
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 October 2017
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.01993
This article is part of the Research Topic Molecular Self-assembly Inspired Advanced Materials Against Antibiotic-resistant Pathogens View all 6 articles
 Gabriel R. Fernandes1†
Gabriel R. Fernandes1† Aulus E. A. D. Barbosa1†
Aulus E. A. D. Barbosa1† Renan N. Almeida1
Renan N. Almeida1 Fabíola F. dos S. Castro2,3
Fabíola F. dos S. Castro2,3 Marina de C. P. da Ponte2
Marina de C. P. da Ponte2 Celio Faria-Junior4Fernanda M. P. Müller4
Celio Faria-Junior4Fernanda M. P. Müller4 Antônio A. B. Viana1
Antônio A. B. Viana1 Dario Grattapaglia1,5
Dario Grattapaglia1,5 Octavio L. Franco1*
Octavio L. Franco1* Sérgio A. Alencar1Simoni C. Dias1
Sérgio A. Alencar1Simoni C. Dias1Streptococcus pyogenes, also known as group A Streptococcus (GAS), is a human pathogen that causes diverse human diseases including streptococcal toxic shock syndrome (STSS). A GAS outbreak occurred in Brasilia, Brazil, during the second half of the year 2011, causing 26 deaths. Whole genome sequencing was performed using Illumina platform. The sequences were assembled and genes were predicted for comparative analysis with emm type 1 strains: MGAS5005 and M1 GAS. Genomics comparison revealed one of the invasive strains that differ from others isolates and from emm 1 reference genomes. Also, the new invasive strain showed differences in the content of virulence factors compared to other isolated in the same outbreak. The evolution of contemporary GAS strains is strongly associated with horizontal gene transfer. This is the first genomic study of a Streptococcal emm 1 outbreak in Brazil, and revealed the rapid bacterial evolution leading to new clones. The emergence of new invasive strains can be a consequence of the injudicious use of antibiotics in Brazil during the past decades.
Streptococcus pyogenes, a group A Streptococcus (GAS), is a human pathogen. This Gram-positive facultative anaerobe bacterium is responsible for many infections, including pharyngitis, scarlet fever, and cellulitis. GAS is also attributed to life-threatening diseases – such as necrotizing fasciitis as streptococcal toxic shock syndrome (STSS) – and post-infection sequelae, such as Rheumatic fever (Al-ajmi et al., 2012; Hondorp et al., 2012). In past decades, Streptococcal infection, an important health problem, with 660,000 new cases every year (Carapetis et al., 2005). The emergence of more virulent strains, antimicrobial resistance, the increase in the immunologically depleted patient population, and socio-demographic status are factors that facilitate bacterial transmission (Martin et al., 2011; Steer et al., 2012).
Some GAS strains associated with invasive infections can produce exotoxins and specific superantigens that lead to systemic inflammatory responses that result in the most severe infections (Unnikrishnan et al., 2002). Many virulence factors are responsible for the pathogenesis
mechanism, such as M-protein that plays a significant role in phagocytosis evasion (Courtney et al., 2006). The two-component system, known as covRS, is responsible for repressing the expression of important virulence factors, such as streptolysin S and streptodornase. Mutations in covRS gene may lead to enhanced virulence (Graham et al., 2002).
The whole genome sequencing of different GAS isolates can help to understand the factors that may lead to invasive infections. The samples were collected from patients during an outbreak of invasive S. pyogenes that occurred in the city of Brasília, Brazil. Four strains of S. pyogenes were isolated from blood of patients with flu-like symptoms, such as high fever, tonsillitis, respiratory failure, and petechiae; and one strain was isolated from the nasal cavity of a patient with pharyngitis. Comparative analysis of the assembled genomes allows the identification of the main virulence factors that could be related to the invasive infection. The data presented here reports the first invasive S. pyogenes genomic analysis in South America up to date.
Bacterial samples were collected during an outbreak of invasive S. pyogenes infection that occurred in the city of Brasília, in Brazil, in the period from August to December in 2011, when 101 cases were reported and 26 resulted in deaths. Four samples were isolated from the blood of patients who died due to infection, cultivated in 5% defibrinated sheep blood agar and underwent 24 h incubation 36 ± 1°C with 5% CO2. The bacterial species were identified using automated method (MicroScan WalkAway, Siemens Healthcare Systems) according to manufacturer’s instructions. Standard biochemical tests were also used to confirm the identification of bacterial species. Isolates were frozen at -70°C.
The antimicrobial susceptibility testing was also performed according to manufacturer’s instructions (MicroScan WalkAway, Siemens Healthcare Systems) and by the Kirby-Bauer disk diffusion using CLSI procedures. In order to determine the MIC for vancomycin, E-test method (bioMerieux Inc., Hazelwood, MO, United States) was used in accordance to the manufacturer’s instructions. The E-test MIC readings were performed to conform the dilution scale established by the Clinical and Laboratory Standards Institute for the broth microdilution method. Quality control of susceptibility testing panels from MicroScan was done by testing Streptococcus pneumoniae ATCC49619. The antibiotic testing results were interpreted in their susceptibility using the CLSI. S. pyogenes strains were typed according to their susceptibility to ampicillin, penicillin, ceftriaxone, cefepime, clindamycin, erythromycin, tetracycline, and vancomycin.
Patients were hospitalized in three different hospitals in Brasília. Table 1 describes the symptoms manifested by each patient. Patients were anonymized and informed consent was not required. This study was approved by the research ethics committee and registered with the number 16131213.0.0000.5553.

TABLE 1. Description of the symptoms observed in patients infected with invasive and non-invasive S. pyogenes.
Four selected S. pyogenes samples (Sp1–Sp4) had their genomic DNA extracted using a CTAB protocol described previously (Clarke, 2009). The 10 ml of overnight cultures were centrifuged and suspended in 300 μl of CTAB lysis buffer (2% CTAB, 1.4 M NaCl, 100 mM Tris-HCl pH 8.0, 20 mM EDTA and 0.2% mercaptoethanol). The suspensions were left for 30 min in a 65°C dry bath incubator and, after cell lysis, one volume of chloroform: isoamyl alcohol (24:1) was added to the samples. The samples were centrifuged at 10.000 RPM for 10 min and the aqueous phase were transferred to new microtubes. The chromosomal DNAs were precipitated with 0.6 volumes of isopropanol, centrifuged at 10.000 RPM for 5 min, washed with 70% ethanol, and resuspended in 100 μl TE buffer. All procedures were performed in a biosafety level 2 lab. Before DNA sequencing the samples were additionally purified with PowerClean DNA Clean-Up Kit (Mobio).
The sequencing libraries were constructed using the TruSeq PE Cluster Kit, following the manufacturer instructions. The whole genome of each sample was sequenced with paired-end libraries (2 × 75 bp) to high depth (all above 300×) using TruSeq SBS kit (Illumina) in the Illumina GA II platform at the Federal District High-throughput Genomic Center (Brasília, Brazil).
Data pre-processing was carried out by removing low-quality reads using the Trimmomatic tool (Bolger et al., 2014). Leading and trailing nucleotides with quality below eight were excluded, as well as ‘N’ bases. A 4-base wide sliding window was set to scan the reads and cut when the average quality per base dropped below 20. All pre-processed reads below 36 bases long were removed.
The high-quality paired-end reads resulted from pre-processing were then used for de novo genome assembly of each sample separately using SPAdes (Bankevich et al., 2012). The software Quast (Gurevich et al., 2013) was used to assess the assembly stats and quality.
The assembled genomes were aligned to the Streptococcus pyogenes MGAS5005 (Accession CP000017.2) and M1 (Accession AE004092.2) genomes to identify structural variation. The alignment and visualization were performed using the BRIG (Alikhan et al., 2011) tool.
Multiple genome alignments were performed using the DNA sequences of the S. pyogenes available in Ensembl Bacteria database and our assembled contigs. The alignment was carried out using Mauve (Darling et al., 2010) and the phylogenetic tree is an output of the progressive alignment performed with the default parameters.
The raw sequence data used for the assembly of the genome of each S. pyogenes sample (Sp1, Sp2, Sp3, Sp4, and Sp5) have been deposited at NCBI SRA under accession number SRP040580.
The genes were predicted using a locally installed version of GeneMark (Besemer and Borodovsky, 2005). GeneMark suite was used to build the HMM model for prokaryotes using the assembled genomes as references. The produced model was used to generate the GFF file, protein and CDS FASTA files for each genome.
A local orthology clustering was also done using the OrthoMCL software (Li et al., 2003). All predicted proteins from each genome were used as input to perform the analysis, and the procedure was run with default parameters.
The predicted proteins were functionally annotated using a BLAST search in a KEGG Orthology database enriched with UniProt entries (Fernandes et al., 2008).
The marker genes for MLST analysis were obtained in the MLST Streptococcus pyogenes database1 (Jolley and Maiden, 2010). Allele sequences were downloaded and formatted for BLAST (Altschul et al., 1990) search. The assembled contigs were aligned against the alleles database. The MLST was determined by the combination of the best hit with 100% of identity for each marker gene.
A BLAST alignment identified the emm gene type among the predicted genes. We used emm sequences downloaded from the Centers for Disease Control and Prevention website2 as the reference for the search. The best alignment hit with 100% of coverage and identity was used to assign the emm type.
Virulence factors were identified in the predicted proteome using a BLAST search. The reference database was downloaded from VFDB (Chen et al., 2005) and the best alignment for each protein was used as virulence signature.
Five S. pyogenes samples were collected, isolated, and submitted to sequencing. The information about the pathogens, as well as their hosts, is summarized in Table 1. The DNA was sequenced, and produced reads covered, in average, 340 times the genome length. The assembled genomes were around 1.8 Mb long. The number of predicted coding genes is around 1800 on the invasive strains. Table 2 reports the assembly quality measures.
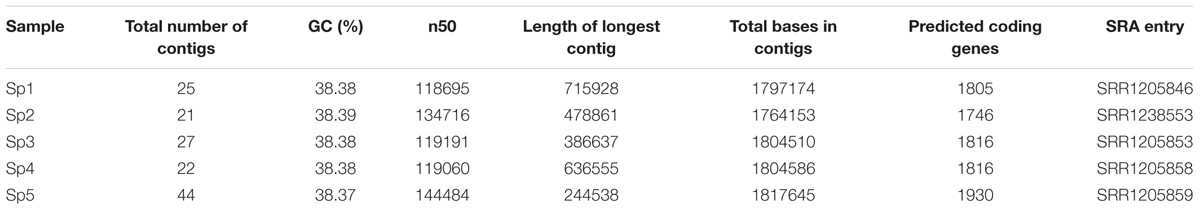
TABLE 2. Assembled genome metrics for each S. pyogenes sample.
All the predicted proteins from invasive isolates, combined with the proteins from MGAS5005 and M1 GAS, were arranged into 1918 orthologous groups. Clusters comprising proteins from all six isolates summed 1486. One hundred and seventy-two groups contained proteins only from the MGAS5005-like invasive stains. Figure 1 shows the number of genes present in different genomes. All the non-core genes were aligned to visualize exclusive groups and sequences absent in only one genome. As show in Figure 2, there are several exclusive genes from M1 GAS genome, as well as sequences that are missing only in this strain. We could identify 44 genes present exclusively in MGAS5005 genome. Among them, we can identify some genes related to genetic processing: transposase, integrase, relaxase, transcriptional regulator, ribosomal assembly, and energetic metabolism. In isolate Sp2 there were 51 absent genes, most of them are from prophage origin, including the streptodornase spd3 gene and an antirepressor. The gene products are listed in Supplementary Tables S1 and S2, for MGAS5005 and Sp2 respectively.
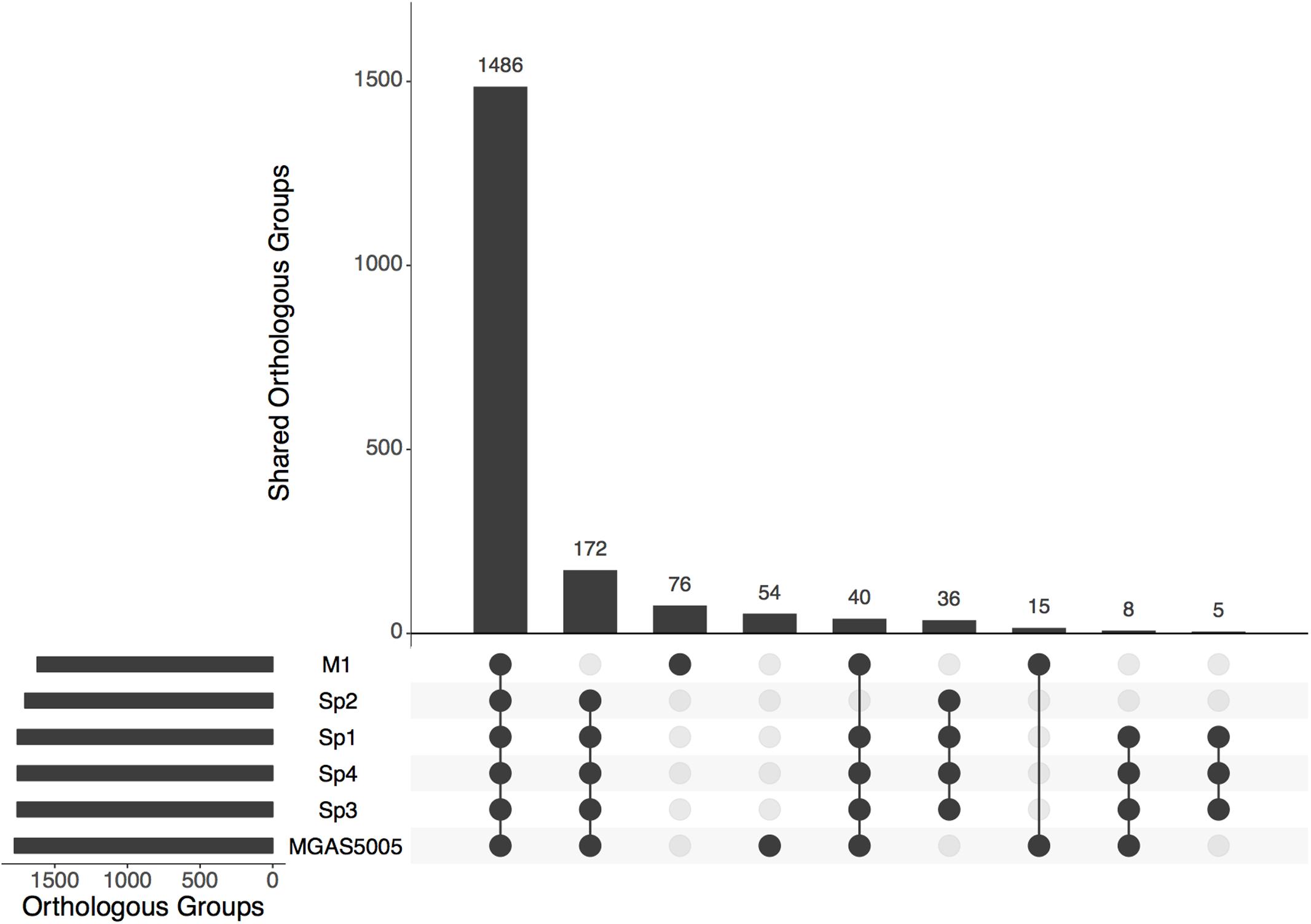
FIGURE 1. Number of orthologous genes shared six compared strains. Vertical bars show the number of genes shared by the combination of genomes represented in the horizontal axis.
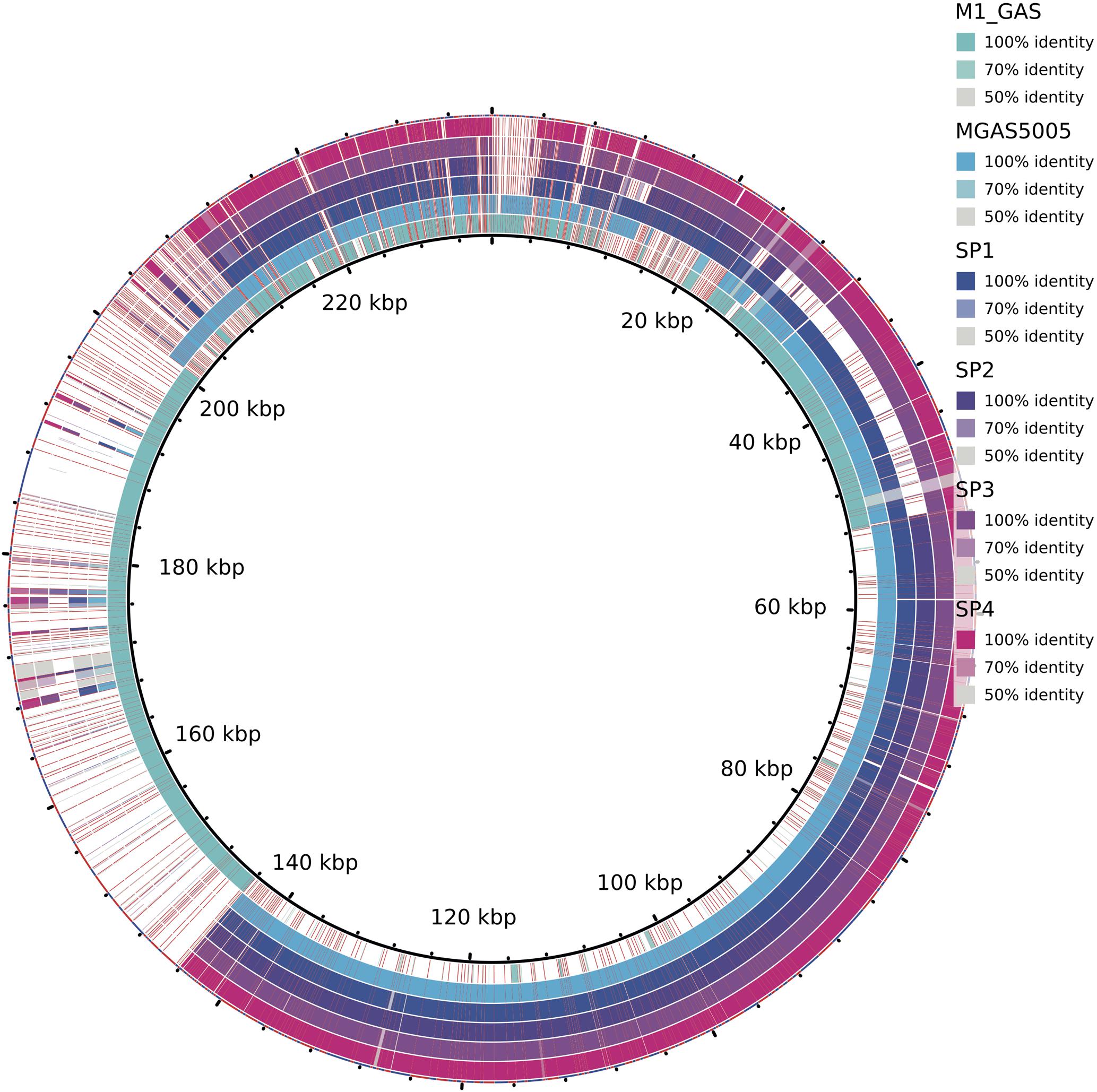
FIGURE 2. Alignment of non-core genes in Sp1–Sp4, MGAS5005, and M1 GAS strains. Colors represent the alignment identity. The white spaces mean the absence of the gene in the respective genome.
The phylogenetic analysis compared the isolated strains with reference Streptococcus pyogenes reference genomes from ENSEMBL Bacteria. The whole-genome comparison revealed that Sp1, Sp3, and Sp4 share a common ancestor with the reference strain GA41345 (Supplementary Figure S1). A high similarity can be observed in the cDNA content as well. The Brazilian outbreak isolates Sp1, Sp3, and Sp4 show more similar gene content among each other than when compared to Sp2 (Supplementary Figure S2). The non-invasive strain, Sp5, shares a recent common ancestor with GA19681, an emm6 strain.
The whole genome alignment, using Streptococcus pyogenes MGAS5005 as the reference, showed that Sp2 is missing a 30 kb region located next to the 1,200,000 bp position of the reference genome (Figure 3). This missing fragment has the same genomic location and content as the previously described prophages Φ5005.2 and Φ370.3 (Sumby et al., 2005).
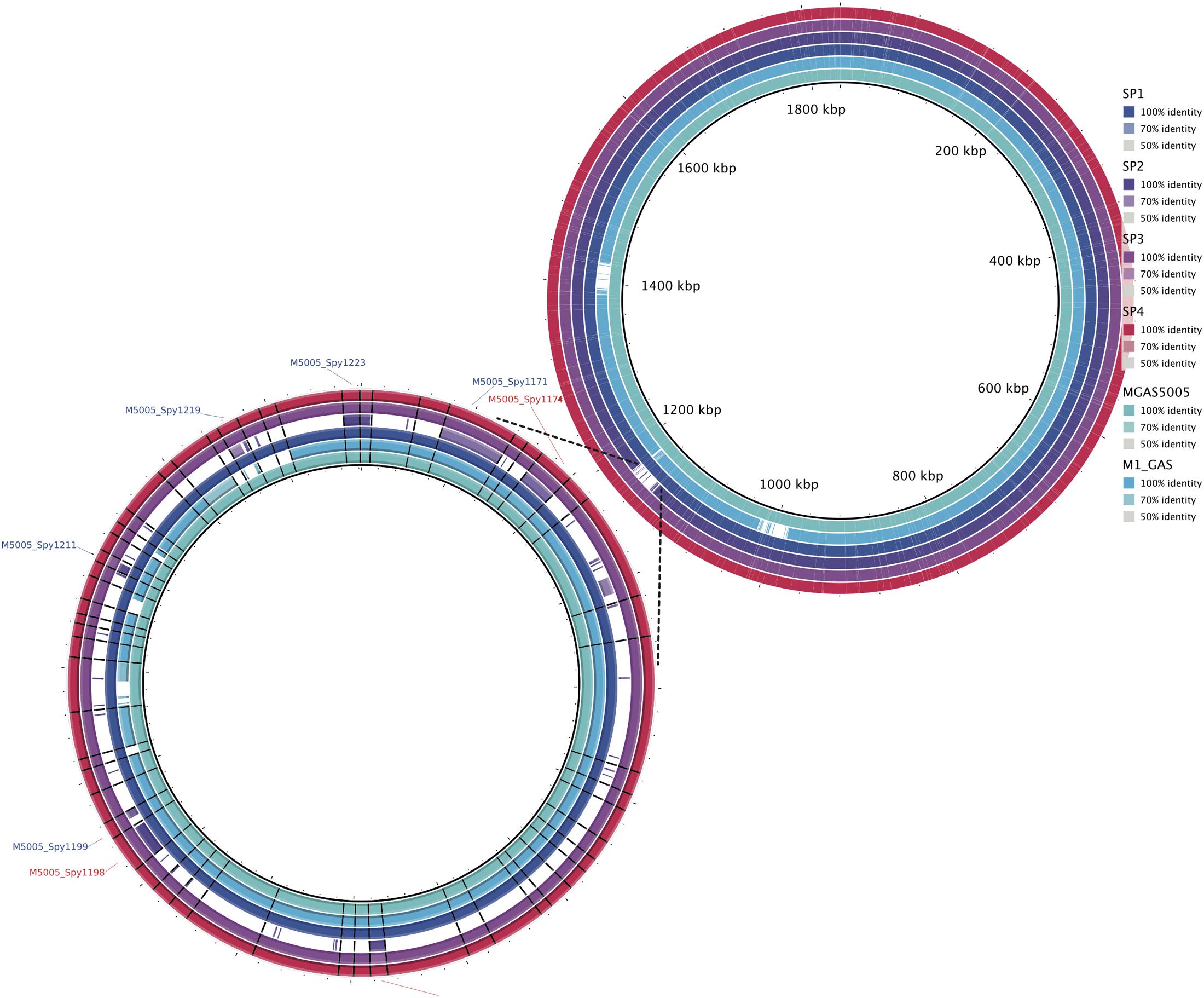
FIGURE 3. Genomic alignment of invasive strains (Sp1–Sp4) and M1 GAS using MGAS5005 as reference. A 30 kb deletion, at the position 1,200,000 bp, in Sp2 strain corresponds to the Φ5005.2 prophage. Similar deletion can be observed at 1,000,000 bp and 1,400,000 bp positions, in M1 GAS strain, referring to Φ5005.1 and Φ5005.3, respectively. The bottom-left circle expands the Φ5005.2 deletion to highlight the missing genes (white spaces) in Sp2.
The multilocus sequencing typing (MLST) approach revealed that Sp1–Sp4, as well as the reference emm1 strains, belong to the same allele type – Sequence type 28 – for the seven genes used: gki 4, gtr 3, muri 4, muts 4, recp 4, xpt 2, yqil 4. The Sp5 strain is the same sequencing type as other five emm6 strains (Figure 4).

FIGURE 4. Multilocus sequencing typing (MLST) dendrogram. The invasive outbreak isolates belong to the same sequencing type, which correspond to emm type 1 strains.
Analyzing the content of virulence factors, we observed that most of the genes are present in all invasive samples (Figure 5). The strain Sp2 is slightly different from others due to the absence of two factors: streptodornase and mitogen factor 3 (MF3). Another difference is the presence, only in Brazilian samples, of LPXTG-2 gene that produces an extracellular matrix binding protein.
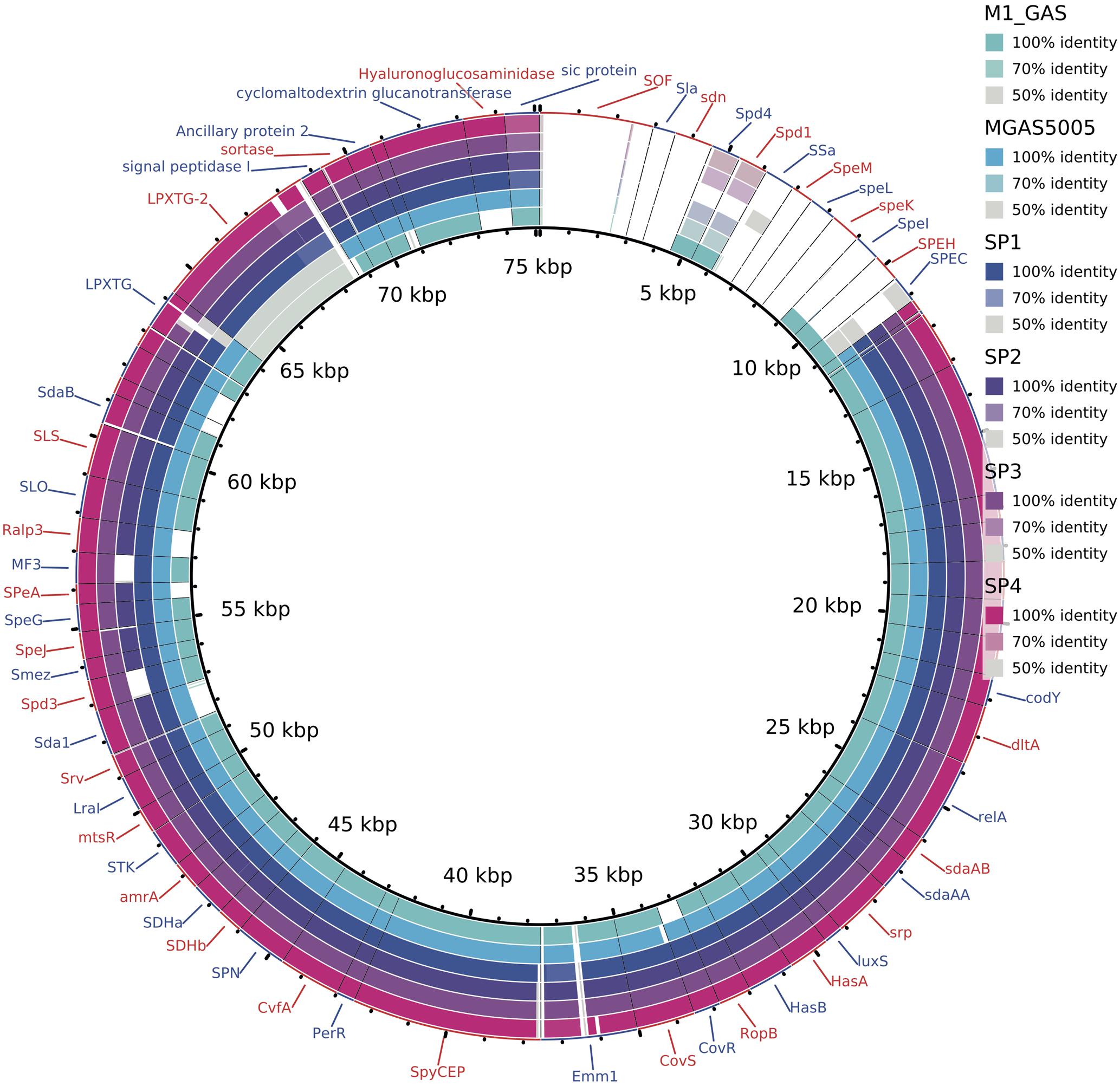
FIGURE 5. Alignment of predicted genes to virulence factors sequences. Colors, according to the legend, reflect the alignment identity, while white spaces report the absence of that gene. Two genes are missing exclusively in Sp2 genome: MF3 and spd3. LPXTG-2 proteins are conserved exclusively in the outbreak clones, while in M1GAS and MGAS5005 genomes it presents low identity to the reference sequence.
Group A Streptococci infections have been reported as a worldwide public health problem. Several isolates were sequenced and analyzed to better understand the infection mechanism, evolution and to suggest alternatives to slow down the spreading (Nasser et al., 2014). This is the first report of this kind of outbreak in South America.
The information brought from the genome sequencing shows some differences between invasive and non-invasive strains (Nasser et al., 2014). As expected, the pharynx isolate, Sp5, has a different gene content and genomic structure when compared to the invasive isolates. This dissimilarity corroborates with the emm type and MLST classification. Moreover, our invasive isolates share a very similar genomic content, with the exception of a 30 kb deletion observed in Sp2. Associated with host factors that are not considered in this study, these genomics structures can be related to the lethality of infection.
The characterization of the outbreak strain, based on comparison with other emm1 strains, MGAS5005 (Sumby et al., 2005) and M1 GAS (Ferretti et al., 2001), brings some information about the gene content of the clone that generated the Brazilian outbreak. As shown in Figures 1, 2, our strains have a similar functional repertoire when compared to the contemporary emm1 reference. This group, when compared to M1 GAS, acquired two large regions (Figure 3). These regions correspond to the prophages Φ5005.1, at position 1,000,000bp, and Φ5005.3, at position 1,400,000 bp (Sumby et al., 2005). Lateral gene transfer is known as a source of innovation in bacteria (Ochman et al., 2000). This event brings ecological adaptation and is related to pathogenesis mechanism. The prophages are linked to the emergence of new invasive strains (Nakagawa et al., 2003). Prophage Φ5005.2 deletion in Sp2 suggests the rise of new GAS clone (Figure 3). Pieces of evidence of this episode have recently been reported in six clones among 3,615 sequenced (Nasser et al., 2014), supporting that this is a rare variation. The Φ5005.2 prophage deletion does not strongly change the cellular function since most of the information is phage-related proteins. However, one virulence factor is missing due to this event: streptodornase spd3. This gene encodes protein harboring DNAse activity to break the DNA framework of neutrophil extracellular traps (NETs) (Walker et al., 2007; Uchiyama et al., 2012). Another important protein missing is a phage antirepressor that relieves the inhibition promoted by cl-like repressor proteins. It binds to the repressor after initiation of SOS response (Davis et al., 2002). The virulence factor MF3 is also absent in Sp2 strain (Figure 5). The MF3 performs endonuclease activity. Digesting DNA released from dead cells, the enzyme reduces the viscosity of pus and allows the organism greater motility, contributing to the invasion (Bisno et al., 2003). The presence of SdaB, another DNase with the same function, balances the absence of MF3.
Brazilian isolates contain a protein not identified in previous GAS emm1 references: LPXTG-2, which is a cell wall anchor domain protein. This extracellular-matrix-binding protein contains eight copies of a conserved domain DUF1542 (pfam07564) associated with antibiotic resistance and cellular adhesion (Clarke et al., 2002). The cited domain is also present in five other Streptococcus pyogenes strains: M1, M12, M28, M4, and M49), and in Streptococcus and Staphylococcus species, that are present in throat infections as well. The cellular adhesion promoted by the LPxTG proteins is essential for a successful infection (Lofling et al., 2011). The LPxTG sequence motif allows covalent linkage of extracellular-matrix proteins with the bacterial peptidoglycan via the activity of sortases (Kreikemeyer, 2004). These proteins are potential targets for drugs and prevention (Flock, 1999).
Although all the emm1 genomes studied belong to the same MLST group, they have some gene content variation. MGAS5005 has a group of genes that is not present in Brazilian isolates. Our clones miss genes related to genetic information processing and bacterial evolution, such as transposases, relaxase, integrase, ribosomal protein, and chaperone. These genes are not essential to bacterial survival, or are overrepresented in the genome (Jordan et al., 2002). The ribosomal protein L33 is related to 70S ribosome assembly, although its absence does not compromise the ribosomal function (Maguire and Wild, 1997). 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase is an essential enzyme for the isoprenoids biosynthesis. In Mycobacterium tuberculosis, isoprenoids play a role in the biosynthesis of structural components of the cell wall, being a potential target for antimicrobial drugs (Andreassi et al., 2004; Kang et al., 2011). The usage of antibiotics increase in Brazil during the past decades is 76%, while the world average is 36% (Van Boeckel et al., 2014). This indiscriminate consumption may have led to a strain resistant to these isoprenoids targeted drugs. Other changes are related to energetic metabolism. Tagatose-6-phosphate kinase plays a role in catabolism of lactose and galactose. It also acts as a metabolic sensor, regulating the gene expression by metabolic enzymes, allowing essential transcription programs to be coupled with perceived nutritional status (Loughman and Caparon, 2006). Another missing gene codifies the pyruvate phosphate dikinase. This enzyme is partially deleted in MGAS5005 genome, producing a 69aa product, whereas the protein is 881aa long in Streptococcus agalactiae 2603. This full deletion reflects the adaptation in energy metabolism since the bacteria is even more related and adapted to the host.
Regardless of the gain and gene loss events, the isolates maintained their invasive phenotype. This can be explained by the conservation of many important virulence factors with different functions. Some of them are transcription regulators: Mga, Mga2 (Hondorp et al., 2012), Ralp3 (Kwinn et al., 2007), and CovR (Alam et al., 2013). These transcriptional factors are responsible for transcriptional activation of virulence factors such as Sic, M protein, ScpA, hyaluronic acid capsule, and Sda (Kwinn et al., 2007; Hause and McIver, 2012; Alam et al., 2013). The sequenced covRS genes had no variations when compared to the reference genomes. In addition, Ralp3 may also act in epithelial cell invasion and bloodstream survival (Siemens et al., 2012; Alam et al., 2013). In M49 S. pyogenes strains, the inactivation of Ralp3 reduces bacteria attachment and internalization into human keratinocyte. This transcriptional factor controls the expression of several metabolic functions and virulence factors (Siemens et al., 2012). These additional transcriptional factors could provide the invasive strains with higher levels of expression of essential virulence factors in the processes of toxic shock syndrome and, thus, increase their ability to invade other tissues causing invasive infection. In our patients, S. pyogenes was isolated directly from blood, indicating a severe invasive infection. The enzyme hyaluronoglucosaminidase catalyzes the breakdown of hyaluronic acid in the host, increasing the permeability in tissues to large molecules (Starr and Engleberg, 2006). SpeA is a superantigen that induces the release of pro-inflammatory cytokines by T cells. This high cytokine release is related to the symptoms of Streptococcal toxic shock syndrome (Michaelsen et al., 2011). In addition, SpeA is able to deflect the host immune response through the activation a range of T-cell subsets, facilitating the establishment of a invasive infection (Maamary et al., 2012). Sda1 is the main streptodornase of invasive S. pyogenes and degrades DNA-based NETs (Walker et al., 2007).
As previously cited, the indiscriminate antibiotics consumption in Brazil increased twice more when compared to worldwide rise. (Van Boeckel et al., 2014). This behavior can explain the rapid evolution and emergence of such different strains in 5 months interval. When compared to MGAS5005, gene loss and prophage deletion can be a consequence of changes to adapt to a specific host and optimize the energy metabolism. In some Streptococci strains there is a gene content decay, most of them are not involved in basic cellular processes and tend to fit the metabolism to the available carbon source (Bolotin et al., 2004).
Genomic data reported here have shed some light on the outbreaks caused by S. pyogenes. While four strains have virulence factors content highly similar to the MGAS5005 strain, the Sp2 isolate has different genomic organization and virulence factors composition. This suggests the emergence of new S. pyogenes clones due to rapid evolution. The MGAS5005 strain is invasive and dispersed worldwide, and this was the first time that a 5005-like outbreak is genetically characterized in South America. Additionally, the great capacity for dispersal of this aggressive bacterium is a serious public health issue. The stress caused by the unselective usage of antimicrobial drugs, which is very common in Brazil, could have led to the rapid evolution and adaptation of this pathogen. The presence of a new LPXTG protein, not observed in MGAS5005 and M1 GAS, is an evidence of a common gain of function for better interaction with the host. This protein can be used as marker to identify these modern 5005-like infections. The genome sequence can be used to develop specific drugs to control the infections. In summary, an improvement in surveillance systems is extremely urgent, so that outbreaks of invasive S. pyogenes may be identified and contained in the near future.
AB, RA, SA, AV, DG, and GF performed the genomic analysis. FC, MP, FM, and CF-J collected the samples. GF, AB, SD, and OF wrote the manuscript. All authors designed the experiments, read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors gratefully thank the Federal District Research Foundation (FAPDF – Fundação de Apoio a Pesquisa do Distrito Federal) for the financial support through grant GENOMICA-DF (193.000.158/2008). The authors thank the Brazilian Council for Scientific and Technological Development (CNPq) for its continued support through research fellowships.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.01993/full#supplementary-material
Al-ajmi, J. A., Hill, P., Boyleb, C. O., Garcia, M. L., Malkawi, M., George, A., et al. (2012). Group A Streptococcus toxic shock syndrome: an outbreak report and review of the literature. J. Infect. Public Health 5, 388–393. doi: 10.1016/j.jiph.2012.07.006
Alam, F. M., Turner, C. E., Smith, K., Wiles, S., and Sriskandan, S. (2013). Inactivation of the CovR/S virulence regulator impairs infection in an improved murine model of Streptococcus pyogenes naso-pharyngeal infection. PLOS ONE 8:e61655. doi: 10.1371/journal.pone.0061655
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andreassi, J. L., Dabovic, K., and Leyh, T. S. (2004). Streptococcus pneumoniae isoprenoid biosynthesis is downregulated by diphosphomevalonate: an antimicrobial target. Biochemistry 43, 16461–16466. doi: 10.1021/bi048075t
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Besemer, J., and Borodovsky, M. (2005). GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454. doi: 10.1093/nar/gki487
Bisno, A. L., Brito, M. O., and Collins, C. M. (2003). Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3, 191–200. doi: 10.1016/S1473-3099(03)00576-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolotin, A., Quinquis, B., Renault, P., Sorokin, A., Ehrlich, S. D., Kulakauskas, S., et al. (2004). Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat. Biotechnol. 22, 1554–1558. doi: 10.1038/nbt1034
Carapetis, J. R., Steer, A. C., Mulholland, E. K., and Weber, M. (2005). The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694. doi: 10.1016/S1473-3099(05)70267-X
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Clarke, J. D. (2009). Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harb. Protoc. 2009:pdb.prot5177. doi: 10.1101/pdb.prot5177
Clarke, S. R., Harris, L. G., Richards, R. G., and Foster, S. J. (2002). Analysis of Ebh, a 1.1-megadalton cell wall-associated fibronectin-binding protein of Staphylococcus aureus. Infect. Immun. 70, 6680–6687. doi: 10.1128/IAI.70.12.6680-6687.2002
Courtney, H. S., Hasty, D. L., and Dale, J. B. (2006). Anti-phagocytic mechanisms of Streptococcus pyogenes: binding of fibrinogen to M-related protein. Mol. Microbiol. 59, 936–947. doi: 10.1111/j.1365-2958.2005.04977.x
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Davis, B. M., Kimsey, H. H., Kane, A. V., and Waldor, M. K. (2002). A satellite phage-encoded antirepressor induces repressor aggregation and cholera toxin gene transfer. EMBO J. 21, 4240–4249. doi: 10.1093/emboj/cdf427
Fernandes, G. R., Barbosa, D. V. C., Prosdocimi, F., Pena, I. A., Santana-Santos, L., Coelho Junior, O., et al. (2008). A procedure to recruit members to enlarge protein family databases–the building of UECOG (UniRef-Enriched COG Database) as a model. Genet. Mol. Res. 7, 910–924.
Ferretti, J. J., McShan, W. M., Ajdic, D., Savic, D. J., Savic, G., Lyon, K., et al. (2001). Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. U.S.A. 98, 4658–4663. doi: 10.1073/pnas.071559398
Flock, J. I. (1999). Extracellular-matrix-binding proteins as targets for the prevention of Staphylococcus aureus infections. Mol. Med. Today 5, 532–537.
Graham, M. R., Smoot, L. M., Migliaccio, C. A. L., Virtaneva, K., Sturdevant, D. E., Porcella, S. F., et al. (2002). Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U.S.A. 99, 13855–13860. doi: 10.1073/pnas.202353699
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hause, L. L., and McIver, K. S. (2012). Nucleotides critical for the interaction of the Streptococcus pyogenes Mga virulence regulator with Mga-regulated promoter sequences. J. Bacteriol. 194, 4904–4919. doi: 10.1128/JB.00809-12
Hondorp, E. R., Hou, S. C., Hempstead, A. D., Hause, L. L., Beckett, D. M., and McIver, K. S. (2012). Characterization of the Group A Streptococcus Mga virulence regulator reveals a role for the C-terminal region in oligomerization and transcriptional activation. Mol. Microbiol. 83, 953–967.
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Jordan, I. K., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2002). Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 12, 962–968. doi: 10.1101/gr.87702
Kang, H. J., Coulibaly, F., Proft, T., and Baker, E. N. (2011). Crystal structure of Spy0129, a Streptococcus pyogenes class B sortase involved in pilus assembly. PLOS ONE 6:e15969. doi: 10.1371/journal.pone.0015969
Kreikemeyer, B. (2004). The intracellular status of Streptococcus pyogenes: role of extracellular matrix-binding proteins and their regulation. Int. J. Med. Microbiol. 294, 177–188. doi: 10.1016/j.ijmm.2004.06.017
Kwinn, L. A., Khosravi, A., Aziz, R. K., Timmer, A. M., Doran, K. S., Kotb, M., et al. (2007). Genetic characterization and virulence role of the RALP3/LSA locus upstream of the streptolysin s operon in invasive M1T1 Group A Streptococcus. J. Bacteriol. 189, 1322–1329. doi: 10.1128/JB.01256-06
Li, L., Stoeckert, C. J. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lofling, J., Vimberg, V., Battig, P., and Henriques-Normark, B. (2011). Cellular interactions by LPxTG-anchored pneumococcal adhesins and their streptococcal homologues. Cell Microbiol. 13, 186–197. doi: 10.1111/j.1462-5822.2010.01560.x
Loughman, J. A., and Caparon, M. G. (2006). A novel adaptation of aldolase regulates virulence in Streptococcus pyogenes. EMBO J. 25, 5414–5422. doi: 10.1038/sj.emboj.7601393
Maamary, P. G., Ben Zakour, N. L., Cole, J. N., Hollands, A., Aziz, R. K., Barnett, T. C., et al. (2012). Tracing the evolutionary history of the pandemic group A streptococcal M1T1 clone. FASEB J. 26, 4675–4684. doi: 10.1096/fj.12-212142
Maguire, B. A., and Wild, D. G. (1997). The roles of proteins L28 and L33 in the assembly and function of Escherichia coli ribosomes in vivo. Mol. Microbiol. 23, 237–245.
Martin, J., Murchan, S., O’Flanagan, D., and Fitzpatrick, F. (2011). Invasive Group A streptococcal disease in Ireland, 2004 to 2010. Euro Surveil. 16:19988.
Michaelsen, T. E., Andreasson, I. K. G., Langerud, B. K., and Caugant, D. A. (2011). Similar superantigen gene profiles and superantigen activity in norwegian isolates of invasive and non-invasive group A streptococci. Scand. J. Immunol. 74, 423–429. doi: 10.1111/j.1365-3083.2011.02594.x
Nakagawa, I., Kurokawa, K., Yamashita, A., Nakata, M., Tomiyasu, Y., Okahashi, N., et al. (2003). Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 13, 1042–1055. doi: 10.1101/gr.1096703
Nasser, W., Beres, S. B., Olsen, R. J., Dean, M. A., Rice, K. A., Long, S. W., et al. (2014). Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc. Natl. Acad. Sci. U.S.A. 111, E1768–E1776. doi: 10.1073/pnas.1403138111
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Siemens, N., Fiedler, T., Normann, J., Klein, J., Munch, R., Patenge, N., et al. (2012). Effects of the ERES pathogenicity region regulator Ralp3 on Streptococcus pyogenes serotype M49 virulence factor expression. J. Bacteriol. 194, 3618–3626. doi: 10.1128/JB.00227-12
Starr, C. R., and Engleberg, N. C. (2006). Role of hyaluronidase in subcutaneous spread and growth of group A streptococcus. Infect. Immun. 74, 40–48. doi: 10.1128/IAI.74.1.40-48.2006
Steer, A. C., Lamagni, T., Curtis, N., and Carapetis, J. R. (2012). Invasive group a streptococcal disease: epidemiology, pathogenesis and management. Drugs 72, 1213–1227. doi: 10.2165/11634180-000000000-00000
Sumby, P., Porcella, S. F., Madrigal, A. G., Barbian, K. D., Virtaneva, K., Ricklefs, S. M., et al. (2005). Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192, 771–782. doi: 10.1086/432514
Uchiyama, S., Andreoni, F., Schuepbach, R. A., Nizet, V., and Zinkernagel, A. S. (2012). DNase Sda1 allows invasive M1T1 Group A Streptococcus to prevent TLR9-dependent recognition. PLOS Pathog. 8:e1002736. doi: 10.1371/journal.ppat.1002736
Unnikrishnan, M., Altmann, D. M., Proft, T., Wahid, F., Cohen, J., Fraser, J. D., et al. (2002). The bacterial superantigen streptococcal mitogenic exotoxin Z is the major immunoactive agent of Streptococcus pyogenes. J. Immunol. 169, 2561–2569.
Van Boeckel, T. P., Gandra, S., Ashok, A., Caudron, Q., Grenfell, B. T., Levin, S. A., et al. (2014). Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect. Dis. 14, 742–750. doi: 10.1016/S1473-3099(14)70780-7
Keywords: Streptococcus pyogenes, streptococcal toxic shock syndrome, invasive infection outbreak, emm, virulence factor, prophage
Citation: Fernandes GR, Barbosa AEAD, Almeida RN, Castro FFS, da Ponte MCP, Faria-Junior C, Müller FMP, Viana AAB, Grattapaglia D, Franco OL, Alencar SA and Dias SC (2017) Genomic Comparison among Lethal Invasive Strains of Streptococcus pyogenes Serotype M1. Front. Microbiol. 8:1993. doi: 10.3389/fmicb.2017.01993
Received: 15 November 2016; Accepted: 28 September 2017;
Published: 23 October 2017.
Edited by:
Martin G. Klotz, Washington State University Tri-Cities, United StatesReviewed by:
Stefania Stefani, Università degli Studi di Catania, ItalyCopyright © 2017 Fernandes, Barbosa, Almeida, Castro, da Ponte, Faria-Junior, Müller, Viana, Grattapaglia, Franco, Alencar and Dias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Octavio L. Franco, b2NmcmFuY29AZ21haWwuY29t
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.