 Carolina Ramírez
Carolina Ramírez Jaime Romero
Jaime Romero- 1Laboratorio de Biotecnología de los Alimentos, Instituto de Nutrición y Tecnología de los Alimentos, Universidad de Chile, Santiago, Chile
- 2Doctorado en Acuicultura, Programa Cooperativo Universidad de Chile, Universidad Católica del Norte, Pontificia Universidad Católica de Valparaíso, Santiago, Chile
Seriola lalandi is an economically important species that is globally distributed in temperate and subtropical marine waters. Aquaculture production of this species has had problems associated with intensive fish farming, such as disease outbreaks or nutritional deficiencies causing high mortalities. Intestinal microbiota has been involved in many processes that benefit the host, such as disease control, stimulation of the immune response, and the promotion of nutrient metabolism, among others. However, little is known about the potential functionality of the microbiota and the differences in the composition between wild and aquacultured fish. Here, we assayed the V4-region of the 16S rRNA gene using high-throughput sequencing. Our results showed that there are significant differences between S. lalandi of wild and aquaculture origin (ANOSIM and PERMANOVA, P < 0.05). At the genus level, a total of 13 genera were differentially represented between the two groups, all of which have been described as beneficial microorganisms that have an antagonistic effect against pathogenic bacteria, improve immunological parameters and growth performance, and contribute to nutrition. Additionally, the changes in the presumptive functions of the intestinal microbiota of yellowtail were examined by predicting the metagenomes using PICRUSt. The most abundant functional categories were those corresponding to the metabolism of cofactors and vitamins, amino acid metabolism and carbohydrate metabolism, revealing differences in the contribution of the microbiota depending on the origin of the animals. To our knowledge, this is the first study to characterize and compare the intestinal microbiota of S. lalandi of wild and aquaculture origin using high-throughput sequencing.
Introduction
Seriola lalandi (yellowtail kingfish) is a marine, pelagic, and carnivorous fish found globally in subtropical and temperate waters of the Pacific and Indian Oceans (Fowler et al., 2003). This species is important for aquaculture in Australia (Hutson et al., 2007), New Zealand (Moran et al., 2008), Japan (Nakada, 2002), and Chile, where it is part of the Chilean aquaculture diversification Program (Programa de Diversificación de la Acuicultura Chilena, PDACH). This species has excellent attributes that promote its cultivation, including high rates of growth and market acceptance (Poortenaar et al., 2001). However, disease outbreaks have increased with the intensification of aquaculture, especially during the early stages of fish development; an example of this problem in larval culture is vibriosis (Toranzo et al., 2005). Nutritional deficiencies are another important point to consider among the causes of mortalities in aquacultured fish species (Chen et al., 2007). Therefore, protecting cultured fish from diseases is essential for the sustainability of the aquaculture industry. The control of bacterial diseases in fish culture is traditionally countered by the use of antibiotics. Several authors have warned about the negative effects of the excessive use of antibiotics, which can lead to the selection of antibiotic-resistant bacteria (Dang et al., 2009; Romero et al., 2012). Therefore, the manipulation of the intestinal microbiota through the supplementation of beneficial microbes is presented as an alternative to overcome the adverse effects of antibiotics and drugs (Nayak, 2010).
The intestinal microbiota is considered important for the host, since the microorganisms that reside in the digestive tract may influence many biological processes that generate benefits to the host. Examples of which include providing enzymes to complement the digestion processes; supplying vitamins to enhance nutrition; preventing colonization by pathogens, competing for nutrients and adhesion sites; producing antimicrobial substances; and modulating the host immune system (Verschuere et al., 2000; Rawls et al., 2004; Chabrillón et al., 2005; Ringø et al., 2006; Hovda et al., 2007). This microbial community can be subcategorized into two groups. One group simply passes through the lumen with food or digesta—the allochthonous microbiota, and the other group is potentially resident and intimately associated with host tissues—the autochthonous microbiota (Gajardo et al., 2016; Tarnecki et al., 2017). The use of beneficial bacteria in aquaculture has been recently revised by Akhter et al. (2015). Several studies reported that indigenous bacteria are more beneficial than bacteria isolated from other sources (Nayak, 2010; Mills et al., 2011). This may be due at least in part to the specificity in colonization by the host-strain relationship (Ying et al., 2007).
Sullam et al. (2012) identified a significant association between the intestinal microbiota composition and fish taxonomy, suggesting a potential co-evolution of fishes and their gut microbiota. However, there are few studies that report the impact of fish microbiota under aquaculture conditions. In this context, the aim of this study is to evaluate possible differences in the microbiota associated to the intestinal contents of S. lalandi of wild and aquaculture origin using next generation sequencing (NGS) and to explore the potential importance of these differences to the host.
Materials and Methods
Sample Collection
Intestinal content samples were collected from aquaculture and wild S. lalandi and were immediately stored at −20°C. Yellowtail kingfish specimens from an aquaculture facility were collected from the Universidad Católica del Norte (Coquimbo, Chile; latitude S 29.966; longitude W 71.751), which were reared in a land based recirculating aquaculture system (RAS), using conditions as described by Aguilera et al. (2013) and diet described in Table S4. Five animals (roughly 3–5 kg) without deformities or apparent illnesses were used for feces collection. The intestinal contents were obtained with the help of a catheter and performing soft massage on the abdomen of fish that had been anesthetized with DOLICAL® 80% (Centrovet, Santiago, Chile). Yellowtail kingfish specimens from the wild environment were collected from latitude S 30.104; longitude W 71.377 to latitude S 30.302; longitude W 70.608, during December 2015. After caught, fish were kept in ice until processing, and five animals (roughly 3–4 kg) were included in this study. The intestinal contents were obtained as described above. This study followed the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Committee on the Ethics of Animal Experiments of the INTA Universidad de Chile approved the protocol.
DNA Extraction and Sequencing
DNA was extracted from the intestinal content samples (0.25 g) using the MO BIO PowerFecal®DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer's protocol. DNA concentrations were measured fluorometrically using the High Sensitivity (HS) kit on the Qubit Fluorometer 3.0 (Invitrogen Co., Carlsbad, USA). The V4 region of the 16S rRNA gene was amplified by the fusion primer method using the primers 515F and 806R described by Caporaso et al. (2011). Variable region 4 was selected because of its high coverage, low error rate and minimum loss of taxonomic resolution (Kuczynski et al., 2012; Lokesh and Kiron, 2016). In addition, the resulting amplicons are of suitable length for use with the Ion Torrent™ sequencing platform (Life Technologies). All PCR reactions were performed as described by Lokesh and Kiron (2016), except volume reaction was 25 μL; each reaction mixture containing 22.5 μL of Platinium PCR SuperMix High Fidelity (Life Technologies), 0.25 μL of primer mix (200 nM) and 2.25 μL of the template DNA (~20 ng). A negative PCR control without the DNA template was also included in the run. The PCR conditions included an initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, and extension at 68°C for 45 s. After the cycling procedure, the amplicons from each sample were pooled and run on a 1% agarose gel. Subsequently, the amplicons were purified with the QIAquick PCR Purification kit (Qiagen, Valencia, CA). DNA sequencing was performed via Ion Torrent Personal Genome Machine system (Life Technologies, California) using a 318 chip at the facilities of the University of Plymouth Enterprise Ltd.
Bioinformatics Analyses
Sequencing reads of the 16S rRNA gene were processed and analyzed using UPARSE (Edgar, 2013) and QIIME (Caporaso et al., 2010), as described previously by Ramírez and Romero (2017), except that the sequences were trimmed to 270 bp. Sequences assigned to chloroplast and unclassified at the kingdom level, were removed. OTUs containing < 10 sequences were also removed from the dataset. The analyses of diversity indexes included Good's coverage, alpha diversity indexes comprising community diversity (Simpson and Shannon index), richness (Chao-1) and phylogeny-based metrics (PD Whole Tree), which were calculated using QIIME. Principal coordinates analysis (PCoA) was used to evaluate the Beta diversity obtained by unweighted UniFrac and weighted UniFrac analyses, using the “beta_diversity.py” QIIME script. EMPeror was used to visualize the PCoA plots from the unweighted and weighted UniFrac metrics. The inferred metagenomics and predicted functional analysis was performed using PICRUSt as described previously by Ramírez and Romero (2017), and it included all the readings (325,039 from wild; 314,323 from aquaculture). The accuracy of the predictions of the metagenomes was assessed by computing the NSTI (Nearest Sequenced Taxon Index), which is an index that indicates the relationship of the microbes in a particular sample to the bacterial genomes in a database (Table S3). The associated metabolic pathways were deciphered by employing HUMAnN2 (The HMP Unified Metabolic Analysis Network) with the default settings.
Statistical Analyses
Shannon Diversity index, Simpson index, richness and PD Whole Tree were included to examine the differences in alpha diversity between the wild and aquacultured yellowtail kingfish. The normality was tested with Shapiro and Wilk test (Shapiro and Wilk, 1965). The comparisons were done with student t-test or Mann-Whitney test when distributions of data were normal or non-normal, respectively. The calculations were made in GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) with P < 0.05 considered statistically significant. The UniFrac distance matrices were analyzed by Analysis of similarities (ANOSIM) and Permutation multivariate analysis of variance (PERMANOVA) with 9999 permutations. For this purpose, the dissimilarity matrix for the unweighted and weighted UniFrac were analyzed in QIIME, with P < 0.05 considered statistically significant. To identify differentially abundant OTUs between two origins, the DESeq2 software package was employed considering P < 0.05 (Love et al., 2014). The t-test was used to evaluate bacterial functional pathways that were differentially abundant in intestinal microbiota of wild yellowtail and aquaculture yellowtail kingfish. All P-values were corrected with the Benjamini–Hochberg false discovery rate method.
Data Deposition
Raw sequences from 16S rRNA gene profiling are accessible through the SRA study accession numbers: SRS2110405, SRS2110403, SRS2110404, SRS2110402, SRS2110401, SRS2110400, SRS2110398, SRS2110397, SRS2110399, SRS2110396.
Results
Sequencing Depth
Intestinal contents were collected from wild (n = 5) and aquacultured (n = 5) S. lalandi. The microbiota composition was analyzed using barcoded sequencing of the V4 region of the 16S rRNA gene. After filtering as described above, 632,079 reads remained; 329,314 reads from wild S. lalandi with an average of 65,863 ± 10,706 reads per individual sample. And 302,765 reads from aquacultured S. lalandi with an average of 60,553 ± 5,663 reads per individual sample. These 270 bp sequences were assigned to 683 operational taxonomic units (OTUs) based on 97% similarity using QIIME. Good's coverage estimators for all samples were >0.99, indicating that sufficient sequencing coverage was achieved and that the OTUs detected in the samples are representative of the sampled population (Table S1). Rarefaction curves based on alpha diversity metrics (Chao1 and PD whole tree) reached the saturation phase in wild and aquaculture yellowtail at 30,000 sequence readings (Supplementary Figures S1, S2).
Diversity Analysis of Microbiota of Wild and Aquacultured Yellowtail
The alpha diversity indexes, Chao1, Shannon and Simpson, were found to be significantly higher for the microbiota of aquacultured samples (Figure 1). Beta diversity analysis are illustrated as principal coordinates plots in Figure 2, showing notable differentiation among bacterial communities, as they grouped by origin (wild or aquacultured animals). The graphical representation for unweighted UniFrac analyses highlights the variability in the microbiota composition between wild and aquacultures fish (Figure 2A). Furthermore, PERMANOVA and ANOSIM tests (P < 0.05) confirmed the significant differences in the composition of the microbiota depending on the origin of the fish (Table 1).
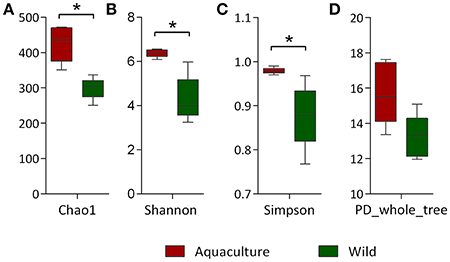
Figure 1. Comparison of alpha diversity indexes between wild and aquaculture yellowtail kingfish (Seriola lalandi). Diversity in the gut bacterial community was measured using Chao-1 (A), Shannon index (B), Simpson index (C), and phylogeny-based metrics (D). The asterisks indicate significant differences in the alpha diversity between the wild and aquaculture yellowtail (P < 0.05). Chao-1 and PD whole tree was evaluated using t-test. Shannon and Simpson index were evaluated using Mann-Whitney.
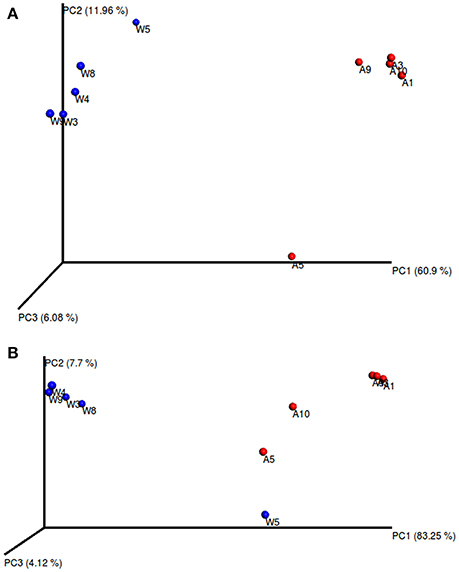
Figure 2. Principal coordinates analysis (PCoA) of the bacterial communities derived from the unweighted (A) and weigthed (B) UniFrac distance matrix. Circles represent individual samples from S. lalandi intestinal microbiota. Red circles correspond to samples derived from aquaculture, and blue circles correspond to samples from wild fish.
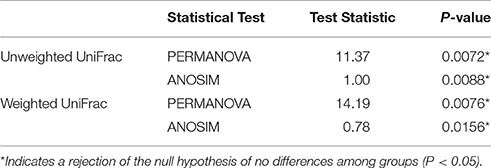
Table 1. Comparison of similarities in microbiota composition between wild and aquaculture yellowtail kingfish.
Composition of Microbial Communities
The taxonomic compositions of the S. lalandi intestinal microbiota of wild and aquacultured origin primarily present representatives of the phyla Proteobacteria, Firmicutes, and Actinobacteria followed by Bacteroidetes, Planctomycetes, Chloroflexi, and Cyanobacteria with low relative abundance (Figure 3). However, differences in relative abundances of each of these phyla were observed depending on the wild or aquacultured origin of the samples. In wild yellowtail, the most abundant phyla were Proteobacteria (83%), Actinobacteria (8%), and Firmicutes (7%). In aquacultured yellowtail, the most abundant phyla were Firmicutes (61%), Proteobacteria (20%), and Actinobacteria (14%). At the class level, the wild yellowtail microbiota consisted of Gammaproteobacteria (80%), Actinobacteria (8%), Bacilli (7%), and Alphaproteobacteria (2%) followed by the Betaproteobacteria and Clostridia in low relative abundance; whereas aquacultured yellowtail presented Bacilli (54%), Actinobacteria (14%), Gammaproteobacteria (11%), Clostridia (7%), Alphaproteobacteria (5%), and Betaproteobacteria (3%). At the order level, the following bacteria were observed in the wild yellowtail samples: Pseudomonadales (52%), Alteromonadales (23%), Actinomycetales (8%), Vibrionales (5%), Bacillales (3%), and Lactobacillales (3%); whereas aquacultured yellowtail presented Bacillales (34%), Lactobacillales (20%), Actinomycetales (14%), Pseudomonadales (10%), Clostridiales (6%), and Sphingomonadales (4%) (Table S2).
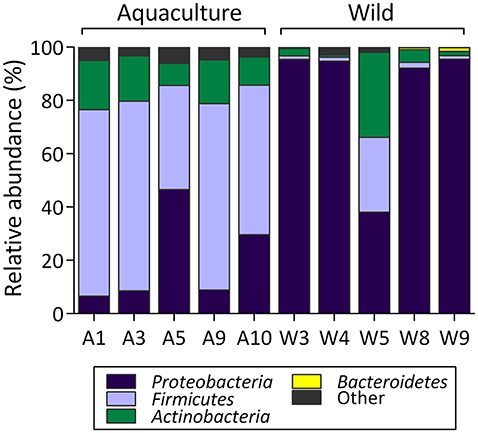
Figure 3. Relative abundance (percentage) at phylum level for each sample in the intestinal microbiota from wild and aquaculture S. lalandi. In the figure, A corresponds to aquaculture fish (Aquaculture S. lalandi) and W corresponds to individual wild fish (Wild S. lalandi).
Differential Abundances of the Bacterial Populations Associated with Origin
Specific taxa that were differentially distributed between the wild and aquacultured yellowtail were identified using DESeq2 software package. This approach allows significant differences in the abundances of each OTU to be identified based on statistical tools. The results are shown in Table 2, which depicts each phylum and genus presenting a significant difference between the two origins, aquaculture (positive) and wild (negative). In order to visualize these differences, Figure 4 was structured using the relative abundance of these OTUs as taxa. Figure 4A shows the bacterial components are more abundant in aquacultured yellowtail, starting at the phylum level, in this case corresponding to Firmicutes and Actinobacteria. In more detail, Figure 4B shows the differences at the genus level within those phyla, highlighting Staphylococcus, Clostridium, Aerococcus, Jeotgalicoccus. In contrast, in wild yellowtail, Proteobacteria was the most abundant phyla as shown in Figure 4C, which highlights the abundance of Shewanella, Psychrobacter, and Pseudomonas (Figure 4D).
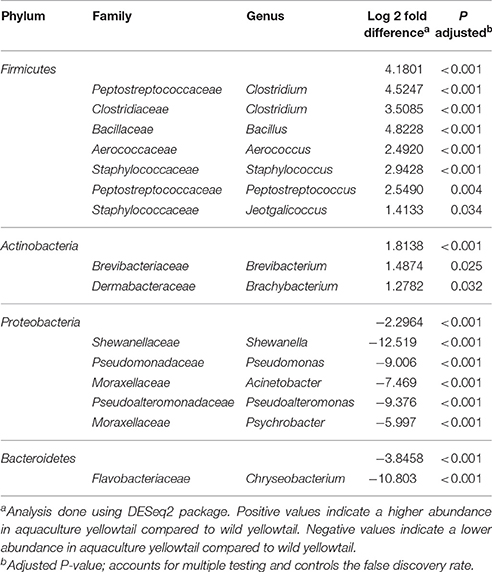
Table 2. Summary of differential abundance at phylum and genus level between wild and aquaculture yellowtail kingfish.
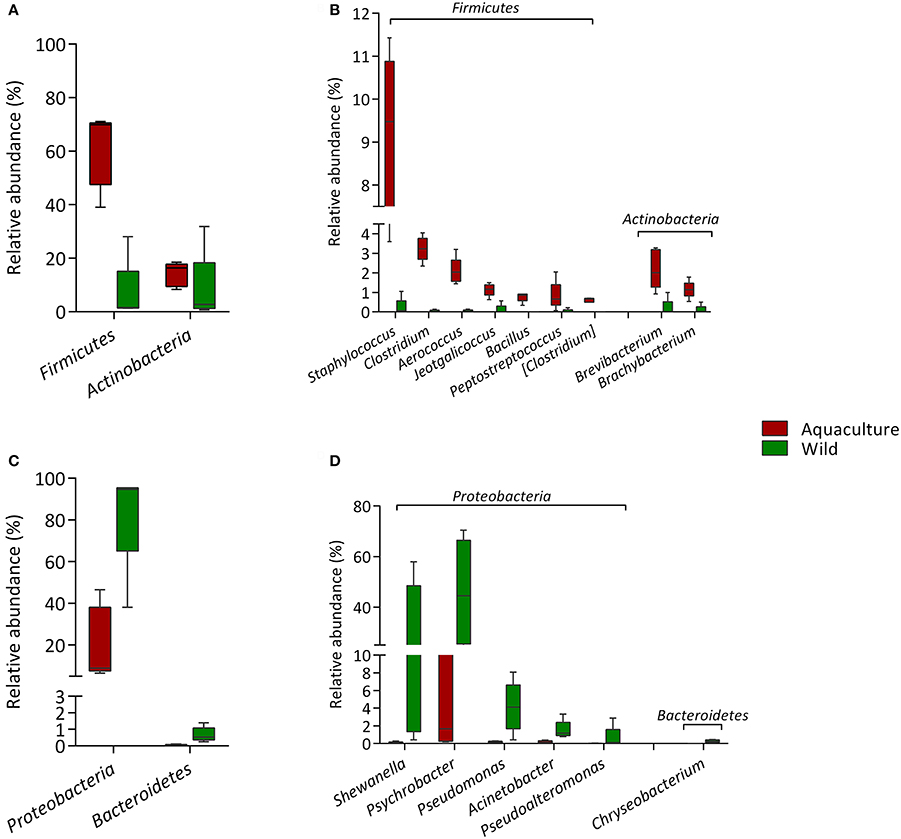
Figure 4. Comparison of the intestinal microbiota between wild and aquaculture yellowtail. This figure illustrates the differences in the microbiota in terms of relative abundance of taxa (Phylum; genus). (A) This section highlights the most abundant phyla in aquaculture yellowtail kingfish; (B) this shows most abundant genera in aquaculture yellowtail; (C) this section highlights the most abundant phyla in wild yellowtail kingfish; (D) this shows most abundant genera in wild yellowtail. Only statistical significant taxa are represented, according to DESeq2 analysis.
Potential Functions of Yellowtail Metagenome Showing Significant Differences
The changes in the presumptive functions of the intestinal microbiota of yellowtail were examined by predicting the metagenomes using PICRUSt. The accuracy of the prediction was evaluated by computing the Nearest Sequenced Taxon Index (NSTI), and the mean of the samples was 0.048 ± 0.005, indicating a relatively good match to reference genomes (ideal NSTI ≤ 0.03; Langille et al., 2013). Figure 5 depicts the general metabolic pathways, comparing microbiota functions from animals of both origins, which highlights the significant differential distribution of pathways, including those related to amino acid metabolism, carbohydrate metabolism and nucleotide metabolism. Furthermore, 19 functional pathways were found to be more highly abundant in wild yellowtail, including pathways related to the biodegradation of xenobiotics, and the metabolism of terpenoids and polyketides. In the case of aquacultured yellowtail, 31 pathways were found to be more highly abundant, including those related to amino acid metabolism, carbohydrate metabolism, and nucleotide metabolism. The significant differences in bacterial function between wild and aquacultured yellowtail is detailed in Table 3.
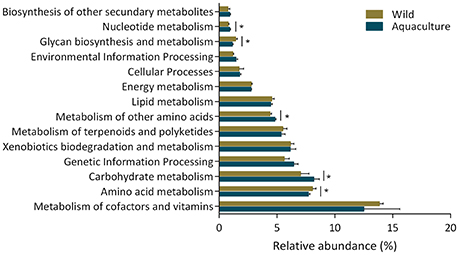
Figure 5. The general metabolic pathways of the intestinal microbiota from wild and aquaculture S. lalandi. The asterisks indicate significant differences in pathways of the bacterial components between wild and aquaculture yellowtail kingfish, this was assessed using t-test, P-values were corrected with the Benjamini–Hochberg false discovery rate method. Those values were considered significant P < 0.05.
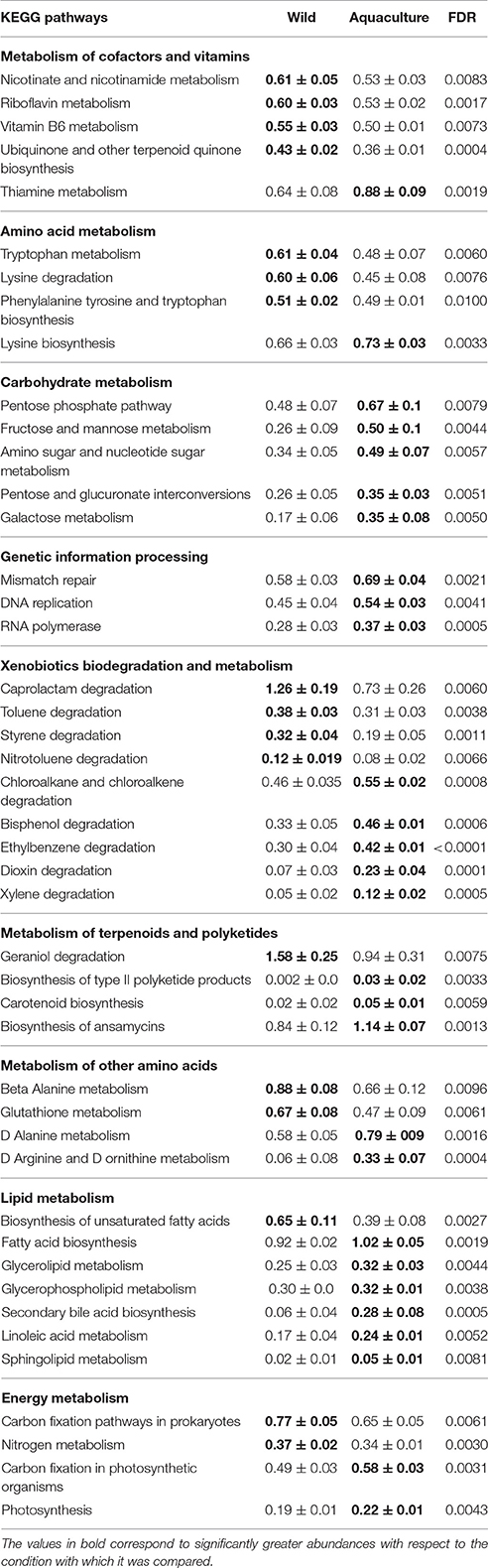
Table 3. Summary of differences in pathways between predicted metagenomes.
Discussion
To our knowledge, this is the first study to characterize and compare the intestinal microbiota of wild and aquacultured S. lalandi using high-throughput sequencing. A previous study by Aguilera et al. (2013) examined the culture dependent microbiota of yellowtail juveniles in an aquaculture system. The authors described that in an aquaculture system the microbiota was represented by the phylum Proteobacteria. These results contrast with those obtained in our study, where the intestinal microbiota of aquacultured yellowtail was dominated by the phylum Firmicutes, exhibiting 61% of relative the abundance, whereas the phylum Proteobacteria only reached a 20% relative abundance (Figure 4A). The differences of our results with respect to Aguilera et al. (2013) could be attributed to the methodological differences since the microbiota composition obtained using culture-dependent approaches is highly influenced by the culture medium used to perform the isolation of microorganisms. In contrast, culture-independent approaches, such as NGS, exceed the limits of bacterial recovery from the culture medium, which in marine environments does not exceed 1% of cultivable bacteria (Amann et al., 1995). In addition, NGS strategies allow for a more complete view of the composition of the microbiota, with high and deep coverage, allowing the taxonomic classification of bacteria using thousands of reads (Whiteley et al., 2012). This approach has improved our view of the fish microbiome and several reviews have been published recently (Llewellyn et al., 2014; Ghanbari et al., 2015; Tarnecki et al., 2017). These compilations indicate that structure of fish microbiomes could be more similar to the microbiome of their environments than those of mammals; in fact, previous reports addressing gut microbiota in fish have showed that gut microbiota composition was different from those found in the surrounding environment such as feed or water (Kormas et al., 2014; Bakke et al., 2015; Estruch et al., 2015; Li et al., 2015). Therefore, fish microbiome structure are not simply a reflection of the environment, but are a result of both phylogenetic factors and host ecology (Ghanbari et al., 2015).
Our results indicate that wild S. lalandi has a different microbiota compared to the aquaculture animals (Figure 2, Table 1). These results could be associated with differences in fish feeding depending on their origin. Gajardo et al. (2017) reported that there are significant changes in beta diversity statistics associated with the Salmo salar intestinal microbiota when fed with different diets. Similar to results were described by Schmidt et al. (2016), who suggested that the microbiota compositions were modulated by the diets under a RAS environment, using an ANOSIM test. These authors indicated that the salmon microbiota fed with two diets (fishmeal-based and fishmeal-free) were dominated by Lactobacillales, Aeromonadales, and Actinomycetales, followed by Sphingomonadales and Clostridiales; but at the genus level, the microbiota showed significant differences depending on the diet. Similar results were described in Sparus aurata (Estruch et al., 2015), where total fishmeal replacement had an important impact on microbial profiles with Streptococcus was highly represented in fish fed with fishmeal diets and microbial composition of the RAS was totally different to that of the sea bream gastrointestinal tract. These match with our results where the S. lalandi microbiota of aquacultured fish under RAS was dominated by bacteria of the order Bacilales.
The differences in relative abundances of the components of the intestinal microbiota of S. lalandi, shown in Figure 4, indicate that in the case of wild yellowtail the predominant phylum is Proteobacteria. This phylum has been described as the most predominant in freshwater wild fish by Liu et al. (2016), as well as in wild marine fish by Star et al. (2013) and Ramírez and Romero (2017). In those reports, the most abundant class identified within this phylum was Gammaproteobacteria, similar to the results of our study. The order Pseudomonadales was the most abundant and contained differentially represented genera in wild yellowtail, such as Psychrobacter, Pseudomonas, and Acinetobacter.
Psychrobacter sp. has been described in the intestinal microbiota of several fish, such as Atlantic cod (Ringø et al., 2006), grouper (Sun et al., 2009; Yang et al., 2011) and fine flounder (Ramírez and Romero, 2017). Psychrobacter sp. has previously showed an in vitro antagonistic effect against a number of pathogenic species, such as S. aureus, Vibrio harveyi, Vibrio metschnikovii, and Vibrio alginolyticus (Sun et al., 2009). This may be an important observation because vibriosis has been reported as a primary disease in hatcheries and cultures of marine fish (Reid et al., 2009). More recently, Makled et al. (2017) reported that the dietary administration of a strain of Psychrobacter improved immunological parameters and growth performance in tilapia. Similarly, Pseudomonas is a very diverse group showing adaptability to a range of environmental niches and a broad ecological distribution. Pseudomonas strains have a wide variety of metabolic abilities and some of them have been used as probiotics in aquaculture, improving the response of different hosts to pathogens (Alavandi et al., 2004; Korkea-aho et al., 2011; Giri et al., 2014). In contrast, Acinetobacter has been reported in the microbiota of several fish, such as rainbow trout (Spanggaard et al., 2001), Atlantic salmon (Navarrete et al., 2009) and yellowtail kingfish (Aguilera et al., 2013). This bacterial genus has been reported to inhabit soil and aquatic environments, including freshwater ecosystems, raw sewage and wastewater treatment plants, as well as activated sludge (Doughari et al., 2011). They are primarily free-living saprophytes that are found ubiquitously in nature and have a variety of metabolic capabilities, such as the degradation of aromatic compounds (Mazzoli et al., 2007). Another genus with significant relative abundance in wild yellowtail was Shewanella; several strains of this genus have previously been reported to produce polyunsaturated fatty acids (Hirota et al., 2005; Bianchi et al., 2014) including some strains retrieved from microbiota of freshwater fish Dailey et al. (2016). Shewanella strains have shown probiotic properties, improving survival against vibriosis in Solea senegalensis (Tapia-Paniagua et al., 2014) and enhancing immune parameters in Sparus aurata L (Cordero et al., 2015).
In contrast to wild yellowtail, the statistically dominant phylum in aquaculture specimens was Firmicutes (Figure 4A). This phylum has been described as the most predominant in the intestinal content of aquacultured fishes such as Siberian sturgeon (Geraylou et al., 2013), grass carp (Wu et al., 2012) and Atlantic salmon (Schmidt et al., 2016). At the genus level, Staphylococcus, Clostridium, Aerococcus, Brevibacterium, were the most abundant (Figure 4C). Bacteria of these genera have been described as beneficial bacteria for hosts, such as Brevibacterium spp., and Staphylococcus spp., and they may contribute to nutritional processes in Arctic charr (Ringø et al., 1995). Furthermore, oral administration of Clostridium butyricum to rainbow trout enhanced their resistance to vibriosis by increasing the phagocytic activity of leucocytes (Sakai et al., 1995). Some strains of Aerococcus have also been described as an antagonist to opportunist pathogens (Burbank et al., 2012; Valchuk et al., 2015).
The PICRUSt analysis was used to infer functional capabilities of the microbial communities. This approach predicts functional potentials of a community by comparing its metagenome with reference genomes (Langille et al., 2013). According to metagenome prediction, we inferred that the most abundant functional categories were those corresponding to the functions of the metabolism of cofactors and vitamins, amino acid metabolism, and carbohydrate metabolism (Figure 5) and found significant differences in KEGG pathways between wild and aquacultured yellowtail (Table 2). For example, pathways relating to the metabolism of group B vitamins and ubiquinone biosynthesis were more abundant in the wild yellowtail microbiota. Ubiquinone has been described as having bioactivities related to energy metabolism, immunological competence, and antioxidation (Pravst et al., 2010). Similarly, KEGG pathways related to amino acid metabolism, such as tryptophan and phenylalanine, were significantly associated with wild yellowtail, whereas lysine biosynthesis was associated with aquacultured fish. These amino acids are commonly mentioned as essential nutritional requirements in several fish (Wilson and Halver, 1986). In terms of carbohydrate metabolism, the pentose and galactose pathways were more significantly represented in aquacultured yellowtail. This observation could be related to the artificial diet (Table S4), because it contains a larger amount of carbohydrates (>12%) with respect to common prey for wild yellowtail, such as fish, shrimp, and squid (< 6%; Sidwell et al., 1974). In contrast, lipopolysaccharide biosynthesis was significantly associated with wild yellowtail, and this is coincident with a larger abundance of Proteobacteria in these fish, which are gram-negative bacteria harboring LPS genes. Another important KEGG pathway significantly present in wild yellowtail was the biosynthesis of unsaturated fatty acids, which include essential omega-3 fatty acids (eicosapentaenoic acid and docosahexaenoic acid). As mentioned before, Gammaproteobacteria was dominant in wild fish, and this class includes the previously reported EPA/DHA bacterial producers (Dailey et al., 2016; Ramírez and Romero, 2017). However, PICRUSt is a predictor of potential functions within a metagenome; metabolomic approaches could be used to identify differences in the metabolic functions in new environments such as the microbiota of wild and aquaculture fish (Gajardo et al., 2016).
In conclusion, this study reveals the differences in the composition of the intestinal microbiota of wild origin and aquacultured S. lalandi. These differences likely result in different potential contributions of the microbiota to the host. These results indicate a strong influence of host feeding on the composition and diversity of the intestinal microbiota. The bacterial genera differentially represented between fish of both origins present positive characteristics for the host, especially those associated with wild yellowtail. Therefore, it would be interesting to evaluate the activity of isolates of these genera as potential probiotics for their use in S. lalandi aquaculture. Our findings could provide a promising direction for the healthy aquaculture of S. lalandi.
Author Contributions
CR Performed DNA extraction; data collection; data analysis, and interpretation; wrote the initial version of the manuscript. JR Conception or design of the work; contributed to the discussion and writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CR would like to acknowledge the National Commission of Scientific and Technologic Research (CONICYT) for the funding through the National PhD funding program. We thank Rodrigo Rojas from Laboratorio de Patología Acuática, Universidad Católica del Norte for the supplying the intestinal contents of fish. This project was funded by a research grant from FONDECYT 1140734 and 1171129 (Fondo Nacional de Desarrollo Científico y Tecnológico, Chile), and supported by project Aquapacifico 15PCTI-46284 from Corfo.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01844/full#supplementary-material
References
Aguilera, E., Yany, G., and Romero, J. (2013). Cultivable intestinal microbiota of yellowtail juveniles (Seriola lalandi) in an aquaculture system. Lat. Am. J. Aquat. Res. 41, 395–403.
Akhter, N., Wu, B., Mahmood, A., and Mohsin, M. (2015). Probiotics and prebiotics associated with aquaculture: a review. Fish Shellfish Immunol. 45, 733–741. doi: 10.1016/j.fsi.2015.05.038
Alavandi, S., Vijayan, K., Santiago, T., Poornima, M., Jithendran, K., Ali, S., et al. (2004). Evaluation of pseudomonas sp. PM 11 and Vibrio fluvialis PM 17 on immune indices of tiger shrimp, Penaeus monodon. Fish Shellfish Immunol. 17, 115–120. doi: 10.1016/j.fsi.2003.11.007
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Bakke, I., Coward, E., Andersen, T., and Vastein, O. (2015). Selection in the host structures the microbiota associated with developing cod larvae (Gadus morhua). Environ. Microbiol. 17, 3914–3924. doi: 10.1111/1462-2920.12888
Bianchi, A., Olazábal, L., Torre, A., and Loperena, L. (2014). Antarctic microorganisms as source of the omega-3 polyunsaturated fatty acids. World J. Microbiol. Biotechnol. 30, 1869–1878. doi: 10.1007/s11274-014-1607-2
Burbank, D., Lapatra, S., Fornshell, G., and Cain, K. (2012). Isolation of bacterial probiotic candidates from the gastrointestinal tract of rainbow trout, Oncorhynchus mykiss (Walbaum), and screening for inhibitory activity against Flavobacterium psychrophilum. J. Fish Dis. 35, 809–816. doi: 10.1111/j.1365-2761.2012.01432.x
Caporaso, J., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J., Lauber, C., Walters, W., Berg-Lyons, D., Lozupone, C., Turnbaugh, P., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chabrillón, M., Rico, R., Arijo, S., Diaz-Rosales, P., Balebona, M., and Morinigo, M. (2005). Interactions of microorganisms isolated from gilthead sea bream, Sparus aurata L., on Vibrio harveyi, a pathogen of farmed Senegalese sole, Solea senegalensis (Kaup). J. Fish Dis. 28, 531–537. doi: 10.1111/j.1365-2761.2005.00657.x
Chen, B. N., Qin, J. G., Carragher, J. F., Clarke, S. M., Kumar, M. S., and Hutchinson, W. G. (2007). Deleterious effects of food restrictions in yellowtail kingfish Seriola lalandi during early development. Aquaculture 271, 326–335. doi: 10.1016/j.aquaculture.2007.04.016
Cordero, H. F. A., Guardiola, F. S. T., Tapia-Paniagua, S. A., Cuesta, A. J., Meseguer, J., Balebona, M., et al. (2015). Modulation of immunity and gut microbiota after dietary administration ofalginate encapsulated Shewanella putrefaciens Pdp11 to gilthead seabream (Sparus aurata L.). Fish Shellfish Immunol. 45, 608–618. doi: 10.1016/j.fsi.2015.05.0010
Dailey, F. E., McGraw, J. E., Jensen, B. J., Bishop, S. S., Lokken, J. P., Dorff, K. J., et al. (2016). The microbiota of freshwater fish and freshwater niches contain omega-3 fatty acid-producing Shewanella species. Appl. Environ. Microbiol. 82, 218–231. doi: 10.1128/AEM.02266-15
Dang, H., Zhao, J., Song, L., Chen, M., and Chang, Y. (2009). Molecular characterizations of chloramphenicol- and oxytetracycline-resistant bacteria and resistance genes in mariculture waters of China. Mar. Pollut. Bull. 58, 987–994. doi: 10.1016/j.marpolbul.2009.02.016
Doughari, H. J., Ndakidemi, P. A., Human, I. S., and Benade, S. (2011). The ecology, biology and pathogenesis of Acinetobacter spp.: an overview. Microbes Environ. 26, 101–112. doi: 10.1264/jsme2.ME10179
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Estruch, G., Collado, M. C., Pe-aranda, D. S., Tomás Vidal, A., Jover Cerdá, M., Pérez Martínez, G., et al. (2015). Impact of fishmeal replacement in diets for gilthead sea bream (Sparus aurata) on the gastrointestinal microbiota determined by pyrosequencing the 16S rRNA gene. PLoS ONE 10:e0136389. doi: 10.1371/journal.pone.0136389
Fowler, A. J., Ham, J. M., and Jennings, P. R. (2003). Discriminating between Cultured and Wild Yellowtail Kingfish (Seriola lalandi) in South Australia. Report of SARDI Aquatic Sciences. Adelaide, SA: SARDI.
Gajardo, K., Jaramillo-Torres, A., Kortner, T., Merrifield, D., Tinsley, J., Bakke, A., et al. (2017). Alternative protein sources in the diet modulate microbiota and functionality in the distal intestine of Atlantic salmon (Salmo salar). Appl. Environ. Microbiol. 83:e02615-16. doi: 10.1128/AEM.02615-16
Gajardo, K., Rodiles, A., Kortner, T. M., Krogdahl, Å., Bakke, A. M., Merrifield, D. L., et al. (2016). A high-resolution map of the gut microbiota in Atlantic salmon (Salmio salar): a basis for comparative gut microbial research. Sci. Rep. 6:30893. doi: 10.1038/srep30893
Geraylou, Z., Souffreau, C., Rurangwa, E., Maes, G. E., Spanier, K. I., Courtin, C. M., et al. (2013). Prebiotic effects of arabinoxylan oligosaccharides on juvenile Siberian sturgeon (Acipenser baerii) with emphasis on the modulation of the gut microbiota using 454 pyrosequencing. FEMS Microbiol. Ecol. 86, 357–371. doi: 10.1111/1574-6941.12169
Ghanbari, M., Kneifel, W., and Domig, K. J. (2015). A new view of the fish gut microbiome: advances from next-generation sequencing. Aquaculture 448, 464–475. doi: 10.1016/j.aquaculture.2015.06.033
Giri, S., Sukumaran, V., Sen, S., and Jena, P. (2014). Effects of dietary supplementation of potential probiotic Bacillus subtilis VSG1 singularly or in combination with Lactobacillus plantarum VSG3 or/and Pseudomonas aeruginosa VSG2 on the growth, immunity and disease resistance of Labeo rohita. Aquacult. Nutr. 20, 163–171. doi: 10.1111/anu.12062
Hirota, K., Nodasaka, Y., Orikasa, Y., Okuyama, H., and Yumoto, I. (2005). Shewanella pneumatophori sp. nov., an eicosapentaenoic acid-producing marine bacterium isolated from the intestines of Pacific mackerel (Pneumatophorus japonicus) Int. J. Syst. Evol. Microbiol. 55, 2355–2359. doi: 10.1099/ijs.0.63804-0
Hovda, M., Lunestad, B., Fontanillas, R., and Rosnes, J. (2007). Molecular characterization of the intestinal microbiota of farmed Atlantic salmon (Salmo salar L.). Aquaculture 272, 581–588. doi: 10.1016/j.aquaculture.2007.08.045
Hutson, K. S., Ernst, I., and Whittington, I. (2007). Risk assessment for metazoan parasites of yellowtail kingfish Seriola lalandi (Perciformes: Carangidae) in South Australian sea-cage aquaculture. Aquaculture 271, 85–99. doi: 10.1016/j.aquaculture.2007.03.020
Korkea-aho, T., Heikkinen, J., Thompson, K., Wright, A., and Austin, B. (2011). Pseudomonas sp. M174 inhibits the fish pathogen Flavobacterium psychrophilum. J. Appl. Microbiol. 111, 266–277. doi: 10.1111/j.1365-2672.2011.05044.x
Kormas, K. A., Meziti, A., Mente, E., and Frentzos, A. (2014). Dietary differences are reflected on the gut prokaryotic community structure of wild and commercially reared sea bream (Sparus aurata). Microbiologyopen 3, 718–728. doi: 10.1002/mbo3.202
Kuczynski, J., Lauber, C., Walters, W., Wegener, L., Clemente, J., Gevers, D., et al. (2012). Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 13, 47–58. doi: 10.1038/nrg3129
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, T., Long, M., Gatesoupe, F.-J., Zhang, Q., Li, A., and Gong, X. (2015). Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microb. Ecol. 69, 25–36. doi: 10.1007/s00248-014-0480-8
Liu, H., Guo, X., Gooneratne, R., Lai, R., Zeng, C., Zhan, F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6:24340. doi: 10.1038/srep24340
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H., and Derome, N. (2014). Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5:207. doi: 10.3389/fmicb.2014.00207
Lokesh, J., and Kiron, V. (2016). Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Sci. Rep. 6:19707. doi: 10.1038/srep19707
Love, M., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Makled, S. O., Hamdan, A. M., El-Sayed, A.-F. M., and Hafez, E. E. (2017). Evaluation of marine psychrophile, Psychrobacter namhaensis SO89, as a probiotic in Nile tilapia (Oreochromis niloticus) diets. Fish Shellfish Immunol. 61, 194–200. doi: 10.1016/j.fsi.2017.01.001
Mazzoli, R., Pessione, E., Giuffrida, M. G., Fattori, P., Barello, C., Giunta, C., et al. (2007). Degradation of aromatic compounds by Acinetobacter radioresistens S13: growth characteristics on single substrates and mixtures. Arch. Microbiol. 188, 55–68. doi: 10.1007/s00203-007-0223-z
Mills, S., Stanton, C., Fitzgerald, G., and Ross, P. (2011). Enhancing the stress responses of probiotics for a lifestyle from gut to product and back again. Microb. Cell Fact. 10, 1–19. doi: 10.1186/1475-2859-10-S1-S19
Moran, D., Wells, R., and Pether, S. (2008). Low stress response exhibited by juvenile yellowtail kingfish (Seriola lalandi Valenciennes) exposed to hypercapnic conditions associated with transportation. Aquacult. Res. 39, 1399–1407. doi: 10.1111/j.1365-2109.2008.02009.x
Nakada, M. (2002). Yellowtail culture development and solutions for the future. Rev. Fish. Sci. 10, 559–575. doi: 10.1080/20026491051794
Navarrete, P., Espejo, R. T., and Romero, J. (2009). Molecular analysis of microbiota along thedigestive tract of juvenile Atlantic salmon (Salmo salar L.). Microb. Ecol. 57, 550–561. doi: 10.1007/s00248-008-9448-x
Nayak, S. (2010). Probiotics and immunity: A fish perspective. Fish Shellfish Immunol. 29, 2–14. doi: 10.1016/j.fsi.2010.02.017
Poortenaar, C. W., Hooker, S. H., and Sharp, N. (2001). Assessment of yellowtail kingfish (Seriola lalandi lalandi) reproductive physiology, as a basis for aquaculture development. Aquaculture 201, 271–286. doi: 10.1016/S0044-8486(01)00549-X
Pravst, I., Žmitek, K., and Žmitek, J. (2010). Coenzyme Q10 Contents in foods and fortification strategies. Crit. Rev. Food Sci. Nutr. 50, 269–280. doi: 10.1080/10408390902773037
Ramírez, C., and Romero, J. (2017). Fine flounder (Paralichthys adspersus) microbiome showed important differences between wild and reared specimens. Front. Microbiol. 8:271. doi: 10.3389/fmicb.2017.00271
Rawls, J., Samuel, B., and Gordon, J. (2004). Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. U.S.A. 101, 4596–4601. doi: 10.1073/pnas.0400706101
Reid, H., Treasurer, J., Adam, B., and Birkbeck, H. (2009). Analysis of bacterial populations in the gut of developing cod larvae and identification of Vibrio logei, Vibrio anguillarum and Vibrio splendidus as pathogens of cod larvae. Aquaculture 288, 36–43. doi: 10.1016/j.aquaculture.2008.11.022
Ringø, E., Sperstad, S., Myklebust, R., Refstie, S., and Krogdahl, Å. (2006). Characterisation of the microbiota associated with intestine of Atlantic cod (Gadus morhua L.): the effect of fish meal, standard soybean meal and a bioprocessed soybean meal. Aquaculture 261, 829–841. doi: 10.1016/j.aquaculture.2006.06.030
Ringø, E., Strøm, E., and Tabachek, T. (1995). Intestinal microflora of salmonids: a review. Aquacult. Res. 26, 773–789. doi: 10.1111/j.1365-2109.1995.tb00870.x
Romero, J., Feijoo, C. G., and Navarrete, P. (2012). “Antibiotics in aquaculture – use, abuse and alternatives,” in Health and Environment in Aquaculture, ed E. Carvalho (Rijeka: Intech), 159–198.
Sakai, M., Yoshida, T. S., Atsuta, S., and Kobayas, M. (1995). Enhancement of resistance to vibriosis in rainbow trout, Oncorhynchus mykiss (Walbaum), by oral administration of Clostridium butyricum bacterin. J. Fish Dis. 18, 187–190. doi: 10.1111/j.1365-2761.1995.tb00276.x
Schmidt, V., Amaral-Zettler, L., Davidson, J., Summerfelt, S., and Good, C. (2016). The influence of fishmeal-free diets on microbial communities in Atlantic salmon Salmo salar recirculation aquaculture systems. Appl. Environ. Microbiol. 82, 4470–4481. doi: 10.1128/AEM.00902-16
Shapiro, S. S., and Wilk, M. B. (1965). An analysis of variance test for normality (complete samples). Biometrika 52, 591–611. doi: 10.1093/biomet/52.3-4.591
Sidwell, V., Foncannon, P., Moore, N., and Bonnet, J. (1974). Composition of the edible portion of raw (fresh or frozen) crustaceans, finfish, and mollusks.1. protein, fat, moisture, ash, carbohydrate, energy values, and cholesterol. Mar. Fish. Rev. 36, 21–35.
Spanggaard, B., Huber, I., Nielsen, J., Sick, E. B., Pipper, C. B., Martinussen, T., et al. (2001). The probiotic potential against vibriosis of the indigenous microflora of rainbow trout. Environ. Microbiol. 3, 755–765. doi: 10.1046/j.1462-2920.2001.00240.x
Star, B., Haverkamp, T. H., Jentoft, S., and Jakobsen, K. S. (2013). Next generation sequencing shows high variation of the intestinal microbial species composition in Atlantic cod caught at a single location. BMC Microbiol. 13:248. doi: 10.1186/1471-2180-13-248
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O'Connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sun, Y., Yang, H., Ling, Z., Chang, J., and Ye, J. (2009). Gut microbiota of fast and slow growing grouper Epinephelus coioides. Afr. J. Microbiol. Res. 3, 637–640
Tapia-Paniagua, S., Vidal, C., Lobo, M. J., Prieto-Álamo, J., Jurado, H., Cordero, H., et al. (2014). The treatment with the probiotic Shewanella putrefaciens Pdp11 of specimens of Solea senegalensis exposed to high stocking densities to enhance their resistance to disease. Fish Shellfish Immunol. 41, 209–221. doi: 10.1016/j.fsi.2014.08.019
Tarnecki, A. M., Burgos, F. A., Ray, C. L., and Arias, C. R. (2017). Fish intestinal microbiome : diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol 123, 2–17. doi: 10.1111/jam.13415
Toranzo, A., Magari-os, B., and Romalde, J. (2005). A review of the main bacterial fish diseases in mariculture systems. Aquaculture 246, 37–61. doi: 10.1016/j.aquaculture.2005.01.002
Valchuk, S. I., Stepansky, D. A., Shevchenko, T. N., Koshevaya, I. P., Ryzhenko, S. A., and Kremenchutsky, G. N. (2015). Biological properties of aerococci and bacilli as a component of new associate-probiotic complex. Visn. Dnipropetr. Univ. Biol. Med. 6, 57–62. doi: 10.15421/021511
Verschuere, L., Rombaut, G., Sorgeloos, P., and Verstraete, W. (2000). Probiotic bacteria as biological control agents in aquaculture. Microbiol. Mol. Biol. Rev. 64, 651–671. doi: 10.1128/MMBR.64.4.655-671.2000
Whiteley, A. S., Jenkins, S., Waite, I., Kresoje, N., Payne, H., Mullan, B., et al. (2012). Microbial 16S rRNA ion tag and community metagenome sequencing using the ion torrent (PGM) platform. J. Microbiol. Methods 91, 80–88. doi: 10.1016/j.mimet.2012.07.008
Wilson, R., and Halver, J. (1986). Protein and amino acid requirements of fishes. Annu. Rev. Nutr. 6, 225–244.
Wu, S., Wang, G., Angert, E. R., Wang, W., Li, W., and Zou, H. (2012). Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS ONE 7:e30440. doi: 10.1371/journal.pone.0030440
Yang, H., Sun, Y., Ma, R., Li, J., and Huang, K. (2011). Probiotic Psychrobacter sp. improved the autochthonous microbial diversity along the gastrointestinal tract of grouper Epinephelus coioides. J. Aquac. Res. Dev. S1:001. doi: 10.4172/2155-9546.S1-001
Keywords: microbiota, high-throughput sequencing, yellowtail, Seriola
Citation: Ramírez C and Romero J (2017) The Microbiome of Seriola lalandi of Wild and Aquaculture Origin Reveals Differences in Composition and Potential Function. Front. Microbiol. 8:1844. doi: 10.3389/fmicb.2017.01844
Received: 12 May 2017; Accepted: 08 September 2017;
Published: 26 September 2017.
Edited by:
Télesphore Sime-Ngando, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
José Manuel Mazón-Suástegui, Centro de Investigaciones Biológicas del Noroeste, MexicoEric E. Allen, Scripps Institution of Oceanography, United States
Miguel Ignacio Uyaguari-Diaz, University of British Columbia, Canada
Copyright © 2017 Ramírez and Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaime Romero, jromero@inta.uchile.cl