, M., et al. (2013). Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286. doi: 10.1126/science.1239181
Picardeau, M. (2017). Virulence of the zoonotic agent of leptospirosis: still terra incognita? Nat. Rev. Microbiol. 15, 297–307. doi: 10.1038/nrmicro.2017.5
Pop, M. (2009). Genome assembly reborn: recent computational challenges. Brief. Bioinformatics 10, 354–366. doi: 10.1093/bib/bbp026
Pourcel, C., Salvignol, G., and Vergnaud, G. (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. doi: 10.1099/mic.0.27437-0
Rao, C. T., Guyard, C., Pelaz, C., Wasserscheid, J., Bondy-Denomy, J., Dewar, K., et al. (2016). Active and adaptive Legionella CRISPR-Cas reveals a recurrent challenge to the pathogen. Cell. Microbiol. 18, 1319–1338. doi: 10.1111/cmi.12586
Ratzow, S., Gaia, V., Helbig, J. H., Fry, N. K., and Luck, P. C. (2007). Addition of neuA, the gene encoding N-acylneuraminate cytidylyl transferase, increases the discriminatory ability of the consensus sequence-based scheme for typing Legionella pneumophila serogroup 1 strains. J. Clin. Microbiol. 45, 1965–1968. doi: 10.1128/JCM.00261-07
Ren, S. X., Gang, F., Jiang, X. G., Zeng, R., Miao, Y. G., Xu, H., et al. (2003). Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 422, 888–893. doi: 10.1038/nature01597
Rodriguez-Martinez, S., Sharaby, Y., Pecellin, M., Brettar, I., Hofle, M., and Halpern, M. (2015). Spatial distribution of Legionella pneumophila MLVA-genotypes in a drinking water system. Water Res. 77, 119–132. doi: 10.1016/j.watres.2015.03.010
Sampson, S. L. (2011). Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin. Dev. Immunol. 2011:497203. doi: 10.1155/2011/497203
Sangwan, N., Xia, F. F., and Gilbert, J. A. (2016). Recovering complete and draft population genomes from metagenome datasets. Microbiome 4:8. doi: 10.1186/s40168-016-0154-5
Schmidt, T. M., and Schaechter, M. (2012). Topics in Ecological and Environmental Microbiology. New York, NY: Academic Press.
Schmitz-Esser, S., Linka, N., Collingro, A., Beier, C. L., Neuhaus, H. E., Wagner, M., et al. (2004). ATP/ADP translocases: a common feature of obligate intracellular amoebal symbionts related to chlamydiae and rickettsiae. J. Bacteriol. 186, 683–691. doi: 10.1128/JB.186.3.683-691.2004
Schroeder, G. N., Petty, N. K., Mousnier, A., Harding, C. R., Vogrin, A. J., Wee, B., et al. (2010). Legionella pneumophila strain 130b possesses a unique combination of Type IV secretion systems and novel Dot/Icm secretion system effector proteins. J. Bacteriol. 192, 6001–6016. doi: 10.1128/JB.00778-10
Segata, N., Bornigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304. doi: 10.1038/ncomms3304
Shariat, N., and Dudley, E. G. (2014). CRISPRs: molecular signatures used for pathogen subtyping. Appl. Environ. Microbiol. 80, 430–439. doi: 10.1128/AEM.02790-13
Soderberg, M. A., Dao, J., Starkenburg, S. R., and Cianciotto, N. P. (2008). Importance of type II secretion for survival of Legionella pneumophila in tap water and in amoebae at low temperatures. Appl. Environ. Microbiol. 74, 5583–5588. doi: 10.1128/AEM.00067-08
Stelma, G. N., Lye, D. J., Smith, B. G., Messer, J. W., and Payment, P. (2004). Rare occurrence of heterotrophic bacteria with pathogenic potential in potable water. Int. J. Food Microbiol. 92, 249–254. doi: 10.1016/j.ijfoodmicro.2003.08.011
Sun, C. L., Thomas, B. C., Barrangou, R., and Banfield, J. F. (2016). Metagenomic reconstructions of bacterial CRISPR loci constrain population histories. ISME J. 10, 858–870. doi: 10.1038/ismej.2015.162
Tamaki, H., Zhang, R., Angly, F. E., Nakamura, S., Hong, P. Y., Yasunaga, T., et al. (2012). Metagenomic analysis of DNA viruses in a wastewater treatment plant in tropical climate. Environ. Microbiol. 14, 441–452. doi: 10.1111/j.1462-2920.2011.02630.x
Tennessen, K., Andersen, E., Clingenpeel, S., Rinke, C., Lundberg, D. S., Han, J., et al. (2016). ProDeGe: a computational protocol for fully automated decontamination of genomes. ISME J. 10, 269–272. doi: 10.1038/ismej.2015.100
Thomas, V., Loret, J. F., Jousset, M., and Greub, G. (2008). Biodiversity of amoebae and amoebae-resisting bacteria in a drinking water treatment plant. Environ. Microbiol. 10, 2728–2745. doi: 10.1111/j.1462-2920.2008.01693.x
Tilney, L. G., Harb, O. S., Connelly, P. S., Robinson, C. G., and Roy, C. R. (2001). How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114, 4637–4650.
Tortora, G., Funke, B., and Case, C. (2013). Microbiology: An Introduction, 11th Edn. Yorkshire: Pearson.
Vaerewijck, M. J. M., Huys, G., Palomino, J. C., Swings, J., and Portaels, F. (2005). Mycobacteria in drinking water distribution systems: ecology and significance for human health. FEMS Microbiol. Rev. 29, 911–934. doi: 10.1016/j.femsre.2005.02.001
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74. doi: 10.1126/science.1093857
Voth, D. E., Broederdorf, L. J., and Graham, J. G. (2012). Bacterial Type IV secretion systems: versatile virulence machines. Future Microbiol. 7, 241–257. doi: 10.2217/fmb.11.150
Wadowsky, R. M., Butler, L. J., Cook, M. K., Verma, S. M., Paul, M. A., Fields, B. S., et al. (1988). Growth-supporting activity for Legionella-pneumophila in tap water cultures and implication of hartmannellid amebas as growth-factors. Appl. Environ. Microbiol. 54, 2677–2682.
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wilson, J. W., Schurr, M. J., LeBlanc, C. L., Ramamurthy, R., Buchanan, K. L., and Nickerson, C. A. (2002). Mechanisms of bacterial pathogenicity. Postgrad. Med. J. 78, 216–224. doi: 10.1136/pmj.78.918.216
Wilson, R. W., Steingrube, V. A., Bottger, E. C., Springer, B., Brown-Elliott, B. A., Vincent, V., et al. (2001). Mycobacterium immunogenum sp nov., a novel species related to Mycobacterium abscessus and associated with clinical disease, pseudo-outbreaks and contaminated metalworking fluids: an international cooperative study on mycobacterial taxonomy. Int. J. Syst. Evol. Microbiol. 51, 1751–1764. doi: 10.1099/00207713-51-5-1751
Wu, H. J., Wang, A. H. J., and Jennings, M. P. (2008). Discovery of virulence factors of pathogenic bacteria. Curr. Opin. Chem. Biol. 12, 93–101. doi: 10.1016/j.cbpa.2008.01.023
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Yu, V. L., Plouffe, J. F., Pastoris, M. C., Stout, J. E., Schousboe, M., Widmer, A., et al. (2002). Distribution of Legionella species and serogroups isolated by culture in patients with sporadic community-acquired legionellosis: an international collaborative survey. J. Infect. Dis. 186, 127–128. doi: 10.1086/341087
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, Y., Oh, S., and Liu, W.-T. (2017). Impact of drinking water treatment and distribution on the microbiome continuum: an ecological disturbance's perspective. Environ. Microbiol. 19, 3163–3174. doi: 10.1111/1462-2920.13800
Keywords: virulence, genomic analysis, drinking water distribution systems, Legionella, Mycobacterium, Parachlamydia, Leptospira, CRISPR
Citation: Zhang Y, Kitajima M, Whittle AJ and Liu W-T (2017) Benefits of Genomic Insights and CRISPR-Cas Signatures to Monitor Potential Pathogens across Drinking Water Production and Distribution Systems. Front. Microbiol. 8:2036. doi: 10.3389/fmicb.2017.02036
Received: 27 June 2017; Accepted: 05 October 2017;
Published: 19 October 2017.
Edited by:
Timothy R. Julian, Swiss Federal Institute of Aquatic Science and Technology, SwitzerlandReviewed by:
Claire Bertelli, Simon Fraser University, CanadaCopyright © 2017 Zhang, Kitajima, Whittle and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Tso Liu, d3RsaXVAaWxsaW5vaXMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.