 Wayne Dawson
Wayne Dawson Jens Hör3†
Jens Hör3† Mark van Kleunen
Mark van Kleunen Michael Pester
Michael Pester- 1Department of Biosciences, Durham University, Durham, United Kingdom
- 2Department of Biology, University of Konstanz, Konstanz, Germany
- 3Faculty of Medical and Life Sciences, Institute of Precision Medicine, Furtwangen University, Villingen-Schwenningen, Germany
Plant growth can be affected by soil bacteria. In turn, plants are known to influence soil bacteria through rhizodeposits and changes in abiotic conditions. We aimed to quantify the phylotype richness and relative abundance of rhizosphere bacteria that are actually influenced in a plant species-specific manner and to determine the role of the disproportionately large diversity of low-abundance bacteria belonging to the rare biosphere (<0.1 relative abundance) in this process. In addition, we aimed to determine whether plant phylogeny has an influence on the plant species-specific rhizosphere bacterial community. For this purpose, 19 herbaceous plant species from five different plant orders were grown in a common soil substrate. Bacterial communities in the initial soil substrate and the established rhizosphere soils were compared by 16S rRNA gene amplicon sequencing. Only a small number of bacterial operational taxonomic units (OTUs, 97% sequence identity) responded either positively (ca. 1%) or negatively (ca. 1%) to a specific plant species. On average, 91% of plant-specific positive response OTUs comprised bacteria belonging to the rare biosphere, highlighting that low-abundance populations are metabolically active in the rhizosphere. In addition, low-abundance OTUs were in terms of their summed relative abundance major drivers of the bacterial phyla composition across the rhizosphere of all tested plant species. However, no effect of plant phylogeny could be observed on the established rhizosphere bacterial communities, neither when considering differences in the overall established rhizosphere communities nor when considering plant species-specific responders only. Our study provides a quantitative assessment of the effect of plants on their rhizosphere bacteria across multiple plant orders. Plant species-specific effects on soil bacterial communities involved only 18–111 bacterial OTUs out of several 1000s; this minority may potentially impact plant growth in plant–bacteria interactions.
Introduction
The performance and growth of plants is tightly connected to chemical, physical, and biological properties of the inhabited soil. In turn, plants directly influence abiotic and biotic soil properties (e.g., van der Putten et al., 2013). For example, plants invest 11–40% of their photosynthetically fixed carbon and 10–16% of total plant nitrogen into rhizodeposits that modulate the microbiota in their close root vicinity (Berendsen et al., 2012; Bulgarelli et al., 2013). Such rhizodeposits include inorganic ions, organic acids, sugars, purines, and nucleosides but also secondary metabolites like vitamins, phytosiderophores, antibiotics, and quorum-sensing mediating compounds (Berendsen et al., 2012; Bulgarelli et al., 2013). In addition, plant roots modulate their microbiota by influencing the soil pH, soil structure and oxygen availability of their surrounding soil (Marschner et al., 1986; Dennis et al., 2010).
Through these mechanisms, several plant species including Arabidopsis thaliana (thale cress), Zea mays (maize), Solanum tuberosum (potato), and Hordeum vulgare (barley) (Weinert et al., 2011; Bulgarelli et al., 2012, 2015; Lundberg et al., 2012; Peiffer et al., 2013) have been shown to modify the composition and activity of the soil microbiota in close proximity to their roots (rhizosphere) and directly on or inside their roots (Bulgarelli et al., 2013; Philippot et al., 2013). For example, symbionts such as the N2-fixing Bradyrhizobium japonicum and pathogens such as Phytophthora sojae are attracted by specific plant flavonoids (Philippot et al., 2013). In more general terms, a step-wise selection model has been proposed for root microbiota differentiation (Bulgarelli et al., 2013; Reinhold-Hurek et al., 2015). In the initial soil, edaphic factors (e.g., soil pH and structure) determine the composition of the soil microbial community. The first selection step takes place in the rhizosphere, the thin soil layer directly surrounding the roots, where rhizodeposits and root cell wall features promote the growth of mainly organotrophic microorganisms and thereby initiate a shift in the soil microbiome. In a second and third step, a host genotype-dependent selection takes place close to and within the root corpus, respectively, and thereby fine-tunes community profiles thriving on the rhizoplane (i.e., the root surface) and within plant roots (Bulgarelli et al., 2013; Reinhold-Hurek et al., 2015).
The bacterial domain harbors an exceptionally high species richness among soil microorganisms (Curtis et al., 2002; Schloss et al., 2016) as compared to archaea (Schloss et al., 2016) and fungi (Bridge and Spooner, 2001; Fierer et al., 2007). The question whether plant species differentiate their root bacteria in a species-specific manner has been addressed in various studies over the past decades. Here, cultivation efforts as well as molecular fingerprinting techniques such as DGGE (denaturing gradient gel electrophoresis), T-RFLP (terminal restriction fragment length polymorphism) or PLFA (phospholipid-derived fatty acids) analysis revealed that different plant species can indeed select for specific rhizosphere bacteria (e.g., Miethling et al., 2000; Smalla et al., 2001; Garbeva et al., 2008; Berg and Smalla, 2009; Bezemer et al., 2010; Pendergast et al., 2013). However, only few studies extended this knowledge so far to a more complete census of the plant species-specific rhizosphere bacterial community using modern next-generation amplicon sequencing approaches that target the phylogenetic breadth of most bacteria at sufficient sampling depth (Rodríguez-Echeverría et al., 2013; Rosenzweig et al., 2013; Hortal et al., 2015). Furthermore, studies on the role of low-abundance bacterial populations in shaping the plant species-specific root bacteriome are still scarce (Dohrmann et al., 2013; Nuccio et al., 2016; Saleem et al., 2016; Shi et al., 2016).
Next-generation amplicon sequencing can generate several millions of sequences over several 100 multiplexed samples (Caporaso et al., 2011, 2012). This approach was a breakthrough to address microbial ecology questions in a (semi-)quantitative and well-replicated manner, especially for highly complex bacterial communities such as those inhabiting soil environments. Soils are typically inhabited by several 1000s of bacterial species, which exhibit a continuous but highly skewed rank abundance (e.g., Lauber et al., 2009; Nemergut et al., 2010; Hausmann et al., 2016; Wörner et al., 2016). A minor proportion of these taxa are very or moderately abundant (≥1 and ≥0.1% relative abundance, respectively) and are typically thought to be responsible for major ecosystem functions. The remaining major part of these species have an individual relative abundance of <0.1% and are defined as the rare biosphere (Sogin et al., 2006), which is typically thought of as a ‘seed’ bank for the recruitment of dormant cells to become metabolically active and numerically dominant after environmental conditions change (Pedrós-Alió, 2012; Lynch and Neufeld, 2015). However, not all low-abundance bacteria are metabolically inactive and there are a few documented cases where they even can fulfill important ecosystem functions, e.g., as N2-fixing bacteria in the ocean (Großkopf et al., 2012) or as sulfate-reducing bacteria in peatland soils (Pester et al., 2010; Hausmann et al., 2016). For soil environments, additional ecosystem functions have been demonstrated for bacterial populations of low relative abundance (reviewed in Jousset et al., 2017). For example, removal of low-abundance species from bulk soil using dilution-to-extinction had a positive effect on the establishment of newly introduced species, suggesting that low-abundance species pre-occupy ecological niches and thus slow down invasion by incoming species (van Elsas et al., 2012; Vivant et al., 2013; Mallon et al., 2015). Soil microorganisms of low relative abundance were also shown to play a role in community-wide species interactions, e.g., by being involved in the production of antifungal compounds that protect plants from pathogens (Hol et al., 2015). However, how taxa within this rare biosphere respond to growth of different plant species is not well-understood.
To assess how the composition of the rhizosphere bacterial community is shaped by different plant species, we used 19 herbaceous plant species grown independently in a common soil substrate. Our objectives were to (i) quantify how many species-level bacterial taxa respond in a plant-species specific manner, (ii) address the role of the rare biosphere in shaping the rhizosphere bacterial community, and (iii) determine whether plant phylogeny has an influence on the rhizosphere bacterial community.
Materials and Methods
Plant Mesocosms
We used 19 herbaceous, mostly perennial grassland species (Table 1) that had been previously analyzed in a plant-soil feedback experiment for their relationship between specific growth rates and biotic soil effects by Lemmermeyer et al. (2015). Seeds were collected in 2012 in Switzerland and southern Germany (both in the vicinity of Konstanz) from 3 to 10 mother plants per species originating from one population. Equal numbers of seeds per mother plant were then pooled to get one bulk sample per species.
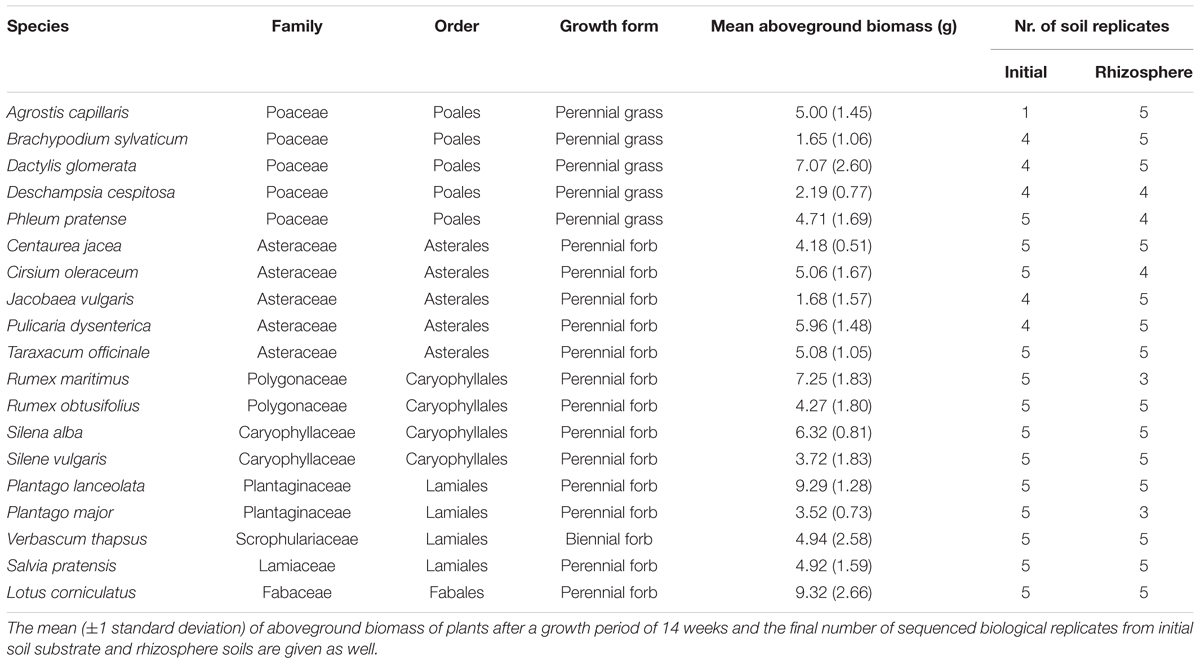
TABLE 1. Overview on the taxonomy and growth form of the 19 studied grassland plant species.
In May 2013, 100 seeds were sown in five 4.5-L pots per species, giving five independent replicates per plant species. The substrate in the pots consisted of 4 L of a mixture of topsoil (‘Rasenerde,’ Ökohum GmbH, Herbertingen, Germany), vermiculite (type ‘palabora,’ 0–1 mm, Deutsche Vermiculite Dämmstoff GmbH, Sprockhövel, Germany) and washed sand (Type 22200, 0.3–0.8 mm, Emil Steidle GmbH & Co. KG, Sigmaringen, Germany) (ratio of 1:1:1 by volume). With the aim of providing the plants with an inoculum of a soil microbial community native to the plant species, we mixed in 200 ml of homogenized soil collected from seven grassland areas in the vicinity of Konstanz (coordinates: 47°41′26″ N, 9°11′27″ E; 47°41′18″ N, 9°11′28″ E; 47°41′16″ N, 9°11′34″ E; 47°41′13″ N, 9°11′18″ E; 47°41′10″ N, 9°11′26″ E; 47°40′59″ N, 9°11′25″ E; 47°40′39″ N, 9°11′46″ E). The soils in this region are brown earths, and predominantly loamy in texture (Hartwich et al., 2013). Here, soil of approximately 0–10 cm depth was obtained from 12 to 20 points located at regular intervals (5 m) along line transects in each grassland area. This yielded a total of ∼40 L of soil, which was bulked, sieved (using a 5-mm mesh to remove plant roots and stones) and mixed thoroughly before mixing it with the rest of the substrate in the pots. This gave a total of 95 pots, all of which initially received the same homogenized soil substrate mixture. Before sowing seeds of the 19 species into five replicate pots each, every pot was assigned to one of the species as a replicate, and a sample of the prepared soil substrate was taken from the center of each pot to a depth of 10 cm using a sterile 2-cm diameter soil corer. Each of these initial soil substrate samples was stored individually at -80°C until DNA extraction (see below). The hole was then filled in with surrounding soil substrate, and seeds were sown.
After germination, seedlings were thinned to a constant density of five plants per pot. Resulting plants were then grown outside in the botanical garden of the University of Konstanz initially for 10 weeks. Natural rainfall was supplemented with watering during dry periods. For an additional 4 weeks, pots were placed in a glasshouse to promote further growth until 17–18 September 2013. Throughout this period, newly germinating seedlings of target and non-target plants were constantly removed. At the end of the experiment, the aboveground biomass per pot was harvested, and pot contents were removed immediately. Aboveground biomass was dried at 70°C for 72 h and later weighed to an accuracy of 1 mg. We carefully separated the roots from the bulk of the soil substrate, then gently shook roots to dislodge attached soil (rhizosphere soil), and collected the rhizosphere-soil samples (one per individual pot, 10–20 ml in volume) into sterile 50-mL conical centrifuge tubes (Carl Roth GmbH, Karlsruhe, Germany). These were stored at -80°C until required for DNA extraction.
DNA Extraction and Amplicon Sequencing
We extracted DNA from 0.25 g of each initial soil substrate and rhizosphere soil sample using the PowerSoil DNA Isolation Kit (MOBIO Laboratories, Inc., Carlsbad, CA, United States), following the manufacturer’s instructions. To characterize the bacterial community of each sample, 16S rRNA genes were amplified according to the manual of the NEXTflexTM 16S V1–V3 amplicon sequencing kit (Bioo Scientific, Austin, TX, United States), which included the preparation of a multiplexed amplicon library for paired-end sequencing on the Illumina® MiSeq platform. In brief, 25 ng of total extracted DNA were used as template for amplifying the V1–V3 region of bacterial 16S rRNA genes using the universal primers 8f (5′-AGAGTTTGATCCTGGCTCAG-3′) and 536r (5′-GTATTACCGCGGCTGCTGG-3′) that were elongated by oligonucleotide adaptors for subsequent PCR barcoding. This PCR consisted of an initial denaturation at 98°C for 4 min followed by 10 cycles of denaturation at 98°C for 30 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s with a final elongation for 4 min at 72°C. PCR products were purified using Agencourt AMPure XP Magnetic beads (Beckman Coulter, Indianapolis, IN, United States) and used as template in a subsequent PCR with 14 cycles to introduce barcodes and Illumina sequencing adaptors according to the manufacturer’s instructions. PCR conditions were the same as outlined for the first PCR. Amplicon sequencing was performed at Furtwangen University on the Illumina MiSeq platform using paired-end sequencing (2 × 300 bp) based on the MiSeq Reagents kit v3.
Amplicon reads were processed in mothur v.1.34.4 (Schloss et al., 2009) including quality control, removal of unique singletons, de novo chimera filtering using UCHIME (Edgar et al., 2011), de novo clustering of operational taxonomic units (OTUs) at 97% sequence identity (approximate species level), and removal of OTUs with fewer than five reads to discriminate against sequencing artifacts and sample bleeding (Mitra et al., 2015). This resulted in 6.1 million high-quality paired-end reads forming 18838 OTUs. The library sizes had on average 34552 paired-end reads per sample, with 90% of all samples having library sizes between 22514 and 53707 paired-end reads (5 and 95% quantiles, respectively). Good’s coverage (Good, 1953) of processed reads was on average 95%, with 90% of all samples having a coverage between 94 and 98% (5 and 95% quantiles, respectively). Individual sequences were classified with the Bayesian classifier and the RDP 16S rRNA training set 14 (Cole et al., 2014) as implemented in mothur using a k-mer size of 8 and a confidence threshold of 80. Illumina sequences were deposited at the Sequence Read Archive at NCBI under accession number SRP081003.
Quantitative PCR
A quantitative PCR (qPCR) assay targeting most bacteria and archaea was performed following the procedure described by Hausmann et al. (2016) using the universal primers 1389F (5′-TGYACACACCGCCCGT-3′) and modified 1492R (5′-NTACCTTGTTACGACT-3′). qPCRs were performed in 20-μl reactions on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) using the GoTaq qPCR Master Mix (Promega, Madison, WI, United States), bovine serum albumine (10 ng μl-1), 0.8–6.3 ng of template DNA and the following primer end-concentrations: 1389F (0.75 μM) and 1492R (1.00 μM). Thermal cycling was carried out by an initial denaturation step at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and elongation at 72°C for 60 s. A standard curve with a purified 16S rRNA gene-PCR product of a Nitrosomonas urea-clone was generated from 1.84 × 102 to 1.84 × 109 template copies per assay (R2 = 0.993–0.998) in a background of 5 ng of purified λ-phage DNA cI857 Sam7 (Promega). PCR efficiencies of the assays ranged from 83 to 91%. qPCR products were regularly checked for specificity by (i) comparative melting-curve analyses of the sample-derived PCR products and the respective reference-derived PCR products and (ii) agarose gel electrophoresis. Partial inhibition of the qPCR was not evident when testing the serial dilution of DNA extracted from a representative soil sample. To estimate the size of overall bacterial and archaeal populations inhabiting the analyzed soils, the retrieved 16S rRNA gene copies per ng total DNA were normalized against the amount of DNA extracted from 250 mg of soil. We assumed a DNA extraction efficiency of 100%.
Statistical Analysis
All analyses were performed using the program R, versions 3.1.1 and 3.3.0 (R Core Team, 2015). Only OTUs that were detected by at least three reads in three different soil replicates were used to further discriminate against sequencing artifacts and to enable meaningful statistical analysis. Alpha diversity metrics were calculated in the R package phyloseq 1.8.2 (McMurdie and Holmes, 2013) based on uniform library sizes of 16417 reads as obtained by rarefaction back-sampling. Differences in bacterial community composition were visualized using non-metric multidimensional scaling (NMDS) plots made in phyloseq using Bray–Curtis dissimilarities and relative abundance data normalized by the number of reads per soil replicate. Variation in bacterial community composition explained by plant-species identity, sampling time (initial soil substrate or rhizosphere soil), and the interaction between them was tested for significance using the Bray–Curtis dissimilarities and permutational analysis of variance (PERMANOVA, function ‘adonis’ in the R package ‘vegan’ version 2.4-1; Oksanen et al., 2013). The level of dissimilarity among sample communities was compared to a distribution of dissimilarities obtained from 9999 randomisations. Agrostis capillaris was excluded from this analysis due to an insufficient number of soil replicates from the initial soil substrate. To assess whether greater plant growth and biomass accumulation resulted in greater shifts in rhizosphere bacterial community composition, we performed another PERMANOVA using Bray–Curtis dissimilarities as above, but only for the rhizosphere soils, and with aboveground biomass as the explanatory variable and species as a random effect.
Differences in variability of bacterial community compositions among samples within groups (initial soil substrate or rhizosphere soils of each species) were assessed using the ‘betadisper’ function in ‘vegan.’ This was done by measuring the distance in multivariate space between each group’s sample community compositions and the group’s centroid. The amount of heterogeneity among plant species in their bacterial community dispersion as well as between initial soil substrate and rhizosphere soils was compared to that obtained from 9999 randomisations using the function ‘permutest.’ A significant p-value (p < 0.05) would indicate that the within-group variation of bacterial community composition differs significantly among groups.
To test for differential abundance of OTUs in the initial soil substrate versus rhizosphere soils, we used the general linear model approach in combination with quasi-likelihood shrunken dispersion estimates of the R package edgeR (Lund et al., 2012). This analysis was based on RLE (relative log expression) normalized abundance data (Anders and Huber, 2010). OTUs were considered to respond significantly in terms of their relative abundance change if their false detection rate-corrected p-value was below 0.01 (i.e., positive and negative responses increased and decreased significantly in relative abundance, respectively). These tests effectively represent paired tests comparing OTU abundances in initial soil substrate versus rhizosphere soils from the same pots. We repeated the PERMANOVA described above for the subsets of bacterial OTUs that responded either commonly to all plant species or specifically to one plant species only using the rhizosphere soils. We did this to assess how much variation in these subsets was explained by plant identity.
Finally, in order to assess whether similarity in rhizosphere bacterial community composition among samples was correlated with phylogenetic similarity among plants, we applied a Mantel test (with a Spearman’s correlation coefficient) on the phylogenetic correlation matrix of the plants, and a Bray–Curtis dissimilarity matrix of the rhizosphere bacterial community. The phylogeny was created by pruning the aged phylogeny of European plant species (Durka and Michalski, 2012), and by adding extra branches representing the replicate samples within each species. Two Mantel tests were performed: one with a rhizosphere bacterial community dissimilarity matrix based on all OTUs, and a second based only on the plant species-specific responding OTUs. This tests involved 9999 random permutations of rows in the dissimilarity matrix, producing a null distribution of correlations to assess observed correlation significance. The phylogeny was built using the package ‘ape’ version 3.5 (Paradis et al., 2004), and the Mantel test was performed using the function ‘mantel’ in the package ‘vegan’ version 2.4-1 (Oksanen et al., 2013).
Results
Plant Species Identity Is a Minor Driver of Bacterial Rhizosphere Communities
We used next-generation amplicon sequencing of bacterial 16S rRNA genes (V1–V3 region) to assess the influence of 19 different herbaceous plant species on their own rhizosphere bacterial community. This was contrasted with the initial soil substrate that was used to grow these plants. To maximize detection of plant species influence and to provide equal starting conditions to all plants, this initial soil substrate was made of two components. The major component was a mixture of commercial topsoil, vermiculite and washed sand (ratio 1:1:1), which was foreign to all plant species and used as a background substrate. The minor component was a small inoculum of homogenized soil native to the investigated plant species.
Bacterial alpha diversity in the rhizosphere of plants was clearly different from that in the initial soil substrate irrespective of the plant species (Supplementary Figure S1). When rarefied to an equal sampling depth of 16417 reads per replicate, soils harbored on average 2181 ± 246 (mean ± SD) Chao1-estimated OTUs (97% sequence identity) in the initial soil substrate, which slightly but significantly increased to an average of 2727 ± 286 OTUs in rhizosphere soils (ANOVA: F1,136 = 194.97, p < 0.001). Incubation time explained 58% of the observed variation in OTU richness, while 13% of the variation could be attributed to individual plant species (ANOVA: F17,136 = 2.41, p < 0.01). There was no significant interaction between incubation per se and plant species. There was also a trend toward higher bacterial diversity in rhizosphere soils compared to the initial soil substrate as indicated by the Shannon–Wiener and Simpson’s indices (Supplementary Figure S1). The observed increase in alpha diversity was paralleled by a trend toward larger bacterial and archaeal 16S rRNA gene copy numbers in the rhizosphere soils (Supplementary Table S1), as revealed by qPCR analysis across seven selected plant species representing the five analyzed plant orders.
The initial soil substrate and rhizosphere soils were dominated by bacteria belonging to the phyla Proteobacteria (classes Alpha-, Beta-, Gamma-, and Deltaproteobacteria), Bacteroidetes, Chloroflexi, Parcubacteria, Actinobacteria, Acidobacteria, Verrucomicrobia, Planctomycetes or unclassified bacteria, with each of these phyla constituting more than 1% relative abundance of the detected 16S rRNA genes (Figure 1 and Supplementary Figure S2). Major relative abundance differences in the rhizosphere soils as compared to the initial soil substrate were evident in a massive decrease of Bacteroidetes (from 31.4 ± 3.6% to 9.7 ± 1.0%) and Tenericutes (from 2.4 ± 1.0% to 0.01 ± 0.00%), which was paralleled by an increase in Acidobacteria (from 1.4 ± 0.3% to 3.6 ± 0.3%), Chloroflexi (from 2.7 ± 0.5% to 8.0 ± 0.8%), Parcubacteria (from 1.1 ± 0.8% to 8.8 ± 1.7%), Planctomycetes (from 1.1 ± 0.1% to 2.2 ± 0.3%), and unclassified bacteria (from 5.5 ± 0.9% to 14.4 ± 1.1%). Interestingly, these increases in relative abundance were not solely driven by normalized sequence counts of abundant OTUs (≥0.1% relative abundance per OTU) but to a substantial part also by the sum of normalized sequence counts of low-abundance OTUs (<0.1% relative abundance per OTU). For example, the summed relative abundance of low-abundance OTUs within the Acidobacteria made up 1.2% (± 0.4%) and 2.9% (± 0.2%) of communities in the initial soil substrate and rhizosphere soils, respectively. In contrast, the summed relative abundance of abundant Acidobacteria OTUs made up only 0.2% (± 0.1%) and 0.8% (±0.2%) of communities in the initial soil substrate and rhizosphere soils, respectively. Among the 35 bacterial phyla and proteobacterial classes that were identified, 15 showed an increase in relative abundance that was dominated by the sum of normalized sequence counts of low-abundance OTUs (Figure 1). This included numerically dominant phyla such as the Acidobacteria and Planctomycetes as well as phyla that represented only a minority of the bacterial community such as Lenthisphaerae or Deinococcus-Thermus (Figure 1).
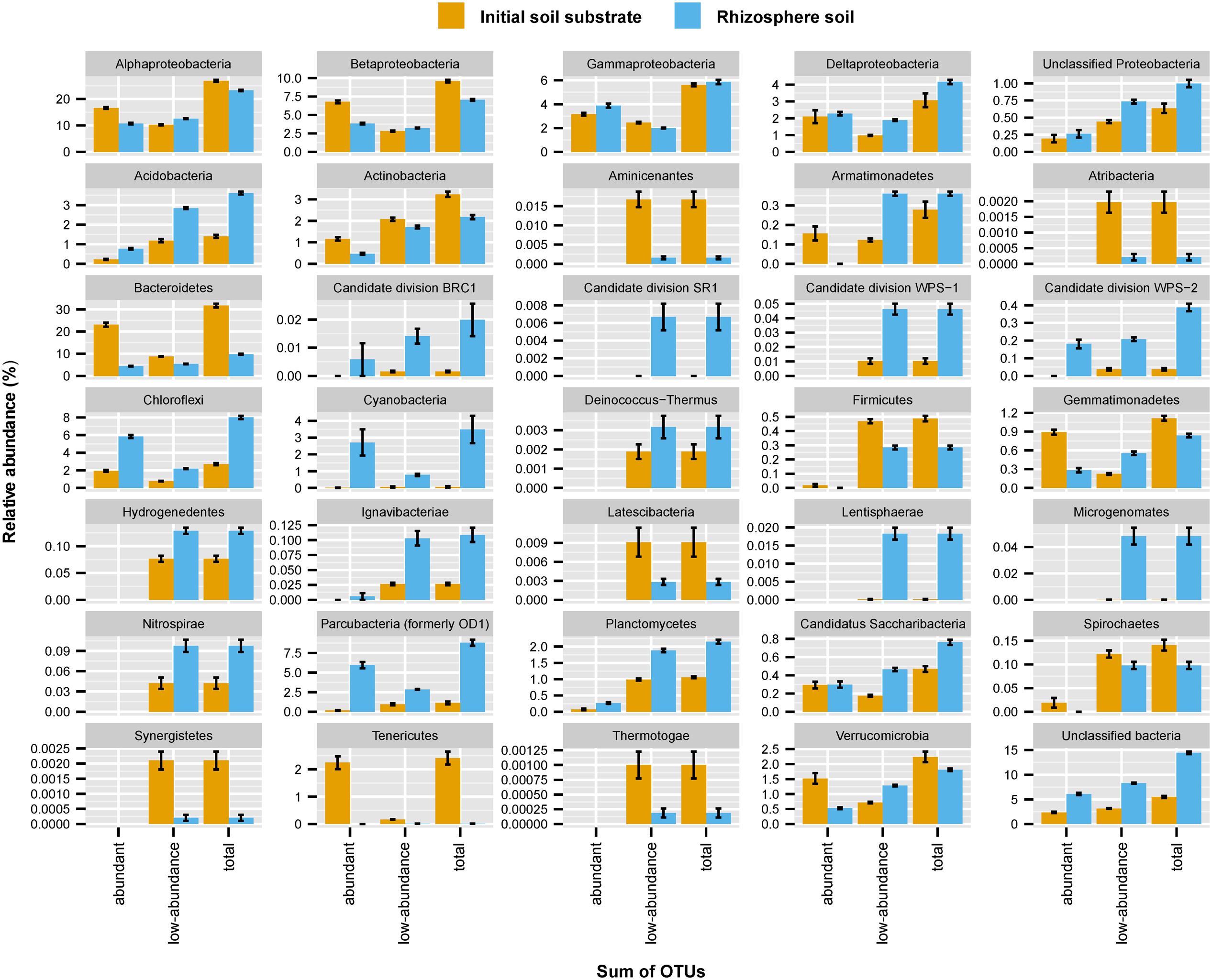
FIGURE 1. Mean relative abundance of bacterial phyla or proteobacterial classes across the initial soil substrate and rhizosphere soils as averaged across all analyzed plant species (n = 19). Error bars represent one standard deviation. The contribution of on average abundant (≥0.1%) and low-abundance (<0.1%) OTUs to changes between soils as well as their sum are displayed separately. A detailed distribution of bacterial phyla per plant species is given in Supplementary Figure S2.
The large difference in bacterial beta diversity between the initial soil substrate and the rhizosphere soils was also evident when analyzed at the level of individual OTUs. Here, a clear separation of replicated initial soil substrate and rhizosphere soils could be observed in a NMDS plot irrespective of the analyzed plant species (Figure 2). This shift was highly significant as determined by a PERMANOVA analysis (Table 2). 55% of the variation in bacterial community composition among soil samples was explained by the incubation over 14 weeks. In addition, variation in bacterial community composition among plant species was also significant but much smaller (7%). A significant interaction between plant-species identity and incubation indicated that bacterial communities responded differently depending upon plant species, and this interaction explained 7% of the variation (Table 2). Since this result may reflect different starting communities among species and soils, we also tested for dispersion of communities among samples. Dispersion did not differ significantly among species (F17,154 = 0.494, p = 0.957). However, it did differ significantly between the initial soil substrate and rhizosphere soils (F1,170 = 10.607, p = 0.001), which reflects the less variable bacterial community composition in rhizosphere soils as compared to the initial soil substrate (Figure 2).
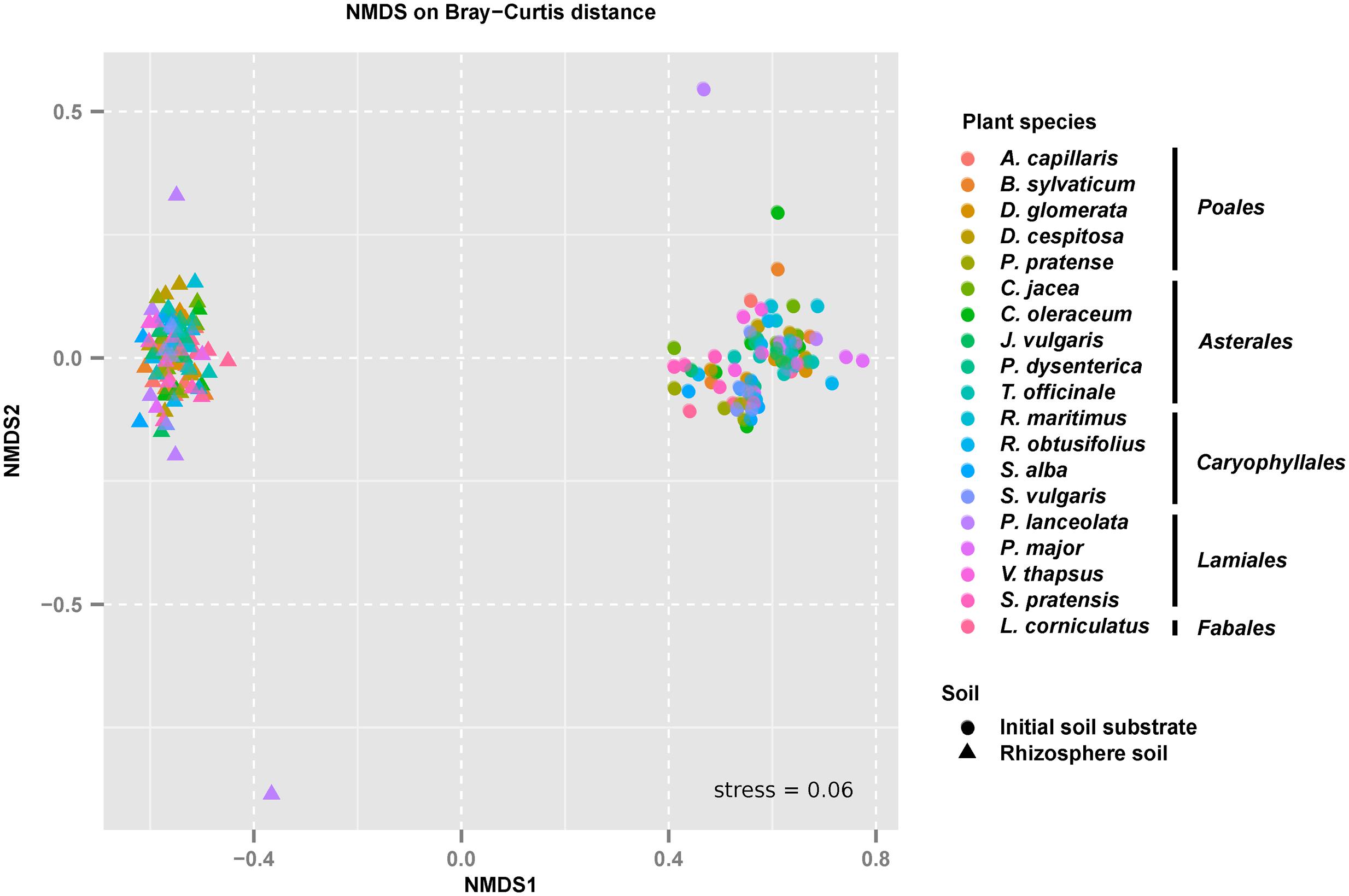
FIGURE 2. Beta-diversity of bacterial communities in the initial soil substrate and the corresponding rhizosphere soils of 19 different grassland plant species. Separation of bacterial communities in the analyzed soils is shown according to a non-metric multidimensional scaling (NMDS) analysis based on Bray–Curtis distances. Individual points represent sequenced soil replicates.
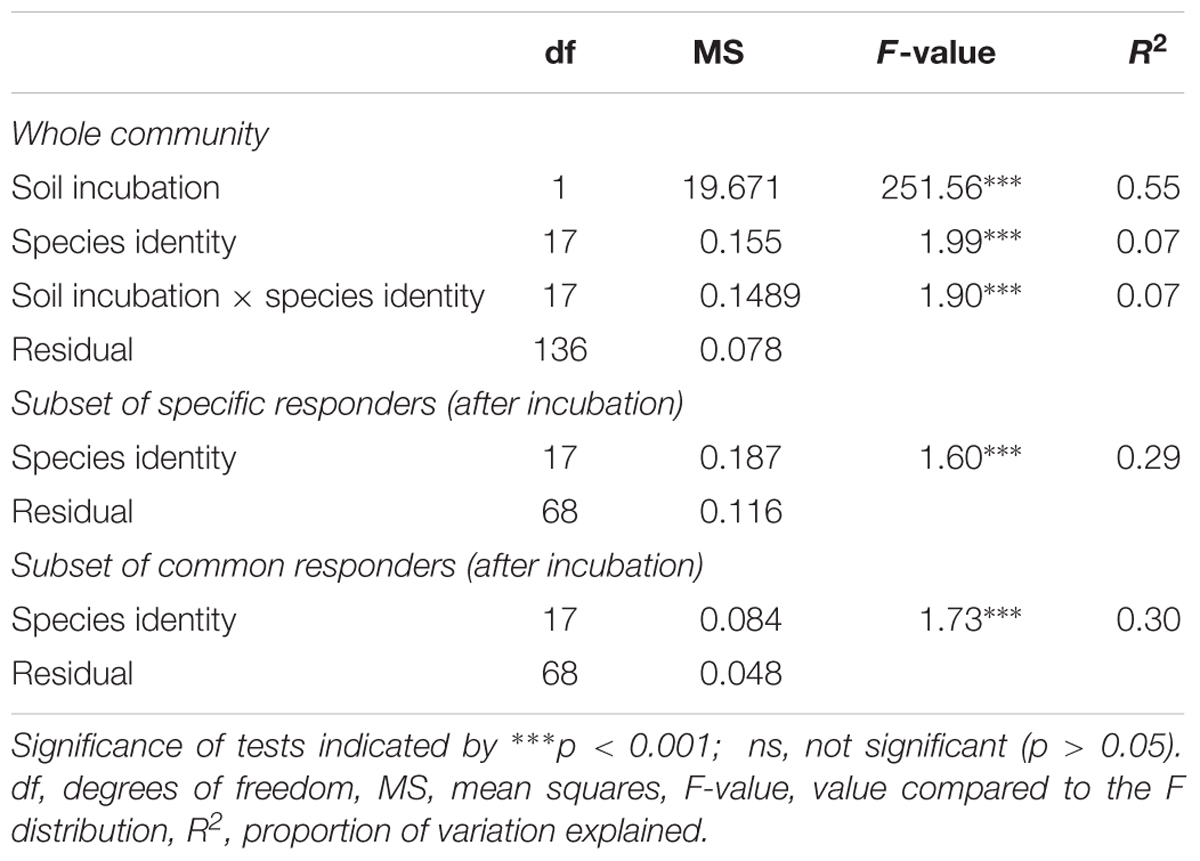
TABLE 2. Permutational analysis of variance (PERMANOVA) assessing dissimilarity of rhizosphere bacterial communities according to soil incubation and plant species identity (using a Bray–Curtis dissimilarity matrix).
Interestingly, there was no significant correlation between phylogenetic distance among plant species and rhizosphere bacterial community dissimilarity (Mantel ρ = -0.09, n = 86, p = 0.99). This strongly suggests that bacterial communities were no more dissimilar between distantly related (spanning five different plant orders), than they were between more closely related plant species (e.g., of the same genus). Variation in bacterial community composition in the rhizosphere soils was also not significantly explained by the amount of biomass accumulated by plants during the incubation period of 14 weeks (F1,84 = 1.684, p = 0.082, R2 = 0.02).
Positive Plant-specific Responders Constitute on Average 1% of the Bacterial OTU Richness in the Rhizosphere
Each plant species had a small subset of 18–111 OTUs that responded specifically (either positive or negative) to that plant species only (Figure 3). The number of specific positive responders ranged from only 1 OTU up to 71 OTUs, which averaged 0.9% of observed OTUs per plant (range: 0.03–2.3%). On average, 91% of these positively responding OTUs were of low relative abundance (range: 60–100%). These low-abundance OTUs represented in 12 of 18 analyzed plant species also the first or second strongest positive responder in terms of log-fold change (e.g., unclassified Betaproteobacteria for Brachypodium sylvaticum and Deschampsia cespitosa or an unclassified Sinobacteraceae for Centaurea jacea) (Supplementary Table S2). This was also reflected in their summed relative abundance. Here, the sum of normalized sequence counts of positively responding low-abundance OTUs dominated numerically over positively responding abundant OTUs in 12 of the 18 analyzed plant species (Supplementary Table S2).
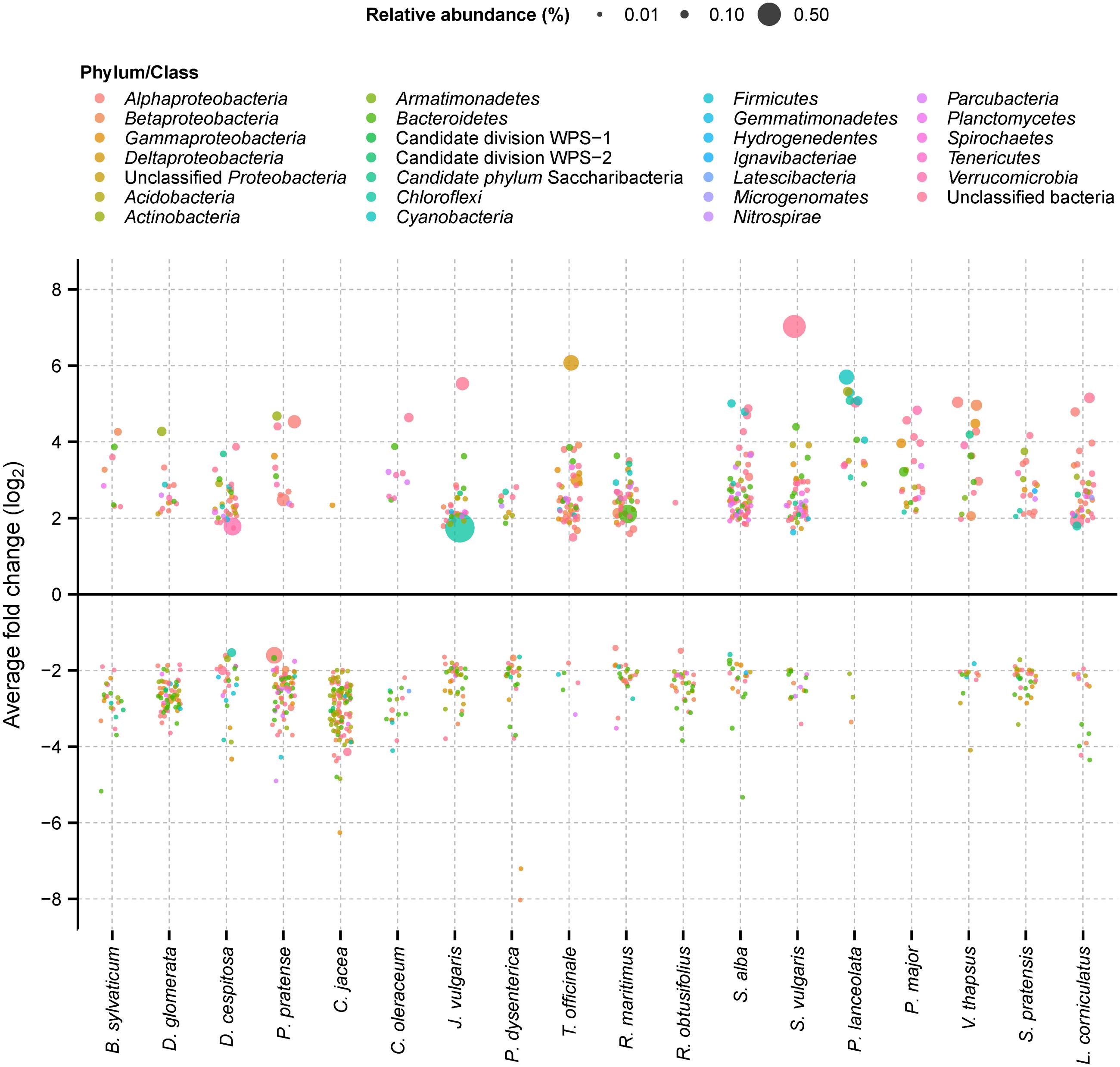
FIGURE 3. Bacterial OTUs (97% sequence identity) that responded significantly by relative abundance change according to one plant species only (plant-specific responders). Each point represents an OTU with the size of the points being proportional to the mean relative abundance of OTUs in the plant-specific rhizosphere soils. Point colors indicate the taxonomic affiliation of plant-specific responders. Further details are provided in Supplementary Table S2.
Plant-specific positive responders were mainly affiliated to the Alphaproteobacteria, unclassified bacteria, Bacteroidetes, Parcubacteria, Betaproteobacteria, or Verrucomicrobia (in descending order, ≥20 responders). However, there was no clear trend toward a certain phylogenetic lineage among the top or overall positive responding OTUs per plant species. For example, commonly observed members of the Rhizobiales (Alphaproteobacteria) were present only in 11 out of 18 plant species among the positive responding OTUs and members of the Flavobacterales (Bacteroidetes) in 10 plant species only (Supplementary Table S2).
The number of plant-species specific negative responders ranged from 0 to 110 OTUs per plant species (Figure 3), which averaged 1.0% of observed OTUs per plant (range: 0.0–3.6%). Again, their summed relative abundance was dominated by low-abundance OTUs in 13 of 18 analyzed plant species (Supplementary Table S2). The majority of these OTUs were affiliated to the Bacteroidetes, Alphaproteobacteria, unclassified bacteria, Betaproteobacteria, Gammproteobacteria, and Acidobacteria (in descending order, ≥20 responders). An overwhelmingly large fraction of these OTUs were of low relative abundance in the rhizosphere soils (average: 99%, range: 92–100%) (Supplementary Table S2).
These results were corroborated by a PERMANOVA analysis, showing a significant effect of plant-species identity on the observed plant-specific responders (Table 2). Interestingly, plant-species identity explained only 29% of total variation within plant-specific responders (both positive and negative). This indicates that while all these OTUs responded significantly to only one plant species, there was still a large amount of variation in abundance of these OTUs among soils of the other plant species. This also likely explains why there was no clear separation of the positive plant-specific responders according to plant phylogeny in a NMDS analysis (Supplementary Figure S3). This conclusion was further supported by a Mantel test on plant species-specific responses. Here, no significant correlation was observed between plant phylogeny and bacterial community dissimilarity of plant species-specific responders (Mantel ρ = -0.02, n = 86, p = 0.70).
Common Responders Are Dominated by Chloroflexi, Parcubacteria, and Proteobacteria
A subset of 340 OTUs (9–13% of observed OTUs per soil replicate) differed significantly between the initial soil substrate and rhizosphere soils across all plant species. This included 127 and 213 OTUs that responded by increasing and decreasing in relative abundance, respectively (Supplementary Figure S4). Among the 21 phyla or proteobacterial classes that harbored common responders, 9 contained positive as well as negative responders with the Alphaproteobacteria and the Bacteroidetes being the most prominent ones. Phyla or classes that harbored exclusively positive responders were dominated by the Chloroflexi and Parcubacteria (out of seven phyla/classes in total), while exclusively negative responders were restricted to the Tenericutes, Spirochaetes, Hydrogenedetes, and unclassified Proteobacteria (Supplementary Figure S4).
In rhizosphere soils, the sum of normalized sequence counts of all common positive responders corresponded on average to a relative abundance of 26.1% of the total bacterial community (in comparison to 0.3% in the initial soil substrate). Roughly half (55%) of the positively responding OTUs were abundant, while the other half (45%) were of low-abundance (Supplementary Table S3). The most abundant positively responding OTUs belonged to the Chloroflexi (OTU4, 1.8% relative abundance), Parcubacteria (OTU7, 1.3% relative abundance), Cyanobacteria (OTU11, 1.0% relative abundance), and Gammaproteobacteria (OTU15, 1.0% relative abundance) (Supplementary Figure S4 and Table S3). In contrast, normalized sequence counts of all common negative responders corresponded in sum to only 1.3% relative abundance of the total bacterial community in rhizosphere soils (in comparison to 44.4% in the initial soil substrate) and belonged exclusively to low-abundance OTUs. The largest relative abundance declines among negative responders were observed among members of the Bacteroidetes (OTUs 1, 8, 9, 10) and Tenericutes (OTU3), which were all present at 1% to 4% relative abundance in the initial soil substrate (Supplementary Table S3).
Interestingly, a significant amount (30%) of the variation in bacterial community composition of the common responders was explained by plant-species identity (Table 2). This suggests that even though these OTUs responded in the rhizosphere of all plants in a similar way, the magnitude of those responses differed according to plant species.
Discussion
Plant-specific Rhizosphere Bacteria belong Mainly to the Rare Biosphere
Plant-species identity is one of several factors that shapes the rhizosphere bacterial community (e.g., Miethling et al., 2000; Smalla et al., 2001; Garbeva et al., 2008; Berg and Smalla, 2009; Bezemer et al., 2010; Pendergast et al., 2013). For example, a comparison of plants belonging to different plant orders like oilseed rape (Brassica napus, order Brassicales), potato (Solanum tuberosum, order Solanales) and strawberry (Fragaria ananassa, order Rosales) revealed obvious differences in DGGE-fingerprint profiles of the bacterial community inhabiting the rhizosphere (Smalla et al., 2001; Costa et al., 2006). The same was true when comparing plants belonging to the same order-like rank like the herbeceous species hound’s tongue (Cynoglossum officinale) and spear thistle (Cirsium vulgare) (both Asterids) (Kowalchuk et al., 2002) or the same family like the grass species Indian ricegrass (Stipa hymenoides), James’ galleta (Hilaria jamestii), and drooping brome (Bromus tectorum) (all belonging to the Poaceae) (Kuske et al., 2002). The majority of such studies used fingerprinting techniques, which allowed differentiation of abundant bacteria at low phylogenetic resolution. In our study, we expanded investigation of the root bacteriome to the large bacterial diversity represented by low-abundance bacteria (Dohrmann et al., 2013; Nuccio et al., 2016; Saleem et al., 2016; Shi et al., 2016) and analyzed how this rare biosphere shapes the plant species-specific root bacteriome.
Already at the level of individual bacterial phyla (or proteobacterial classes), it was evident that changes in the rare biosphere drive a substantial part of the bacterial community shift between the initial soil substrate and rhizosphere soils. Approximately 40% of the detected bacterial phyla and proteobacterial classes showed an increase in relative abundance that was dominated by the sum of normalized sequence counts of low-abundance OTUs (Figure 1). This was equally true for phyla that represented a numerically small proportion of the bacterial population (e.g., the Lenthisphaerae) as for numerically abundant phyla (e.g., the Acidobacteria). Contrary to one of our hypotheses, the composition of established rhizosphere communities did not show a significant imprint of host plant phylogeny (see Mantel test results). Indeed, only a small subset of the rhizosphere bacterial community was plant-species specific. This included both abundant OTUs, as was already known from previous studies (e.g., Miethling et al., 2000; Smalla et al., 2001; Garbeva et al., 2008; Berg and Smalla, 2009; Bezemer et al., 2010; Pendergast et al., 2013), but to a large extent also low-abundance OTUs. The significant positive response of low-abundance OTUs to a specific host plant indicates population growth and metabolic activity, especially in the context of increasing overall bacterial and archaeal populations (Figure 3 and Supplementary Table S1) Thus, our results contribute to a growing body of evidence that bacterial populations that belong to the rare biosphere are not just part of a dormant ‘seed’ bank that can be ‘activated’ upon environmental change but can actually directly contribute to ongoing processes in the soil and other environments (Pester et al., 2010; Großkopf et al., 2012; Lynch and Neufeld, 2015; Hausmann et al., 2016; Jousset et al., 2017). Especially in the highly structured root environment with secondary and primary roots including root hairs and caps, it can easily be envisaged that such overall low-abundance populations may constitute on a microscale locally abundant bacteria that could have an important effect on plant growth.
On average, roughly 1% of rhizosphere bacterial OTUs were promoted by the specific plant-host with an equally large part being suppressed (Figure 3 and Supplementary Table S2). However, plant species explained only 29% of total variation within these plant-specific responders (Table 2). This is likely a result of most of these plant-specific responders sustaining individual population sizes of low relative abundance. While for their specific host plant the response was significant, these OTUs were likely at the detection limit of our sequencing effort for the other plants, with rather sporadic and inconsistent encounters that resulted in high variance among samples within species. Nonetheless, these results contribute to the evidence that plant species leave a discernible imprint on their respective rhizosphere soil.
In our study, rhizosphere soil was defined as soil attached to the roots. As such, we integrated over a gradient of soil that had a variable impact by plant roots (from strong to weak). Rhizosphere soil that is in close proximity to plant roots (a few millimeter) can be expected to harbor more plant-specific bacteria at potentially higher relative abundance. However, the small numbers of plant-specific responders in our study was comparable to studies that analyzed effects of different cultivars of the same plant species. Among different A. thaliana cultivars, 1.5% of observed bacterial OTUs in the endophyte-compartment were genotype-specific (Lundberg et al., 2012) and in Solanum tuberosum about 4% of rhizosphere- and root-inhabiting OTUs were genotype-dependent across two different analyzed soils (Weinert et al., 2011). Thus, the number of bacteria responding to specific plants does not appear to increase from genotypes of the same species (Weinert et al., 2011; Lundberg et al., 2012) through to species of different orders (this study). If these small subsets of responding bacteria influence plant performance, then these findings would agree with a recent meta-analysis, which found no plant phylogenetic signal in the strength of plant-soil feedback effects (Mehrabi and Tuck, 2015).
The Rhizosphere Bacterial Community is Strongly Influenced by Common Responders
The observed community shifts between the initial soil substrate and rhizosphere soils (Figure 2) were strongly influenced by a roughly equally large number of abundant and low-abundance OTUs that significantly responded in the presence of all tested plant species (Supplementary Figure S4 and Table S3). These common responders were represented by 340 OTUs, which constituted in terms of their summed relative abundance one quarter of the total bacterial community across the rhizosphere of all analyzed plant species. Due to our experimental design, we cannot differentiate between OTUs that responded to plant growth only as opposed to those that responded solely due to abiotic conditions, e.g., recolonization of provided substrate such as vermiculite or washed sand. However, since it is known that plants strongly influence their rhizosphere (Marschner et al., 1986; Dennis et al., 2010; Berendsen et al., 2012; Bulgarelli et al., 2013; van der Putten et al., 2013), at least part of these common responders likely responded to plant growth.
Among the common positive responders, the two most abundant OTUs (>1.3% relative abundance) belonged to the little-explored bacterial phyla Chloroflexi and Parcubacteria (formerly candidate phylum OD1). All 11 Chloroflexi OTUs among the common responders, including the most abundant common responder OTU, were affiliated to the Anaerolineaceae (subphylum 1) within the Chloroflexi. Cultivated representatives of the Anaerolineaceae have a heterotrophic lifestyle, being able to degrade carbohydrates and/or peptides and have been isolated from habitats like rice paddy soil, tundra meadow soil and anaerobic sludge (Costello and Schmidt, 2006; Yamada and Sekiguchi, 2009). The 11 Chloroflexi common responders of this study changed by 10–200 fold and constituted per OTU a relative abundance of 0.06–1.79% of the final rhizosphere bacterial community, with seven of these OTUs having relative abundancies of >0.1% (Supplementary Figure S4). As such, they may represent major generalists driving organic carbon mineralisation in the rhizosphere.
The second most abundant common responder OTU belonged to the Parcubacteria, with 17 additional Parcubacteria OTUs responding positively during the incubation irrespective of the host plant (8–200 fold changes; Supplementary Figure S4). All known members of this phylum possess very small and streamlined genomes lacking biosynthesis pathways for nucleotides, amino acids, fatty acids and co-factors like quinones and flavins. Therefore, a mutualistic or parasitic lifestyle has been proposed for these bacteria (Wrighton et al., 2012; Kantor et al., 2013; Rinke et al., 2013; Nelson and Stegen, 2015). This could also be the case in our study although we cannot exclude a direct association of these bacteria with plant roots. It is intriguing that 12 of these common Parcubacteria responders had a moderate or even high relative abundance (>0.1% relative abundance) with one of them being the second most abundant common responder (1.3% relative abundance, Supplementary Figure S4). Such large bacterial populations are generally postulated to be directly involved in major ecosystem functions like the carbon and energy flow (Pedrós-Alió, 2012; Lynch and Neufeld, 2015). Our results suggest that symbionts associated with microorganims can hitchhike this principle allowing them to build up large populations without being directly involved in the degradation of plant-derived material in the rhizosphere.
Conclusion
Plants are known to modify the bacterial community in their close root vicinity (Berg and Smalla, 2009; Bulgarelli et al., 2013; Philippot et al., 2013). Our study shows that plant species identity is only a minor driver in this process with no significant imprint of plant phylogeny. The small number of rhizosphere bacterial OTUs that actually responded in a plant-species specific manner was dominated by members that sustain populations of low relative abundance. In addition, populations of low-abundance bacteria were in sum the major drivers of common responses at the phylum level. This adds to a growing body of evidence that the rare biosphere is not only a dormant seed bank but also harbors species that display enhanced metabolic activity, increase functional diversity, or increase community-wide species interactions (Jousset et al., 2017). Future studies need to demonstrate whether and how such low-abundance populations play an important role in affecting plant growth.
Author Contributions
WD designed and performed experiments, analyzed the data and wrote the manuscript. MP designed experiments, analyzed the data and wrote the manuscript. JH performed experiments and contributed to writing the manuscript. MvK and ME contributed to designing experiments and to writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MP and WD were funded by the Zukunftskolleg of the University of Konstanz. MP was funded by the German Research Foundation (PE2147/1-1). WD was funded by the University of Konstanz Young Scholar Fund (ZUK 52/2) and the German Research Foundation (AZ DA 1502/1-1). We are grateful to the Genomics Centre of the University of Konstanz and Daniel M. Parera for preparing the Illumina sequencing library. We further thank B. Rüter, K. Mamanova-Stift, and S. Glöckner for their great technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00975/full#supplementary-material
FIGURE S1 | Alpha diversity of analyzed samples when rarefied to an equal sampling depth of 16417 reads per replicate. Observed number of bacterial OTUs, Chao1 richness estimate, Shannon–Wiener and Simpson’s Index for the initial soil substrate and respective rhizosphere soils are displayed for each plant species.
FIGURE S2 | Mean relative abundance of bacterial phyla, and in case of Proteobacteria classes, across the initial soil substrate and rhizosphere soils for each plant species.
FIGURE S3 | Beta-diversity of bacteria in rhizosphere soils that responded positively to one plant species only. Separation of bacterial subcommunities in the analyzed soils is shown according to a non-metric multidimensional scaling (NMDS) analysis based on Bray–Curtis distances (stress = 0.19). Individual points represent sequenced soil replicates. Agrostis capillaris was excluded from statistical analysis due to an insufficient number of initial soil substrate replicates and is therefore not shown.
FIGURE S4 | Bacterial OTUs (97% sequence identity) that significantly responded by relative abundance change between the initial soil substrate and respective rhizosphere soils across all plant species (common responders). OTUs are arranged by phyla, and in case of Proteobacteria by classes. Each point represents an OTU with the size of the points being proportional to the mean relative abundance of OTUs across all plant soil replicates in the rhizosphere soils.
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berg, G., and Smalla, K. (2009). Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 68, 1–13. doi: 10.1111/j.1574-6941.2009.00654.x
Bezemer, T. M., Fountain, M. T., Barea, J. M., Christensen, S., Dekker, S. C., Duyts, H., et al. (2010). Divergent composition but similar function of soil food webs of individual plants: plant species and community effects. Ecology 91, 3027–3036. doi: 10.1890/09-2198.1
Bridge, P., and Spooner, B. (2001). Soil fungi: diversity and detection. Plant Soil 232, 147–154. doi: 10.1023/A:1010346305799
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Bulgarelli, D., Rott, M., Schlaeppi, K., van Themaat, E. V. L., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Themaat, E. V. L. V., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Costa, R., Götz, M., Mrotzek, N., Lottmann, J., Berg, G., and Smalla, K. (2006). Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol. Ecol. 56, 236–249. doi: 10.1111/j.1574-6941.2005.00026.x
Costello, E. K., and Schmidt, S. K. (2006). Microbial diversity in alpine tundra wet meadow soil: novel Chloroflexi from a cold, water-saturated environment. Environ. Microbiol. 8, 1471–1486. doi: 10.1111/j.1462-2920.2006.01041.x
Curtis, T. P., Sloan, W. T., and Scannell, J. W. (2002). Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. U.S.A. 99, 10494–10499. doi: 10.1073/pnas.142680199
Dennis, P. G., Miller, A. J., and Hirsch, P. R. (2010). Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 72, 313–327. doi: 10.1111/j.1574-6941.2010.00860.x
Dohrmann, A. B., Kuting, M., Junemann, S., Jaenicke, S., Schluter, A., and Tebbe, C. C. (2013). Importance of rare taxa for bacterial diversity in the rhizosphere of Bt- and conventional maize varieties. ISME J. 7, 37–49. doi: 10.1038/ismej.2012.77
Durka, W., and Michalski, S. G. (2012). Daphne: a dated phylogeny of a large European flora for phylogenetically informed ecological analyses. Ecology 93, 2297–2297. doi: 10.1890/12-0743.1
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fierer, N., Breitbart, M., Nulton, J., Salamon, P., Lozupone, C., Jones, R., et al. (2007). Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 73, 7059–7066. doi: 10.1128/AEM.00358-07
Garbeva, P., van Elsas, J. D., and van Veen, J. A. (2008). Rhizosphere microbial community and its response to plant species and soil history. Plant Soil 302, 19–32. doi: 10.1038/srep45318
Good, I. J. (1953). The population frequencies of species and the estimation of population parameters. Biometrika 40, 237–264. doi: 10.1093/biomet/40.3-4.237
Großkopf, T., Mohr, W., Baustian, T., Schunck, H., Gill, D., Kuypers, M. M. M., et al. (2012). Doubling of marine dinitrogen-fixation rates based on direct measurements. Nature 488, 361–364. doi: 10.1038/nature11338
Hartwich, R., Hasse, G., Richter, A., Roeschmann, G., and Schmidt, R. (2013). Bodenübersichtskarte der Bundesrepublik Deutschland 1:1000000. Hannover: Bundesanstalt für Geowissenschaften und Rohstoffe.
Hausmann, B., Knorr, K.-H., Schreck, K., Tringe, S. G., Glavina del Rio, T., Loy, A., et al. (2016). Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms. ISME J. 10, 2365–2375. doi: 10.1038/ismej.2016.42
Hol, W. H. G., Garbeva, P., Hordijk, C., Hundscheid, M. P. J., Gunnewiek, P. J. A. K., van Agtmaal, M., et al. (2015). Non-random species loss in bacterial communities reduces antifungal volatile production. Ecology 96, 2042–2048. doi: 10.1890/14-2359.1
Hortal, S., Bastida, F., Moreno, J. L., Armas, C., García, C., and Pugnaire, F. I. (2015). Benefactor and allelopathic shrub species have different effects on the soil microbial community along an environmental severity gradient. Soil Biol. Biochem. 88, 48–57. doi: 10.1016/j.soilbio.2015.05.009
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.1174
Kantor, R. S., Wrighton, K. C., Handley, K. M., Sharon, I., Hug, L. A., Castelle, C. J., et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio 4, e00708–e00713. doi: 10.1128/mBio.00708-13
Kowalchuk, G. A., Buma, D. S., de Boer, W., Klinkhamer, P. G. L., and van Veen, J. A. (2002). Effects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms. Antonie Van Leeuwenhoek 81, 509–520. doi: 10.1023/A:1020565523615
Kuske, C. R., Ticknor, L. O., Miller, M. E., Dunbar, J. M., Davis, J. A., Barns, S. M., et al. (2002). Comparison of soil bacterial communities in rhizospheres of three plant species and the interspaces in an arid grassland. Appl. Environ. Microbiol. 68, 1854–1863. doi: 10.1128/AEM.68.4.1854-1863.2002
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Lemmermeyer, S., Lörcher, L., van Kleunen, M., and Dawson, W. (2015). Testing the plant growth-defense hypothesis belowground: do faster-growing herbaceous plant species suffer more negative effects from soil biota than slower-growing ones? Am. Natur. 186, 264–271. doi: 10.1086/682005
Lund, S., Nettleton, D., McCarthy, D., and Smyth, G. (2012). Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Stat. Appl. Genet. Mol. B. 11:8. doi: 10.1515/1544-6115.1826
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Lynch, M. D. J., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Mallon, C. A., Poly, F., Le Roux, X., Marring, I., van Elsas, J. D., and Salles, J. F. (2015). Resource pulses can alleviate the biodiversity–invasion relationship in soil microbial communities. Ecology 96, 915–926. doi: 10.1890/14-1001.1
Marschner, H., Römheld, V., Horst, W. J., and Martin, P. (1986). Root-induced changes in the rhizosphere: importance for the mineral nutrition of plants. J. Plant. Nutr. Soil Sci. 149, 441–456. doi: 10.1002/jpln.19861490408
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Mehrabi, Z., and Tuck, S. L. (2015). Relatedness is a poor predictor of negative plant-soil feedbacks. New Phytol. 205, 1071–1075. doi: 10.1111/nph.13238
Miethling, R., Wieland, G., Backhaus, H., and Tebbe, C. C. (2000). Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33. Microb. Ecol. 40, 43–56. doi: 10.1007/s002480000021
Mitra, A., Skrzypczak, M., Ginalski, K., and Rowicka, M. (2015). Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using Illumina platform. PLoS ONE 10:e0120520. doi: 10.1371/journal.pone.0120520
Nelson, W. C., and Stegen, J. C. (2015). The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 6:713. doi: 10.3389/fmicb.2015.00713
Nemergut, D. R., Costello, E. K., Hamady, M., Lozupone, C., Jiang, L., Schmidt, S. K., et al. (2010). Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13, 135–144. doi: 10.1111/j.1462-2920.2010.02315.x
Nuccio, E. E., Anderson-Furgeson, J., Estera, K. Y., Pett-Ridge, J., de Valpine, P., Brodie, E. L., et al. (2016). Climate and edaphic controllers influence rhizosphere community assembly for a wild annual grass. Ecology 97, 1307–1318. doi: 10.1890/15-0882.1
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R., et al. (2013). Vegan: Community Ecology Package. R Package Version 2.0-10. Available at: http://vegan.r-forge.r-project.org/
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pedrós-Alió, C. (2012). The rare bacterial biosphere. Annu. Rev. Mar. Sci. 4, 449–466. doi: 10.1146/annurev-marine-120710-100948
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Pendergast, T. H., Burke, D. J., and Carson, W. P. (2013). Belowground biotic complexity drives aboveground dynamics: a test of the soil community feedback model. New Phytologist 197, 1300–1310. doi: 10.1111/nph.12105
Pester, M., Bittner, N., Pinsurang, D., Wagner, M., and Loy, A. (2010). A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 4, 1591–1602. doi: 10.1038/ismej.2010.75
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and van der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
R Core Team. (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available at: http://www.R-project.org
Reinhold-Hurek, B., Bünger, W., Burbano, C. S., Sabale, M., and Hurek, T. (2015). Roots shaping their microbiome: global hotspots for microbial activity. Annu. Rev. Phytopathol. 53, 403–424. doi: 10.1146/annurev-phyto-082712-102342
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Rodríguez-Echeverría, S., Armas, C., Pistón, N., Hortal, S., and Pugnaire, F. I. (2013). A role for below-ground biota in plant–plant facilitation. J. Ecol. 101, 1420–1428. doi: 10.1111/nph.14499
Rosenzweig, N., Bradeen, J. M., Tu, Z. J., McKay, S. J., and Kinkel, L. L. (2013). Rhizosphere bacterial communities associated with long-lived perennial prairie plants vary in diversity, composition, and structure. Can. J. Microbiol. 59, 494–502. doi: 10.1139/cjm-2012-0661
Saleem, M., Law, A. D., and Moe, L. A. (2016). Nicotiana roots recruit rare rhizosphere taxa as major root-inhabiting microbes. Microb. Ecol. 71, 469–472. doi: 10.1007/s00248-015-0672-x
Schloss, P. D., Girard, R. A., Martin, T., Edwards, J., and Thrash, J. C. (2016). Status of the archaeal and bacterial census: an update. mBio 7:e201-e216. doi: 10.1128/mBio.00201-00216
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shi, S., Nuccio, E. E., Shi, Z. J., He, Z., Zhou, J., and Firestone, M. K. (2016). The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol. Lett. 19, 926–936. doi: 10.1111/ele.12630
Smalla, K., Wieland, G., Buchner, A., Zock, A., Parzy, J., Kaiser, S., et al. (2001). Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67, 4742–4751. doi: 10.1128/AEM.67.10.4742-4751.2001
Sogin, M. L., Morrison, H. G., Huber, J. A., Mark Welch, D., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
van der Putten, W. H., Bardgett, R. D., Bever, J. D., Bezemer, T. M., Casper, B. B., Fukami, T., et al. (2013). Plant–soil feedbacks: the past, the present and future challenges. J. Ecol. 101, 265–276. doi: 10.1111/1365-2745.12054
van Elsas, J. D., Chiurazzi, M., Mallon, C. A., Elhottovâ, D., Krištùfek, V., and Salles, J. F. (2012). Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. U.S.A. 109, 1159–1164. doi: 10.1073/pnas.1109326109
Vivant, A.-L., Garmyn, D., Maron, P.-A., Nowak, V., and Piveteau, P. (2013). Microbial diversity and structure are drivers of the biological barrier effect against Listeria monocytogenes in soil. PLoS ONE 8:e76991. doi: 10.1371/journal.pone.0076991
Weinert, N., Piceno, Y., Ding, G.-C., Meincke, R., Heuer, H., Berg, G., et al. (2011). PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 75, 497–506. doi: 10.1111/j.1574-6941.2010.01025.x
Wörner, S., Zecchin, S., Dan, J., Todorova, N. H., Loy, A., Conrad, R., et al. (2016). Gypsum amendment to rice paddy soil stimulated bacteria involved in sulfur cycling but largely preserved the phylogenetic composition of the total bacterial community. Environ. Microbiol. Rep. 8, 413–423. doi: 10.1111/1758-2229.12413
Wrighton, K. C., Thomas, B. C., Sharon, I., Miller, C. S., Castelle, C. J., VerBerkmoes, N. C., et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665. doi: 10.1126/science.1224041
Keywords: microbiome, next-generation amplicon sequencing, rhizosphere, 16S rRNA gene, rare biosphere
Citation: Dawson W, Hör J, Egert M, van Kleunen M and Pester M (2017) A Small Number of Low-abundance Bacteria Dominate Plant Species-specific Responses during Rhizosphere Colonization. Front. Microbiol. 8:975. doi: 10.3389/fmicb.2017.00975
Received: 07 March 2017; Accepted: 15 May 2017;
Published: 29 May 2017.
Edited by:
Stéphane Hacquard, Max Planck Institute for Plant Breeding Research, GermanyReviewed by:
Christoph C. Tebbe, Thünen Institut - Federal Research Institute for Rural Areas, Forestry and Fisheries, GermanyMadhaiyan Munusamy, Temasek Life Sciences Laboratory, Singapore
Gera Hol, Netherlands Institute of Ecology (NIOO-KNAW), Netherlands
Copyright © 2017 Dawson, Hör, Egert, van Kleunen and Pester. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Pester, bWljaGFlbC5wZXN0ZXJAdW5pLWtvbnN0YW56LmRl
†Present address: Jens Hör, RNA Biology Group, Institute for Molecular Infection Biology, University of Würzburg, Josef-Schneider-Straße 2-4, Würzburg, Germany