 Chrizelle W. Beukes1Marike Palmer1Puseletso Manyaka1Wai Y. Chan1Juanita R. Avontuur1Elritha van Zyl1Marcel Huntemann2
Chrizelle W. Beukes1Marike Palmer1Puseletso Manyaka1Wai Y. Chan1Juanita R. Avontuur1Elritha van Zyl1Marcel Huntemann2 Alicia Clum2Manoj Pillay2Krishnaveni Palaniappan2Neha Varghese2
Alicia Clum2Manoj Pillay2Krishnaveni Palaniappan2Neha Varghese2 Natalia Mikhailova2Dimitrios Stamatis2T. B. K. Reddy2Chris Daum2Nicole Shapiro2Victor Markowitz2
Natalia Mikhailova2Dimitrios Stamatis2T. B. K. Reddy2Chris Daum2Nicole Shapiro2Victor Markowitz2 Natalia Ivanova2
Natalia Ivanova2 Nikos Kyrpides2
Nikos Kyrpides2 Tanja Woyke2
Tanja Woyke2 Jochen Blom3William B. Whitman4
Jochen Blom3William B. Whitman4 Stephanus N. Venter1*Emma T. Steenkamp1
Stephanus N. Venter1*Emma T. Steenkamp1- 1Department of Microbiology and Plant Pathology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa
- 2DOE Joint Genome Institute, Walnut Creek, CA, United States
- 3Bioinformatics and Systems Biology, Justus-Liebig-University Giessen, Giessen, Germany
- 4Department of Microbiology, University of Georgia, Athens, GA, United States
Although the taxonomy of Burkholderia has been extensively scrutinized, significant uncertainty remains regarding the generic boundaries and composition of this large and heterogeneous taxon. Here we used the amino acid and nucleotide sequences of 106 conserved proteins from 92 species to infer robust maximum likelihood phylogenies with which to investigate the generic structure of Burkholderia sensu lato. These data unambiguously supported five distinct lineages, of which four correspond to Burkholderia sensu stricto and the newly introduced genera Paraburkholderia, Caballeronia, and Robbsia. The fifth lineage was represented by P. rhizoxinica. Based on these findings, we propose 13 new combinations for those species previously described as members of Burkholderia but that form part of Caballeronia. These findings also suggest revision of the taxonomic status of P. rhizoxinica as it is does not form part of any of the genera currently recognized in Burkholderia sensu lato. From a phylogenetic point of view, Burkholderia sensu stricto has a sister relationship with the Caballeronia+Paraburkholderia clade. Also, the lineages represented by P. rhizoxinica and R. andropogonis, respectively, emerged prior to the radiation of the Burkholderia sensu stricto+Caballeronia+Paraburkholderia clade. Our findings therefore constitute a solid framework, not only for supporting current and future taxonomic decisions, but also for studying the evolution of this assemblage of medically, industrially and agriculturally important species.
Introduction
The genus Burkholderia was originally introduced to accommodate an assemblage of seven Pseudomonas species (Yabuuchi et al., 1992), two of which were later transferred to Ralstonia (Gillis et al., 1995; Yabuuchi et al., 1995). Since then, the number of Burkholderia species has grown substantially, to about 108 in 2015 (Estrada-de los Santos et al., 2016), spanning a range of human, animal and plant pathogens, as well as numerous strains with significant biotechnological potential (Depoorter et al., 2016; Estrada-de los Santos et al., 2016). The latter includes the so-called plant beneficial and environmental (PBE) species (Suárez-Moreno et al., 2012), many of which are plant-associated (e.g., those with plant growth promoting activities, the symbiotic diazotrophs and free-living species with diazotrophic, bioremedial and antibiotic activities) (Depoorter et al., 2016; Estrada-de los Santos et al., 2016). Because of this heterogeneity, new genera [e.g., ‘Caballeronia’ (Gyaneshwar et al., 2011) and ‘Paraburkholderia’ (Sawana et al., 2014)] has been introduced to accommodate most of the PBE species (Oren and Garrity, 2015a,b, 2017), while retaining the pathogens in Burkholderia sensu stricto. Most recently, a third genus, Robbsia was introduced to accommodate the phytopathogen previously referred to as B. andropogonis (Lopes-Santos et al., 2017).
Overall, the taxonomy of Burkholderia sensu lato remains in significant flux (Estrada-de los Santos et al., 2016). With their review of the group, Estrada-de los Santos et al. (2016) recognized two monophyletic groups [Groups A and B; A consists of Caballeronia and Paraburkholderia as circumscribed by Gyaneshwar et al. (2011) and Sawana et al. (2014), respectively, while Group B includes most of the notable human, animal and plant pathogens, as well as the so-called “B. cepacia complex”]. They showed that B. andropogonis (now Robbsia andropogonis) is separated into its own group, and they designated two so-called “Transition Groups” (i.e., 1 and 2; neither were supported as monophyletic and both contained mainly environmental species). Since then, Dobritsa and Samadpour (2016) have proposed the transfer of species in Transition Group 2 to a new genus. However, to complicate the issue, this new genus was named “Caballeronia” although its proposed usage is not synonymous with the one previously proposed by Gyaneshwar et al. (2011) for accommodating all the PBE isolates.
The proposals to split Burkholderia sensu lato were based almost entirely on evidence from 16S ribosomal RNA (rRNA) phylogenetic trees with limited and in some cases no statistical support (Gyaneshwar et al., 2011; Dobritsa and Samadpour, 2016; Eberl and Vandamme, 2016). Even phylogenies based on conventional multilocus sequence analysis (MLSA) using the combined sequence information for 4–7 genes (Gevers et al., 2005) produced phylogenies in which the major groups were not supported as monophyletic (Estrada-de los Santos et al., 2013). Also, the most comprehensive phylogenetic hypothesis to date (based on 21 conserved gene sequences) lacked sufficient representation across this diverse assemblage (Sawana et al., 2014). Thus, uncertainties remain regarding the genomic and evolutionary coherence of Burkholderia sensu lato and its lineages. This, in turn, blurs the boundaries of the Burkholderia sensu lato genera currently recognized and also casts doubt on the appropriateness and legitimacy of their taxonomic circumscriptions.
In this study, we aimed to resolve the relationships within Burkholderia sensu lato, particularly those pertaining to Paraburkholderia and Caballeronia, by making use of whole genome sequence data. For this purpose, we utilized all of the sequences for type strains (or appropriate representatives) available in the public domain. To increase representation of the so-called environmental species, we also determined the sequences for eight additional taxa via Phase III of the GEBA (Genomic Encyclopedia of Bacterial and Archaeal type strains) project (Whitman et al., 2015). These included the rhizobial species P. aspalathi (Mavengere et al., 2014) and P. diazotrophica (Sheu et al., 2013), and the soil bacteria P. hospita (Goris et al., 2002), P. phenazinium (Viallard et al., 1998), P. sartisoli (Vanlaere et al., 2008), P. terricola (Goris et al., 2002), as well as the plant-associated diazotrophic species P. caballeronis (Martínez-Aguilar et al., 2013) and P. tropica (Reis et al., 2004).
Materials and Methods
Whole-Genome Sequencing of Eight Paraburkholderia Type Strains
The eight type strains (P. aspalathi LMG 27731T, P. hospita LMG 20598T, P. diazotrophica LMG 206031T, P. phenazinium LMG 2247T, P. sartisoli LMG 24000T, P. terricola LMG 20594T, P. tropica LMG 22274T and P. caballeronis LMG 26416T) were obtained from the Belgian Coordinated Collections of Microorganisms (University of Gent, Belgium). Routine growth of these bacteria in the laboratory and extraction of high quality genomic DNA were completed as described previously (Steenkamp et al., 2015). Whole genome sequencing was performed by the Joint Genome Institute (JGI) following standard protocols1 and using the Illumina HiSeq-2500 1TB platform with an Illumina 300 base pair (bp) insert standard shotgun library.
All raw sequences were filtered using BBDuk (Bushnell, 2017), which removes known Illumina artifacts, and PhiX. Reads with more than one “N” or with quality scores (before trimming) averaging less than 8 or reads shorter than 51 bp (after trimming) were discarded. The remaining reads were mapped to masked versions of human, cat and dog reference sequences using BBMap (Bushnell, 2017) and discarded if identity values exceeded 93%. The remaining reads were then assembled into contigs using Velvet version 1.2.07 (Zerbino and Birney, 2008) (the settings used were velveth: 63 –shortPaired and velvetg: –very clean yes –exportFiltered yes –min contig lgth 500 –scaffolding no –cov cutoff 10). The Velvet contigs were then used to generate 1–3 kbp simulated paired end reads using wgsim version 0.3.02 (the settings used were –e 0 –1 100 –2 100 –r 0 R 0 –X 0). We then assembled the quality filtered Illumina reads with the simulated read pairs using Allpaths-LG version r46652 (Gnerre and MacCallum, 2011) (the settings used were PrepareAllpathsInputs: PHRED 64 = 0 PLOIDY = 1 FRAG COVERAGE = 125 JUMP COVERAGE = 25 LONG JUMP COV = 50 and RunAllpathsLG: THREADS = 8 RUN = std shredpairs TARGETS = standard VAPI WARN ONLY = True OVERWRITE = True).
The standard JGI microbial genome annotation pipeline (Huntemann et al., 2015) was used to predict and annotate genes in each of the eight assembled genomes. For this purpose, we specifically used the Prodigal algorithm to identify protein-coding genes (Hyatt et al., 2010). Additional annotation was performed using JGI’s Integrated Microbial Genomes (IMG) system (Markowitz et al., 2014).
Sequence Datasets and Multiple Alignments
Protein-coding gene datasets were generated for the eight bacteria sequenced here, as well as all the Burkholderia sensu lato type strains (or suitable conspecific strains) for which whole genome sequences were available (Supplementary Table S1). This was achieved by using the EDGAR (Efficient Database framework for comparative Genome Analyses using BLAST score Ratios) server3 (Blom et al., 2016) to identify single-copy orthologous genes shared among all of the genomes examined. The respective amino acid and nucleotide sequences for each gene dataset were then batch-aligned using the Multiple Sequence Comparison by Log-Expectation (MUSCLE) (Edgar, 2004) iteration-based alignment tool implemented in CLC Main Workbench 7.6 (CLC Bio).
Individual alignments were manually curated in BioEdit version 7.2.5 (Hall, 2011), during which we discarded those genes for which one or more taxa contained more than 5% missing data. The pair-wise protein similarity for the remaining genes (i.e., those for which the datasets were ≥95% complete) were individually determined with Geneious v. 6.1 (Biomatters Limited4), followed by concatenation with FASconCAT-G v. 1.02 (Kuck and Longo, 2014). The total pair-wise similarity among the various taxa included in the study was also calculated by making use of the concatenated nucleotide and amino acid datasets using Geneious v. 6.1.
We also evaluated the genomic distribution and functional roles for the genes with ≥95% complete sequence data. The putative function of each gene product was inferred using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) databases and the GhostKoala mapping tool5 (Kanehisa et al., 2016), as well as through comparison with the annotated genome of the type species Burkholderia cepacia ATCC 25416T (Yabuuchi et al., 1992). This genome was also used to determine the relative genomic position of each gene used in our dataset. This was done by making use of Geneious v. 6.1 and the publicly available annotations of the ATCC 25416T genome on the National Center for Biotechnology Information (NCBI6) website.
The level of substitution saturation in the various nucleotide and amino acid datasets were evaluated as described before (Palmer et al., 2017). For this purpose, distances based on actual substitutions (p-distance) were compared to those inferred using an appropriate substitution model (Jeffroy et al., 2006; Philippe et al., 2011). The modeled distances for the nucleotide data were inferred using the General Time Reversible (GTR) substitution model (Tavaré, 1986) and the minimum-evolution distance algorithm (Desper and Gascuel, 2002). Both the p- and GTR-distances were determined in DAMBE v. 6.0.1 (Xia and Xie, 2001) and were calculated for the full nucleotide datasets and for the third codon positions only. For the amino acid datasets, MEGA v.6.06 (Tamura et al., 2013) was used to calculate the p-distances and those based on the Jones-Taylor-Thornton (JTT) model (Jones et al., 1992). Graphical representations of the correlation between the respective distances for each dataset were constructed in Microsoft Excel 2013, followed by linear regression analyses.
Phylogenetic Analyses
The respective nucleotide and amino acid alignments for the ≥95% complete protein-coding genes were concatenated and subjected to maximum likelihood phylogenetic analyses with RAxML v. 8.2.1 (Stamatakis, 2014). For this purpose, the sequences were concatenated and partitioned using FASconCAT-G. For the amino acid data, each partition employed the best-fit substitution model as indicated by ProtTest v. 3.4 (Abascal et al., 2005). For the nucleotide data, we used the GTR model with independent parameter estimation for each partition. Branch support was estimated in RAxML using the estimated model parameters, the rapid hill-climbing algorithm and non-parametric bootstrap analyses of 1000 repetitions.
Results
Whole-Genome Sequences for Eight Type Strains of Paraburkholderia
Illumina sequencing allowed assembly of high-coverage (i.e., 67.4 to 119.3 X) draft genomes for the type strains of eight Paraburkholderia species (Table 1). The number of contigs for each genome ranged from 22 to 188 where more than 50% of the individual genomes were incorporated into relatively large contigs (i.e., respective N50-values ranged from 144482 to 573607). The assembled genomes ranged in size from 5.9 for P. sartisoli LMG 24000T to 11.2 million bases for P. hospita LMG 20498T. The number of genes predicted for each genome also corresponded well with their overall sizes (e.g., 5407 genes were predicted for P. sartisoli LMG 24000T and 10534 for P. hospita LMG 20498T). The G+C content for the eight species ranged from 61.09% for P. aspalathi to 67.03% for P. caballeronis. The assembled genome sequences for all eight species are available from NCBI (see Table 1 for accession numbers). Overall the sizes and GC content were comparable to previously sequenced genomes of other Burkholderia sensu lato species (Table 2).
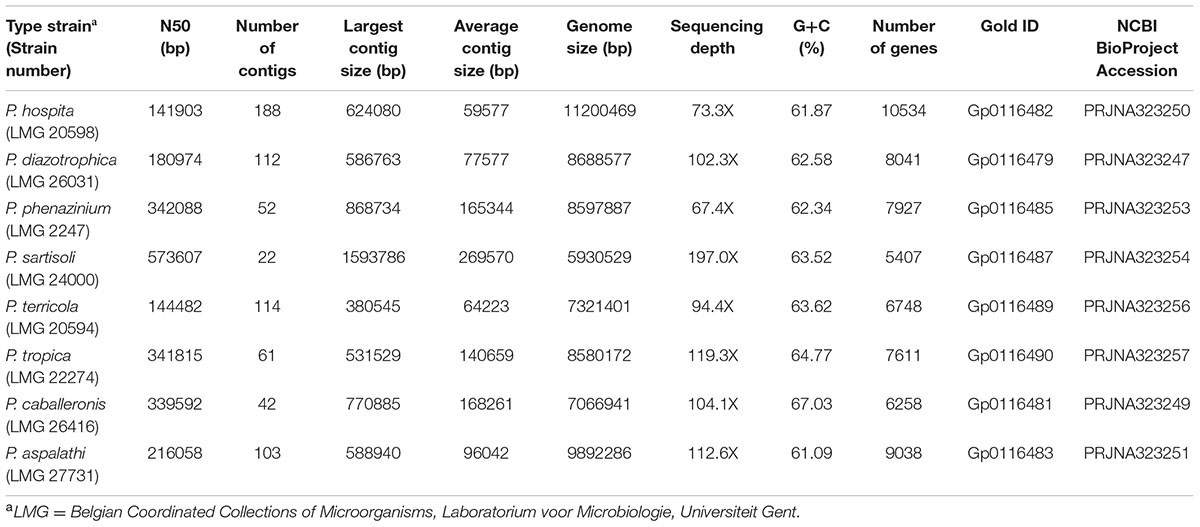
TABLE 1. Details regarding the eight Paraburkholderia type strains sequenced at JGI for the GEBA Phase III project.
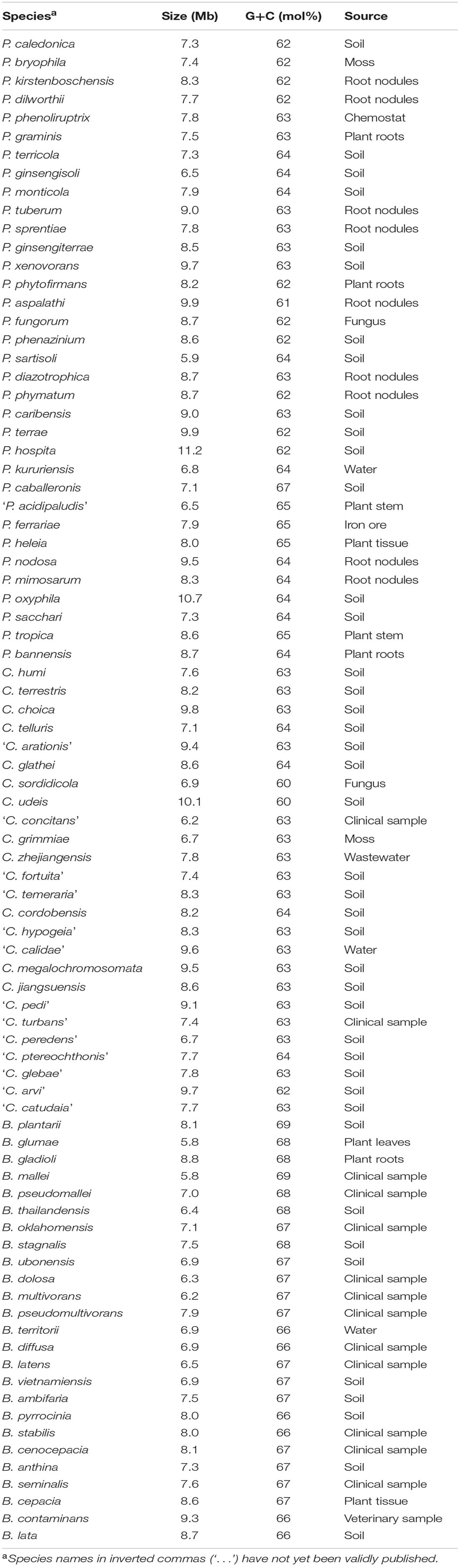
TABLE 2. Genome properties for all the investigated species forming part of Burkholderia sensu stricto, Caballeronia and Paraburkholderia.
Sequence Datasets and Multiple Alignments
A set of 106 genes with ≥95% complete sequences were identified among the genomes of 86 Burkholderia sensu lato species and the 6 outgroup taxa. The 106 genes were identified using a strict orthology estimation performed in EDGAR (Blom et al., 2016). Only those sequences with a mean % identity of 60.22 (median 54.63%) and a mean Expect(E)-value of 6.494625e-09 (median 1.00e-101) of the accepted BLAST hits were included in the final datasets. Although the full set of shared genes among these taxa would be considerably larger, the examined genomes differed substantially in their level of completeness and the annotation approaches utilized. Our conservative approach for generating these datasets therefore attempted to avoid inadvertently including phylogenetic noise caused by potential sequencing and annotation inconsistencies.
The concatenated dataset for the 106 genes consisted of 92 taxa with 25499 residues in the amino acid alignment and 80027 bases in the nucleotide alignment. The amino acid dataset consisted of 99.1% coding characters with 0.9% of the dataset consisting of alignment gaps, while the nucleotide dataset consisted of 98.6% coding characters with 1.4% of the dataset consisting of alignment gaps. Neither of these datasets included any poorly aligned regions because of the absence of more divergent sequences. For example, the amino acid and nucleotide similarity across the entire dataset (including Ralstonia and Cupriavidus outgroups) were >77% and >73%, respectively (Figure 1 and Supplementary File S1). Within each of the main phylogenetic clades inferred from the data (see below), these values were generally >92% and >84%, respectively.
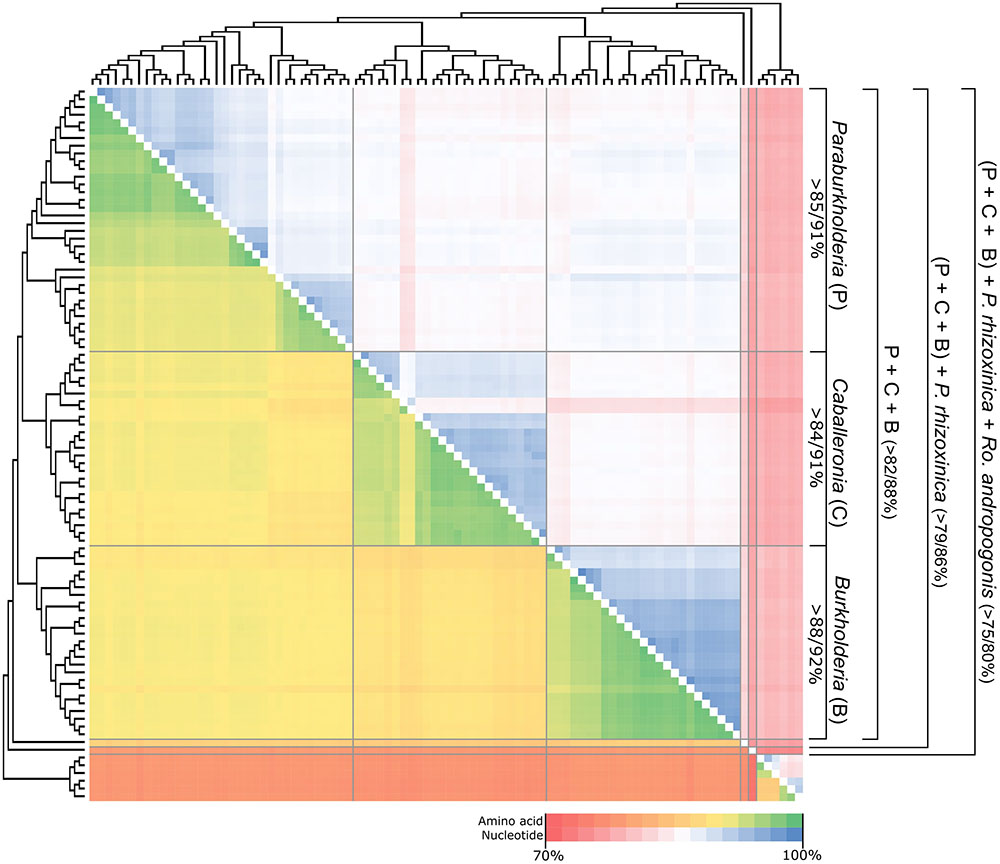
FIGURE 1. A heat map depicting the sequence similarity of the concatenated sequence of the conserved 106 genes used for phylogenetic analysis. The cladogram indicating the various intra- and intergeneric relationships were inferred from the amino acid based ML topology. Nucleotide similarity values are indicated in the upper triangle of the map, with amino acid similarity values indicated in the lower triangle of the map. A summary of the similarity values for the 5 lineages of interest are indicated for each group (nucleotide/amino acid %), in the panel on the right. For specific values, refer to Supplementary File S1.
Despite the high-level of conservation observed in the 106 genes, both the nucleotide and the amino acid data were free from significant levels of substitution saturation (Supplementary File S2). For both datasets, this was evident from the slope of the linear regression line for the plot between actual and modeled distances. However, compared to the nucleotide dataset, the amino acid dataset was least saturated, as the slope of its regression line was closest to 1. Our results also suggest the limited saturation present in the nucleotide data may be ascribed to multiple substitutions primarily occurring at third codon positions (Supplementary File S2).
We investigated the genomic distribution of the 106 genes by mapping them to those in the annotated genome of strain ATCC 25416T of Burkholderia cepacia, which is also the type species for Burkholderia (Yabuuchi et al., 1992). These analyses showed that 101 of the genes mapped to chromosome 1 of this species (Supplementary Figure S1), where they appeared to be scattered throughout the replicon (see Supplementary Table S2 for the nucleotide positions and orientation of the respective genes). The remaining five genes mapped to chromosome 2 (Supplementary Figure S2 and Table S2).
Analysis of the putative functions of the 106 genes revealed that they are likely involved in a multitude of diverse functions. Based on both the original annotations for B. cepacia ATCC 25416T and the KEGG analysis with GhostKOALA, only four of the 106 gene were classified as having unknown or hypothetical functions (Supplementary Table S3). About 44% of the remaining 102 genes represented “informational genes” (sensu Jain et al., 1999) and encoded products involved in processes relating to nucleotide synthesis, DNA replication and repair, transcription, translation and related processes. A further 35% of the genes encoded products involved in carbohydrate, lipid and amino acid metabolism, while the remaining 21% encoded products involved in diverse functions (e.g., signal transduction, membrane transport, iron scavenging, etc.) (Supplementary Table S3).
Phylogenetic Analyses
Because of the limited substitution saturation detected in the concatenated amino acid and nucleotide datasets, both datasets were subjected to maximum likelihood phylogenetic analysis in RAxML “as is” (i.e., no attempt was made to exclude saturated sites). However, these analyses were conducted using substitution models specific for each gene, which in all cases accounted for invariable sites and included gammaa correction to account for among site rate variation. Although the nucleotide data partitions utilized the GTR model, each partition used independent model parameters (i.e., each gene partition utilized the six nucleotide substitution rates specific to it) (see Supplementary Table S2 for details on the substitution models used for the respective amino acid partitions).
Highly similar and congruent topologies were inferred from the amino acid and nucleotide data for the 106 genes included in this study (Figure 2 and Supplementary Figure S3). All of the branches in the two trees further received bootstrap support values exceeding 90% (with most supported by values of 100%). The only differences observed between the two trees were in terms of the placement of some species within certain terminal clades (e.g., in the nucleotide phylogeny P. ginsengisoli forms a distinct lineage within a larger clade containing P. caledonica, P. bryophila, P. kirstenboschensis, P. dilworthii, P. phenoliruptrix, P. graminis, P. terricola, P. aspalathi, P. fungorum, P. ginsengiterrae, P. phytofirmans, P. xenovorans, P. monticola, P. tuberum, and P. sprentiae but in the amino acid tree it is basal to a smaller clade consisting of P. monticola, P. tuberum, and P. sprentiae). These small topological differences probably reflect limited phylogenetic signal in the datasets for resolving more recent divergences. No disparities were observed regarding the composition of the main clades recovered from the two datasets.
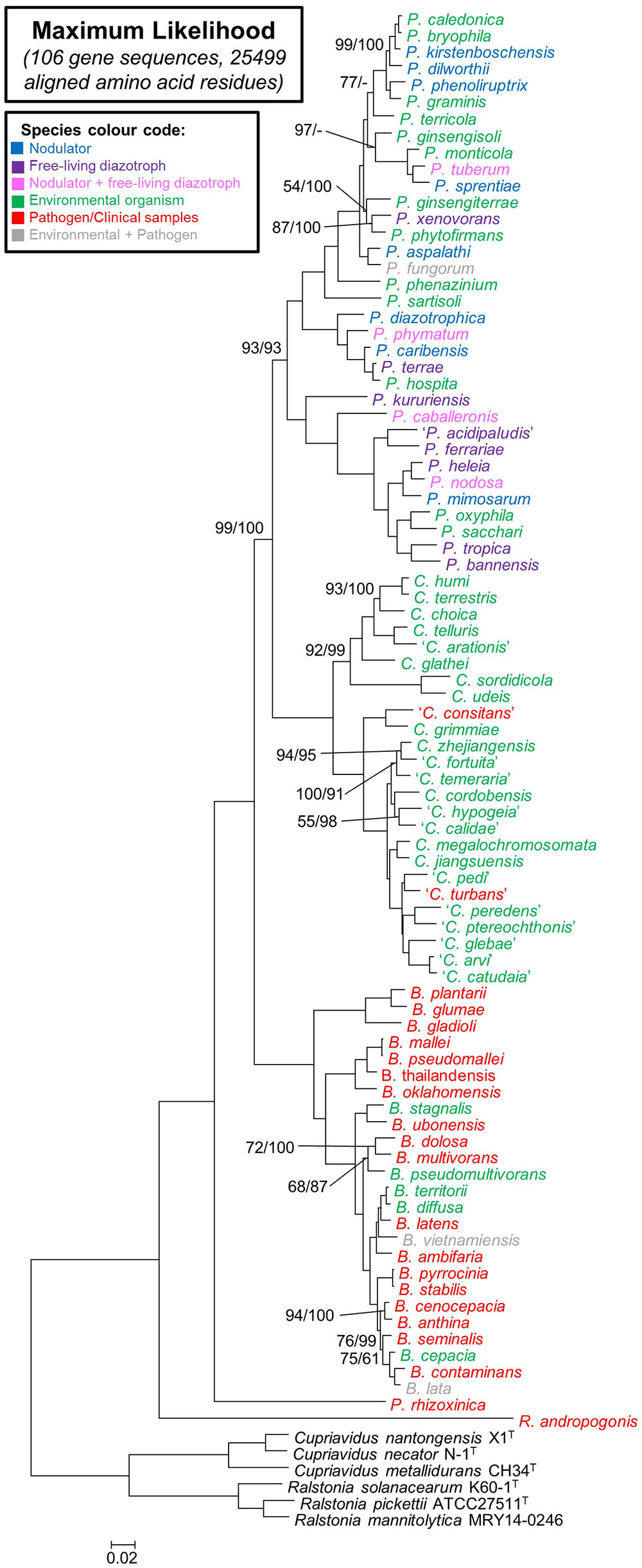
FIGURE 2. A maximum-likelihood phylogeny of the amino acid sequences of 106 concatenated genes for the 92 strains used in this study. A similar topology was obtained using the nucleotide sequences for these genes (Supplementary Figure S3). New combinations that have not yet been validated are indicated in inverted commas. General species substrates and origins are color coded according to the key provided. The majority of branches received 100% bootstrap in both the amino acid and nucleotide phylogenies and therefore only those branches in which 100% was not calculated for both analyses are indicated. Support is indicated in the order amino acid/nucleotide. The scale bar indicates the number of changes per site.
In terms of the phylogenetic relationships among the taxa, both trees separated the Burkholderia sensu lato species into five distinct lineages (Figure 1 and Supplementary Figure S3). Three of these corresponded to clades, respectively, representing Paraburkholderia, Caballeronia, and Burkholderia sensu stricto. The remaining two lineages were represented by R. andropogonis and P. rhizoxinica. Within this phylogeny, Paraburkholderia and Caballeronia were recovered as sister groups that shared an origin with Burkholderia sensu stricto. In turn, these three clades shared a most recent common ancestor with the lineage represented by P. rhizoxinica. Based on our analyses, the lineage represented by R. andropogonis is the most basal taxon in the Burkholderia sensu lato tree.
The Paraburkholderia clade consisted of 34 species. Of these, 33 were recently formally transferred to Paraburkholderia and the new combinations have been validated. Our data show that the novel combination (suggested by Sawana et al., 2014) requires P. acidipaludis still awaits validation. A similar situation exists for the Caballeronia clade. Of the 25 species it included, 12 were recently formally transferred to Caballeronia, but our results suggest that a further 13 (recently accepted as Burkholderia species) also need to be incorporated in this genus (Table 3).
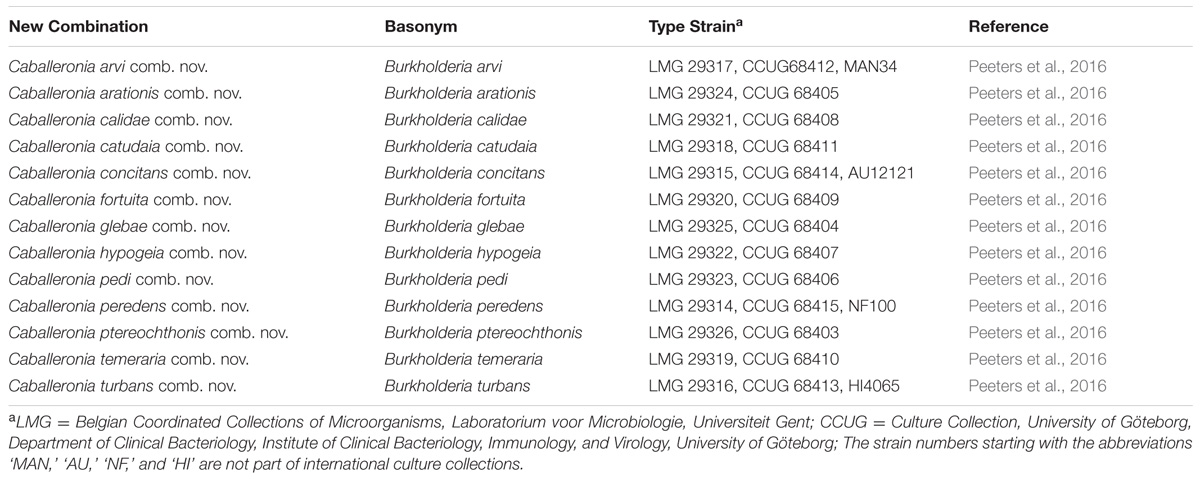
TABLE 3. Summary of the novel combinations proposed for 13 species of Caballeronia.
Discussion
To achieve our primary goal of resolving the generic boundaries and relationships within Burkholderia sensu lato, we endeavored to use as wide a taxon selection as possible. Therefore, to complement the genome data already in the public domain for 78 species in this assemblage, we determined the whole genome sequences for an additional eight PBE species. The genomes for these species exhibited similar characteristics as those of other members of Burkholderia sensu lato (see Table 2). This was particularly true in terms of genome size and total numbers of genes encoded. Some differences were observed in G+C content. As have been observed before (Gyaneshwar et al., 2011; Estrada-de los Santos et al., 2013; Sawana et al., 2014), the Burkholderia sensu stricto genomes were higher in G+C content than Paraburkholderia and Caballeronia, which were similar in G+C content. Future studies aimed at exploring genome architecture and the functions encoded on these genomes will undoubtedly reveal traits and processes that more clearly characterize the various lineages of this economically important assemblage of bacteria.
For inferring a robust phylogeny that are congruent with the evolutionary history of Burkholderia sensu lato, we attempted to avoid or limit the effect of factors known to negatively impact phylogenetic trees (Philippe et al., 2011). The criteria used for generating the respective datasets therefore focused on the use of orthologous loci and on limiting the effects of non-phylogenetic signal. The former was accomplished by using EDGAR to identify orthologous protein-coding genes (Blom et al., 2016). The orthologous nature of a large proportion of the genes included in our final dataset was also congruent with expectations of the so-called complexity hypothesis (Jain et al., 1999; Cohen et al., 2011). In silico functional analysis showed that about 44% of these genes represented “informational genes” with products that potentially participate in processes related to DNA replication and repair, transcription and translation. Due to the complexity of their interactions with different proteins and other cellular constituents, these genes are typically less prone to horizontal gene transfer (Jain et al., 1999; Cohen et al., 2011). Our approach for identifying suitable gene sequences from which to infer the phylogeny thus lessened the chances considerably of accidentally using paralogous or xenologous gene copies (Koonin, 2005).
To limit the amount of non-phylogenetic signal in the data, a three-tiered approach was used. [i] The final dataset was large, almost devoid of missing sites (i.e., where genes in some taxa were not sequenced in their entirety) and consisted of the sequences for 106 genes common to Burkholderia sensu lato and its Ralstonia and Cupriavidus outgroups. Such large datasets typically outperform smaller datasets that only contain the sequences for one or a few genes (Daubin et al., 2002; Coenye et al., 2005; Galtier and Daubin, 2008; Bennet et al., 2012; Chan et al., 2012). This is because the “true” phylogenetic signal inherent to orthologs included in such a large dataset will dominate the analysis and typically attenuate or dilute the effects of spurious non-phylogenetic signal associated with one or a few genes (Daubin et al., 2002; Andam and Gogarten, 2011). [ii] Lack of evolutionary independence among loci may contribute to non-phylogenetic signal during tree inference (Gevers et al., 2005). For example, genes that are clustered or whose products are involved in similar or linked processes typically experience similar evolutionary forces, which is accordingly also reflected in their phylogenies (i.e., these reflect the linked evolutionary history of the genes and not the evolutionary history of the species or genus). However, the 106 genes used for resolving Burkholderia sensu lato were not significantly clustered (see Supplementary Figures S1, S2), while their inferred products were predicted to participate in diverse functions (see Supplementary Table S3). [iii] Substitution saturation is another important source of non-phylogenetic signal (Philippe and Forterre, 1999; Xia et al., 2003; Jeffroy et al., 2006; Philippe et al., 2011), and to compensate for its limited occurrence in our datasets, all phylogenetic analyses utilized independent substitution models for each gene partition. This approach proved fairly successful as both the nucleotide and amino acid data supported congruent trees with highly similar topologies.
Our maximum likelihood analyses of the aligned amino acid and nucleotide sequences for 106 genes produced a highly supported phylogeny for Burkholderia sensu lato (see Figure 2). Most of the branches on this 92-taxon phylogeny received full (100%) bootstrap support. The generation of such a well-resolved phylogeny is, however, not unusual when large datasets containing the information of numerous genes are used. Various previous studies have shown the value of this approach for resolving systematic questions at taxonomic ranks from the genus level and up (e.g., Zhang et al., 2011; Richards et al., 2014; Ormeno-Orrillo et al., 2015; Rahman et al., 2015). Our study thus adds to the growing body of work demonstrating how genome-informed taxonomic decisions represent more robust solutions than those based solely on 16S rRNA or conventional MLSA.
Based on our results, boundaries can for the first time be confidently demarcated for Burkholderia sensu stricto, Caballeronia and Paraburkholderia. These three genera, respectively, represent three of the five distinct lineages recovered among the Burkholderia sensu lato species. Burkholderia sensu stricto is represented by a large clade that includes the B. cepacia complex as well as the B. pseudomallei group, and consists primarily of pathogenic species, as suggested previously (Gyaneshwar et al., 2011; Sawana et al., 2014; Estrada-de los Santos et al., 2016). The Caballeronia clade includes environmental species that initially formed part of Transition Group 2 of Estrada-de los Santos et al. (2016) and that were transferred to the genus Caballeronia by Dobritsa and Samadpour (2016). This clade also includes all 13 of the recently described and validated Burkholderia glathei-like species (Oren and Garrity, 2016; Peeters et al., 2016). Based on these findings, we propose the formal inclusion of these species in the genus Caballeronia (sensu Dobritsa and Samadpour, 2016) (see Table 3 for details of the proposed new combinations). The inclusion of these taxa into Caballeronia raises the number of species to 25. Based on our analyses of their genomes, these species do not encode common nod or nif and fix loci, suggesting that none of the current Caballeronia species represent rhizobia or diazotrophs.
The Paraburkholderia clade is represented by diverse species, including both free-living and symbiotic diazotrophs, as well as environmental species. Although most of the taxa in this clade have already been formally transferred to Paraburkholderia (Sawana et al., 2014) and the novel combinations have been validated (Oren and Garrity, 2015a,b), this genus should also clearly include ‘P. acidipaludis’ (Aizawa et al., 2010) isolated from water chestnut as suggested by Sawana et al. (2014). This novel combination, however, still awaits validation. Interestingly, Paraburkholderia separates into two fully supported sub-clades, one including at least 23 species (spanning from P. caledonica to P. hospita in Figure 2) and the other including 11 species (P. kururiensis to P. sacchari in Figure 2). Although we could not identify any obvious reason for this split, future studies should explore its possible biological and taxonomic significance.
The two remaining lineages of Burkholderia sensu lato is represented by R. andropogonis [a pathogen of sorghum (Lopes-Santos et al., 2017)] and P. rhizoxinica [a member of Transition Group 1 of Estrada-de los Santos et al. (2016)]. Various previous studies have pointed out that these species should be excluded from Burkholderia sensu stricto, Caballeronia and/or Paraburkholderia (e.g., Estrada-de los Santos et al., 2013, 2016; Dobritsa and Samadpour, 2016). In fact, they have been suggested to represent new genera (Estrada-de los Santos et al., 2013; Dobritsa and Samadpour, 2016). This debate ultimately culminated in the introduction of the new genus Robbsia to accommodate R. andropogonis (Lopes-Santos et al., 2017). Based on our findings, the taxonomy of P. rhizoxinica requires similar revision. This species is definitely not a member of Paraburkholderia despite having been moved there from Burkholderia by Sawana et al. (2014). Both R. andropogonis and P. rhizoxinica currently represent the only members of their respective lineages for which whole genome sequences are available. Future studies should therefore seek to identify their respective congeneric species [some of which will likely include those in Transition Group 1 (Estrada-de los Santos et al., 2016)] and to understand the biological and evolutionary properties underlying these two lineages.
In addition to allowing unambiguous demarcation of the genera in Burkholderia sensu lato, this study also revealed, for the first time, the relationships among these taxa. Burkholderia sensu stricto has a well-supported sister group relationship with the clade containing Caballeronia, and Paraburkholderia. P. rhizoxinica is sister to the Burkholderia sensu stricto+Caballeronia+Paraburkholderia clade, while R. andropogonis occupies the most basal position in the tree. Knowledge about these relationships could inform hypotheses regarding the biology and evolution of these bacteria, especially in terms of virulence and pathogenicity. For example, Burkholderia sensu stricto primarily includes human and animal pathogens, while P. rhizoxinica and Robbsia are also represented by pathogens (Estrada-de los Santos et al., 2016; Lopes-Santos et al., 2017). Moreover, certain Caballeronia and Paraburkholderia species have also been isolated from clinical samples [e.g., ‘C. consitans’ and ‘C. turbans’ (Peeters et al., 2016) and P. fungorum (Coenye et al., 2001), and P. tropica (Deris et al., 2010), respectively]. The availability of a robust phylogenetic framework for these taxa would thus be invaluable for deciphering the processes and mechanisms involved in the evolution of these species.
Description of New Species Combinations
Description of Caballeronia arvi comb. nov.
Caballeronia arvi (ar’vi. L. gen. n. arvi of a field).
Basonym: Burkholderia arvi Peeters et al., 2016.
The description is as provided in Peeters et al. (2016). Analysis of 106 conserved protein-coding sequences have shown that this species is placed in the genus Caballeronia with very high support.
The type strain is LMG 29317T (= CCUG 68412T = MAN34T).
Description of Caballeronia arationis comb. nov.
Caballeronia arationis (a.ra.ti.o’nis. L. gen. n. arationis from a field).
Basonym: Burkholderia arationis Peeters et al., 2016.
The description is as provided in Peeters et al. (2016). Phylogenetic analysis of 106 conserved protein-coding loci clearly showed that there is high support for the placement of this species in Caballeronia.
The type strain is LMG 29324T (=CCUG 68405T).
Description of Caballeronia calidae comb. nov.
Caballeronia calidae (ca’li.dae. L. gen. n. calidae from warm water, because this strain was isolated from pond water in a tropical garden).
Basonym: Burkholderia calidae Peeters et al., 2016.
The description is as provided in Peeters et al. (2016). Phylogenetic analysis of 106 conserved protein-coding loci showed (with a high degree of certainty) that this species belongs in the genus Caballeronia.
The type strain is LMG 29321T (=CCUG 68408T).
Description of Caballeronia catudaia comb. nov.
Caballeronia catudaia (ca.tu.da’ia. Gr. adj. catudaios subterraneous; N. L. fem. adj. catudaia, earth-born).
Basonym: Burkholderia catudaia Peeters et al., 2016.
The description is as provided in Peeters et al. (2016). Our analyses of 106 conserved protein-coding loci clearly indicate that this species has high support for being included in Caballeronia.
The type strain is LMG 29318T (=CCUG 68411T).
Description of Caballeronia concitans comb. nov.
Caballeronia concitans (con.ci’tans. L. fem. part. pres. concitans disturbing, upsetting; because the isolation of this bacterium from human sources, including blood, further disturbs the image of this lineage of Burkholderia species as benign bacteria).
Basonym: Burkholderia concitans Peeters et al., 2016.
The description is as provided in Peeters et al. (2016). Analysis of 106 conserved protein-coding loci showed that this species has high support for belonging to the genus Caballeronia.
The type strain is LMG 29315T (=CCUG 68414T = AU12121T).
Description of Caballeronia fortuita comb. nov.
Caballeronia fortuita (for.tu.i’ta. L. fem. adj. fortuita accidental, unpremeditated; referring to its fortuitous isolation when searching for Burkholderia caledonica endophytes).
Basonym: Burkholderia fortuita Peeters et al., 2016.
The description is as described in Peeters et al. (2016). Our analysis of 106 conserved protein-coding loci clearly show this species is included in the genus Caballeronia.
The type strain is LMG 29320T (=CCUG 68409T).
Description of Caballeronia glebae comb. nov.
Caballeronia glebae (gle’bae. L. gen. n. glebae from a lump or clod of earth, soil).
Basonym: Burkholderia glebae Peeters et al., 2016.
The description appears in Peeters et al. (2016). Analysis of 106 conserved protein-coding loci shows high support for the placement of this species in the genus Caballeronia.
The type strain is LMG 29325T (=CCUG 68404T).
Description of Caballeronia hypogeia comb. nov.
Caballeronia hypogeia (hy.po.ge’ia. Gr. adj. hypogeios subterraneous; N. L. fem. adj. hypogeia, subterraneous, earth-born).
Basonym: Burkholderia hypogeia Peeters et al., 2016.
The description appears in Peeters et al. (2016). Our analysis of 106 conserved protein-coding loci supports the inclusion of this species into the genus Caballeronia.
The type strain is LMG 29322T (=CCUG 68407T).
Description of Caballeronia pedi comb. nov.
Caballeronia pedi (pe’di. Gr. n. pedon soil, earth; N. L. gen. n. pedi, from soil).
Basonym: Burkholderia pedi Peeters et al., 2016.
The description is listed in Peeters et al. (2016). The analysis of 106 conserved protein-coding loci, showed that this species is placed in Caballeronia.
The type strain is LMG 29323T (=CCUG 68406T).
Description of Caballeronia peredens comb. nov.
Caballeronia peredens (per.e’dens. L. fem. part. pres. peredens consuming, devouring; referring to the capacity of this bacterium to degrade fenitrothion).
Basonym: Burkholderia peredens Peeters et al., 2016.
The description is as discussed in Peeters et al. (2016). Our analysis of 106 conserved protein-coding loci clearly shows that this species should be included in the genus Caballeronia.
The type strain is LMG 29314T (=CCUG 68415T = NF100T).
Description of Caballeronia ptereochthonis comb. nov.
Caballeronia ptereochthonis (pte.re.o.chtho’nis Gr. n. pteris fern; Gr. n. chthon soil; N. L. gen. n. ptereochthonis, from fern soil).
Basonym: Burkholderia ptereochthonis Peeters et al., 2016.
The description appears in Peeters et al. (2016). The analysis of 106 conserved protein-coding loci clearly shows that this species should be included in Caballeronia.
The type strain is LMG 29326T (=CCUG 68403T).
Description of Caballeronia temeraria comb. nov.
Caballeronia temeraria (te.me.ra’ri.a. L. fem. adj. temeraria accidental, inconsiderate; referring to its accidental isolation when searching for Burkholderia caledonica endophytes).
Basonym: Burkholderia temeraria Peeters et al., 2016.
The description of this species appears in Peeters et al. (2016). The analysis of 106 conserved protein-coding loci here, shows that this species is included in Caballeronia with high support.
The type strain is LMG 29319T (=CCUG 68410T).
Description of Caballeronia turbans comb. nov.
Caballeronia turbans (tur’bans. L. fem. part. pres. turbans disturbing, agitating, because the isolation of this bacterium from human pleural fluid further disturbs the image of this lineage of Burkholderia species as benign bacteria).
Basonym: Burkholderia turbans Peeters et al., 2016.
The original species description appears in Peeters et al. (2016). Our analysis of 106 conserved protein-coding loci shows that this species forms part of Caballeronia.
The type strain is LMG 29316T (=CCUG 68413T = HI4065T).
Author Contributions
CB, MPa, PM, SV, and ES: Original concept; analyses; interpretation of results, writing and proofreading. WC, JA, and EvZ: Analyses, writing and proofreading. MH, AC, MPi, KP, NV, NM, DS, TR, CD, NS, VM, NI, NK, and TW: Genome analyses; interpretation of results; proofreading. JB and WW Genome analyses; interpretation of results; proofreading.
Funding
We thank the South African National Research Foundation and the Department of Science and Technology for the funding received via the Centre of Excellence programme. The work conducted by the United States Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the United States Department of Energy under Contract No. DE-AC02-05CH11231.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We also acknowledge the Bioinformatics and Computational Biology Unit of the University of Pretoria for access to their computational infrastructure.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01154/full#supplementary-material
Footnotes
- ^http://www.jgi.doe.gov
- ^https://github.com/lh3/wgsim
- ^https://edgar.computational.bio.uni-giessen.de
- ^http://www.geneious.com
- ^www.kegg.jp/blastkoala/
- ^https://www.ncbi.nlm.nih.gov/
References
Abascal, F., Zardoya, R., and Posada, D. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Aizawa, T., Ve, N. B., Vijarnsorn, P., Nakajima, M., and Sunairi, M. (2010). Burkholderia acidipaludis sp. nov., aluminium-tolerant bacteria isolated from Chinese water chestnut (Eleocharis dulcis) growing in highly acidic swamps in South-East Asia. Int. J. Syst. Evol. Microbiol. 60, 2036–2041. doi: 10.1099/ijs.0.018283-0
Andam, C. P., and Gogarten, J. P. (2011). Biased gene transfer in microbial evolution. Nat. Rev. Microbiol. 9, 543–555. doi: 10.1038/nrmicro2593
Bennet, J. S., Jolley, K. A., Earle, S. G., Corton, C., Bentley, S. D., Parkhill, J., et al. (2012). A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158, 1570–1580. doi: 10.1099/mic.0.056077-0
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bushnell, B. (2017). BBTools Software Package. Available at: http://sourceforge.net/projects/bbmap
Chan, J. Z.-M., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiology 12:302. doi: 10.1186/1471-2180-12-302
Coenye, T., Gevers, D., Van de Peer, Y., Vandamme, P., and Swings, J. (2005). Towards a prokaryotic genomic taxonomy. FEMS Microbiol. Rev. 29, 147–167. doi: 10.1016/j.fmrre.2004.11.004
Coenye, T., Laevens, S., Willems, A., Ohlén, M., Hannant, W., Govan, J. R. W., et al. (2001). Burkholderia fungorum sp. nov. and Burkholderia caledonica sp. nov., two new species isolated from the environment, animals and human clinical samples. Int. J. Syst. Evol. Microbiol. 51, 1099–1107. doi: 10.1099/00207713-51-3-1099
Cohen, O., Gophna, U., and Pupko, T. (2011). The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 28, 1481–1489. doi: 10.1093/molbev/msq333
Daubin, V., Gouy, M., and Perrière, G. (2002). A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 12, 1080–1090. doi: 10.1101/gr.187002
Depoorter, E., Bull, M. J., Peeters, C., Coenye, T., Vandamme, P., and Mahenthiralingam, E. (2016). Burkholderia: and update on taxonomy and biotechnological potential as antibiotic producers. Appl. Microbiol. Biotechnol. 100, 5215. doi: 10.1007/s00253-016-7520-x
Deris, Z. Z., Van Rostenberghe, H., Habsah, H., Noraida, R., Tan, G. C., Chan, Y. Y., et al. (2010). First isolation of Burkholderia tropica from a neonatal patient successfully treated with imipenem. Int. J. Infect. Dis. 14, e73–e74. doi: 10.1016/j.ijid.2009.03.005
Desper, R., and Gascuel, O. (2002). Fast and accurate phylogeny reconstruction algorithms based on the Minimum-Evolution Principle. J. Comput. Biol. 9, 687–705. doi: 10.1089/106652702761034136
Dobritsa, A. P., and Samadpour, M. (2016). Transfer of eleven Burkholderia species to the genus Paraburkholderia and proposal of Caballeronia gen. nov., a new genus to accommodate twelve species of Burkholderia and Paraburkholderia. Int. J. Syst. Evol. Microbiol. 66, 2836–2846. doi: 10.1099/ijsem.0.001065
Eberl, L., and Vandamme, P. (2016). Members of the genus Burkholderia: good and bad guys. F1000Res. 5:1007. doi: 10.12688/f1000research.8221.1
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Estrada-de los Santos, P., Rojas-Rojas, F. U., Tapia-García, E. Y., Vásquez-Murrieta, M. S., and Hirsch, A. M. (2016). To split or not to split: an opinion on dividing the genus Burkholderia. Ann. Microbiol. 66, 1303–1314. doi: 10.1007/s13213-015-1183-1
Estrada-de los Santos, P., Vinuesa, P., Martínez-Aguilar, L., Hirsch, A. M., and Caballero-Mellado, J. (2013). Phylogenetic analysis of Burkholderia species by multilocus sequence analysis. Curr. Microbiol. 67, 51–60. doi: 10.1007/s00284-013-0330-9
Galtier, N., and Daubin, V. (2008). Dealing with incongruence in phylogenomic analyses. Philos. Tans. R. Soc. B 363, 4023–4029. doi: 10.1098/rstb.2008.0144
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Gillis, M., Van Van, T., Bardin, R., Goor, M., Hebbar, P., Willems, A., et al. (1995). Polyphasic taxonomy in the genus Burkholderia leading to an emended description of the genus and proposition of Burkholderia vietnamiensis sp. nov. for N2-fixing isolates from rice in Vietnam. Int. J. Syst. Bacteriol. 45, 274–289. doi: 10.1099/00207713-45-2-274
Gnerre, S., and MacCallum, I. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Goris, J., Dejonghe, W., Falsen, E., De Clerck, E., Geeraerts, B., Willems, A., et al. (2002). Diversity of transconjugants that acquired plasmid pJP4 or pEMT1 after inoculation of a donor strain in the A- and B-horizon of an agricultural soil and description of Burkholderia hospita sp. nov. and Burkholderia terricola sp. nov. Syst. Appl. Microbiol. 25, 340–352. doi: 10.1078/0723-2020-00134
Gyaneshwar, P., Hirsch, A. M., Moulin, L., Chen, W.-M., Elliott, G. N., Bontemps, C., et al. (2011). Legume-nodulating Betaproteobacteria: diversity, host range, and future prospects. Mol. Plant Microbe Interact. 24, 1276–1288. doi: 10.1094/MPMI-06-11-0172
Huntemann, M., Ivanova, N. N., Mavromatis, K., Tripp, H. J., Paez-Espino, D., Palaniappan, K., et al. (2015). The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4.). Stand. Genomic Sci. 10, 86. doi: 10.1186/S40793-015-0077-y
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Lar-imer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jain, R., Rivera, M. C., and Lake, J. A. (1999). Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. U.S.A. 96, 3801–3806. doi: 10.1073/pnas.96.7.3801
Jeffroy, O., Brinkmann, H., Delsuc, F. D. R., and Philippe, H. (2006). Phylogenomics: the beginning of incongruence? Trends Genet. 22, 225–231. doi: 10.1016/j.tig.2006.02.003
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKoala: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Koonin, E. V. (2005). Orthologs, paralogs and evolutionary genomics. Annu. Rev. Genet. 39, 309–338. doi: 10.1146/annurev.genet.39.073003.114725
Kuck, P., and Longo, G. (2014). FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 11:81. doi: 10.1186/s12983-014-0081-x
Lopes-Santos, L., Castro, D. B. A., Ferreira-Tonin, M., Corrêa, D. B. A., Weir, B. S., Park, D., et al. (2017). Reassessment of the taxonomic position of Burkholderia andropogonis and description of Robbsia andropogonis gen. nov., comb. nov. Antonie Van Leeuwenhoek 110, 727–736. doi: 10.1007/s10482-017-0842-6
Markowitz, V. M., Chen, I. M., Palaniappan, K., Chu, K., Szeto, E., Pillay, M., et al. (2014). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567. doi: 10.1093/nar/gkt963
Martínez-Aguilar, L., Salazar-Salazar, C., Méndez, R. D., Caballero-Mellado, J., Hirsch, A. M., Vásquez-Murrieta, M. S., et al. (2013). Burkholderia caballeronis sp. nov., a nitrogen fixing species isolated from tomato (Lycopersicon esculentum) with the ability to effectively nodulate Phaseolus vulgaris. Antonie Van Leeuwenhoek 104, 1063–1071. doi: 10.1007/s10482-013-0028-9
Mavengere, N. R., Ellis, A. G., and Le Roux, J. J. (2014). Burkholderia aspalathi sp. nov., isolated from root nodules of the South African legume Aspalathus abietina Thunb. Int. J. Syst. Evol. Microbiol. 64, 1906–1912. doi: 10.1099/ijs.0.057067-0
Oren, A., and Garrity, G. M. (2015a). List of new names and new combinations previously effectively, but not validly, published. Validation List no. 164. Int. J. Syst. Evol. Microbiol. 65, 2017–2025. doi: 10.1099/ijs.0.000317
Oren, A., and Garrity, G. M. (2015b). List of new names and new combinations previously effectively, but not validly, published. Validation List no. 165. Int. J. Syst. Evol. Microbiol. 66, 2777–2783. doi: 10.1099/ijsem.0.000464
Oren, A., and Garrity, G. M. (2016). List of new names and new combinations previously effectively, but not validly, published. Validation List No. 171. Int. J. Syst. Evol. Microbiol. 66, 3761–3764. doi: 10.1099/ijsem.0.001321
Oren, A., and Garrity, G. M. (2017). List of new names and new combinations previously effectively, but not validly, published. Validation List No. 173. Int. J. Syst. Evol. Microbiol. 67, 1–3. doi: 10.1099/ijsem.0.001733
Ormeno-Orrillo, E., Servín-Garcidueñas, L. E., Rogel, M. A., González, V., Peralta, H., Mora, J., et al. (2015). Taxonomy of rhizobia and agrobacteria from the Rhizobiaceae family in light of genomics. Syst. Appl. Microbiol. 38, 287–291. doi: 10.1016/j.syapm.2014.12.002
Palmer, M., Steenkamp, E. T., Coetzee, M. P. A., Chan, W.-Y., van Zyl, E., De Maayer, P., et al. (2017). Phylogenomic resolution of the bacterial genus Pantoea and its relationship with Erwinia and Tatumella. Antonie Van Leeuwenhoek doi: 10.1007/s10482-017-0852-4
Peeters, C., Meier-Kolthoff, J. P., Verheyde, B., De Brandt, E., Cooper, V. S., and Vandamme, P. (2016). Phylogenomic study of Burkholderia glathei-like organisms, proposal of 13 novel Burkholderia species and emended descriptions of Burkholderia sordidicola, Burkholderia zhejiangensis, and Burkholderia grimmiae. Front. Microbiol. 7:877. doi: 10.3389/fmicb.2016.00877
Philippe, H., Brinkmann, H., Lavrov, D. V., Littlewood, D. T. J., Manuel, M., Wörheide, G., et al. (2011). Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9:e1000602. doi: 10.1371/journal.pbio.1000602
Philippe, H., and Forterre, P. (1999). The rooting of the universal tree of life is not reliable. J. Mol. Evol. 49, 509–523. doi: 10.1007/PL00006573
Rahman, N. A., Parks, D. H., Vanwonterghem, I., Morrison, M., Tyson, G. W., and Hugenholtz, P. (2015). A phylogenomic analysis of the bacterial Phylum Fibrobacteres. Front. Microbiol. 6:1469. doi: 10.3389/fmicb.2015.01469
Reis, V. M., Estrada-de los Santos, P., Tenorio-Salgado, S., Vogel, J., Stoffels, M., Guyon, S., et al. (2004). Burkholderia tropica sp. nov., a novel nitrogen-fixing, plant-associated bacterium. Int. J. Syst. Evol. Microbiol. 54, 2155–2162. doi: 10.1099/ijs.0.02879-0
Richards, V. P., Palmer, S. R., Bitar, P. D. P., Qin, X., Weinstock, G. M., Highlander, S. K., et al. (2014). Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Boil. Evol. 6, 741–753. doi: 10.1093/gbe/evu048
Sawana, A., Adeolu, M., and Gupta, R. S. (2014). Molecular signatures and phylogenomic analysis of the genus Burkholderia: proposal for division of this genus into the emended genus Burkholderia containing pathogenic organisms and a new genus Paraburkholderia gen. nov. harbouring environmental species. Front. Genet. 5:429. doi: 10.3389/fgene.2014.00429
Sheu, S.-Y., Chou, J.-H., Bontemps, C., Elliott, G. N., Gross, E., dos Reis Junior, F. B., et al. (2013). Burkholderia diazotrophica sp. nov., isolated from root nodules of Mimosa spp. Int. J. Syst. Evol. Microbiol. 63, 435–441. doi: 10.1099/ijs.0.039859-0
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steenkamp, E. T., van Zyl, E., Beukes, C. W., Avontuur, J. R., Chan, W. Y., Palmer, M., et al. (2015). Burkholderia kirstenboschensis sp. nov. nodulates papilionoid legumes indigenous to South Africa. Syst. Appl. Microbiol. 38, 545–554. doi: 10.1016/j.syapm.2015.09.003
Suárez-Moreno, Z. R., Caballero-Mellado, J., Coutinho, B. G., Mendonça-Previato, L., James, E. K., and Venturi, V. (2012). Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb. Ecol. 63, 249–266. doi: 10.1007/s00248-011-9929-1
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tavaré, S. (1986). “Some probabilistic and statistical problems in the analysis of DNA sequences,” in Lectures on Mathematics in the Life Sciences, Vol. 17ed. R. M. Miura (Providence: American Mathematical Society), 57–86.
Vanlaere, E., van der Meer, J. R., Falsen, E., Salles, J. F., de Brandt, E., and Vandamme, P. (2008). Burkholderia sartisoli sp. nov., isolated from a polycyclic aromatic hydrocarbon-contaminated soil. Int. J. Syst. Evol. Microbiol. 58, 420–423. doi: 10.1099/ijs.0.65451-0
Viallard, V., Poirier, I., Cournoyer, B., Haurat, J., Wiebkin, S., Ophel-Keller, K., et al. (1998). Burkholderia graminis sp. nov., a rhizospheric Burkholderia species, and reassessment of [Pseudomonas] phenazinium, [Pseudomonas] pyrrocinia and [Pseudomonas] glathei as Burkholderia. Int. J. Syst. Bacteriol. 48, 549–563. doi: 10.1099/00207713-48-2-549
Whitman, W. B., Woyke, T., Klenk, H.-P., Zhou, Y., Lilburn, T. G., Beck, B. J., et al. (2015). Genomic encyclopedia of bacterial and archaeal type strains, phase III: the genomes of soil and plant-associated and newly described type strains. Stand. Genomic Sci. 10:26. doi: 10.1186/s40793-015-0017-x
Xia, X., and Xie, Z. (2001). DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373. doi: 10.1093/jhered/92.4.371
Xia, X., Xie, Z., Salemi, M., Chen, L., and Wang, Y. (2003). An index of substitution saturation and its application. Mol. Phylogenet. Evol. 26, 1–7. doi: 10.1016/S1055-7903(02)00326-3
Yabuuchi, E., Kosako, Y., Oyaizu, H., Yano, I., Hotta, H., Hashimoto, Y., et al. (1992). Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol. Immunol. 36, 1251–1275. doi: 10.1111/j.1348-0421.1992.tb02129.x
Yabuuchi, E., Kosako, Y., Yano, I., Hotta, H., and Nishiuchi, Y. (1995). Transfer of two Burkholderia and an Alcaligenes species to Ralstonia gen. nov.: Proposal of Ralstonia pickettii (Ralston, Palleroni and Doudoroff 1973) comb. nov., Ralstonia solanacearum (Smith 1896) comb. nov. and Ralstonia eutropha (Davis 1969) comb. nov. Microbiol. Immunol. 39, 897–904. doi: 10.111/j.1348-0421.1995.tb03275.x
Zerbino, D., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Burkholderia, Paraburkholderia, Caballeronia, phylogenomics, Robbsia andropogonis, Burkholderia rhizoxinica
Citation: Beukes CW, Palmer M, Manyaka P, Chan WY, Avontuur JR, van Zyl E, Huntemann M, Clum A, Pillay M, Palaniappan K, Varghese N, Mikhailova N, Stamatis D, Reddy TBK, Daum C, Shapiro N, Markowitz V, Ivanova N, Kyrpides N, Woyke T, Blom J, Whitman WB, Venter SN and Steenkamp ET (2017) Genome Data Provides High Support for Generic Boundaries in Burkholderia Sensu Lato. Front. Microbiol. 8:1154. doi: 10.3389/fmicb.2017.01154
Received: 20 April 2017; Accepted: 07 June 2017;
Published: 26 June 2017.
Edited by:
Sabela Balboa Méndez, Universidade de Santiago de Compostela, SpainReviewed by:
Paulina Estrada De Los Santos, Instituto Politécnico Nacional, MexicoRadhey S. Gupta, McMaster University, Canada
Copyright © 2017 Beukes, Palmer, Manyaka, Chan, Avontuur, van Zyl, Huntemann, Clum, Pillay, Palaniappan, Varghese, Mikhailova, Stamatis, Reddy, Daum, Shapiro, Markowitz, Ivanova, Kyrpides, Woyke, Blom, Whitman, Venter and Steenkamp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanus N. Venter, ZmFudXMudmVudGVyQHVwLmFjLnph