 Aaron Lerner
Aaron Lerner Marina Arleevskaya
Marina Arleevskaya Andreas Schmiedl2
Andreas Schmiedl2- 1The Ruth and Bruce Rappaport Faculty of Medicine, Technion Israel Institute of Technology, Haifa, Israel
- 2Department of Research, AESKU.KIPP Institute, Wendelsheim, Germany
- 3Central Research Laboratory, Kazan State Medical Academy Kazan, Kazan, Russia
The links between microorganisms/viruses and autoimmunity are complex and multidirectional. A huge number of studies demonstrated the triggering impact of microbes and viruses as the major environmental factors on the autoimmune and inflammatory diseases. However, growing evidences suggest that infectious agents can also play a protective role or even abrogate these processes. This protective crosstalk between microbes/viruses and us might represent a mutual beneficial equilibrium relationship between two cohabiting ecosystems. The protective pathways might involve post-translational modification of proteins, decreased intestinal permeability, Th1 to Th2 immune shift, induction of apoptosis, auto-aggressive cells relocation from the target organ, immunosuppressive extracellular vesicles and down regulation of auto-reactive cells by the microbial derived proteins. Our analysis demonstrates that the interaction of the microorganisms/viruses and celiac disease (CD) is always a set of multidirectional processes. A deeper inquiry into the CD interplay with Herpes viruses and Helicobacter pylori demonstrates that the role of these infections, suggested to be potential CD protectors, is not as controversial as for the other infectious agents. The outcome of these interactions might be due to a balance between these multidirectional processes.
Introduction
Infection and Autoimmunity
The relationship between infections and autoimmunity is complex. Microbial and viral infections might act as environmental triggers inducing or propagating autoimmune and inflammatory processes, resulting in symptomatic presentation of a disease in genetically high risk individuals (Lerner, 2015; Arleevskaya et al., 2017). An autoimmune disease onset following an infectious agent exposure has been well-documented (Pordeus et al., 2008; Bogdanos et al., 2015; Sakkas and Bogdanos, 2016). At least, for CD, the following infections were suggested to be associated with the disease: viruses: enterovirus, Epstein-Barr virus (EBV), Cytomegalovirus (CMV), hepatitis C virus (HCV), hepatitis B virus (HBV), and rotavirus, microbes: Bacteroides species, Campylobacter jejuni, Pneumococcus, Mycobacterium tuberculosis, and Helicobacter pylori (Lerner, 2015). However, recent serological evidence suggests the opposite outcome, which is the protection against autoimmune conditions following bacterial/viral exposure (Christen and von Herrath, 2005). At least the suggestive protector agents for CD were CMV, EBV, Rubella, and Herpes simplex type 1 virus (HSV1) when compared to healthy people (Plot and Amital, 2009; Jansen et al., 2016). According to the “hygiene hypothesis,” the excessively sterile environment leads to the enhanced incidence of autoimmune disorders, asthma, and allergies, thus, associating surge of CD incidence with decreased infectious environment (Lerner et al., 2015b,e; Bloomfield et al., 2016).
Celiac Disease
Celiac disease is a life-long autoimmune disease (Lerner et al., 1996) mainly of the proximal intestine, affecting genetically predisposed individuals. Gluten, the storage protein of wheat, is the environmental inducer of the disease in addition to other structurally related molecules found in barley, rye, and oat (Lerner, 2014). Many environmental factors were suggested to induce or enhance the disease: multiple infections (Lerner and Reif, 2015), early infections (Myléus et al., 2012), early gastrointestinal infections (Beyerlein et al., 2017), lack of breast feeding (Lerner and Matthias, 2016c), time and amount of gluten consumption (Chmielewska et al., 2015), microbiome/dysbiome repertoire (Lerner et al., 2015a; Lerner and Matthias, 2017a,b), mode of delivery (Decker et al., 2011), early vaccination (Kemppainen et al., 2017) or early consumption of antibiotics (Canova et al., 2014) and geo-epidemiological influences (Lerner, 1994, 2015; Reif and Lerner, 2004b; Lerner and Matthias, 2015a). The abnormal immune response is directed, in particular, against tissue transglutaminase (tTG), representing the autoantigen, (Reif and Lerner, 2004a; Lerner et al., 2015c) and the two main autoantibodies, anti-endomysium and anti-tTG antibodies, are the most prevalent serological markers used to screen for the condition (Shamir et al., 2002; Lerner and Matthias, 2015d). Recently, the list of CD serological markers was expanded by two additional autoantibodies: anti-deamidated gliadin peptide and anti- tTG neo-epitope antibodies, found to be reliable for CD diagnosis (Rozenberg et al., 2012; Lerner and Blank, 2014; Lerner et al., 2015e). As yet, HLA-DQ2 and HLA-DQ8 are known predisposing genetic factors. The sequential events in disease progression were unraveled in the last years and gave rise to multiple future therapeutic strategies (Lerner, 2010). Notably, its epidemiological, incidental, and clinical presentation are changing continuously, and new clinical pictures are reported and expand the abundance of clinical variance of the disease (Lerner et al., 2015d). In fact, age of disease onset increases and the traditional enteric presentation is more and more replaced by extraintestinal manifestations. Skin (Lerner et al., 2015d), endocrine (Lerner and Matthias, 2016d; Lerner et al., 2017), hepatic (Anania et al., 2015), metabolic (Eliyah Livshits et al., 2017), skeletal (Lerner and Matthias, 2016a), rheumatic (Lerner and Matthias, 2015b), geriatric (Lerner and Matthias, 2015c), hematological (Branski et al., 1992), neurological (Zelnik et al., 2004; Lerner et al., 2012), gynecological and infertility (Mårild et al., 2012; Casella et al., 2016), oral and dental (Cantekin et al., 2015), hypercoagulability (Lerner and Blank, 2014), cardiac (Lerner et al., 2015d), and behavioral abnormalities (Zelnik et al., 2004) are often described. Those epidemiological and clinical changes can explain why the disease is diagnosed during the whole human life-span including in the elderly (Lerner and Matthias, 2015c). There is no doubt that in the last decades its incidence is constantly increasing, ranging between 1 and 3% nowadays (Lerner, 2014; Lerner et al., 2015b). The present review will concentrate, expand and update on the multiple faces of the inductive/protective roles that infectious agents might play in CD pathogenesis. This aspect is further interesting since pathogens are the major drivers of human selective genetic adaptation during evolution (Vatsiou et al., 2016), and the question of microbes that are bugging the celiac patient “are they friends or foes?” is the subject of the current review.
Infections and CD
It should be clarified that, although the trigger role of microorganisms and viruses in the CD development was undoubtedly traced in numerous investigations, it substantially differs from other immune pathogenesis like rheumatoid arthritis (as a classic model of an autoimmune disease; Arleevskaya et al., 2016; Kemppainen et al., 2017).
The induction of rheumatoid arthritis most likely occurs under the influence of the burden of many trivial infections, influencing the patient's immune system due to the frequent and prolonged infectious episodes (Arleevskaya et al., 2014). Individuals at CD risk apparently do not have such features in their mucosal immunity, nor significant defects in systemic anti-infective protection, impacting infection susceptibility. Since all the CD studies are focused only on the disease link with various gastrointestinal infections, such association is different from what was shown in rheumatoid arthritis (Riddle et al., 2012).
The number of infectious agents related to CD is continuously increasing (Figure 1). Examples for viruses enterovirus, EBV, Cytomegalovirus (CMV), HCV, HBV, and rotavirus And for microbes Bacteroides species, C. jejuni, Pneumococcus, M. tuberculosis, and H. pylori (Lerner, 2015). However, links between CD and infections were more associative and less causative, thus, far from being elucidated. Moreover, the mutually exclusive hypotheses about the provocative and protective role of a particular microorganism/virus in CD pathogenesis were suggested and discussed in various publications.
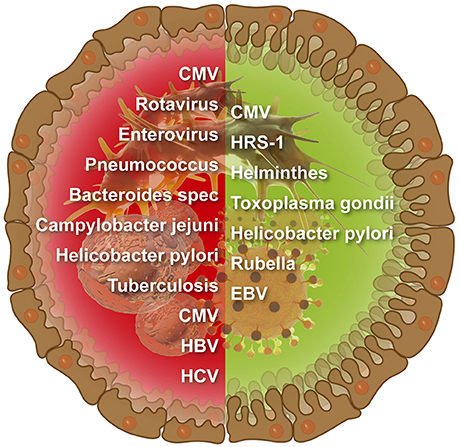
Figure 1. Infectious agents that were suggested to induce (left side) or protect against (right side) intestinal celiac disease.
It appears that the essential condition for the CD induction is a gastrointestinal infection (Kemppainen et al., 2017). Apparently, the other major condition is an early childhood—namely immature gastrointestinal tract, immature immune system, and gastrointestinal microbiome at the early phase of formation. The beginning of gut colonization by microorganisms set the stage for the cross talks between the epithelium, enteric lymphoid tissue, and microflora, all together establishing the intestinal barrier and as a consequence, a strictly dosed delivery of macromolecules into the internal environment and shaping of mucosal tolerance to food antigens and normal flora (Makarova et al., 2014).
So, in the vulnerable infant period any gastrointestinal infection, even a transitory one, is potentially able to disturb these processes of gut microbiome maturation and the establishment of local immunity and immune tolerance, including that against food and microbial antigens. Apparently, deleterious coincidence of these circumstances leads to an error in the negative selection of gluten-reactive lymphocyte clones.
Thus, the community of microorganisms, being extremely vulnerable during the ripening period, appears to be an inert system in adults. For example, in adults it needs no more than 30–60 days to restore gut microflora after the exposure to antibiotics (Spanhaak et al., 1998; Tannock et al., 2000). At the same time, a weekly clindamycin treatment of a newborn reduces Bacteroides diversity for the next 2 years (Jernberg et al., 2007). The same infections, capable of altering the fate of a sick infant, are merely a light ripples on the ocean surface for an adult microbiome.
In conclusion, there is an undoubted link between CD development and microorganisms, and this link looks to be rather specific. Gastrointestinal infections in predisposed infants with an immature gastrointestinal tract and immune system might shape gut microbiome in the immature and therefore labile circumstances. Such an unfortunate combination could trigger early CD development or becomes a ticking time bomb, represented by the structural features of the gut microbiome and persistence of gluten–reactive lymphocyte clones with latent basal cell proliferation without overt disruptive inflammatory activation.
Gut Microbiome Signature in Celiac Disease
Gut microbiome analysis in the healthy adult human populations revealed about 1150 bacterial species, the majority (50–75%) being represented by Firmicutes, and then Bacteroidetes (10–50%), Actinobacteria (1–10%), with fewer than 1% being Proteobacteria (Manichanh et al., 2012). Apparently, the HLA system, to a certain extent, shapes microbiome structure. Besides, polymorphisms of some other non-HLA genes were found to correlate with a certain microbiome structure (Spor et al., 2011). Interestingly, in addition to the HLA system, microbiome composition may be due to CD-associated polymorphisms of defensin, some molecules of Toll-like receptor signaling pathways and vitamin D receptor genes (San-Pedro et al., 2005; Fernandez-Jimenez et al., 2010; Wang et al., 2016). Whole genome study of 93 individuals and 16S rRNA gene pyrosequencing of their body microflora revealed 83 alliances between genetic variance in host sequence and plethora of specific microbial taxa (Blekhman et al., 2015). In particular, the links with CD-associated host genes were revealed. In addition to the host genes related to immunity, a link was found between the microbiome composition and SNP of the genes not related to immunity. For example, the authors revealed an interesting correlation between the abundance of Bifidobacterium in the gastrointestinal tract and host genetic variation in LCT gene, encoding the lactase enzyme hydrolyzing dietary lactose. This gene SNPs are known to be associated with lactose intolerance, which is frequently associated with celiac disease (Ojetti et al., 2005). Bifidobacterium is able to metabolize lactose, and there are some strains preferring lactose instead of glucose. The authors suggested that the problems with individual's consumption of milk products might impact the richness of Bifidobacterium in the gastrointestinal tract.
The results of gut microbiome structure investigations in the infants at CD risk as well as in the therapy-naïve patients at the disease onset are somewhat contradictory. Besides the bulk of the results was obtained by study of feces, while the principal for CD microbe community in the small intestine boundary layers has its own peculiarities, although in a certain extent it is associated with fecal microbes. However, a certain tendency can be traced. A single and rather limited study of mucosa-associated microbiota in the proximal gut—using enteric samples from 45 children with CD and 18 clinical controls born during the “Swedish CD epidemic”—demonstrated only marginally differences between the groups. Enrichment with Clostridium, Prevotella, and Actinomyces was revealed in the most of the CD samples (Ou et al., 2009). Feces studies demonstrated, that infants genetically predisposed to CD, had significantly higher abundance of Firmicutes (Clostridium) and Proteobacteria (Escherichia/Shigella) and desreased proportions of Actinobacteria (Bifidobacterium) and Bacteroidetes compared to low-risk infants (Sellitto et al., 2012; Olivares et al., 2015). In a proof of concept study, Sellitto and co-workers have traced the longitudinal changes in the microbial populations colonizing from birth to 24 months in 30 genetically predisposed infants. They demonstrated that even at 2 years of age their microbiota do not resemble that of adults, while in not at-risk infants the maturation was complete at 1 year of age. The group was divided into an early and a late gluten exposure groups (17 and 13 infants, respectively). The authors showed that genetically susceptible infants may benefit from delayed gluten exposure not before 12 months of age. A hypothesis was forwarded that lack of maturity of the enteric microbiota faced with early gluten consumption can induce or accelerate the autoimmunogenetic process. Not less interesting was the infant's metabolome. When solid food was introduced at 6 months of age, succinate, acetate, propionate, and butyrate accumulated in their stools. However, by 2 years of age, butyrate, and acetate were the dominant short-chain fatty acids (SCFA). Since bacteroidetes are associated most strongly with propionate, while Firmicuters are negatively correlated to SCFAs production (Koenig et al., 2011), Sellitto's group envisioned that the high Firmicutes and low Bactriodetes abundance in CD infants results in down production of those protective SCFAs, thereby abrogating enteric health and predisposing them to autoimmune diseases. The decrease in Lactobacillus spp. associated with lower lactate production, observed in between 6 and 12 months of age, accompanied by decreased SCFAs' feces repertoire, during a vulnerable time of mucosal immune maturation and microbiome compositional changes might lead to loss of tolerance to non-self-antigens like gluten in those CD genetically predisposed infants (Sellitto et al., 2012). Once again it points to the important relations connecting nutrition to microbiome composition and diversity, its metabolome and the local maturation and functioning of the immune system.
From this perspective, it is interesting to compare the composition of the gut flora in infants at CD risk and premature babies with a priori immature gut. Arboleya and coauthors compared the gradual establishment of the intestinal microbiome in very-low birth-weight preterm infants with that of healthy full-term, vaginally born, breast-fed neonates using 16S rRNA gene profiling and quantitative PCR for the various microbial taxa. It was demonstrated that preterm neonates sheltered a higher relative proportion of Firmicutes at 2 days of age, and of Proteobacteria in the later sampling times, compared to control babies. Prematurity reflected reduced levels of Bacteroidetes at day 2 and as well as in later sampling times together with Actinobacteria (Arboleya et al., 2016). In addition, very-low birth-weight preterm infants frequently displayed a lag in establishing an adult microbiota compared to full-term children (Weng and Walker, 2013). So, the parallels to the peculiarities of the gut flora in CD prone individuals seem to be obvious. Combining the results of Arboleya et al. and Sellitto et al. about the enteric mucosal immaturity and the unbalanced microbiome found early in life and in CD high-risk infants, it can be suggested that by ingesting gluten peptides, the disease will progress in these individuals, unlike in non-CD high-risk premature infants, that will never develop CD. The features of CD gut microbiome, incorporated in early childhood seem to persist in the adulthood even despite gluten withdrawal (Nistal et al., 2012; Wacklin et al., 2014).
Caminero and co-workers demonstrated that gluten amounts in feces of healthy volunteers, CD patients and individuals under risk receiving gluten-free or normal diet depended on gluten intake. The greatest amount of gluten was found in fecal samples from healthy volunteers being on normal diet, with a significant decrease in the untreated CD patients and the individuals under risk. It is noteworthy that in all the groups fecal peptidase activity against the gluten-derived peptide 33-mer inversely correlated with gluten amount in the samples (Caminero et al., 2015). It looks like the increased functional proteolytic activity of gut microflora in CD patients can affect gluten excretion. The same research group isolated 144 strains belonging to 35 microbial species that might be involved in gluten digestion in the human intestine. Most of the strains were part of the phyla Firmicutes and Actinobacteria, mainly from the genera Lactobacillus, Streptococcus, Staphylococcus, Clostridium and Bifidobacterium (Caminero et al., 2014). Ninety-four of these strains were capable to metabolize gluten, 61 of them showed an extracellular proteolytic activity of gluten proteins, and several strains showed a peptidase activity toward the “supra-molecule” 33-mer peptide, the luminal immunogenic molecule in CD patients (Caminero et al., 2014). At the end of the day, there is a certain difference in gluten proteolysis by various bacteria, and the immunogenicity of the generated peptide fragments might be different. It should be noted that these studies were carried out in cultures, in which the glutenase activity of aggregate microbial community as a whole (biofilm) in the gut boundary layer might significantly differ from the isolated activity of the individual members of this community.
It is known that the microbiome impact gastrointestinal and systemic functions by its metabolome, the most studied one being the short chain fatty acids (SCFA). Most recently, the topic of nutrition, microbiome, and SCFA associations in CD was updated (Lerner et al., 2016). Multiple beneficial effects to the host were attributed to them. Changes in microbiota and their SCFA production is clearly related to the pathogenesis of CD. Interestingly, peculiar dysbiosis and significant changes in stool SCFA profile were described in several autoimmune diseases, one of those is in Behcet's disease where decreased butyrate production was suggested to play a role in its pathogenesis (Consolandi et al., 2015).
Taken together, the microbiota/dysbiota disbalance may present a risk factor for CD either directly by influencing the mucosal immune responses or by intensifying inflammatory responses to gluten. In contrary, several microbial species are capable to break down gliadin and perhaps therefore decrease the immunopathogenicity of consumed gliadin (Sjöberg et al., 2013; Carding et al., 2015; Lerner and Matthias, 2015a; Rostami-Nejad et al., 2015).
Coexistence of Certain Infections and CD: A Critical Look at the Issue
It is of interest how CD affected individuals survived in the presence of harmful conditions (increased gluten content and toxicity in wheat, increased gluten consumption worldwide; Lerner et al., 2017) and despite them, thrived and expanded? (Lerner, 2011; Lerner et al., 2015b). Despite being underprivileged with nutritional deficiencies, failure to thrive and high morbidity and mortality, a substantial increase in the disease incidence is observed in the last decades. There are several theories explaining this paradox. One is positive evolutionary selection, in which the celiac patient accumulates protective genes (Lerner, 2011). The other is due to some pathogens which are the major drivers of human selective genetic adaptation (Vatsiou et al., 2016) that could have been beneficial environmental factors, protecting the CD populations. Indeed, in contrast to the observations that infections may induce CD, some infection agents were assumed to have a protective impact (Christen and von Herrath, 2005; Gaisford and Cooke, 2009; Kivity et al., 2009; Plot et al., 2009).
This assumption was based in particular on a lower incidence of the serum antibodies against Cytomegalovirus (CMV), EBV, and Rubella in CD patients when compared to healthy people (Plot and Amital, 2009). Another argument for the protective effect is the inverse correlation between serum anti-CMV, -EBV, and/or -Herpes simplex type 1 virus (HSV1) IgG levels and anti-tTG antibodies (Jansen et al., 2016).
Viruses
It should be specified that Jansen and coauthors determined the antiviral antibody levels in the sera samples of 6-year old children (Jansen et al., 2016). CMV single infection and combined CMV, EBV, and/or herpes simplex virus type 1 infection antibodies were inversely associated with strongly tTg-IgA positivity. The authors suggested that the serological profile may indicate a protective effect of herpesvirus infections in the pathogenesis of celiac disease autoimmunity. It is accepted that by this age IgG and IgM production is close to that of the adults. However, the immune system is still immature and its reaction is not fully functional at this age even in healthy children, whereas the immune system of CD prone children, at least, the local immune defense, is somewhat delayed in the maturation. Thus, Jansen et al. suggested explanation should be taken with a grain of salt.
In Plot's publication, the age profile of the studied cohorts was not presented (Plot and Amital, 2009). There are some curious details in this publication which need to be interpreted. First, while the prevalence of the anti-EBV capsid antigen and anti-EBV nuclear antigen IgG in the CD patients was significantly lower than that in the controls, the prevalence of the anti-EBV early antigen IgG was comparable in the two groups and the prevalence of anti-EBV capsid antigen IgM, though unreliably, was more than twice higher in the CD group. In general, the anti-EBV capsid antigen IgMs are known to be produced during the acute phase (the first days—6 months from the onset of the disease) or during acute exacerbation of chronic EBV infection (http://www.tiensmed.ru/news/epstein-barr-bc1.html#nov1). In addition, if the anti-EBV early antigen IgG is not concurrently revealed, it may indicate incubation period or the very beginning of the infection (up to 1 week of symptoms). So, either in the CD patients this generally latent infection is apt to more frequent exacerbations, or, given the reduced frequency of the studied IgG antiviral antibodies, the existence of some features of the antiviral antibody production in CD can be assumed. Secondly, revealed by PCR, CMV DNA presence in the samples is not always accompanied by the presence of serum specific antibodies. This situation is typical in particular for the infants with immature immune system (Dong et al., 2004, 2005).
A number of publications on the HBV vaccine non-responsiveness in CD might attest for possible unique features of the antiviral antibody response formation (Noh et al., 2003; Park et al., 2007; Urganci and Kalyoncu, 2013), further criticizing the assumption that decrease antibodies activities in CD patients represent a protective effect.
As for the inverse correlation of the antiviral and anti-tTG antibodies demonstrated by Jansen et al. (2016), similar patterns are not uncommon in autoimmune diseases, and this fact by no means demonstrates a lesser exposure to the infections. For example, high levels of autoantibodies against double-stranded DNA, reflecting the activity and severity of systemic lupus erythematosus, are quite often combined with lower levels of antibodies to particular bacterial DNA (Pisetsky and Drayton, 1997). The inverse correlation of those indexes is usually explained by a distorted immune humoral response.
Additionally, it should be specified that, in general, in Herpesvirus infections and viral hepatitis the specific antibodies are not the principal players in the antiviral defense but are accepted to be the reliable serological markers for an infection (Grinde, 2013). It is advisable to note that caution should be used for the interpretation of the protective role of these infections in CD, relying only on data on the incidence of the corresponding antibodies.
As for Rubella infection, the specific antibodies are the major antiviral response players. However, the incidence of CD in children vaccinated with inactivated rubella virus as part of polio vaccine was close to that in the unvaccinated children (Myléus et al., 2012). Thus, a reduced incidence of the anti-rubella IgG antibodies demonstrated by Plot and co-workers can mean an equally probable lower exposure of CD patients to Rubella and the above-discussed features of the specific IgG antibody production. Moreover, since the antiviral immune response is always multi-componental, the disturbed antibody formation, although being a weak link of an antiviral defense, does not necessarily entail an increased susceptibility to Rubella.
The data which might testify for the direct and reverse links of CD to Herpes virus infection are summarized in Table 1. The analysis of the data demonstrates that the links of CD and herpes infections are multi-directional. On one hand there are some peculiarities, which can promote the viral infecting: (1) CD-associated DQA1*0501/DQB1*0201 genotype, which is also due to the imperfect response against Herpes viral infection; (2) the typical CD immature gastro—intestinal tract and the delayed process of microbiome maturation, which might be risk factors for the virus infecting; (3) the typical CD mucosal overexpression of epidermal growth factor receptors, by which Herpes viruses enter the cells; (4) increased expression of IL-33, suppressing local antiviral immunity in CD patients. On the other hand, the increased levels of several humoral factors with the antiviral activity; increased expression of some cytokines, which promote mucosa maturation and thus increase its' resistance to the viruses; as well as some potential features of CD microbiome that might indicate a backward link between CD and Herpes viral infection. The protective effect of the infection on atopic manifestations was demonstrated in the case of the early (infancy or early childhood) EBV exposure, while the later infection predisposes to the atopic disease (Nilsson et al., 2005, 2009). So, if to extrapolate the data on the links of herpes and atopic diseases to CD, it is likely that the early (infancy or early childhood) EBV exposure might play a protective role, while the later infection might trigger CD or have no impact on it at all.

Table 1. Direct (⇑⇑) and reverse (⇑⇓) links of CD and Herpes virus infections.
Helicobacter pylori (Hp)
The permanent interest in CD and Hp infection coexistence is quite natural, due to the gut-stomach axis (Lerner and Matthias, 2016b). The infectious inflammatory process directly in the gastrointestinal tract—CD epicenter, which might shape the local immune system and microbiota, might obviously play a role in CD pathogenesis. In addition, both CD and Hp infection in a number of cases are associated with the diffuse lymphocytic gastroenteropathy (Lynch et al., 1995; Broide et al., 2007; Pai, 2014). However, diffuse lymphocytic gastroenteropathy is far from being obligatory attributed only to both entities (Wu and Hamilton, 1999; Nielsen et al., 2014). Besides, lymphocytic gastritis and a subsequently villous atrophy are accepted to be a non-specific manifestation of many pathological conditions in the gastrointestinal tract, due to a wide variety of infectious, immunologic or any inflammatory stimuli raising intraepithelial lymphocyte numbers. Lymphocytic duodenitis and increased intraepithelial lymphocytosis are known to be associated with diseases that are completely different in their pathogenesis, such as autoimmune disorders like CD (Broide et al., 2007; Rostami et al., 2010), tropical sprue, food protein intolerance, Hp-induced duodenitis, peptic duodenitis, parasitic, and viral infections, intestinal lymphoma (Chang et al., 2005; Brown et al., 2006; Pallav et al., 2012; Rosinach et al., 2012; Shmidt et al., 2014) drugs' induced duodenitis (non-steroidal anti-inflammatory drug; Shmidt et al., 2014) and small-intestine bacterial overgrowth (Lappinga et al., 2010). The differential diagnosis of lymphocytic gastritis is not less restricted. CD and HP are not the only ones. Various non-HP infections, inflammatory conditions and several non-celiac autoimmune diseases were described (Broide et al., 2007; Polydorides, 2014).
The interest in the CD—HP infection link is fueled by the well-known data, indicating that childhood infection with HP could protect against the development of Crohn's disease, severe gastric-reflux disease, Barrett's esophagus and esophagus adenocarcinoma (Chen and Blaser, 2008). Yet, as for the protective role of Hp in CD, the available data are limited and quite contradictory. The prevalence of CD among Hp-positive adults was 0.05% compared with 0.09% among Hp-negative individuals (statistically non-significant) while the prevalence of Crohn's disease among Hp–positive patients was 0.07% compared with 0.24% among Hp-negative patients (Bartels et al., 2016). Based on these data, at least in adults, the protective effect of Hp on CD is minimal, if at all existing. However, given the fact that in childhood gastrointestinal infection appears to be a more important condition for CD triggering than in adults, this conclusion is not necessarily true in the case of early exposure to Hp (Kemppainen et al., 2017). Unfortunately, we failed to find the similar data on the CD frequency in children infected with Hp early in life.
The incidences of Hp and CD worldwide vary enormously (9–100%, 0.3–3.9%, respectively; Rostami-Nejad et al., 2016). Most of the studies aimed to determine the ratio of Hp infected individuals in CD and non-CD control groups. In our opinion, this study design gives less information about the inductive/protective effect of Hp infection on the CD development as on the susceptibility of the CD patients for the infection. The results of various studies on Hp both in children and adults are contradictory (Table 2). The spread in the ratios of both Hp infected CD patients and non-CD controls in these publications might be due to the national and age-related characteristics of the studied cohorts (Eusebi et al., 2014). As for the diametrically opposed regularities of HP incidence in CD and non-CD groups, a meticulously analysis of these publications, did not lead us to any reason for the conflicting results, but to the possible differences in the poorly described clinical characteristics of the controls. In these studies, the authors examined the collections of endoscopically obtained biopsies and sera allocating the cases into CD and non-CD (control) groups. It is obvious that the persons accepted as controls underwent the relevant examination because they had any gastroenterological problems associated or not associated with HP infection.
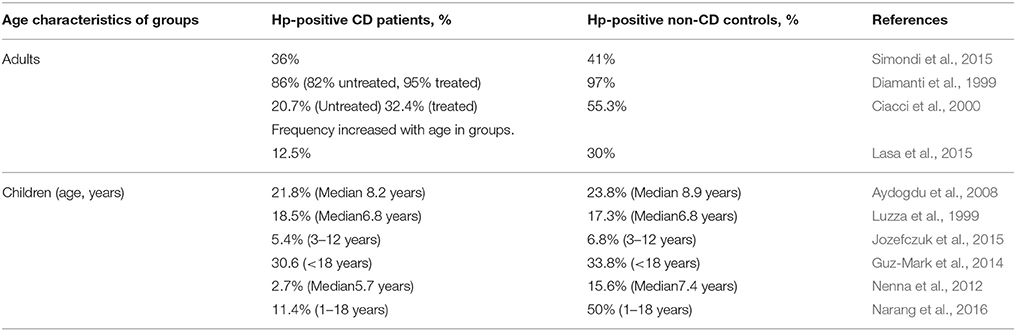
Table 2. The incidence of Helicobacter pylori infection (Hp-positive, %) in CD patients and non-CD controls.
The analysis of the literature data which might testify the direct and reverse links of CD and Hp infection (Table 3) shows that the interaction of the two diseases represents an interweaving of differently directed processes. Perhaps the end result might depend on the balance of these processes, being deeply individual in each specific case. Important is the assumption that the direct or reverse CD/HP link may depend on the age at which the encounter with the bacterium occurred. At least, it is important for allergies–Hp links. Our attempt to test this hypothesis failed, because the literary data accumulated to the present moment are largely insufficient.
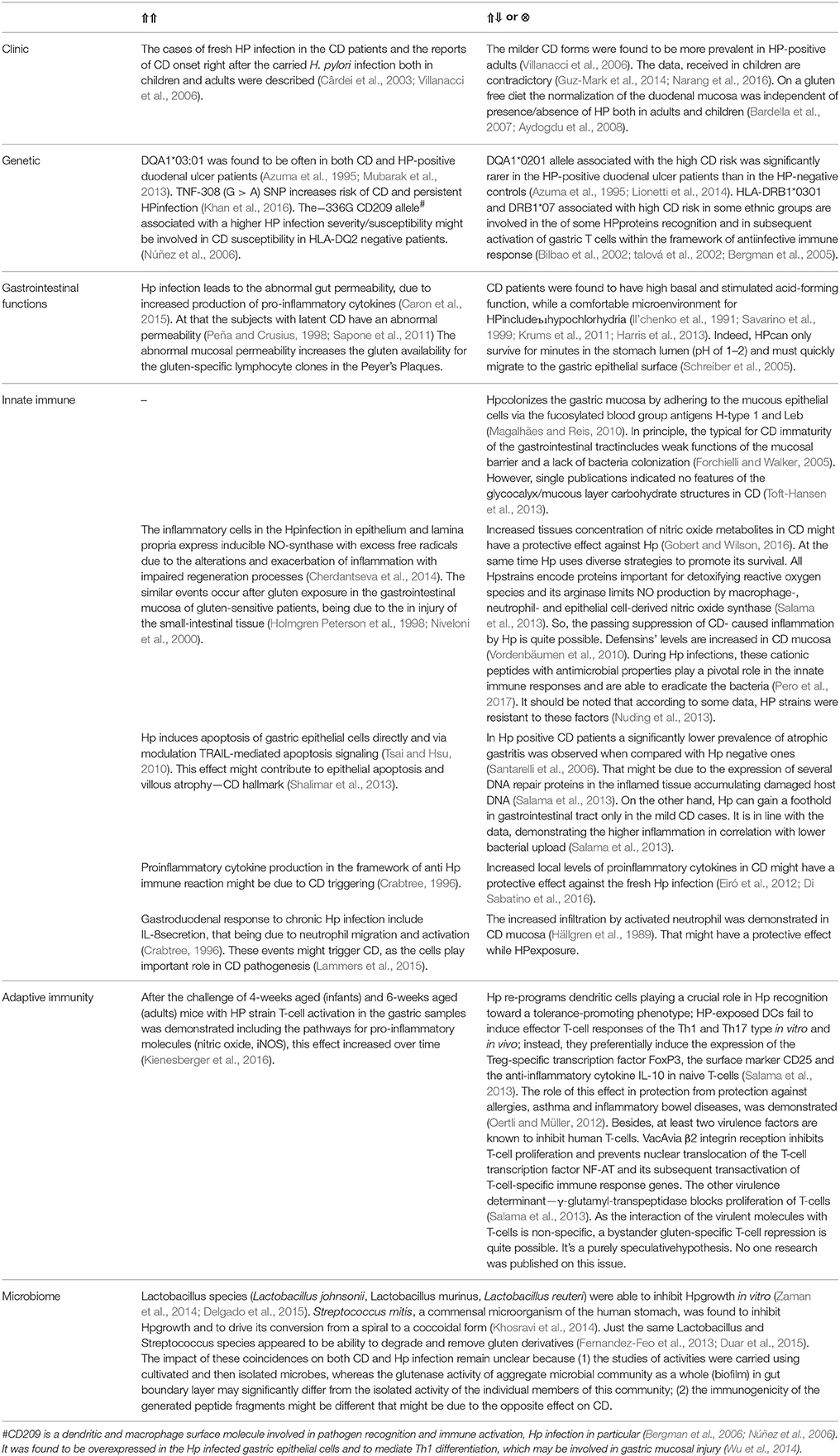
Table 3. Direct (⇑⇑), reverse (⇑⇓) or no (⊗) links of CD and Hp infection.
Potential Mechanisms for Beneficial Bugs' Effects in Celiac Gut
Multiple potential pathophysiological avenues were suggested to understand the microbial-gut cross-talks in CD.
Post-translational Modification of Protein (PTMP) from Non-self- to Self-proteins
Endogenous and microbial enzymes are capable to generate intestinal enzymatic neo-antigens via PTMP. The modifications taking place in the intestinal lumen include peptides crosslinking, de/amination/deamidation by the transglutaminases, de/phosphorylation, a/deacetylation, de/tyrosination, and many other enzymatic modifications exist (Lerner et al., 2016). Related to the present topic, the human endogenous intestinal enzymes, tTG and its family member, the exogenous microbial transglutaminases, induce multiple neo-epitopes on the TG-gliadin cross-linked complex resulting in the formation of antibodies against the complexes in CD. CD is a classical disease where luminal PTMP is driving the disease. It seems logical that a microbial agent might modify non-self-peptide to self-one—reducing its immunogenicity. Additionally, in CD, some microbe strains might modify gluten in the lumen, thus preventing or aggravating the inflammatory cascade and the intestinal damage progression via PTMP (Caminero et al., 2016).
Horizontal Gene Transfer in the Human Gut Lumen
Given the extensive influence of the microbiota on human health, the gut-microbiome integrity is of prime importance for host health and survival. In this regards, our bodies' “second genome” cohabit with the human one to form a stable equilibrium for the two kingdom's long term survival. As opposed to long-term evolutionary events, newer genetic manipulations with microorganisms, plants, animals, or nutrients, applying new food technologies and/or microbial engineered delivery systems or novel mode of therapies are rapidly evolving.
Due to the close relationship and intimate cross-talks between the human and the gut's biospheres, consumption of the modified genetic cargo into the human intestinal ecosystem might occur. It was hypothesized that modern probiotic ingestion, genetically manipulated food consumption and genetically manipulated microorganism usage are potential genetic driving forces for changing the evolutionary equilibrium established during the last millions of years (Cho and Blaser, 2012). Horizontal gene exchange is the ability to transfer genetic material between contacting biological domains, including eukaryote (plants, animals, and man), prokaryote (microbes), and viruses (Aminov, 2011; Ruggiero et al., 2015). Despite not being investigated in CD, various virulent genes, the most studied one is the antibiotic resistance gene, were described to be laterally transferred. It is hypothesized that the opposite might occur. This infectious genetic cargo might include anti-inflammatory/pro-apoptotic/Treg or other immune-modulatory genes, attenuating or abrogating autoimmunity.
Infections as Tight Junction Closure Enhancers
The tight junction protein, Zonulin, is involved in the regulation of the intestinal permeability between gut epithelial cells. Several clinical trials with Zonulin antagonist (Larazotide acetate, AT-1001, Alba Therapeutics, USA) demonstrated the promising therapeutic effect in CD (Lerner, 2010; Khaleghi et al., 2016). Larazotide acetate—an octa-peptide derived from a cholera toxin ZO, antagonizing zonulin via receptor blockade—is aimed to decrease the paracellular transport caused by gluten and thus to suppress the activation of the pathological immune cascade. In addition to the above mentioned cholera toxin derivative, many other factors produced by microorganisms can improve tight junction performance, modulating intestinal permeability. Salmonella enterica serovar, Escherichia coli, and C. jejuni modulated enteric epithelial barrier functions in chickens (Awad et al., 2012, 2014, 2015), and the probiotics Lactobacillus casei DN-114001 and E. coli strain Nissle 1917 decreased intestinal epithelium permeability in human intestinal originated cell lines (Parassol et al., 2005; Zyrek et al., 2007; Trebichavsky et al., 2010). Taken together, modulation of gut permeability by infectious agent, counteracting the breached tight junction integrity in CD, might represent a protective pathway.
Molecular Mimicry between Infectious Agents and Self-antigens
Generally, molecular mimicry between foreign (infectious/environmental) and self-antigens is a well-described pathway of autoimmune disease induction. Recently, it was suggested that antigen mimicry between foreign and self-antigens might be due to the long-term regulation of inflammation (Pontes-de-Carvalho et al., 2013). In a cohort of African patients, infected with Schistosoma it was found that the parasite inhibited production of anti-nuclear antibodies (Mutapi et al., 2011). More recently, apparent effectiveness of rotavirus vaccination was found to prevent the onset of CD autoimmunity (Silvester and Leffler, 2016). This finding gives indirect evidence for the persistence of regulatory cells in the lack of stimulation of the immune system by pathogen-derived processes. The strength of the immune adjustment, however, may increase with the uninterrupted presence of the pathogen or its antigens. Thus, molecular mimicry can represent a protective mechanism of autoimmunity.
Th2 to Th1 Shift
Inflammation, given rise by microbes, viruses and especially by parasites such as helminths, can shift the Th1 pathway to Th2 one, resulting in a more immunosuppressive state where regulatory T cells might be induced or be activated (Shor et al., 2013). Recent studies have supplied such an evidence for pathogen-specific regulatory cells in Leishmania major, Herpes simplex virus, and Friend retrovirus (murine leukemia virus) infections (Christen and von Herrath, 2005). During the acute phase of the infection this Th2 profile counter-regulates Th1-driven autoimmune pathologies. Along the chronic stage of infection, immune-regulatory networks arise, mainly led by regulatory T cells. These cells produce IL-10 and TGFβ, which has observative effect on Th1-related autoimmune diseases, such type 1 diabetes mellitus or CD. In fact, several helminths were tried in CD patients with encouraging results (Croese et al., 2015). Treatment with helminthes or helminthes ova ameliorated the clinical pictures of several autoimmune conditions in patients as well as in animal models (Smallwood et al., 2017). A major recent contribution to the field is the helminth phosphorylcholine proved to be an immunemodulatory molecule. Most recently, tuftsin-phosphorylcholine, a novel helminth-based compound was shown to reduce pro-inflammatory cytokine production and induced anti-inflammatory cytokine expression and Treg and Breg cell expansion in mouse models of rheumatoid arthritis, lupus nephritis, and colitis (Bashi et al., 2015b, 2016; Shor et al., 2015).
It is conceivable, that the ability of helminthic parasites to attenuate host immune responses into an anti-inflammatory/regulatory phenotype is attributed to the endogenous component that the parasites secrete and/or excrete interacting with immune effector cells to regulate their function (Lund et al., 2014; Selmi, 2016).
An additional mechanism was suggested for the helminth's immunomodulation of autoimmunity, in addition to the Th1 to Th2 shift. Accelerated T and B regulatory phenotypes, decreased levels of the inflammatory cytokines like IFNγ and Il-17 or vice versa, promoting IL-4, IL-10, and TGF-β release (Bashi et al., 2015a). Since CD is a Th1 profile disease, shifting the immune pathway to Th2 profile might reduce the intestinal damage (Lerner, 2010).
Immune Activation Induced Cell Death
Inflammation can cause a substantial hyperactivation of auto-aggressive lymphocytes, leading to activation-induced cell death and attenuate the systemic load of aggressive T cells. It seems that repeated encounter with powerful antigenic stimuli leading to restriction of an immune response is well-established in viral infections, where the primary response undergoes a major restriction after antigen elimination. EBV, HBV, and CMV infections are some of the examples. Similarly, administration of mycobacterial products, such as bacilli Calmette-Guérin, prevented the onset and recurrence of type 1 diabetes mellitus in NOD mice by inducing apoptosis of autoreactive T cells (Christen and von Herrath, 2005). In view of the fact that viruses are inducers of immune cells apoptosis while sparing the Treg cells (Che et al., 2015) and apoptosis is enhanced in CD (Shalimar et al., 2013), it is suggested that viruses, by abrogating immune activation, might attenuate intestinal autoimmune progression in CD.
Infection at Another Location might Keep Auto-Aggressive Cells from Reaching the Site of Autoimmune Destruction
As suggested by Christen (Christen and von Herrath, 2005), an infectious inflammation elsewhere in the body might keep auto-aggressive cells from arriving into the sites of autoimmune destruction, that might be due to the abrogation of type 1 diabetes in NOD mice after LCMV infection. The authors suggested that this occurred because the “abrogative” virus grew predominantly in peripheral lymphoid organs and other sites rather than the pancreas or its islets themselves. Thus, the sites of severe inflammation might act as a filter for auto-aggressive T cells removing them from the circuit and depriving them from homing the pancreatic islets. Similar scenarios might operate where infection with B. coxsackievirus or Salmomella typhi murium protected against autoimmunity (Tracy et al., 2002; Raine et al., 2006).
Immunosuppression by Extracellular Vesicles
Release of extracellular vesicles is a natural phenomenon of almost all cell types. They derive either from multivesicular bodies or from the cellular plasma membrane. Those vesicles contain a subset of cell derived proteins, lipids, including nucleic acids. Extracellular vesicles regulate immune responses against pathogens, as well as autoimmunity. It is suggested that these suppressive vesicles would prevent peripheral self-antigens and commonly encountered foreign antigens from causing chronic inflammation and autoimmunity. Following this lines, it is hypothesized that various infectious agents can induce those regulatory extracellular vesicles, counteracting autoimmune pathways, playing a protective anti-autoimmune role (Robbins and Morelli, 2014; Robbins et al., 2016).
Infectious Agents' Secretion of Anti-Autoreactive T Cells Proteins
Infections with helminths can prevent or attenuate auto-inflammatory/immune diseases. In addition to their Th1 to Th2 shift, most recently, Helminth secreted proteins were shown to prevent autoimmunity. The excretory/secretory products of Fasciola hepatica contain immune-modulatory molecules that arbitrate protection from autoimmune diabetes via the activation and provision of a regulatory immune environment (Lund et al., 2014). Such a mechanism was not studied in CD, but might explain the new potential therapeutic strategy to treat CD with Necator Americanus larvae (Croese et al., 2015; Giacomin et al., 2015).
Conclusions
The cross-talks between infections and autoimmunity are complex (Figure 1). Most of the data indicate that microbes and viruses are major environmental factors in autoimmunity induction. However, growing evidences conversely suggest that infectious agent can abrogate or protect against autoimmunity. This protective evolutionary cross-talks between microbes/viruses and us might represent a mutual beneficial equilibrium relationship between two cohabiting ecosystems. The protective pathways might involve PTMP, decreased intestinal permeability, Th1 to Th2 immune shift, induction of inflammatory immune cell apoptosis, auto-aggressive cells relocation from the target organ, immunosuppressive extracellular vesicles and anti-autoreactive cell immune-regulatory proteins.
Yet, our analysis demonstrates that the interaction of the microorganisms /viruses and CD is always a set of multi-directional processes. With a detailed consideration of possible mechanisms of CD and CMV, EBV, Herpes simplex type 1, Rubella, H. pylori, it can be assumed that the role of these infections suggested to be potential CD protectors infections, is not so unambiguous positive and the outcome of this interactions might be due to a balance between these multi-directional processes. In summary, there are more publications on the inducer role of infections in CD, and the few ones advocating the protective role should be further explored. The present review expend on several avenues that can be studied to understand the protective cross-talks between infectious agents and CD. Apprehending them can potentially suggest new therapeutic strategies for CD.
Author Contributions
AL: designed, wrote, edited, submitted, MA: designed, wrote, AS: literature search, revised, reviewed, TM: literature search, edited, and revised.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Mr. Alf Neu for the artistic design of the figure.
References
Aminov, R. (2011). Horizontal gene exchange in environmental microbiota. Front. Microbiol. 2:158. doi: 10.3389/fmicb.2011.00158
Anania, C., De Luca, E., De Castro, G., Chiesa, C., and Pacifico, L. (2015). Liver involvement in pediatric celiac disease. World J. Gastroenterol. 21, 5813–5822. doi: 10.3748/wjg.v21.i19.5813
Arboleya, S., Sánchez, B., Solís, G., Fernández, N., Suárez, M., Hernández-Barranco, A. M., et al. (2016). Impact of prematurity and perinatal antibiotics on the developing intestinal microbiota: a functional inference study. Int. J. Mol. Sci. 17:E649. doi: 10.3390/ijms17050649
Arleevskaya, M. I., Gabdoulkhakova, A. G., Filina, Y. V., Miftakhova, R. R., Bredberg, A., and Tsybulkin, A. P. (2014). A transient peak of infections during onset of rheumatoid arthritis: a 10-year prospective cohort study. BMJ Open 4:e005254. doi: 10.1136/bmjopen-2014-005254
Arleevskaya, M. I., Kravtsova, O. A., Lemerle, J., Renaudineau, Y., and Tsibulkin, A. P. (2016). How rheumatoid arthritis can result from provocation of the immune system by microorganisms and viruses. Front. Microbiol. 7:1296. doi: 10.3389/fmicb.2016.01296
Arleevskaya, M. I., Manukyan, G., Inoue, R., and Aminov, R. (2017). Editorial: Microbial and environmental factors in autoimmune and inflammatory diseases. Front. Immunol. 8:243. doi: 10.3389/fimmu.2017.00243
Awad, W. A., Aschenbach, J. R., Khayal, B., Hess, C., and Hess, M. (2012). Intestinal epithelial responses to Salmonella enterica serovar Enteritidis: effects on intestinal permeability and ion transport. Poult Sci. 91, 2949–2957. doi: 10.3382/ps.2012-02448
Awad, W. A., Hess, C., Khayal, B., Aschenbach, J. R., and Hess, M. (2014). In vitro exposure to Escherichia coli decreases ion conductance in the jejunal epithelium of broiler chickens. PLoS ONE 9:e92156. doi: 10.1371/journal.pone.0092156
Awad, W. A., Molnár, A., Aschenbach, J. R., Ghareeb, K., Khayal, B., Hess, C., et al. (2015). Campylobacter infection in chickens modulates the intestinal epithelial barrier function. Innate Immun. 21, 151–160. doi: 10.1177/1753425914521648
Aydogdu, S., Cakir, M., Yuksekkaya, H. A., Tumgor, G., Baran, M., and Arikan, C. (2008). Helicobacter pylori infection in children with celiac disease. Scand. J. Gastroenterol. 43, 1088–1093. doi: 10.1080/00365520802101846
Azuma, T., Ito, Y., Miyaji, H., Dojyo, M., Tanaka, Y., Hirai, M., et al. (1995). Immunogenetic analysis of the human leukocyte antigen DQA1 locus in patients with duodenal ulcer or chronic atrophic gastritis harbouring Helicobacter pylori. Eur. J. Gastroenterol. Hepatol. 7(Suppl. 1), S71–S73.
Bardella, M. T., Velio, P., Cesana, B. M., Prampolini, L., Casella, G., Di Bella, C., et al. (2007). Coeliac disease: a histological follow-up study. Histopathology 50, 465–471. doi: 10.1111/j.1365-2559.2007.02621.x
Barone, M. V., Gimigliano, A., Castoria, G., Paolella, G., Maurano, F., Paparo, F., et al. (2007). Growth factor-like activity of gliadin, an alimentary protein: implications for coeliac disease. Gut 56, 480–488. doi: 10.1136/gut.2005.086637
Bartels, L. E., Jepsen, P., Christensen, L. A., Gerdes, L. U., Vilstrup, H., and Dahlerup, J. F. (2016). Diagnosis of Helicobacter Pylori infection is associated with lower prevalence and subsequent incidence of Crohn's Disease. J. Crohns Colitis 10, 443–448. doi: 10.1093/ecco-jcc/jjv229
Bashi, T., Bizzaro, G., Ben-Ami Shor, D., Blank, M., and Shoenfeld, Y. (2015a). The mechanisms behind helminth's immunomodulation in autoimmunity. Autoimmun. Rev. 14, 98–104. doi: 10.1016/j.autrev.2014.10.004
Bashi, T., Blank, M., Ben-Ami Shor, B. A., Fridkin, M., Gendelman, O., Versini, M., et al. (2015b). Successful modulation of murine lupus nephritis with tuftsin-phosphorylcholine. J. Autoimmun. 59, 1–7. doi: 10.1016/j.jaut.2015.03.001
Bashi, T., Shovman, O., Fridkin, M., Volkov, A., Barshack, I., Blank, M., et al. (2016). Novel therapeutic compound tuftsin-phosphorylcholine attenuates collagen-induced arthritis. Clin. Exp. Immunol. 184, 19–28. doi: 10.1111/cei.12745
Bergman, M. P., Vandenbroucke-Grauls, C. M., Appelmelk, B. J., D'Elios, M. M., Amedei, A., Azzurri, A., et al. (2005). The story so far: Helicobacter pylori and gastric autoimmunity. Int. Rev. Immunol. 24, 63–91. doi: 10.1080/08830180590884648
Bergman, M., Del Prete, G., van Kooyk, Y., and Appelmelk, B. (2006). Helicobacter pylori phase variation, immune modulation and gastric autoimmunity. Nat. Rev. Microbiol. 4, 151–159. doi: 10.1038/nrmicro1344
Beyerlein, A., Donnachie, E., and Ziegler, A. -G. (2017). Infections in early life and development of celiac disease. Am. J. Epidemiol. doi: 10.1093/aje/kwx190. [Epub ahead of print].
Bilbao, J. R., Martín-Pagola, A., Vitoria, J. C., Zubillaga, P., Ortiz, L., and Castaño, L. (2002). HLA-DRB1 and MHC class 1 chain-related A haplotypes in Basque families with celiac disease. Tissue Antigens 60, 71–76. doi: 10.1034/j.1399-0039.2002.600109.x
Blekhman, R., Goodrich, J. K., Huang, K., Sun, Q., Bukowski, R., Bell, J. T., et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. doi: 10.1186/s13059-015-0759-1
Bleotu, C., Chifiriuc, M. C., Lazăr, V., Drăguşel, R., Matei, L., Aldea, I. M., et al. (2015). Influence of Staphylococcus aureus attachment to the herpes simplex virus infected cells. Rom. J. Morphol. Embryol. 56, 433–437.
Bloomfield, S. F., Rook, G. A., Scott, E. A., Shanahan, F., Stanwell-Smith, R., and Turner, P. (2016). Time to abandon the hygiene hypothesis: new perspectives on allergic disease, the human microbiome, infectious disease prevention and the role of targeted hygiene. Perspect. Public Health. 136, 213–224. doi: 10.1177/1757913916650225
Bogdanos, D. P., Smyk, D. S., Rigopoulou, E. I., Sakkas, L. I., and Shoenfeld, Y. (2015). Infectomics and autoinfectomics: a tool to study infectious-induced autoimmunity. Lupus 24, 364–373. doi: 10.1177/0961203314559088
Branski, D., Ashkenazy, A., Frier, S., and Lerner, A. (1992). “Extra intestinal manifestation and associated disorders of celiac disease,” in Gluten-Sensitive Enteropathy, Front Gastrointest Res, eds D. Branski, P. Rozen, and M. F. Kaganoff (Basel: Karger), 164–175.
Broide, E., Sandbank, J., Habler, L., Scapa, E., Kimchi, N. A., Shapiro, M., et al. (2007). The immunohistochemistry profile of lymphocytic gastritis in celiac disease and Helicobacter pylori infection- Interplay between infection and inflammation. Mediators Inflamm. 2007:81838. doi: 10.1155/2007/81838
Brottveit, M., Beitnes, A. C., Tollefsen, S., Bratlie, J. E., Jahnsen, F. L., Johansen, F. E., et al. (2013). Mucosal cytokine response after short-term gluten challenge in celiac disease and non-celiac gluten sensitivity. Am. J. Gastroenterol. 108, 842–850. doi: 10.1038/ajg.2013.91
Brown, I., Mino-Kenudson, M., Deshpande, V., and Lauwers, G. Y. (2006). Intraepithelial lymphocytosis in architecturally preserved proximal small intestinal mucosa: an increasing diagnostic problem with a wide differential diagnosis. Arch. Pathol. Lab. Med. 130, 1020–1025. doi: 10.1043/1543-2165(2006)130[1020:]ILIAPP2.0.CO;2
Caminero, A., Galipeau, H. J., McCarville, J. L., Johnston, C. W., Bernier, S. P., Russell, A. K., et al. (2016). Duodenal bacteria from patients with celiac disease and healthy subjects distinctly affect gluten breakdown and immunogenicity. Gastroenterology 151, 670–683. doi: 10.1053/j.gastro.2016.06.041
Caminero, A., Herrán, A. R., Nistal, E., Pérez-Andrés, J., Vaquero, L., Vivas, S., et al. (2014). Diversity of the cultivable human gut microbiome involved in gluten metabolism: isolation of microorganisms with potential interest for coeliac disease. FEMS Microbiol. Ecol. 88, 309–319. doi: 10.1111/1574-6941.12295
Caminero, A., Nistal, E., Herrán, A. R., Pérez-Andrés, J., Ferrero, M. A., Vaquero Ayala, L., et al. (2015). Differences in gluten metabolism among healthy volunteers, coeliac disease patients and first-degree relatives. Br. J. Nutr. 114, 1157–1167. doi: 10.1017/S0007114515002767
Canova, C., Zabeo, V., Pitter, G., Romor, P., Baldovin, T., Zanotti, R., et al. (2014). Association of maternal education, early infections, and antibiotic use with celiac disease: a population-based birth cohort study in northeastern Italy. Am. J. Epidemiol. 180, 76–85. doi: 10.1093/aje/kwu101
Cantekin, K., Arslan, D., and Delikan, E. (2015). Presence and distribution of dental enamel defects, recurrent aphthous lesions and dental caries in children with celiac disease. Pak. J. Med. Sci. 31, 606–609. doi: 10.12669/pjms.313.6960
Cârdei, E., Moraru, D., Trandafir, L., Bozomitu, L., and Mihăilă, D. (2003). Celiac disease associated with Helicobacter pylori infection. Rev. Med. Chir. Soc. Med. Nat. Iasi. 107, 633–635.
Carding, S., Verbeke, K., Vipond, D. T., Corfe, B. M., and Owen, L. J. (2015). Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 26:26191. doi: 10.3402/mehd.v26.26191
Caron, T. J., Scott, K. E., Fox, J. G., and Hagen, S. J. (2015). Tight junction disruption: Helicobacter pylori and dysregulation of the gastric mucosal barrier. World, J. Gastroenterol. 21, 11411–11427. doi: 10.3748/wjg.v21.i40.11411
Casella, G., Orfanotti, G., Giacomantonio, L., Bella, C. D., Crisafulli, V., Villanacci, V., et al. (2016). Celiac disease and obstetrical-gynecological contribution. Gastroenterol. Hepatol. Bed Bench. 9, 241–249.
Chang, F., Mahadeva, U., and Deere, H. (2005). Pathological and clinical significance of increased intraepithelial lymphocytes (IELs) in small bowel mucosa. APMIS 113, 385–399. doi: 10.1111/j.1600-0463.2005.apm_204.x
Che, J. W., Kraft, A. R., Selin, L. K., and Welsh, R. M. (2015). Regulatory T cells resist virus infection-induced apoptosis. J. Virol. 89, 2112–2120. doi: 10.1128/JVI.02245-14
Chen, Y., and Blaser, M. J. (2008). Helicobacter pylori colonization is inversely associated with childhood asthma. J. Infect. Dis. 198, 553–560. doi: 10.1086/590158
Cherdantseva, L. A., Potapova, O. V., Sharkova, T. V., Belyaeva, Y. Y., and Shkurupiy, V. A. (2014). Association of Helicobacter pylori and iNOS production by macrophages and lymphocytes in the gastric mucosa in chronic gastritis. J. Immunol. Res. 2014:762514. doi: 10.1155/2014/762514
Chmielewska, A., Pieścik-Lech, M., Szajewska, H., and Shamir, R. (2015). Primary prevention of celiac disease: environmental factors with a focus on early nutrition. Ann. Nutr. Metab. 67, 43–50. doi: 10.1159/000440992
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. doi: 10.1038/nrg3182
Christen, U., and von Herrath, M. G. (2005). Infections and autoimmunity—good or bad? J. Immunol. 174, 7481–7486. doi: 10.4049/jimmunol.174.12.7481
Ciacci, C., Squillante, A., Rendina, D., Limauro, S., Bencivenga, C., Labanca, F., et al. (2000). Helicobacter pylori infection and peptic disease in coeliac disease. Eur. J. Gastroenterol. Hepatol. 12, 1283–1287. doi: 10.1097/00042737-200012120-00004
Consolandi, C., Turroni, S., Emmi, G., Severgnini, M., Fiori, J., Peano, C., et al. (2015). Behçet's syndrome patients exhibit specific microbiome signature. Autoimmun. Rev. 14, 269–276. doi: 10.1016/j.autrev.2014.11.009
Conti, C., Malacrino, C., and Mastromarino, P. (2009). Inhibition of herpes simplex virus type 2 by vaginal lactobacilli. J. Physiol. Pharmacol. 60(Suppl. 6), 19–26.
Crabtree, J. E. (1996). Immune and inflammatory responses to Helicobacter pylori infection. Scand. J. Gastroenterol. Suppl. 215, 3–10. doi: 10.3109/00365529609094526
Croese, J., Giacomin, P., Navarro, S., Clouston, A., McCann, L., Dougall, A., et al. (2015). Experimental hookworm infection and gluten microchallenge promote tolerance in celiac disease. J. Allergy Clin. Immunol. 135, 508–516. doi: 10.1016/j.jaci.2014.07.022
Decker, E., Hornef, M., and Stockinger, S. (2011). Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Gut Microbes. 2:91–98. doi: 10.4161/gmic.2.2.15414
Delgado, S., Leite, A. M., Ruas-Madiedo, P., and Mayo, B. (2015). Probiotic and technological properties of Lactobacillus spp. strains from the human stomach in the search for potential candidates against gastric microbial dysbiosis. Front. Microbiol. 5:766. doi: 10.3389/fmicb.2014.00766
Di Sabatino, A., Giuffrida, P., Fornasa, G., Salvatore, C., Vanoli, A., Naviglio, S., et al. (2016). Innate and adaptive immunity in self-reported nonceliac gluten sensitivity versus celiac disease. Dig. Liver Dis. 48, 745–752. doi: 10.1016/j.dld.2016.03.024
Di Sabatino, A., Pickard, K. M., Gordon, J. N., Salvati, V., Mazzarella, G., Beattie, R. M., et al. (2007). Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology 133, 1175–1187. doi: 10.1053/j.gastro.2007.08.018
Diamanti, A., Maino, C., Niveloni, S., Pedreira, S., Vazquez, H., Smecuol, E., et al. (1999). Characterization of gastric mucosal lesions in patients with celiac disease: a prospective controlled study. Am. J. Gastroenterol. 94, 1313–1319. doi: 10.1111/j.1572-0241.1999.01082.x
Dong, G. P., Shang, S. Q., Du, L. Z., Yu, X. L., Xu, Y. P., and Wu, X. J. (2004). Detection of four human herpesviruses DNA and virus-specific IgM antibody in blood specimens of infants. Zhonghua Er Ke Za Zhi 42, 367–370.
Dong, G., Shang, S., Liang, L., and Yu, X. (2005). Determination of the six major human herpesviruses in cerebrospinal fluid and blood specimens of children. Acta Paediatr. 94, 38–43. doi: 10.1080/08035250410022477
Duar, R. M., Clark, K. J., Patil, P. B., Hernández, C., Brüning, S., Burkey, T. E., et al. (2015). Identification and characterization of intestinal lactobacilli strains capable of degrading immunotoxic peptides present in gluten. Appl. Microbiol. 118, 515–527. doi: 10.1111/jam.12687
Eiró, N., González-Reyes, S., González, L., González, L. O., Altadill, A., Andicoechea, A., et al. (2012). Duodenal expression of Toll-like receptors and interleukins are increased in both children and adultceliac patients. Dig. Dis. Sci. 57, 2278–2285. doi: 10.1007/s10620-012-2184-6
Eliyah Livshits, O., Shauol, R., Reifen, R., Matthias, T., and Lerner, A. (2017). Can celiac disease present along with childhood obesity? Int. J. Celiac Dis. 5, 19–23. doi: 10.12691/ijcd-5-1-7
Eusebi, L. H., Zagari, R. M., and Bazzoli, F. (2014). Epidemiology of Helicobacter pylori infection. Helicobacter 19(Suppl. 1), 1–5. doi: 10.1111/hel.12165
Fernandez-Feo, M., Wei, G., Blumenkranz, G., Dewhirst, F. E., Schuppan, D., Oppenheim, F. G., et al. (2013). The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clin. Microbiol. Infect. 19, E386–E394. doi: 10.1111/1469-0691.12249
Fernandez-Jimenez, N., Castellanos-Rubio, A., Plaza-Izurieta, L., Gutierrez, G., Castaño, L., Vitoria, J. C., et al. (2010). Analysis of beta-defensin and Toll-like receptor gene copy number variation in celiac disease. Hum. Immunol. 71, 833–836. doi: 10.1016/j.humimm.2010.05.012
Forchielli, M. L., and Walker, W. A. (2005). The effect of protective nutrients on mucosal defense in the immature intestine. Acta Paediatr. Suppl. 94, 74–83. doi: 10.1080/08035320510043592
Forsberg, G., Fahlgren, A., Hörstedt, P., Hammarström, S., Hernell, O., and Hammarström, M. L. (2004). Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am. J. Gastroenterol. 99, 894–904. doi: 10.1111/j.1572-0241.2004.04157.x
Gaisford, W., and Cooke, A. (2009). Can infections protect against autoimmunity? Curr. Opin. Rheumatol. 21, 391–396. doi: 10.1097/BOR.0b013e32832c2dee
Giacomin, P., Zakrzewski, M., Croese, J., Su, X., Sotillo, J., McCann, L., et al. (2015). Experimental hookworm infection and escalating gluten challenges are associated with increased microbial richness in celiac subjects. Sci. Rep. 5:13797. doi: 10.1038/srep13797
Gobert, A. P., and Wilson, K. T. (2016). The immune battle against Helicobacter pylori infection: NO offense. Trends Microbiol. 24, 366–376. doi: 10.1016/j.tim.2016.02.005
Grinde, B. (2013). Latency and reactivation - viral strategies and host response. J. Oral. Microbiol. 5, 1–9. doi: 10.3402/jom.v5i0.22766
Guz-Mark, A., Zevit, N., Morgenstern, S., and Shamir, R. (2014). Duodenal intraepithelial lymphocytosis is common in children without coeliac disease, and is not meaningfully influenced by Helicobacter pylori infection. Aliment. Pharmacol. Ther. 39, 1314–1320. doi: 10.1111/apt.12739
Hällgren, R., Colombel, J. F., Dahl, R., Fredens, K., Kruse, A., Jacobsen, N. O., et al. (1989). Neutrophil and eosinophil involvement of the small bowel in patients with celiac disease and Crohn's disease: studies on the secretion rate and immunohistochemical localization of granulocyte granule constituents. Am. J. Med. 86, 56–64. doi: 10.1016/0002-9343(89)90230-1
Harris, P. R., Serrano, C. A., Villagrán, A., Walker, M. M., Thomson, M., Duarte, I., et al. (2013). Helicobacter pylori-associated hypochlorhydria in children, and development of iron deficiency. J. Clin. Pathol. 66, 343–347. doi: 10.1136/jclinpath-2012-201243
Hazrati, E., Galen, B., Lu, W., Wang, W., Ouyang, Y., Keller, M. J., et al. (2006). Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 177, 8658–8666. doi: 10.4049/jimmunol.177.12.8658
Holmgren Peterson, K., Fälth-Magnusson, K., Magnusson, K. E., Stenhammar, L., and Sundqvist, T. (1998). Children with celiac disease express inducible nitric oxide synthase in the small intestine during gluten challenge. Scand. J. Gastroenterol. 33, 939–943. doi: 10.1080/003655298750026958
Il'chenko, A. A., Korshunova, V. M., Aruin, L. I., Zhukhovitskiĭ, V. G., Dugasheva, L. G., and Radakova, E. D. (1991). The intestinal microflora and acid-forming function of the stomach in peptic ulcer patients with Helicobacter pylori bacteriosis. Z. Mikrobiol. Epidemiol. Immunobiol. 10, 17–19.
Jansen, M. A., van den Heuvel, D., van der Zwet, K. V., Jaddoe, V. W., Hofman, A., Escher, J. C., et al. (2016). Herpesvirus infections and transglutaminase type 2 antibody positivity in childhood: the generation R study. J. Pediatr. Gastroenterol. Nutr. 63, 423–430. doi: 10.1097/MPG.0000000000001163
Jernberg, C., Lofmark, S., Edlund, C., and Jansson, J. K. (2007). Long-term ecological impacts of antibiotic administration on the human Intestinal microbiota. ISME J. 1, 56–66. doi: 10.1038/ismej.2007.3
Jozefczuk, J., Bancerz, B., Walkowiak, M., Glapa, A., Nowak, J., Piescikowska, J., et al. (2015). Prevalence of Helicobacter pylori infection in pediatric celiac disease. Eur. Rev. Med. Pharmacol. Sci. 19, 2031–2035.
Juuti-Uusitalo, K., Lindfors, K., Mäki, M., Patrikainen, M., Isola, J., and Kaukinen, K. (2009). Inhibition of epithelial growth factor receptor signalling does not preserve epithelial barrier function after in vitro gliadin insult. Scand. J. Gastroenterol. 44, 820–825. doi: 10.1080/00365520902898119
Kemppainen, K. M., Lynch, K. F., Liu, E., Lönnrot, M., Simell, V., Briese, T., et al. (2017). Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin. Gastroenterol. Hepatol. 15, 694.e5–702.e5. doi: 10.1016/j.cgh.2016.10.033
Khaleghi, S., Ju, J. M., Lamba, A., and Murray, J. A. (2016). The potential utility of tight junction regulation in celiac disease: focus on larazotide acetate. Ther. Adv. Gastroenterol. 9, 37–49. doi: 10.1177/1756283X15616576
Khan, S., Mandal, R. K., Jawed, A., Wahid, M., Panda, A. K., Areeshi, M. Y., et al. (2016). TNF-α−308 G > A (rs1800629) Polymorphism is associated with celiac disease: a meta-analysis of 11 case-control studies. Sci. Rep. 6:32677. doi: 10.1038/srep32677
Khani, S., Motamedifar, M., Golmoghaddam, H., Hosseini, H. M., and Hashemizadeh, Z. (2012). In vitro study of the effect of a probiotic bacterium Lactobacillus rhamnosus against herpes simplex virus type 1. Braz. J. Infect. Dis. 16, 129–135. doi: 10.1016/S1413-8670(12)70293-3
Khosravi, Y., Dieye, Y., Loke, M. F., Goh, K. L., and Vadivelu, J. (2014). Streptococcus mitis induces conversion of Helicobacter pylori to coccoid cells during co-culture in vitro. PLoS ONE 9:e112214. doi: 10.1371/journal.pone.0112214
Kienesberger, S., Cox, L. M., Livanos, A., Zhang, X. S., Chung, J., Perez-Perez, G. I., et al. (2016). Gastric Helicobacter pylori infection affects local and distant microbial populations and host responses. Cell. Rep. 14, 1395–1407. doi: 10.1016/j.celrep.2016.01.017
Kivity, S., Agmon-Levin, N., Blank, M., and Shoenfeld, Y. (2009). Infections and autoimmunity–friends or foes? Trends Immunol. 30, 409–414. doi: 10.1016/j.it.2009.05.005
Koelle, D. M., Johnson, M. L., Ekstrom, A. N., Byers, P., and Kwok, W. W. (1997). Preferential presentation of herpes simplex virus T-cell antigen by HLA DQA1*0501/DQB1*0201 in comparison to HLA DQA1*0201/DQB1*0201. Hum. Immunol. 53, 195–205. doi: 10.1016/S0198-8859(97)00034-7
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108 (Suppl. 1), 4578–4585. doi: 10.1073/pnas.1000081107
Kotalová, R., Vraná, M., Dobrovolná, M., Nevoral, J., and Loudová, M. (2002). [HLA-DRB1/DQA1/DQB1 alleles and haplotypes in Czech children with celiac sprue]. Cas. Lek. Cesk. 141, 518–522.
Krums, L. M., Sabel'nikova, E. A., and Parfenov, A. I. (2011). Functional condition of the stomach, pancreas, liver and gallbladder in celiac disease. Ter. Arkh. 83, 20–24.
Lammers, K. M., Chieppa, M., Liu, L., Liu, S., Omatsu, T., Janka-Junttila, M., et al. (2015). Gliadin induces neutrophil migration via engagement of the formyl peptide receptor, FPR1. PLoS ONE 10:e0138338. doi: 10.1371/journal.pone.0138338
Lappinga, P. J., Abraham, S. C., Murray, J. A., Vetter, E. A., Patel, R., and Wu, T. T. (2010). Small intestinal bacterial overgrowth: histopathologic features and clinical correlates in an underrecognized entity. Arch Pathol. Lab Med. 134, 264–270.
Lasa, J., Zubiaurre, I., Dima, G., Peralta, D., and Soifer, L. (2015). HELICOBACTER PYLORI PREVALENCE IN PATIENTS WITH CELIAC DISEASE: results from a cross-sectional study. Arq. Gastroenterol. 52, 139–142. doi: 10.1590/s0004-28032015000200012
Lerner, A. (1994). Factors affecting the clinical presentation and time diagnosis of celiac disease: The Jerusalem and the West Bank-Gaza experience (editorial). Isr. J. Med. Sci. 30, 294–295.
Lerner, A. (2010). New therapeutic strategies for celiac disease. Autoimmun. Rev. 9, 144–147. doi: 10.1016/j.autrev.2009.05.002
Lerner, A. (2011). The last two millennias eco-catastrophes are the driving forces for thepotential genetic advantage mechanisms in celiac disease. Med. Hypotheses 77, 773–776. doi: 10.1016/j.mehy.2011.07.034
Lerner, A. (2014). Serological diagnosis of celiac disease –moving beyond the tip of the iceberg. Int. J. Celiac Dis. 2, 64–66. doi: 10.12691/ijcd-2-2-8
Lerner, A. (2015). “Non nutritional environmental factors associated with celiac disease: infections and vaccinations,” in Vaccines and Autoimmunity, eds Y. Shoenfeld, N. Agmon-Levin, and L. Tomljenovic (Hoboken, NJ: Wiley Blackwell), 301–306.
Lerner, A., Aminov, R., and Matthias, T. (2016). Dysbiosis may trigger autoimmune diseases via inappropriate posttranslational modification of host proteins. Front. Microbiol. 7:84. doi: 10.3389/fmicb.2016.00084
Lerner, A., and Blank, M. (2014). Hypercoagulability in celiac disease-an update. Autoimmun. Rev. 13, 1138–1141. doi: 10.1016/j.autrev.2014.07.004
Lerner, A., and Matthias, T. (2015a). Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 14, 479–489. doi: 10.1016/j.autrev.2015.01.009
Lerner, A., and Matthias, T. (2015b). Rheumatoid arthritis-celiac disease relationship: joints get that gut feeling. Autoimmun. Rev. 14, 1038–1047. doi: 10.1016/j.autrev.2015.07.007
Lerner, A., and Matthias, T. (2015c). Increased knowledge and awareness of celiac disease will benefit the elderly. Int. J. Celiac Dis. 3, 112–114. doi: 10.12691/ijcd-3-3-6
Lerner, A., and Matthias, T. (2015d). Editorial: Celiac disease: intestinal, heart and skin inter-connections. Int. J. Celiac Dis. 3, 28–30. doi: 10.12691/ijcd-3-1-6
Lerner, A., and Matthias, T. (2016a). Gut- bone cross talks and implications in celiac disease. Int. J. Celiac Dis. 4, 19–23. doi: 10.12691/ijcd-4-1-4
Lerner, A., and Matthias, T. (2016b). The gut-stomach axis: Helicobacter pylori and celiac disease. Int. J. Celiac Dis. 4, 77–79.
Lerner, A., and Matthias, T. (2016c). The jigsaw of breast feeding and celiac disease. Int. J Celiac Dis. 4, 87–89. doi: 10.12691/ijcd-3-3-4
Lerner, A., and Matthias, T. (2016d). Autoimmune thyroid diseases in celiac disease: if and when to screen? Int. J Celiac Dis. 4, 124–126. doi: 10.12691/ijcd-4-4-10
Lerner, A., and Matthias, T. (2017a). Extraintestinal manifestations of CD: common pathways in the gut-remote organs' axes. Int. J. Celiac Dis. 5, 24–27. doi: 10.12691/ijcd-5-1-5
Lerner, A., and Matthias, T. (2017b). Gluten free diet- tough alley in torrid time. Int. J. Celiac Dis. 5, 50–55. doi: 10.12691/ijcd-5-2-4
Lerner, A., and Reif, S. (2015). “Chapter 50: Nonnutritional environmental factors associated with Celiac disease: the infectome,” in Infections and Autoimmunity, 2nd Edn., eds Y. Shoenfeld, N. Agmon-Levine, and N. R. Rose (Amsterdam: Elsevier), 829–837.
Lerner, A., Blank, M., and Shoenfeld, Y. (1996). Celiac disease and autoimmunity. Isr. J. Med. Sci. 32, 33–36.
Lerner, A., Jeremias, P., and Matthias, T. (2015a). The world incidence and prevalence of autoimmune diseases is increasing: a review. Int. J. Celiac Dis. 3, 151–155. doi: 10.12691/ijcd-3-4-8
Lerner, A., Jeremias, P., and Matthias, T. (2017). The gut-thyroid axis: beyond the thyroid: what's happening in the gut. Endocrinol. Cennections 6, R52–R58. doi: 10.1530/EC-17-0021
Lerner, A., Jermias, P., and Matthias, T. (2015b). The world incidence of celiac disease is increasing: a review. Int. J. Recent Sci. Res. 7, 5491–5496.
Lerner, A., Makhoul, B., and Eliakim, R. (2012). Neurological manifestations of celiac disease in children and adults. Eur. Neurolog. J. 4, 15–20. doi: 10.1016/j.autrev.2014.11.009
Lerner, A., Neidhöfer, S., and Matthias, T. (2015c). Transglutaminase 2 and anti transglutaminase 2 autoantibodies in celiac disease and beyond: Part A: TG2 double-edged sword: gut and extraintestinal involvement. Immunome Res. 11, 101–105. doi: 10.4172/1745-7580.10000101
Lerner, A., Neidhöfer, S., and Matthias, T. (2015d). Transglutaminase 2 and anti transglutaminase 2 autoantibodies in celiac disease and beyond. Part B: Anti- Transglutaminase 2 autoantibodies: friends or enemies. Immunome Res. 11, 3–7.
Lerner, A., Neidhöfer, S., and Matthias, T. (2015e). Serological markers and/or intestinal biopsies in the case-finding of celiac disease. Int. J. Celiac Dis. 3, 53–55.
Lionetti, E., Castellaneta, S., Francavilla, R., Pulvirenti, A., Tonutti, E., Amarri, S., et al. (2014). SIGENP (Italian Society of Pediatric Gastroenterology, Hepatology, and Nutrition) Working Group on Weaning and CD Risk. Introduction of gluten, HLA status, and the risk of celiac disease in children. N. Engl. J. Med. 371, 1295–1303. doi: 10.1056/NEJMoa1400697
López-Casado, M. A., Lorite, P., Palomeque, T., and Torres, M. I. (2015). Potential role of the IL-33/ST2 axis in celiac disease. Cell. Mol. Immunol. 14, 285–292. doi: 10.1038/cmi.2015.85
Lund, M. E., O'Brien, B. A., Hutchinson, A. T., Robinson, M. W., Simpson, A. M., Dalton, J. P., et al. (2014). Secreted proteins from the helminth Fasciola hepatica inhibit the initiation of autoreactive T cell responses and prevent diabetes in the NOD mouse. PLoS ONE 9:e86289. doi: 10.1371/journal.pone.0086289
Luzza, F., Mancuso, M., Imeneo, M., Mesuraca, L., Contaldo, A., Giancotti, L., et al. (1999). Helicobacter pylori infection in children with celiac disease: prevalence and clinicopathologic features. J. Pediatr. Gastroenterol. Nutr. 28, 143–146. doi: 10.1097/00005176-199902000-00009
Lynch, D. A., Sobala, G. M., Dixon, M. F., Gledhill, A., Jackson, P., Crabtree, J. E., et al. (1995). Lymphocytic gastritis and associated small bowel disease: a diffuse lymphocytic gastroenteropathy? J. Clin. Pathol. 48, 939–945. doi: 10.1136/jcp.48.10.939
Magalhães, A., and Reis, C. A. (2010). Helicobacter pylori adhesion to gastric epithelial cells is mediated by glycan receptors. Braz. J. Med. Biol. Res. 43, 611–618. doi: 10.1590/S0100-879X2010007500049
Maheshwari, A. (2004). Role of cytokines in human intestinal villous development. Clin. Perinatol. 31, 143–155. doi: 10.1016/j.clp.2004.03.003
Makarova, S. G., Boldyreva, M. N., Lavrova, T., and Ye Petrovskaya, M. I. (2014). Intestinal Microbiocenosis, Food Tolerance and Food Allergy. Current State of a Problem. Voprosy Sovremennoj Pediatrii 13, 21–29. doi: 10.15690/vsp.v13i3.1024
Manichanh, C., Borruel, N., Casellas, F., and Guarner, F. (2012). The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 9, 599–608. doi: 10.1038/nrgastro.2012.152
Mårild, K., Stephansson, O., Montgomery, S., Murray, J. A., and Ludvigsson, J. F. (2012). Pregnancy outcome and risk of celiac disease in offspring: a nationwide case-control study. Gastroenterology. 142, 39.e3–45.e3. doi: 10.1053/j.gastro.2011.09.047
Meresse, B., Korneychuk, N., Malamut, G., and Cerf-Bensussan, N. (2015). Interleukin-15, a master piece in the immunological jigsaw of celiac disease. Dig. Dis. 33, 122–130. doi: 10.1159/000369521
Mubarak, A., Spierings, E., Wolters, V., van Hoogstraten, I., Kneepkens, C. M., and Houwen, R. (2013). Human leukocyte antigen DQ2.2 and celiac disease. J. Pediatr. Gastroenterol. Nutr. 56, 428–430. doi: 10.1097/MPG.0b013e31827913f9
Mutapi, F., Imai, N., Nausch, N., Bourke, C. D., Rujeni, N., Mitchell, K. M., et al. (2011). Schistosome infection intensity is inversely related to auto-reactive antibody levels. PLoS ONE. 6:e19149. doi: 10.1371/journal.pone.0019149
Myléus, A., Stenlund, H., Hernell, O., Gothefors, L., Hammarström, M. L., Persson, L. Å., et al. (2012). Early vaccinations are not risk factors for celiac disease. Pediatrics 130, e63–e70. doi: 10.1542/peds.2011-2806
Naing, Z., Rayner, B., Killikulangara, A., Vunnam, K., Leach, S., McIver, C. J., et al. (2013). Prevalence of viruses in stool of premature neonates at a neonatal intensive care unit. J. Paediatr. Child Health 49, E221–E226. doi: 10.1111/jpc.12113
Narang, M., Puri, A. S., Sachdeva, S., Singh, J., Kumar, A., and Saran, R. K. (2016). Celiac disease and H. Pylori infection in children: is there any association? J. Gastroenterol. Hepatol. J Gastroenterol. Hepatol. 32, 1178–1182. doi: 10.1111/jgh.13654
Nenna, R., Magliocca, F. M., Tiberti, C., Mastrogiorgio, G., Petrarca, L., Mennini, M., et al. (2012). Endoscopic and histological gastric lesions in children with celiac disease: mucosal involvement is not only confined to the duodenum. J. Pediatr. Gastroenterol. Nutr. 55, 728–732. doi: 10.1097/MPG.0b013e318266aa9e
Nielsen, J. A., Roberts, C. A., Lager, D. J., Putcha, R. V., Jain, R., and Lewin, M. (2014). Lymphocytic gastritis is not associated with active Helicobacter pylori infection. Helicobacter 19, 349–355. doi: 10.1111/hel.12139
Nilsson, C., Larsson Sigfrinius, A. K., Montgomery, S. M., Sverremark-Ekström, E., Linde, A., Lilja, G., et al. (2009). Epstein-Barr virus and cytomegalovirus are differentially associated with numbers of cytokine-producing cells and early atopy. Clin. Exp. Allergy 39, 509–517. doi: 10.1111/j.1365-2222.2008.03147.x
Nilsson, C., Linde, A., Montgomery, S. M., Gustafsson, L., Näsman, P., Blomberg, M. T., et al. (2005). Does early EBV infection protect against IgE sensitization? J. Allergy Clin. Immunol. 116, 438–444. doi: 10.1016/j.jaci.2005.04.027
Nistal, E., Caminero, A., Vivas, S., Ruiz de Morales, J. M., Sáenz de Miera, L. E., Rodríguez-Aparicio, L. B., et al. (2012). Differences in faecal bacteria populations and faecal bacteria metabolism in healthy adults and celiac disease patients. Biochimie 94, 1724–1729. doi: 10.1016/j.biochi.2012.03.025
Niveloni, S., Weksler-Zangen, S., Pedreira, S., Dezi, R., Granot, E., Doldan, I., et al. (2000). Time course of nitric oxide synthase generation after gluten exposure in the rectal mucosa of gluten-sensitive patients. Scand. J. Gastroenterol. 35, 1150–1156. doi: 10.1080/003655200750056619
Noh, K. W., Poland, G. A., and Murray, J. A. (2003). Hepatitis B vaccine nonresponse and celiac disease. Am. J. Gastroenterol. 98, 2289–2292. doi: 10.1111/j.1572-0241.2003.07701.x
Nuding, S., Gersemann, M., Hosaka, Y., Konietzny, S., Schaefer, C., Beisner, J., et al. (2013). Gastric antimicrobial peptides fail to eradicate Helicobacter pylori infection due to selective induction and resistance. PLoS ONE 8:e73867. doi: 10.1371/journal.pone.0073867
Núñez, C., Rueda, B., Martínez, A., Maluenda, C., Polanco, I., López-Nevot, M. A., et al. (2006). A functional variant in the CD209 promoter is associated with DQ2-negative celiac disease in the Spanish population. World J. Gastroenterol. 12, 4397–4400. doi: 10.3748/wjg.v12.i27.4397
Oertli, M., and Müller, A. (2012). Helicobacter pylori targets dendritic cells to induce immune tolerance, promote persistence and confer protection against allergic asthma. Gut Microbes 3, 566–571. doi: 10.4161/gmic.21750
Oh, J. E., Kim, B. C., Chang, D. H., Kwon, M., Lee, S. Y., Kang, D., et al. (2016). Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc. Natl. Acad. Sci. U.S.A. 113, E762–E771. doi: 10.1073/pnas.1518589113
Ojetti, V., Nucera, G., Migneco, A., Gabrielli, M., Lauritano, C., Danese, S., et al. (2005). High prevalence of celiac disease in patients with lactose intolerance. Digestion 71, 106–110. doi: 10.1159/000084526
Olivares, M., Neef, A., Castillejo, G., Palma, G. D., Varea, V., Capilla, A., et al. (2015). The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 64, 406–417. doi: 10.1136/gutjnl-2014-306931
Ou, G., Hedberg, M., Hörstedt, P., Baranov, V., Forsberg, G., Drobni, M., et al. (2009). Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 104, 3058–3067. doi: 10.1038/ajg.2009.524
Pai, R. K. A. (2014). Practical approach to small bowel biopsy interpretation: celiac disease and its mimics. Semin. Diagn. Pathol. 31, 124–136. doi: 10.1053/j.semdp.2014.02.006
Pallav, K., Leffler, D. A., Tariq, S., Kabbani, T., Hansen, J., Peer, A., et al. (2012). Noncoeliac enteropathy: the differential diagnosis of villous atrophy in contemporary clinical practice. Aliment. Pharmacol. Ther. 35, 380–390. doi: 10.1111/j.1365-2036.2011.04938.x
Parassol, N., Freitas, M., Thoreux, K., Dalmasso, G., Bourdet-Sicard, R., and Rampal, P. (2005). Lactobacillus casei DN-114 001 inhibits the increase in paracellular permeability of enteropathogenic Escherichia coli-infected T84 cells. Res. Microbiol. 156, 256–262. doi: 10.1016/j.resmic.2004.09.013
Park, S. D., Markowitz, J., Pettei, M., Weinstein, T., Sison, C. P., Swiss, S. R., et al. (2007). Failure to respond to hepatitis B vaccine in children with celiac disease. J. Pediatr. Gastroenterol. Nutr. 44, 431–435. doi: 10.1097/MPG.0b013e3180320654
Peña, A. S., and Crusius, J. B. (1998). Food allergy, coeliac disease and chronic inflammatory bowel disease in man. Vet. Q. 20 (Suppl. 3), S49–S52.
Pero, R., Coretti, L., Nigro, E., Lembo, F., Laneri, S., Lombardo, B., et al. (2017). β-defensins in the fight against Helicobacter pylori. Molecules 22:424. doi: 10.3390/molecules22030424
Pisetsky, D. S., and Drayton, D. M. (1997). Deficient expression of antibodies specific for bacterial DNA by patients with systemic lupus erythematosus. Proc. Assoc. Am. Phys. 109, 237–244.
Plot, L., Amital, H., Barzilai, O., Ram, M., Bizzaro, N., and Shoenfeld, Y. (2009). Infections may have a protective role in the etiopathogenesis of celiac disease. Ann. N. Y. Acad. Sci. 1173, 670–674. doi: 10.1111/j.1749-6632.2009.04814.x
Plot, L., and Amital, H. (2009). Infectious associations of Celiac disease. Autoimmun Rev. 8, 316–319. doi: 10.1016/j.autrev.2008.10.001
Polydorides, A. D. (2014). Pathology and differential diagnosis of chronic, noninfectious gastritis. Semin. Diagn. Pathol. 31, 114–123. doi: 10.1053/j.semdp.2014.02.008
Pontes-de-Carvalho, L., Mengel, J., Figueiredo, C. A., and Social Changes, Asthma Allergy in Latin America (SCAALA)'s Study Group, Alcântara-Neves, N. M. (2013). Antigen mimicry between infectious agents and self or environmental antigens may lead to long-term regulation of inflammation. Front. Immunol. 4:314. doi: 10.3389/fimmu.2013.00314
Pordeus, V., Szyper-Kravitz, M., Levy, R. A., Vaz, N. M., and Shoenfeld, Y. (2008). Infections and autoimmunity: a panorama. Clin. Rev. Allergy Immunol. 34, 283–99. doi: 10.1007/s12016-007-8048-8
Raine, T., Zaccone, P., Mastroeni, P., and Cooke, A. (2006). Salmonella typhimurium infection in nonobese diabetic mice generates immunomodulatory dendritic cells able to prevent type 1 diabetes. J. Immunol. 177, 2224–2233. doi: 10.4049/jimmunol.177.4.2224
Ramanathan, J., Rammouni, M., Baran, J. Jr., and Khatib, R. (2000). Herpes simplex virus esophagitis in the immunocompetent host: an overview. Am. J. Gastroenterol. 95, 2171–2176. doi: 10.1111/j.1572-0241.2000.02299.x
Reichstetter, S., Kwok, W. W., and Nepom, G. T. (1999). Impaired binding of a DQ2 and DQ8-binding HSV VP16 peptide to a DQA1*0501/DQB1*0302 trans class II heterodimer. Tissue Antigens 53, 101–105. doi: 10.1034/j.1399-0039.1999.530111.x
Reif, S., and Lerner, A. (2004b). “Celiac disease and infection,” in Infections and Autoimmunity, eds Y. Shoenfeld and N. Rose (Amsterdam: Elsevier), 689–692.
Reif, S., and Lerner, A. (2004a). Tissue transglutaminase—the key player in celiac disease: a review. Autoimmun. Rev. 3, 40–45. doi: 10.1016/S1568-9972(03)00065-X
Riddle, M. S., Murray, J. A., and Porter, C. K. (2012). The incidence and risk of celiac disease in a healthy US adult population. Am. J. Gastroenterol. 107, 1248–1255. doi: 10.1038/ajg.2012.130
Robbins, P. D., Dorronsoro, A., and Booker, C. N. (2016). Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Invest. 126, 1173–1180. doi: 10.1172/JCI81131
Robbins, P. D., and Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208. doi: 10.1038/nri3622
Roblin, X., Pillet, S., Oussalah, A., Berthelot, P., Del Tedesco, E., Phelip, J. M., et al. (2011). Cytomegalovirus load in inflamed intestinal tissue is predictive of resistance to immunosuppressive therapy in ulcerative colitis. Am. J. Gastroenterol. 106, 2001–2008. doi: 10.1038/ajg.2011.202
Rollwagen, F. M., Yu, Z. Y., Li, Y. Y., and Pacheco, N. D. (1998). IL-6 rescues enterocytes from hemorrhage induced apoptosis in vivo and in vitro by abcl-2 mediated mechanism. Clin. Immunol. Immunopathol. 89, 205–213. doi: 10.1006/clin.1998.4600
Rosinach, M., Esteve, M., González, C., Temiño, R., Mariné, M., Monzón, H., et al. (2012). Lymphocytic duodenosis: aetiology and long-term response to specific treatment. Dig. Liver Dis. 44, 643–648. doi: 10.1016/j.dld.2012.03.006
Rostami, K. A., Al Dulaimi, D., Rostami Nejad, M., Villanacci, V., and Danciu, M. (2010). Microscopic enteritis and pathomechanism of malabsorption. Auto Immun. Highlights 1, 37–38. doi: 10.1007/s13317-010-0006-4
Rostami-Nejad, M., Ishaq, S., Al Dulaimi, D., Zali, M. R., and Rostami, K. (2015). The role of infectiousmediators and gut microbiome in the pathogenesis of celiac disease. Arch. Iran Med. 18, 244–249.
Rostami-Nejad, M., Javad Ehsani-Ardakani, M., Assadzadeh, H., Shahbazkhani, B., Ierardi, E., Losurdo, G., et al. (2016). Pathological and clinical correlation between celiac disease and Helicobacter pylori infection; a review of controversial reports. Middle East J. Dig. Dis. 8, 85–92. doi: 10.15171/mejdd.2016.12
Rozenberg, O., Lerner, A., Pacht, A., Grinberg, M., Reginashvili, D., Henig, C., et al. (2012). Novel algorithm for childhood celiac disease serological diagnosis based upon intestinal biopsies. Crit. Rev. Allerg. Immunol. 42, 331–341. doi: 10.1007/s12016-010-8250-y
Ruggiero, M. A., Gordon, D. P., Orrell, T. M., Bailly, N., et al. (2015). A higher level classification of all living organisms. PLoS ONE 10:e0119248. doi: 10.1371/journal.pone.0119248
Sakkas, L. I., and Bogdanos, D. P. (2016). Infections as a cause of autoimmune rheumatic diseases. Autoimmun. Highlights 7, 13. doi: 10.1007/s13317-016-0086-x
Salama, N. R., Hartung, M. L., and Müller, A. (2013). Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 11, 385–399. doi: 10.1038/nrmicro3016
San-Pedro, J. I., Bilbao, J. R., Perez de Nanclares, G., Vitoria, J. C., Martul, P., and Castaño, L. (2005). Heterogeneity of vitamin D receptor gene association with celiac disease and type 1 diabetes mellitus. Autoimmunity 38, 439–444. doi: 10.1080/08916930500288455
Santarelli, L., Gabrielli, M., Santoliquido, A., Cuoco, L., Cazzato, A., Candelli, M., et al. (2006). Interaction between Helicobacter pylori infection and untreated coeliac disease on gastric histological pattern. Scand. J. Gastroenterol. 41, 532–535. doi: 10.1080/00365520500349549
Sapone, A., Lammers, K. M., Casolaro, V., Cammarota, M., Giuliano, M. T., De Rosa, M., et al. (2011). Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: celiac disease and gluten sensitivity. BMC. Med. 9:23. doi: 10.1186/1741-7015-9-23
Savarino, V., Mela, G. S., Zentilin, P., Lapertosa, G., Bisso, G., Mele, M. R., et al. (1999). Histological and functional recovery in patients with multifocal atrophic gastritis after eradication of Helicobacter pylori infection. Ital. J. Gastroenterol. Hepatol. 31, 4–8.
Schreiber, S., Bücker, R., Groll, C., Azevedo-Vethacke, M., Garten, D., Scheid, P., et al. (2005). Rapid loss of motility of Helicobacter pylori in the gastric lumen in vivo. Infect. Immun. 73, 1584–1589. doi: 10.1128/IAI.73.3.1584-1589.2005
Sellitto, M., Bai, G., Serena, G., Fricke, W. F., Sturgeon, C., Gajer, P., et al. (2012). Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE 7:e33387. doi: 10.1371/journal.pone.0033387
Selmi, C. (2016). Are helminths to be trusted as allies in the war against autoimmunity and chronic inflammation? Isr. Med. Assoc. J. 18, 139–140.
Shalimar, D. M., Das, P., Sreenivas, V., Gupta, S. D., Panda, S. K., and Makharia, G. K. (2013). Mechanism of villous atrophy in celiac disease: role of apoptosis and epithelial regeneration. Arch. Pathol. Lab. Med. 13, 1262–1269. doi: 10.5858/arpa.2012-0354-OA
Shamir, R., Eliakim, R., Lahat, N., Sobel, E., and Lerner, A. (2002). ELISA assay of anti endomysial antibodies in the diagnosis of celiac disease: comparison with immunofluorescence assay of anti endomysial antibodies and tissue transglutaminase antibodies. Isr. Med. Assoc. J. 4, 594–596.
Shmidt, E., Smyrk, T. C., Boswell, C. L., Enders, F. T., and Oxentenko, A. S. (2014). Increasing duodenal intraepithelial lymphocytosis found at upper endoscopy: time trends and associations. Gastrointest. Endosc. 80, 105–111. doi: 10.1016/j.gie.2014.01.008
Shor, B.-A. D., Bashi, T., Lachnish, J., Fridkin, M., Bizzaro, G., Barshak, I., et al. (2015). Phosphorylcholine-tuftsin compound prevents development of dextransulfate-sodium-salt induced murine colitis: implications for the treatment of human inflammatory bowel disease. J. Autoimmun. 56, 111–117. doi: 10.1016/j.jaut.2014.11.001
Shor, B.-A. D., Harel, M., Eliakim, R., and Shoenfeld, Y. (2013). The hygiene theory harnessing helminths and their ova to treat autoimmunity. Clin. Rev. Allergy Immunol. 45, 211–216. doi: 10.1007/s12016-012-8352-9
Silvester, J. A., and Leffler, D. A. (2016). Is autoimmunity infectious? the effect of gastrointestinal viral infections and vaccination on risk of celiac disease autoimmunity. Clin. Gastroenterol. Hepatol. 15, 703–705. doi: 10.1016/j.cgh.2016.12.014
Simondi, D., Ribaldone, D. G., Bonagura, G. A., Foi, S., Sapone, N., Garavagno, M., et al. (2015). Helicobacter pylori in celiac disease and in duodenal intraepithelial lymphocytosis: active protagonist or innocent bystander? Clin. Res. Hepatol. Gastroenterol. 39, 740–745. doi: 10.1016/j.clinre.2015.03.005
Sjöberg, V., Sandström, O., Hedberg, M., Hammarström, S., Hernell, O., and Hammarström, M. L. (2013). Intestinal T-cell responses in celiac disease — impact of celiac disease associated bacteria. PLoS ONE 8:e53414. doi: 10.1371/journal.pone.0053414
Smallwood, T. B., Giacomin, P. R., Loukas, A., Mulvenna, J. P., Clark, R. J., and Miles, J. J. (2017). Helminth Immunomodulation in Autoimmune Disease. Front Immunol. 8:453. doi: 10.3389/fimmu.2017.00453
Spanhaak, S., Havenaar, R., and Schaafsma, G. (1998). The effect of consumption of milk fermented by Lactobacillus casei strain Shirota on the intestinal microflora and immune parameters in humans. Eur. J. Clin. Nutr. 52, 899–907. doi: 10.1038/sj.ejcn.1600663
Spor, A., Koren, O., and Ley, R. (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 9, 279–290. doi: 10.1038/nrmicro2540
Sütas, Y., Autio, S., Rantala, I., and Isolauri, E. (1997). IFN-gamma enhances macromolecular transport across Peyer's patches in suckling rats: implications for natural immune responses to dietary antigens early in life. J. Pediatr. Gastroenterol. Nutr. 24, 162–169. doi: 10.1097/00005176-199702000-00009
Tannock, G. W., Munro, K., Harmsen, H. J., et al. (2000). Analysis of the fecal microflora of human subjects consuming a probiotic product containing Lactobacillus rhamnosus DR20. Appl. Environ. Microbiol. 66, 2578–2588. doi: 10.1128/AEM.66.6.2578-2588.2000
Toft-Hansen, H., Nielsen, C., Biagini, M., Husby, S., and Lillevang, S. T. (2013). Lectin staining shows no evidence of involvement of glycocalyx/mucous layer carbohydrate structures in development of celiac disease. Nutrients 5, 4540–4552. doi: 10.3390/nu5114540
Tracy, S., Drescher, K. M., Chapman, N. M., Kim, K. S., Carson, S. D., Pirruccello, S., et al. (2002). Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J. Virol. 76, 12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002
Trebichavsky, I., Splichal, I., Rada, V., and Splichalova, A. (2010). Modulation of natural immunity in the gut by Escherichia coli strain Nissle 1917. Nutr. Rev. 68, 459–464. doi: 10.1111/j.1753-4887.2010.00305.x
Tsai, H. F., and Hsu, P. N. (2010). Interplay betweenHelicobacter pylori and immune cells in immunepathogenesis of gastric inflammation and mucosal pathology. Cell Mol. Immunol. 7, 255–259. doi: 10.1038/cmi.2010.2
Urganci, N., and Kalyoncu, D. (2013). Response to hepatitis A and B vaccination in pediatric patients with celiac disease. J. Pediatr. Gastroenterol. Nutr. 56, 408–411. doi: 10.1097/MPG.0b013e31827af200
Vatsiou, A. I., Bazin, E., and Gaggiotti, O. E. (2016). Changes in selective pressures associated with human population expansion may explain metabolic and immune related pathways enriched for signatures of positive selection. BMC. Genomics. 17:504. doi: 10.1186/s12864-016-2783-2
Villanacci, V., Bassotti, G., Liserre, B., Lanzini, A., Lanzarotto, F., and Genta, R. M. (2006). Helicobacter pylori infection in patients with celiac disease. Amer. J. Gastroenterol. 101, 1880–1885. doi: 10.1111/j.1572-0241.2006.00621.x
Vordenbäumen, S., Pilic, D., Otte, J. M., Schmitz, F., and Schmidt-Choudhury, A. (2010). Defensin-mRNA expression in the upper gastrointestinal tract is modulated in children with celiac disease and Helicobacter pylori-positive gastritis. J. Pediatr. Gastroenterol. Nutr. 50, 596–600. doi: 10.1097/MPG.0b013e3181cd26cd
Wacklin, P., Laurikka, P., Lindfors, K., Collin, P., Salmi, T., Lähdeaho, M. L., et al. (2014). Altered duodenal microbiota composition in celiac disease patients suffering from persistent symptoms on a long-term gluten-free diet. Am. J. Gastroenterol. 109, 1933–1941. doi: 10.1038/ajg.2014.355
Wang, J., Thingholm, L. B., Skiecevicienė, J., Rausch, P., Kummen, M., Hov, J. R., et al. (2016). Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48, 1396–1406. doi: 10.1038/ng.3695
Weng, M., and Walker, W. A. (2013). The role of gut microbiota in programming the immune phenotype. J. Dev. Orig. Health Dis. 4, 203–214. doi: 10.1017/S2040174412000712
Wu, J., Lin, K., Zeng, J., Liu, W., Yang, F., Wang, X., et al. (2014). Role of DC-SIGN in Helicobacter pylori infection of gastrointestinal cells. Front. Biosci. 19, 825–834. doi: 10.2741/4250
Wu, T. T., and Hamilton, S. R. (1999). Lymphocytic gastritis: association with etiology and topology. Am. J. Surg. Pathol. 23, 153–158. doi: 10.1097/00000478-199902000-00003
Zaman, C., Osaki, T., Hanawa, T., Yonezawa, H., Kurata, S., and Kamiya, S. (2014). Analysis of the microbial ecology between Helicobacter pylori and the gastric microbiota of Mongolian gerbils. J. Med. Microbiol. 63, 129–137. doi: 10.1099/jmm.0.061135-0
Zeissig, S., and Blumberg, R. S. (2014). Commensal microbial regulation of natural killer T cells at the frontiers of the mucosal immune system. FEBS Lett. 588, 4188–4194. doi: 10.1016/j.febslet.2014.06.042
Zelnik, N., Pacht, A., Obeid, R., and Lerner, A. (2004). Range of neurological disorders in patients with celiac disease. Pediatrics 113, 1672–1676. doi: 10.1542/peds.113.6.1672
Zyrek, A. A., Cichon, C., Helms, S., Enders, C., Sonnenborn, U., and Schmidt, M. A. (2007). Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell. Microbiol. 9, 804–816. doi: 10.1111/j.1462-5822.2006.00836.x
Keywords: celiac disease, bacteria, viruses, gut, microbiome, environmental inducer, environmental protectors
Citation: Lerner A, Arleevskaya M, Schmiedl A and Matthias T (2017) Microbes and Viruses Are Bugging the Gut in Celiac Disease. Are They Friends or Foes? Front. Microbiol. 8:1392. doi: 10.3389/fmicb.2017.01392
Received: 15 May 2017; Accepted: 10 July 2017;
Published: 02 August 2017.
Edited by:
Kuldeep Dhama, Indian Veterinary Research Institute (IVRI), IndiaReviewed by:
Mario M. D'Elios, University of Florence, ItalyMaryam Dadar, Razi Vaccine and Serum Research Institute, Iran
Copyright © 2017 Lerner, Arleevskaya, Schmiedl and Matthias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron Lerner, YWFyb25sZXJuZXIxOTQ4QGdtYWlsLmNvbQ==