
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 October 2017
Sec. Fungi and Their Interactions
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.01924
This article is part of the Research TopicTropical Fungal DiseasesView all 10 articles
 Vania A. Vicente1,2†
Vania A. Vicente1,2† Vinícius A. Weiss3,4†
Vinícius A. Weiss3,4† Amanda Bombassaro1
Amanda Bombassaro1 Leandro F. Moreno1,5,6Flávia F. Costa2Roberto T. Raittz3
Leandro F. Moreno1,5,6Flávia F. Costa2Roberto T. Raittz3 Aniele C. Leão2,3,4Renata R. Gomes1
Aniele C. Leão2,3,4Renata R. Gomes1 Anamelia L. Bocca7Gheniffer Fornari1
Anamelia L. Bocca7Gheniffer Fornari1 Raffael J. A. de Castro7Jiufeng Sun8Helisson Faoro4Michelle Z. Tadra-Sfeir4Valter Baura4Eduardo Balsanelli4Sandro R. Almeida9
Raffael J. A. de Castro7Jiufeng Sun8Helisson Faoro4Michelle Z. Tadra-Sfeir4Valter Baura4Eduardo Balsanelli4Sandro R. Almeida9 Suelen S. Dos Santos9Marcus de Melo Teixeira7,10Maria S. Soares Felipe11
Suelen S. Dos Santos9Marcus de Melo Teixeira7,10Maria S. Soares Felipe11 Mariana Machado Fidelis do Nascimento1
Mariana Machado Fidelis do Nascimento1 Fabio O. Pedrosa4Maria B. Steffens3,4
Fabio O. Pedrosa4Maria B. Steffens3,4 Derlene Attili-Angelis12Mohammad J. Najafzadeh13Flávio Queiroz-Telles1,14
Derlene Attili-Angelis12Mohammad J. Najafzadeh13Flávio Queiroz-Telles1,14 Emanuel M. Souza3,4*
Emanuel M. Souza3,4* Sybren De Hoog1,5,6*
Sybren De Hoog1,5,6*Fonsecaea and Cladophialophora are genera of black yeast-like fungi harboring agents of a mutilating implantation disease in humans, along with strictly environmental species. The current hypothesis suggests that those species reside in somewhat adverse microhabitats, and pathogenic siblings share virulence factors enabling survival in mammal tissue after coincidental inoculation driven by pathogenic adaptation. A comparative genomic analysis of environmental and pathogenic siblings of Fonsecaea and Cladophialophora was undertaken, including de novo assembly of F. erecta from plant material. The genome size of Fonsecaea species varied between 33.39 and 35.23 Mb, and the core genomes of those species comprises almost 70% of the genes. Expansions of protein domains such as glyoxalases and peptidases suggested ability for pathogenicity in clinical agents, while the use of nitrogen and degradation of phenolic compounds was enriched in environmental species. The similarity of carbohydrate-active vs. protein-degrading enzymes associated with the occurrence of virulence factors suggested a general tolerance to extreme conditions, which might explain the opportunistic tendency of Fonsecaea sibling species. Virulence was tested in the Galleria mellonella model and immunological assays were performed in order to support this hypothesis. Larvae infected by environmental F. erecta had a lower survival. Fungal macrophage murine co-culture showed that F. erecta induced high levels of TNF-α contributing to macrophage activation that could increase the ability to control intracellular fungal growth although hyphal death were not observed, suggesting a higher level of extremotolerance of environmental species.
Melanized fungi belonging to the order Chaetothyriales are clinically relevant as agents of a gamut of diseases in humans and animals, varying in severity from superficial to systemic and fatal infections (De Hoog et al., 2004; Badali et al., 2008; Seyedmousavi et al., 2011). A large number of species have complex life cycles, indicating dynamic niches or vectored transmission (Sudhadham et al., 2008). In the environment, they occupy adverse micro-habitats, which is probably stimulated by their low competitive ability toward co-occurring microorganisms, judged from the fact that their isolation is enhanced significantly by the use of selective methods (Vicente et al., 2014). Many species of Chaetothyriales cause implantation diseases from an environmental source. One of the common disorders is chromoblastomycosis, a mutilating and recalcitrant skin disease eventually leading to emerging eruptions. Fungal cells in host tissue provoke as an inflammatory granulomatous disease (De Hoog et al., 2007; de Azevedo et al., 2015a; Queiroz-Telles, 2015). Agents of chromoblastomycosis are traumatically inoculated from environmental sources such as plant thorns or wooden splinters carrying the respective opportunist (Salgado et al., 2004; De Hoog et al., 2007; Vicente et al., 2014). Those species are mainly found in Fonsecaea and Cladophialophora, of which F. pedrosoi, F. monophora, and Cladophialophora carrionii are recurrently recovered from patients in tropical and semi-arid climate zones, respectively around the globe (Xi et al., 2009; Queiroz-Telles, 2015). Recently, less common agents were described, such as F. nubica (Najafzadeh et al., 2010), F. pugnacius (de Azevedo et al., 2015b), and C. samoensis (Badali et al., 2010). The genera Fonsecaea and Cladophialophora are morphologically classified by differences in their conidial apparatus; however, DNA polymorphisms suggest that both genera are polyphyletic (De Hoog et al., 2007), as they are distributed within “bantiana-clade” and “carrionii-clade” groups of the family Herpotrichiellaceae (Chaetothyriales) (Vicente et al., 2014; de Azevedo et al., 2015b).
Closely related species of Fonsecaea differ significantly in their ecology and ability to cause infection in humans and animals (Vicente et al., 2014); virulence genes seem to be unequally distributed among members of the bantiana-clade. Fonsecaea pedrosoi and F. nubica are strictly associated to chromoblastomycosis, while F. monophora also causes primary brain disease (Surash et al., 2005; Takei et al., 2007; Koo et al., 2010). Fonsecaea multimorphosa and F. brasiliensis were isolated from disseminated infections in animal hosts (Najafzadeh et al., 2011; Vicente et al., 2012) and the environmental species F. erecta and F. minima were described from plants and no report from clinical cases (Vicente et al., 2014) has as yet been published.
Therefore, the central question of the present study concerns the difference in infectious potential between closely related members of Fonsecaea. Agents of chromoblastomycosis upon tissue invasion show dimorphism to muriform cells, while this behavior is not known from most plant debris-inhabiting siblings (Queiroz-Telles, 2015). Comparative genomic analysis of Fonsecaea pathogenic and non-pathogenic siblings was applied to highlight genes involved in fungal adaptation from a plant debris- to an animal-associated life style. To this aim, a de novo assembly of F. erecta and recently published black yeast genomes (Bombassaro et al., 2016; Costa et al., 2016; Leão et al., 2017; Teixeira et al., 2017) was included in the analysis.
A large-scale DNA extraction was conducted based on a method as described by Vicente et al. (2008). The strains were grown in Sabouraud broth for 7 days. The DNA was extracted by the cetyltrimethylammonium bromide (CTAB) method based and phenol-chloroform/isoamyl alcohol. Total DNA was purified with the Microbial DNA UltraClean™ kit. The genome was sequenced on MiSeq (Illumina™) sequencer using paired-end and mate-paired libraries and on Ion Proton (Thermo Fisher Scientific™) sequencer using single-end approaches. The library construction was done with Ion Plus Fragment Library Kit (Thermo Fisher Scientific™) and Nextera XT (Illumina™) following the manufacturer instructions. The read quality analysis was performed with FastQC and reads with quality below PHRED20 were removed (Andrews, 2016). The reads were assembled de novo using SPAdes v3.6.2 (Bankevich et al., 2012). The gap closure was performed with FGAP (Piro et al., 2014) and scaffolding with SSPACE (Boetzer et al., 2010). The mitochondrial genomes were assembled in Fonsecaea species by extracting reads using the complete mtDNA of Exophiala dermatitidis as reference. The reads were mapped using Bowtie2 (Langmead and Salzberg, 2012) and the mapped reads assembled with SPAdes v3.6.2 (Bankevich et al., 2012).
Protein-coding genes were predicted with GeneMark-ES v4.39 (Besemer, 2001). The automatic annotation was done by RAFTS3 (Vialle et al., 2016) best hits comparison with self-score cutoff of 0.5 using a black yeast protein database available on (www.broadinstitute.org/annotation/genome/Black_Yeasts/). Protein domain families and functional annotation was accessed using InterProScan5 (Quevillon et al., 2005). The tRNAs annotation used the ARAGORN software (Laslett and Canback, 2004). Putative enzymes and peptidases coding genes using CAZY (Cantarel et al., 2009) and peptidases coding genes using MEROPS database (Rawlings et al., 2015) and putative pathogen host interaction genes using PHI base (Winnenburg et al., 2007). Pathogen Host interacting (PHI) partners were identified by subjecting the predicted proteomes to BLASTp against the PHI database v4.2 with an E-value threshold of 10–5 with best hits.
We compared the genome of F. erecta CBS 125763T to 5 Fonsecaea species, including F. monophora, CBS 269.37T (Bombassaro et al., 2016), F. nubica CBS 269.64T (Costa et al., 2016), F. multimorphosa CBS 980.96T (Leão et al., 2017), Fonsecaea multimorphosa CBS 102226 (Teixeira et al., 2017), and F. pedrosoi CBS 271.37T (Teixeira et al., 2017) in addition to other 6 black yeast-like fungi belonging to the order Chaetothyriales (Teixeira et al., 2017): Cladophialophora carrionii CBS 160.54, Cladophialophora yegresii, Capronia semiimmersa, Cladophialophora bantiana, Cladophialophora psammophila, Cladophialophora immunda, and Rhinocladiella mackenziei (Table 2).
Protein sequences were compared using an all-vs.-all similarity search and self-score cutoff of 0.5 using RAFTS3 (Vialle et al., 2016). The clustering was done when at least one protein was shared amidst clusters. After the clustering verification step was done K-means and the cluster vectors where split into new clusters using the ratio of the cluster size and the number of organisms present in the analysis. For the resulting clusters it was calculated a centroid for each vector and chosen the best gene that represents each cluster based on the shortest distance. For both K-means and centroid analysis, a vectorial representation for the genes was created based on sparse k-mers sequences. A final clustering step was done using RAFTS all-vs.-all similarity searches with self-score of 90 (Vialle et al., 2016). The amino-acid sequences of each family were aligned with Muscle (Edgar, 2004) and poorly aligned regions were automatically removed using GBLOCKS (Talavera and Castresana, 2007). A maximum likelihood tree was done using PHYML (Guindon et al., 2005) and 1,000 bootstraps were used to infer branch support.
To identify functional expansions and contractions, InterPro domains were predicted using InterProScan5 (Quevillon et al., 2005) for 12 strains of black yeast species: 6 Fonsecaea species, 5 Cladophialophora species and 1 Coniosporium apollinis as outgroup (Table 2). Gene family evolution was estimated with CAFE version 3.0 (De Bie et al., 2006) using significance family-wide p-values threshold of <0.05 and VITERBI p < 0.001. To search for BIRTH (λ) values we run the program with the “-s” option. Two files were used as input in CAFE analyses: a table containing organism number of copies of each InterPro domain and an ultrametric tree.
In order to research genes related to pathogenicity through analysis of the core, clusters and perform a correlation analysis, initially were rescued the 22 yeast genomes available at Broad Institute (http://www.broadinstitute.org/annotation/genome/Black_Yeasts/GenomesInde) and including the Fonsecaea sibling associated to (sub)cutaneous and systemic infection, totaling 26 genomes. They were all (re) annotated using toolbox RAFTS3 (Vialle et al., 2016). The next step was to vectorize and cluster each gene, which generated 28,355 gene clusters equal or bigger the 50% of identity between them. This analysis was sized in an array with 26 rows × 28,355 columns, the rows being the organisms and the columns all the genes of all clustered organisms.
To perform the correlation analysis between the clusters using Point-biserial correlation to each of 28,355 clusters, we selected a set of genes that was used as reference to access a set of already known pathogenic genes, being them: Cell Division Control Protein 42 (KIV82855.1), Cytochrome P450 (KIW97819.1), Thioredoxin (XP_013289847.1), HSP60-like protein (KIW92920.1), HSP90-like protein (AYO21_00238), Homogentisate 1,2-dioxygenase (KIW31930.1), and two hypothetical proteins (AYO21_05248 and KIW22607.1) which were present in the same clusters of paraoxonases.
The correlations of each cluster with the frequency of these pathogenic set genes in the organisms were calculated according to formula (Figure 1), which was filtered in 1,803 clusters of genes. Analyzing the gene families within organism related to systemic infection (F. multimorphosa, C. bantiana, F. monophora, and R. mackenziei) and subcutaneous infection (F. pedrosoi, C. carrionii, F. monophora, and F. nubica) only 280 showed positive correlation above 80% with 5.5 × 10−7 of max e-value.
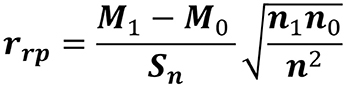
Figure 1. The point-biserial correlation coefficient: measure of the relationship between a continuous and a binary variable. For each protein of the 26 analyzed genomes, 0 and 1 scores correspond to the presence or absence of a protein of the binary variable, respectively. M1 is the mean of the presence of proteins and M0 is the mean of the missing proteins. The value “n” represents the total number of the proteins, where n1 is the total of proteins present and n0 are the total of the missing proteins. Sn is the standard deviation of the continuous variable.
The strains F. pedrosoi ATCC 46428, F. pedrosoi CBS 271.37, F. erecta CBS 125763 and F. monophora CBS 102248 were selected for this study. The yeast strains were grown on Sabouraud Glucose Agar (SGA; Himedia, Mumbai, India) at 28°C for 7 days, transferred to Potato Dextrose Broth (PDB; Himedia) and incubated under agitation (150 rpm) at 37°C. After 5 days, the cultures were allowed to settle in order to decant the larger particles such as hyphae and conidia. Fungal cells were separated by filtration through a 40 μm cell strainer, (BD) washed with PBS 1 × three times. Finally, the cells were re-suspended at 1 × 106 cells/mL for F. pedrosoi ATCC 46428, F. pedrosoi CBS 271.37, and F. erecta CBS 125763, or at 1 × 105 cells/mL for F. monophora CBS 102248.
Galleria mellonella larvae were selected using as criteria similar size and weight ranging 0.10–0.15 (g). For the survival experiments, each group consisted of 20 larvae. The selected larvae were inoculated by injecting 10 μL of the different inoculum in the last left pro-leg with a Hamilton syringe (0.75 mm diameter needle) according to Fuchs et al. (2010). The control group was inoculated with PBS and the same number of larvae. The following control groups were used in the experiment. The first group included the larvae that received 10 μL of PBS to monitoring survival mortality related to trauma. A second group of larvae (SHAM) received no injection and no injury. All larvae were placed in sterile Petri dishes and kept in the dark at 37°C. Mortality was monitored once per day. The death of the larvae was assessed by the lack of movement, no response to stimulation and discoloration of the cuticle. Melanization was checked every 24 h with a NIKON D3100 camera and images were analyzed. Survival curves were plotted and statistical analyses were performed using the Log-rank (Mantel-Cox) test with Graph Pad Prism software. Statistical differences were set at p < 0.05.
Larvae were fixed by immersion in Carnoy (60% methanol, 30% chloroform and 10% acetic acid) 7 days after infection. After 48 h the larvae were immersed in 70% ethanol. Then the sections were then embedded in paraffin wax, sectioned and stained with Periodic Acid-Schiff (PAS) for microscopic examination. The photomicrographs were obtained from Olympus BX41 microscope coupled with digital camera Olympus SC30.
The strains F. pedrosoi CBS 271.37 and F. erecta CBS 125763 were cultivated on Sabouraud Glucose Agar (SGA, Himedia) supplemented with 100 mg/L−1 chloramphenicol at 37°C. To obtain purified conidia and hyphae, fungi propagules were grown in PDB supplemented with 100 mg/L−1 chloramphenicol, in a rotary shaker (120 rpm) at 30°C. Fungal purification was performed according to Siqueira et al. (2017). Briefly, 15 days suspension containing conidia and hyphal fragments was subjected to successive filtrations through 70 μm and 40 μm cell strainers (BD). Hyphae retained on the 40 μm cell strainer were re-suspended in phosphate buffered saline (PBS), centrifuged at 1,000 × g and re-suspended in PBS. This process was repeated twice. Ninety-eight percent of 98% of this suspension consisted of hyphae. The 40 μm cell strainer filtrate containing conidia and small hyphal fragments was subjected to a filtration through 14 μm filter paper (J. Prolab, Brazil), washed twice in PBS, and recovered by centrifugation at 3,000 × g. This fraction contained conidia more than 98% pure.
Macrophage infection assays were adapted from Hayakawa et al. (2006), Palmeira et al. (2008) and Siqueira et al. (2017). Mouse macrophages (J774 cell line) were plated in DMEM (Dulbecco's Modified Eagle's medium, Sigma) supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and infected with conidia or hyphae of the Fonsecaea species at a multiplicity of infection (MOI) of 1. After 3 h of infection, non-phagocytized fungi were washed, fresh medium was added and co-culture was allowed to proceed for 24 h. For fungal burden determination (number of fungal cells in macrophages), macrophages were lysed with 0.05% SDS solution. Intracellular fungi were quantified by plating serial dilutions of cell lysates onto SDA medium, supplemented with 100 mg/L−1 chloramphenicol and cultivated at 30°C for 7 days. Fungal burden was then measured by counting fungi colony-forming units (CFU). The cell culture supernatants were used to determine tumor necrosis factor-α (TNF-α) levels by enzyme-linked immunosorbent assay (ELISA), in accordance with manufacture's (eBioscience) instructions. As positive control to TNF-α production, 500 ng/mL LPS (Escherichia coli serotype 0111:B, Sigma-Aldrich) was used. Results were expressed as number of CFU or pg/mL of cytokine ± standard deviation (SD).
BALB/c mice were maintained under standard laboratory conditions. Mice (10–12 weeks old males) were inoculated by injecting 50 μL (per hind footpad) of PBS containing 1 × 106 F. pedrosoi or F. erecta propagules obtained by mixing the purified hyphae and conidia in the proportion of 3:1, respectively. Five animals per group were euthanized with CO2 in an appropriate chamber at 7 and 14 days post-infection. The mice footpad were photographed, removed, weighed and thereafter homogenized in tubes with steel beads on a Precellys homogenizer. For fungal burden determination, homogenized tissue were diluted and plated as above mentioned. Results were expressed as number of CFU ± standard error of mean (SEM) per gram of fresh tissue. Cytokine production was measured from homogenized tissue obtained from infected and non-infected animals (healthy) by ELISA. The cytokines interleukin-1β (IL-1β), TNF-α, interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1/Ccl2) were measured with kits purchased from eBioscience and used according the manufacturer's instructions. Results were expressed as pg of cytokine ± standard error of mean (SEM) per 100 milligrams of tissue. All animal experimentation in this study were approved by the Federal University of Paraná Ethics Committee (approval certificate 1002) and performed according to the Committee's recommendations.
Whole genome sequencing of Fonsecaea erecta CBS 125763T was performed using Illumina Hiseq 2000 and yielded 1,534,038 paired-end reads with average insert size of 1 Kb ± 1 Kb and 3,133 mate-paired reads with average insert size of 5 Kb ± 4 Kb. To increase sequence coverage, two additional Ion Torrent shotgun libraries were sequenced generating 5.4 Gb and 25,219,375 single reads. The final high quality draft genome of Fonsecaea erecta comprised 57 scaffolds. The genome size was estimated to be 34.75 Mb, with average coverage of 60X and G+C content of 53%. Protein coding regions account for 18,279,031 bp, corresponding to 12,327 genes. A total of 12,090 proteins encoding genes, one rRNA multi-copy segment and 30 tRNA genes were predicted (Table 1).

Table 1. Fonsecaea erecta genome data assembly and quality.
Comparing F. erecta with related species of Chaetotryriales it was observed that all genomes used in this study including the reference species are similar in size. The genome size of Fonsecaea species varied between 33.39 and 35.23 Mb. The F. monophora genome is nearly 1.84 Mb larger in size following of the plant associate species F. erecta with 34.75, while the reference genome of the species plant-associated Cladophialophora yegresii presented a reduced size of 27.9 Mb. The total number of initial predicted genes in Fonsecaea varied between 11,681 in F. nubica to 12,527 in F. pedrosoi; between 10,944 (93.69%) and 11,948 (96.59%) of the genes identified as conserved hypothetical proteins. In addition, repetitive element identification was considered to be low in Fonsecaea siblings, ranging from 1.06 to 1.13% in F. multimorphosa to 1.93% in F. monophora (Table 2).
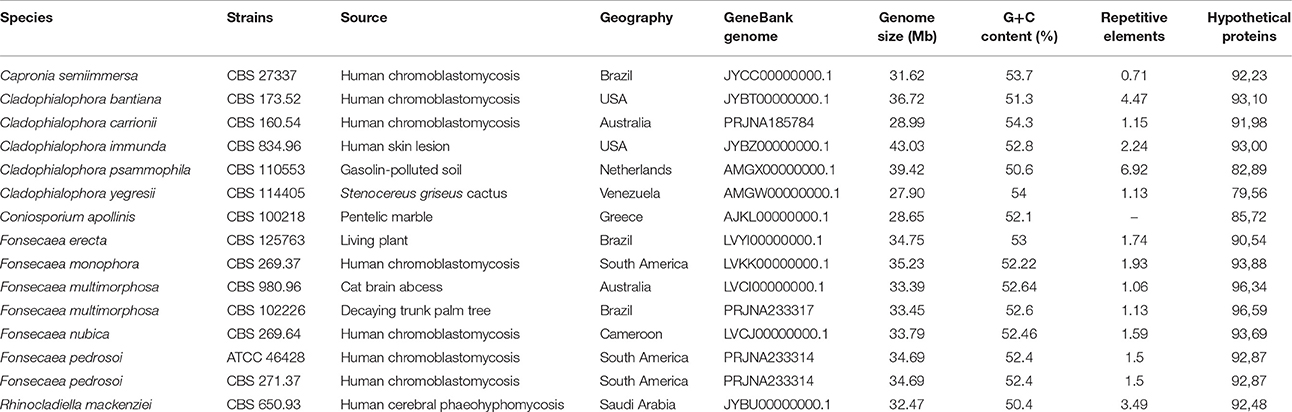
Table 2. Genome studied.
Fonsecaea erecta CBS 125763T and F. pedrosoi CBS 271.37T mtDNA was assembled in one contig, measuring 25.7 and 25 Kb, respectively. The mtDNA of F. monophora assembled into eight contigs comprising 24.7 Kb, the mtDNA of F. nubica resulted in a single contig with 24.5 Kb and the mtDNA of F. multimorphosa CBS 980.96T resulted in seven contigs with a size of 26.4 Kb. Although the gene composition of all mitochondrial genomes analyzed shared 16 protein coding genes involved in the respiratory chain and ATP synthesis, the synteny of these genes is not conserved, showing rearrangements when compared between Fonsecaea species and with the reference sequence of E. dermatitidis (Figure 2).
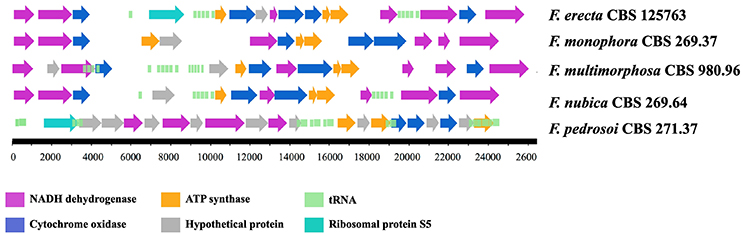
Figure 2. Mitochondrial genomes of Fonsecaea sibling species.
In order to study gene family evolution in Fonsecaea, we identified 20,713 orthologous gene clusters across 17 fungal genomes of environmental species and agents of subcutaneous and disseminated infection in animals and humans, including 9 species from the bantiana-clade and 2 from the carrionii-clade using Coniosporium apollinis, Exophiala aquamarina, and Phialophora attae as outgroup. The comparison showed a total of 5,727 of gene family clusters shared among Fonsecaea species, which was used to build a phylogenomic tree (Figure 3A).
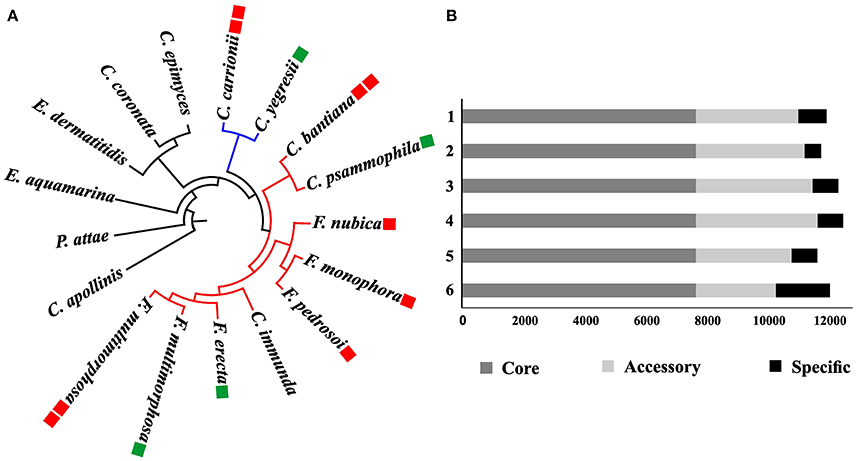
Figure 3. Phylogenomic analysis among Fonsecaea sibling species. (A) Phylogenetic tree based on the orthologous gene clusters. The bantiana-clade is indicated by a red line and the carrionii-clade by a blue line. Species names in green boxes are environmental strains, in red boxes agents of chromoblastomycosis and cerebral infection. (B) Gene cluster distribution in Fonsecaea siblings. Clusters of orthologous genes are showed in colors: dark gray core genes; light gray accessory genes, black specific genes. 1. F. monophora CBS 269.37, 2. F. multimorphosa CBS 980.96. 3. F. multimorphosa CBS 102226, 4. F. pedrosoi CBS 271.37, 5. F. nubica CBS 269.64, 6. F. erecta CBS 125763.
The analysis showed strong support for published phylogenetic relationships between bantiana-and carrionii-clades in the order Chaetothyriales. Agents of chromoblastomycosis were distributed in the separate clades, with species associated with vertebrate infection clearly being distinct from environmental siblings. Fonsecaea pedrosoi and F. nubica causing chromoblastomycosis were closely affiliated with F. monophora involved in the same disease and as well as in brain infection. The plant-associated F. erecta is distinct from the clinical species. In addition, F. multimorphosa, an environmental species that once caused a feline cerebral abscess, formed a separate cluster with an environmental sibling (Figure 3A).
Cluster analysis was performed in the protein set of four Fonsecaea species: F. erecta CBS 125763T, F. multimorphosa CBS 980.96T (Leão et al., 2017), F. monophora CBS 269.37T (Bombassaro et al., 2016) and F. nubica CBS 269.64T (Costa et al., 2016). The core genome comprised almost 70% (~8,000) of the genes (Table S1); the number of accessory genes (Table S2) in F. erecta was less than 20%, with specific genes (Table S3) lower than 10% (~2,000) (Figure 3B).
The gene core set was annotated with Eukaryotic Orthologous Group (Table S1). A total of 6,775 genes families clusters were redundantly assigned into KOG classifications, of which 2,259 genes were annotated as poorly characterized proteins, 1,382 assignments were in the category of Information Storage and Processing, 2,128 in the Cellular Processes and Signaling category and 3,265 in the Metabolism category (Figure 4). Of the eight KOG sub-classifications allocated in the Metabolism category, 718 genes were annotated to the Secondary Metabolites Biosynthesis, Transport and Catabolism [Q], 566 genes annotated to Lipid Transport and Metabolism [I] and 552 genes annotated to Energy Production and Conversion [C], composing the three more relevant classifications in this category. Moreover, in the Cellular Process and Signaling category, 631 genes were annotated with 51% proteins distributed in the Post-translational Modification Protein Turnover Chaperones class [O] with 450 genes annotated and the class Signal Transduction Mechanisms [T] and 326 genes in the Intracellular Trafficking Secretion and Vesicular Transport [U], composing the top three classifications in this category to each species analyzed (Figures 5A,B).
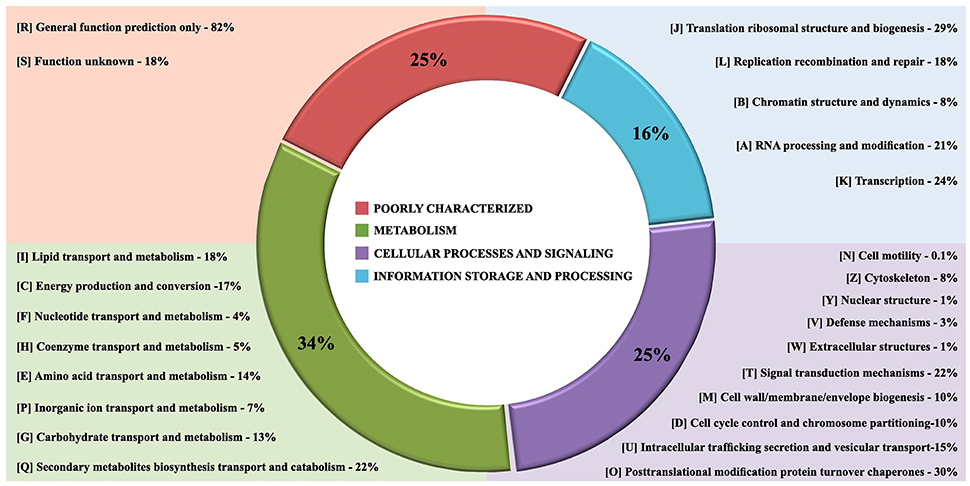
Figure 4. The core gene annotation from Eukaryotic Orthologous Group (KOG) in Fonsecaea species. KOG annotation by categories: In red poorly characterized proteins; in green metabolism category, in purple cellular processes and signaling category and processing and in blue category of information storage.
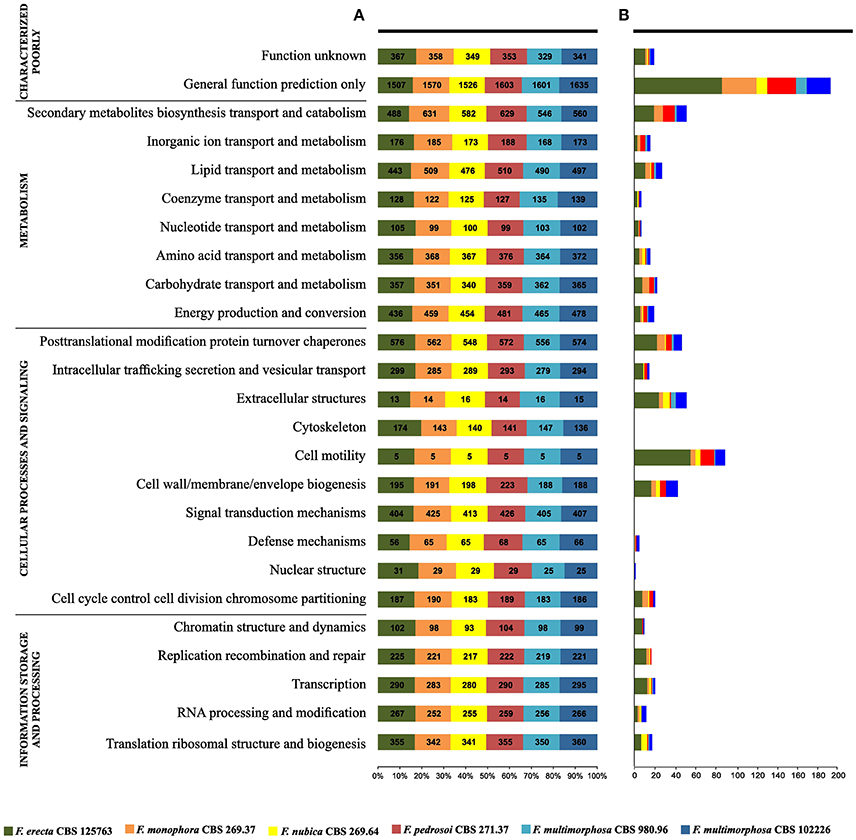
Figure 5. Gene families shared between Fonsecaea siblings based on annotation from Eukaryotic Orthologous Group (KOG). (A) The siblings are summarized in colored boxes. (B) Specific gene annotation among Fonsecaea siblings.
In the Q class (Figure 4) significant domains were identified as being common among Fonsecaea clinical and plant-associated species. The catalytic domain related to zinc-containing alcohol dehydrogenase (ADH) (IPR002328, IPR013149, IPR013154) is the most abundant domain in the Fonsecaea siblings which has been implicated in many biochemical pathways with a role in pathogenicity, stress and intoxication and were previously reported to be expanded in black yeast species (Das and Vasudevan, 2007; Grahl et al., 2011; Strommer, 2011; Teixeira et al., 2017). In addition, flavin-containing monooxygenases (FMOs) (IPR000960) constitute a family of xenobiotic-metabolizing enzymes that are widely present in Fonsecaea species. Moreover, related to metabolic processes, some protein domains in Q class were related to the biosynthesis of melanin and were widely found among Fonsecaea siblings: IPR011141, IPR019587, and IPR004235 with a role in DHN-melanin pathway, IPR002227 related to DOPA-melanin pathway, and IPR005708, IPR005955, and IPR005956 related to L-tyrosine degradation pathway. Some key enzymes involved in melanin biosynthesis were observed, including the family multicopper oxidases (MCOs) (IPR011706, IPR011707, and IPR001117). This family includes laccases, ferroxidases, bilirubin oxidases and ascorbate oxidases (Hoegger et al., 2006). Likewise, the superfamily aldehyde dehydrogenase (ALDH) (IPR015590, IPR008274) represents a divergently related group of enzymes that metabolize a wide variety of endogenous and exogenous aldehydes (Vasiliou et al., 2000).
In the T class (Figure 4) a caspase-like domain (IPR029030) and peptidase C14A, caspase non-catalytic subunit p10 (IPR002138), which are broadly classified as cysteine peptidases, were identified among the Fonsecaea species studied. Cysteine-dependent aspartyl-specific protease is mainly involved in mediating cell death processes, while caspases also have roles other than in apoptosis, e.g., caspase-1 (interleukin-1 beta convertase), which is involved in inflammatory processes (Abraham and Shaham, 2004; Lamkanfi et al., 2007). In the T class, small GTPases family (IPR001806), an independent superfamily within the larger class of regulatory GTP hydrolases (Bourne et al., 1990), was observed in Fonsecaea siblings ARF type (IPR02456), SAR1 type (IPR006687), ARF/SAR1 type (IPR006689) and Small GTPase, Ras type (IPR020849).
Within class O, post-translational modification protein turnover chaperones are involved in folding, maintenance, intracellular transport and degradation of proteins as well as in facilitating cell signaling. Many heat shock protein (Hsp) families have been identified in this study, such as Hsp20-like chaperone (IPR008979), Hsp40/DnaJ peptide-binding (IPR008971), Heat shock protein Hsp90, N-terminal (IPR020575), Heat shock protein Hsp90 family (IPR001404), Heat shock protein Hsp90, conserved site (IPR019805).
Likewise, using the functional annotation based on Gene Ontology (GO), a total of 7,392 genes were assigned (Figure S1). Of these genes, 409 were redundantly assigned into Cellular Component Ontology, 4,752 into Molecular Function Ontology and 2,231 into Biological Process Ontology. Most of the genes were annotated to binding (434) and oxyreductase (277) and methyltransferase (187) activity in the Molecular Function Ontology. In the Biological Process the overrepresented functions were metabolic processes (495), transport (239), and biosynthetic processes (195). The less represented ontology classes were membrane (87), protein complex (69) and chromosome (32) in Cellular Component Ontology.
Judging from these analyses, the presence and abundance of these functional domains could be related to the ecology of this agents, i.e., Siderophore iron transporter (GO:0015892, IPR010573) recovered by cells either by the reductive system or by specific transporters able to internalize the siderophore-iron complex (Itoh et al., 2000) and the Quinoprotein amine dehydrogenase (IPR011044), which is related to extremotolerance, this domain has been shown to interact with series of metal ions in anhydrous organic media (Matthews and Sunde, 2002).
In addition, GO enrichment analyses were used to determine the functional characteristics in the studied species (Table S4). It was observed that F. erecta presents a mechanism to regulate the use of nitrogen (GO: 0006808), a feature that may favor its survival in plants. Likewise, F. erecta and C. yegresii shared GO enrichments related to degradation of phenolic compounds such as L-phenylalanine catabolic process (GO: 0006559), tyrosine catabolic process (GO: 0006572) and homogentisate 1,2-dioxygenase activity (GO: 0004411). On the other hand, C. carrionii and F. pedrosoi exhibited an overrepresentation of the following GO terms: cellular response to iron ion starvation (GO: 0010106), siderophore biosynthetic process (GO: 0019290) and ferric triacetylfusarinine C transport (GO: 0015686) that are involved in iron metabolism.
The gene families deduced by KOG annotations for each Fonsecaea species studied were presented in the Figure 5A. Considering the specific genes observed in each species (Figure 5B), the plant-associated F. erecta presented the largest number of genes related to General function prediction and Secondary metabolites biosynthesis transport and catabolism. The species had a Leucine-rich repeat variant (IPR004830, PF13855) and Universal stress protein signature (USP) (PF00582) domains. Moreover, the family fungal Fucose-specific lectin (IPR012475) and the domain Jacalin-type lectin domain profile (IPR001229) involved in metabolism of lectins also were found in this specie (Table S3). Zinc finger domains (Znf) are shared by Fonsecaea sibling, but some domains are present in specific proteins to some species (Table S3). The Znf are relatively small protein motifs, which contain multiple finger-like protrusions that make tandem contacts with their target molecules. There are many superfamilies of Znf motifs, varying in both sequence and structure (Matthews and Sunde, 2002).
Using PHI (Pathogen Host Interactions data base) (Winnenburg et al., 2007), genes of species belonging to the bantiana-and carrionii-clades were classified into categories considering virulence and pathogenicity. In total 3,865 genes were divided into 3 categories: Lethal 2,801 genes, Increased virulence (hypervirulence) 1,013 genes and Effector (plant avirulence determinant) 51 genes (Table S5). According to this analysis, 3,614 genes were present in all species studied that includes different transcription factors. On the other hand, 251 genes were limited to one or more species such as Xeg1 observed in F. erecta and Gr-VAP gene in C. yegresii and F. erecta. In the class hypervirulence, all the analyzed species share the transcription factor Amr1, a gene described in Alternaria brassicicola this is an obligate plant pathogen as an inducer of melanin biosynthesis and associated with pathogenesis, suggesting that it is an important gene for the species to be a competitive saprophytic and also as a parasite (Cho et al., 2012). Moreover, the RpfF gene involved in pathogenesis of opportunistic pathogens (Suppiger et al., 2016) was observed in F. multimorphosa, F. pedrosoi, F. monophora, and F. nubica from the bantiana-clade and it was absent in the species from carrionii-clade (Table 3).
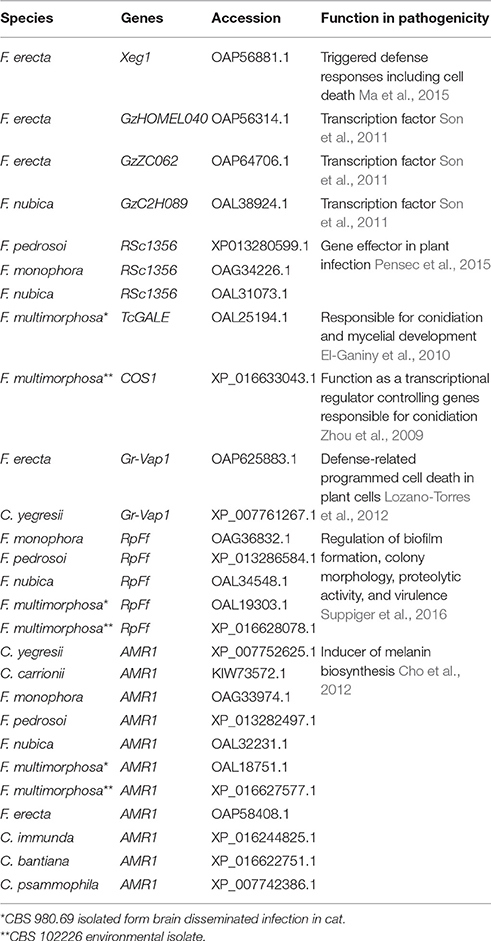
Table 3. Fonsecaea and Cladophialophora specific genes annotated in the PHI base.
The bantiana-and carrionii-clades were annotated with carbohydrate-active enzymes (CAZymes) database resulting in 5,058 genes encoding putative CAZymes, comprising 723 auxiliary activities (AA), 155 carbohydrate binding module (CBM), 1,318 carbohydrate esterases (CE), 1,800 glycoside hydrolases (GH), 1,061 glycosyl transferases (GT) and 1 polysaccharide lyase (Figure 6). The phylogenomic tree showing the carbohydrate and peptidase metabolism content in bantiana-and carrionii-clades was presented in the Figure 6A.
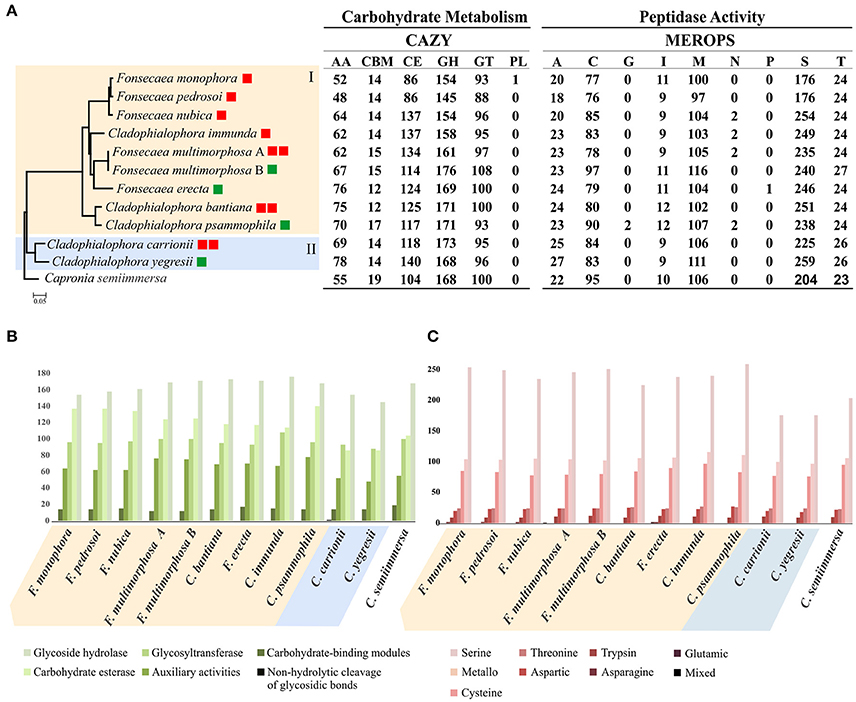
Figure 6. Analysis of carbohydrate and peptidase metabolism content in bantiana-and carrionii-clades. (A) Phylogenomic tree of bantiana- (I) and carrionii-clades (II) Fonsecaea multimorphosa (A) CBS 980.69. Fonsecaea multimorphosa. (B) CBS 102.226. All nodes on the phylogeny were supported by bootstrap values of 100% and the letters indicate ancestors. Species names followed by boxes: in green environmental strains, in red agents of chromoblastomycosis and cerebral infection. The ratio of MEROPS enzymes to CAZY enzymes for each genome is shown in the last column. (B) CAZY annotation: categories include AA (auxiliary activities), CBM (carbohydrate-binding modules), CE (carbohydrate esterase), GH (glycoside hydrolase), GT (glycosyltransferase) and PL (non-hydrolytic cleavage of glycosidic bonds). (C) MEROPS annotation: categories include A (aspartic), C (cysteine), G (glutamic), I (trypsin), M (metallo), N (asparagine), P (mixed), S (serine), and T (threonine).
In both clades CAZymes associated with degradation of polysaccharides such as chitin, hemicellulose, glucans and pectin were the largest enzyme families. The number of glycoside hydrolase 43 (GH43) enzyme family related to pectin and hemicellulose degradation was higher in F. erecta than the other species (Table S6). In addition, in the bantiana-clade the activity carbohydrate esterase (CE) was higher than in the carrionii-clade, while no polysaccharide lyase (PL) was identified in Fonsecaea sibling genomes, only one in C. carrionii (Figure 6B).
Peptidase-encoding genes were predicted using the MEROPS database (Rawlings et al., 2015) (Table S7). The bantiana- and carrionii-clades were predicted to produce a wide repertoire of different endo- and exopeptidases. We identified for both clades 5,266 peptidases, of which the largest families were serine peptidases (2,549), metallo peptidases (1,155) and cysteine peptidases (912), wherein peptidase family S1 containing Serine endopeptidase was higher in the bantiana-clade (Figure 6C). The relation between CAZY and MEROPS) in bantiana- and carrionii-clades is shown in Figure 6 (family-specific classification shown in Tables S6, S7). The species in bantiana- and carrionii-clades showed similarity in gene contents, sharing different carbohydrate metabolism and peptidase genes (p = 0.00398; Mann-Whitney U test), suggesting that these fungi are able to degrade plant and animal substrates.
Protein domain expansions and contractions were inferred from the abundance of protein domains predicted by InterProScan searches and statistically tested by CAFE v3.0 (De Bie et al., 2006) (Table S8); letters indicating values for ancestors are shown in Figure 7 and Table S8. The adaptive functions of organism gene contents were evaluated by expansion and contraction of the protein family in Fonsecaea siblings in the bantiana-clade compared with species from the carrionii-clade. Several domains were expanded in the bantiana-clade, while in the carrionii-clade there is contraction demonstrating that different clades related to chromoblastomycosis are evolved in different directions due to ecological preferences (Figure 7).
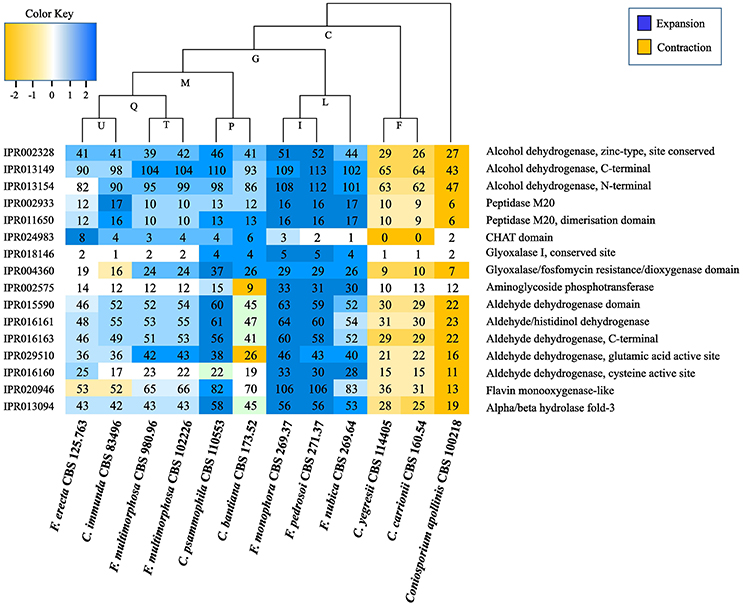
Figure 7. Interpro domains in Fonsecaea siblings. The letters indicate values for ancestry. The species are organized in phylogenetic order.
Many domains and sites (IPR015590, IPR016161, IPR016163, IPR029510, and IPR016160) related to superfamily aldehyde dehydrogenase (ALDH) and alcohol dehydrogenase zinc type conserved site (IPR002328) showed an expansion in the ancestor of F. pedrosoi, F. monophora and F. nubica. Similarly, expansions in F. erecta and C. bantiana were observed in the protein family peptidase M20 (IPR002933 and IPR011650) and in the CHAT domain (IPR024983) related to caspases (Figure 7, Table S8).
Moreover, two domains associated with the glyoxal pathway, the glyoxalase I (IPR018146) and the glyoxalase/fosfomycin resistance/dioxygenase (IPR004360) are expanded in the ancestor of F. pedrosoi, F. monophora, and F. nubica. This enzyme catalyzes the first step of the glyoxal pathway, and in addition to its role in detoxifying glyoxal it may have other roles in stress response (Kim et al., 1993; Yim et al., 2001). Further this ancestor showed expansion in the aminoglycoside phosphotransferase (IPR002575) domain (Figure 7, Table S8); this domain consists of bacterial antibiotic resistance proteins, which confers resistance to various aminoglycosides (Trower and Clark, 1990; Chow, 2000).
Survival tests using Galleria mellonella larvae as a model infected with F. pedrosoi ATCC 46428 and CBS 271.37T, F. erecta CBS 125763T, and F. monophora CBS 102248 showed a low larvae survival rate when compared to control groups. Larvae infected with F. erecta had a lower survival rate (Figure 8A). The infected larvae from the tested groups developed varying degrees of melanization, whereas the controls (PBS and SHAM) (Thomaz et al., 2013) did not present any melanization (Figure 8B1–6). Melanization could be observed from the first day post-inoculation in all tested groups. Histopathology (Figure 8C) showed the presence of pigmented nodules and granulomata in the tissue of infected larvae. Based on these results it was concluded that F. erecta CBS 125763T is potentially able to infect animal hosts. In order to find out how F. pedrosoi and F. erecta conidia and hyphae trigger macrophage activation, microbicidal activity and TNF-α production was measured. Fonsecaea erecta hyphae are more resistant to macrophage destruction than F. pedrosoi after 24 h of co-culturing with macrophages in vitro (Figure 9). No difference was observed when macrophages were co-cultured with conidia from both Fonsecaea species (Figure 9A). Only hyphae from both Fonsecaea species were able to induce TNF-α secretion (Figure 9B).
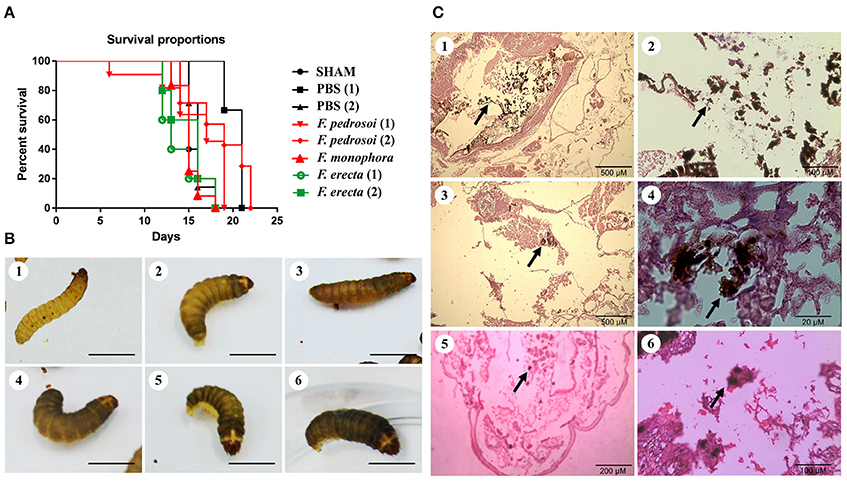
Figure 8. Virulence of Fonsecaea siblings using Galleria mellonella larvae. (A) Survival of G. mellonella larvae infected with Fonsecaea species. (B) Melanization of G. mellonella larvae infected with Fonsecaea species: (1, 2). Controls: SHAM and PBS; (3). Fonsecaea erecta CBS 125763; (4). Fonsecaea monophora; (5). Fonsecaea pedrosoi CBS 271.37; (6). Fonsecaea pedrosoi ATCC 46428. (C) Histology of infected tissue of G. mellonella with Fonsecaea species. The internal structures were fixed, embedded in paraffin and stained with PAS. Black arrows show hyphae spreading through the larva tissue. (1, 2). Fonsecaea erecta CBS 125763; (3, 4). Fonsecaea pedrosoi CBS 271.37; (5, 6). Fonsecaea monophora CBS 102248.
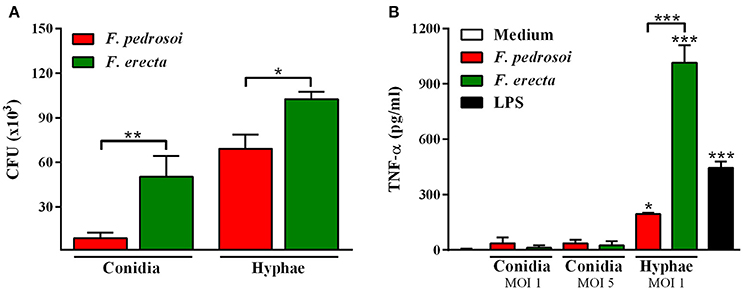
Figure 9. Fungal burden and production of pro-inflammatory cytokines of F. pedrosoi and F. erecta. (A) J774 murine macrophages (J774) were co-cultured with conidia or hyphal fragments, in the MOI 1 during 24 h. CFU data showed faster clearance of inoculated conidia from F. pedrosoi than F. erecta. (B) High levels of TNF-α were observed in macrophage co-culture with hypha, but not with conidia. Data were analyzed by one-way ANOVA with Tukey's post-hoc test. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with F. pedrosoi; or compared with non-infected macrophages.
In vivo assay using BALB/c mice as a model infected by pathogenic species F. pedrosoi and plant associated species F. erecta showed ulcerative and plaque type lesions. However, the F. pedrosoi infected mice showed lesions with dark plaque while F. erecta infected mice presented higher areas of edema (Figure 10A). All groups showed high levels of fungal burden at 7 dpi followed by reduction of viable fungi in the injured area (Figure 10B). The levels of cytokines were similar in both groups, except to IL-1β levels in F. erecta infected mice that increase lately when compared with F. pedrosoi infected mice (Figures 10C–F).
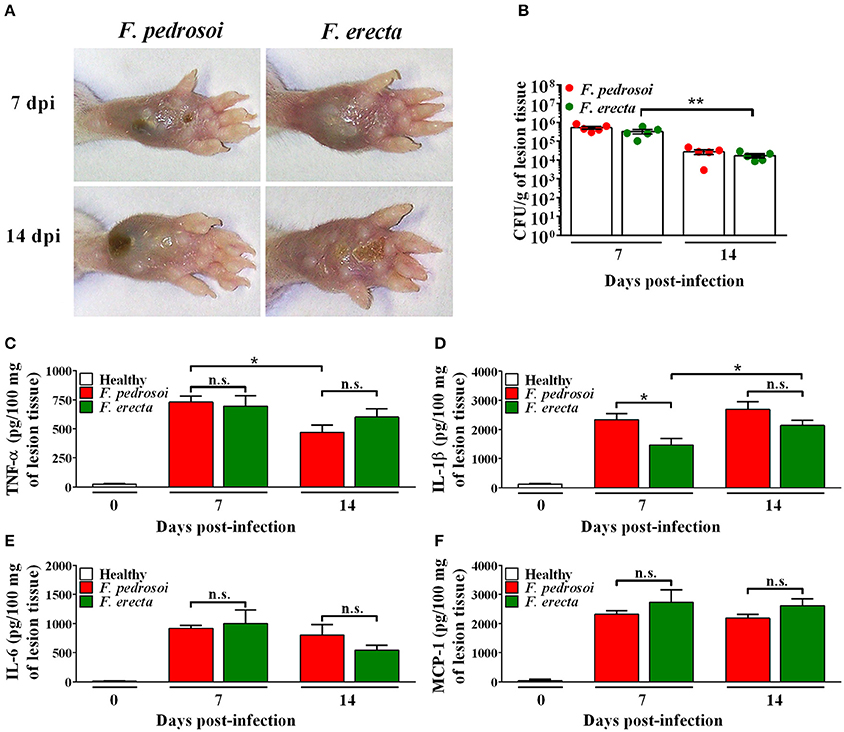
Figure 10. Virulence and immunostimulatory potential test of Fonsecaea sibling species using BALB/c mice as a model. (A) Macroscopic aspect of the disease. (B) CFU data showed a high tissue fungal burden of F. erecta which decline over the course of the infection. (C–F) At 7 and 14 days post-infection, high levels of TNF-α, IL-1β, IL-6 and MCP-1 were observed similarly in the footpad of mice infected with F. pedrosoi or F. erecta. Data were analyzed by two-way ANOVA with Tukey's post-hoc test. *p < 0.05 and **p < 0.01 between groups indicated by brackets; n.s.: not significantly.
An analysis of common genes related to the casual agents of disease showed 43 gene clusters shared by agents of systemic infection and 32 gene clusters related to (sub)cutaneous infection. Annotation of these clusters in Fonsecaea revealed 33 domains related to disseminated infection and 24 domains related to subcutaneous infection (Table 4).

Table 4. Prediction of virulence domains related to systemic and (sub)cutaneous infection.
In the agents of systemic infection a high frequency was noted of the domains DJ-1/PfpI family (IPR002818) and glyoxalase/fosfomycin resistance protein/dioxygenase superfamily (IPR004360) related to glyoxal pathway, which have important roles in detoxifying glyoxals (Lee et al., 2013) and domains related to flavin proteins, such as monooxygenase-like flavin-binding (IPR020946) and flavin-containing amine oxidoreductase (IPR002937), and the family multicopper oxidases (MCOs) (IPR011706, IPR011707, and IPR001117) that includes laccases, ferroxidases, bilirubin oxidases and ascorbate oxidases (Hoegger et al., 2006).
The subcutaneous agents shared a number of domains related to enzymes and transporters (Table 4), such as major facilitator, sugar transporter-like (IPR005828) and major facilitator superfamily IPR011701), aggregating several families of transporters. Among them a siderophore transporter, RhtX/FptX family and the yellow-like family is a gene class characterized by the presence of a major royal jelly protein (MRJP) domain (Ferguson et al., 2011) with multiple physiological and development functions in insects (Drapeau et al., 2003), such as synthesis of melanin pigments and sex-specific reproductive maturation. A large variety of enzymes such as acyltransferases (IPR002656), hydrolases (IPR013094 and IPR000757), proteases, lipases, permeases (IPR004841) and dehydrogenases (IPR002347) were observed. Nevertheless, also domains related to invasion of the plant tissue were observed, such as cutinase (IPR000675) (Dickman et al., 1989) and LysM domain related to chitinase (IPR018392) (Gruber and Seidl-Seiboth, 2012).
The genus Fonsecaea is located in the Chaetothyriales, family Herpotrichiellaceae and contains species of environmental saprobes on plants or plant debris and pathogenic species, associated with subcutaneous and deep infections in human and animal hosts. The environmental species are consistently distinct from the Fonsecaea spp. commonly founded as agents of disease. The same phenomenon of environmental next to pathogenic species is known in Cladophialophora (Vicente et al., 2014).
Phylogenomic analysis shows that Fonsecaea is intermingled with some Cladophialophora species, while Cladophialophora agents of chromoblastomycosis are located in a separate carrionii-clade (Figure 3A). The invasive potential of black yeast-like fungi is known to differ significantly between species (Badali et al., 2008; Seyedmousavi et al., 2011). Both clades with agents of chromoblastomycosis also contain non-pathogenic representatives. Formation of muriform cells in host tissue is considered as the hallmark of the disease, but some environmental species are able to produce such cells in vitro (Badali et al., 2008).
Genomes of species of Fonsecaea under study are quite similar. An abundance of domains related to zinc-containing alcohol dehydrogenase (ADH) was observed (Figure 4), which have multiple biological functions such as detoxification. Similar results have been reported for other black yeasts (Teixeira et al., 2017), suggesting that tolerance of extreme and toxic environmental habitats is a mainstay in the ecology of black yeast-like fungi. Likewise, the superfamily ALDH represents enzymes that metabolize a wide variety of endogenous and exogenous aldehydes (Lindahl, 1992; Vasiliou et al., 2000). The expansion of ALDH in the ancestor of Fonsecaea (Figure 7) indicates metabolic plasticity explaining a dual ecological ability, surviving hostile environments as well as mammal host tissue. Also Flavin-containing monooxygenases (FMOs) play a role in a wide variety of processes such as the detoxification of drugs, biodegradation of aromatic compounds, siderophore biosynthesis and biosynthesis of antibiotics (van Berkel et al., 2006), which could thus be further key executors in processes related to extremotolerance.
Gene Ontology (GO) annotation indicated presence of siderophore iron transporters in the core genome of Fonsecaea (Figure S1). Most fungi are able to produce and utilize intracellular siderophores as an iron storage compound (Eisendle et al., 2006). In Candida albicans siderophore transporter-defective mutants were clearly compromised in invading keratinocyte layers suggesting that siderophore uptake is required to epithelial invasion and penetration (Leon-Sicairos et al., 2015). The clinical species C. carrionii and F. pedrosoi show gene enrichment in cellular response to siderophore biosynthetic process and ferric triacetylfusarinine C transport which might play a role in virulence of these agents, as these genes were not enriched in the environmental species (Table S4).
Fonsecaea siblings produce melanin via different pathways demonstrating that in these fungi the pathways are conserved (Chen et al., 2014; Teixeira et al., 2017). Melanin and carotenoids deposited in the cell walls are considered a putative virulence factor in established human pathogens such as Histoplasma capsulatum, Sporothrix schenckii, and Cryptococcus neoformans. Melanin is associated to the survival and competitive abilities of fungi in hostile environments (Nosanchuk et al., 2002; Mednick et al., 2005; Nosanchuk, 2005; Taborda et al., 2008) and also enhances tolerance of oxygenic burst in macrophages. Key enzymes involved in melanin biosynthetic pathways belong to the family Multicopper oxidases (MCOs) including laccases, ferroxidases, bilirubin, oxidases and ascorbate oxidases that catalyze the oxidation of a variety of substrates and mainly aromatic compounds (Hoegger et al., 2006). Laccases form the largest subgroup and are considered key enzymes of the DOPA melanin pathway (Walton et al., 2005). They are abundant in fungal genomes, related to their divergent physiological roles and differential regulation upon changing environmental conditions (Cañero and Roncero, 2008; Giardina et al., 2010). Teixeira et al. (2010) suggested that melanin pigments protect the fungus from the mammalian host's innate immune responses providing resistance to oxidizing agents and fungal cell death during phagocytosis. Melanin is important because has a role in the protection against antifungal drugs (van den Sande et al., 2007) and is significant in differentiation of the muriform cell, the invasive form of chromoblastomycosis.
The glyoxalase system consists of two consecutive enzymatic reactions (Glyoxalase I and II) with the terminal product D-lactate by metabolism of the physiological substrate methylglyoxal. The widespread distribution of glyoxalase in prokaryotic and eukaryotic cells suggests it fulfills a function of fundamental importance to life. Inhibition of the glyoxalase system leads to methylglyoxal accumulation to toxic levels. It has been implicated in control of cell growth and proliferation, and detoxification of methylglyoxal (Thornalley, 1993). Glyoxalase enzymes are modified during phagocytosis, and the enzymatic reaction has been implicated as a virulence factor for neutralization of the immune response during invasion (Gillespie, 1978, 1981). It has been reported in sugar-limited environments, the fungus relies on fatty acid metabolism for growth (Sexton and Howlett, 2006). Accumulation of lipid in thick-walled resting cells at the expense of sugars is a key mechanism in yeast-to-mold conversion in black yeasts (Oujezdsky et al., 1973). Glyoxalase might thus play a central role in the response to varying conditions. In Fonsecaea the pathway is expanded in the clinical species of the bantiana-clade (Figure 7) suggesting that the glyoxal pathway cycle might be required for virulence during invasion, in addition to its role in surviving extreme environmental conditions (Teixeira et al., 2017).
Caspases occur in the Fonsecaea core genome where they are synthesized as pro-proteins, possessing weak proteolytic activity (Madeo et al., 2002; Cheng et al., 2003; Abraham and Shaham, 2004) and also induce apoptosis enhancing pathogenesis (Douglas et al., 2015). Activation of apoptosis may lead to caspase-1 activation, providing a link between apoptosis and inflammation (Schumann et al., 1998; Lamkanfi et al., 2007). In the T class, a small GTPase family and an independent superfamily of GTP-binding proteins share enzymatic activity and play pivotal roles in cell division, protein synthesis and signaling (Paduch et al., 2001). Small GTPase—Ras type (IPR020849) is the most represented domain regulating cell growth, proliferation and differentiation (Table S1). The Ras family of GTPase proteins has been shown to control morphogenesis in many organisms, including pathogenic fungi such as Cryptococcus neoformans (RAS1) (Alspaugh et al., 2000), Candida albicans (CaRAS1 and CaRSR1) (Leberer et al., 2001) and Aspergillus fumigatus (rhbA) (Panepinto et al., 2003).
Heat shock proteins (Hsp) in the core genome (Table S1) are essential eukaryotic molecular chaperones, being first proteins that are up-regulated under conditions of elevated temperature (Lund, 2001). Especially, Hsp90 chaperones are unique in their ability to regulate a specific subset of cellular signaling proteins that have been implicated in disease, including intracellular protein kinases, steroid hormone receptors and growth factor receptors (Tamayo et al., 2013), which are likely mechanism of mammal infection where elevated temperature is an essential condition (Vicente et al., 2012).
The above discussed domains may explain the capacity to survive extreme conditions, of which living mammal tissue is one, but do not explain differences between species of the same clade. The significant predisposition observed in agents of chromoblastomycosis (Vicente et al., 2001, 2008, 2014) probably rests upon diversity enzymatic reactions. CAFE analysis of Fonsecaea pedrosoi and Cladophialophora carrionii, both causing this disease, evolve in opposite directions, as several domains expanded in the bantiana-clade appeared to be contracted in the carrionii-clade (Figure 7). This may demonstrate that members of different clades causing chromoblastomycosis have evolved in different directions due to clade-specific ecological preferences, or perhaps more likely that the displayed domains in toto reflect phylogeny rather than ecology.
The CAZy Database is a powerful reporter of fungal lifestyles once the fungi degrade an enormous functional and structural diversity of complex plant polysaccharides. Zhao et al. (2013) revealed that most fungi that lack PL and tend to lose CE8, CE11, GH6, GH73, GH80, and GH82 families are saprobes; these were also observed in bantiana- and carrionii-clades. A wide variety of extracellular peptidases is produced to degrade a gamut of environmental substrate complexes, indicative for a less specialized nutritional status (da Silva et al., 2006; Sriranganadane et al., 2010). Species of the bantiana-and carrionii-clades produce enzymes involved in plant cell wall pectin and hemicellulose degradation. Besides, in both clades a significant similarity was observed among gene content related to carbohydrate metabolism and peptidase (Figure 6). It suggests that these fungi are able to degrade plant and animal substrates demonstrating a duality in lifestyle that could enable Chaetothyriales pathogenic species to transfer from environmental niches to animal material. The similarity of carbohydrate-active and protein degrading enzymes associated to the occurrence of additional virulence factors, which may support the tolerance to extreme environmental niches of the fungus (Teixeira et al., 2017), suggests an opportunistic tendency of Fonsecaea sibling species.
Primary fungal pathogens attempt to disrupt host cell homeostasis while avoiding and/or suppressing host recognition. In opportunists these mechanisms are not sophisticated and probably have emerged due to flexibility in nutrient acquisition (Dickman and Figueired, 2011) and extremotolerance (Moreno et al., 2017). Prenafeta-Boldú et al. (2006) and Casadevall (2007) suggested that this unfocused virulence explains the “dual use” determinants in unexpected agents of disease. It is likely that this principle also holds true for most black yeast-like fungi. However, as some common agents of chromoblastomycosis seem to have a significant predilection for this disease and are rarely found in the environment (Vicente et al., 2014) a certain degree of pathogenic adaptation cannot be excluded.
The subcutaneous infections by Fonsecaea and Cladophialophora species frequently result from a trauma from environmental sources. The muriform cell, considered to be a chromoblastomycosis-specific tissue form in humans, has been observed in cactus thorns infected with C. yegresii (De Hoog et al., 2007) and also in vitro in several environmental species (Badali et al., 2008). The RSc1356 effector is involved in plant infection (Pensec et al., 2015) and its presence in Fonsecaea might support this hypothesis, although plant- and human-pathogenicity are almost mutually exclusive (De Hoog et al., 2000). The use of nitrogen and degradation of phenolic compounds that are also enriched in environmental F. erecta and C. yegresii (Table S7) are more likely causes of opportunism. The class of protein lectins (Table S3), which is implicated in many essential cellular and molecular recognition processes (Varrot et al., 2013) was present F. erecta isolated from plant material. De Hoog et al. (2004) stated that pathology on humans is coincidental, humans not being the primary hosts of these fungi.
The above hypothesis is partially supported by results of virulence testing using G. mellonella larvae as a model. Larvae infected by environmental F. erecta had a lower survival than those infected by clinical strains of F. pedrosoi (ATCC 46428 and CBS 271.37T) and F. monophora (CBS 102248). In addition, F. erecta hyphae induced high levels of TNF-α (Figure 9), contributing to macrophage activation after phagocytosis. Macrophages activated by TNF-α increase their ability to control intracellular fungal growth, stimulate recruitment of inflammatory cells and stimulate the formation and maintenance of granulomata (Algood et al., 2005; Juhász et al., 2013; Gyurkovska and Ivanovska, 2016). Although F. erecta hyphae induced high levels of TNF-α, hyphal death was not observed, suggesting a higher level of extremotolerance. The higher virulence of strictly environmental Fonsecaea species does however not explain which the species which are commonly found on human hosts show lower virulence in the Galleria model.
Furthermore, based on PHI database the genes classified as lethal are mostly transcription factors (TFs) that orchestrate gene expression which determines life and functionality of the cell (Shelest, 2008) by controlling cellular signaling pathways and thus are key mediators of cellular function of fungi (Shelest, 2008; Wang et al., 2011).
The high number of domains related to enzymes and transporters reveals important mechanisms of nutrient acquisition and extremotolerance, constituting a genomic machinery that allows hydrolysis of recalcitrant components present in plant debris and suggests multiple survival strategies including mammal infection. Explanation of the observed differences in prevalence in the human host between closely related species appeared nevertheless impossible, as illustrated by the lower Galleria survival rate after infection with the non-pathogenic F. erecta. The large number of proteins (Figure 4, Table 4) with unknown function demands further investigation of these genes and their potential role in survival; small but crucial differences between these closely related fungi may have been concealed in the present set of proteins studied. Despite the close relationship in classical marker genes, Fonsecaea species are surprisingly different in their mitochondrial genomes, which in most fungi are highly conserved (Torriani et al., 2014; Jelen et al., 2016). Differences in routes of transmission allowing passage of properties acquired in the host to a next generation and thus, allowing evolutionary adaptation (Queiroz-Telles et al., 2017) are not easily revealed.
Conceived and designed the experiments: VV, VW, LM, RR, ALB, SA, ES, SDH. Generation sequence data: HF, MZTS, VB, EB. Performed the experiments: GF, ALB, RdC, SA, SDS, MMFdN. Analyzed the data: VV, VW, AB, LM, FC, RR, AL. Contributed reagents/materials/analysis tools: RR, VV, FP, ES. Contributed to preparing the manuscript and revising it critically: VV, VW, AB, FC, AL, RG. Annotation and analysis of data; preparation, creation and/or presentation of the tables; graphics and figures: VW, AB, LM, FC, RR, AL, RG. Strains offered and/or Substantial contributions to the work FQ, ALB, SA, MMFN, SDH. Conceived and revised paper: JS, MT, MSF, MS, DA, MJN, VV, ES, SDH. Conception and design of the work and writing the manuscript. VV, VW, AB, ES, SDH.
This work was supported by Brazilian Federal Agency for Support and Evaluation of Graduate: Education Coordination for the Improvement of Higher Education Personnel—CAPES (PVE project grant number 0592012) (www.capes.gov.br) and National Counsel of Technological and Scientific Development (http://cnpq.br/), Brazil; and by The National Institute of Science and Technology of Biological Nitrogen Fixation/CNPq/MCT (grant number 573828/2008-3). VV received fellowships from National Council for Scientific and Technological Development (CNPq), Brasilia, Brazil.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was developed in collaboration with the Broad Institute and the Bioinformatics Laboratory from Federal University of Parana. The authors are grateful to the INCT-BNF for providing computing resources.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.01924/full#supplementary-material
Abraham, M. C., and Shaham, S. (2004). Death without caspases, caspases without death. Trends Cell Biol. 14, 184–193. doi: 10.1016/j.tcb.2004.03.002
Algood, H. M., Lin, P. L., and Flynn, J. L. (2005). Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 1(Suppl. 3), S189–S193. doi: 10.1086/429994
Alspaugh, J. A., Cavallo, L. M., Perfect, J. R., and Heitman, J. (2000). RAS1 regulates filamentation, mating and growth at high temperature of Cryptococcus neoformans. Mol. Microbiol. 36, 352–365. doi: 10.1046/j.1365-2958.2000.01852.x
Andrews, S. (2016). FastQC: A Quality Control Tool for High Throughput Sequence Data; 1. Available online at: from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Badali, H., de Hoog, G. S., Curfs-Breuker, I., Klaassen, C. H. W., and Meis, J. F. (2010). Use of amplified fragment length polymorphism to identify 42 Cladophialophora strains related to cerebral phaeohyphomycosis with in vitro antifungal susceptibility. J. Clin. Microbiol. 48, 2350–2356. doi: 10.1128/JCM.00653-10
Badali, H., Gueidan, C., Najafzadeh, M. J., Bonifaz, A., Gerrits van den Ende, A. H., and de Hoog, G. S. (2008). Biodiversity of the genus Cladophialophora. Stud. Mycol. 61, 175–191. doi: 10.3114/sim.2008.61.18
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Besemer, J. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Boetzer, M., Henke, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2010). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bombassaro, A., de Hoog, S., Weiss, V. A., Souza, E. M., Leão, A. C. R., Costa, F. F., et al. (2016). Draft genome sequence of Fonsecaea monophora strain CBS 269.37, an agent of human chromoblastomycosis. Genome Announc. 4, 731–716. doi: 10.1128/genomeA.00731-16
Bourne, H. R., Sanders, D. A., and McCormick, F. (1990). The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132. doi: 10.1038/348125a0
Cañero, D. C., and Roncero, M. I. (2008). Functional analyses of laccase genes from Fusarium oxysporum. Phytopathology 98, 509–518. doi: 10.1094/PHYTO-98-5-0509
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Casadevall, A. (2007). Determinants of virulence in the pathogenic fungi. Fung. Biol. Rev. 21, 130–132. doi: 10.1016/j.fbr.2007.02.007
Chen, Z., Martinez, D. A., Gujja, S., Sykes, S. M., Zeng, Q., Szaniszlo, P. J., et al. (2014). Comparative genomic and transcriptomic analysis of Wangiella dermatitidis, a major cause of phaeohyphomycosis and a model black yeast human pathogen. G3 4, 561–578. doi: 10.1534/g3.113.009241
Cheng, J., Park, T. S., Chio, L. C., Fischl, A. S., and Ye, X. S. (2003). Induction of apoptosis by sphingoid long-chain bases in Aspergillus nidulans. Mol. Cell. Biol. 23, 163–177. doi: 10.1128/MCB.23.1.163-177.2003
Cho, Y., Srivastava, A., Ohm, R. A., Lawrence, C. B., Wang, K. H., Grigoriev, I. V., et al. (2012). Transcription factor amr1 induces melanin biosynthesis and suppresses virulence in Alternaria brassicicola. PLoS Pathog. 8:e1002974. doi: 10.1371/journal.ppat.1002974
Chow, J. W. (2000). Aminoglycoside resistance in enterococci. Clin. Infect. Dis. 31, 586–589. doi: 10.1086/313949
Costa, F. F., de Hoog, S., Raittz, R. T., Weiss, V. A., Leão, A. C., Bombassaro, A., et al. (2016). Draft genome sequence of Fonsecaea nubica strain CBS 269.64, causative agent of human chromoblastomycosis. Genome Announc. 4, 735–716. doi: 10.1128/genomeA.00735-16
Das, S. K., and Vasudevan, D. M. (2007). Alcohol-induced oxidative stress. Life Sci. 81, 177–187. doi: 10.1016/j.lfs.2007.05.005
de Azevedo, C. M., Gomes, R. R., Vicente, V. A., Santos, D. W., Marques, S. G., do Nascimentob, M. M., et al. (2015b). Fonsecaea pugnacius, a novel agent of disseminated chromoblastomycosis. J. Clin. Microbiol. 53, 2674–2685. doi: 10.1128/JCM.00637-15
de Azevedo, C. M., Marques, S. G., Santos, D. W., Silva, R. R., Silva, N. F., Santos, D. A., et al. (2015a). Squamous cell carcinoma derived from chronic chromoblastomycosis in Brazil. Clin. Infect. Dis. 60, 1500–1504. doi: 10.1093/cid/civ104
De Hoog, G. S., Attili-Angelis, D., Vicente, V. A., Gerrits van den Ende, A. H., and Queiroz-Telles, F. (2004). Molecular ecology and pathogenic potential of Fonsecaea species. Med. Mycol. 42, 405–416. doi: 10.1080/13693780410001661464
De Hoog, G. S., Nishikaku, A. S., Fernandez-Zeppenfeldt, G., Padín-González, C., Burger, E., Badali, H., et al. (2007). Molecular analysis and pathogenicity of the Cladophialophora carrionii complex, with the description of a novel species. Stud. Mycol. 58, 219–234. doi: 10.3114/sim.2007.58.08
De Hoog, G. S., Queiroz-Telles, F., Haase, G., Fernandez-Zeppenfeldt, G., Attili Angelis, D., Gerrits van den Ende, A. H., et al. (2000). Black fungi: clinical and pathogenic approaches. Med. Mycol. 38(Suppl. 1), 243–250. doi: 10.1080/mmy.38.s1.243.250
Dickman, M. B., and Figueired, P. (2011). Comparative pathobiology of fungal pathogens of plants and animals. PLoS Pathog. 7:e1002324. doi: 10.1371/journal.ppat.1002324
Dickman, M. B., Podila, G. K., and Kolattukudy, P. E. (1989). lnsertion of cutinase gene into a wound pathogen enables it to infect intact host. Nature 342, 446–448. doi: 10.1038/342446a0
Douglas, T., Champagne, C., Morizot, A., Lapointe, J. M., and Saleh, M. (2015). The inflammatory caspases-1 and−11 mediate the pathogenesis of dermatitis in sharpin-deficient mice. J. Immun. 195, 2365–2373. doi: 10.4049/jimmunol.1500542
Drapeau, M. D., Radovic, A., Wittkopp, P. J., and Long, A. D. (2003). A gene necessary for normal male courtship, yellow, acts downstream of fruitless in the Drosophila melanogaster larval brain. J. Neurobiol. 55, 53–72. doi: 10.1002/neu.10196
Edgar, R. C. (2004). Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Eisendle, M., Schrett, M., Krag, C., Muller, D., Illmer, P., and Haas, H. (2006). The intracellular siderophore ferricrocin is involved in iron storage, oxidative-stress resistance, germination, and sexual development in Aspergillus nidulans. Eukaryot Cell. 5, 1596–1603. doi: 10.1128/EC.00057-06
El-Ganiny, A. M., Sheoran, I., Sanders, D. A., and Kaminskyi, S. G. (2010). Aspergillus nidulans UDP-glucose-4-epimerase UgeA has multiple roles in wall architecture, hyphal morphogenesis, and asexual development. Fungal Genet. Biol. 47, 629–635. doi: 10.1016/j.fgb.2010.03.002
Ferguson, L. C., Green, J., Surridge, A., and Jiggins, C. D. (2011). Evolution of the insect yellow gene family. Mol. Biol. Evol. 28, 257–272. doi: 10.1093/molbev/msq192
Fuchs, B. B., Eby, J., Nobile, C. J., El Khoury, J. B., Mitchell, A. P., and Mylonakis, E. (2010). Role of filamentation in Galleria mellonella killing by Candida albicans. Microbes Infect. 12, 488–496. doi: 10.1016/j.micinf.2010.03.001
Giardina, P., Faraco, V., Pezzella, C., Piscitelli, A., Vanhulle, S., and Sannia, G. (2010). Laccases: a never-ending story. Cell. Mol. Life Sci. 67, 369–385. doi: 10.1007/s00018-009-0169-1
Gillespie, E. (1978). Concanavalin A increases glyoxalase enzyme activities in polymorphonuclear leukocytes and lymphocytes. J. Immunol. 121, 923–925.
Gillespie, E. (1981). The tumor promoting phorbol diester, 12-O-tetradecanoylphorbol-13-acetate (TPA) increases glyoxalase I and decreases glyoxalase II activity in human polymorphonuclear leucocytes. Biochem. Biophys. Res. Commun. 98, 463–470. doi: 10.1016/0006-291X(81)90862-7
Grahl, N., Puttikamonkul, S., Macdonald, J. M., Gamcsik, M. P., Ngo, L. Y., Hohl, T. M., et al. (2011). In vivo hypoxia and fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary Aspergillosis. PLoS Pathog. 7:e1002145. doi: 10.1371/journal.ppat.1002145
Gruber, S., and Seidl-Seiboth, V. (2012). Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology 158, 26–34. doi: 10.1099/mic.0.052613-0
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML Online-a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, 557–559. doi: 10.1093/nar/gki352
Gyurkovska, V., and Ivanovska, N. (2016). Distinct roles of TNF-related apoptosis-inducing ligand (TRAIL) in viral and bacterial infections: from pathogenesis to pathogen clearance. Inflamm. Res. 65, 427–437. doi: 10.1007/s00011-016-0934-1
Hayakawa, M., Ghosn, E. E., da Gloria Teixeria de Sousa, M., and Ferreira, K. S. (2006). Phagocytosis, production of nitric oxide and pro-inflammatory cytokines by macrophages in the presence of dematiaceous fungi that cause chromoblastomycosis. Scand. J. Immunol. 64, 382–387. doi: 10.1111/j.1365-3083.2006.01804.x
Hoegger, P. J., Kilaru, S., James, T. Y., Thacker, J. R., and Kües, U. (2006). Phylogenetic comparison and classification of laccase and related multicopper oxidase protein sequences. FEBS J. 273, 2308–2326. doi: 10.1111/j.1742-4658.2006.05247.x
Itoh, S., Taniguchi, M., Takada, N., Nagatomo, S., Kitagawa, T., and Fukuzumi, S. (2000). Effects of metal ions on the electronic, redox, and catalytic properties of cofactor TTQ of quinoprotein amine dehydrogenases. J. Am. Chem. Soc. 122, 12087–12097. doi: 10.1021/ja0020207
Jelen, V., de Jonge, R., Van de Peer, Y., Javornik, B., and Jakše, J. (2016). Complete mitochondrial genome of the Verticillium-wilt causing plant pathogen Verticillium nonalfalfae. PLoS ONE 11:e0148525. doi: 10.1371/journal.pone.0148525
Juhász, K., Buzás, K., and Duda, E. (2013). Importance of reverse signaling of the TNF superfamily in immune regulation. Expert Rev. Clin. Immunol. 9, 335–348. doi: 10.1586/eci.13.14
Kim, N. S., Umezawa, Y., Ohmura, S., and Kato, S. (1993). Human glyoxalase I: cDNA cloning, expression, and sequence similarity to glyoxalase I from Pseudomonas putida. J. Biol. Chem. 268, 11217–11221.
Koo, S., Klompas, M., and Marty, F. M. (2010). Fonsecaea monophora cerebral phaeohyphomycosis: case report of successful surgical excision and voriconazole treatment and review. Med. Mycol. 48, 769–774. doi: 10.3109/13693780903471081
Lamkanfi, M., Kanneganti, T. D., Franchi, L., and Núñez, G. (2007). Caspase-1 inflammasomes in infection and inflammation. J. Leukoc. Biol. 82, 220–225. doi: 10.1189/jlb.1206756
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359. doi: 10.1038/nmeth.1923
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Leão, A. C. R., Weiss, V. A., Vicente, V. A., Costa, F. F., Bombassaro, A., Raittz, R. T., et al. (2017). Genome sequence of type strain Fonsecaea multimorphosa CBS 980.96T, a causal agent of feline cerebral phaeohyphomycosis. Genome Announc. 5, 1–2. doi: 10.1128/genomeA.01666-16
Leberer, E., Harcus, D., Dignard, D., Johnson, L., Ushinsky, S., Thomas, D. Y., et al. (2001). Ras links cellular morphogenesis to virulence by regulation of the MAP kinase and cAMP signaling pathways in the pathogenic fungus Candida albicans. Mol. Microbiol. 42, 673–687. doi: 10.1046/j.1365-2958.2001.02672.x
Lee, C., Kim, I., and Park, C. (2013). Glyoxal detoxification in Escherichia coli K-12 by NADPH dependent aldo-keto reductases. J. Microbiol. 51, 527–530. doi: 10.1007/s12275-013-3087-8
Leon-Sicairos, N., Reyes-Cortes, R., Guadrón-Llanos, A. M., Madueña-Molina, J., Leon-Sicairos, C., and Canizalez-Román, A. (2015). Strategies of intracellular pathogens for obtaining iron from the environment. Biomed Res. Int. 2015, 1–17. doi: 10.1155/2015/476534
Lindahl, R. (1992). Aldehyde dehydrogenases and their role in carcinogenesis. Crit. Rev. Biochem. Mol. Biol. 27, 283–335. doi: 10.3109/10409239209082565
Lozano-Torres, J. L., Wilbers, R. H., Gawronski, P., Boshoven, J. C., Finkers-Tomczak, A., Cordewener, J. H., et al. (2012). Dual disease resistance mediated by the immune receptor Cf-2 in tomato requires a common virulence target of a fungus and a nematode. Proc. Natl. Acad. Sci. U.S.A. 109, 10119–10124. doi: 10.1073/pnas.1202867109
Lund, P. A. (2001). Microbial molecular chaperones. Adv. Microb. Physiol. 44, 93–140. doi: 10.1016/S0065-2911(01)44012-4
Ma, Z., Song, T., Zhu, L., Ye, W., Wang, Y., Shao, Y., et al. (2015). A Phytophthora sojae Glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell. 27, 2057–2072. doi: 10.1105/tpc.15.00390
Madeo, F., Herker, E., Maldener, C., Wissing, S., Lächelt, S., Herlan, M., et al. (2002). A caspase-related protease regulates apoptosis in yeast. Mol. Cell. 9, 911–917. doi: 10.1016/S1097-2765(02)00501-4
Matthews, J. M., and Sunde, M. (2002). Zinc fingers—folds for many occasions. IUBMB Life 54, 351–355. doi: 10.1080/15216540216035
Mednick, A. J., Nosanchuk, J. D., and Casadevall, A. (2005). Melanization of Cryptococcus neoformans affects lung inflammatory responses during cryptococcal infection. Infect. Immun. 73, 2012–2019. doi: 10.1128/IAI.73.4.2012-2019.2005
Moreno, L. F., Feng, P., Weiss, V. A., Vicente, V. A., Stielow, J. B., et al. (2017). Phylogenomic analyses reveal the diversity of laccase-coding genes in Fonsecaea genomes. PLoS ONE 12:e0171291. doi: 10.1371/journal.pone.0171291
Najafzadeh, M. J., Sun, J., Vicente, V. A., Xi, L., van den Ende, A. H., and de Hoog, G. S. (2010). Fonsecaea nubica sp. nov, a new agent of human chromoblastomycosis revealed using molecular data. Med. Mycol. 48, 800–806. doi: 10.3109/13693780903503081
Najafzadeh, M. J., Vicente, V. A., Sun, J., Meis, J. F., and de Hoog, G. S. (2011). Fonsecacea multimorphosa sp. nov, a new species of Chaetothyriales isolated from a feline cerebral abscess. Fungal Biol. 115, 1066–1076. doi: 10.1016/j.funbio.2011.06.007
Nosanchuk, J. D. (2005). Protective antibodies and endemic dimorphic fungi. Curr. Mol. Med. 5, 435–442. doi: 10.2174/1566524054022530
Nosanchuk, J. D., Gomez, B. L., Youngchim, S., Diez, S., Aisen, P., Zancopé-Oliveira, R. M., et al. (2002). Histoplasma capsulatum synthesizes melanin-like pigments in vitro and during mammalian infection. Infect. Immun. 70, 5124–5131. doi: 10.1128/IAI.70.9.5124-5131.2002
Oujezdsky, K. B., Grove, S. N., and Szaniszlo, P. J. (1973). Morphological and structural changes during the yeast-to-mold conversion of Phialophora dermatitidis. J. Bact. 113, 468–477.
Paduch, M., Jelen, F., and Otlewski, J. (2001). Structure of small G proteins and their regulators. Acta Biochimic Pol. 48, 829–850.
Palmeira, V. F., Kneipp, L. F., Rozental, S., Alviano, C. S., and Santos, A. L. (2008). Beneficial effects of HIV peptidase inhibitors on Fonsecaea pedrosoi: promising compounds to arrest key fungal biological processes and virulence. PLoS ONE 3:e3382. doi: 10.1371/journal.pone.0003382
Panepinto, J. C., Oliver, B. G., Fortwendel, J. R., Smith, D. L., Askew, D. S., and Rhodes, J. C. (2003). Deletion of the Aspergillus fumigatus gene encoding the Ras-related protein RhbA reduces virulence in a model of invasive pulmonary aspergillosis. Infect. Immun. 71, 2819–2826. doi: 10.1128/IAI.71.5.2819-2826.2003
Pensec, F., Lebeau, A., Daunay, M. C., Chiroleu, F., Guidot, A., and Wicker, E. (2015). Towards the identification of type III effectors associated with Ralstonia solanacearum virulence on tomato and eggplant. Phytopathology 105, 1529–1544. doi: 10.1094/PHYTO-06-15-0140-R
Piro, V. C., Faoro, H., Weiss, V. A., Steffens, M. B., Pedrosa, F. O., Souza, E. M., et al. (2014). FGAP: an automated gap closing tool. BMC Res. Notes. 7:371. doi: 10.1186/1756-0500-7-371
Prenafeta-Boldú, F. X., Summerbell, R., and de Hoog, S. G. (2006). Fungi growing on aromatics hydrocarbons: biotechnology's unexpected encounter with biohazard? FEMS Microbiol. Rev. 30, 109–130 doi: 10.1111/j.1574-6976.2005.00007.x
Queiroz-Telles, F. (2015). Chromoblastomycosis: a neglected tropical disease. Rev. Inst. Med. Trop. 57, 46–50. doi: 10.1590/S0036-46652015000700009
Queiroz-Telles, F., de Hoog, S. G., Santos, D. W. C. L., Salgado, C. G., Vicente, V. A., Bonifaz, A., et al. (2017). Chromoblastomycosis. Clin. Microbiol. Rev. 30, 233–276. doi: 10.1128/CMR.00032-16
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, 116–120. doi: 10.1093/nar/gki442
Rawlings, N. D., Barrett, A. J., and Finn, R. (2015). Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44, 343–350. doi: 10.1093/nar/gkv1118
Salgado, C. G., da Silva, J. P., Diniz, J. A., Silva, M. B., Costa, P. F., Teixeira, C., et al. (2004). Isolation of Fonsecaea pedrosoi from thorns of Mimosa pudica, a probable natural source of chromoblastomycosis. Rev. Inst. Med. Trop. 46, 33–36. doi: 10.1590/S0036-46652004000100006
Schumann, R. R., Belka, C., Reuter, D., Lamping, N., Kirschning, C. J., et al. (1998). Lipopolysaccharide activates caspase-1 (interleukin-1–converting enzyme) in cultured monocytic and endothelial cells. Blood 91, 577–584.
Sexton, A. C., and Howlett, B. J. (2006). Parallels in fungal pathogenesis on plant and animal hosts. Eukaryotic Cell 5, 1941–1949. doi: 10.1128/EC.00277-06
Seyedmousavi, S., Badali, H., Chlebicki, A., Zhao, J., Prenafeta-Boldú, F. X., and De Hoog, G. S. (2011). Exophiala sideris, a novel black yeast isolated from environments polluted with toxic alkyl benzenes and arsenic. Fung. Biol. 115, 1030–1037. doi: 10.1016/j.funbio.2011.06.004
Shelest, E. (2008). Transcriptional factors in fungi. FEMS Microbiol. Lett. 286, 145–151. doi: 10.1111/j.1574-6968.2008.01293.x
da Silva, B. A., dos Santos, A. L., Barreto-Bergter, E., and Pinto, M. R. (2006). Extracellular peptidase in the fungal pathogen Pseudallescheria boydii. Curr. Microbiol. 53, 18–12. doi: 10.1007/s00284-005-0156-1
Siqueira, I. M., de Castro, R. J. A., Leonhardt, L. C. M., Jerônimo, M. S., Soares, A. C., Raiol, T., et al. (2017). Modulation of the immune response by Fonsecaea pedrosoi morphotypes in the course of experimental chromoblastomycosis and their role on inflammatory response chronicity. PLoS Negl. Trop. Dis. 11:e0005461. doi: 10.1371/journal.pntd.0005461
Son, H., Sep, Y. S., Min, K., Park, A. R., Lee, J., Jin, J. M., et al. (2011). A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog. 7:e1002310. doi: 10.1371/journal.ppat.1002310
Sriranganadane, D., Waridel, P., Salamin, K., Reichard, U., Grouzmann, E., Neuhaus, J. M., et al. (2010). Aspergillus protein degradation pathways with different secreted protease sets at neutral and acidic pH. J. Proteome Res. 9, 3511–3519. doi: 10.1021/pr901202z
Strommer, J. (2011). The plant ADH gene family. Plant J. 66, 128–142. doi: 10.1111/j.1365-313X.2010.04458.x
Sudhadham, M., Prakitsin, S., Sivichai, S., Chaiyarat, R., Dorrestein, G. M., Menken, S. B. J., et al. (2008). The neurotropic black yeast Exophiala dermatitidis has a possible origin in the tropical rain forest. Stud. Mycol. 61, 145–155. doi: 10.3114/sim.2008.61.15
Suppiger, A., Eshwar, A. K., Stephan, R., Kaever, V., Eberl, L., and Lehner, A. (2016). The DSF type quorum sensing signalling system RpfF/R regulates diverse phenotypes in the opportunistic pathogen Cronobacter. Sci. Rep. 6:18753. doi: 10.1038/srep18753
Surash, S., Tyagi, A., de Hoog, G. S., Zeng, J. S., Barton, R. C., and Hobson, R. P. (2005). Cerebral phaeohyphomycosis caused by Fonsecaea monophora. Med. Mycol. 43, 465–472. doi: 10.1080/13693780500220373
Taborda, C. P., da Silva, M. B., Nosanchuk, J. D., and Travassos, L. R. (2008). Melanin as a virulence factor of Paracoccidioides brasiliensis and other dimorphic pathogenic fungi: a minireview. Mycopathologia 165, 331–339. doi: 10.1007/s11046-007-9061-4
Takei, H., Goodman, J. C., and Powell, S. Z. (2007). Cerebral phaeohyphomycosis caused by Cladophialophora bantiana and Fonsecaea monophora: report of three cases. Clin. Neuropathol. 26, 21–27. doi: 10.5414/NPP26021
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systemat Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tamayo, D., Muñoz, J. F., Torres, I., Almeida, A. J., Restrepo, A., McEwen, J. G., et al. (2013). Involvement of the 90 kDa heat shock protein during adaptation of Paracoccidioides brasiliensis to different environmental conditions. Fungal Genet. Biol. 51, 34–41. doi: 10.1016/j.fgb.2012.11.005
Teixeira, M. M., Moreno, L. F., Stielow, B. J., Muszewska, A., Hainaut, M., Gonzaga, L., et al. (2017). Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud. Mycol. 86, 1–28. doi: 10.1016/j.simyco.2017.01.001
Teixeira, P. A., De Castro, R. A., Ferreira, F. R., Cunha, M. M., Torres, A. P., Penha, C. V., et al. (2010). L-DOPA accessibility in culture medium increases melanin expression and virulence of Sporothrix schenckii yeast cells. Med. Mycol. 48, 687–695. doi: 10.3109/13693780903453287
Thomaz, L., Garcia-Rodas, R., Guimarães, A. J., Taborda, C. P., Zaragoza, O., and Nosanchuk, J. D. (2013). Galleria mellonella as a model host to study Paracoccidioides lutzii and Histoplasma capsulatum. Virulence 4:2. doi: 10.4161/viru.23047
Thornalley, P. J. (1993). The glyoxalase system in health and disease. Elsevier 14, 287–371. doi: 10.1016/0098-2997(93)90002-U
Torriani, S. F., Penselin, D., Knogge, W., Felder, M., Taudin, S., Platzer, M., et al. (2014). Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fung. Genet. Biol. 62, 34–42. doi: 10.1016/j.fgb.2013.11.001
Trower, M. K., and Clark, K. G. (1990). PCR cloning of a streptomycin phosphotransferase (aphE) gene from Streptomyces griseus ATCC 12475. Nucleic Acids Res. 18:4615. doi: 10.1093/nar/18.15.4615
van Berkel, W. J., Kamerbeek, N. M., and Fraaije, M. W. (2006). Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124, 670–689. doi: 10.1016/j.jbiotec.2006.03.044
van den Sande, W. W., de Kat, J., Coppens, J., Ahmed, A. O., Fahal, A., Verbrugh, H., et al. (2007). Melanin biosynthesis in Madurella mycetomatis and its effect on susceptibility to itraconazole and ketoconazole. Microb. Infect. 9, 1114–1123. doi: 10.1016/j.micinf.2007.05.015
Varrot, A., Basheer, S. M., and Imberty, A. (2013). Fungal lectins: structure, function and potential applications. Curr. Opin. Struct. Biol. 23, 678–685. doi: 10.1016/j.sbi.2013.07.007
Vasiliou, V., Pappa, A., and Petersen, D. R. (2000). Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem. Biol. Interact. 129, 1–19. doi: 10.1016/S0009-2797(00)00211-8
Vialle, R. A., Pedrosa, F. O., Weiss, V. A., Guizelini, D., Tibaes, J. H., Marchaukoski, J. N., et al. (2016). RAFTS3: Rapid alignment-free tool for sequence similarity search. BioRxiv. 1–32. doi: 10.1101/055269
Vicente, V. A., Atilli-Angellis, D., Pie, M. R., Queiroz-Telles, F., Cruz, L. M., Najafzadeh, M. J., et al. (2008). Environmental isolation of black yeast-like fungi involved in human infection. Stud. Mycol. 61, 137–144. doi: 10.3114/sim.2008.61.14
Vicente, V. A., Attili-Angelis, D., Queiróz-Telles, F., and Pizzirani-Kleiner, A. A. (2001). Isolation of herpotrichiellacious fungi from the environment. Br. J. Microbiol. 32, 47–51. doi: 10.1590/S1517-83822001000100011
Vicente, V. A., Najafzadeh, M. J., Sun, J., Gomes, R. R., Robl, D., Marques, S. G., et al. (2014). Environmental siblings of black agents of human chromoblastomycosis. Fung. Div. 65, 47–63. doi: 10.1007/s13225-013-0246-5
Vicente, V. A., Orélis-Ribeiro, R., Najafzadeh, M. J., Sun, J., Guerra, R. S., Miesch, S., et al. (2012). Black yeast-like fungi associated with Lethargic Crab Disease (LCD) in the mangrove-land crab, Ucides cordatus (Ocypodidae). Vet. Microbiol. 158, 109–122. doi: 10.1016/j.vetmic.2012.01.031
Walton, F. J., Idnurm, A., and Heitman, J. (2005). Novel gene functions required for melanization of the human pathogen Cryptococcus neoformans. Mol. Microbiol. 57, 1381–1396. doi: 10.1111/j.1365-2958.2005.04779.x
Wang, Y., Liu, W., Hou, Z., Wang, C., Zhou, X., Jonkers, W., et al. (2011). A novel transcriptional factor important for pathogenesis and ascosporogenesis in Fusarium graminearum. Mol. Plant Microbe Interact. 24, 118–128. doi: 10.1094/MPMI-06-10-0129
Winnenburg, R., Urban, M., Beacham, A., Baldwin, T. K., Holland, S., Lindeberg, M., et al. (2007). PHI-base update: additions to the pathogen host interaction database. Nucleic Acids Res. 36, 572–576. doi: 10.1093/nar/gkm858
Xi, L., Sun, J., Lu, C., Liu, H., Xie, Z., Fukushima, K., et al. (2009). Molecular diversity of Fonsecaea (Chaetothyriales) causing chromoblastomycosis in southern China. Med. Mycol. 47, 27–33. doi: 10.1080/13693780802468209
Yim, M. B., Yim, H. S., Lee, C., Kang, S. O., and Chock, P. B. (2001). Protein glycation: creation of catalytic sites for free radical generation. Ann. N.Y. Acad. Sci. 928, 48–53. doi: 10.1111/j.1749-6632.2001.tb05634.x
Zhao, Z., Liu, H., Wang, C., and Xu, J. R. (2013). Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 14:274. doi: 10.1186/1471-2164-14-274
Keywords: Fonsecaea species, black yeast, genomics, chromoblastomycosis, comparative genomics, Fonsecaea erecta
Citation: Vicente VA, Weiss VA, Bombassaro A, Moreno LF, Costa FF, Raittz RT, Leão AC, Gomes RR, Bocca AL, Fornari G, de Castro RJA, Sun J, Faoro H, Tadra-Sfeir MZ, Baura V, Balsanelli E, Almeida SR, Dos Santos SS, Teixeira MdM, Soares Felipe MS, do Nascimento MMF, Pedrosa FO, Steffens MB, Attili-Angelis D, Najafzadeh MJ, Queiroz-Telles F, Souza EM and De Hoog S (2017) Comparative Genomics of Sibling Species of Fonsecaea Associated with Human Chromoblastomycosis. Front. Microbiol. 8:1924. doi: 10.3389/fmicb.2017.01924
Received: 02 August 2017; Accepted: 21 September 2017;
Published: 09 October 2017.
Edited by:
Hector Mora Montes, Universidad de Guanajuato, MexicoReviewed by:
Sonia Rozental, Federal University of Rio de Janeiro, BrazilCopyright © 2017 Vicente, Weiss, Bombassaro, Moreno, Costa, Raittz, Leão, Gomes, Bocca, Fornari, de Castro, Sun, Faoro, Tadra-Sfeir, Baura, Balsanelli, Almeida, Dos Santos, Teixeira, Soares Felipe, do Nascimento, Pedrosa, Steffens, Attili-Angelis, Najafzadeh, Queiroz-Telles, Souza and De Hoog. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanuel M. Souza, c291emFlbUB1ZnByLmJy
Sybren De Hoog, cy5ob29nQHdlc3RlcmRpamtpbnN0aXR1dGUubmw=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.