 Blair Ney1,2
Blair Ney1,2 Carlo R. Carere3
Carlo R. Carere3 Richard Sparling3,4
Richard Sparling3,4 Thanavit Jirapanjawat1
Thanavit Jirapanjawat1 Matthew B. Stott3Colin J. Jackson5John G. Oakeshott2
Matthew B. Stott3Colin J. Jackson5John G. Oakeshott2 Andrew C. Warden2*
Andrew C. Warden2* Chris Greening1,2*
Chris Greening1,2*- 1School of Biological Sciences, Monash University, Clayton, VIC, Australia
- 2Land and Water Flagship, The Commonwealth Scientific and Industrial Research Organisation, Acton, ACT, Australia
- 3GNS Science, Wairakei Research Centre, Lower Hutt, New Zealand
- 4Department of Microbiology, University of Manitoba, Winnipeg, MB, Canada
- 5Research School of Chemistry, Australian National University, Acton, ACT, Australia
F420 is a microbial cofactor that mediates a wide range of physiologically important and industrially relevant redox reactions, including in methanogenesis and tetracycline biosynthesis. This deazaflavin comprises a redox-active isoalloxazine headgroup conjugated to a lactyloligoglutamyl tail. Here we studied the catalytic significance of the oligoglutamate chain, which differs in length between bacteria and archaea. We purified short-chain F420 (two glutamates) from a methanogen isolate and long-chain F420 (five to eight glutamates) from a recombinant mycobacterium, confirming their different chain lengths by HPLC and LC/MS analysis. F420 purified from both sources was catalytically compatible with purified enzymes from the three major bacterial families of F420-dependent oxidoreductases. However, long-chain F420 bound to these enzymes with a six- to ten-fold higher affinity than short-chain F420. The cofactor side chain also significantly modulated the kinetics of the enzymes, with long-chain F420 increasing the substrate affinity (lower Km) but reducing the turnover rate (lower kcat) of the enzymes. Molecular dynamics simulations and comparative structural analysis suggest that the oligoglutamate chain of F420 makes dynamic electrostatic interactions with conserved surface residues of the oxidoreductases while the headgroup binds the catalytic site. In conjunction with the kinetic data, this suggests that electrostatic interactions made by the oligoglutamate tail result in higher-affinity, lower-turnover catalysis. Physiologically, we propose that bacteria have selected for long-chain F420 to better control cellular redox reactions despite tradeoffs in catalytic rate. Conversely, this suggests that industrial use of shorter-length F420 will greatly increase the rates of bioremediation and biocatalysis processes relying on purified F420-dependent oxidoreductases.
Introduction
Diverse enzymes employ flavins and similar cofactors to mediate biological redox reactions (Leys and Scrutton, 2016). In addition to using the universal flavin cofactors FAD and FMN, some bacteria and archaea employ the deazaflavin cofactor F420 (Ney et al., 2017). In its enzyme-unbound state, this redox cofactor has unique redox properties compared to free FMN and FAD, namely a lower standard redox potential (-340 mV) and exclusive two-electron reactivity (Walsh, 1986; Greening et al., 2016). Due to these properties, F420 can mediate a wide range of otherwise challenging redox transformations, including the one-carbon reactions of methanogenesis (Thauer et al., 2008; Greening et al., 2016). In mycobacteria and streptomycetes, the cofactor has been shown to be important for central metabolism (Bashiri et al., 2008; Ahmed et al., 2015), secondary metabolite biosynthesis (Ikeno et al., 2006; Wang et al., 2013), cell wall production (Purwantini and Mukhopadhyay, 2013; Purwantini et al., 2016), and biodegradation pathways (Taylor et al., 2010; Jirapanjawat et al., 2016). Beyond its physiological importance, F420 has received recent attention for its potential industrial applications. Notably, actinobacterial F420H2-dependent reductases catalyze the penultimate step of tetracycline antibiotic biosynthesis (Wang et al., 2013), the reductive activation of the clinically approved antituberculosis prodrug delamanid (Cellitti et al., 2012), and the biodegradation of environmental contaminants such as nitroaromatic explosives (Ebert et al., 1999) and arylmethane dyes (Jirapanjawat et al., 2016). F420 has also been identified as a promising next-generation cofactor to mediate in vitro and in vivo biocatalytic cascades (Taylor et al., 2013; Greening et al., 2017).
Nevertheless, there remains an incomplete understanding of how the chemical structure of F420 relates to its physiological function and industrial application. Structurally, the cofactor comprises two major components (Figure 1A): (i) a redox-active headgroup comprising a modified isoalloxazine tricycle and (ii) a catalytically-inactive side chain comprising a ribitylphospholactyl moiety and an oligoglutamate chain of variable length (Eirich et al., 1978; Ashton et al., 1979). Fo (8-hydroxy-5-deazaflavin), a chromophore used by DNA photolyases, serves as the biosynthetic precursor to F420 (Graupner and White, 2001). The phospholactyl and oligoglutamate constituents are added to this precursor by three dedicated biosynthetic enzymes (CofC, CofD, CofE) (Nocek et al., 2007; Forouhar et al., 2008; Grochowski et al., 2008; Bashiri et al., 2016). It is well-established that key chemical substitutions in the isoalloxazine group confer the unique redox properties of deazaflavins over flavins (Walsh, 1986; Greening et al., 2016). However, it remains to be understood why organisms have selected to incorporate the lactyloligoglutamate side chain. It also remains elusive why the length of the oligoglutamate chain varies between organisms: two to three residues in methanogens without cytochromes, three to six in Proteobacteria and methanogens with cytochromes (Methanosarcinales), and five to eight in Actinobacteria and Chloroflexi (Gorris and van der Drift, 1994; Bair et al., 2001; Ney et al., 2017). The side chain does not significantly affect the chemical reactivity or redox properties of F420 relative to its precursor Fo (Greening et al., 2016); indeed, previous studies have shown that the redox potential of F420 is the same as Fo (-340 mV) and hence is not modulated by the oligoglutamate tail (Jacobson and Walsh, 1984). Moreover, while the charged nature of F420 ensures it does not diffuse from the cell in contrast to its precursor Fo (Ney et al., 2017), this does not explain why organisms selected to synthesize a polyanionic rather than monoanionic cofactor.
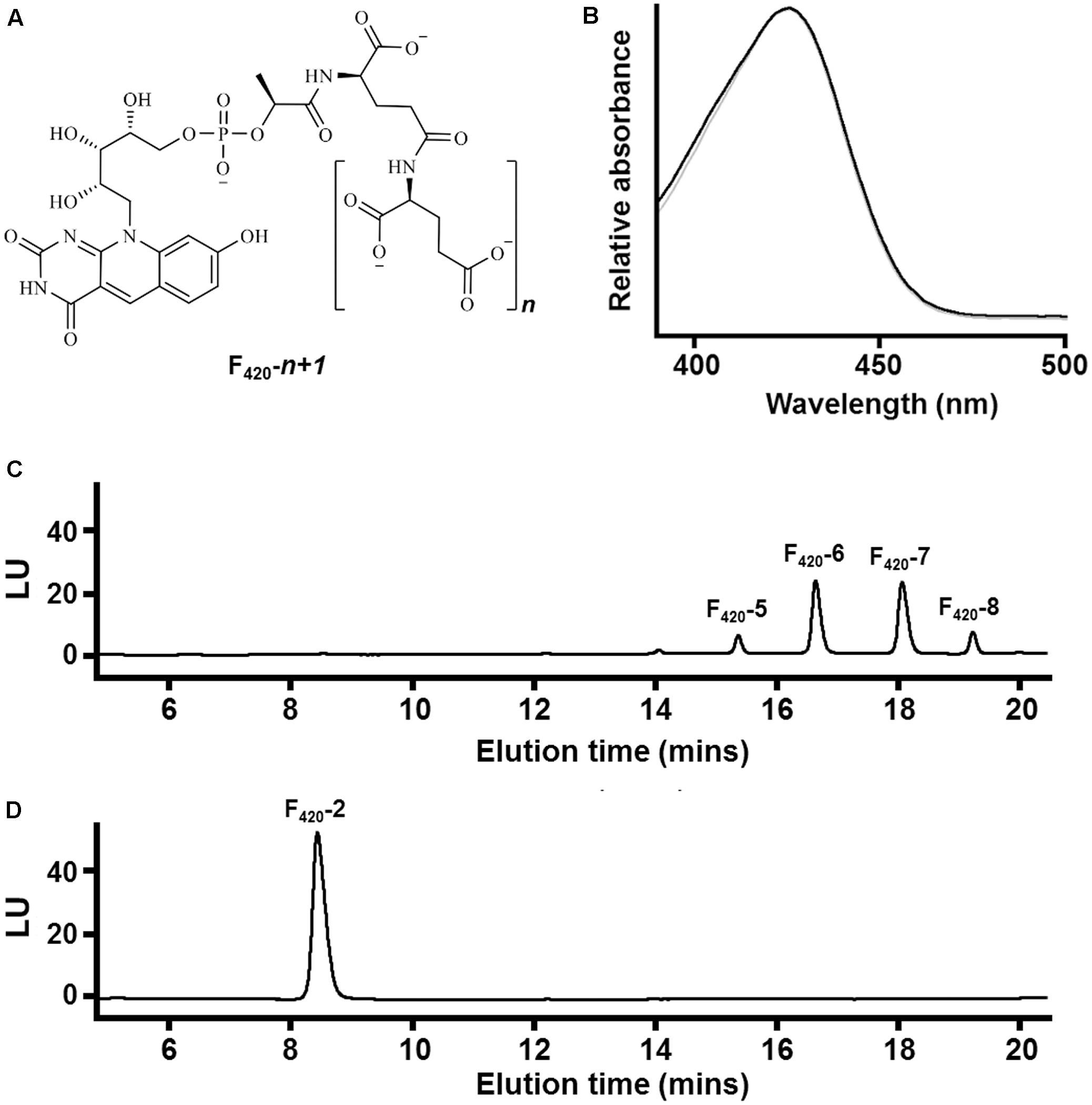
FIGURE 1. Chemical composition of F420 purified from different sources. (A) Chemical structure of F420 showing the redox-active isoalloxazine headgroup and lactyloligoglutamyl tail. The number of glutamate residues (n) varies between bacteria and archaea. (B) Absorbance spectrum of F420 purified from Mycobacterium smegmatis mc24517 (black) and Methanothermobacter marburgensis A60 (gray). (C) HPLC trace showing F420 purified from M. smegmatis mc24517 varies in chain length between five to eight glutamates. (D) HPLC trace showing F420 purified from Mtb. marburgensis A60 predominantly contains two glutamate residues.
We recently hypothesized that catalytic constraints may have driven the synthesis of the side chain in F420 (Ney et al., 2017). Specifically, the oligoglutamate chain may facilitate higher-affinity electrostatic interactions between enzyme and cofactor. We propose that, in addition to driving specific F420-dependent reactions, such high-affinity interactions may be crucial for maintaining redox homeostasis and discriminating between cofactor pools (Ney et al., 2017). In support of this observation, two cofactor-bound crystal structures suggest that the oligoglutamate chain can interact with surface cationic residues of F420-dependent oxidoreductases (Cellitti et al., 2012; Ahmed et al., 2016), though the significance of this has not been considered. Other structural analyses have proposed that, while the ribityl and phosphate groups of F420 make hydrogen bonds with surrounding residues, the oligoglutamate tail instead extends into the solvent phase without contributing to binding (Bashiri et al., 2008). In this work, we addressed the effect of the oligoglutamate side chain on the catalytic activity of F420-dependent oxidoreductases. To do this, we purified F420 from two sources: short-chain F420 from a methanogen and long-chain F420 from a mycobacterium. We subsequently studied the cofactor binding affinities and substrate consumption kinetics of mycobacterial F420-dependent oxidoreductases in the presence of these different F420 variants. We focused on a representative from each of the three main superfamilies of F420-dependent oxidoreductases found in bacteria (Selengut and Haft, 2010), namely the luciferase-like hydride transferases (LLHTs; TIM barrel fold) (Bashiri et al., 2008; Greening et al., 2016) and flavin/deazaflavin oxidoreductase superfamilies A (FDOR-As; monomeric split β-barrel proteins) and B (FDOR-Bs; dimeric split β-barrel proteins) (Ahmed et al., 2015; Greening et al., 2016).
Materials and Methods
F420 Production
Long-chain F420 was recombinantly overproduced in Mycobacterium smegmatis mc24517 cells harboring an inducible pYUBDuet shuttle vector encoding the F420 biosynthesis genes cofC, cofD, and cofE (Bashiri et al., 2010). Cultures were grown in twenty 2 L Erlenmeyer flasks each containing 500 mL LB broth supplemented with 0.05% Tween 80 (LBT), 50 μg mL-1 hygromycin B and 20 μg mL-1 kanamycin. The cultures were grown to stationary-phase in a rotary incubator (200 rpm) at 37°C for 5 days before harvesting. Short-chain F420 was extracted from a thermophilic methanogen strain, Methanothermobacter marburgensis A60. We isolated the strain by repeated serial dilution of geothermally heated sediments from Ngatamariki, New Zealand. 16S rRNA gene sequencing of genomic DNA extracts (NucleoSpin Tissue Kit, Macherey-Nagel) using the archaeal-specific primer set 109f/912r confirmed the strain shared 99% sequence identity with the well-studied laboratory strain Methanothermobacter marburgensis MarburgT (Liesegang et al., 2010). For F420 production, the strain was cultured in thirty 1 L bottles each containing 400 mL of a previously defined media supplemented with 29 mM sodium formate (Sparling et al., 1993) and a H2/CO2 atmosphere (80:20 v/v). Cultures were grown to stationary-phase in a rotary incubator (100 rpm) at 60°C for 3 days with periodic gas feeding before harvesting.
F420 Purification
F420 was harvested from the mycobacterial and methanogen cultures through variations on an existing protocol (Isabelle et al., 2002). The cells were harvested by centrifugation at 10,000 × g for 20 min, the resultant pellets were washed, and the cultures were resuspended in 20 mM TrisHCl (pH 7.5) at a ratio of 1 g per 10 mL. The cells were autoclaved at 121°C to release F420, a heat-stable cofactor, into the buffer. The cell debris was removed by centrifugation at 18,000 × g for 20 min and the supernatant was decanted and vacuum-filtrated through 0.45 μm filter paper. The F420 was isolated by FPLC (fast protein liquid chromatography) with a Macro-prep High Q Resin anion exchange column (Bio-Rad). A gradient of buffer A (20 mM TrisHCl, 100 mM NaCl, pH 7.5) and buffer B (20 mM TrisHCl, 1 M NaCl, pH 7.5) was applied, with buffer B increasing from 0 to 100% over 10 column volumes. Fractions containing F420 were identified via analysis of absorbance spectra on a SpectraMax® M3 Multi-Mode Microplate Reader (Bio-Strategy, Australia). Fractions containing F420 were pooled. The F420 solution was further purified and concentrated by hydrophobic interaction chromatography through a high capacity C18 column equilibrated in H2O. F420 was eluted in 2 mL fractions in 20% methanol, dried by rotary evaporation, and stored at -20°C.
HPLC and LC/MS Analysis
An ion-paired reverse phase HPLC (high-performance liquid chromatography) protocol was used to determine the oligoglutamate chain length of F420 purified from mycobacterial and methanogen sources. An Agilent 1200 series system equipped with an Agilent Poroshell 120 EC-C18 2.1 × 50 mm 2.7 μm column and diode array detector was used. The F420 species were separated at a flow rate of 0.3 mL min-1 using a gradient of two buffers, namely A (20 mM ammonium phosphate, 10 mM tetrabutylammonium phosphate, pH 7.0) and B (100% acetonitrile). A gradient was run from 25 to 40% buffer B as follows: 0–1 min 25%, 1–10 min 25–35%, 10–13 min 35%, 13–16 min 35–40%, 16–19 min 40–25%. F420 absorbance was measured at 420 nm using a diode array detector. This system was also used to execute an absorbance scan from 400 to 600 nm on the sole F420-2 peak of the methanogen F420 and the prominent F420-6 peak of the mycobacterial F420. The length of the F420 oligoglutamate tails were verified by a reverse phase LC/MS (liquid chromatography / mass spectrometry) protocol with an Agilent 1100 series LC/MSD TOF equipped with a Poroshell 120 EC-C18 2.1 × 100 mm 2.7 μm column. A gradient protocol comprising of Buffer A (20 mM ammonium acetate pH 6.8) and Buffer B (100% acetonitrile) was applied as follows: Held from 0 – 1 min at 5% B; 1 – 10 min from 5 – 20% B. Negative mode ESI was used with a capillary voltage of 2500 V and gas temperature of 300°C. The system was run at a flow rate of 0.2 mL min-1 and chemical species were scanned from 150 – 1500 m/z.
Enzymatic Assays
The F420-reducing glucose 6-phosphate dehydrogenase (Fgd; MSMEG locus 0777) and two F420H2-dependent reductases (FDORs; MSMEG loci 2027, 3380) from M. smegmatis mc2155 were recombinantly overexpressed in Escherichia coli BL21(DE3) using previously described vectors and protocols (Taylor et al., 2010; Ahmed et al., 2015). Cells were harvested by centrifugation, resuspended in lysis buffer, and lysed in an EmulsiFlex-C3 homogenizer (ATA Scientific, Australia) according to previously described protocols (Greening et al., 2017). Enzymes were purified from soluble extracts by Ni-nitrilotriacetic acid (NTA) affinity chromatography using gravity columns as previously described (Taylor et al., 2010; Ahmed et al., 2015) and stored in elution buffer (50 mM NaH2PO4 300 mM NaCl, 250 mM imidazole, pH 8.0) until use in assays. The high purity of the proteins was confirmed by running the fractions on NuPAGE Novex 10% Bis-Tris gels (Invitrogen, Australia) and staining with Coomassie Brilliant Blue. We measured the activities of the enzymes by monitoring the rates of F420 reduction or F420H2 oxidation in the presence of different substrate concentrations; this serves as a reliable measure of substrate transformation given F420-dependent oxidoreductases directly mediate hydride transfer between cofactor and substrate in an equimolar manner (Greening et al., 2016; Jirapanjawat et al., 2016). Enzyme activities were measured in 96-well plates containing degassed TrisHCl buffer [200 mM TrisHCl, 0.1% (w/v) Triton X-100, pH 8.0] sequentially supplemented with substrate at the specified concentration, 50 μM of the relevant cofactor, and 100 nM of the relevant enzyme. Reaction rates were measured by monitoring the initial linear change of absorbance of the reaction mixture at 420 nm using a SpectraMax® M3 Multi-Mode Microplate Reader (Molecular Devices); loss of absorbance was observed due to Fgd-mediated reduction of F420, whereas gain of absorbance occurred due to FDOR-mediated reoxidation of F420H2. Prior to measurement of FDOR activity, F420 was enzymatically reduced with 1 μM Fgd in a nitrogen glovebox for 4 h and purified by spin filtration as previously described (Ahmed et al., 2015). Reaction velocities were calculated by subtracting rates of no-enzyme controls from the initial linear rates of F420 reduction or F420H2 reoxidation measured.
Intrinsic Tryptophan Fluorescence Quenching
F420 dissociation constants were calculated by monitoring the decrease of intrinsic tryptophan fluorescence upon gradual titration of F420 as previously described (Ahmed et al., 2015). A SpectraMax® M3 Multi-Mode Microplate Reader (Molecular Devices) with a quartz cuvette containing 500 nM of protein in 20 mM TrisHCl, pH 8.0 at 24°C was used. Samples were excited at 290 nm and emission was monitored at 340 nm. One microliter aliquots of F420 standards in the same buffer were added to produce a solution with final concentrations ranging from 0 to 12.3 μM F420, with the concentration recalculated for the incremental increase in volume. The fractional saturation (F/Fmax) was plotted against the concentration of free F420, and the Kd derived from fitting the data points to the function: F/Fmax = Fmax∗[Free F420] / (Kd + [Free F420]).
Molecular Dynamics Simulations
Molecular dynamics simulations used the 1.5 Å resolution crystal structure of MSMEG_2027 [PDB: 4Y91 (Ahmed et al., 2015)] and 1.2 Å resolution structure of MSMEG_3380 [PDB: 3F7E (Taylor et al., 2010)]. The structure of the 26 residues missing from the MSMEG_2027 crystal structure was predicted by homology modeling in Phyre2 (intensive mode) (Kelley et al., 2015) using M. tuberculosis Rv3547/Ddn [PDB: 3RZ (Cellitti et al., 2012)] as the template. Simulations were visualized in the VMD: Visual Molecular Dynamics software (Humphrey et al., 1996) and calculations were performed using Amber16 (University of California San Francisco) employing the ff14SB forcefield (Maier et al., 2015). The Antechamber module within Amber16 was used to parameterize the F420-2 and F420-6 moieties with the GAFF2 forcefield and mulliken charges, and ionsjc_TIP3P parameters were used for the Na+ counterions. F420-2 and F420-6 were modeled into the structure of MSMEG_3380 and docked into the structure of MSMEG_2027 using AutoDock Vina (Morris et al., 2009). For MSMEG_3380, the positions of the cofactor up to the first glutamate residue were based on the cofactor position in the homologous protein Rv1155 [PDB: 4QVB (Mashalidis et al., 2015)]. Diglutamate and hexaglutamate tails were manually constructed and initial geometry optimisations were performed in Discovery Studio 3.5 (Accelrys). The headgroup was constructed in its deprotonated F420H- form (Mohamed et al., 2016a) and all carboxylate groups were modeled in deprotonated form. Protein-cofactor complexes were solvated in an octahedral TIP3P water box with a minimum periodic boundary distance of 10.0 Å from the solute. Each system was relaxed for a maximum of 25,000 steps of steepest descent and 25,000 steps of conjugate gradient whilst constraining the protein atoms, after which a production run of 400 ns was performed at 298 K and 1 bar with a pressure relaxation time of 2.0 ps. Langevin dynamics was employed with a collision frequency of 5.0 and SHAKE constraints were applied to all hydrogen atoms. The ribityl-bearing nitrogen of the headgroup of each F420 moiety had light positional restraints enforced (10 kcal mol-1 Å2) to prevent the headgroup from leaving the active site and to allow maximal rotational freedom within the active site so as minimize bias on the interaction energy. MMPBSA (Molecular Mechanics Poisson Boltzmann Surface Area) calculations were carried out on 2,000 frames of each 400 ns simulation employing an ionic strength of 0.15 mM and fillratio setting of 4.0.
Comparative Structural Analysis
For comparative structural analysis, protein sequences of F420-dependent oxidoreductases from different subgroups within the LLHT, FDOR-A, and FDOR-B superfamilies were retrieved from the NCBI database. Multiple sequence alignments were constructed with Clustal Omega (Sievers et al., 2011). Homology models of MSMEG_0777 and Rv0132c were constructed in RaptorX (Källberg et al., 2012) using M. tuberculosis Rv0407/Fgd as the template (Bashiri et al., 2008). Protein structures were visualized in UCSF Chimera (Pettersen et al., 2004).
Results
The F420 Oligoglutamate Chain Influences Cofactor-Binding Affinity and Reaction Kinetics of F420-Dependent Oxidoreductases
At present, no chemical syntheses or enzymatic cascades have been developed for cell-free production of F420. We therefore obtained sufficient F420 for this study through large-scale cultivation of two F420-producing strains, namely the new methanogen isolate Methanothermobacter marburgensis A60 and a previously described F420 overproduction strain of Mycobacterium smegmatis mc24517 (Bashiri et al., 2010), under conditions that would promote high-level F420 production. F420 was purified from these strains through a sequence of anion-exchange chromatography, hydrophobic interaction chromatography, and rotary evaporation. We detected the eponymous absorbance peak of F420 in the purified fractions (Figure 1B). To confirm chain length, we separated the purified F420 on a HPLC equipped with an anion-exchange column and detected the cofactor at 420 nm using a diode array detector. F420 purified from M. smegmatis contained between five to eight glutamates (Figure 1C), consistent with previous mass validation (Bashiri et al., 2010; Ney et al., 2017). In contrast, HPLC traces showed that all detectable F420 purified from Mtb. marburgensis contained two glutamate residues (Figure 1D). The mass of the dominant chemical species was validated by LC/MS (Supplementary Figure S1).
We used intrinsic fluorescence quenching to determine the binding affinities of the two purified F420 variants for three F420-dependent oxidoreductases from M. smegmatis: the F420-dependent glucose 6-phosphate dehydrogenase MSMEG_0777 (LLHT family), the F420H2-dependent quinone reductase MSMEG_2027 (FDOR-A family), and a promiscuous F420H2-dependent reductase of unknown function MSMEG_3380 (FDOR-B family). We observed that long-chain mycobacterial F420 bound the enzymes with nanomolar affinities (Kd values) of 650 nM, 190 nM, and 54 nM respectively (Figure 2 and Supplementary Figure S2), similar to values derived from previous enzymatic studies (Bashiri et al., 2008; Ahmed et al., 2015). In contrast, short-chain methanogen F420 bound with six- to ten-fold lower affinities, i.e., 4.1 μM, 1.4 μM, and 570 nM respectively (Figure 2 and Supplementary Figure S2). We observed no significant binding of the biosynthetic precursor Fo, at concentrations up to 50 μM, for any of the three enzymes. This finding suggests that interactions between the F420 oligoglutamate chain and mycobacterial F420-dependent oxidoreductases are crucial for high-affinity cofactor-enzyme associations.
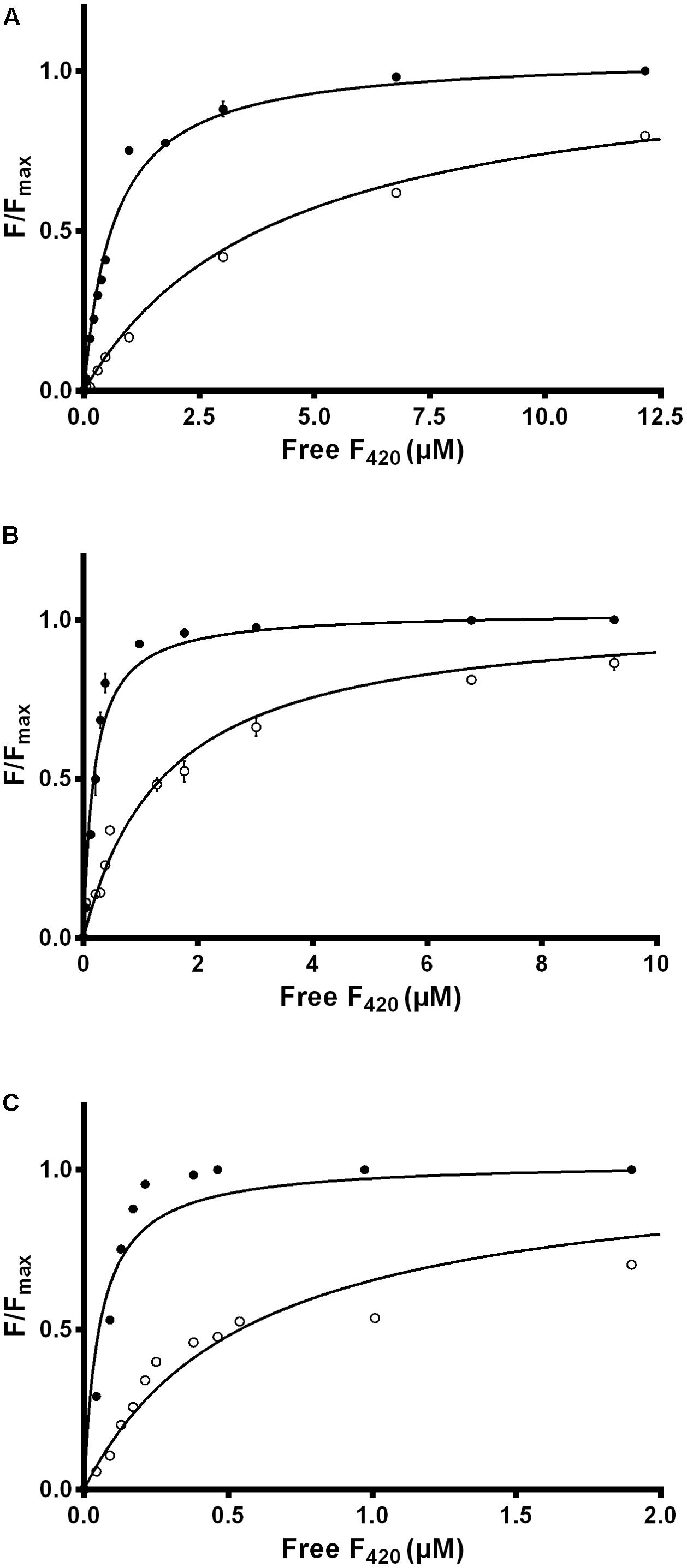
FIGURE 2. Affinity of F420 for F420-dependent oxidoreductases depends on oligoglutamate chain length. The intrinsic fluorescence quenching caused by the binding of the cofactor was measured with the (A) F420-dependent glucose 6-phosphate dehydrogenase MSMEG_0777, (B) F420H2-dependent reductase MSMEG_2027, and (C) F420H2-dependent reductase MSMEG_3380. Quenching is shown with long-chain mycobacterial F420 (●) and short-chain methanogen F420 (○). Supplementary Figure S2 shows additional data points for the methanogen F420 that are omitted here. Error bars show standard deviations from three independent replicates.
We compared the reaction kinetics of the three enzymes in the presence of the different F420 variants. The enzymes were catalytically active in the presence of F420 purified from both sources. Consistent with previous findings (Bashiri et al., 2008; Taylor et al., 2010; Ahmed et al., 2015), reaction kinetics of all three enzymes followed Michaelis–Menten models (Figure 3), though the kinetic parameters differed depending on the length of the F420 oligoglutamate chain. Observed substrate turnover rates (kcat) were between 1.9 and 3.7 times greater in the presence of short-chain F420 compared to long-chain F420 (Table 1); such enhancements of initial rate were observed irrespective of the F420 concentration used (Supplementary Figure S3). A decrease in Km was also observed in the presence of the long-chain species, with differences ranging from 3.5-fold for MSMEG_2027 to a modest 1.5-fold for MSMEG_0777 and MSMEG_3380 (Table 1). Hence, high-affinity cofactor binding results in both enhanced substrate binding and decreased reaction turnover.
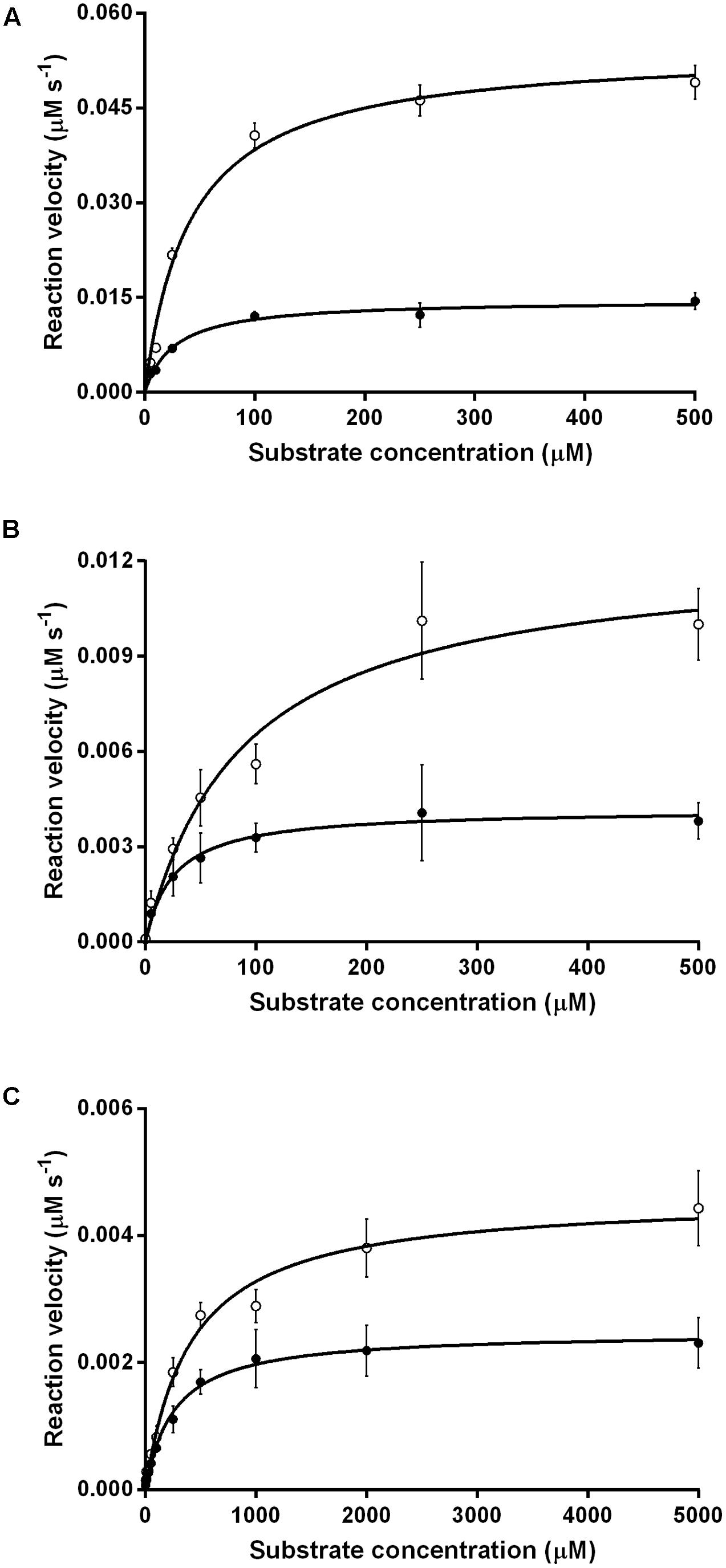
FIGURE 3. Kinetics of substrate oxidation/reduction of F420-dependent oxidoreductases depends on F420 oligoglutamate chain length. Three activities were measured, namely (A) F420-dependent oxidation of glucose 6-phosphate by native Fgd activity, (B) F420H2-dependent reduction of menadione by native MSMEG_2027 activity, and (C) F420H2-dependent reduction of cyclohexenone by promiscuous MSMEG_3380. Activities are shown with long-chain bacterial F420 (●) and short-chain methanogen F420 (○). Error bars show standard deviations from three independent replicates.
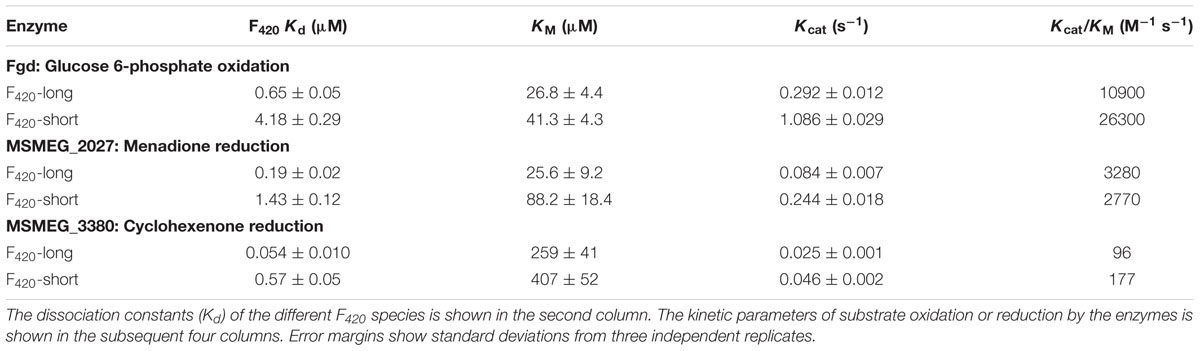
TABLE 1. Kinetic parameters of F420-dependent oxidoreductases in the presence of long-chain F420 and short-chain F420.
The Oligoglutamate Chain Makes Multiple Electrostatic Interactions with Surface Anionic Residues of FDORs
To determine the structural and mechanistic basis for these differences, we used molecular dynamics simulations to compare the binding of long-chain (F420-6) and short-chain (F420-2) variants of F420 to the available crystal structures of the FDOR-A MSMEG_2027 (Ahmed et al., 2015) and the FDOR-B MSMEG_3380 (Taylor et al., 2010). An overarching characteristic of all simulations was that, following equilibration of the position of the cofactor tail, the glutamate residues made multiple transient electrostatic contacts with specific arginine and lysine residues on the surfaces of the oxidoreductases. The first two glutamates of both F420-2 and F420-6 made interactions with residues Lys73, Arg49, Lys44, and sporadically Lys68 of MSMEG_2027 (Figures 4A,B). These glutamate residues also interacted with Arg205, Arg23, and Arg54 of MSMEG_3380, but these interactions were more transient (Figures 4C,D). In both cases, the terminal four glutamates of F420-6 made multiple transient electrostatic interactions with surface cationic residues, but these residues were highly dynamic and displayed little specificity in the various interactions they formed. These additional interactions also appeared to stabilize the core interactions made by the cofactor, for example between the phosphate group and Lys53 of MSMEG_3380 (Figures 4B,D). The multiple alternative tail conformations observed remained favorable even though the headgroup was weakly restrained within the active site (Supplementary Figure S4).
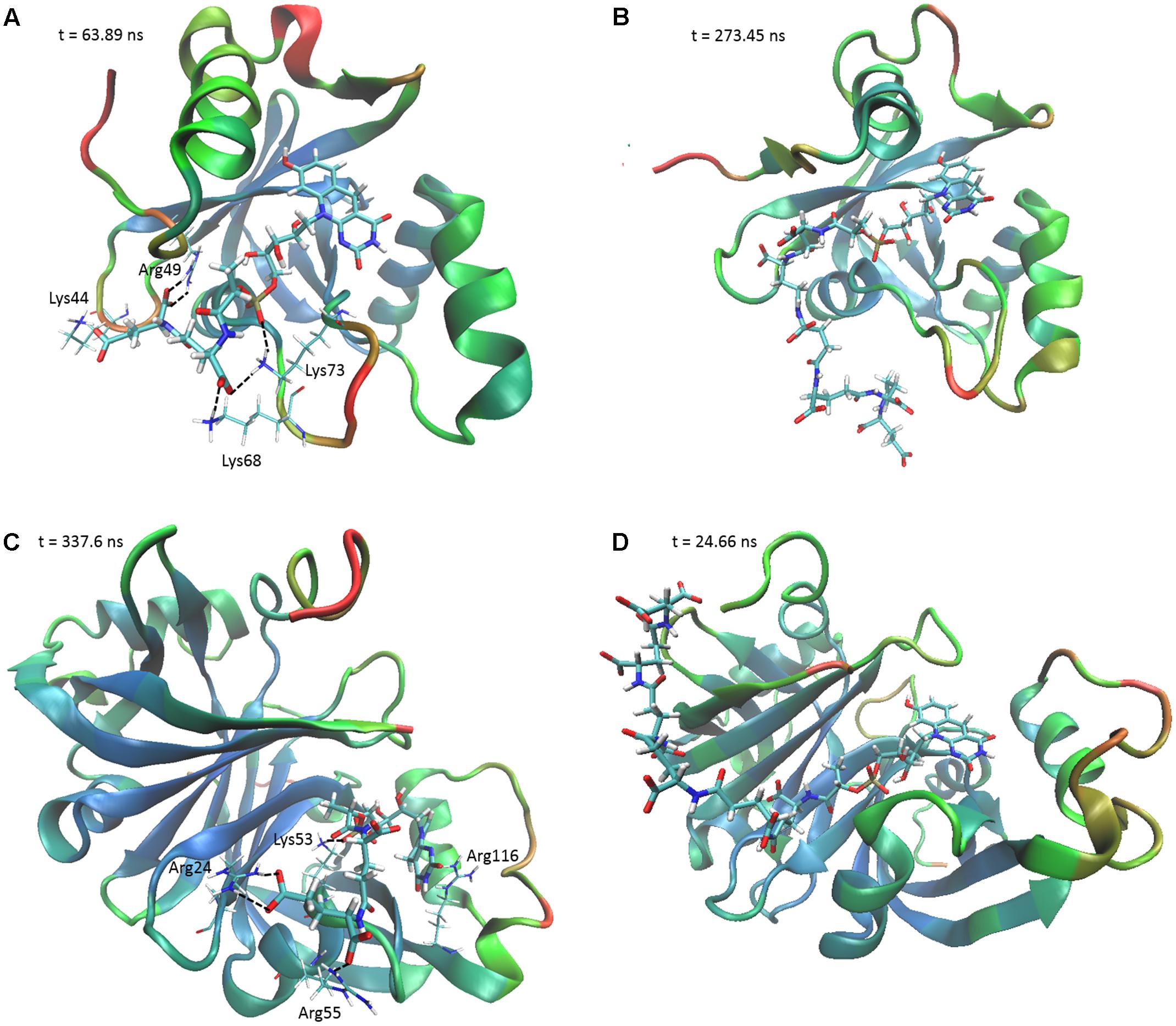
FIGURE 4. The F420 oligoglutamate chain makes electrostatic interactions with FDORs. Snapshots are shown of the molecular dynamics simulations of (A) MSMEG_2027 with F420-2 (t = 63.89 ns), (B) MSMEG_2027 with F420-6 (t = 273.45 ns), (C) MSMEG_3380 with F420-2 (t = 337.6 ns), and (D) MSMEG_3380 with F420-6 (t = 24.66 ns) with some of the more stable interactions observed. The colors on the secondary structure of the enzyme represent flexibility as calculated by the backbone RMSD over the course of the whole simulation, with red representing high mobility and blue representing high stability. Black dashed lines indicate electrostatic interactions. The cofactor is depicted in stick representation with thicker bonds, whereas the relevant residues are depicted as sticks with thinner bonds. Interacting residues are not shown for F420-6 for clarity.
We subsequently evaluated the cofactor-enzyme binding energies for the trajectories using MMPBSA analysis, which confirmed that the oligoglutamate chain modulated binding. While the cofactor-enzyme contacts reduced solvation energy, this was offset by the combined energies from 1 to 4 electrostatic and other non-bonded interactions, resulting in a lower total ΔGbinding for the complex. Consistent with the additional dynamic interactions observed, F420-6 reached lower ΔGbinding values than F420-2 in simulations with both MSMEG_2027 and MSMEG_3380 (Figures 5A,B). Such observations are consistent with the lower Kd values determined in the tryptophan fluorescence quenching experiments (Figure 2 and Supplementary Figure S2). Loss of multiple electrostatic interactions between the enzyme and cofactor tail increased the solvation energy and often pushed ΔGbinding into positive territory (Figure 5B). The internal bond, angle, and dihedral energies of the cofactor were essentially unaffected by the various binding modes (Supplementary Figure S4), emphasizing that most binding energy changes occur due to flexibility of the oligoglutamate chain rather than the isoalloxazine ring. These findings in turn suggests that there are no combinations of interactions that would release the headgroup from the active site and instead cofactor dissociation may be driven by tail solvation.
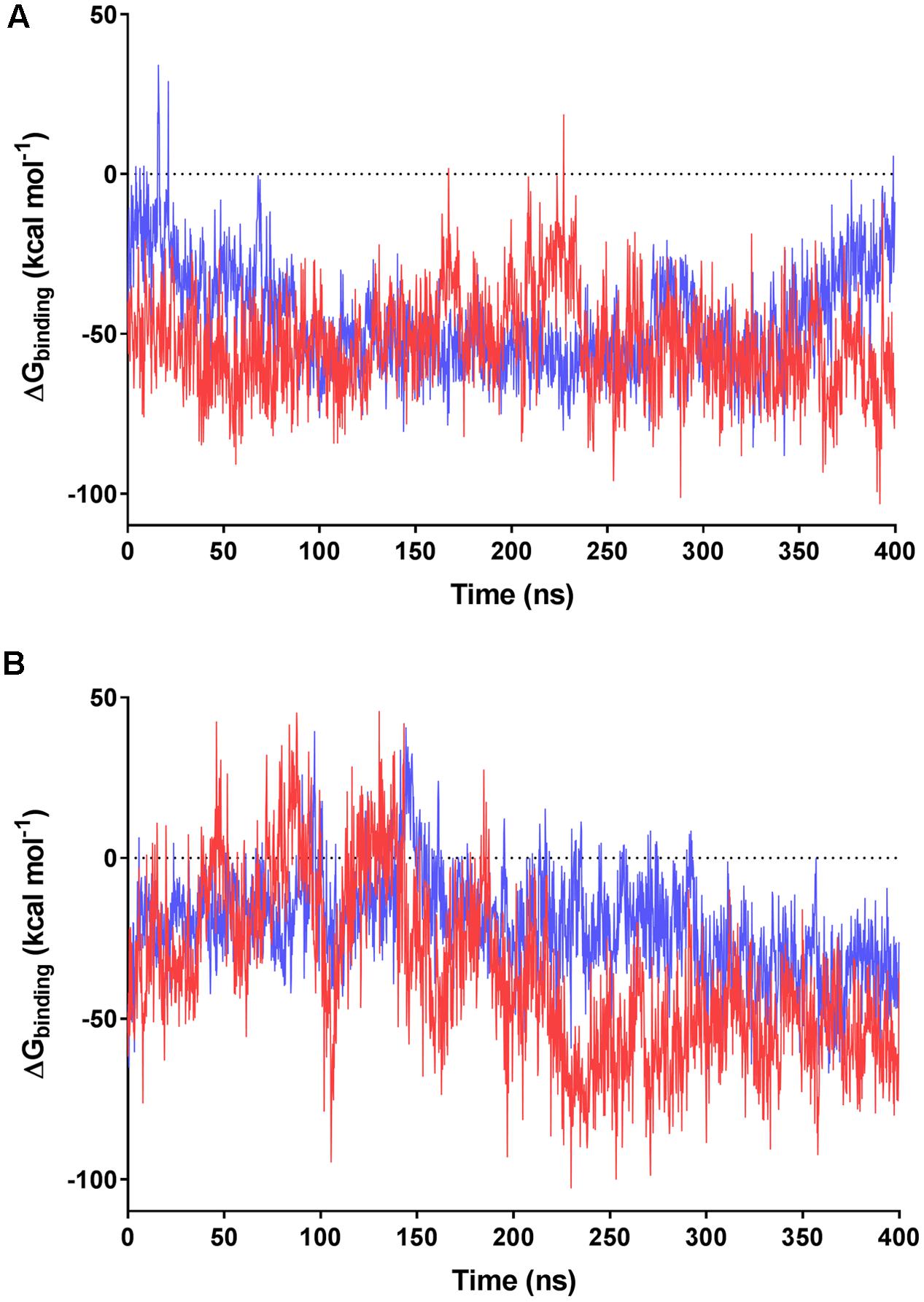
FIGURE 5. MMPBSA-calculated binding energies of F420 variants with F420-dependent oxidoreductases. Data based on the 400 ns trajectories for the molecular dynamics simulations with (A) MSMEG_2027 and (B) MSMEG_3380. The calculated binding energies are shown for F420-2 in blue and F420-6 in red.
Interestingly, we observed that the monomeric enzyme MSMEG_2027 exhibited markedly different cofactor binding modes compared to the dimeric enzyme MSMEG_3380. While most of the interacting positive charges were accommodated on the flexible loop regions of MSMEG_2027 (Lys44, Lys68, Lys73), the main contacting residues of MSMEG_3380 (Arg205, Arg23, Arg54) all lie on highly stable β-sheet and helical secondary structural regions. Multiple sequence alignments and comparative structural analysis suggest that these cationic residues were highly conserved within their respective FDOR-A and FDOR-B superfamilies (Supplementary Figures S5, S6). Most notably, the sequence motif Gx[KR]xG[QKE]xR occurs in all enzymes in the FDOR-A superfamily, among them enzymes sharing less than 25% identity. This sequence forms a loop joining two β-strands (Ahmed et al., 2015); the Gly residues likely contribute to the flexibility of the loop and the cationic residues serve as the main site of sustained interaction with the oligoglutamate chain (Figure 4). Conserved cationic surface residues are also proximal to the F420 oligoglutamate chain in the LLHT superfamily (Supplementary Figures S5, S6), providing further support that electrostatic interactions generally occur between bacterial F420-dependent oxidoreductases and the F420 oligoglutamate chain.
Discussion
Since the structure of F420 was proposed in Eirich et al. (1978), studies on its catalytic behavior have focused on its redox-active headgroup (Walsh, 1986; Greening et al., 2016) and the role of its side chain has not been addressed. In this study, we reveal that the F420 oligoglutamate chain modulates catalysis in bacterial F420-dependent oxidoreductases. Our experimental findings demonstrate that synthesis of long-chain F420 results in higher-affinity enzyme-cofactor interactions. Molecular dynamics simulations focused on FDOR-A and FDOR-B representatives provide a rationale for these findings by showing that the F420 tail electrostatically interacts with conserved cationic residues on the surface of mycobacterial F420-dependent oxidoreductases; while the diglutamate chain can make sustained electrostatic interactions, the multiple additional transient interactions made by oligoglutamate chain offsets solvation energy and increases binding energy. We made compatible findings across three different protein families in the presence of both physiological and non-physiological substrates. It is therefore probable that the oligoglutamate chain of F420 is of general relevance to catalysis of bacterial F420-dependent oxidoreductases.
We also observed that higher-affinity cofactor binding modulates reaction kinetics by increasing substrate affinity but decreasing turnover. Such findings likely reflect that mycobacterial F420-dependent oxidoreductases mediate catalysis through a ternary complex, with hydride transfer occurring directly between the isoalloxazine headgroup of the cofactor and the substrate (Greening et al., 2016; Mohamed et al., 2016b). If cofactor dissociation is the rate-limiting step in the catalytic cycle of these oxidoreductases, higher-affinity cofactor-enzyme interactions may result in lower cofactor dissociation rates (koff) and hence reduced substrate turnover. It is also plausible that conformational changes caused by the electrostatic interactions are transmitted to the adjoined isoalloxazine- and substrate-binding sites, thereby modulating substrate affinity; this may be particularly important in FDOR-A proteins, where interactions between the terminal glutamate residues and the lysine-rich loop region may stabilize the split β-barrel fold and in turn the substrate-binding site. The finding that substrate turnover of F420-dependent oxidoreductases is accelerated in the presence of short-chain F420 is important for biotechnological reasons: it suggests that F420 purified from methanogen sources will result in higher turnovers in the various bioremediation and biocatalysis processes for which F420-dependent oxidoreductases have been advocated (Taylor et al., 2013; Greening et al., 2016, 2017). A tradeoff would be the reduction in substrate affinity, but this is likely to be negligible for biocatalytic applications given they rely on high substrate concentrations. Obstacles in metabolic engineering must be overcome, however, if short-chain F420 is to be heterologously produced at industrially-relevant scales.
Future studies are required to determine whether the observed tradeoffs between affinity and turnover are physiologically relevant. We hypothesize that the oligoglutamate chain ensures the affinity of interactions between cofactor and enzyme remain in the physiologically desirable nanomolar range. In turn, this may increase the substrate specificity of the oxidoreductases that bind the cofactor. In bacterial cells, loss of this chain may compromise specific F420-dependent reactions and have wider effects on redox homeostasis and cofactor partitioning. Consistently, studies on nitroimidazole resistance suggest that the enzyme responsible for oligoglutamate chain elongation, CofE (F420-0:γ-glutamyl ligase), is required for optimal functionality of F420 in mycobacterial cells, though is less important than the other F420 biosynthetic enzymes (Haver et al., 2015). In contrast, most methanogens appear to suffice with a diglutamate- rather than oligoglutamate-containing side chain. One explanation is that F420-dependent enzymes in such organisms may be less kinetically constrained, given F420 serves as the primary catabolic cofactor and is generally present at higher concentrations than in bacterial cells (Thauer et al., 2008). However, further studies are required to understand the significance of the F420 diglutamate chain in the catalysis of F420-dependent oxidoreductases in methanogens and why Methanosarcinales synthesize longer-chain F420 variants (Gorris and van der Drift, 1994).
The observed differences between bacterial and archaeal F420 may also be relevant for understanding the evolution of the biosynthesis of deazaflavins. The F420 biosynthesis pathway appears to have undergone a complex evolutionary trajectory, with phylogenetic evidence unable to resolve whether the cofactor originated in bacteria, archaea, or the last universal common ancestor (Nelson-Sathi et al., 2015; Weiss et al., 2016; Ney et al., 2017). We recently proposed that Fo served as the primordial cofactor in deazaflavin-dependent enzymes, but selective pressure to produce a membrane-impermeable derivative resulted in the evolution of F420 biosynthetic enzymes (CofC, CofD, CofE) and in turn the production of short-chain F420 (Ney et al., 2017). We propose here that the synthesis of longer-chain derivatives was driven by selection pressure for higher-affinity cofactor-enzyme interactions or more controlled redox homeostasis. This was likely mediated through evolution of the CofE, which is a single-domain enzyme in short-chain F420 producers but is fused with an FMN reductase domain in most long-chain producers (Ney et al., 2017); recent structural and kinetic studies on mycobacterial CofE have demonstrated this second domain is essential for elongation of the oligoglutamate chain (Bashiri et al., 2016). It is plausible that the three families of bacterial F420-dependent oxidoreductases co-evolved with CofE, resulting in higher-affinity cofactor-enzyme interactions.
Author Contributions
CG and AW conceived the study. CG, AW, BN, CC, RS, TJ, JO, and MS designed experiments. BN, TJ, AW, CG, CC, RS, and MS performed experiments. CG and CJ supervised students. CG, AW, BN, JO, TJ, and CJ interpreted data. CG, AW, and BN wrote the paper.
Funding
This work was supported by a CSIRO Office of the Chief Executive Postdoctoral Fellowship and an ARC DECRA Fellowship (DE170100310) awarded to CG, a Marsden Grant (GNS-035) awarded to CC, and Australian Research Council grants (DE120102673, DP130102144) awarded to CJ.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01902/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmed, F. H., Carr, P. D., Lee, B. M., Afriat-Jurnou, L., Mohamed, A. E., Hong, N.-S., et al. (2015). Sequence-structure-function classification of a catalytically diverse oxidoreductase superfamily in mycobacteria. J. Mol. Biol. 427, 3554–3571. doi: 10.1016/j.jmb.2015.09.021
Ahmed, F. H., Mohamed, A. E., Carr, P. D., Lee, B. M., Condic-Jurkic, K., O’Mara, M. L., et al. (2016). Rv2074 is a novel F420H2-dependent biliverdin reductase in Mycobacterium tuberculosis. Protein Sci. 25, 1692–1709. doi: 10.1002/pro.2975
Ashton, W. T., Brown, R. D., Jacobson, F., and Walsh, C. (1979). Synthesis of 7,8-didemethyl-8-hydroxy-5-deazariboflavin. J. Am. Chem. Soc. 101, 4419–4420. doi: 10.1021/ja00509a083
Bair, T. B., Isabelle, D. W., and Daniels, L. (2001). Structures of coenzyme F420 in Mycobacterium species. Arch. Microbiol. 176, 37–43. doi: 10.1007/s002030100290
Bashiri, G., Rehan, A. M., Greenwood, D. R., Dickson, J. M. J., and Baker, E. N. (2010). Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLOS ONE 5:e15803. doi: 10.1371/journal.pone.0015803
Bashiri, G., Rehan, A. M., Sreebhavan, S., Baker, H. M., Baker, E. N., and Squire, C. J. (2016). Elongation of the poly-gamma-glutamate tail of F420 requires both domains of the F420:gamma-glutamyl ligase (FbiB) of Mycobacterium tuberculosis. J. Biol. Chem. 291, 6882–6894. doi: 10.1074/jbc.M115.689026
Bashiri, G., Squire, C. J., Moreland, N. J., and Baker, E. N. (2008). Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J. Biol. Chem. 283, 17531–17541. doi: 10.1074/jbc.M801854200
Cellitti, S. E., Shaffer, J., Jones, D. H., Mukherjee, T., Gurumurthy, M., Bursulaya, B., et al. (2012). Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 20, 101–112. doi: 10.1016/j.str.2011.11.001
Ebert, S., Rieger, P.-G., and Knackmuss, H.-J. (1999). Function of coenzyme F420 in aerobic catabolism of 2,4,6-trinitrophenol and 2,4-dinitrophenol by Nocardioides simplex FJ2-1A. J. Bacteriol. 181, 2669–2674.
Eirich, L. D., Vogels, G. D., and Wolfe, R. S. (1978). Proposed structure for coenzyme F420 from Methanobacterium. Biochemistry 17, 4583–4593. doi: 10.1021/bi00615a002
Forouhar, F., Abashidze, M., Xu, H., Grochowski, L. L., Seetharaman, J., Hussain, M., et al. (2008). Molecular insights into the biosynthesis of the F420 coenzyme. J. Biol. Chem. 283, 11832–11840. doi: 10.1074/jbc.M710352200
Gorris, L. G., and van der Drift, C. (1994). Cofactor contents of methanogenic bacteria reviewed. Biofactors 4, 139–145.
Graupner, M., and White, R. H. (2001). Biosynthesis of the phosphodiester bond in coenzyme F420 in the methanoarchaea. Biochemistry 40, 10859–10872. doi: 10.1021/bi0107703
Greening, C., Ahmed, F. H., Mohamed, A. E., Lee, B. M., Pandey, G., Warden, A. C., et al. (2016). Physiology, biochemistry, and applications of F420- and Fo-dependent redox reactions. Microbiol. Mol. Biol. Rev. 80, 451–493. doi: 10.1128/MMBR.00070-15
Greening, C., Jirapanjawat, T., Afroze, S., Ney, B., Scott, C., Pandey, G., et al. (2017). Mycobacterial F420H2-dependent reductases promiscuously reduce diverse compounds through a common mechanism. Front. Microbiol. 8:1000. doi: 10.3389/fmicb.2017.01000
Grochowski, L. L., Xu, H., and White, R. H. (2008). Identification and characterization of the 2-phospho-L-lactate guanylyltransferase involved in coenzyme F420 biosynthesis. Biochemistry 47, 3033–3037. doi: 10.1021/bi702475t
Haver, H. L., Chua, A., Ghode, P., Lakshminarayana, S. B., Singhal, A., Mathema, B., et al. (2015). Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 5316–5323. doi: 10.1128/AAC.00308-15
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Ikeno, S., Aoki, D., Hamada, M., Hori, M., and Tsuchiya, K. S. (2006). DNA sequencing and transcriptional analysis of the kasugamycin biosynthetic gene cluster from Streptomyces kasugaensis M338-M1. J. Antibiot. 59, 18–28. doi: 10.1038/ja.2006.4
Isabelle, D., Simpson, D. R., and Daniels, L. (2002). Large-scale production of coenzyme F420-5,6 by using Mycobacterium smegmatis. Appl. Environ. Microbiol. 68, 5750–5755. doi: 10.1128/AEM.68.11.5750-5755.2002
Jacobson, F., and Walsh, C. (1984). Properties of 7,8-didemethyl-8-hydroxy-5-deazaflavins relevant to redox coenzyme function in methanogen metabolism. Biochemistry 23, 979–988. doi: 10.1021/bi00300a028
Jirapanjawat, T., Ney, B., Taylor, M. C., Warden, A. C., Afroze, S., Russell, R. J., et al. (2016). The redox cofactor F420 protects mycobacteria from diverse antimicrobial compounds and mediates a reductive detoxification system. Appl. Environ. Microbiol. 82, 6810–6818. doi: 10.1128/AEM.02500-16
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., et al. (2012). Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522. doi: 10.1038/nprot.2012.085
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Leys, D., and Scrutton, N. S. (2016). Sweating the assets of flavin cofactors: new insight of chemical versatility from knowledge of structure and mechanism. Curr. Opin. Struct. Biol. 41, 19–26. doi: 10.1016/j.sbi.2016.05.014
Liesegang, H., Kaster, A.-K., Wiezer, A., Goenrich, M., Wollherr, A., Seedorf, H., et al. (2010). Complete genome sequence of Methanothermobacter marburgensis, a methanoarchaeon model organism. J. Bacteriol. 192, 5850–5851. doi: 10.1128/JB.00844-10
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi: 10.1021/acs.jctc.5b00255
Mashalidis, E. H., Gittis, A. G., Tomczak, A., Abell, C., Barry, C. E., and Garboczi, D. N. (2015). Molecular insights into the binding of coenzyme F420 to the conserved protein Rv1155 from Mycobacterium tuberculosis. Protein Sci. 24, 729–740. doi: 10.1002/pro.2645
Mohamed, A. E., Ahmed, F. H., Arulmozhiraja, S., Lin, C. Y., Taylor, M. C., Krausz, E. R., et al. (2016a). Protonation state of F420H2 in the prodrug-activating deazaflavin dependent nitroreductase (Ddn) from Mycobacterium tuberculosis. Mol. Biosyst. 12, 1110–1113. doi: 10.1039/c6mb00033a
Mohamed, A. E., Condic-Jurkic, K., Ahmed, F. H., Yuan, P., O’Mara, M. L., Jackson, C. J., et al. (2016b). Hydrophobic shielding drives catalysis of hydride transfer in a family of F420H2-dependent enzymes. Biochemistry 55, 6908–6918.
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi: 10.1002/jcc.21256
Nelson-Sathi, S., Sousa, F. L., Roettger, M., Lozada-Chavez, N., Thiergart, T., Janssen, A., et al. (2015). Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517, 77–80. doi: 10.1038/nature13805
Ney, B., Ahmed, F. H., Carere, C. R., Biswas, A., Warden, A. C., Morales, S. E., et al. (2017). The methanogenic redox cofactor F420 is widely synthesized by aerobic soil bacteria. ISME J. 11, 125–137. doi: 10.1038/ismej.2016.100
Nocek, B., Evdokimova, E., Proudfoot, M., Kudritska, M., Grochowski, L. L., White, R. H., et al. (2007). Structure of an amide bond forming F420:γ-glutamyl ligase from Archaeoglobus fulgidus - a member of a new family of non-ribosomal peptide synthases. J. Mol. Biol. 372, 456–469. doi: 10.1016/j.jmb.2007.06.063
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Purwantini, E., Daniels, L., and Mukhopadhyay, B. (2016). F420H2 is required for phthiocerol dimycocerosate synthesis in mycobacteria. J. Bacteriol. 198, 2020–2028. doi: 10.1128/JB.01035-15
Purwantini, E., and Mukhopadhyay, B. (2013). Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLOS ONE 8:e81985. doi: 10.1371/journal.pone.0081985
Selengut, J. D., and Haft, D. H. (2010). Unexpected abundance of coenzyme F420-dependent enzymes in Mycobacterium tuberculosis and other actinobacteria. J. Bacteriol. 192, 5788–5798. doi: 10.1128/JB.00425-10
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Sparling, R., Blaut, M., and Gottschalk, G. (1993). Bioenergetic studies of Methanosphaera stadtmanae, an obligate H2–methanol utilising methanogen. Can. J. Microbiol. 39, 742–748. doi: 10.1139/m93-109
Taylor, M. C., Jackson, C. J., Tattersall, D. B., French, N., Peat, T. S., Newman, J., et al. (2010). Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol. Microbiol. 78, 561–575. doi: 10.1111/j.1365-2958.2010.07356.x
Taylor, M. C., Scott, C., and Grogan, G. (2013). F420-dependent enzymes - potential for applications in biotechnology. Trends Biotechnol. 31, 63–64. doi: 10.1016/j.tibtech.2012.09.003
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Walsh, C. (1986). Naturally occurring 5-deazaflavin coenzymes: biological redox roles. Acc. Chem. Res. 19, 216–221. doi: 10.1021/ar00127a004
Wang, P., Bashiri, G., Gao, X., Sawaya, M. R., and Tang, Y. (2013). Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc. 135, 7138–7141. doi: 10.1021/ja403516u
Keywords: F420, redox, biocatalysis, biodegradation, mycobacterium, actinobacteria, cofactor
Citation: Ney B, Carere CR, Sparling R, Jirapanjawat T, Stott MB, Jackson CJ, Oakeshott JG, Warden AC and Greening C (2017) Cofactor Tail Length Modulates Catalysis of Bacterial F420-Dependent Oxidoreductases. Front. Microbiol. 8:1902. doi: 10.3389/fmicb.2017.01902
Received: 21 July 2017; Accepted: 15 September 2017;
Published: 27 September 2017.
Edited by:
Dirk Tischler, Freiberg University of Mining and Technology, GermanyReviewed by:
Alberto A. Iglesias, National University of the Littoral, ArgentinaMatthias Boll, Albert Ludwigs University of Freiburg, Germany
Copyright © 2017 Ney, Carere, Sparling, Jirapanjawat, Stott, Jackson, Oakeshott, Warden and Greening. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris Greening, Y2hyaXMuZ3JlZW5pbmdAbW9uYXNoLmVkdQ== Andrew C. Warden, YW5kcmV3LndhcmRlbkBjc2lyby5hdQ==