
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 April 2017
Sec. Extreme Microbiology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00591
 Sarita Pudasaini1
Sarita Pudasaini1 John Wilson1
John Wilson1 Mukan Ji1Josie van Dorst1Ian Snape2Anne S. Palmer2
Mukan Ji1Josie van Dorst1Ian Snape2Anne S. Palmer2 Brendan P. Burns1
Brendan P. Burns1 Belinda C. Ferrari1*
Belinda C. Ferrari1*Browning Peninsula is an ice-free polar desert situated in the Windmill Islands, Eastern Antarctica. The entire site is described as a barren landscape, comprised of frost boils with soils dominated by microbial life. In this study, we explored the microbial diversity and edaphic drivers of community structure across this site using traditional cultivation methods, a novel approach the soil substrate membrane system (SSMS), and culture-independent 454-tag pyrosequencing. The measured soil environmental and microphysical factors of chlorine, phosphate, aspect and elevation were found to be significant drivers of the bacterial community, while none of the soil parameters analyzed were significantly correlated to the fungal community. Overall, Browning Peninsula soil harbored a distinctive microbial community in comparison to other Antarctic soils comprised of a unique bacterial diversity and extremely limited fungal diversity. Tag pyrosequencing data revealed the bacterial community to be dominated by Actinobacteria (36%), followed by Chloroflexi (18%), Cyanobacteria (14%), and Proteobacteria (10%). For fungi, Ascomycota (97%) dominated the soil microbiome, followed by Basidiomycota. As expected the diversity recovered from culture-based techniques was lower than that detected using tag sequencing. However, in the SSMS enrichments, that mimic the natural conditions for cultivating oligophilic “k-selected” bacteria, a larger proportion of rare bacterial taxa (15%), such as Blastococcus, Devosia, Herbaspirillum, Propionibacterium and Methylocella and fungal (11%) taxa, such as Nigrospora, Exophiala, Hortaea, and Penidiella were recovered at the genus level. At phylum level, a comparison of OTU's showed that the SSMS shared 21% of Acidobacteria, 11% of Actinobacteria and 10% of Proteobacteria OTU's with soil. For fungi, the shared OTUs was 4% (Basidiomycota) and <0.5% (Ascomycota). This was the first known attempt to culture microfungi using the SSMS which resulted in an increase in diversity from 14 to 57 microfungi OTUs compared to standard cultivation. Furthermore, the SSMS offers the opportunity to retrieve a greater diversity of bacterial and fungal taxa for future exploitation.
Browning Peninsula is a remote and understudied area at the Southern end of the Windmill Islands, Eastern Antarctica. It lies 20 km away from the well-studied Casey station (Chong et al., 2009), and is described as an ice-free desert landscape comprised of barren rocks (Figure 1) with low diversity of invertebrates and vascular plant life (Azmi and Seppelt, 1998; Stewart et al., 2011). The mean annual temperature is −9.3°C with the average temperature rising above freezing point (+0.2°C) only in January (Beyer and Bolter, 2000), and annual precipitation is approximately 176 mm falling primarily as snow (Beyer and Bolter, 2000). The entire valley is described as a polar desert, comprised of frost boils ranging from 2 to 10 m in diameter (Stewart et al., 2011; Ferrari et al., 2015).

Figure 1. (A) Location of Browning Peninsula, Windmill Islands, Eastern Antarctica. (B) The spatially explicit design of sampling along three parallel transects, each 2 m apart. The sampling points were taken at 0, 2, 100, 102, 200, and 202 m distances along all three transects. (C) The photograph depicts a close up of Browning Peninsula.
Frost boils are upheavals of mud that occur through frost heave and freeze-thaw processes common in permafrost regions (Walker et al., 2004). In the Arctic, frost boils have been reported to exhibit a higher accumulation of organic materials within undisturbed inter-boil areas, leading to a systematic variation of vegetation growth (Walker et al., 2004). As a result of frost heave, soil biomass can increase from approximately 183 to 831 g m−2 and this effect is highest within frost boils that are comprised of fine grain sediments. Due to the extreme temperatures, limited water availability, reduced growing season, unstable substrates and severe local climatic conditions (Melick et al., 1994), no vegetation or bryophytes have been observed on Browning Peninsula (Stewart et al., 2011) and the effect on other biomass is unknown. However, the soil organic content has been found to be lower than that in Arctic soils, with Browning Peninsula soils classified as nutrient-poor and low in total carbon, nitrogen and moisture (Ferrari et al., 2015).
Cold-adapted microbes have great potential for the biotechnological industry. The biomass produced via the cultivation of microbes from extreme environments has led to the discovery of extracellular substances with antibacterial and/or antifungal properties (Gesheva, 2010; Gesheva and Negoita, 2011; Tomova et al., 2015). Additionally, the production of surfactants (glycolipids), the ability to grow on paraffin and naphthalene (Gesheva and Negoita, 2011) and their resistance to heavy metals (Tomova et al., 2015), highlights the potential for cold-adapted microbes to emulsify contaminants, such as hydrocarbons. Continued isolation of rare polar species could provide access to further strains for biotechnological applications, such as bioremediation and discovery of novel bioactive compounds, such as antibiotics (Tosi et al., 2010; Tytgat et al., 2014).
It is well established that a very small percentage of bacterial and fungal species are able to grow under traditional culture conditions (Handelsman, 2004; Ferrari et al., 2008, 2011). It is now known that the application of diverse sample dilutions, culture media, pH, incubation temperature and atmospheres, increases the metabolic diversity of species that can be cultivated from a single sample (Donachie et al., 2007). Novel methods have also been developed to increase the microbial biota representation under culture conditions and as a result select for a greater range of oligotroph. Some extend incubation times (Davis et al., 2005), or use dilute media (Ferrari et al., 2011), while others mimic the natural environment, for example implementing a diffusion chamber (Ferrari et al., 2005; Bollmann et al., 2007, 2010) or an in-situ trap (Gavrish et al., 2008). The excessive supply of nutrients in the artificial media often selects for fast-growing fungi and bacteria and to address this problem in soil, a simulated environment that mimics the natural soil environment called the soil slurry membrane system (SSMS) was developed (Ferrari et al., 2005). The diffusion chamber approach relies on the use of soil as the substrate for growth and has been found to enrich for members from a diversity of cultured and uncultured bacterial phyla, including the Saccharibacteria, better known as Candidate division TM7 (van Dorst et al., 2016). While the approach has been adopted to enrich for bacteria, the suitability of the method to select for fungi is not yet known.
Modern molecular phylogenetic and high throughput DNA sequencing have provided a comprehensive culture-independent analysis of soil microbial diversity (Margulies et al., 2005; Cowan et al., 2014), thereby accelerating the rate of gene discovery and giving rise to many applications in biotechnology (Handelsman, 2004; Jansson and Tas, 2014). Recently, NGS tools have been used to uncover bacterial (Tytgat et al., 2014; van Dorst et al., 2014; Ji et al., 2015) and fungal diversity (Kochkina et al., 2012; Ji et al., 2015) from Antarctic soil sources. This not only revealed previously unclassified bacterial diversity but also increased the size of microbial diversity datasets, improving access to vast microbial communities within a range of biospheres (Tytgat et al., 2014; Ji et al., 2015).
Limited studies have been conducted on Browning Peninsula. Studies to date have compared bacterial biomass between soil types (with and without penguin colonies) (Roser et al., 1993), isolated soil microfungi (Azmi and Seppelt, 1998), surveyed bacterial diversity using denaturing gradient gel electrophoresis (Chong et al., 2009) and examined the soil physical, chemical and microbial properties (Stewart et al., 2011). Most recently, analysis of environmental drivers of polar soil diversity and structure across the poles showed bacterial and fungal communities are driven primarily by soil fertility and pH (Siciliano et al., 2014). Microbial community connectivity within soils at Browning Peninsula was found to be highly fragmented and this fragmentation or dis-connectivity mirrored the fragmented nature of the frost boil landscape (Ferrari et al., 2015).
Several microbial ecology studies have compared the recovery of bacterial and fungal diversity using culture-dependent and culture-independent techniques, with only minor overlaps in microbial taxa observed (Donachie et al., 2007; Kochkina et al., 2012; Tytgat et al., 2014; Stefani et al., 2015). In these cases, traditional cultivation methods using standard cultivation media and different incubation temperatures were used and selected primarily for R-strategists (Watve et al., 2000; Ferrari et al., 2011). In this study, our aim was to use a traditional cultivation, novel micro-cultivation (SSMS), and tag pyrosequencing to uncover the total bacterial and fungal diversity present within soils of Browning Peninsula. To our knowledge, this research is the first report of enriching fungi, particularly microfungi using the SSMS.
The soil was sampled from browning Peninsula (Figure 1) in 2005 as a part of larger biodiversity project (Siciliano et al., 2014). Due to limitations of sampling site, such as erratic boulders, frost wedges, melt ponds and unstable slopes, the spatially explicit design uses three parallel transects which together provide a reasonable sample representation across the site (Siciliano et al., 2014). A total of 18 samples was selected for this investigation, from three transects, 2 m apart, with samples taken at variable lag distances of 0, 2, 100, 102, 200, and 202 m (Figure 1C). The soil samples (1 kg) were taken from top 10 cm of the soil profile, aseptically sieved and stored at −80°C until analyzed. Samples were taken from a number of different frost boils and all frost boils that were sampled from were given arbitrary number from 1 to 42.
Comprehensive physical and chemical properties of all 18 soil samples were obtained using standard approaches (van Dorst et al., 2014). Briefly, variables, such as location, elevation, slope and aspect were obtained using a combination of global positioning system (GPS), geographic information system (GIS) and site digital elevation models. Physical variables, such as grain size was measured by sieving soil particles larger than 2 mm and separating other particles based on grain size into mud (<63 μm) and sand (63–2000 μm). Chemical analysis, such as conductivity, pH, , , and was obtained using 1 in 5 dilution of soil on the dry mass basis (mg kg−1). Total carbon (TC), total phosphorus (TP), and total nitrogen (TN) were measured by combustion and NDIR gas analysis (Rayment and Lyons, 2011). The measurement of other elements concentration, such as SO3,P2O5, and Cl were determined using X-Ray Fluorescence (XFR) analysis.
The SSMS setup was performed following the protocol described by Ferrari et al. (2008) with slight modifications. Briefly, approximately 3–5 g of soil was placed into a sterile Tissue Culture Insert (TCI) (Merck Millipore; MA, USA) and the soil slurry was prepared by adding a few drops of water into the TCI and mixing well using a vortex. An inoculum was prepared by adding 1 g of the same soil to 10 ml of water and mixing well using vortex. The inoculum was then allowed to stand for 5 min to settle heavy particles and 100 μl of inoculum was filtered onto a 0.22 μm, 25 mm PolyCarbonate (PC) membrane (Merck Millipore; MA, USA) using filtration manifold. The PC membrane was then placed on top of pre-prepared TCI, within a six-well plate and incubated at 8°C for 21 days.
After 21 days of incubation, the presence of adequate growth of the micro-colonies on SSMS membranes was confirmed using epi-fluorescence microscopy following SYBR green staining (Invitrogen; Paisley UK). A quarter slice of a growth membrane was cut using a sterile scalpel blade and forceps and a thin layer of heated 1% agarose was then poured into a glass slide. The slice was inverted onto the agarose and allowed to dry at 30°C for 15 min. The growth membrane was taken from the slide and a drop of Vectashield mounting medium containing DAPI (Vector laboratories Inc, Burlingame) was added onto the remaining agarose. Subsequently, 10 μl of 1:100 dilutions of SYBR green was added to the agar. To stain fungi, 10 μl of 2X Calcofluor white stain (Sigma-Aldrich, Australia) was also added to the agarose and a coverslip applied. The microbial growth was then visualized using an Olympus BX61 microscope (Olympus, North Ryde Australia), with appropriate filters for green (Bacteria) and UV (fungi) fluorescence.
The prepGEM DNA extraction protocol was used as described in Ferrari et al. (2008) with some modifications. Firstly a quarter slice of each growth membrane was placed in a PCR tube with 99 μl of 1X Buffer (diluted from 10X) and 1 μl of prepGEM enzyme. Samples were mixed and placed in a thermocycler (Bioteke corporation), following the program of 37°C for 15 min, 75°C for 15 min, and 95°C for 15 min. The microfuge tube was centrifuged at 14,000 rpm for 3 min. The membrane slice was then removed with the help of sterile tweezers and 80 μl of DNA lysate was transferred to a new PCR tube. The genomic DNA was quantified using Quanti-iT™ Picogreen® dsDNA reagent (Invitrogen, Paisley UK) following manufacturer's instructions and stored at −20°C until used.
Genomic DNA was extracted in triplicate from the soil samples using the MP FastDNA™ SPIN Kit for soil (MP Biomedicals, NSW Australia), as described (van Dorst et al., 2014). The DNA extracts were quantified using Quanti-iT™ Picogreen® dsDNA reagent (Invitrogen, Paisley UK) on a black 96 well plate at an absorbance of 520 nm using a fluorescence plate reader (SpectraMax M3 Multi-Mode Microplate Reader; Molecular Devices, CA). The DNA lysate concentrations produced ranged from 0.21 to 0.37 ng μl−1 and were stored at −80°C until used further.
Bacterial ARISA was performed to allow for the selection of replicates for tag sequencing. In comparison with the metabarcoding approach, ARISA is a cheap and rapid alternative, ideal for ensuring appropriate replication in large microbial ecology studies (van Dorst et al., 2014). All soil DNA was extracted in triplicate and then ARISA was used to evaluate technical replicates and ecological patterns. One of each of the technical replicates was then selected for sequencing. Briefly, ARISA PCR was carried out targeting the bacterial ITS region using the universal primers 1392f; 5′ GYACACACCGCCCGT 3′and 5′ MAX labeled 23Sr 5′GGGTTBCCCCATTCRG3′ (Fisher and Triplett, 1999; Hewson and Fuhrman, 2006). The PCR reaction mix included 0.5 μM of forward (1392) and reverse primer (5′ MAX labeled 23Sr), 1X buffer, 2.5 mM MgCl2 (Promega), 0.25 mM of each dNTP (Promega), and 0.8 μg μl−1 of BSA (Promega), 1 unit Taq polymerase with addition of water to final reaction volume of 25 μl. The cycling temperature was 94°C for 2 min denaturation temperature followed by 30 cycles of amplification at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s with a final extension 72°C for 5 min. The fragment separation was performed at Macquarie University on an Applied Bio-system 3730 fragment analyzer (Life technology), which was further interpreted via GENEMAPPER software (Life Technology). The sequences obtained were further filtered by removing the background fluorescence. Fragments were assigned to appropriate bins and final data were transformed into peak area verses sample matrix where the single peak represented single OTU (van Dorst et al., 2014). The samples were then compared for shared and unique bacterial OTUs using the Primer 6 version 6.1.13 and PERMANOVA version 1.0.3 software and a nonmetric multidimensional scaling (nMDS) plot and a cumulative rank abundance was generated using a Bray-Curtis similarity matrix (Clarke and Gorley, 2006; van Dorst et al., 2014).
After confirming a strong correlation between technical replicates using ARISA (van Dorst et al., 2014), a representative gDNA extract from each replicate was sent for barcode tag pyrosequencing to an external facility (MR DNA, Molecular Research Laboratory, TX USA) which was performed on the Roche 454 FLX titanium platform. The research laboratory followed a single step 30 cycle PCR using Hot StartTaq Plus Master Mix Kit (Qiagen, Valencia, CA) with the following conditions: 94°C for 3 min denaturation, followed by 28 cycle of 94°C for 30 s, an annealing temperature of 53°C for 40 s and elongation at 72°C for 1 min with a final extension step at 72°C for 5 min (Dowd et al., 2008). The bacterial universal primers, 28F and 519R were used to amplify the bacterial DNA (Dowd et al., 2008). For fungi, the ITS1 and ITS2 region was targeted with the primers ITS1 and ITS4 (Gardes and Burns, 1993). Following PCR, all amplicons from different samples were mixed in equal concentration and purified using Agencourt Ampure (Agencourt Bioscience Corporation; MA, USA).
Raw 454 pyrosequenced data were received in the form of a standard flowgram format (sff) file. Flowgrams were processed according to Schloss et al. (2011) using MOTHUR software Version 1.32.1. Initially, bacterial 16S rRNA reads were denoised by the implementation of PyroNoise component ensuring all reads were 200 bp long. This step included removal of the barcode, primer sequences, sequences with homo-polymers longer than 8 bp and retrieving the reverse complement of each sequence (Quince et al., 2011). An alignment of sequences was generated against the SILVA-compatible alignment database (Pruesse et al., 2007) and chimeras and other contaminant sequences were removed (Schloss et al., 2009). The sequences were pre-clustered at 1% to account for 454's titanium instrument error (Huse et al., 2010). Aligned sequences were then clustered into OTUs at 4% divergence (96% similarity) for the best definition of species level (van Dorst et al., 2014). In addition to this, all singletons generated were removed (van Dorst et al., 2014). A sample by OTU abundance matrix was generated using the MOTHUR software pipeline (Schloss et al., 2009) and the taxonomic assignment was determined against the Greengenes database (2013 May version) (Kim et al., 2011).
Quality filtering was carried out using MOTHUR software Version 1.32.1 in a similar way to Bacteria. Chimeras were removed from the sequence data using UCHIME (Edgar et al., 2011). The sequence grouping was completed by adding the nucleotide sequences of tag barcodes and identified sequences using the SEED platform (Overbeek et al., 2014). The barcode sequences were removed using SEED. The ITS1 region was then detected and extracted with the ITSx (an improved software), as the ITSx tool has the capacity to extract higher proportions of true positives and remove non-ITS sequences from data sets (Bengtsson-Palme et al., 2013). The obtained reads were then clustered together at 97% similarity to closely define species using USEARCH, representative sequences were identified using mafft and singletons were removed (Caporaso et al., 2010). The sequences were then compared against the UNITE fungal ITS database (Koljalg et al., 2005). A sample by OTU abundance matrix was generated using the SEED package (Overbeek et al., 2014).
Diversity indices were generated within MOTHUR (Schloss et al., 2009) and utilized as part of the multivariate analysis. The bacterial and fungal OTU sequence datasets were transformed to square root, standardized and (dis) similarity matrix was generated using Bray-Curtis coefficient (Clarke and Gorley, 2006; Anderson et al., 2008). Using a Primer 6 version 6.1.13 and PERMANOVA version 1.0.3 software PCO plots were configured for visualization and overlaid with normalized environmental data using Person correlation (0.5) (Clarke and Gorley, 2006).
Following analysis of total microbial diversity, six of the 18 soils, were selected for cultivation experiments. The selection was based on the nMDS plot generated and the clustering of bacterial OTU abundances was performed at community similarities of 40% (samples similarity threshold within frost boils). Each represented a major cluster of community and a different frost boils across the sample landscape (Supplementary Figure 2). The matrix generated for the nMDS plot was also used for analysis of similarities (one-way ANOSIM) and factor frost boil was tested for its significance against soil environment, bacterial communities, and fungal communities.
The environmental parameters were combined into a matrix and visualized with a draftmans plot. The right skewed variables, such as elevation, slope, TN, sand, mud, NO2, NO3, TKP, pH, TC, PO4, SO3, SO4, P2O5 and conductivity were log transformed then all variables were normalized. The normalized environmental variables were used to create a resemblance matrix based on the Euclidean distance between samples (Supplementary Tables 2, 3). The environmental resemblance matrix was then used for a DistLM analysis in Primer and PERMANOVA (Clarke and Gorley, 2006; Ferrari et al., 2015). The selected criteria included adjusted R2 and step-wise procedures.
Fungi and bacteria were isolated by suspending 1 g of soil with 10 ml of ultrapure milliQ water and further diluted to between 102 and 104. Aliquots of 100 μl were transferred onto various media including BG11 (Sigma-Aldrich Castle Hill Australia), NA (Nutrient Agar, Sigma-Aldrich Castle Hill Australia), SCA (Starch Casein Agar- US Biological, Swampscott USA) and RAVAN (Watve et al., 2000) for bacterial isolation. For fungal isolation, PDA (Potato dextrose agar- Sigma-Aldrich Castle Hill Australia), CRBA (Crook Rose Bengal Agar) and MEA (Malt Extract Agar-Sigma-Aldrich Castle Hill Australia) were used. All media were prepared to 1X and 0.1X concentrations and incubated at 8°C for 1–2 months or RT (20 ± 1°C) for 7–15 days. Bacterial isolates were sub-cultured at least three times onto NA plates and fungi were sub-cultured on PDA plates. The use of 0.1X media and SSMS were to select for “K” strategists.
Following the isolation of pure cultures, genomic DNA was extracted from bacterial isolates using a simplified FastPrep bead beating method previously applied for fungal DNA extraction (Ferrari et al., 2011). For all fungal isolates, the Fast DNA® SPIN Kit (MP Biomedicals, NSW Australia) was used following the manufacturer's instructions.
All primers required for bacterial and fungal PCR amplification were obtained from Integrated DNA Technology (IDT; MCLeans Ridge, Australia). The reaction components used for bacterial 16S rRNA gene PCR were 10 μl of 5X Go Taq buffer (Promega; Madison, USA), 4 μl MgCl2 (25 mM), 1 μl dNTP (10 mM), 40 mM 1 μl each Forward Primer (356F) and Reverse Primer (1064R) (Winsley et al., 2012), 0.25 μl (5 U μl−1) Taq polymerase, 2 μl DNA, 5 ml BSA (0.1 μg μl−1), 24.75 μl water (Nuclease-free water). The PCR program consisted of an initial denaturation of 95°C for 3 min, 35 cycles of 95°C for 35 s, annealing temperature of 60°C for 30 s and an extension step at 72°C for 5 min.
Fungal internal transcribed spacer (ITS) region genes were amplified using ITS1 forward primer and ITS4 reverse primer (Gardes and Burns, 1993) as described by Ferrari et al. (2011) with slight modification. The PCR reaction mixture consisted of 10 μl of 5X Go Taq buffer, 4 μl MgCl2 (25 mM), 1 μl dNTP (10 mM), 40 mM 1 μl each Forward Primer (ITS1) and Reverse Primer (ITS4) (Gardes and Burns, 1993), 0.25 μl (5 U μl−1) Taq polymerase, 2 μl DNA, 5 μL BSA (0.1 μg μl−1) and 24.75 μl water (Nuclease-free water). The PCR program consisted of a 94°C initial denaturation step for 2 min, followed by 30 cycles of 94°C for 45 s, an annealing temperature of 55°C for 30 s and a final extension step at 72°C for 5 min. PCR amplicons were then visualized on 2% agarose gel stained with SybrSafe (Invitrogen, Paisley UK). The PCR product was treated with two restriction enzymes, Hinfl (Promega, Madison, USA) with the cleavage site 5′-G↓ANT C-3′, 3′-C TNA↑G-5′ and Rsal (Promega, Madison, USA) with cleavage site 5′↓GTAC-3′-3′ CATG↑ 5′. The reaction component included 0.17 μl restriction enzyme (Hinfl or Rsal), 2 μl 10X buffer B for Hinfl and Buffer C for Rsal, 0.2 μl 100X BSA, 2.63 μl water, and 15 μl of PCR product. The mixture was incubated overnight at 37°C and the digest was loaded onto 2% agarose gel and analyzed for band sizes. From selected isolates, PCR products (bacteria and fungi) were then purified by using the QIAquick PCR purification kit from Qiagen following the manufacturer's instructions. The purified PCR amplicons were sequenced at the Ramaciotti Center for Gene Function Analysis (UNSW, Australia) for Sanger sequencing using ABI 3730 Capillary sequencer. The data received were used for isolate identification by blasting (BLASTn) sequences against NCBI database (Johnson et al., 2008).
To compare the OTUs recovered using traditional cultivation and the SSMS from soil samples, the bacterial and fungal OTUs recovered using both cultivation methods were combined with the OTUs identified from total soil community sequencing. For cultured bacteria, the 16S rRNA gene sequences obtained from Sanger sequencing were realigned and trimmed to the same length as the tag sequences obtained from the soil and SSMS enrichments using the MOTHUR pipeline. The common bacterial and fungal OTUs were identified by clustering sequences at 97% identity using USEARCH (Edgar, 2010).
Bacterial and fungal sequences obtained from the SSMS have been deposited in NCBI under accession number PRJNA350563. For cultured bacterial isolates the sequences were deposited into NCBI under accession numbers KY432693-KY432726, and fungal isolates under KY432734-KY432749 and KY608091. Pyrosequencing data for the soil is available at https://data.aad.gov.au/metadata/records/soil_bacteria_fungi.
A total of 57,893 16S rRNA and 62,547 ITS quality checked bacterial and fungal reads were recovered after processing the raw datasets from total soil gDNA. Using a similarity threshold of 96% to cluster bacterial sequences and 97% to cluster fungal sequences (Siciliano et al., 2014), a total of 2063 bacterial and 279 fungal OTUs were obtained. Overall bacterial diversity was much greater than fungal diversity (Supplementary Table 1). Rarefaction curves revealed that fungal richness reached asymptote, whilst the bacterial OTU richness was still increasing (Supplementary Figure 1). Soil bacterial diversity detected by tag pyrosequencing spanned 36 phyla, 84 classes, 214 families and 407 genera, whilst fungal diversity was limited to four phyla, spanning 14 classes, 59 families, 104 genera and one unclassified fungi.
Actinobacteria dominated the bacterial communities present, comprising >36% of the total relative abundance across the site. Chloroflexi was the next most abundant phyla, followed by Acidobacteria, Cyanobacteria and Proteobacteria; together accounting for over 54% of total relative abundance within the entire 18 samples analyzed (Supplementary Figure 3). For fungal diversity, Ascomycota dominated all soil samples analyzed, accounting for 93% of the total relative abundance, followed by Basidiomycota (5%) and Zygomycota (1%) (Supplementary Figure 4).
We selected six of the soils from the dataset for a comparison against the SSMS and traditional culturing. In total, 31,797 bacterial quality checked reads representing 401 bacterial OTUs were obtained from the SSMS dataset. Together, the SSMS enrichments recovered 19 phyla (52% of total relative abundance), 61 classes, and 234 (48%) of the genera detected in soil directly. This is 15 times higher than by cultivation only (Figure 4), as traditional culturing using artificial media recovered just 4 (11%) of the bacterial phyla and 15 (3%) of the genera originally detected in the soil gDNA sequencing dataset. Despite the large number of quality checked reads for the fungal ITS region, the fungal community, as expected, represented a limited diversity covering just two phyla, 9 classes, and 30 genera.
The relative abundance of major bacterial phyla present in the enriched SSMS varied considerably to the total soil communities (Figure 2). Instead of a dominance of Actinobacteria, Proteobacteria (58%) was most dominant followed by Actinobacteria (36%), Firmicutes (2%), and Acidobacteria (1%). Of major interest was the presence of a further 15 bacterial phyla, such as Bacteroidetes, Chloroflexi, Gemmatimonadetes, TM7, Planctomycetes and Verrucomicrobia representing only 3% of total relative abundance recovered from SSMS enrichments. Of these, Bacteroidetes and Gemmatimonadetes were most dominant; both were present in soil at 2 and 6% of the total relative abundance. There was a 2-fold decrease in the total relative abundance of Bacteroidetes and 6-fold decrease in the total relative abundance of Gemmatimonadetes.
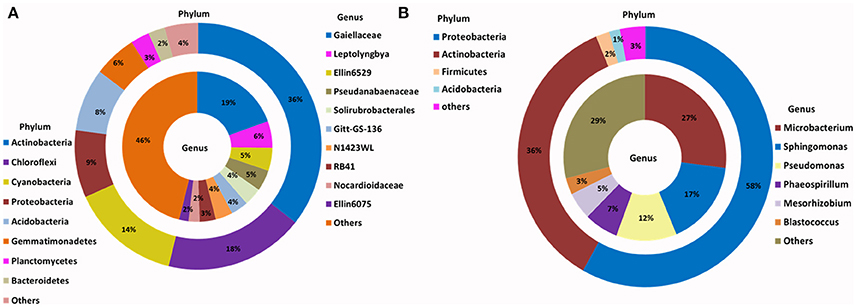
Figure 2. Phylogenetic distribution of bacteria across (A) Browning Peninsula soil: data from metabarcoding and (B) SSMS soil enrichments: data from metabarcoding. (A) Actinobacteria and Chloroflexi were the most dominant bacterial phyla while, unclassified Gaiellaceae and unclassified Ellin6529 were most dominant at the genus level. (B) Most abundant bacterial phyla in SSMS enrichments were Proteobacteria followed by Actinobacteria and the dominant genera were Microbacterium and Sphingomonas. Others represent a sum of relative abundance of <2% phyla or genera present in all soils or SSMS enrichment used.
Within Actinobacteria, while the relative abundance was similar between the soil and SSMS enrichments, large shifts in the recovered community were observed at the genus level (Figure 2). For example, unclassified Gaiellaceae, the dominant genera in Browning soil attributing >19% of total relative abundance, was replaced by Microbacterium representing 27% of total relative abundance in the SSMS enrichments (Figure 2). While a few reads related to unclassified Gaiellaceae were also retrieved from the SSMS pyrosequencing dataset, the relative abundance was negligible. Other dominant genera in the soil gDNA dataset including Ellin6529 (Chloroflexi; 5%), RB41 (Acidobacteria; 3%), and Solirubrobacterales (Actinobacteria; 4%) were retrieved only as a minor fraction (<0.05%) of the community in the enrichments (Figure 2). The well characterized cyanobacterium, Leptolyngbya present at 6% of the relative abundance in the soil dataset was not recovered in the SSMS. Importantly, a further 400 minor genera, for examples Microbacterium, Sphingomonas, Pseudomonas, Phaeospirillum and Blastococcus which together accounted for just 7% of total relative abundance in soil thrived in the SSMS enrichment reaching up to 67% of the total relative abundance. Among these isolates Microbacterium, Sphingomonas, and Pseudomonas were endemic to Antarctica.
Following traditional cultivation, 380 bacterial strains were isolated to purity and 207 were successfully extracted and amplified for identification. After screening using RFLP, in total, 34 bacterial strains distributed among 15 bacterial genera within four phyla were identified; Actinobacteria (99–100% identity), Proteobacteria (96–100%), Firmicutes (96–100%) and Bacteroidetes (99% identity) (Table 1). These strains comprised the genera Kribbella, Mycobacterium, Paenisporosarcina, Sphingomonas, Arthrobacter, Streptomyces, and Caulobacter with >99% identity to known species (Table 1). The most common bacterial isolates matched with Streptomyces fildesensis (100% identity) previously isolated from rhizosphere of moss and Arthrobacter scleromae (>99% identity) previously isolated from a human specimen. Other bacterial genera isolated were most similar to strains previously isolated from clinical samples, such as Mycobacterium fluoranthenivorans (99% identity) and Dyella japonica (99% identity).
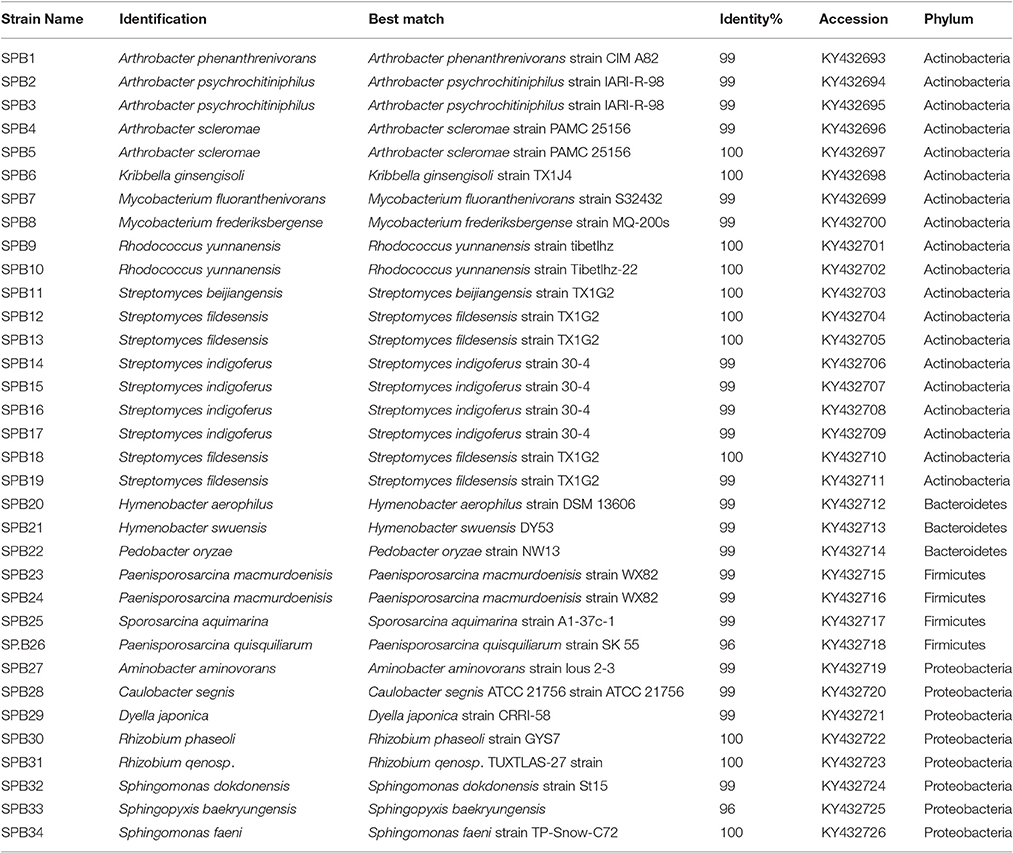
Table 1. Bacteria recovered from different culture media from Browning Peninsula soils.
The measured chemical and physical properties for all 18 soils showed the carbon content across the sampling site ranged from 2.79 to 3.5% w/w (log transformed) while the pH ranged from 6.38 to 6.75 (Supplementary Table 2). It was expected that the distribution of frost boils across the Browning Peninsula would influence the localized soil chemical and physical environment, and subsequently the microbial diversity distribution. This was found to be the case when tested with an analysis of similarity (ANOSIM), with the frost boils significantly correlated to both the local soil physical and chemical parameters (P < 0.001, a global R-value of 0.726) and the distribution of the bacterial community (P < 0.001, a global R-value of 0.922). However, the frost boil distribution had no significant correlation with the fungal community composition (P > 0.1, a global R-value of −0.215).
In this study, the environmental factors chlorine, phosphate, aspect and elevation were observed to be significantly affecting bacterial community distribution across the site. However, fungal distributions were not affected by any of the environmental variables analyzed. Chlorine content was observed to be most significant predictor variable with pseudo F = 3.75, P < 0.001, and the biological variance of 19% (Table 3). Furthermore, aspect degree, phosphate and elevation added up to 25% of the biological variance. For other variables, such as soil conductivity (18%), grain size (sand and mud together 21%), and total nitrogen (9%), the variables were only statistically significant when analyzed individually (Table 3).
For the SSMS enrichments, 110,851 fungal quality checked reads representing 86 fungal OTUs were obtained following tag pyrosequencing. It was demonstrated here that in addition to bacteria various fungi, particularly microfungi (Cladosporium, Malassezia, Exophiala, Horteae, and Rhodotorula) were successfully enriched for using the SSMS (Figure 3). While three phyla dominated the soil dataset, following SSMS enrichments, only two were remained with the relative abundance of Basidiomycota increasing from 2 to 17% (Figure 3).
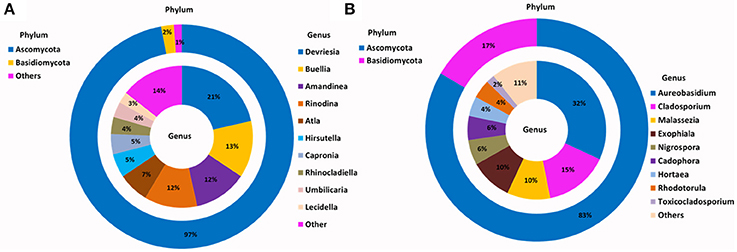
Figure 3. Phylogenetic distribution of fungi across (A) Browning Peninsula soil: data from metabarcoding and (B) SSMS soil enrichments: data from metabarcoding. (A) Dominant fungal phyla in Browning Peninsula soil were Ascomycota followed by Basidiomycota and at the genera level Devriesia and Buellia were most abundant. (B) Only Ascomycota and Basidiomycota were enriched in the SSMS, with Aureobasidium the most dominant fungal genera recovered. Others represent a sum of <2% relative abundance of other phyla or genera present in the soils or SSMS enrichments.
At the genus level, Devriesia (Ascomycota), which represented >30% of total relative abundance of soils, as well as Buellia (Ascomycota) which represented >13% of relative abundance and Amandinae, Capronia, Rinodina, Atla, and Hirsutella (together representing 41% of relative abundance) were not detected in the enrichments. Instead, Aureobasidium (Ascomycota; 32%) appeared as the most dominant fungi in the SSMS enrichment. As with bacteria, 96 minor genera present in the soil gDNA dataset, which accounted for just 14% of the total relative abundance (ie. Cladosporium, Malassezia and Rhodotorula), were selected for in the SSMS enrichments, reaching up to 29% of the total relative abundance (Figure 3). Among these Malassezia and Rhodotorula belonged to the Basidiomycota.
Using artificial media, 80 fungal strains were isolated to purity. After RFLP, 17 strains were selected for sequencing. In total, 17 strains spanning two phyla, Ascomycota (>96% identity) and Basidiomycota (>98% identity), were identified including Cryptococcus, Phoma, Engyodontium, Thelobolus and Cladosporium (Table 2). Almost all fungal strains isolated from Browning Peninsula were microfungi with >98% similarity to previously isolated fungi species (Table 2). The most abundant microfungi strain recovered belonged to Geomyces pannorum (99% identity), previously isolated from the cyst of a nematode (Globodera pallida) and Holtermanniella watticus (>99% identity), previously isolated from King George Island, Chile (Table 2).
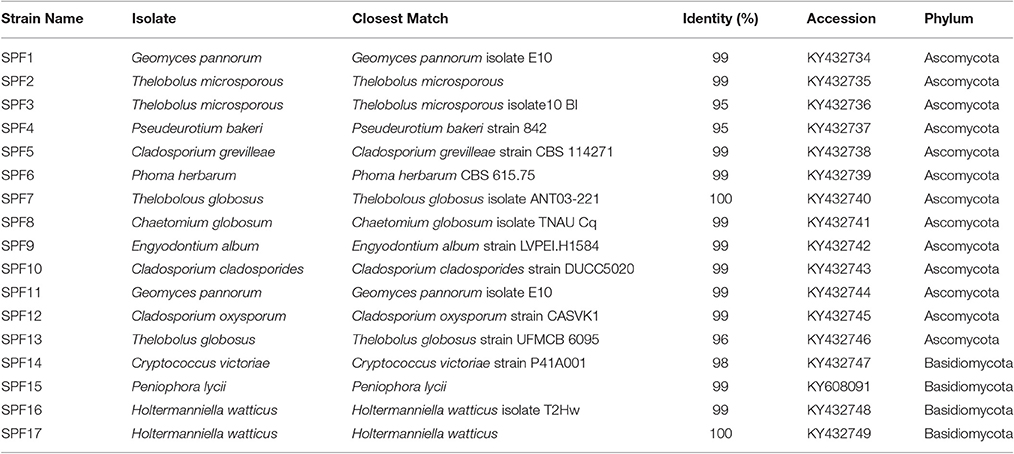
Table 2. Fungi recovered from different cultivation media from Browning Peninsula soil.
A large proportion of bacterial and fungal OTUs were not shared between culture dependent and independent techniques (Figure 4). For example, 135 OTUs detected were common in soil and SSMS datasets; only four OTUs were common in SSMS and artificial media and one OTU was present in the artificial media and soil datasets (Figure 4). In fact, there were more unique bacterial genera retrieved than shared between techniques. For instance, 824 bacterial OTUs were not retrieved from culture-dependent techniques. Overall, only 13 unique OTUs were obtained from artificial-cultivation. Surprisingly, 306 bacterial OTUs recovered from SSMS were also not detected in the other two approaches suggesting minor taxa are enriched for using the SSMS (Figure 4).
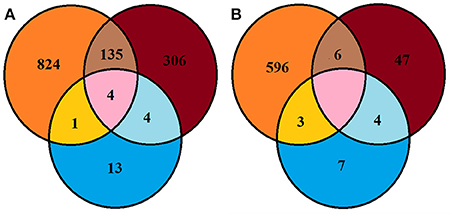
Figure 4. Venn diagram representing Browning Peninsula microbial diversity at the OTU level recovered from all three methods used in this study (A) bacteria (B) fungi. Orange circles indicate OTUs recovered from the soil datasets, blue represents the recovery of OTUs from artificial cultivation and brown represents OTUs recovered from the SSMS. The pink area represents common OTUs recovered from the soil, SSMS microcultivation and artificial cultivation.
The Shannon diversity index for fungi retrieved from the soil gDNA dataset was 3.51 markedly lower than for bacteria (5.5) (Supplementary Table 1). A similar recovery pattern of bacterial and fungal communities was observed from the SSMS enrichments with a Shannon index of 3.15 for bacteria and 2.57 for fungi. Combined, a total of 663 fungal OTUs were recovered using three approaches, yet no common OTU was recovered from all three methods used. Altogether, six of the fungal OTUs recovered were common in the soil and SSMS enrichments, four OTUs were common in enrichments and artificial media, while three OTUs were present in the soil and artificial media datasets. Similar to the bacterial data, 596 unique fungal OTUs were retrieved from the soil dataset only, 47 OTUs from SSMS enrichments and seven OTUs from cultivation (Figure 4).
Comparing recovery of individual phylum level OTUs from both soil and SSMS, SSMS recovered 39.5% of unique Proteobacteria, 23% of Actinobacteria, 27% of Bacteroidetes and 24% of Firmicutes (Supplementary Table 4). The shared OTUs were 10% for Proteobacteria, 11% for Actinobacteria, 12% for Bacteroidetes and 3.5% for Firmicutes. The recovery of fungal unique Ascomycota OTUs from soil was 90% and SSMS was 9%. For Basidiomycota unique OTUs obtained were 87% from soil and 7.23% from SSMS (Supplementary Table 5).
The soils of Browning Peninsula harbor a bacterial community dominated by Actinobacteria (up to a 36% relative abundance), followed by Chloroflexi, Cyanobacteria, Proteobacteria and Acidobacteria. The high abundance of Actinobacteria is in stark contrast to Arctic and other Antarctic sites commonly dominated by Proteobacteria, Cyanobacteria, and Acidobacteria (Steven et al., 2007, 2013; Koyama et al., 2014). For example, the McMurdo Dry Valleys, a well-studied site of Antarctica, has been found to be dominated by Proteobacteria (Clocksin et al., 2007; Cary et al., 2010); Miers Valley, is dominated by Bacteriodetes, Proteobacteria, and Actinobacteria (Stomeo et al., 2012), and Victoria Land is comprised of a heterogeneous bacterial community encompassing Proteobacteria, Acidobacteria, Actinobacteria, Chloroflexi, and Cyanobacteria (Kim et al., 2015). The variability in diversity structures in each of these sites may be due to variable organic matter content (Takebayashi et al., 2007; Bell et al., 2013; Ji et al., 2015). Previously, the dominance of Proteobacteria was linked with areas rich in higher organic matter (Bell et al., 2013) and moisture content (Horn et al., 2014; Niederberger et al., 2015), while dominance of Actinobacteria has been documented in arid soils (Niederberger et al., 2015), and linked to low moisture and nutrients (Takebayashi et al., 2007). Further biotic and abiotic factors, such as plants, penguins, birds, seal carcasses, and human activity are also reported to affect microbial community distributions across Antarctica (Cowan et al., 2014; Wang et al., 2015).
Although fungi are more tolerant of freezing and thawing compared to bacteria (Sharma et al., 2006), the fungal diversity of Browning soil was markedly lower than that of the bacterial community. Of those fungi present, Browning soil was dominated by Ascomycota (>97% of the total relative abundance) and was limited to four phyla only. In contrast, nearby Eastern Antarctic sites that do not contain frost boils harbor at least 60% more fungal richness than Browning Peninsula (Ferrari et al., 2015). A lack of tolerance of extreme variation in the salinity, combined with continuous mixing of soil due to cryoturbation, may have limited the fungal diversity at Browning Peninsula (Ferrari et al., 2015). Dispersal limitation across the site due to the presence of hard-edge barriers surrounding frost boils, may also contribute to the reduced fungal richness observed compared to highly connected Antarctic sites, as the larger size of fungi limits their dispersal compared to bacteria (Rao et al., 2011; Ferrari et al., 2015). Moreover, the lack of vegetation (Stewart et al., 2011), and fungal vectors including insects and animals could contribute to reduced fungal diversity in Antarctica compared with those of the Arctic. Fungi has been shown to have symbiotic association with plants (Geml et al., 2012; Timling et al., 2012) and no plant life has been documented at Browning Peninsula (Stewart et al., 2011).
The environmental factors chlorine, phosphate, aspect and elevation were observed to affect the bacterial community at Browning, whilst none of the analyzed environmental parameters were observed to be statistically significant to affect fungal community variation. Of the environmental parameters analyzed, chlorine was primarily correlated to the bacterial community composition, contributing 19% of the total biological variance. Chlorine was not significant to fungal community variation here, but has been previously observed to have strong effects on polar fungi (Siciliano et al., 2014). Elevation was also observed to affect the bacterial distribution and several in-depth studies of effect of elevation on microbial community have revealed dramatic shifts in the microbial taxa along elevation transects (Bryant et al., 2008; Cowan et al., 2010; Shen et al., 2013). It is also well established that the soil heterogeneity of Arctic and Antarctic soil depends on organic matter (total carbon) and nitrate (total nitrogen) availability (Michaelson et al., 2012; Shen et al., 2013; Siciliano et al., 2014). The reduced bacterial and fungal diversity of Browning Peninsula soil may, therefore, be a result of the relatively poor nutrient content of these soils (Chong et al., 2009). The phosphate content and aspect were also correlated to the bacterial community contributing to 19% of the total variation (Table 3). In contrast, fungal diversity distributions have been previously linked with pH (Connell et al., 2006; Arenz and Blanchette, 2011), conductivity (Arenz and Blanchette, 2011) and moisture content (Connell et al., 2006). However, none of the environmental factors were significantly correlated with the fungal community distribution here, which may be due to the unique landscape and limited diversity present.
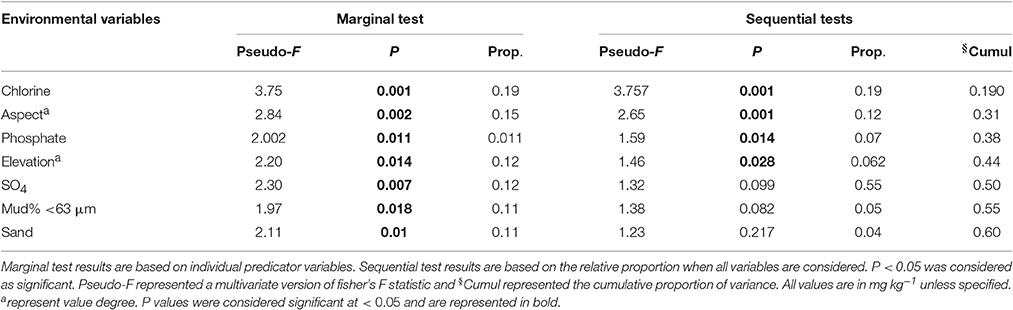
Table 3. DistLM results indicating the correlation between environmental parameters as a predictor of the microbial community distribution.
The present investigation was the first study to culture fungi using the SSMS, with 30 non-lichenized microfungi including Toxicocladosporium, Exophiala, Hortaea, and Cadophora species selected for using the approach. From the two phyla recovered, Ascomycota members enriched for included Aureobasidium, Cladosporium, Nigrosphora and Cadophora, while Basidiomycota members included Malassezia, Hortaea, and Rhodotorula (Figure 3). Most of these fungal genera have been previously isolated from soils, plants and animals. However, the SSMS appeared to facilitate the cultivation of a wide range of microfungi, as demonstrated by the recovery of the endemic mesophilic psychrotolerant microfungi, such as Aureobasidium and Cladosporium (Zucconi et al., 2011) to host specific genera, such as Nigrosphora, Malassezia genera; a plant and animal pathogen (Hudson, 1963; Velegraki et al., 2015), Cadophora; a “soft rot” wood decomposer isolated from Antarctic hut wood (Blanchette et al., 2004; Ludley and Robinson, 2008), Rhodotorula; previously isolated from Windmill Island, Antarctica soils (Gesheva, 2010) and Hortaea; a halotolerant black yeast (Plemenitas et al., 2008). For bacteria, the SSMS enrichments led to an increase in Proteobacteria relative abundances up to 58%, highlighting the system's selective bias toward heterotrophs (Ferrari et al., 2008). Under the conditions used, the SSMS failed to enrich for Cyanobacteria, Chlorobi and Chloroflexi. Further modifications to include a light source may aid in the isolation of these phototrophic taxa.
The most commonly isolated bacteria Arthrobacter and Streptomyces were previously isolated from Antarctic cold deserts (Smith et al., 2006), post-glacial soils (Zdanowski et al., 2012) and Princess Elisabeth Station soil (Peeters et al., 2011), temperate soils and variety of other sources (Kaewkla and Franco, 2013). For fungal isolates, Geomyces, Thelobolus and Phoma have been previously recovered from Antarctic soils (Frate and Caretta, 1990; Azmi and Seppelt, 1998; Arenz et al., 2006; Connell et al., 2006; Arenz and Blanchette, 2011; Godinho et al., 2013). Interestingly, Chrysosporium sp., Mortierella gamsii, Mycelia sterilia, Phoma sp., and Thelobolus microspores have also been previously isolated from Browning Peninsula (Azmi and Seppelt, 1998). Similar to Devriesia the requirement for a heat pre-treatment to germinate cells before culturing into artificial media (Onofri et al., 2011) may have been a limiting factor in the diversity of cultured members. The recovery of cold-adapted bacterial strains, such as Dyella have been proposed to degrade single molecules, such as N-Acylhomoserine Lactones (Chen and Chan, 2012), and UV radiation-resistant fungi, such as Phoma (Hughes et al., 2003) may have significant biotechnological potential that warrants further investigation.
Of the 1284 bacterial OTUs and 663 fungal OTUs detected using multiple approaches here, tag pyrosequencing from soil gDNA detected 75% of the total bacterial and 91% of total fungal community diversity, which is in line with previous studies reporting ≤60% of the total bacterial diversity detected using sequencing alone (Vaz-Moreira et al., 2011; Stefani et al., 2015). It is well established that very low percentages of bacteria have been recovered into pure culture (Handelsman, 2004; Ferrari et al., 2008), and here 1.7% of the total bacteria diversity was recovered into artificial media. This recovery increased to 34% within the SSMS enrichments, highlighting the potential of this approach to recover novel species (van Dorst et al., 2016). This is comparable to the recovery of 50% of total bacterial diversity using the ichip method (a similar culturing technique) described by Nichols et al. (2010).
Only four of the total bacterial OTUs detected were present in all three techniques used. The absence of consistency between techniques, or a “culture clash” has been consistently observed between clone libraries and cultivation attempts (Maturrano et al., 2006). Previously, higher community diversity (2.4% of bacterial and 8.2% of fungal communities) has been shared between artificial media and tag pyrosequencing from hydrocarbon contaminated soils(Stefani et al., 2015; van Dorst et al., 2016). The rapid growth of microfungi, particularly yeasts on the SSMS may have occurred following the addition of water to the soils (Vishniac, 1996) with freely available water potentially aiding the germination of spores.
Microfungi are difficult to culture and enrichment of microfungi using the SSMS offers a new opportunity to harness the biotechnological potential of cold-adapted Antarctic fungi. Despite inherent bias associated with the techniques used, such as selection of phenotypic characteristics of colonies from artificial media (Tytgat et al., 2014), DNA extraction (Evans and Seviour, 2012; Plassart et al., 2012), PCR amplification (Schloss et al., 2011) and inadequate sequence-depth when carrying out 454 tag pyrosequencing (Kim et al., 2011; Kumar et al., 2011; Lagier et al., 2012), multiple approaches delivered a comprehensive view of the soil microbial community of Browning Peninsula, unveiling an ecosystem dominated by rare unclassified bacterial taxa combine with extremely low fungal diversity.
All authors contributed to the study design. SP, MJ, JV, and AP carried out the experimental procedure and data analysis of the present study. SP prepared the draft of the manuscript. SP, JW, MJ, JV, IS, AP, and BF finalized the manuscript. All authors read and approved the manuscript. JW and BF coordinated this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the Australian Antarctic Division and field team for collecting soil samples from Browning Peninsula, Antarctica and UNSW Australia and Sydney for financial support.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00591/full#supplementary-material
Anderson, M. J., Gorley, M. J., and Clarke, K. R. (2008). PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods. Plymouth: PRIMER-E.
Arenz, B. E., and Blanchette, R. A. (2011). Distribution and abundance of soil fungi in Antarctica at sites on the Peninsula, Ross Sea Region and McMurdo Dry Valleys. Soil Biol. Biochem. 43, 308–315. doi: 10.1016/j.soilbio.2010.10.016
Arenz, B. E., Held, B. W., Jurgens, J. A., Farrell, R. L., and Blanchette, R. A. (2006). Fungal diversity in soils and historic wood from the Ross Sea Region of Antarctica. Soil Biol. Biochem. 38, 3057–3064. doi: 10.1016/j.soilbio.2006.01.016
Azmi, O. R., and Seppelt, R. D. (1998). The broad-scale distribution of microfungi in the Windmill Islands region, continental Antarctica. Polar Biol. 19, 92–100. doi: 10.1007/s003000050219
Bell, T. H., Yergeau, E., Maynard, C., Juck, D., Whyte, L. G., and Greer, C. W. (2013). Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J. 7, 1200–1210. doi: 10.1038/ismej.2013.1
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4, 914–919. doi: 10.1111/2041-210x.12073
Beyer, L., and Bolter, M. (2000). Chemical and biological properties, formation, occurance and classification of Spodic Cryosol in terrestrail ecosystem of East Antarctica (Wilkes Land). CATENA 39, 95–119. doi: 10.1016/S0341-8162(99)00089-2
Blanchette, R. A., Held, B. W., Jurgens, J. A., McNew, D. L., Harrington, T. C., Duncan, S. M., et al. (2004). Wood-destroying soft rot fungi in the historic expedition huts of Antarctica. Appl. Environ. Microbiol. 70, 1328–1335. doi: 10.1128/AEM.70.3.1328-1335.2004
Bollmann, A., Lewis, K., and Epstein, S. S. (2007). Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Appl. Environ. Microbiol. 73, 6386–6390. doi: 10.1128/AEM.01309-07
Bollmann, A., Palumbo, A. V., Lewis, K., and Epstein, S. S. (2010). Isolation and physiology of bacteria from contaminated subsurface sediments. Appl. Environ. Microbiol. 76, 7413–7419. doi: 10.1128/AEM.00376-10
Bryant, J. A., Lamanna, C., Morlon, H., Kerkhoff, A. J., Enquist, B. J., and Green, J. L. (2008). Colloquium paper: microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. U.S.A. 105 (Suppl. 1), 11505–11511. doi: 10.1073/pnas.0801920105
Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Cary, S. C., McDonald, I. R., Barrett, J. E., and Cowan, D. A. (2010). On the rocks: the microbiology of Antarctic Dry Valley soils. Nat. Rev. Microbiol. 8, 129–138. doi: 10.1038/nrmicro2281
Chen, J. W., and Chan, K. G. (2012). Genome sequence of Dyella japonica strain A8, a quorum-quenching bacterium that degrades N-acylhomoserine lactones, isolated from Malaysian tropical soil. J. Bacteriol. 194:6331. doi: 10.1128/jb.01637-12
Chong, C. W., Annie Tan, G. Y., Wong, R. C. S., Riddle, M. J., and Tan, I. K. P. (2009). DGGE fingerprinting of bacteria in soils from eight ecologically different sites around Casey Station, Antarctica. Polar Biol. 32, 853–860. doi: 10.1007/s00300-009-0585-6
Clocksin, K. M., Jung, D. O., and Madigan, M. T. (2007). Cold-active chemoorganotrophic bacteria from permanently ice-covered Lake Hoare, McMurdo Dry Valleys, Antarctica. Appl. Environ. Microbiol. 73, 3077–3083. doi: 10.1128/AEM.00085-07
Connell, L., Redman, R., Craig, S., and Rodriguez, R. (2006). Distribution and abundance of fungi in the soils of Taylor Valley, Antarctica. Soil Biol. Biochem. 38, 3083–3094. doi: 10.1016/j.soilbio.2006.02.016
Cowan, D. A., Makhalanyane, T. P., Dennis, P. G., and Hopkins, D. W. (2014). Microbial ecology and biogeochemistry of continental Antarctic soils. Front. Microbiol. 5:154. doi: 10.3389/fmicb.2014.00154
Cowan, D. A., Pointing, S. B., Stevens, M. I., Craig Cary, S., Stomeo, F., and Tuffin, I. M. (2010). Distribution and abiotic influences on hypolithic microbial communities in an Antarctic Dry Valley. Polar Biol. 34, 307–311. doi: 10.1007/s00300-010-0872-2
Davis, K. E., Joseph, S. J., and Janssen, P. H. (2005). Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Appl. Environ. Microbiol. 71, 826–834. doi: 10.1128/AEM.71.2.826-834.2005
Donachie, S. P., Foster, J. S., and Brown, M. V. (2007). Culture clash: challenging the dogma of microbial diversity. ISME J. 1, 97–99. doi: 10.1038/ismej.2007.22
Dowd, S. E., Callaway, T. R., Wolcott, R. D., Sun, Y., McKeehan, T., Hagevoort, R. G., et al. (2008). Evaluation of the bacterial diversity in the feces of cattle using 16S rRNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 8:125. doi: 10.1186/1471-2180-8-125
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Evans, T. N., and Seviour, R. J. (2012). Estimating biodiversity of fungi in activated sludge communities using culture-independent methods. Microb. Ecol. 63, 773–786. doi: 10.1007/s00248-011-9984-7
Ferrari, B. C., Binnerup, S. J., and Gillings, M. (2005). Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Appl. Environ. Microbiol. 71, 8714–8720. doi: 10.1128/AEM.71.12.8714-8720.2005
Ferrari, B. C., Bissett, A., Snape, I., van Dorst, J., Palmer, A. S., Ji, M., et al. (2015). Geological connectivity drives microbial community structure and connectivity in polar, terrestrial ecosystems. Environ. Microbiol. 18, 1834–1849. doi: 10.1111/1462-2920.13034
Ferrari, B. C., Winsley, T., Gillings, M., and Binnerup, S. (2008). Cultivating previously uncultured soil bacteria using a soil substrate membrane system. Nat. Protoc. 3, 1261–1269. doi: 10.1038/nprot.2008.102
Ferrari, B. C., Zhang, C., and van Dorst, J. (2011). Recovering greater fungal diversity from pristine and diesel fuel contaminated sub-antarctic soil through cultivation using both a high and a low nutrient media approach. Front. Microbiol. 2:217. doi: 10.3389/fmicb.2011.00217
Fisher, M. M., and Triplett, E. W. (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 65, 4630–4636.
Frate, G. D., and Caretta, G. (1990). Fungi isolated from Antarctic material. Polar Biol. 11, 1–7. doi: 10.1007/BF00236515
Gardes, M., and Burns, T. D. (1993). ITS primers with enhanced specificity for basidomycetes-application to the identification of mycorrhizae and rust. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Gavrish, E., Bollmann, A., Epstein, S., and Lewis, K. (2008). A trap for in situ cultivation of filamentous actinobacteria. J. Microbiol. Methods 72, 257–262. doi: 10.1016/j.mimet.2007.12.009
Geml, J., Timling, I., Robinson, C. H., Lennon, N., Nusbaum, H. C., Brochmann, C., et al. (2012). An arctic community of symbiotic fungi assembled by long-distance dispersers: phylogenetic diversity of ectomycorrhizal basidiomycetes in Svalbard based on soil and sporocarp DNA. J. Biogeogr. 39, 74–88. doi: 10.1111/j.1365-2699.2011.02588.x
Gesheva, V. (2010). Production of antibiotics and enzymes by soil microorganisms from the windmill islands region, Wilkes Land, East Antarctica. Polar Biol. 33, 1351–1357. doi: 10.1007/s00300-010-0824-x
Gesheva, V., and Negoita, T. (2011). Psychrotrophic microorganism communities in soils of Haswell Island, Antarctica, and their biosynthetic potential. Polar Biol. 35, 291–297. doi: 10.1007/s00300-011-1052-8
Godinho, V. M., Furbino, L. E., Santiago, I. F., Pellizzari, F. M., Yokoya, N. S., Pupo, D., et al. (2013). Diversity and bioprospecting of fungal communities associated with endemic and cold-adapted macroalgae in Antarctica. ISME J. 7, 1434–1451. doi: 10.1038/ismej.2013.77
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685. doi: 10.1128/MMBR.68.4.669-685.2004
Hewson, I., and Fuhrman, J. A. (2006). Improved strategy for comparing microbial assemblage fingerprints. Microb. Ecol. 51, 147–153. doi: 10.1007/s00248-005-0144-9
Horn, D. J. V., Okle, J. G., Buelow, H. N., Gooseff, M. N., Barrett, J. E., and Takacs-Vesbach, C. D. (2014). Soil microbial responses to increased moisture and organic resources along a salinit gradient in a polar desert. Appl. Environ. Microbiol. 80, 3034–3043. doi: 10.1128/AEM.03414-13
Hudson, H. J. (1963). The perfect state of Nigrospora oryzae. Trans. Br. Mycol. Soc. 46, 355–360. doi: 10.1016/S0007-1536(63)80027-3
Hughes, K. A., Lawley, B., and Newsham, K. K. (2003). Solar UV-B radiation inhibits the growth of Antarctic terrestrial fungi. Appl. Environ. Microbiol. 69, 1488–1491. doi: 10.1128/AEM.69.3.1488-1491.2003
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Jansson, J. K., and Tas, N. (2014). The microbial ecology of permafrost. Nat. Rev. Microbiol. 12, 414–425. doi: 10.1038/nrmicro3262
Ji, M., van Dorst, J., Bissett, A., Brown, M. V., Palmer, A. S., Snape, I., et al. (2015). Microbial diversity at Mitchell Peninsula, Eastern Antarctica: a potential biodiversity “hotspot”. Polar Biol. 39, 237–249. doi: 10.1007/s00300-015-1776-y
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9. doi: 10.1093/nar/gkn201
Kaewkla, O., and Franco, C. M. (2013). Rational approaches to improving the isolation of endophytic actinobacteria from Australian native trees. Microb. Ecol. 65, 384–393. doi: 10.1007/s00248-012-0113-z
Kim, M., Cho, A., Lim, H. S., Hong, S. G., Kim, J. H., Lee, J., et al. (2015). Highly heterogeneous soil bacterial communities around Terra Nova Bay of Northern Victoria Land, Antarctica. PLoS ONE 10:e0119966. doi: 10.1371/journal.pone.0119966
Kim, M., Morrison, M., and Yu, Z. (2011). Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 84, 81–87. doi: 10.1016/j.mimet.2010.10.020
Kochkina, G., Ivanushkina, N., Ozerskaya, S., Chigineva, N., Vasilenko, O., Firsov, S., et al. (2012). Ancient fungi in Antarctic permafrost environments. FEMS Microbiol. Ecol. 82, 501–509. doi: 10.1111/j.1574-6941.2012.01442.x
Koljalg, U., Larsson, K. H., Abarenkov, K., Nilsson, R. H., Alexander, I. J., Eberhardt, U., et al. (2005). UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166, 1063–1068. doi: 10.1111/j.1469-8137.2005.01376.x
Koyama, A., Wallenstein, M. D., Simpson, R. T., and Moore, J. C. (2014). Soil bacterial community composition altered by increased nutrient availability in Arctic tundra soils. Front. Microbiol. 5:516. doi: 10.3389/fmicb.2014.00516
Kumar, P. S., Brooker, M. R., Dowd, S. E., and Camerlengo, T. (2011). Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS ONE 6:e20956. doi: 10.1371/journal.pone.0020956
Lagier, J.-C., Armougom, F., Million, M., Hugon, P., Pagnier, I., Robert, C., et al. (2012). Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 18, 1185–1193. doi: 10.1111/1469-0691.12023
Ludley, K. E., and Robinson, C. H. (2008). ‘Decomposer’ basidiomycota in Arctic and Antarctic ecosystems. Soil Biol. Biochem. 40, 11–29. doi: 10.1016/j.soilbio.2007.07.023
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Maturrano, L., Santos, F., Rossello-Mora, R., and Anton, J. (2006). Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl. Environ. Microbiol. 72, 3887–3895. doi: 10.1128/AEM.02214-05
Melick, D. R., Hovenden, M. J., and Seppelt, R. D. (1994). Phytogeography of bryophyte and lichen vegetation in the Windmill Island, Wilkes Land, Continental Antarctica. Vegetatio 111, 71–87.
Michaelson, G. J., Ping, C. L., and Walker, D. A. (2012). Soils associated with biotic activity on frost boils in Arctic Alaska. Soil Sci. Soc. Am. J. 76:2265. doi: 10.2136/sssaj2012.0064
Nichols, D., Cahoon, N., Trakhtenberg, E. M., Pham, L., Mehta, A., Belanger, A., et al. (2010). Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 76, 2445–2450. doi: 10.1128/AEM.01754-09
Niederberger, T. D., Sohm, J. A., Gunderson, T. E., Parker, A. E., Tirindelli, J., Capone, D. G., et al. (2015). Microbial community composition of transiently wetted Antarctic Dry Valley soils. Front. Microbiol. 6:9. doi: 10.3389/fmicb.2015.00009
Onofri, S., Anastasi, A., Del Frate, G., Di Piazza, S., Garnero, N., Guglielminetti, M., et al. (2011). Biodiversity of rock, beach and water fungi in Italy. Plant Biosyst. 145, 978–987. doi: 10.1080/11263504.2011.633117
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Peeters, K., Ertz, D., and Willems, A. (2011). Culturable bacterial diversity at the Princess Elisabeth Station (Utsteinen, Sor Rondane Mountains, East Antarctica) harbours many new taxa. Syst. Appl. Microbiol. 34, 360–367. doi: 10.1016/j.syapm.2011.02.002
Plassart, P., Terrat, S., Thomson, B., Griffiths, R., Dequiedt, S., Lelievre, M., et al. (2012). Evaluation of the ISO standard 11063 DNA extraction procedure for assessing soil microbial abundance and community structure. PLoS ONE 7:e44279. doi: 10.1371/journal.pone.0044279
Plemenitas, A., Vaupotic, T., Lenassi, M., Kogej, T., and Gunde-Cimerman, N. (2008). Adaptation of extremely halotolerant black yeast Hortaea werneckii to increased osmolarity: a molecular perspective at a glance. Stud. Mycol. 61, 67–75. doi: 10.3114/sim.2008.61.06
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Quince, C., Lanzen, A., Davenport, R. J., and Turnbaugh, P. J. (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38. doi: 10.1186/1471-2105-12-38
Rao, S., Chan, Y., Lacap, D. C., Hyde, K. D., Pointing, S. B., and Farrell, R. L. (2011). Low-diversity fungal assemblage in an Antarctic Dry Valleys soil. Polar Biol. 35, 567–574. doi: 10.1007/s00300-011-1102-2
Rayment, R. E., and Lyons, D. J. (2011). Soil Chemical Methods-Australasia. Collingwood, VIC: CSIRO Publishing
Roser, D. J., Seppelt, R. D., and Ashboli, N. (1993). Microbiology of orthogenic soils from the Windmill Island, Budd Coast, continental Antarctica: microbial biomass distribution. Soil Biol. Biochem. 25, 165–175. doi: 10.1016/0038-0717(93)90023-5
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310. doi: 10.1371/journal.pone.0027310
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sharma, S., Szele, Z., Schilling, R., Munch, J. C., and Schloter, M. (2006). Influence of freeze-thaw stress on the structure and function of microbial communities and denitrifying populations in soil. Appl. Environ. Microbiol. 72, 2148–2154. doi: 10.1128/AEM.72.3.2148-2154.2006
Shen, C., Xiong, J., Zhang, H., Feng, Y., Lin, X., Li, X., et al. (2013). Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 57, 204–211. doi: 10.1016/j.soilbio.2012.07.013
Siciliano, S. D., Palmer, A. S., Winsley, T., Lamb, E., Bissett, A., Brown, M. V., et al. (2014). Soil fertility is associated with fungal and bacterial richness, whereas pH is associated with community composition in polar soil microbial communities. Soil Biol. Biochem. 78, 10–20. doi: 10.1016/j.soilbio.2014.07.005
Smith, J. J., Tow, L. A., Stafford, W., Cary, C., and Cowan, D. A. (2006). Bacterial diversity in three different Antarctic Cold Desert mineral soils. Microb. Ecol. 51, 413–421. doi: 10.1007/s00248-006-9022-3
Stefani, F. O., Bell, T. H., Marchand, C., de la Providencia, I. E., El Yassimi, A., St-Arnaud, M., et al. (2015). Culture-dependent and -independent methods capture different microbial community fractions in hydrocarbon-contaminated soils. PLoS ONE 10:e0128272. doi: 10.1371/journal.pone.0128272
Steven, B., Briggs, G., McKay, C. P., Pollard, W. H., Greer, C. W., and Whyte, L. G. (2007). Characterization of the microbial diversity in a permafrost sample from the Canadian high Arctic using culture-dependent and culture-independent methods. FEMS Microbiol. Ecol. 59, 513–523. doi: 10.1111/j.1574-6941.2006.00247.x
Steven, B., Lionard, M., Kuske, C. R., and Vincent, W. F. (2013). High bacterial diversity of biological soil crusts in water tracks over permafrost in the high arctic polar desert. PLoS ONE 8:10. doi: 10.1371/journal.pone.0071489
Stewart, K. J., Snape, I., and Siciliano, S. D. (2011). Physical, chemical and microbial soil properties of frost boils at Browning Peninsula, Antarctica. Polar Biol. 35, 463–468. doi: 10.1007/s00300-011-1076-0
Stomeo, F., Makhalanyane, T. P., Valverde, A., Pointing, S. B., Stevens, M. I., Cary, C. S., et al. (2012). Abiotic factors influence microbial diversity in permanently cold soil horizons of a maritime-associated Antarctic Dry Valley. FEMS Microbiol. Ecol. 82, 326–340. doi: 10.1111/j.1574-6941.2012.01360.x
Takebayashi, S., Narihiro, T., Fujii, Y., and Haraishi, A. (2007). Water availability is a critical determinant of a population shift from proteobacteria to actinobacteria during start-up operation of mesophilic fed-batch composting. Microb. Environ. 22, 279–289. doi: 10.1264/jsme2.22.279
Timling, I., Dahlberg, A., Walker, D. A., Gardes, M., Charcosset, J. Y., Welker, J. M., et al. (2012). Distribution and drivers of ectomycorrhizal fungal communities across the North American Arctic. Ecosphere 3:111. doi: 10.1890/ES12-00217.1
Tomova, I., Stoilova-Disheva, M., Lazarkevich, I., and Vasileva-Tonkova, E. (2015). Antimicrobial activity and resistance to heavy metals and antibiotics of heterotrophic bacteria isolated from sediment and soil samples collected from two Antarctic islands. Front. Life Sci. 8:1044130. doi: 10.1080/21553769.2015.1044130
Tosi, S., Kostadinova, N., Krumova, E., Pashova, S., Dishliiska, V., Spassova, B., et al. (2010). Antioxidant enzyme activity of filamentous fungi isolated from Livingston Island, Maritime Antarctica. Polar Biol. 33, 1227–1237. doi: 10.1007/s00300-010-0812-1
Tytgat, B., Verleyen, E., Obbels, D., Peeters, K., De Wever, A., D'Hondt, S., et al. (2014). Bacterial diversity assessment in Antarctic terrestrial and aquatic microbial mats: a comparison between bidirectional pyrosequencing and cultivation. PLoS ONE 9:e97564. doi: 10.1371/journal.pone.0097564
van Dorst, J., Bissett, A., Palmer, A. S., Brown, M., Snape, I., Stark, J. S., et al. (2014). Community fingerprinting in a sequencing world. FEMS Microbiol. Ecol. 89, 316–330. doi: 10.1111/1574-6941.12308
van Dorst, J. M., Hince, G., Snape, I., and Ferrari, B. C. (2016). Novel culturing techniques select for heterotrophs and hydrocarbon degraders in a subantarctic soil. Sci. Rep. 6:36724. doi: 10.1038/srep36724
Vaz-Moreira, I., Egas, C., Nunes, O. C., and Manaia, C. M. (2011). Culture-dependent and culture-independent diversity surveys target different bacteria: a case study in a freshwater sample. Antonie Van Leeuwenhoek 100, 245–257. doi: 10.1007/s10482-011-9583-0
Velegraki, A., Cafarchia, C., Gaitanis, G., Latta, R., and Boekhout, T. (2015). Malassezia infection in human and animals: pathophysiology, detection, and treatment. PLoS Pathog 11:e1004523. doi: 10.1371/journal.ppat.1004523
Vishniac, H. (1996). Biodiversity of yeast and filamentous microfungi in terrestrial Antarctic ecosystems. Biodivers. Conserv. 5, 1365–1378. doi: 10.1007/BF00051983
Walker, D. A., Epstein, H. E., Gould, W. A., Kelley, A. M., Kade, A. N., Knudson, J. A., et al. (2004). Frost-boil ecosystems: complex interactions between landforms, soils, vegetation and climate. Permafrost Periglacial Process. 15, 171–188. doi: 10.1002/ppp.487
Wang, N. F., Zhang, T., Zhang, F., Wang, E. T., He, J. F., Ding, H., et al. (2015). Diversity and structure of soil bacterial communities in the Fildes Region (maritime Antarctica) as revealed by 454 pyrosequencing. Front. Microbiol. 6:1188. doi: 10.3389/fmicb.2015.01188
Watve, M., Shejval, V., Sonawane, C., Rahalkar, M., Matapurkar, A., Shouche, Y., et al. (2000). The ‘K’ selected oligophilic bacteria: a key to uncultured diversity? Curr. Sci. 78, 1535–1542.
Winsley, T., van Dorst, J. M., Brown, M. V., and Ferrari, B. C. (2012). Capturing greater 16S rRNA gene sequence diversity within the domain bacteria. Appl. Environ. Microbiol. 78, 5938–5941. doi: 10.1128/AEM.01299-12
Zdanowski, M. K., Żmuda-Baranowska, M. J., Borsuk, P., Świątecki, A., Górniak, D., Wolicka, D., et al. (2012). Culturable bacteria community development in post-glacial soils of Ecology Glacier, King George Island, Antarctica. Polar Biol. 36, 511–527. doi: 10.1007/s00300-012-1278-0
Keywords: antarctic soil, frost boils, bacterial diversity, fungal diversity, cultivation
Citation: Pudasaini S, Wilson J, Ji M, van Dorst J, Snape I, Palmer AS, Burns BP and Ferrari BC (2017) Microbial Diversity of Browning Peninsula, Eastern Antarctica Revealed Using Molecular and Cultivation Methods. Front. Microbiol. 8:591. doi: 10.3389/fmicb.2017.00591
Received: 26 September 2016; Accepted: 22 March 2017;
Published: 07 April 2017.
Edited by:
Philippe M. Oger, Institut National des Sciences Appliquées de Lyon, FranceReviewed by:
Arwyn Edwards, Aberystwyth University, UKCopyright © 2017 Pudasaini, Wilson, Ji, van Dorst, Snape, Palmer, Burns and Ferrari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Belinda C. Ferrari, Yi5mZXJyYXJpQHVuc3cuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.