
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 April 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00541
This article is part of the Research Topic Mechanisms of Activation and Evasion of The Complement System by Pathogens View all 13 articles
 Javier Querol-García1†Francisco J. Fernández1,2,3,4†
Javier Querol-García1†Francisco J. Fernández1,2,3,4† Ana V. Marin2,3Sara Gómez1Daniel Fullà5Cecilia Melchor-Tafur1Virginia Franco-Hidalgo1
Ana V. Marin2,3Sara Gómez1Daniel Fullà5Cecilia Melchor-Tafur1Virginia Franco-Hidalgo1 Sebastián Albertí6
Sebastián Albertí6 Jordi Juanhuix5Santiago Rodríguez de Córdoba1
Jordi Juanhuix5Santiago Rodríguez de Córdoba1 José R. Regueiro2,3
José R. Regueiro2,3 M. Cristina Vega1*
M. Cristina Vega1*The Gram-positive anaerobic human pathogenic bacterium Atopobium vaginae causes most diagnosed cases of bacterial vaginosis as well as opportunistic infections in immunocompromised patients. In addition to its well-established role in carbohydrate metabolism, D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Streptococcus pyogenes and S. pneumoniae have been reported to act as extracellular virulence factors during streptococcal infections. Here, we report the crystal structure of GAPDH from A. vaginae (AvGAPDH) at 2.19 Å resolution. The refined model has a crystallographic Rfree of 22.6%. AvGAPDH is a homotetramer wherein each subunit is bound to a nicotinamide adenine dinucleotide (NAD+) molecule. The AvGAPDH enzyme fulfills essential glycolytic as well as moonlight (non-glycolytic) functions, both of which might be targets of chemotherapeutic intervention. We report that AvGAPDH interacts in vitro with the human C5a anaphylatoxin and inhibits C5a-specific granulocyte chemotaxis, thereby suggesting the participation of AvGAPDH in complement-targeted immunoevasion in a context of infection. The availability of high-quality structures of AvGAPDH and other homologous virulence factors from Gram-positive pathogens is critical for drug discovery programs. In this study, sequence and structural differences between AvGAPDH and related bacterial and eukaryotic GAPDH enzymes are reported in an effort to understand how to subvert the immunoevasive properties of GAPDH and evaluate the potential of AvGAPDH as a druggable target.
Atopobium vaginae is a Gram-positive anaerobic bacterium of the Coriobacteriaceae family that is held responsible for about 80% of diagnosed cases of bacterial vaginosis (BV) in humans, a condition that can lead to obstetric complications and gynecological disorders (Polatti, 2012; Datcu, 2014; Xiao et al., 2016). Further, complications arising from A. vaginae infections might range from predisposing to opportunistic infections by HIV, type II Herpes simplex virus, and Chlamydia trachomatis infections (Fredricks et al., 2005; Livengood, 2009) to late abortion (Donati et al., 2010), premature rupture of the amniotic membrane (McDonald et al., 2007), chorio-amnionitis (Fahey, 2008), post-partum endometritis (Sweet, 1995; Hillier et al., 1996; Pellati et al., 2008), and failure of in vitro fertilization and embryo transfer (Fredricks et al., 2005; Livengood, 2009). Since an effective and long-lasting treatment for BV is not available, with relapses occurring in about 30% of cases within a month after treatment completion, new therapeutic approaches are a necessity. Although the molecular basis for A. vaginae infection, survival to currently used antibiotic treatments, or the mechanisms behind the acquisition of resistance to metronidazole showed by most strains (Chan et al., 2012; Machado and Cerca, 2015) are not yet fully understood, they are likely to involve many different genes encoding for biofilm formation activities as well as for proteins with virulence and immunoevasive properties.
Interestingly, phylogenetically diverse bacteria have been shown to encode virulence factors that interfere with the proper function of the human complement system, one of the first barriers of defense against bacterial infections from the innate immune system. One such virulence factor is the glycolytic enzyme D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, E.C. 1.2.1.12). To perform its glycolytic activity GAPDH requires the nicotinamide adenine dinucleotide (NAD+) cofactor that remains tightly bound in the active site. The active site of GAPDH contains also two structurally distinct phosphate-binding sites, the Pi and Ps sites, where inorganic phosphate and the phosphate group from the glyceraldehyde-3-phosphate (G3P) substrate bind, respectively, during reaction (Skarzyński et al., 1987; Yun et al., 2000). In fact, there are two different Pi sites depending on the precise location within the active site of the inorganic phosphate moiety, which are termed the “classical” and “new” Pi sites (Yun et al., 2000; Mukherjee et al., 2010).
The anaerobic lifestyle of A. vaginae implies that a functional glycolytic pathway might be important to sustain infection, therefore making A. vaginae (Av) GAPDH an interesting target for the design of potent inhibitors. This proposal parallels previous studies on GAPDH from trypanosomatid parasites such as Leishmania mexicana (the causative agent of lepromatous leprosy), Trypanosoma cruzi (Chagas' disease) and T. brucei (African sleeping sickness). These protozoan parasites cause severely impairing diseases with a heavy death toll worldwide, and the lack of wholly effective treatments for these diseases and the problems associated with current drugs (toxicity effects and resistance) have spurred the study of glycosomal trypanosomatid GAPDH. The rationale for targeting trypanosomatid GAPDH for drug discovery relies in the almost exclusive reliance of trypanosomatids on the glycolysis to produce ATP. Indeed, the structures of glycosomal GAPDH from trypanosomatid parasites have been identified as potential drug targets because there are substantial differences in the cavity forming the binding site of the adenine ring of NAD+ cofactor between the human and glycosomal GAPDH and in nearby hydrophobic cavities (Suresh et al., 2001). Drugs targeting those hydrophobic cavities could inhibit GAPDH through allosteric effects.
In addition to the known roles of GAPDH in the glycolytic pathway, in the last years there has been an increasing awareness that mammalian and bacterial GAPDH enzymes display many other diverse functions. In mammals, GAPDH participates in membrane fusion, microtubule bundling, nuclear RNA export, DNA replication and repair and apoptosis, and it is in these non-glycolytic roles that mammalian GAPDH has been associated with various diseases including cancer (Zhang et al., 2015), viral pathogenesis (Allonso et al., 2015), Huntington's disease (Hwang et al., 2015; Mikhaylova et al., 2016), Parkinson's disease (Liu et al., 2015) and Alzheimer's disease (El Kadmiri et al., 2014). Bacterial GAPDH enzymes have also shown a wide functional repertoire (Henderson and Martin, 2014). In bacterial pathogens encoding type III secretion systems, GAPDH has been shown to be transported from the bacterial cell to the mammalian host cell through the syringe system (Aguilera et al., 2012), where GAPDH perturbs the redox balance of the attacked host cell leading to its demise or to the creation of more favorable environmental conditions for the invasive bacterial cells. It is significant that GAPDH from many different bacteria is exported to the bacterial cell wall and the extracellular space, where it seems to acquire functions that facilitate infection through direct binding to innate immune proteins. This function has been reported for S. pyogenes and S. pneumoniae through the interaction of GAPDH with the human C5a anaphylatoxin, which is implicated in inflammatory processes contributing to both the innate and adaptive immune response, and with the C1q complement factor that mediates interactions with IgG and IgM (Terrasse et al., 2012), respectively. In the former case, a solvent-exposed segment of S. pyogenes GAPDH spanning the N-terminal 1−165 amino acid residues was identified as the likely docking sequence for C5a (Terao et al., 2006). Other glycolytic enzymes have also been demonstrated to undergo translocation to the extracellular space where they acquire radically different functions, oftentimes pro-infection roles (Agarwal et al., 2012). It is intriguing to consider the possibility that AvGAPDH might also be involved in fostering A. vaginae infection after its secretion or translocation to the outside of the bacterial cells by diminishing the ability of neutrophils to locate and destroy the pathogen, lowering its antibody-mediated clearance, or perturbing the normal immune response by other unexpected pathways. Providing answers to these open questions will hinge on a more thorough understanding of the structure of AvGAPDH and its moonlighting functions in an infection context.
Therapeutic action on the glycolytic or virulence-associated functions of GAPDH represents an attractive route to help curb bacterial infections, while it depends on the availability of specific drugs that can distinguish human GAPDH from the targeted pathogen. The determination of the crystal structure of AvGAPDH to 2.19-Å resolution provides a detailed description of the overall structure of this enzyme as well as a high-quality view into the active site. By comparing the AvGAPDH structure with those of other pathogens and the available human GAPDH structures, we have observed differences that provide insights into the design of selective inhibitors. Furthermore, the analysis of the peptide segment that corresponds to the C5a-binding motif described for S. pyogenes GAPDH hinted at the possibility that GAPDH enzymes from other Gram-positive pathogenic bacteria could participate in the same strategy of infection regardless of the exact colonization environment in the host organism (e.g., throat, lungs, vaginal flora). Binding assays with recombinant C5a and cell migration inhibition assays by surface-immobilized AvGAPDH of granulocytes challenged with zymosan-activated serum (where native C5a is produced) have confirmed that AvGAPDH could functionally act as a virulence factor, a feature that might contribute to the infectivity and survival of A. vaginae.
Genomic DNA from an A. vaginae type strain isolated from the human vaginal flora of a healthy individual was purchased from the DSMZ (DSM-15829). The gene encoding the 356-amino-acid residue AvGAPDH (UniProt Accession No. F1T6A5) was constructed by a two-step PCR protocol designed to incorporate in the PCR amplicon an additional N-terminal hexahistidine tag and a tobacco etch virus cleavage (TEV) site in frame with the AvGAPDH gene. This PCR fragment was then inserted into pET21b (Novagen) by restriction-ligation cloning. In addition to the full-length construct, a PCR-amplified DNA sequence encoding only the N-terminal fragment of AvGAPDH (residues 1−185) was subcloned in pGEX-6P-1 (GE Healthcare) fused in-frame with the glutathione-S-transferase (GST) gene and a 3C protease cleavage site. Expression plasmids for hexahistidine-tagged S. pyogenes (Sp) GAPDH (UniProt Accession No. P68777) and C. perfringens (Cp) GAPDH (UniProt Accession No. Q0STD4) were constructed similarly to that of full-length AvGAPDH.
The expression plasmids were transformed into E. coli Rossetta(DE3)pLysS cells (Novagen) and transformants isolated in selective LB-agar plates. Transformants from a freshly prepared plate or from a bacterial glycerol stock were used to inoculate a 40-ml overnight starter culture from which a larger culture (2 l) was initiated the next morning. The culture was allowed to grow at 37°C in LB or Power Broth (Athena) media supplemented with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol to an absorbance of 0.6−0.8 at 600 nm and the temperature was lowered to 20°C for 1 h before induction. The culture was then induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 18 h. Cells were harvested by centrifugation at 6,000 g for 20 min at 4°C and either used immediately or stored at −80°C until use.
For the hexahistidine-tagged full-length AvGAPDH, the cell pellet from 2 l of culture was resuspended in 40 ml of a buffer containing 50 mM Tris−HCl pH 8.0, 500 mM NaCl, 20 mM imidazole and 1 mM phenylmethylsulfonyl fluoride (PMSF), and cells were lysed by sonication. The lysate was centrifuged at 20,000 × g for 30 min at 4°C and clarified further by filtration through a 0.22 μm membrane. The purification procedure consisted of a capture step by affinity chromatography in which the clarified lysate containing N-terminal hexahistidine AvGAPDH was loaded on a 5-ml HisTrap column (GE Healthcare) charged with nickel chloride. AvGAPDH was eluted using a linear gradient of increasing imidazole concentration (250 mM) over 20 column volumes. Fractions containing AvGAPDH were pooled and dialyzed against a buffer containing 50 mM Tris−HCl pH 8.0, 150 mM NaCl. Finally, AvGAPDH was subjected to gel filtration chromatography over a HiLoad 16/60 Superdex 200 equilibrated in a buffer consisting in 10 mM Tris−HCl pH 7.5, 150 mM NaCl. We typically obtained 40 mg/l culture of pure AvGAPDH with high purity, >95% pure on a Coomassie brilliant blue-stained SDS-PAGE gel (Supplementary Figure 6A). Comparison of the elution volume of AvGAPDH with a calibration curve constructed using high and low molecular weight calibration kits (GE Healthcare), following the recommended calibration mixtures following the manufacturer's instructions, corroborated that the quaternary structure of AvGAPDH corresponds to a homotetramer (Supplementary Figure 6B). Finally, AvGAPDH was dialyzed against a salt-free buffer (10 mM Tris−HCl pH 7.5), concentrated to 30 mg/ml, dispensed in 30-μl aliquots, snap-frozen in liquid nitrogen and stored away at −80°C. Expression (50 ml) and purification of hexahistidine-tagged full-length SpGAPDH and CpGAPDH was carried out as indicated for AvGAPDH.
For the GST-3C-fused AvGAPDH truncated protein (residues 1−185), the cell pellet from 100 ml of culture was resuspended in 10 ml of a buffer containing 50 mM Tris−HCl pH 8.0, 150 mM NaCl, 10 mM EDTA, and 1 mM PMSF, and cells were lysed by sonication. The lysate was centrifuged at 20,000 × g for 30 min at 4°C and clarified further by filtration through a 0.22 μm membrane. The clarified lysate was then loaded onto a column packed with 3 ml Glutathione Sepharose 4B beads (GE Healthcare), washed thoroughly with binding buffer to baseline absorbance, and on-column tag cleavage was then performed by overnight incubation with 3C protease at 4°C. Purified protein was recovered in the flow-through, concentrated, and applied onto a Superdex 200 10/300GL Increase gel filtration column equilibrated in a buffer consisting in 10 mM HEPES−NaOH pH 7.4, 150 mM NaCl, 3.4 mM EDTA. Peak fractions were analyzed by SDS-PAGE electrophoresis and those fractions corresponding to the estimated molecular mass for monomeric AvGAPDH 1−185 were pooled together for further analyses.
The recombinant 74-amino-acid residue human C5a anaphylatoxin (including the C-terminal Arg74) used in this study can be obtained from Crelux GmbH.
Before attempting crystallization of AvGAPDH the required number of aliquots was quickly thawed and centrifuged at 10,000 × g for 10 min at 4°C to remove any potential aggregates that might have resulted from a freezing-thawing cycle. A commercial Pre-Crystallization Test (Hampton Research) was used to adjust the protein concentration to a suitable concentration for more extensive crystallization screenings, which was finally set to 7.5 mg/ml. The full JCSG-plus sparse matrix and the PACT premier systematic PEG/Ion/pH screenings (Molecular Dimensions) were performed by the sitting-drop vapor-diffusion method using drops containing 1 μl AvGAPDH at 7.5 mg/ml supplemented with 0.6 mM NAD+ and 1 μl crystallization condition at 20°C. Moderately sized, plate or cube-shaped crystals of AvGAPDH (100−200 μm in their longest dimension) appeared within 1 week under several conditions from both commercial screenings. Two conditions were selected for diffraction studies, condition 1 (0.1 M sodium citrate pH 5.5, 20% PEG 3000) and condition 2 (0.2 M sodium nitrate, 0.1 M Bis-Tris propane pH 8.5, 20% PEG 3350). Crystals were then cryoprotected with 20% (v/v) sterile glycerol, mounted in standard MicroMount (MiTeGen) and flash frozen in liquid nitrogen.
Diffraction data were collected from flash-frozen crystals at 100 K using a photon-counting Pilatus 6M (DECTRIS Ltd, Baden, Switzerland) at the BL13-XALOC beamline (ALBA, Barcelona, Spain; Juanhuix et al., 2014). Crystals grown in either condition 1 or 2 diffracted to moderate-to-high resolution (2.19−2.5 Å) and complete data sets were collected from both crystal forms, indexed and processed with XDS (Kabsch, 2014) and scaled and merged with Aimless (Evans and Murshudov, 2013) from the CCP4 software suite (Winn et al., 2011). Crystals grown under either condition were highly isomorphic, belonging to space group P21, with unit-cell dimensions a = 71.4, b = 132.1, c = 85.3 Å, and β = 104.9°. Based on the data merging statistics we selected condition 1 for structure determination. The Matthews coefficient, VM, is 2.43 Å3/Da, indicates an approximate solvent content of 49.3%. Merging of symmetry-related reflections resulted in 77,410 unique reflections and a multiplicity of 6.7 (6.4 in the last shell, 2.27−2.19 Å) with an Rmeas value of 0.126 (0.904) and CC1/2 of 0.997 (0.881). The completeness of the data to 2.19 Å was 98% (89%). Data collection and processing statistics are summarized in Table 1.
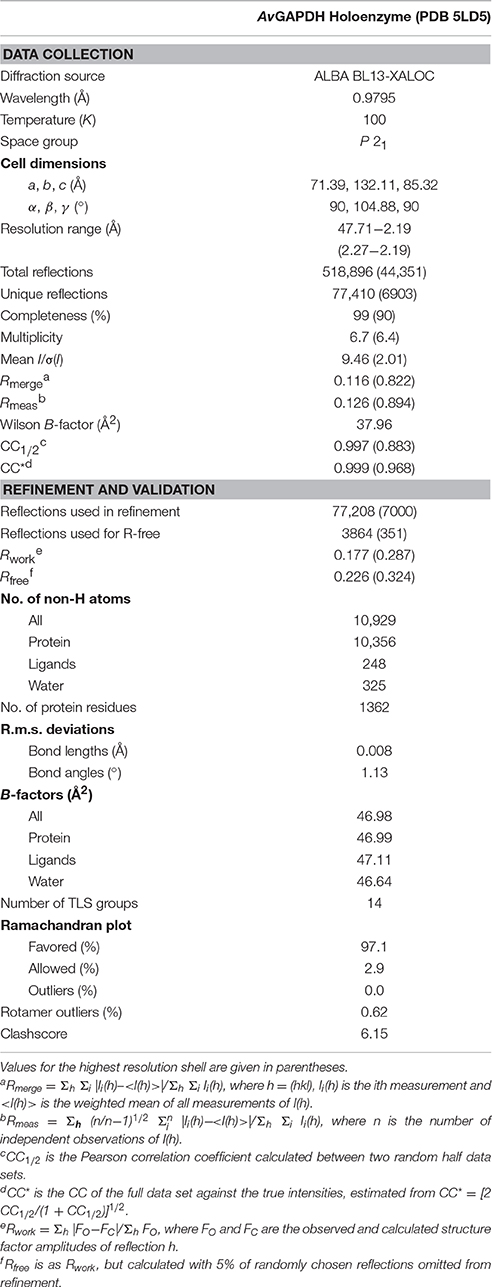
Table 1. Crystallographic data collection and refinement statistics.
The structure was determined by the molecular replacement method using the program PHASER (McCoy et al., 2007) from the PHENIX program suite (Adams et al., 2011). Either the full homotetramer (the biological unit) or a dimer of Staphylococcus aureus GAPDH devoid of NAD+ (SaGAPDH; PDB ID 3LVF; Mukherjee et al., 2010) were used as search models after mutation of the PDB file according to a sequence alignment (63% identity) with CHAINSAW (Stein, 2008). Using the SaGAPDH tetramer a single solution was found in the rotation and translation searches with RFZ and TFZ of 30.0 and 15.4, respectively. Rigid-body refinement as implemented in PHASER elicited only minor improvements in correlation coefficient (CC) and R factors. In contrast, using a dimer as the search model led to the identification and subsequent use by PHASER of a significant peak in the Patterson map indicating the presence of pseudo translational symmetry. PHASER then switched on the tNCS-enabled MR search that successfully identified two independent solutions that together assembled the biological homotetramer, with RFZ and TFZ scores of 19.2 and 13.2 for the first dimer plus the second, translationally symmetric, dimer. The solutions were improved by rigid-body refinement as implemented in PHASER, with R/Rfree factors of 0.39/0.40 for the tetrameric model and 0.40/0.40 for the solution obtained with two dimeric models. At this stage both solutions were indistinguishable (root-mean-square displacement, r.m.s.d., of 0.001 Å over 1,014 superimposed Cα atoms), hence we selected the solution obtained with a full biological unit for further refinement.
An omit map calculated from the model phases before (rigid) refinement or model building showed electron density corresponding to four NAD+ molecules, one per protomer. The complete homotetramer was then used for rigid-body and maximum likelihood refinement in the resolution range 47.71−2.19 Å within phenix.refine (Afonine et al., 2012) setting aside 5% of the reflections (selected randomly) to create a data set of test reflections for cross-validation of the refinement procedure. Refinement cycles were interspersed with cycles of manual building (first, placing NAD+ and then solvent molecules) and validation with Coot (Emsley et al., 2010). Torsion-angle non-crystallographic symmetry restraints were applied during the initial refinement but were removed during the final refinement stages. Upon convergence, the refined AvGAPDH model had Rwork/Rfree = 0.177/0.226 with good stereochemistry. The final model consists of 340 residues of each subunit, four NAD+ molecules, 325 water molecules and 12 glycerol molecules (from the cryoprotectant solution). Crystallographic refinement statistics are summarized in Table 1.
The AvGAPDH enzyme activity was followed spectrophotometrically by the change in absorbance at 340 nm due to NADH formation, according to a previously described method (Ferdinand, 1964). Assays were performed in an Eppendorf BioSpectrometer spectrophotometer at 25°C. A standard assay was carried out in a final volume of 0.15 ml in the presence of 40 mM Tris-HCl pH 8.5 and 2 mM ethylenediaminetetraacetic acid (EDTA), 0.1 μM of purified enzyme, and indicated concentrations of substrates: nicotinamide adenine dinucleotide (NAD+), glyceraldehyde-3-phosphate (G3P) and inorganic phosphate (Pi). NAD+ concentration was varied between 0.03−2 mM, G3P concentration between 0.6−15 mM, and Pi concentration between 0.65−24 mM. Michaelis–Menten parameters, KM and Vmax were obtained by nonlinear regression fitting of the kinetic data using SigmaPlot v13.0 (Systat Software Inc.).
GAPDH sequences of known structure and with a significant degree of sequence similarity to AvGAPDH were found with BLAST (Camacho et al., 2009) using sequences stored in the PDB, aligned with MUSCLE and used to reconstruct a maximum likelihood phylogenetic tree with PhyML (Guindon et al., 2010).
A ligand blotting assay was set up to detect the interaction between AvGAPDH and C5a essentially as described before (Terao et al., 2006). Briefly, C5a was biotinylated using the ECL Protein Biotinylation System (Amersham Biosciences) following the manufacturer's instructions. Then, different concentrations of purified AvGAPDH were resolved on a 15% SDS-PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked overnight at 4°C with 10% membrane blocking agent and treated with 50 μg/ml biotinylated C5a for 1 h. After extensively washing the membranes and incubating with horseradish peroxidase (HRP)-labeled streptavidin at room temperature for 1 h, C5a-containing bands were revealed with luminol and hydrogen peroxide and subsequently visualized by chemiluminescence on a Luminescent Image Analyzer LAS-3000 at room temperature.
Specific cross-linking assays with AvGAPDH were performed with bis(maleimido)ethane (BMOE; Pierce 22322), a 7-atom, 8.0-Å short spacer arm cross-linking reagent that generates non-cleavable cross-links between sulfhydryl groups that are in close proximity. Cross-linking reactions were performed in assay buffer (10 mM HEPES−NaOH pH 7.4, 150 mM NaCl, 3.4 mM EDTA) containing 0.3 mM BMOE, incubated overnight at 4°C, and visualized by SDS-PAGE electrophoresis and Coomassie-brilliant blue (CBB) staining.
The C5a interaction with AvGAPDH was studied also with an enzyme-linked immunosorbent assay (ELISA). The wells of a polysterene microtiter plate (F96 Maxisorp, Nunc) were coated with 100 μl of 10 μg/ml of purified AvGAPDH in 50 mM Tris−HCl pH 8.5 at 4°C overnight. Wells were emptied and washed with TBS-T (Tris-buffered saline pH 7.4 with 0.05% w/v Tween-20) and the free sites on the plastic surface were blocked with 1% BSA (bovine serum albumin) in TBS-T for 2 h at 37°C. Uncoated wells and wells coated with AvGAPDH were also blocked. After washing extensively with TBS-T, various dilutions of C5a in TBS (1–100 μg/ml) were added to the AvGAPDH-coated wells. The plate was incubated for 3 h at 37°C and then washed 4 times with TBS-T. A mouse anti-human C5a antibody was added at a 1:2,500 dilution in TBS-T (100 μl/well) and the plate was incubated for 1 h at 37°C and washed as before. Next, a goat anti-mouse IgG-HRP conjugate (1:1000 dilution) was added and the plate was incubated at 37°C for 1 h. The unbound conjugate was discarded and the plate was washed 5 times with TBS-T. OPD (o-phenylenediamine dihydrochloride) substrate tables were dissolved in double distilled water and added immediately (100 μl/well) to the plate. The absorbance was measured at 495 nm in a microplate reader.
Purified full-length of AvGAPDH, SpGAPDH, and CpGAPDH, and the 185-amino-acid N-terminal fragment of AvGAPDH were immobilized by multipoint covalent binding on to highly functionalized aminoethyl agarose (AM-4) beads (ABT, 40−60 μmol aminoethyl/ml gel, 4% agarose), thereby allowing protein immobilization through coupling with carboxylate groups from exposed aspartate and glutamate residues and the C terminus. The amount of covalently crosslinked GAPDH was calculated from equation 1 below, where C0 is the protein concentration at starting immobilization time and CF is the protein concentration at final immobilization time:
According to manufacturer instructions, 50 μl of AM-4 resin was rinsed with water and equilibrated with a buffer consisting of 10 mM Tris-HCl pH 7.6. The equilibrated resin was then activated with 60 mM N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide (EDC) and next various amounts of protein were incubated with the treated resin at room temperature on an orbital shaker with gentle stirring. At different times aliquots were removed to monitor the amount of unreacted protein until it reached a plateau (which occurred at about 3 h), indicating the saturation of the accessible functional carboxylate groups on the resin. Next, the suspension was centrifuged (13,500 rpm for 2 min) and the pelleted resin was carefully washed with water, 1 M NaCl, and water again.
After obtaining written informed consent from healthy donors, human granulocytes were isolated from EDTA venous blood samples by centrifugation on a Histopaque-1070/1119 gradient (Sigma-Aldrich). These studies were performed according to the principles expressed in the Declaration of Helsinki and approved by the relevant Institutional Research Ethics Committee. Residual erythrocytes were depleted by incubation with 0.5 ml Ammonium-Chloride-Potassium Lysing Buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.2) for 2.5 min at 25°C. Cells were washed twice with PBS (400 × g for 6 min at 25°C) and used at 2 × 105 cell/ml in RPMI-1640 (Sigma-Aldrich). Chemotactic migration was evaluated in 24-well transwell plates with 5 μm pore size polycarbonate filters (Corning). 0.2 × 106 cells were loaded in the upper chambers, while lower chambers contained 1 nM C5a (diluted from 48 nM C5a from zymosan-activated human serum, measured by ELISA) pre-incubated for 1 h at RT with 15 μl of either the indicated GAPDH-linked or untreated resin beads in RPMI-1640. Plates were then incubated for 30 min at 37°C with 5% CO2. Using forward and side scatter gating, the number of granulocytes recovered from lower chambers was quantified by flow cytometry for 1.5 min (constant flux acquisition, FACSCalibur, BD Biosciences). Normal migration to 1 nM C5a preincubated with untreated resin beads was 5,841 ± 2,141 cells, background migration to EDTA-containing human serum with undetectable C5a by ELISA was 510 ± 245 cells. Migration after preincubation with GAPDH resin beads was expressed relative to normal migration after preincubation with the same amount of untreated resin beads. Statistical analysis and graphical representation were done using the R software.
The crystal structure of AvGAPDH has been solved at 2.19 Å resolution and a complete macromolecular model has been built and refined with correct stereochemical parameters. Upon convergence, the crystallographic R factors were Rwork/Rfree = 0.177/0.226. Table 1 contains the X-ray diffraction data collection and refinement statistics. The overall structure and domain architecture of AvGAPDH closely resemble those described in previous GAPDH structures. The quaternary structure shows four subunits designated O, P, Q, and R with three non-equivalent intersubunit interfaces related by three mutually perpendicular dyad axes (P, Q, and R; nomenclature consistent with PDB code 1CER; Tanner et al., 1996) and one NAD+ molecule bound per active site, which is clearly defined in electron density (Buehner et al., 1974; Roitel et al., 2003). The four monomers in the GAPDH homotetramer are not necessarily equivalent since previous studies have indicated the presence of negative allosteric effects across subunits (Nagradova, 2001). In the final model of AvGAPDH, the tetramer shows minor deviations of <0.2° from the ideal 222 (D2) molecular symmetry (Figure 1A). The AvGAPDH homotetramer contains three asymmetric interfaces between each protomer and the adjacent subunits. The O-P interface contains the largest buried solvent area (1,830 Å2) and includes about 30 hydrogen bonds and 12 salt bridges. The second largest interface is the O-R interface, with 1,325 Å2, and is formed by 10 hydrogen bonds and 2 salt bridges. Finally, the smallest of the three interfaces, between the O-Q subunits, covers about 500 Å2 and includes 11 hydrogen bonds and 4 salt bridges.
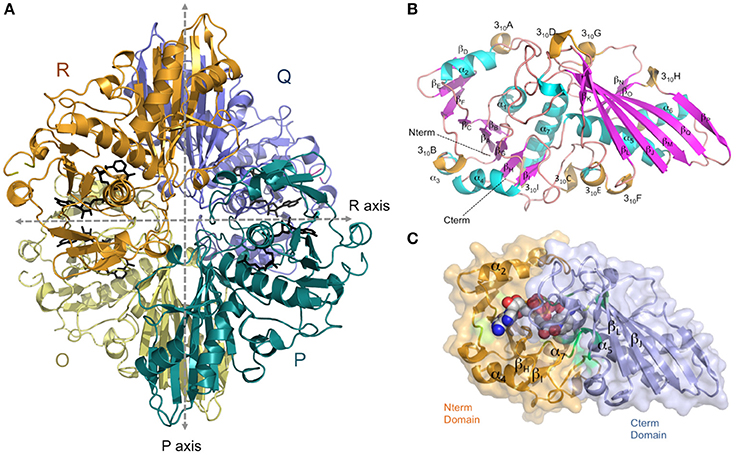
Figure 1. Overall architecture of the homotetrameric AvGAPDH. (A) The AvGAPDH homotetramer subunits O, P, Q, and R are shown in yellow, cyan, violet, and orange, respectively. In the active site of each protomer there is one NAD+ bound (black). Symmetry axes are shown as dashed arrows, with the Q axis shown perpendicular to the plane of the paper. (B) Ribbon representation of the AvGAPDH monomer with secondary structural elements labeled. Helices are colored in cyan, 310 helices in orange, strands in violet, and interconnecting segments in pink. The cofactor molecule is not shown for clarity. (C) Shown in the same orientation as in (B), the two domains of AvGAPDH are mapped in two distinct colors on a molecular surface representation. The N-terminal cofactor-binding domain is mapped in orange and the C-terminal catalytic domain in violet. NAD+ atoms are depicted as spheres in CPK colors. The figure was produced with PyMOL (Schrodinger LLC).
AvGAPDH is characterized by an N-terminal NAD+-binding domain (residues 1−167) and a C-terminal catalytic domain (residues 168−356; Harris and Waters, 1976). The N-terminal NAD+-binding domain features the classical α/β dinucleotide-binding fold or Rossmann fold (Figures 1B,C). It folds into eight β-sheets (βA-βH) connected by helices and short loops. The helices of the N-terminal domain are α1-α2, which are located before and after βB, and three 310 helices: 310A, which immediately precedes βC, βD, and βE, and 310B followed by α3-α4. The third 310 helix (310C) connects an extended section with a central isolated β-bridge centered at Ile147 with the last β-strand (βH) of the NAD+-binding domain. Overall, the structural tandem β-α-β-α-β motif is conserved (Lesk, 1995; Antonyuk et al., 2003). The C-terminal domain consists of a seven-stranded mixed parallel β-sheet (βI-βP), three α-helices (α4-α6) there are six 310 helices that are found immediately before or following β-strands (Figures 1B,C). Helices 310D-F are located between βJ-βK strands, while 310G, 310H and 310I are placed after βJ, βK,and βP-α7, respectively, with α6 between the βL-βM strands. The N-terminal NAD+-binding domain and the C-terminal catalytic domain are bridged at the joint between strand βH and the long helix α4. The catalytically active nucleophile Cys168 and the activating His195 lie at the beginning of helix α5 and the end of the following strand, βI, respectively. The C-terminal α6 helix is inserted into an extensive groove of the N-terminal domain where it forms a significant part of the interdomain interface and contributes to the binding pocket for the nicotinamide ring of the NAD+ cofactor. No traces of a phosphate ion were found bound into the active site.
The distance between the active-site Cys168 sulfhydryl to the Nε2 atom of the active-site His195 is 3.39–3.45 Å in chains O and R and 3.64 Å in chains Q and R, which is consistent with the defined closed conformation for the reported structure of homotetrameric AvGAPDH. Differences in cofactor binding between the subunits of GAPDH from various sources have been previously postulated as the basis for the negative cooperativity observed in NAD+ binding, where the first two NAD+ molecules bind with a higher affinity than the third and fourth NAD+ molecules (100−300-fold less tightly; Nagradova, 2001), although this asymmetry has not been demonstrated for every GAPDH. In particular, no negative co-operativity was observed in trypanosomatid GAPDH enzymes (Lambeir et al., 1991).
The crystal structure of AvGAPDH presents the closed conformation of the enzyme. In support of a closed conformation, the conformation of the NAD+-bound AvGAPDH is significantly more similar to the structure of NAD+-bound Bacillus stearothermophilus (Bs) GAPDH (PDB ID 1GD1) than to the NAD+-free form (PDB ID 2GD1; Skarzyński and Wonacott, 1988) as shown by the corresponding r.m.s.d. values (1.35 Å vs. 1.54 Å, respectively). AvGADPH in solution is likely to be more flexible or have more disordered surface loops than the closed enzyme conformation.
In many GAPDH structures a conserved Val residue adopts a highly strained backbone conformation (Kim et al., 1995), within the most disallowed region of the Ramachandran plot, in order to help maintain a hydrogen bond interaction between a critical Asn and the nicotinamide ring of NAD+ (Souza et al., 1998). In contrast, the equivalent residue in AvGAPDH, Gly257, establishes a hydrogen bond with Asn336 through its main-chain amide that has a similar length (2.9−3.1 Å) and orientation as the more commonly found Val-Asn hydrogen bond. This isofunctional Val to Gly substitution has also been observed in S. aureus and Thermotoga maritima GAPDH (Korndörfer et al., 1995; Mukherjee et al., 2010).
An omit map calculated with molecular replacement phases (prior to refinement) around the catalytic Cys168 of all subunits (Supplementary Figure 1) confirms the absence of covalent oxidation modifications that would inactivate AvGAPDH, as those present in previously reported GAPDH structures, which have been explained as oxidation of the cysteine residue to sulfonic acid or S-oxyl cysteine (Cowan-Jacob et al., 2003). His195, which assists Cys168 by increasing its nucleophilicity, is itself polarized by interactions with the catalytically important residue Arg251 (Nagradova, 2001). The conformation adopted by Arg251 side chain allows it to form hydrogen bonds with Asp200 and the nearby Gln201 side chains, on the opposite side to His195. Similar polarizing interactions between Arg residues with Asp carboxylates have been described to facilitate catalysis by lowering the pKa of Arg251 for the guanidinium side chain from 12.5 to 9.0 (Kuzminskaya et al., 1991; Cowan-Jacob et al., 2003).
NAD+ molecules are tightly bound in an elongated cleft in all four protomers of the homotetrameric AvGAPDH structure (Figures 1A, 2A). The presence and occupancy of NAD+ was assessed with unbiased omit maps (Figure 2B). The nicotinamide group is deeply embedded in the protein, as suggested by the similar or even lower B-factors of the NAD+ molecules (36.8−49.2 Å2) in comparison with the average B-factors for the protein chain (43.0−52.1 Å2). The atomic B-factors for the adenine moiety are between 2.0 Å2 lower and 8.5 Å2 higher than the atomic B-factors for the nicotinamide part of the same NAD+ molecule, thereby indicating a well-defined conformation for the adenine group in all chains (Figures 2, 3). Furthermore, the electron-density real-space correlation coefficient (RSCC) for the NAD+ molecules is 0.94−0.97 (0.90 overall), indicating that the NAD+-binding pocket is fully and equally occupied in all four protomers (Figures 2B, 3A). The relatively low B-factors and near-unity RSCC for the NAD+ molecules argue for a very tight binding of the cofactor into the NAD+-binding pocket.
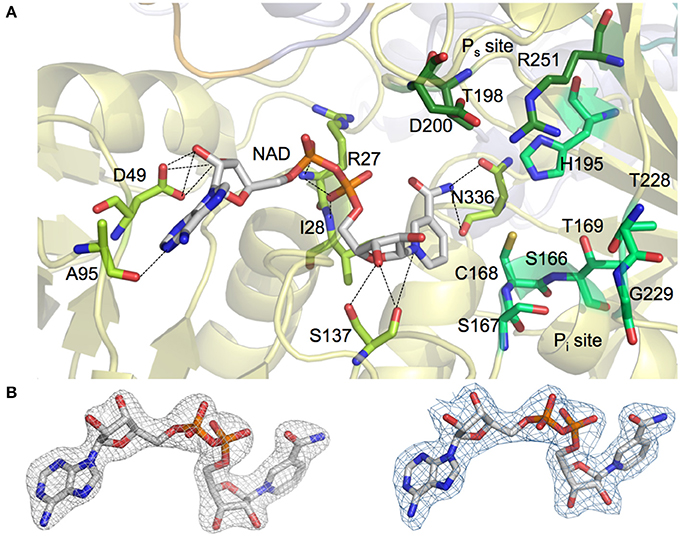
Figure 2. NAD+ and phosphate binding sites in AvGAPDH. (A) Binding pockets for the NAD+ cofactor (shown in sticks with carbon atoms in gray and all other atoms in CPK colors) and for the inorganic and substrate phosphate sites (“new Pi” and Ps sites, labeled). The placement of the Pi site in AvGAPDH as a “new Pi” site is consistent with the S. aureus GAPDH structure (PDB ID 3K73). Enzyme residues in direct contact with NAD+ are represented in sticks (carbon atoms in light green) and polar interactions (hydrogen bonds) are depicted as dotted lines. Residues predicted to participate in the stabilization of the inorganic and substrate phosphate groups (including the catalytic Cys168) are also shown as sticks (with carbon atoms in two different shades of dark green). The AvGAPDH structural scaffold is shown in ribbons in depth-cued light colors. (B) Omit map showing unbiased electron density map of NAD+ cofactor in AvGAPDH. Unbiased omit map calculated with molecular replacement phases (prior to building the cofactor and solvent structures and prior to refinement) contoured at 1.5 σ revealing the presence of the NAD+ cofactor at full occupancy. For comparison, electron density map for the final model of the NAD+ cofactor displayed at the same contour level (right). The figure was produced with PyMOL (Schrodinger LLC).
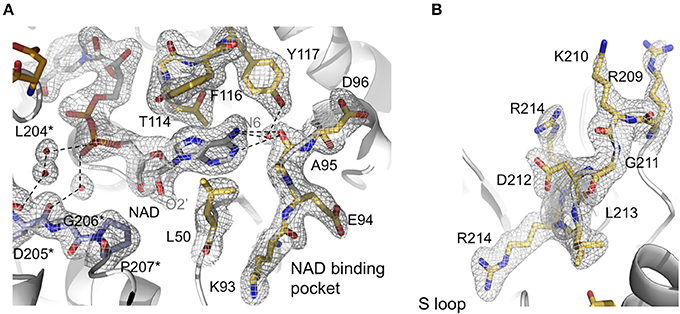
Figure 3. Active site features of AvGAPDH. AvGAPDH is shown in cartoon representation (gray; NAD+ binding site is in gold; and AvGAPDH-specific residues in the S loop in violet). (A) Network of interactions that stabilizes NAD+ binding. The main and side chains of NAD+-binding residues are shown in stick representation. Key water molecules are shown as red spheres. They are overlaid with the σA-weighted (2mFO−DFC) electron density map contoured at 1 σ level. Asterisks denote residues belonging to the neighboring subunit. (B) Continuation of the S loop (residues Ala196-Ile223, in gold) beyond the NAD+ binding site, depicting its elongated, irregular shape. Residues are overlaid with the σA-weighted (2mFO−DFC) electron density map contoured at 1 σ level. The figure was produced with PyMOL (Schrodinger LLC).
Most residues involved in cofactor binding are completely conserved between AvGAPDH and human GAPDH. However, there are several substitutions in the cofactor-binding site, especially in residues located around the entry gateway. Specifically, Pro36, Phe37, and Arg80 in human liver GAPDH are substituted in AvGAPDH by Asp49, Leu50, and Ala95, respectively (Figures 2A, 3A). Structurally, the smaller aliphatic side chain of Leu50 fulfills essentially the same role as Phe37 in the human enzyme, although they build slightly different hydrophobic environments that might be exploited by selective drug targeting. The loss of the positively charged Arg80 present in human GAPDH is likely inconsequential for NAD+ binding, since Arg80, exactly as Ala95 in AvGAPDH, interacts with the cofactor via a main-chain carbonyl hydrogen bond. Interestingly, in Plasmodium falciparum (Satchell et al., 2005) the equivalent position is occupied by a Lys, thereby suggesting that a positively charged side chain might play a secondary functional role in NAD+ and/or substrate binding/release. A network of water molecules further contributes to stabilizing the bound NAD+ conformation in the active site by holding in place the adenosyl and nicotinamide rings and the nucleotide linking phosphate groups (Supplementary Figure 2).
Since the Pi and Ps sites (Supplementary Figure 3) supply essential functions, the GAPDH residues involved in shaping them are highly conserved across very divergent species (Supplementary Figure 4). While the Ps site has been found at an identical location across all known prokaryotic and eukaryotic GAPDH structures, two distinct Pi sites have been described. The so-called “classical Pi” site is modeled after the BsGAPDH co-crystallized with sulfate anions (PDB ID 1GD1, sequence identity: 50.3%; Skarzyński et al., 1987), whereas a “new Pi” site was first characterized in T. maritima (Tm) GAPDH (Korndörfer et al., 1995) and subsequently corroborated in Escherichia coli (Ec) GAPDH and S. aureus (Sa) GAPDH structures (Yun et al., 2000; Mukherjee et al., 2010). Their different locations within the GAPDH active site is explained by the differential placement of the β-strand-loop-α-helix stretch spanning residues 226−231, which is more deeply buried in those structures with a “new Pi” site. The strand-loop-helix motif residues in AvGAPDH were all clearly defined in electron density maps. In the AvGAPDH structure there is no residual electron density at the Pi, “new Pi,” or Ps sites, indicating that they were completely unoccupied in the crystal (Supplementary Figure 4B). By analogy with most GAPDHs of known structure that have been crystallized with inorganic phosphate or sulfate, we could assign the four residues that contribute to the canonical Ps site: Thr195 (Oγ atom), Asp200 (side-chain carboxylate), Arg251 (guanidinium group), and NAD+ (2′ hydroxyl of the nicotinamide-ribose part, O2D; Supplementary Figure 3A). A conformational switch involving Arg251 has been proposed to play an important role in the regulation of the substrate G3P binding and the release of the product, 1,3-bisphosphoglycerate (1,3-BPG; Souza et al., 1998). Inspection of the conformation adopted by the 226−231 loop of AvGAPDH indicates that its Pi site is most likely a “new Pi” site analogous to that of SaGAPDH, which would recruit the side chains of Ser167, His195, Thr228, and Arg251, and the main-chain carbonyl and side-chain hydroxyl of Thr169 (Supplementary Figures 3B, 4). The neighboring residue Arg215 can interact with the side chain of Asp200 to stabilize its position and, conceivably, could also support a product-release mechanism whereby Arg215 would interact with the phosphate moieties of 1,3-BPG thereby displacing the similar interaction with Arg251, which is part of both the “new Pi” and the Ps sites. This displacement would require the side chains of Asp200 and Arg215 to flip in and out according to the substrate binding state of AvGAPDH (Supplementary Figure 4).
An important structural feature of the active site of GAPDH is the S loop, a long winding region folded into an irregular shape along the R dyad groove spanning residues Ala196-Ile223 (Figures 3A,B) around the P dyad (Biesecker et al., 1977; Skarzyński et al., 1987). Residues in the S loop contribute to a complex interaction network that includes hydrophobic and ionic contacts around the R axis and ionic contacts around the P dyad (Skarzyński et al., 1987; Biesecker et al., 1977). Pro207 forms one side of the binding pocket for the adenine-ribose moiety of the NAD+ bound to a neighboring subunit related by the R axis (Figure 3A). Despite the overall conservation of the peculiar S loop sequence in AvGAPDH, there are several substitutions plus a Gly211 insertion at Ile203-Arg217 in the middle of the S loop (Figure 3B). These modifications cause the protrusion of a turn containing both positively charged side chains (Arg209 and Lys210) toward the crevice where the nicotinamide-ribose moiety of NAD+ is located, whereas in most GAPDHs the equivalent turn contains only one conserved Lys (Lys197 in human liver GAPDH; Figure 4A). Electron density for the side chains of Arg209 and Lys210 is weak (Figure 3B), which is attributed to the high thermal parameters of side-chain atoms (backbone atoms have low B-factors). In particular, there is no visible electron density for the Cγ-Nε atoms of Arg209 and Cε-Nζ atoms of Lys210 at 1 σ contour level, although the clearly visible electron density for the first side-chain atoms (Cα-Cβ-Cγ) unambiguously (Figure 3B). The Cα atoms of Arg209 and Lys210 are roughly equidistant (14.1−15.6 Å and 12−13.1 Å) from the N7 atom of the adenine moieties in the cofactors bound to the O and R subunits. The closest distance between either side chain in a fully extended conformation and NAD+ is 7.5 Å. In addition, residues Leu204 and Asp205 are conserved and interact with the cofactor through water mediated and main chain interactions (Figure 4A and Supplementary Figure 3). It is remarkable the conservation between the human and the AvGAPDH enzymes at and around the entry gateway to the adenine ring of NAD+, in particular Val101 (Phe116) and Phe37-Leu50 (Figures 3A, 4C). A Phe in place of Val101 should present steric clashes with Pro82 and Ala97 in AvGAPDH, therefore it appears that this change in sequence has a structural role.
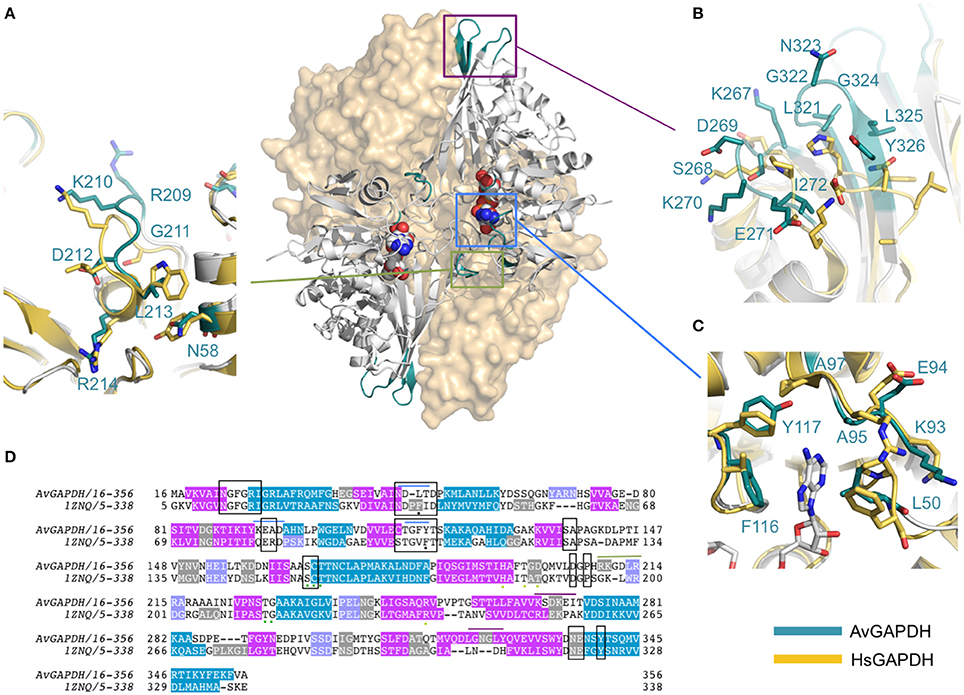
Figure 4. Structural comparison of AvGAPDH and human liver GAPDH. Ribbon representations of the AvGAPDH and human liver GAPDH (PDB ID 1ZNQ) holoenzymes in gray. The molecular surfaces of two subunits are shown in wheat color. The NAD+ cofactor bound to the two subunits represented as ribbons are shown as sphere models in CPK color. Regions where structural differences are more prominent are shown in teal, boxed, and magnified as panels (A–C). (A) S loop. (B) Structurally distinct loop regions on the outer surface of AvGAPDH. (C) Adenine ring subpocket of the active site. Several pharmacologically relevant compounds have been discovered that target the adenine ring subpocket of GAPDH from trypanosomatids. The superposition, molecular surface, and (A–C) were produced with PyMOL (Schrodinger LLC). (D) Sequence alignment including the protein sequences of AvGAPDH and human liver GAPDH.
The closest structural homolog of AvGAPDH is SaGAPDH (PDB ID 3K73; Mukherjee et al., 2010) with an r.m.s.d. value of 1.35 Å when the complete quaternary structures are superimposed (0.96 Å on average when each pair of monomers are superimposed), and 63% sequence identity. The structures of AvGAPDH and SaGAPDH are therefore considerably conserved, with most differences concentrating in loops and connection regions. For instance, the α1-βB junction is different in the two enzymes, leading to a retraction of the peptide chain that causes a broadening of a solvent-exposed groove over the active site in AvGAPDH. In addition, several 310 helices in AvGAPDH are absent in SaGAPDH, which are responsible for some local structural deviations; for example, 310A (residues 69−71), which protrudes toward the interface between the two adjacent subunits, has a maximum displacement to the corresponding segment in SaGAPDH of 4.7 Å. On this face of AvGAPDH, the structural differences at helix 310A are further amplified by additional local changes at the loop connecting the following strands (βB-βC). Likewise, the loop spanning Gly141-Asp143 shifts toward the entryway to the active site, contributing to the closing of the NAD+-binding pocket, with a maximum distance deviation of 5.5 Å at the center of the loop. Finally, concerted changes affecting the βL-α6 loop and the strands βO and βP (spanning residues 266−287 and 319−326, respectively) in a fully solvent exposed area lead to severe main-chain displacements as large as 4.29 Å.
To compare the catalytic efficiency of AvGAPDH with SaGAPDH and other homologous enzymes, we measured its kinetic parameters for the direct reaction by systematically varying NAD+, G3P, and Pi concentrations. The results are shown in Table 2. For G3P, for which there are more complete kinetic data for SaGAPDH, the kcat rate constants of the two enzymes were pretty similar; however, the 10-fold higher KM for G3P determined for AvGAPDH in comparison with that of SaGAPDH resulted in a correspondingly 10-fold lower kcat/KM ratio. It is interesting than the KM for NAD+ is smaller than for the co-substrate, G3P, by a factor of 30.

Table 2. Kinetic parameters.
In addition to the essential glycolytic role of AvGAPDH, we investigated whether AvGAPDH could function as a virulence factor by sequestering the human C5a anaphylatoxin, a complement factor 5 (C5)-derived proteolytic activation fragment which diffuses out of the area wherein C5 is activated to chemoattract neutrophils and macrophages (Marder et al., 1985; Guo and Ward, 2005). This C5a-inhibitory function had been previously observed for S. pyogenes (Sp) GAPDH (Terao et al., 2006), and it requires the translocation of GAPDH to the cell wall or the extracellular medium, a process that has been described for GAPDH enzymes from a variety of prokaryotic and eukaryotic organisms (Aguilera et al., 2012). Although it is still unknown whether AvGAPDH is displayed on A. vaginae cells, it is a conceivable scenario that could play an important function in the long-term survival strategy of the pathogen. Since the AvGAPDH and SpGAPDH sequences share a 60% sequence identity, we investigated whether both proteins could also share the immunoevasive function described for the S. pyogenes enzyme.
In order to test the possible interaction between AvGAPDH and C5a we conducted three sets of experiments. Firstly, we evaluated whether biotinylated C5a could bind to membrane-immobilized AvGAPDH in ligand blotting experiments, which have been used before to demonstrate an interaction between SpGAPDH and C5a (Terao et al., 2006). The results show that biotinylated C5a accumulates rather specifically onto AvGAPDH bands in a quantitative fashion (Figure 5A), thereby indicating that a complex between membrane-immobilized AvGAPDH and biotinylated C5a does indeed form. This is to be expected of an interaction between the fluid-phase C5a and cell wall-associated AvGAPDH. In comparison, it is noteworthy the absence of a signal on the negative control (BSA lane) and the strong signal from the positive control (C5a lane, which contains biotinylated C5a).
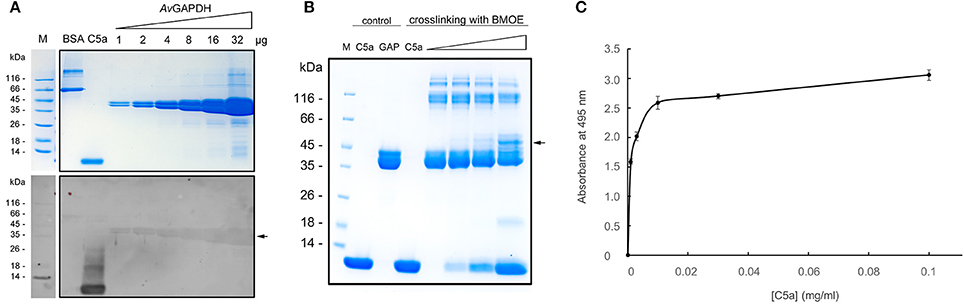
Figure 5. AvGAPDH interacts with human C5a. (A) Detection of C5a interaction by full-length AvGAPDH in ligand blotting experiments. Top, Coomassie brilliant blue (CBB)-stained SDS-PAGE gel where BSA (negative control), biotinylated C5a (positive control, detectable without added biotinylated C5a), and increasing amounts of pure AvGAPDH (1−32 μg) were separated. Bottom, ligand blotting was conducted after the electrotransfer of the same SDS-PAGE gel shown on top onto nitrocellulose membrane by incubating the membrane with biotinylated C5a and developing the blot with horse radish peroxidase (HRP)-conjugated anti-biotin antibody and luminol. (B) Crosslinking of AvGAPDH and C5a with BMOE shown on a CBB-stained SDS-PAGE gel loaded with mock-treated control samples (C5a and AvGAPDH, GAP) and BMOE-treated crosslinked samples (increasing concentrations of C5a for a fixed concentration of AvGAPDH). The two first lanes with added BMOE represent internal controls for C5a (maximum load) and AvGAPDH (no C5a). In (A) and (B) arrowheads point to the bands where AvGAPDH and C5a co-localize. (C) C5a bound to AvGAPDH immobilized on a polystyrene microtiter plate measured by an ELISA assay. The absorbance at 495 nm is plotted against increasing C5a concentrations. Data represents mean ± SD (standard deviation; N = 3).
Secondly, we tested the AvGAPDH-C5a interaction in solution using natively folded AvGAPDH with the highly specific cross-linking reagent BMOE, which tethers together Cys residues that lie in close proximity (typically, within 8 Å). As shown in Figure 5B, cross-linked AvGAPDH-C5a bands developed that prove that a native interaction exists between AvGAPDH and C5a that involves surfaces containing a Cys residue in AvGAPDH and the only free Cys in C5a (Cys704). Treating C5a or AvGAPDH alone with BMOE confirmed that no cross-linking species is generated, thereby ensuring the specificity of the interactions captured by BMOE. However, increasing C5a amounts led to the progressive appearance of a C5a-AvGAPDH complex, which runs with an apparent molecular mass ca. 10 kDa greater than AvGAPDH (Figure 5B, arrow), corresponding to the mass of C5a. As the electrophoretic band containing cross-linked C5a-AvGAPDH species becomes more intense, the amount of free AvGAPDH becomes lower in proportion, further arguing in favor of a quantitative interaction. In parallel, the amount of free biotinylated C5a unreacted with BMOE is significantly lower than in the corresponding BMOE-treated control lane, thus indicating a proportional reduction of both free species as the amount of cross-linked complex increases. While the involvement of Cys704 of C5a uniquely defines the interaction surface for C5a, there remains an ambiguity in assigning the surface area on AvGAPDH engaged for interaction with C5a. While the N-terminal fragment 1−165 of SpGAPDH was sufficient to elicit the interaction with C5a (Terao et al., 2006), an analogous C-terminally truncated version of AvGAPDH (residues 1−185) was only able to bind C5a in the same degree than the full-length enzyme in ligand blotting and cross-linking assays, but not in other functional assays (cf. below). This indicates that the complete AvGAPDH-C5a interaction surface may engage a more extensive region of AvGAPDH than encompassed by the N-terminal deletion construct.
Finally, we also studied the interaction between AvGAPDH and C5a using an enzyme-linked immunosorbent assay (ELISA). In this assay, surface-immobilized AvGAPDH was capable of binding C5a in a concentration-dependent manner (Figure 5C), whereas uncoated wells or AvGAPDH-coated wells that had not been treated with C5a elicited no significant background signal.
To provide further evidence for a potentially immunoevasive function for AvGAPH, we next investigated whether AvGAPDH was able to inhibit granulocyte (mainly neutrophils) motility, a C5a-specific cellular response that is relevant in the context of bacterial infections. Based on the known interaction surface of SpGAPDH and the binding experiments described for AvGAPDH, we would expect that AvGAPDH could bind C5a through surface areas located close to the N terminus of AvGAPDH (Figure 6A). We tested surface-immobilized AvGAPDH by challenging blood-purified human granulocytes in presence of zymosan-activated serum, which contains native human C5a, with increasing concentrations of soluble AvGAPDH and two different amounts of AvGAPDH-bound resin beads, and measuring the inhibition of neutrophil chemotaxis in a cell migration assay (Figure 6B). The resin-immobilized AvGAPDH contained 53 μg/μl full-length AvGAPDH (80−85% immobilization yield), therefore providing a dense surface distribution of AvGAPDH on the resin beads. As positive controls for both experimental designs, we used immobilized SpGAPDH. Lastly, we also used GAPDH from Clostridium perfringens (Cp) as a third related protein to assess the capacity of GAPDH enzymes with varying sequences to inhibit C5a-induced cell migration.
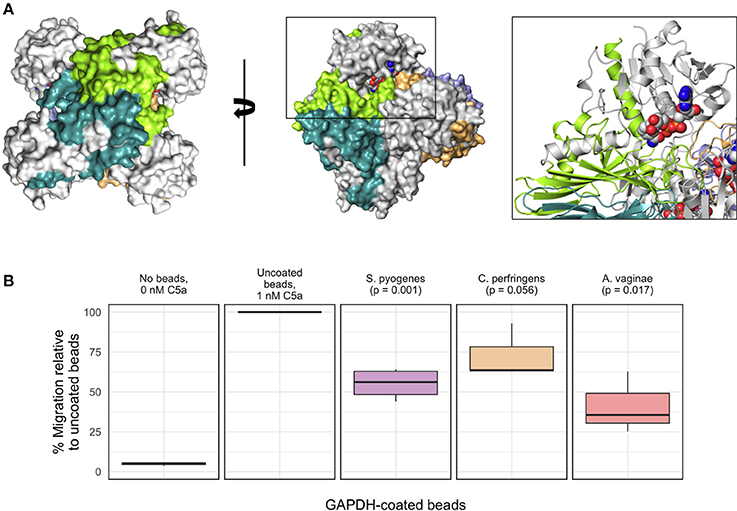
Figure 6. Inhibition of C5a-specific granulocyte chemotaxis by recombinant AvGAPDH variants. (A) Molecular surface of the AvGAPDH tetramer in two views related by a 90° rotation through a vertical axis. Residues 1−185 of AvGAPDH of each protomer, which correspond to the C5a-binding region defined for S. pyogenes GAPDH (Terao et al., 2006), are shown in gray, while the remaining C-terminal sequence is shown in chain-specific colors. The black box surrounding one N terminus (NAD+ atoms are represented as spheres and colored in CPK) is shown as an enlarged view with AvGAPDH structural elements in ribbon representation. (B) The boxplot graph shows the results of a transmigration experiment of human granulocytes towards a C5a-containing lower chamber expressed as percent migration (mean ± SEM of 3−4 independent experiments). All results are normalized to the migration of human granulocytes elicited by 0.96 nM C5a incubated with the corresponding amount of uncoated aminoethyl agarose (AM-4) resin beads, taken as 100%; background signal at 0 nM C5a is 4.8%, shown for comparison. The remaining migration experiments were conducted by pre-incubating 0.96 nM C5a with aminoethyl agarose (AM-4) resin beads, either mock (uncoated resin beads) or with covalently immobilized GAPDH molecules. Besides the C5a control, from left to right, the bar graph shows mock AM-4 beads, AM-4 beads with full-length GAPDH from S. pyogenes, C. perfringens, and A. vaginae. Migration inhibition percentages were normalized to the amount of GAPDH effectively coating the beads. The p-values shown above the boxplots for GAPDH-coated beads were calculated with the R software.
The cell migration assays convincingly showed that resin-bound full-length AvGAPDH could reduce the amount of C5a-mediated transmigration of granulocytes in our cell-based assay (Figure 6B). The marked reduction in cell migration elicited by preincubating beads coated with AvGAPDH (or SpGAPDH) with the activated C5a serum strongly suggested that a direct, physical interaction between AvGAPDH and C5a as the likely mechanism for cell motility arrest. The inhibitory effect of AvGAPDH on granulocyte migration was significant (p = 0.017) and approximately equivalent to that achieved by the same amount of SpGAPDH-immobilized beads (p = 0.001), thereby indicating that the neutralization of C5a by A. vaginae might be as efficient as during S. pyogenes infection. Interestingly, CpGAPDH failed to significantly block granulocyte motility at the concentrations tested (p = 0.056), thereby indicating a sequence-dependent effect of GAPDH-mediated inhibition. The varying degrees of cell migration inhibition, in turn, could reflect upon the varying concentrations of C5a in their respective infection sites in humans and their different mechanisms for infection and immunoevasion. Although the amounts of AvGAPDH used in these assays are larger than in comparable antagonist-based assays (e.g., Huber-Lang et al., 2002), the increase in avidity caused by the cell surface display of AvGAPDH could compensate for the apparent lower binding strength, a binding-potentiating effect that would be even more pronounced during infection, when A. vaginae cells typically grow in a highly concentrated biofilm.
The reported interaction between AvGAPDH and C5a and the inhibitory effect that AvGAPDH has on C5a-induced granulocyte migration may arguably play a role in A. vaginae survival during infection by combating pro-inflammatory defense mechanisms mounted by the human immune system. Specifically, colonization and infection of the vaginal tract by A. vaginae and other pathogenic bacteria such as Gardnerella vaginalis are characterized by triggering an intense inflammatory response with the liberation of interleukins IL-6 and IL-8, which is not elicited by the microbiota of healthy individuals, and that is essential for the pathogenesis (Fichorova et al., 2011; Doerflinger et al., 2014).
GAPDH sequences of known structure and with a significant degree of sequence similarity to AvGAPDH were found with BLAST (Camacho et al., 2009) using sequences stored in the PDB, aligned with MUSCLE and used to reconstruct a maximum likelihood phylogenetic tree with PhyML (Guindon et al., 2010; Figure 7).
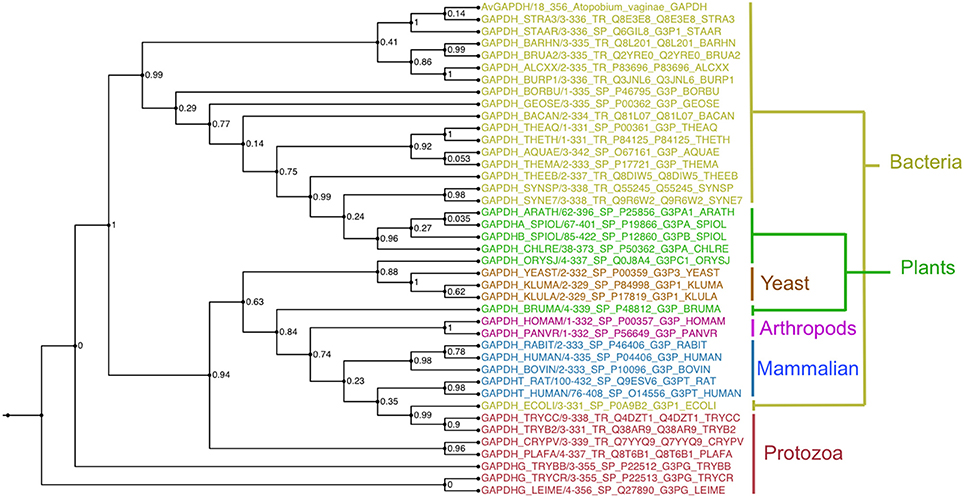
Figure 7. Evolutionary conservation. Unrooted phylogenetic tree of selected GAPDH sequences calculated with the maximum-likelihood software PhyML with branch supports shown at every node. Sequences are colored according to taxonomic grouping, with bacteria in yellow, plants and algae in green, yeasts in orange, arthropods in purple, mammals in blue, and protozoa in red. The multiple sequence alignment is provided in the SI. This figure was produced with FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
In the absence of an efficient treatment for A. vaginae infection and the possibility that AvGAPDH could participate in the onset and maintenance of the pathogen through both metabolic and immunoevasive strategies, we considered whether available GAPDH inhibitors targeting trypanosomatid GAPDH enzymes, which escape binding to human liver GAPDH, could be used to block AvGAPDH. The overall structure of AvGAPDH is similar to those of T. cruzi (PDB ID 1ML3; Castilho et al., 2003), T. brucei (PDB ID 2X0N; Vellieux et al., 1993), and L. mexicana (PDB ID 1GYP; Kim et al., 1995), with r.m.s.d. values of 1.35, 1.30, and 1.30 Å over 320 aligned Cα atoms, respectively. Similarly, superposition of AvGAPDH with the human and rabbit GAPDH enzymes can be achieved with r.m.s.d. values of 1.26 Å (fully occupied by NAD+) and 1.30 Å (devoid of NAD+). While most structural differences are located in flexible surface loops, several unique features stand out when the NAD+-binding site and, especially, the adenosyl-binding pocket, are compared across GAPDH enzymes from different species (Bressi et al., 2001; Kennedy et al., 2001; Figure 8).
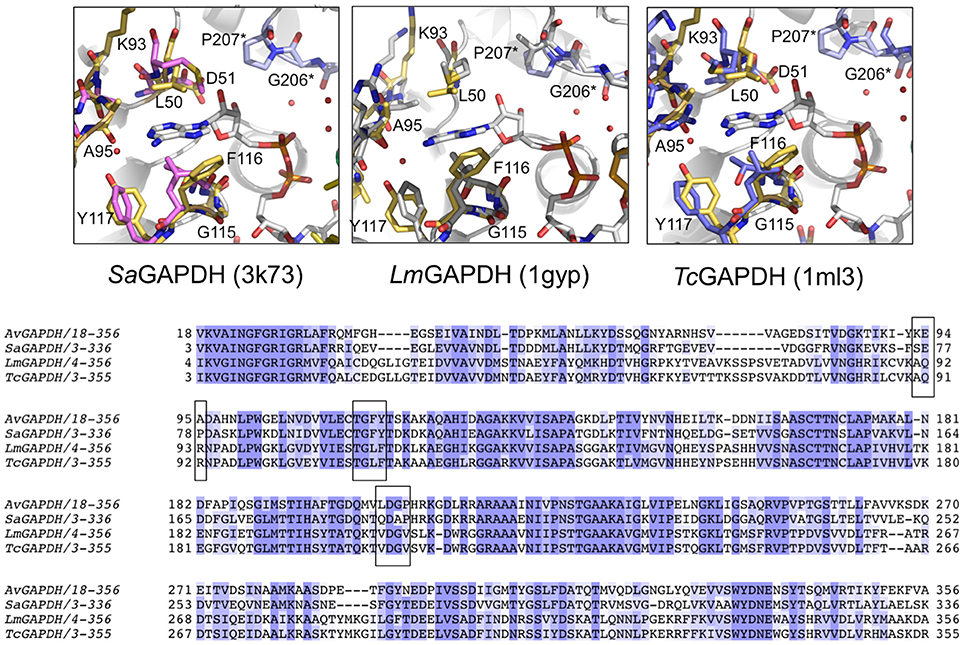
Figure 8. Comparison of the NAD+ adenine ring-binding subsite. Ribbon representation of the NAD+ binding pocket in AvGAPDH with the cofactor and the residues that lock the adenine ring of the cofactor (in gray) shown in sticks (in gold). Residues from the neighboring chain are labeled with an asterisk and their carbon atoms are shown in violet. (A) Comparison with S. aureus SaGAPDH adenosyl-binding pocket (PDB 3K73; pink). (B) L. mexicana LmGAPDH (PDB 1GYP; gray). (C) Trypanosoma cruzi TcGAPDH (PDB 1ML3; blue). The figure was produced with PyMOL (Schrodinger LLC). (D) Multiple sequence alignment of GAPDH from A. vaginae, S. aureus, L. Mexicana, and T. cruzi. Adenosyl-binding residues are boxed.
The most notable difference between AvGAPDH and the human enzyme concerns the anchoring of the adenine ring into the hydrophobic pocket (Figure 4C). It is precisely the less conserved regions between the GAPDH from pathogens and human GAPDH that are appealing for the design of selective inhibitors, which can impair pathogen viability without compromising the host GAPDH function. Thus, for years, changes in the hydrophobicity of the areas adjacent to the N6 position of the adenine ring and the O2′of the ribose of NAD+ molecule have been successfully used as anchor points for selective inhibitors of GAPDH from pathogens such as TmGAPDH, TvGAPDH, and TcGAPDH (Figure 8C). In the crystal structure of AvGAPDH, the side chains of Leu50 and Thr114-Gly115-Phe116-Tyr117 surround the NAD+ binding pocket around the adenine ring (Figure 8). Indeed, the electron density map contoured at 1 σ level confirms the unusual presence of Tyr117 instead of the highly conserved Phe (Figures 3A, 4C, 8) observed in nearly all sequences, with the exception of rat, human sperm and SaGAPDH (Figure 8A). In all cases, a conserved water mediates the interaction between the OH group of Tyr117 side chain with the N5′ atom in the adenine ring (Figures 8A–C). Furthermore, another feature unique to AvGAPDH is the presence of the minimalist loop Ala95-Ala97-Asp96 nearby the position occupied by the adenine ring of NAD+ (Figures 3A, 4C, 8A). The intrusion of this loop facilitates the interaction of Ala95 main-chain with the adenine ring. The tandem Phe116-Tyr117 is also missing along several pathogenic bacteria, both Gram-positive (e.g., Staphylococcus and Streptococcus spp. and Bacillus anthracis) and Gram-negative (e.g., Bartonella, Brucella, Borrelia, and Synechococcus spp.). Also, GAPDH from some thermophilic bacteria (e.g., Thermus, Aquifex, and Thermosynechococcus spp.) lack an equivalent residue to Tyr117. These differences can be exploited for the design of drugs targeting the N6 position of the adenine ring (Figure 7). On the other hand, the ribose ring of the NAD+ cofactor interacts with Pro207, which is conserved in HsGAPDH (Figure 3A). The narrow groove between the Pro207 and Pro36-Phe37 is a distinguishing feature used to search for selective drugs (Figure 3A). A relevant example is 2′-deoxy-2′-(m-methoxybenzamido)adenosine (MBDA), a curved molecule that binds selectively to TcGAPDH while it does not bind to HsGAPDH due to steric hindrance (MBDA; 50). For AvGAPDH, the equivalent residue is Phe30 and Leu50-Thr51, whose Cα's are 0.5 Å are displaced between them. This will probably make NAD+ inhibitors derived with substitutions in O2′ position less effective.
The functional cavities and interfaces of AvGAPDH that are most divergent with respect to the human GAPDHs suggest themselves as potential targets for drug discovery. In AvGAPDH the hydrophobic residues that surround the adenine ring have a distribution resembling more closely the configuration observed in the glycosomal GAPDHs than the human liver enzyme. Specifically, Leu50 occupies in AvGAPDH the position of Pro36-Phe37 in the human liver GAPDH and Thr114, Phe116, and Tyr117 shield the opposite side of the adenine pocket (Figure 4C and Supplementary Figure 5). Based on this observation, other strategies than those used for TvGAPDH, LmGAPDH, and TcGAPDH have to be adopted to inhibit AvGAPDH, and we propose that nucleoside analogs such as the adenosine derivatives like MBDA (MBDA; 50) represent an interesting starting point for future developments.
The crystal structure of AvGAPDH reported herein at 2.19 Å has been refined to convergence with Rwork/Rfree = 0.177/0.226 and the final model exhibits excellent stereochemistry (Table 1). Although the overall fold of AvGAPDH is highly conserved when compared with reported GAPDH structures (Figure 1), there are distinctive structural features in AvGAPDH with important consequences for potential protein binding partners as well as for the discovery of selective inhibitors (Figures 4, 8). The availability of the first high-quality, high-resolution crystal structure of a complement immunoevasive GAPDH can generate insights into the mechanistic basis for complement interference as well as provide a template for inhibitor search.
As previously reported, streptococcal GAPDH can function as an immunoevasion factor by sequestering in-situ produced C5a on the pathogen's surface during the lytic or terminal phase of complement activation (Terao et al., 2006). Terao et al. concluded that the N-terminal region of SpGAPDH (residues 1−165) was the likely C5a-recognition motif (Terao et al., 2006). The homologous region in AvGAPDH spans residues 1−185 and is reasonably well conserved, with 56.4% identity and 66.4% similarity (compare with the full-length sequence alignment, with 64.9% identity and 70.7% similarity). Since the structure of the N-terminal region of GAPDH enzymes is partly defined by the NAD+-binding pocket, these loops share an overall structural organization and they differ mostly by the array of residues that become solvent exposed. In particular, the structure of residues 1−185 in AvGAPDH and the equivalent segment of the closely related SaGAPDH (Mukherjee et al., 2010) trace roughly equivalent spatial trajectories over and above the cofactor-binding site. This is the proposed C5a-docking surface, which provides a relatively flat and flexible platform that could accommodate the binding of the small C5a molecule.
This structure and sequence-backed findings suggest that GAPDH enzymes from pathogenic bacteria located over widely diverse evolutionary distances could however be equipped with conserved C5a-binding surfaces. Since S. pyogenes, A. vaginae, and S. typhimurium are all important human pathogens, the immunoevasive capacity detected for bacterial GAPDH might conceivably have been selected for in the context of infection. If this hypothesis holds true, it would imply that the pattern of C5a sequestration by bacterial GAPDH should be extended to a large diversity of Gram-positive and Gram-negative bacterial pathogens. A second implication would consist in the opening of a window of opportunity for the treatment of antibiotic-resistant bacterial infections with complement stimulatory therapies, e.g., by releasing C5a blockade.
The relatively high sequence identity and similarity shared between SpGAPDH and AvGAPDH suggested that the latter could also elicit C5a sequestration. The experimental evidence presented hereby corroborates a likely role for AvGAPDH as an immunoevasive factor. Of the experiments performed, ligand blotting using biotinylated-C5a (Figure 5A) had been previously used with SpGAPDH to establish its interaction with C5a (Terao et al., 2006). In addition, we have carried out highly specific cross-linking experiments with BMOE to prove a physical interaction between AvGAPDH and C5a (Figure 5B). BMOE crosslinks protein thiol groups <8-Å apart, thereby completely avoiding spurious covalent complexes. Additional ELISA experiments corroborated the significance of the AvGAPDH-C5a interaction originally identified by the ligand blotting and cross-linking experiments. The previous three in vitro approaches converged on a model whereby AvGAPDH and C5a can form a complex. Unfortunately, we could not isolate a stable complex by chromatography to subject it to structural analysis. Given the evidence at hand and that from previous studies (Terao et al., 2006), we attribute this behavior to the complex environment wherein this interaction will occur in vivo, which might potentiate the physical association and/or provide a more favorable context for the interaction (e.g., on the cell-wall surface). Future research should tackle the issue of context and presentation for the host-pathogen interaction described in this work.
To seek independent verification for the AvGAPDH-C5a interaction, we tested the ability of soluble and surface-immobilized AvGAPDH, CpGAPDH, and SpGAPDH to inhibit neutrophil chemoattraction, which was triggered with native C5a generated in situ by incubating C5-containing serum with zymosan (Figure 6B). Since at least AvGAPDH and SpGAPDH are C5a interactors, the most likely mechanism for neutrophil migration blockade is physical sequestration of C5a by its binding partners. The results convincingly show that AvGAPDH and, to a similar extent, SpGAPDH, are capable of reversing the C5a-induced chemoattraction of neutrophils in a cell migration assay. CpGAPDH, in contrast, was less effective than either AvGAPDH or SpGAPDH in sequestering C5a. The observed inhibition was markedly stronger when surface-immobilized AvGAPDH was used as compared with the effect of soluble proteins, thereby indicating that the precise structural-chemical context where the GAPDH-C5a interaction can occur is bound to play a significant biological role. The fact that undecorated agarose beads were incapable of blocking neutrophil migration rules out an inhibitory role for the carrier material.
When combined, the ligand blotting, cross-linking, ELISA interaction assays, and cell migration inhibition assays together offer a coherent view of AvGAPDH (and SpGAPDH) as a virulence factor. In one of its manifold moonlighting functions, secreted and/or surface-immobilized AvGAPDH bears the potential to titrate out serum C5a from the infected areas where the anaphylatoxin is generated. As such, this complement down-regulatory mechanism could be at play in those cases where BV has been associated with a strong pro-inflammatory response as a result of the secretion of IL-6 and IL-8 (Fichorova et al., 2011; Doerflinger et al., 2014).
The two most salient differences between GAPDH from A. vaginae and the human liver enzyme are localized in the adenosine binding subpocket of NAD+ and around the S loop that faces it (Figure 4). These and other differences have been exploited for the successful design of anti-GAPDH compounds against trypanosomatids that escape trapping in the liver. In a similar manner, we reason that the observed differences could be exploited to discover compounds (or reuse anti-trypanosomatid GAPDH drugs) against AvGAPDH. This is a pressing matter owing to the inefficiency of current therapeutic options for the treatment of BV caused by the swift appearance of antibiotic resistant bacterial strains. When the AvGAPDH structure is compared with those of trypanosomatids, the pattern of conserved and diverging structural elements presents differences with those described for the comparison with the human liver enzyme (Figure 8). Interestingly, naïve chemical modeling of known anti-trypanosomatid GAPDH inhibitors into the AvGAPDH active site suggests that these well-studied families of compounds could be effective as inhibitors and, therefore, represent new therapeutic opportunities for BV.
The significance of the high-resolution AvGAPDH structure for drug discovery initiatives targeting human BV might be large if A. vaginae infections rely greatly on a functional glycolytic pathway. In principle, an effective anti-GAPDH agent that binds into the active site could as well disrupt the long loop that covers the NAD+ cofactor and is involved in C5a binding, therefore suggesting the interesting possibility that such an inhibitor could both down-regulate the glycolytic flux and also perturb C5a recognition by AvGAPDH.
Future investigations will address whether compounds related to the available anti-trypanosomatid GAPDH drugs can indeed effectively inhibit AvGAPDH.
There is accumulating persuasive evidence that metabolic enzymes, especially abundant enzymes such as the glycolytic enzymes GAPDH and enolase, are able to perform important functions unrelated to their cognate catalytic activity (Terao et al., 2006; Agarwal et al., 2012; Terrasse et al., 2012; Wang and Jeffery, 2016). These so-called moonlighting functions of predominantly cytosolic enzymes are typically deployed at specific subcellular compartments or locations (e.g., cell nucleus, mitochondria, plasma membrane) or in extracellular contexts. The underlying causes that would explain how glycolytic enzymes came to develop these functions are not yet fully appreciated, although it is now clear that these additional functions have been selected for during evolution and are not merely a side effect of mislocalization or chance occurrence. Most of the hypotheses that have been advanced include the high concentration of these enzymes as a driver factor for their selection to fulfill accessory functions in particular contexts, under the rationale that reassigning existing genes and proteins for new functions is economical and efficient. This strategy appears particularly compelling for genome-encoded virulence factors, where two selective pressures (growth inside the host and immunoevasion) become coupled through a very abundant, multifunctional enzyme like GAPDH.
The involvement of the extracellular GAPDH of S. pyogenes in C5a sequestration (Terao et al., 2006) and of the S. pneumoniae enzyme in C1q binding (Terrasse et al., 2012) provide reference cases for the existence of complement immunoevasion mechanisms in streptococcal infections that depend on the moonlighting action of GAPDH. The extension of these mechanisms to GAPDH from other Gram-positive pathogenic bacteria that share significant sequence similarity with S. pyogenes GAPDH is relevant and intriguing. Firstly, because GAPDH is an essential gene present in all bacteria and could therefore be seen as a universal virulence factor tasked with down-regulating complement-mediated recruitment of neutrophils to sites of infection. Secondly, because sequence similarity across bacterial GAPDH genes cuts across phylogenetic groups and reveals broad patterns of horizontal transfer events, thereby indicating the potential for widespread dispersion of immunoevasive GAPDH sequences. One such example is the high sequence similarity between A. vaginae (a Gram-positive bacterium) and S. aureus (a Gram-negative proteobacterium), which would suggest a potential for C5a sequestration for S. aureus GAPDH. If this were the case, interference with the normal function of the complement anaphylatoxin-mediated signaling could provide a general mechanism for down-modulating the innate immune response. In the context of the infection biology of A. vaginae, deploying molecules capable of depressing the spread of anaphylatoxins away from the site of infection would be advantageous for the bacterium to proliferate (Figure 9). In addition, the depletion of C5a would obviously help the proliferation of the complex community of bacteria that cooperate with A. vaginae in establishing BV, including G. vaginalis and other associated bacteria.
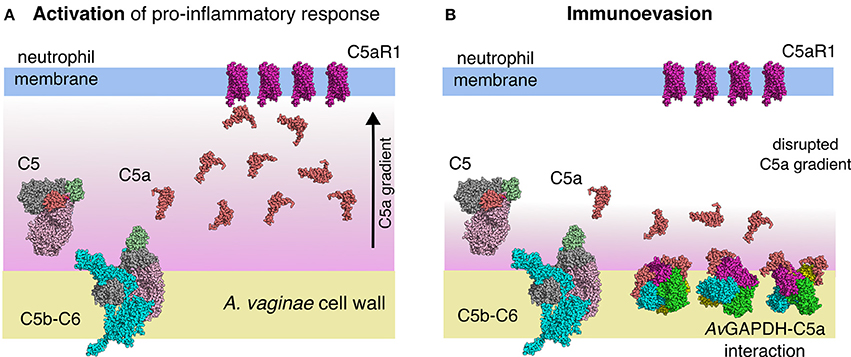
Figure 9. Model for the immunoevasive function of A. vaginae GAPDH through C5a sequestration. (A) A C5a gradient generated by the activation of the lytic phase of complement's alternative pathway leads to the recruitment of neutrophils to the site of infection through the activation of pro-inflammatory signaling cascades via C5a receptor (C5aR1) stimulation. (B) Cell-wall associated AvGAPDH traps in-situ generated C5a, thereby disrupting the chemotactic gradient and effectively “blinding” neutrophils about the presence of the bacterial pathogen. Macromolecular structures are shown to scale and represented as sphere models. C5 (PDB ID 3CU7) (70) is colored according to its domain structure; C345c is shown in pale green, the C5a anaphylatoxin in red, the β-chain in light pink, and the α-chain in gray. In C5b-C6 (PDB ID 4A5W) (71), C5b is colored as C5 (except for the anaphylatoxin domain, which is missing in C5b) and C6 is colored in cyan. C5a (PDB ID 3HQA) (72) is shown in the same color as the anaphylatoxin domain of C5. The transmembrane region of C5aR1 was modeled the crystal structure of ligand-free rhodopsin (PDB ID 3CAP) (73) without loops and colored in violet.
Might then reverting this C5a sequestration mechanism offer therapeutic opportunities against bacterial infections? Bacterial pathogens wreak havoc by the action of specialized virulence factors and by aggressive proliferative behaviors, none of which relies solely on immunoevasive mechanisms. Down-regulating pro-inflammatory responses mediated by C5a can help the pathogen expand at the onset of an infection, whereby escaping the watch of innate immunity may be crucial for its initial survival. Combination therapies attacking other aspects of bacterial pathogenesis while simultaneously freeing the complement system (e.g., C5a and C1q) from interfering influences from bacterial molecules such as GAPDH could result in a more effective strategy, whereby the chance for decimated bacterial populations to endure through the antibiotic treatment and thrive back again as soon as antibiotic treatment stops would be minimized. As further knowledge accumulates as to the potentially beneficial effects of modulating complement protein factors during infections, these strategies should be seriously considered, as their applicability would span a very wide range of bacterial pathogens.
Atomic coordinates and diffraction data of A. vaginae AvGAPDH have been deposited at the Protein Data Bank (PDB) with accession code 5LD5.
CV, JR, SA, and SR conceived the experimental study, designed the experiments, analyzed the data and wrote the manuscript. JQ, FF, CM, and CV expressed and purified AvGAPDH and C5a. JQ and FF crystallized AvGAPDH. FF collected the X-ray data set from AvGAPDH crystals. DF and JJ contributed to the X-ray diffraction data collection. SG measured AvGAPDH kinetics and prepared bead-immobilized GAPDH. AM, SR, CV, and JR conceived, designed and conducted the granulocyte migration experiments and analyzed the data. SA provided materials and helped design the experiments and analyze the data. All authors contributed critically to the manuscript. CV wrote the manuscript.
The research leading to these results has received funding from the Spanish Instituto de Salud Carlos III (PI12/01667 to CV), the Spanish Ministerio de Economía y Competitividad (CTQ2015-66206-C2-2-R and SAF2015-72961-EXP to CV, SAF2015-66287-R (MINECO/FEDER) to SR and SAF2014-54708-R to JR), CSIC (201620E064), the Regional Government of Madrid (S2010/BD-2316 to JR, CV, and SR), and the European Commission (Framework Programme 7 (FP7)) projects ComplexINC (Contract No. 279039 to CV) and EURenOmics (Contract No. 305608 to SR). AVM was supported by the Comunidad de Madrid (S2010/BMD-2316/2326) and the Universidad Complutense de Madrid (CT46/15). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge access to the ALBA Synchrotron Light Source, Barcelona, Spain, for provision of synchrotron radiation facilities at the BL13-XALOC beamline. JQ acknowledges the support of the Ph.D. program in Biology of the Universidad Complutense de Madrid (UCM).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00541/full#supplementary-material
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Echols, N., Headd, J. J., et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. doi: 10.1016/j.ymeth.2011.07.005
Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., et al. (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367. doi: 10.1107/S0907444912001308
Agarwal, V., Hammerschmidt, S., Malm, S., Bergmann, S., Riesbeck, K., and Blom, A. M. (2012). Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J. Immunol. 189, 3575–3584. doi: 10.4049/jimmunol.1102934
Aguilera, L., Ferreira, E., Giménez, R., Fernández, F. J., Taulés, M., Aguilar, J., et al. (2012). Secretion of the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase by the LEE-encoded type III secretion system in enteropathogenic Escherichia coli. Int. J. Biochem. Cell Biol. 44, 955–962. doi: 10.1016/j.biocel.2012.03.002
Allonso, D., Andrade, I. S., Conde, J. N., Coelho, D. R., Rocha, D. C., da Silva, M. L., et al. (2015). Dengue Virus NS1 protein modulates cellular energy metabolism by increasing glyceraldehyde-3-phosphate dehydrogenase activity. J. Virol. 89, 11871–11883. doi: 10.1128/JVI.01342-15
Antonyuk, S. V., Eady, R. R., Strange, R. W., and Hasnain, S. S. (2003). The structure of glyceraldehyde 3-phosphate dehydrogenase from Alcaligenes xylosoxidans at 1.7 A resolution. Acta Crystallogr. D Biol. Crystallogr. 59, 835–842. doi: 10.1107/S0907444903041441
Biesecker, G., Harris, J. I., Thierry, J. C., Walker, J. E., and Wonacott, A. J. (1977). Sequence and structure of D-glyceraldehyde 3-phosphate dehydrogenase from Bacillus stearothermophilus. Nature 266, 328–333. doi: 10.1038/266328a0
Bressi, J. C., Verlinde, C. L., Aronov, A. M., Shaw, M. L., Shin, S. S., Nguyen, L. N., et al. (2001). Adenosine analogues as selective inhibitors of glyceraldehyde-3-phosphate dehydrogenase of Trypanosomatidae via structure-based drug design. J. Med. Chem. 44, 2080–2093. doi: 10.1021/jm000472o
Buehner, M., Ford, G. C., Moras, D., Olsen, K. W., and Rossmann, M. G. (1974). Structure determination of crystalline lobster D-glyceraldehyde-3-phosphate dehydrogenase. J. Mol. Biol. 82, 563–585. doi: 10.1016/0022-2836(74)90249-6
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Castilho, M. S., Pavão, F., Oliva, G., Ladame, S., Willson, M., and Périé, J. (2003). Evidence for the two phosphate binding sites of an analogue of the thioacyl intermediate for the Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase-catalyzed reaction, from its crystal structure. Biochemistry 42, 7143–7151. doi: 10.1021/bi0206107
Chan, J. F., Lau, S. K., Curreem, S. O., To, K. K., Leung, S. S., Cheng, V. C., et al. (2012). First report of spontaneous intrapartum Atopobium vaginae bacteremia. J. Clin. Microbiol. 50, 2525–2528. doi: 10.1128/JCM.00212-12
Cowan-Jacob, S. W., Kaufmann, M., Anselmo, A. N., Stark, W., and Grütter, M. G. (2003). Structure of rabbit-muscle glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 59, 2218–2227. doi: 10.1107/S0907444903020493
Datcu, R. (2014). Characterization of the vaginal microflora in health and disease. Danish Med. J. 61:B4830.
Doerflinger, S. Y., Throop, A. L., and Herbst-Kralovetz, M. M. (2014). Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 209, 1989–1999. doi: 10.1093/infdis/jiu004
Donati, L., Di Vico, A., Nucci, M., Quagliozzi, L., Spagnuolo, T., Labianca, A., et al. (2010). Vaginal microbial flora and outcome of pregnancy. Arch. Gynecol. Obstet. 281, 589–600. doi: 10.1007/s00404-009-1318-3
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Evans, P. R., and Murshudov, G. N. (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214. doi: 10.1107/S0907444913000061
Fahey, J. O. (2008). Clinical management of intra-amniotic infection and chorioamnionitis: a review of the literature. J. Midwifery Womens Health 53, 227–235. doi: 10.1016/j.jmwh.2008.01.001
Ferdinand, W. (1964). The isolation and specific activity of rabbit-muscle glyceraldehyde phosphate dehydrogenase. Biochem. J. 92, 578–585. doi: 10.1042/bj0920578
Fichorova, R. N., Yamamoto, H. S., Delaney, M. L., Onderdonk, A. B., and Doncel, G. F. (2011). Novel vaginal microflora colonization model providing new insight into microbicide mechanism of action. MBio 2, e00168–e00111. doi: 10.1128/mBio.00168-11
Fredricks, D. N., Fiedler, T. L., and Marrazzo, J. M. (2005). Molecular identification of bacteria associated with bacterial vaginosis. N. Engl. J. Med. 353, 1899–1911. doi: 10.1056/NEJMoa043802
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guo, R. F., and Ward, P. A. (2005). Role of C5a in inflammatory responses. Annu. Rev. Immunol. 23, 821–852. doi: 10.1146/annurev.immunol.23.021704.115835
Harris, J. I., and Waters, M. (1976). “Glyceraldehyde-3-phosphate dehydrogenase,” in The Enzymes 3rd Edn., ed P. S. Boyer (New York, NY: Academic Press), 1–49.
Henderson, B., and Martin, A. C. (2014). Protein moonlighting: a new factor in biology and medicine. Biochem. Soc. Trans. 42, 1671–1678. doi: 10.1042/BST20140273
Hillier, S. L., Kiviat, N. B., Hawes, S. E., Hasselquist, M. B., Hanssen, P. W., Eschenbach, D. A., et al. (1996). Role of bacterial vaginosis-associated microorganisms in endometritis. Am. J. Obstet. Gynecol. 175, 435–441. doi: 10.1016/S0002-9378(96)70158-8
Huber-Lang, M. S., Riedeman, N. C., Sarma, J. V., Younkin, E. M., McGuire, S. R., Laudes, I. J., et al. (2002). Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. 16, 1567–1574. doi: 10.1096/fj.02-0209com
Hwang, S., Disatnik, M. H., and Mochly-Rosen, D. (2015). Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington's disease. EMBO Mol. Med. 7, 1307–1326. doi: 10.15252/emmm.201505256
Juanhuix, J., Gil-Ortiz, F., Cuní, G., Colldelram, C., Nicolás, J., Lidón, J., et al. (2014). Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 21, 679–689. doi: 10.1107/S160057751400825X
Kabsch, W. (2014). Processing of X-ray snapshots from crystals in random orientations. Acta Crystallogr. D Biol. Crystallogr. 70, 2204–2216. doi: 10.1107/S1399004714013534
El Kadmiri, N., Slassi, I., El Moutawakil, B., Nadifi, S., Tadevosyan, A., Hachem, A., and Soukri, A. (2014). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer's disease. Pathol. Biol. 62, 333–336. doi: 10.1016/j.patbio.2014.08.002
Kennedy, K. J., Bressi, J. C., and Gelb, M. H. (2001). A disubstituted NAD+ analogue is a nanomolar inhibitor of trypanosomal glyceraldehyde-3-phosphate dehydrogenase. Bioorg. Med. Chem. Lett. 11, 95–98. doi: 10.1016/S0960-894X(00)00608-9
Kim, H., Feil, I. K., Verlinde, C. L., Petra, P. H., and Hol, W. G. (1995). Crystal structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Leishmania mexicana: implications for structure-based drug design and a new position for the inorganic phosphate binding site. Biochemistry 34, 14975–14986. doi: 10.1021/bi00046a004
Korndörfer, I., Steipe, B., Huber, R., Tomschy, A., and Jaenicke, R. (1995). The crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 A resolution. J. Mol. Biol. 246, 511–521. doi: 10.1006/jmbi.1994.0103
Kuzminskaya, E. V., Asryants, R. A., and Nagradova, N. K. (1991). Rabbit muscle tetrameric D-glyceraldehyde-3-phosphate dehydrogenase is locked in the asymmetric state by chemical modification of a single arginine per subunit. Biochim. Biophys. Acta 1075, 123–130. doi: 10.1016/0304-4165(91)90241-8
Lambeir, A. M., Loiseau, A. M., Kuntz, D. A., Vellieux, F. M., Michels, P. A., and Opperdoes, F. R. (1991). The cytosolic and glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei. Kinetic properties and comparison with homologous enzymes. Eur. J. Biochem. 198, 429–435. doi: 10.1111/j.1432-1033.1991.tb16032.x
Lesk, A. M. (1995). NAD-binding domains of dehydrogenases. Curr. Opin. Struct. Biol. 5, 775–783. doi: 10.1016/0959-440X(95)80010-7
Liu, L., Xiong, N., Zhang, P., Chen, C., Huang, J., Zhang, G., et al. (2015). Genetic variants in GAPDH confer susceptibility to sporadic Parkinson's disease in a Chinese Han population. PLoS ONE 10:e0135425. doi: 10.1371/journal.pone.0135425
Machado, A., and Cerca, N. (2015). Influence of biofilm formation by Gardnerella vaginalis and Other Anaerobes on Bacterial Vaginosis. J. Infect. Dis. 212, 1856–1861. doi: 10.1093/infdis/jiv338
Marder, S. R., Chenoweth, D. E., Goldstein, I. M., and Perez, H. D. (1985). Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J. Immunol. 134, 3325–3331.
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi: 10.1107/S0021889807021206
McDonald, H. M., Brocklehurst, P., and Gordon, A. (2007). Antibiotics for treating bacterial vaginosis in pregnancy. Cochrane Database Syst. Rev. CD000262. doi: 10.1002/14651858.CD000262.pub3
Mikhaylova, E. R., Lazarev, V. F., Nikotina, A. D., Margulis, B. A., and Guzhova, I. V. (2016). Glyceraldehyde 3-phosphate dehydrogenase augments the intercellular transmission and toxicity of polyglutamine aggregates in a cell model of Huntington disease. J. Neurochem. 136, 1052–1063. doi: 10.1111/jnc.13463
Mukherjee, S., Dutta, D., Saha, B., and Das, A. K. (2010). Crystal structure of glyceraldehyde-3-phosphate dehydrogenase 1 from methicillin-resistant Staphylococcus aureus MRSA252 provides novel insights into substrate binding and catalytic mechanism. J. Mol. Biol. 401, 949–968. doi: 10.1016/j.jmb.2010.07.002
Nagradova, N. K. (2001). Study of the properties of phosphorylating D-glyceraldehyde-3-phosphate dehydrogenase. Biochem. Mosc. 66, 1067–1076. doi: 10.1023/A:1012472627801
Pellati, D., Mylonakis, I., Bertoloni, G., Fiore, C., Andrisani, A., Ambrosini, G., et al. (2008). Genital tract infections and infertility. Eur. J. Obstet. Gynecol. Reprod. Biol. 140, 3–11. doi: 10.1016/j.ejogrb.2008.03.009
Polatti, F. (2012). Bacterial vaginosis, Atopobium vaginae and nifuratel. Curr. Clin. Pharmacol. 7, 36–40. doi: 10.2174/157488412799218824
Roitel, O., Vachette, P., Azza, S., and Branlant, G. (2003). P but not R-axis interface is involved in cooperative binding of NAD on tetrameric phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. J. Mol. Biol. 326, 1513–1522. doi: 10.1016/S0022-2836(03)00049-4
Satchell, J. F., Malby, R. L., Luo, C. S., Adisa, A., Alpyurek, A. E., Klonis, N., et al. (2005). Structure of glyceraldehyde-3-phosphate dehydrogenase from Plasmodium falciparum. Acta Crystallogr. D Biol. Crystallogr. 61, 1213–1221. doi: 10.1107/S0907444905018317
Skarzyński, T., Moody, P. C., and Wonacott, A. J. (1987). Structure of holo-glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus at 1.8 A resolution. J. Mol. Biol. 193, 171–187. doi: 10.1016/0022-2836(87)90635-8
Skarzyński, T., and Wonacott, A. J. (1988). Coenzyme-induced conformational changes in glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. J. Mol. Biol. 203, 1097–1118. doi: 10.1016/0022-2836(88)90130-1
Souza, D. H., Garratt, R. C., Araújo, A. P., Guimarães, B. G., Jesus, W. D., Michels, P. A., et al. (1998). Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase: structure, catalytic mechanism and targeted inhibitor design. FEBS Lett. 424, 131–135. doi: 10.1016/S0014-5793(98)00154-9
Stein, N. (2008). CHAINSAW : a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643. doi: 10.1107/S0021889808006985
Suresh, S., Bressi, J. C., Kennedy, K. J., Verlinde, C. L., Gelb, M. H., and Hol, W. G. (2001). Conformational changes in Leishmania mexicana glyceraldehyde-3-phosphate dehydrogenase induced by designed inhibitors. J. Mol. Biol. 309, 423–435. doi: 10.1006/jmbi.2001.4588
Sweet, R. L. (1995). Role of bacterial vaginosis in pelvic inflammatory disease. Clin. Infect. Dis. 20(Suppl. 2), S271–S275. doi: 10.1093/clinids/20.Supplement_2.S271
Tanner, J. J., Hecht, R. M., and Krause, K. L. (1996). Determinants of enzyme thermostability observed in the molecular structure of Thermus aquaticus D-glyceraldehyde-3-phosphate dehydrogenase at 25 Angstroms Resolution. Biochemistry 35, 2597–2609. doi: 10.1021/bi951988q
Terao, Y., Yamaguchi, M., Hamada, S., and Kawabata, S. (2006). Multifunctional glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. J. Biol. Chem. 281, 14215–14223. doi: 10.1074/jbc.M513408200
Terrasse, R., Tacnet-Delorme, P., Moriscot, C., Pérard, J., Schoehn, G., Vernet, T., et al. (2012). Human and pneumococcal cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J. Biol. Chem. 287, 42620–42633. doi: 10.1074/jbc.M112.423731
Vellieux, F. M., Hajdu, J., Verlinde, C. L., Groendijk, H., Read, R. J., Greenhough, T. J., et al. (1993). Structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei determined from Laue data. Proc. Natl. Acad. Sci. U.S.A. 90, 2355–2359.
Wang, W., and Jeffery, C. J. (2016). An analysis of surface proteomics results reveals novel candidates for intracellular/surface moonlighting proteins in bacteria. Mol. Biosyst. 12, 1420–1431. doi: 10.1039/C5MB00550G
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242. doi: 10.1107/S0907444910045749
Xiao, B., Niu, X., Han, N., Wang, B., Du, P., Na, R., et al. (2016). Predictive value of the composition of the vaginal microbiota in bacterial vaginosis, a dynamic study to identify recurrence-related flora. Sci. Rep. 6:26674. doi: 10.1038/srep26674
Yun, M., Park, C. G., Kim, J. Y., and Park, H. W. (2000). Structural analysis of glyceraldehyde 3-phosphate dehydrogenase from Escherichia coli: direct evidence of substrate binding and cofactor-induced conformational changes. Biochemistry 39, 10702–10710. doi: 10.1021/bi9927080
Keywords: anaphylatoxin, chemoattraction, complement system, C5a, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), gram-positive pathogen, immunoevasion, x-ray crystallography
Citation: Querol-García J, Fernández FJ, Marin AV, Gómez S, Fullà D, Melchor-Tafur C, Franco-Hidalgo V, Albertí S, Juanhuix J, Rodríguez de Córdoba S, Regueiro JR and Vega MC (2017) Crystal Structure of Glyceraldehyde-3-Phosphate Dehydrogenase from the Gram-Positive Bacterial Pathogen A. vaginae, an Immunoevasive Factor that Interacts with the Human C5a Anaphylatoxin. Front. Microbiol. 8:541. doi: 10.3389/fmicb.2017.00541
Received: 06 November 2016; Accepted: 14 March 2017;
Published: 10 April 2017.
Edited by:
Juarez Antonio Simões Quaresma, Federal University of Pará, BrazilReviewed by:
Lubka T. Roumenina, Cordeliers Research Center (INSERM), FranceCopyright © 2017 Querol-García, Fernández, Marin, Gómez, Fullà, Melchor-Tafur, Franco-Hidalgo, Albertí, Juanhuix, Rodríguez de Córdoba, Regueiro and Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Cristina Vega, Y3ZlZ2FAY2liLmNzaWMuZXM=; Y3Jpc3RpbmEudmVnYUBzdHJ1YmljaWIub3Jn
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.