 Francis Cheng-Hsuan Weng
Francis Cheng-Hsuan Weng Grace Tzun-Wen Shaw
Grace Tzun-Wen Shaw Chieh-Yin Weng1
Chieh-Yin Weng1 Yi-Ju Yang
Yi-Ju Yang Daryi Wang
Daryi Wang- 1Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
- 2Department of Life Science, National Taiwan Normal University, Taipei, Taiwan
- 3Biodiversity Program, Taiwan International Graduate Program, Academia Sinica and National Taiwan Normal University, Taipei, Taiwan
- 4Department of Natural Resources and Environmental Studies, National Dong Hwa University, Hualien, Taiwan
The concerted activity of intestinal microbes is crucial to the health and development of their host organisms. Investigation of microbial interactions in the gut should deepen our understanding of how these micro-ecosystems function. Due to advances in Next Generation Sequencing (NGS) technologies, various bioinformatic strategies have been proposed to investigate these microbial interactions. However, due to the complexity of the intestinal microbial community and difficulties in monitoring their interactions, at present there is a gap between the theory and biological application. In order to construct and validate microbial relationships, we first induce a community shift from simple to complex by manipulating artificial hibernation (AH) in the treefrog Polypedates megacephalus. To monitor community growth and microbial interactions, we further performed a time-course screen using a 16S rRNA amplicon approach and a Lotka-Volterra model. Lotka-Volterra models, also known as predator–prey equations, predict the dynamics of microbial communities and how communities are structured and sustained. An interaction network of gut microbiota at the genus level in the treefrog was constructed using Metagenomic Microbial Interaction Simulator (MetaMIS) package. The interaction network obtained had 1,568 commensal, 1,737 amensal, 3,777 mutual, and 3,232 competitive relationships, e.g., Lactococcus garvieae has a commensal relationship with Corynebacterium variabile. To validate the interacting relationships, the gut microbe composition was analyzed after probiotic trials using single strain (L. garvieae, C. variabile, and Bacillus coagulans, respectively) and a combination of L. garvieae, C. variabile, and B. coagulans, because of the cooperative relationship among their respective genera identified in the interaction network. After a 2 week trial, we found via 16S rRNA amplicon analysis that the combination of cooperative microbes yielded significantly higher probiotic concentrations than single strains, and the immune response (interleukin-10 expression) also significantly changed in a manner consistent with improved probiotic effects. By taking advantage of microbial community shift from simple to complex, we thus constructed a reliable microbial interaction network, and validated it using probiotic strains as a test system.
Introduction
Gut microbes and their hosts exist in a symbiotic relationship. Gut microbes contribute to important host functions, including fermenting unused energy substrates, training the immune system, preventing growth of pathogenic bacteria, and regulating gut development (Hooper et al., 2002; Xu and Gordon, 2003; Li et al., 2008; Perez et al., 2010; Ye et al., 2014). Although recent studies using 16S rRNA amplicon sequencing have emphasized the importance of microbes for their hosts (Manichanh et al., 2006; Peterson et al., 2009; Round and Mazmanian, 2009; Turnbaugh and Gordon, 2009; Turnbaugh et al., 2009; Arumugam et al., 2011), the functional roles of most gut microbes remain unknown. One approach to explore this question is through network inference. This approach has been widely used to explore interactions between various organisms (Newman, 2003; Proulx et al., 2005; Shafiei et al., 2014). To uncover hidden patterns beyond the animal world, the generalized Lotka-Volterra (gLV) equations have recently been adopted as a dynamic model for studying microbial communities (Faust and Raes, 2012; Stein et al., 2013). The gLV model uses non-linear differential equations, which govern prey-predator relations. The gLV equations have been used successfully to predict temporal dynamics of microbiota in the mouse intestine (Stein et al., 2013), and within a cheese-making environment (Mounier et al., 2008), by analyzing microbiome time-series data. Time-series data inherently contain information including the statistical dependency of observations as a function of time. When these features of time-series data are properly modeled, it is possible to gain substantial new insights into the behavior of the system under study. Some studies even suggested that the distribution of interaction pairs (also obtained using a gLV dynamic model) in an ecological system can be used to predict microbiota stability (Coyte et al., 2015).
Network inference methods are commonly distinguished into two groups. The first approach is similarity-based network inference, which assesses the co-occurrence and/or mutual exclusion pattern of two species over multiple samples, using a measure that quantifies the similarity of two species' distributions. However, pairwise relationships do not capture more complex forms of ecological interactions, in which one species is influenced by (or depends on) multiple other species (Faust and Raes, 2012). To infer these types of interactions, the second approach is to apply regression-based networks, in which the abundance of one species is predicted from the combined abundances of other organisms. The latter is more convincing and was used by Stein et al. (2013) to reanalyze the Clostridium difficile infection data generated by Buffie et al. (2012). Marino et al. (2014) also used the gLV equations to model population dynamics of the gut microbiota in mice. However, these works generated interaction networks but did not further validate the inferred relations. To explore how the biological outcomes are directly related to the specific inferred microbial interactions, a well-investigated in vivo system describing the effects of gut microbes on their host must be applied for validation. Conventionally, the prevention and control of aquaculture diseases has focused on the use of vaccines or antibiotics (Pasteris et al., 2009b). However, treating or feeding frogs with antibiotics may cause the development of resistant bacteria (Akinbowale et al., 2006). Further evidence has shown that antibiotics can cause a decrease in the biodiversity of gut bacteria and increase the risk of bacterial infections (Buffie et al., 2012; Taur et al., 2012). Antibiotics and their effects can also persist for several days after the end of treatment (Jernberg et al., 2007; Dethlefsen and Relman, 2011; Buffie et al., 2012). An alternative solution is the use of probiotics (Reid et al., 2003), which are able to inhibit gut colonization by pathogens and to exert inhibitory effects against undesired micro-organisms, as well as to support natural host microbial defense mechanisms (Hernandez et al., 2005). Thus, a wide range of Gram (+) and Gram (−) bacteria, yeast, microalgae, and bacteriophages have been evaluated as probiotics (Pasteris et al., 2009b).
In this study, we highlight the feasibility of conducting network inference at the genus level to decipher the possible interactions within the microbial ecosystem of the amphibian gut. We took advantage of a well-investigated probiotic system to validate the inferred microbial interactions. Compared to the conventional strategy that orally introduces specific single bacterial species as probiotics, we emphasize the power of inferred network by simultaneously applying interacting microbial partners that can be used together to enhance the growth and beneficial effects of target probiotics. In addition to the validation of the inferred network, this test is also the first attempt to use a combination of cooperative strains as probiotics in raniculture.
The innate immune system is the first line of an organism's defense against infection. Probiotics interact with immune cells, such as mono-nuclear phagocytes, polymorphonuclear leukocytes, and natural killer cells, to enhance innate immune responses. Studies have shown that probiotics can increase the numbers of erythrocytes, granulocytes, macrophages, and lymphocytes (Balcazar et al., 2006; Akhter et al., 2015). The most commonly used probiotics in amphibians are lactic acid bacteria (LAB). LAB produce a range of important molecules such as organic acids, hydrogen peroxide, diacetyl, antimicrobial peptides (AMPs), and bacteriocins (Verschuere et al., 2000; Küng et al., 2014). The characterization of these compounds explains the beneficial effects of LAB; thus, some bacterial species with similar functions have been introduced as probiotics to restore beneficial microbial populations (Balcázar et al., 2007a; Pasteris et al., 2009b; Ringø et al., 2010; Mendoza et al., 2012). In this study, we induced a community shift from simple to complex via artificial hibernation (AH) in the treefrog and performed 16S rRNA amplicon analysis on the gut microbiome. Through the use of MetaMIS, a package that employs the gLV model to infer microbial interactions (Shaw et al., 2016), we generated the microbial interaction data and constructed the interaction network. In order to validate the microbial interactions, instead of the conventional strategy that only introduces bacteria with similar functions as probiotics, we selected a target LAB, and selected bacteria of the genera Corynebacterium and Bacillus as functional partners based on the network analysis. Our results showed that the combination of three representatives of these genera, L. garvieae, C. variabile, and B. coagulans, works more efficiently than any single strain, reflecting the reliability of the inferred microbial interaction.
Materials and Methods
Sample Collection
Eighty adult treefrogs (Polypedates megacephalus) were collected from New Taipei City and Taichung City, Taiwan. All animals were housed in 240-l glass tanks at 23°C under a 8:16 h light:dark cycle. Turkestan cockroach nymphs (Finke, 2013) were fed to the treefrogs at a quantity of 10% of treefrog biomass twice a week. The treefrogs were acclimatized for 3 months prior to experiments.
Microbiome 16S rRNA Amplicon Analysis
Fecal samples were collected from treefrog guts within 20 min after euthanasia. To avoid cross-contamination, each sample was collected using a fresh pair of sterile tweezers. The contents of each gut were emptied into a sterile vial and immediately stored at –80°C. DNA was subsequently extracted with the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA). The V4 region (292 bp) of 16S rRNA gene was PCR-amplified with 515F (5′−GTGCCAGCMGCCGCGGTAA−3′) and 12-base barcoded 806R (5′−GGACTACHVGGGTWTCTAAT−3′) primers (Caporaso et al., 2012). Following PCR, samples were gel extracted with the NucleoSpin Gel Extraction kit (Macherey-Nagel, Germany). The purified samples were pooled in equal concentrations and sequenced using an Illumina MiSeq (Illumina, San Diego, CA, USA) with a V2 PE500 cartridge (500 cycles). All datasets have been deposited in GenBank under the BioProject ID PRJNA341914 and BioSample ID SAMN05730167 and SAMN05730170.
All paired-end sequences were merged by FLASH (Magoč and Salzberg, 2011), and all merged sequences were further analyzed by conducting the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (Caporaso et al., 2010b). In the QIIME analysis pipeline, the low-quality sequences (sequences that were <200 bp in length, had a quality score <25, contained ambiguous characters, had an unreadable barcode, or did not contain the primer sequence) were removed using the USEARCH quality filter (Edgar, 2010). The UCLUST (Edgar, 2010) function in QIIME was used to cluster the remaining sequences, with a minimum coverage of 99% and minimum sequence identity of 97%. The longest sequences from each phylotype were selected to perform sequence identification, and PyNAST (Caporaso et al., 2010a) and UCLUST were selected to perform the sequence alignment and taxonomy assignment.
Microbial Community Shift from Simple to Complex and the Generalized Lotka-Volterra Model
A concept for improvement of the investigation of the functional roles of gut microbiota is to explore the microbial world from a simple to complex state, from an initial stage to homeostasis. Hibernation is considered as a survival strategy designed to conserve energy when conditions are harsh. During hibernation, animals can adapt to temperature perturbations and extend their lifespan by slowing their heartbeat, reducing metabolic activity and their energy requirements (Book, 1974). At the same time, most microorganisms in the gut of a host decline in numbers due to extreme temperature and low nutrient supply (Gossling et al., 1982a). Hibernation of treefrogs, therefore, provides a natural model for monitoring microbial growth starting from inoculation (i.e., the beginning of a developing microbial interaction network) to homeostasis, and could provide an opportunity to shed light on our understanding of how microorganisms form their interaction networks.
To stimulate AH, we modified a program previously described in leopard frogs (Rana pipiens) (Gossling et al., 1982a). Fasting is known to reduce microbial complexity in amphibians, as well as fish, reptiles, birds, and mammals (Gossling et al., 1982b; Sonoyama et al., 2009; Costello et al., 2010; Kohl et al., 2014). Therefore, prior to stimulation of AH, food was withheld from the treefrogs kept in the same housing for 7 days to reduce overall diversity. All fasting treefrogs were then transferred into an incubator kept in constant darkness. AH was stimulated initially at 21°C with a relative humidity of 90%. During day 1, the temperature was maintained. During days 2–3, the temperature was gradually reduced to 4°C. Thereafter, treefrogs were housed in the same manner for 7 days. During days 11–12 (i.e., after 7 days of AH described above), the temperature was gradually increased to 21°C, and the treefrogs returned to their active status. Each active treefrog was fed with Turkestan cockroach nymphs (~0.25–0.3 g). Post-feeding samples were collected within 2.5 days. All protocols were approved by the Academia Sinica Biosafety Committee and Institutional Animal Care and Utilization Committee.
To collect time series samples for generating the microbial interaction network, we collected the gut contents of the treefrogs over a total of 12 time points spanning 15 days over the AH period, including day 1 (fasting for 7 days before AH), day 11–11.75 (every 6 h), and days 12–15 (every 12 h). For each time point, three to five treefrogs were euthanized. Details of treefrog body mass, sampling size, and time points of fecal collection over the AH period are presented in Supplementary Table 1.
We used the Metagenomic Microbial Interaction Simulator (MetaMIS) with a user friendly interface (Shaw et al., 2016) to infer microbial relations by introducing a single time-series dataset of microbial composition containing 12 time points. MetaMIS is a tool based on the generalized Lotka-Volterra model (Bhargava, 1989), and designed to infer underlying microbial interactions according to metagenomic abundance profiles. Lotka-Volterra equations have been widely used to infer animal interactions in dynamic systems, and recently have been applied to reveal microbial interacting relationships between operational taxonomic units (OTUs). The detailed algorithms and equations were described by Bucci et al. (Stein et al., 2013). Due to the limitation of computing power (Interl® Core™ i7-4770 CPU @3.40 GHz processor and 32 Gb RAM), it is not feasible to infer interaction network at the species level. Therefore, we assumed that in general, all species in a genus identified in our amplicon study share similar functions. According to the compositional profiles among 12 time points at the genus level, MetaMIS can systematically examine interaction patterns, such as mutualism or competition; only the top 25% of interacting relationships were considered in this study. We used the mfinder tool (Milo et al., 2002) to identify significant 3-node directed motifs that contain two directed edges pointing to the same node. This process was repeated 100 times and all relationships that passed the criteria described above at least 50 times (permutation cutoff > 0.5) were considered to be reliable interacting relationships.
Probiotic Selection and Culturing
There are several genera of bacteria characterized as probiotic in amphibians (Pasteris et al., 2009a,b; Dias et al., 2010; Becker et al., 2011; Mendoza et al., 2012; Bletz et al., 2013). LAB have been considered as the major probiotics for the treatment of raniculture (Dias et al., 2010; Mendoza et al., 2012). In this study, L. garvieae was selected as a probiotic strain because Lactococcus was present in at least 85% of all the samples with a relative abundance of 0.14 ± 0.39%, and L. garvieae has been identified as a biological control agent in bullfrogs (Mendoza et al., 2012). According to the interacting relationships inferred by MetaMIS, Corynebacterium showed beneficial effects on Lactococcus, while Bacillus was found to have a beneficial interacting relationship with Corynebacterium. We decided to use three representative species of the three mentioned genera for further validation. Therefore, L. garvieae, C. variabile, and B. coagulans were chosen to validate the inferred interacting relationships. These three species were selected by their abundance ranking within the genus according to 16S rRNA amplicon data. We reasoned that if the inferred interacting relations were correct, the cooperative combination would yield a higher probiotic concentration than using a single strain.
To prepare for oral administration, L. garvieae and B. coagulans were grown in Tryptic Soy broth at 30 and 55°C respectively. C. variabile was grown in Brain Heart Infusion broth at 30°C. All strains were grown overnight with agitation in a shaking incubator. After incubation, the cells were harvested by centrifugation at 2,500 × g for 20 min at 4°C. The cell pellets were washed twice with 0.9% saline and resuspended using the same buffer. The measured population level of bacteria in the test diet was 107 CFU g−1 (colony-forming unit).
Treefrogs were divided into five groups (N = 8 per group), and acclimated for 1 week before the start of the trial. The trial was conducted for a 2-week period. Each test group of treefrogs was dosed with L. garvieae (G1), C. variabile (G2), or B. coagulans (G3) singly, or with a combination of the three strains (G4) once per day by direct oral gavage. The control (G5) was fed with 0.9% saline during the entire trial period. After the trial, four fecal samples were collected from four treefrogs in each group (Supplementary Table 2) for further 16S rRNA amplicon analysis, and the other four treefrogs were used to perform quantitative PCR for interleukin 10 (IL-10) expression as described in the following section.
Quantitative Real Time PCR for IL-10
In order to test the immune response after oral administration with probiotic, the expression level of IL-10 was measured after LPS (lipopolysaccharide) stimulation (Qi et al., 2015). To characterize the change of treefrog IL-10 expression after LPS stimulation, four treefrogs in each group were injected intraperitoneally (i.p.) with LPS (150 μg/100 g body weight). Animals were anesthetized and euthanized 24 h after injection. All fecal contents were removed and the remaining tissue samples of gut were collected (weight range 0.03 to 0.08 g) (Supplementary Table 3). All tissue samples were homogenized and total RNA was extracted from homogenized samples using Trizol reagent (Invitrogen, USA), quantified using a Nanodrop–1000 spectrophotometer, and reverse transcribed into cDNA using the Superscript II reverse transcription system (Invitrogen, USA) according to the manufacturer's instructions. Quantitative real time PCR was performed using Power SYBR green PCR Mastermix (Applied Biosystems) on a real-time instrument (ABI mode 7300 Sequence Detector) in 96-well reaction plates. The reaction mixture included 10 ml of Power SYBR green PCR Mastermix, 1 μl of forward and reverse primer (10 μM each) and 1 ml of cDNA, and then brought up to a final total volume of 20 μl with ultra pure water. The sequence of IL-10 in P. megacephalus was described in previous study (Huang et al., 2016) and the forward and reverse primers were designed by NCBI Primer-BLAST (Ye et al., 2012). β-actin was used as a housekeeping control. The primer sequences for amplification of IL-10 and β-actin were as follows: (F, 5′-ACGACCCTGCTCACGTTATG-3; R, 5′-TCCGGGATGGAGTAAGAGGG-3′) and (F, 5′-GGTCGCCCAAGACATCAG-3; R, 5′-GCATACAGGGACAACACA-3′) (Hamdan et al., 2016), respectively. The relative expression of IL-10 in gut tissue samples was normalized to the expression of β−actin. The change of gene expression was expressed as fold change (log base 2) and calculated as described (Qi and Nie, 2008; Qi et al., 2010, 2015). A paired Student's t-test was applied to analyze the significance and fold change (log base 2), with a p-value less than 0.05 considered to be statistically significant.
Statistical Analysis
To estimate the change of microbial complexity throughout the AH period, alpha-diversity was determined using the Shannon index, Simpson index, and the Inversed Simpson index. To determine the differences in bacterial community composition during AH, we used the Bray-Curtis similarity index (a taxonomic metric), which provides a measure of phylogenetic distance between communities from individual samples (Lozupone et al., 2007). To test the differences in richness and phylogenetic indices between time points, Student's t-test was used to determine the significance (p-value < 0.05) between time points. For comparison between each bacterial challenge and controls, we determined the fold-change in relative abundance to demonstrate a response to the stimulus relative to the background. Fold-change significantly higher than 2 or smaller than 0.5 was considered to be relevant. Differences between the two groups were analyzed for significance (p-value < 0.05) by Wilcoxon's test.
Results
In this study, we conducted a continuous 15-day time series data collection through communities from simple to complex to infer interacting relationships of microbes in the gut of treefrogs. We obtained an average of 128,608 ± 32,367 high quality, classifiable 16S rRNA gene sequences, with an average count per time point ranging from 91,908 ± 7,767 to 161,142 ± 45,301. At the numbers of reads generated for each sample, the numbers of genera were in the saturation phase (Supplementary Figure 1), indicating that genera from each sample had been sufficiently recovered in MiSeq sequencing. We observed that the phylogenetic indices, including the Shannon index and the Simpson Index, were significantly increased at all time points after day 12 (24 h after AH), compared with day 11.25 (6 h after AH). The Inverse Simpson Index was also significantly increased at days 12, 13.5, 14, 14.5, 15, compared with day 11.25 (Table 1). In addition, three predominant phyla were significantly altered in their relative abundance (Student's t-test, p-value < 0.05) from day 11.25 at all time points after day 13.5 (Figure 1). For example, the increased relative abundance of Bacteroidetes at days 13.5, 14, 14.5, and 15 were 23 ± 4.2%, 24 ± 2.5%, 18 ± 5.6%, and 31 ± 3.6%, compared with 1.7 ± 0.8% at day 11.25. The relative abundance of Firmicutes at day 11.25, 13.5, 14, 14.5, and 15 were 3.9 ± 1.3, 18 ± 3.6, 25 ± 6.5, 53 ± 8.2, and 20 ± 6.2%, while the relative abundance of Proteobacteria at day 11.25, 13.5, 14, 14.5, and 15 were 90 ± 1.6, 49 ± 6.6, 37 ± 6.3, 8.4 ± 1.3, and 34 ± 7.2%. We also found that other phyla were significantly increased, such as Tenericutes and Verrucomicrobia, or significantly decreased, such as Thermi, in relative abundance after day 13.5 compared to day 11.25. These observations suggest that AH successfully reduced the microbial complexity, which may enhance the accuracy of inferred interactions.
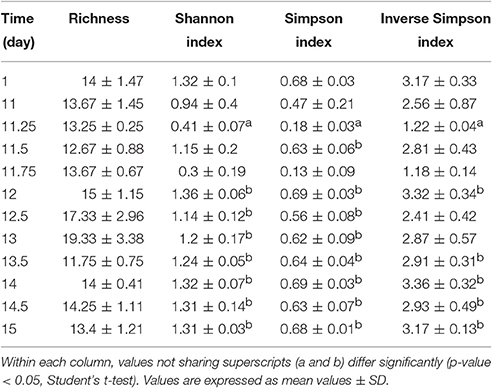
Table 1. Time-dependent phylogenetic diversity spanning 15 days over AH.
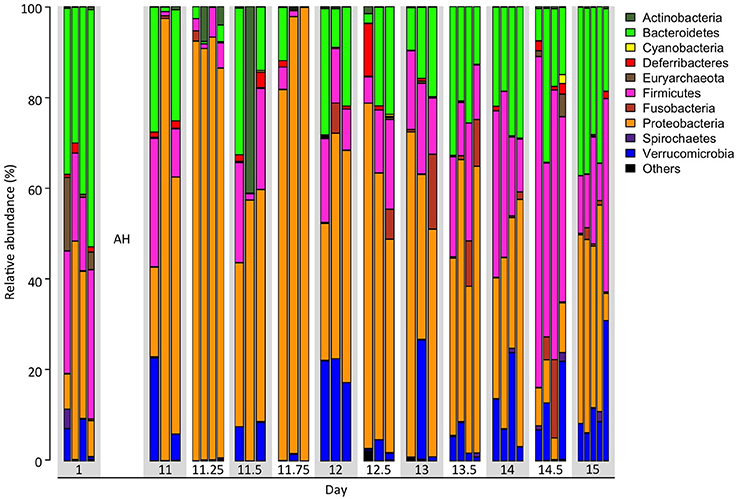
Figure 1. Time dependent taxonomic composition spanning 15 days over AH. Taxonomic composition of fecal microbiota over 12 time points including pre- and post-artificial hibernation (AH) at the phylum level. Each bar represents one individual.
The Inferred Interacting Relations
To infer microbial interactions, we focused on the level of genera. Overall, 325 genera that were characterized via the QIIME pipeline were used to construct the interacting relationships network using MetaMIS. In each run, pairwise interacting relationships with defined criteria (such as commensalism or amensalism) were generated. We obtained interacting paired relationships for 26,320 to 26,327 pairs in 100 permutations. After analysis, 10,314 inferred interaction pairs (IIPs) passed the permutation criterion (cutoff > 0.5 described in Methods), including 1,568 commensal, 1,737 amensal, 3,777 mutual, and 3,232 competitive relationships (Supplementary Table 4). For reference, we also constructed the interaction network at the species level (Supplementary Table 5). Most inferred relations are consistent in both networks generated in the level of genera and species.
To choose probiotic bacteria for the validation of IIPs, we surveyed the literature and selected three genera, i.e., Lactococcus, Lactobacillus, and Pediococcus that have been commonly used in probiotic treatments in raniculture (Dias et al., 2010; Mendoza et al., 2012). In our dataset, there were 84, 64, and 60 IIPs correlated to Lactococcus, Pediococcus, and Lactobacillus, respectively. Therefore, we selected Lactococcus as the target probiotic. In order to select the commensal partners for Lactococcus, we further surveyed the genera that directly imposed beneficial effects on Lactococcus (inferred from the interaction network). Corynebacterium was selected due to the fact it had the highest beneficial effects in the network. Additionally, Bacillus imposed beneficial effects on Corynebacterium and was selected from 69 IIPs of Corynebacterium. Consequently, in this study, we performed oral administration using three representatives of these genera, i.e., L. garvieae, C. variabile, and B. coagulans to validate the inferred interacting relations in gut microbes of the treefrogs.
To further describe probiotic networks, we identified specific IIPs for Lactococcus, Corynebacterium, and Bacillus from 1,012 IIPs to pinpoint the genera that directly or indirectly interact with our selected bacteria (Supplementary Table 6). There were 84 IIPs interacting with Lactococcus, including one commensal, 41 mutual, one amensal, and 41 competitive. From the 84 interacting partners with Lactococcus, three of them were predominant genera (relative abundance on average was larger than 5%), Bacteroides (20.8 ± 16.2%), Citrobacter (6.6 ± 10.4%), and Shewanella (16.3 ± 24.1%), and were all inferred as competing partners, suggesting that a considerable population that colonized the treefrog intestine may intrinsically inhibit the genus Lactococcus. The number of IIPs with Corynebacterium and Bacillus were 69 and 60 respectively. It is worth noting that compared with Lactococcus, Corynebacterium had more mutual relations with other intestinal bacteria; there were no amensal relationships and only 21 of 69 IIPs were competitive. A small group of microbes that interact with Lactococcus, Corynebacterium, and Bacillus is shown in Figure 2.
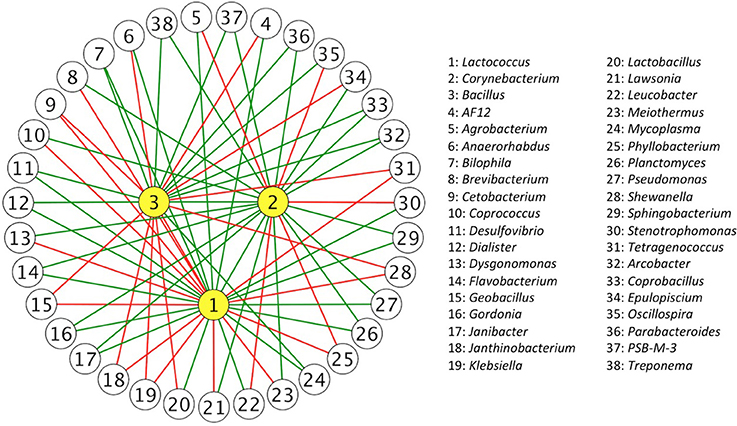
Figure 2. Inferred interaction partners of Bacillus, Corynebacterium, and Lactococcus. Complex relationships were inferred from gut bacterial communities in the 15-day time series data. Each node represents an inferred interaction partner (IIP) and each edge represents an inferred interaction relation between them. The edges in green represent commensal or mutual interactions, and the edges in red represent amensal or competitive interactions. Only the IIPs that contained two inferred interaction relationships are shown.
Oral Administration of L. Garvieae, C. variabile, and B. Coagulans
From the inferred interactions, 84, 69, and 60 IIPs were correlated to Lactococcus, Corynebacterium, and Bacillus, respectively. After oral administration of the three representative probiotics of these genera (L. garvieae, C. variabile, and B. coagulans), 65, 57, and 52 IIPs of Lactococcus, Corynebacterium, and Bacillus were identified in the trial by 16S rRNA amplicon analysis. The IIPs with abundance data of zero were excluded from the following analysis. Oral administration of three distinct bacterial species representative of each genus led to successful gut colonization, and led to a reasonable change in IIPs. In the G1 treatment group (oral administration of L. garvieae), Lactococcus significantly increased in relative abundance 9.88 ± 2.1-fold compared with control (G5 treatment group) (Supplementary Table 6). In the G2 treatment group (oral administration of C. variabile), Corynebacterium significantly increased in relative abundance 160.51 ± 58.54-fold (data insufficient to test the change of Lactococcus). On the other hand, in the G3 treatment group (oral administration of B. coagulans), the relative abundance of Bacillus decreased 0.59 ± 0.49-fold, with no change in Corynebacterium, and a 1.91 ± 0.49-fold increase in Lactococcus. In the G4 treatment group (oral administration of a combination of L. garvieae, C. variabile, and B. coagulans), as expected, Lactococcus significantly increased in relative abundance 26.17 ± 6.96-fold (Wilcoxon's test, p-value < 0.05), 2.64 times greater increase than see for L. garvieae alone, reflecting the positive effects of the commensal partners. Also in the G4 treatment group, Corynebacterium increased 95.99 ± 9.57-fold in relative abundance (Wilcoxon's test, p-value = 0.0625), while Bacillus decreased 0.09 ± 0.04-fold in relative abundance in comparison with controls, reflecting the inhibitory effect of Lactococcus. Figure 3 and Supplementary Table 7 illustrated the relative abundances of microbes that interact with Lactococcus, Corynebacterium, and Bacillus after the two-week oral trials.
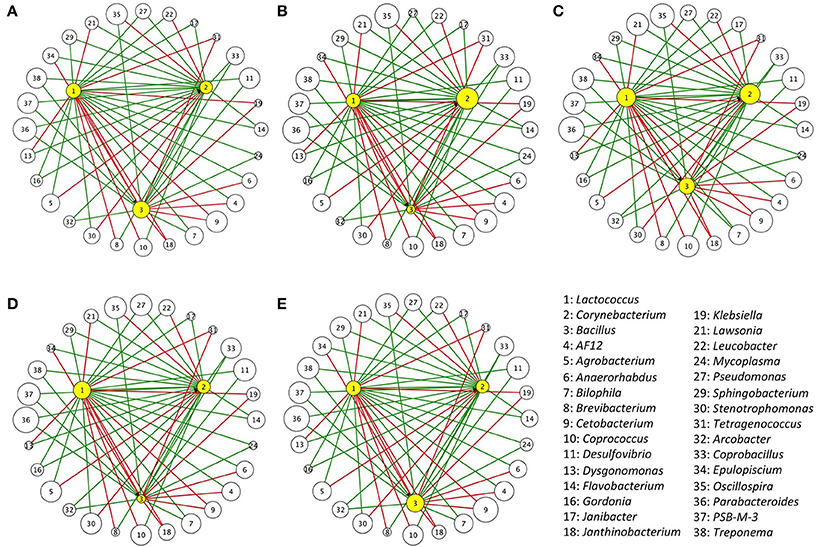
Figure 3. Relative abundance of IIPs of Bacillus, Corynebacterium, and Lactococcus after two-week oral trials. Five oral administrations included (A) G1: L. garvieae (107 CFU g−1) in 0.9% saline, (B) G2: C. variabile (107 CFU g−1) in 0.9% saline, (C) G3: B. coagulans (107 CFU g−1) in 0.9% saline, (D) G4: A combination of B. coagulans, C. variabile, and L. garvieae (each contains 107 CFU g−1) in 0.9% saline, and (E) G5: Control (0.9% saline). Each node represents an IIP that is correlated with Bacillus, Corynebacterium, or Lactococcus, and each edge represents an inferred interaction relation between them. The edges in green represent commensal or mutual interactions, and the edges in red represent amensal or competitive interactions. To better visualize the distribution, the size of each node represents the relative abundance of gut microbes in logarithmic scale.
The inferred network describes the potential interactions among genera, in addition to the interactions mentioned above, more information remained to be discussed (Figure 3). For example, a recent in vitro experiment showed an inhibition of L. garvieae K2 against Klebsiella pneumoniae U11468 (Olaoye, 2016), and our network inference also suggests a similar interaction between these two species. To test the whole immune response after oral probiotic administration, IL-10, an immunoregulatory cytokine involved in immune response in amphibians, was used as an index. The frog IL-10 contains conserved amino acid residues and motifs that are essential for bioactivity. The same residues have been proved to be necessary for immunostimulatory function of human IL-10. Studies have been done using IL-10 expression to examine the host immune response to bacterial infection (Qi et al., 2015). In our study, we found that IL-10 expression variation was consistent with the level of Lactococcus in G1 to G4 treatment groups (Table 2). For example, IL-10 expression was increased in the G1 treatment group, while the corresponding level of Lactococcus in G1 was upregulated by 9.88, and the change was significant compared with controls (Wilcoxon's test, p-value < 0.05). In addition, IL-10 level in the G4 treatment group was significantly higher than in controls, also reflecting the high Lactococcus level in G4. These results also support the idea that a cooperative combination of Lactococcus triggered significantly higher expression of IL-10 than observed when using a single strain.
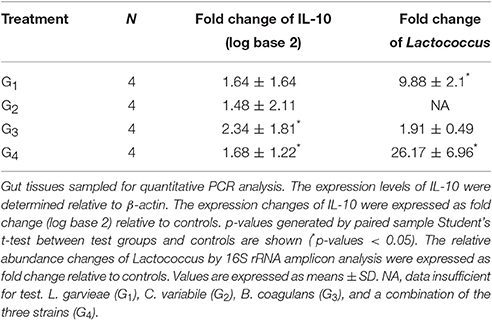
Table 2. Expression analysis of IL-10 and level of Lactococcus.
To validate the inferred interaction network, the corresponding changes of all IIPs were evaluated by calculating the fold changes between the test and control groups. In the G1 group, after oral administration of L. garvieae for 2 weeks, 52.3% (34 out of 65) of IIPs correlated with Lactococcus responded consistently with our inferred relations, including one commensal, 10 competitive, and 23 mutual interactions (Table 3 and Supplementary Table 6). There were 47.4% (27 out of 57) and 46.2% (24 out of 52) of IIPs correlated with Corynebacterium and Bacillus, respectively, which also responded consistently with our inferred relations. However, there were 23.1% (15 out of 65), 31.6% (18 out of 57), and 34.6% (18 out of 52) IIPs that correlated with Lactococcus, Corynebacterium, and Bacillus, presenting a conflicting response with our inferred relations, respectively. Overall, in the G1 treatment group, the ratio of IIPs that responded according to the inferred relationship was 48.9% (85 out of 174), the ratio of IIPs that showed a conflicting response to the inferred relationships was 29.3% (51 out of 174) (Table 3). In the evaluations for the G2, G3, and G4 treatment groups, we found 43.1–54.4% of corresponding changes of IIPs were consistent with the inferred interaction (Table 3).
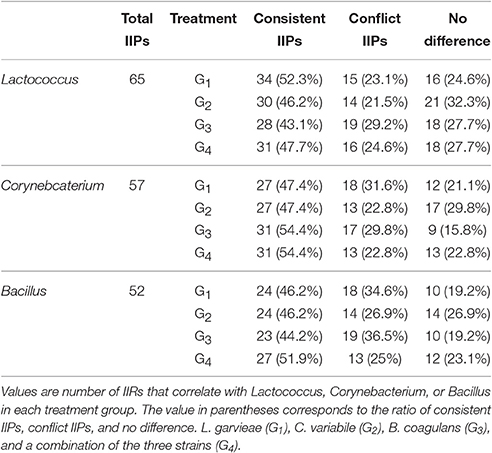
Table 3. Validation of IIPs that correlate with Lactococcus, Corynebacterium, or Bacillus.
Discussion
Recent advances in sequencing technology have created a new opportunity to explore population of microbes and their associations with environmental changes. Combining mathematical and computational models to infer the interacting networks reveals more details of microbe-microbe and microbe-host interactions. For example, Stein et al. (2013) extended generalized Lotka-Volterra equations to study the mechanism of C. difficile colonization in mice. They inferred that the genera Akkermansia, Blautia, and Coprobacillus had inhibitory interactions on C. difficile. In contrast, Enterococcus and Mollicutes could positively affect the growth of C. difficile, while the genus Barnesiella was predicted to inhibit growth of the genus Enterococcus. The results highlight a multi-layered sub-network associated with C. difficile. A study by Trosvik et al. (2010) also inferred microbial interactions in human infant gut using a dynamic systems modeling approach called time-dependent generalized additive models (GAM). They showed an agreement between predictions by dynamic interaction modeling and observed data of Firmicutes and Proteobacteria, suggesting that microbe-microbe interactions were sufficient to explain the growth patterns via modeling from time-series data. However, due to the complexity of microbial composition and difficulty in handling intestinal colonization, there was no experimental data to examine the inferred interactions (Mounier et al., 2008; Trosvik et al., 2010; Stein et al., 2013; Marino et al., 2014). To fill the gap, in this study, we used a user-friendly tool, MetaMIS (Shaw et al., 2016), to apply the Lotka-Volterra equations to infer the microbial interaction. The results were further validated by manipulating the gut bacteria and examine the corresponding changes in microbial composition.
Although MetaMIS is a versatile tool for predicting microbial relationships, Lotka-Volterra equations still have some weaknesses, for example, bias resulting from the complexity of microbiota. For data input, gLV equations require knowledge of the growth rates of all community members, and the complexity of microbiota may cause difficulties in determining the dynamic growth rate calculated from relative abundance, especially in the case of rare species (Pedrós-Alió, 2012). To improve the accuracy of MetaMIS, we implemented a time series data with the microbial communities from simple to complex for network inference. We firstly used AH in the treefrog to reduce the microbial diversity, and consequently less species were involved in the interaction network. After AH, increased temperature triggered bacterial turnover in composition, making it possible to reveal the evolving interactions between bacteria over time. The data also allowed us to collect time-series data after the perturbation to generate more dynamic fluctuations and provide deeper insights, compared with static communities (Holling, 1973; May, 1973, 1974; Ives and Carpenter, 2007). We therefore collected fecal samples in a 15-day time series, and increased the density of sampling points after AH to monitor dynamic changes. The experimental design of the data input could lead to a low ratio of inconsistent IIPs.
To validate the interactions, we focused on a small group of microbes that interact with the probiotic strains, which have been well-studied and applied in raniculture (Dias et al., 2010; Mendoza et al., 2012). The well-known LAB, Bacillus, Enterococcus, and Lactococcus, have been described and applied in many organisms (Balcázar et al., 2007b; Pasteris et al., 2009a; Dias et al., 2010; Nayak, 2010; Mendoza et al., 2012). Their inhibitory mechanisms against pathogenic bacteria have also been discussed (Hyronimus et al., 1998; Payot et al., 1999; Kesarcodi-Watson et al., 2008; Pasteris et al., 2009a). Our results indicated that the inferred network is biologically significant, and the compositional change of probiotics and the immune responses consistently supported our inference. In addition, our inferred relations were supported by the literature. For instance, Enterococcus competed with Citrobacter and Staphylococcus according to the inferred interaction, and these relationships were supported by previous studies showing that Enterococcus spp. 334 maintained its inhibitory effect against Citrobacter freundii and Staphylococcus epidermidis in Lithobates catesbeianus (Mendoza et al., 2012). Another evidence also showed that Enterococcus faecium imposed an inhibitory effect against C. freundii in Rana catesbeiana (Pasteris et al., 2009a). In addition, L. garvieae was found to inhibit C. freundii by the production of organic acid (Mendoza et al., 2012), and this correlation between Citrobacter and Lactococcus is in agreement with our IIPs. Furthermore, the inferred network suggested that Corynebacterium and Lactococcus were mutually beneficial partners. This is consistent with the culture-based studies showing that the culturable microbiota of milk consists of primarily of LAB such as Enterococcus and Lactococcus, and were often accompanied by the presence of Corynebacterium (Coppola et al., 2008).
In this study we generated a microbial interaction network of gut microbiota in the treefrog, and sub-groups of inferred relations were also validated. The experimental approach using probiotic administration indicated that the inferred interactions were reliable, and the results were also supported by the literature. However, there are still some puzzles remaining. For example, in the G2 treatment group, oral administration with C. variabile caused barely any beneficial effects on Lactococcus colonization. Although unexpected changes of gut probiotic levels were commonly observed after probiotic treatment (Hai, 2015; Ramos et al., 2015; Yang et al., 2015), we reasoned that more complex factors derived from other indirect IIPs also imposed various effects (commensalism, mutualism, amensalism, and competition) on these targets. The study is the first attempt to manipulate gut bacteria composition according to the inferred microbial interactions. We demonstrated the possibility that the gut microbiome can be changed accordingly. Moreover, our study provides a new potential strategy for the use of probiotics in raniculture.
Author Contributions
DW and FW designed the analyses. YY, CW, and FW collected the data. FW performed the analyses. GS implemented gLV model. DW and FW wrote the paper. DW was the principle investigator and conceived the analyses. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Ministry of Science and Technology MOST 105-2311-B-001-068.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00525/full#supplementary-material
Supplementary Figure 1. Rarefaction analyses for the observed number of genera from 12 time points. The rarefaction curves for each sample of 12 time points were displayed by different colors.
Supplementary Table 1. Summary of sample information of 12 time points spanning 15 days over AH. Values are expressed as mean values ± SD.
Supplementary Table 2. Summary of sample information of oral administration for bacterial composition. Oral administration of five treatments was carried out for 2 weeks. G1: L. garvieae (107 CFU g−1) in 0.9% saline. G2: C. variabile (107 CFU g−1) in 0.9% saline. G3: B. coagulans (107 CFU g−1) in 0.9% saline. G4: A combination of B. coagulans, C. variabile, and L. garvieae (each contains 107 CFU g−1) in 0.9% saline. G5:Control (0.9% saline). Values are expressed as mean ± SD.
Supplementary Table 3. Summary of sample information of oral administration for quantitative PCR. Oral administration of five treatments was performed over 2 weeks. G1: L. garvieae (107 CFU g−1) in 0.9% saline. G2: C. variabile (107 CFU g−1) in 0.9% saline. G3: B. coagulans (107 CFU g−1) in 0.9% saline. G4: a combination of B. coagulans, C. variabile, and L. garvieae (each contains 107 CFU g−1) in 0.9% saline. G5: Control (0.9% saline). Values are expressed as mean ± SD.
Supplementary Table 4. Ten thousand and three hundred fourteen significant inferred interacting pairs (IIPs) identified by MetaMIS. 10,314 IIPs passed the permutation criterion (cutoff > 0.5 described in Methods), including 1,568 commensal relationships (represented by O), 1,737 amensal relationships (represented by A), 3,777 mutual relationships (represented by M), and 3,232 competitive relationships (represented by C).
Supplementary Table 5. The raw data for the interaction network at the species level. 1,568 IIPs passed the permutation criterion (cutoff > 0.5 described in Methods), including 174 commensal relationships (represented by O), 211 amensal relationships (represented by A), 461 mutual relationships (represented by M), and 722 competitive relationships (represented by C).
Supplementary Table 6. Fold change of the inferred relationships with Lactococcus, Corynebacterium, and Bacillus. Commensalism is represented by O, amensalism by A, mutualism by M, and competition by C. Fold changes are expressed as mean values ± SD. NA, data insufficient for test.
Supplementary Table 7. Relative abundance of IIPs of Bacillus, Corynebacterium, and Lactococcus after 2-week oral trials. Five oral administrations included G1: L. garvieae (107 CFU g−1) in 0.9% saline. G2: C. variabile (107 CFU g−1) in 0.9% saline. G3: B. coagulans (107 CFU g−1) in 0.9% saline. G4: A combination of B. coagulans, C. variabile, and L. garvieae (each contains 107 CFU g−1) in 0.9% saline. G5: Control (0.9% saline). Values are expressed as mean values ± SD.
Abbreviations
IIP, inferred interaction partner; AH, artificial hibernation.
References
Akhter, N., Wu, B., Memon, A. M., and Mohsin, M. (2015). Probiotics and prebiotics associated with aquaculture: a review. Fish Shellfish Immunol. 45, 733–741. doi: 10.1016/j.fsi.2015.05.038
Akinbowale, O. L., Peng, H., and Barton, M. D. (2006). Antimicrobial resistance in bacteria isolated from aquaculture sources in Australia. J. Appl. Microbiol. 100, 1103–1113. doi: 10.1111/j.1365-2672.2006.02812.x
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Balcazar, J. L., de Blas, I., Ruiz-Zarzuela, I., Cunningham, D., Vendrell, D., and Muzquiz, J. L. (2006). The role of probiotics in aquaculture. Vet. Microbiol. 114, 173–186. doi: 10.1016/j.vetmic.2006.01.009
Balcázar, J. L., de Blas, I., Ruiz-Zarzuela, I., Vendrell, D., Calvo, A. C., Márquez, I., et al. (2007a). Changes in intestinal microbiota and humoral immune response following probiotic administration in brown trout (Salmo trutta). Br. J. Nutr. 97, 522–527. doi: 10.1017/S0007114507432986
Balcázar, J. L., De Blas, I., Ruiz-Zarzuela, I., Vendrell, D., Gironés, O., and Muzquiz, J. L. (2007b). Enhancement of the immune response and protection induced by probiotic lactic acid bacteria against furunculosis in rainbow trout (Oncorhynchus mykiss). FEMS Immunol. Med. Microbiol. 51, 185–193. doi: 10.1111/j.1574-695X.2007.00294.x
Becker, M. H., Harris, R. N., Minbiole, K. P., Schwantes, C. R., Rollins-Smith, L. A., Reinert, L. K., et al. (2011). Towards a better understanding of the use of probiotics for preventing chytridiomycosis in Panamanian golden frogs. Ecohealth 8, 501–506. doi: 10.1007/s10393-012-0743-0
Bhargava, S. C. (1989). Generalized Lotka-Volterra equations and the mechanism of technological substitution. Technol. Forecast. Soc. Change 35, 319–326. doi: 10.1016/0040-1625(89)90068-1
Bletz, M. C., Loudon, A. H., Becker, M. H., Bell, S. C., Woodhams, D. C., Minbiole, K. P., et al. (2013). Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16, 807–820. doi: 10.1111/ele.12099
Book, S. G. (1974). Amphibians: Guidelines for the Breeding, Care and Management of Laboratory Animals. Washington, DC: National Academy of Sciences.
Buffie, C. G., Jarchum, I., Equinda, M., Lipuma, L., Gobourne, A., Viale, A., et al. (2012). Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 80, 62–73. doi: 10.1128/IAI.05496-11
Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Coppola, S., Blaiotta, G., and Ercolini, D. (2008). “Dairy products,” Molecular Techniques in the Microbial Ecology of Fermented Foods, eds L. Cocolin and D. Ercolini (Heidelberg: Springer), 31–90.
Costello, E. K., Gordon, J. I., Secor, S. M., and Knight, R. (2010). Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 4, 1375–1385. doi: 10.1038/ismej.2010.71
Coyte, K. Z., Schluter, J., and Foster, K. R. (2015). The ecology of the microbiome: networks, competition, and stability. Science 350, 663–666. doi: 10.1126/science.aad2602
Dethlefsen, L., and Relman, D. A. (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. 108(Suppl. 1), 4554–4561. doi: 10.1073/pnas.1000087107
Dias, D. D. C., De Stéfani, M. V., Ferreira, C. M., França, F. M., Ranzani-Paiva, M. J. T., and Santos, A. A. (2010). Haematologic and immunologic parameters of bullfrogs, Lithobates catesbeianus, fed probiotics. Aquac. Res. 41, 1064–1071. doi: 10.1111/j.1365-2109.2009.02390.x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Finke, M. D. (2013). Complete nutrient content of four species of feeder insects. Zoo Biol. 32, 27–36. doi: 10.1002/zoo.21012
Gossling, J., Loesche, W., and Nace, G. (1982a). Large intestine bacterial flora of nonhibernating and hibernating leopard frogs (Rana pipiens). Appl. Environ. Microbiol. 44, 59–66.
Gossling, J., Loesche, W., and Nace, G. (1982b). Response of intestinal flora of laboratory-reared leopard frogs (Rana pipiens) to cold and fasting. Appl. Environ. Microbiol. 44, 67–71.
Hai, N. V. (2015). Research findings from the use of probiotics in tilapia aquaculture: a review. Fish Shellfish Immunol. 45, 592–597. doi: 10.1016/j.fsi.2015.05.026
Hamdan, A. M., El-Sayed, A. F. M., and Mahmoud, M. M. (2016). Effects of a novel marine probiotic, Lactobacillus plantarum AH 78, on growth performance and immune response of Nile tilapia (Oreochromis niloticus). J. Appl. Microbiol. 120, 1061–1073. doi: 10.1111/jam.13081
Hernandez, D., Cardell, E., and Zarate, V. (2005). Antimicrobial activity of lactic acid bacteria isolated from Tenerife cheese: initial characterization of plantaricin TF711, a bacteriocin-like substance produced by Lactobacillus plantarum TF711. J. Appl. Microbiol. 99, 77–84. doi: 10.1111/j.1365-2672.2005.02576.x
Holling, C. S. (1973). Resilience and stability of ecological systems. Annu. Rev. Ecol. Syst. 4, 1–23. doi: 10.1146/annurev.es.04.110173.000245
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Huang, L., Li, J., Anboukaria, H., Luo, Z., Zhao, M., and Wu, H. (2016). Comparative transcriptome analyses of seven anurans reveal functions and adaptations of amphibian skin. Sci. Rep. 6:24069. doi: 10.1038/srep24069
Hyronimus, B., Le Marrec, C., and Urdaci, M. (1998). Coagulin, a bacteriocin-like-inhibitory substance produced by Bacillus coagulans I. J. Appl. Microbiol. 85, 42–50. doi: 10.1046/j.1365-2672.1998.00466.x
Ives, A. R., and Carpenter, S. R. (2007). Stability and diversity of ecosystems. Science 317, 58–62. doi: 10.1126/science.1133258
Jernberg, C., Löfmark, S., Edlund, C., and Jansson, J. K. (2007). Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 1, 56–66. doi: 10.1038/ismej.2007.3
Kesarcodi-Watson, A., Kaspar, H., Lategan, M. J., and Gibson, L. (2008). Probiotics in aquaculture: the need, principles and mechanisms of action and screening processes. Aquaculture 274, 1–14. doi: 10.1016/j.aquaculture.2007.11.019
Kohl, K. D., Amaya, J., Passement, C. A., Dearing, M. D., and McCue, M. D. (2014). Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 90, 883–894. doi: 10.1111/1574-6941.12442
Küng, D., Bigler, L., Davis, L. R., Gratwicke, B., Griffith, E., and Woodhams, D. C. (2014). Stability of microbiota facilitated by host immune regulation: informing probiotic strategies to manage amphibian disease. PLoS ONE 9:e87101. doi: 10.1371/journal.pone.0087101
Li, M., Wang, B., Zhang, M., Rantalainen, M., Wang, S., Zhou, H., et al. (2008). Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. U.S.A. 105, 2117–2122. doi: 10.1073/pnas.0712038105
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Manichanh, C., Rigottier-Gois, L., Bonnaud, E., Gloux, K., Pelletier, E., Frangeul, L., et al. (2006). Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Marino, S., Baxter, N. T., Huffnagle, G. B., Petrosino, J. F., and Schloss, P. D. (2014). Mathematical modeling of primary succession of murine intestinal microbiota. Proc. Natl. Acad. Sci.U.S.A. 111, 439–444. doi: 10.1073/pnas.1311322111
May, R. M. (1973). Stability and Complexity in Model Ecosystems. Princeton, NJ: Princeton University Press.
May, R. M. (1974). Biological populations with nonoverlapping generations: stable points, stable cycles, and chaos. Science 186, 645–647. doi: 10.1126/science.186.4164.645
Mendoza, G. M., Pasteris, S. E., Ale, C. E., Otero, M. C., Bühler, M. I., and Nader-Macías, M. E. F. (2012). Cultivable microbiota of Lithobates catesbeianus and advances in the selection of lactic acid bacteria as biological control agents in raniculture. Res. Vet. Sci. 93, 1160–1167. doi: 10.1016/j.rvsc.2012.05.007
Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D., and Alon, U. (2002). Network motifs: simple building blocks of complex networks. Science 298, 824–827. doi: 10.1126/science.298.5594.824
Mounier, J., Monnet, C., Vallaeys, T., Arditi, R., Sarthou, A.-S., Hélias, A., et al. (2008). Microbial interactions within a cheese microbial community. Appl. Environ. Microbiol. 74, 172–181. doi: 10.1128/AEM.01338-07
Nayak, S. (2010). Probiotics and immunity: a fish perspective. Fish Shellfish Immunol. 29, 2–14. doi: 10.1016/j.fsi.2010.02.017
Newman, M. E. J. (2003). The structure and function of complex networks. Siam Rev. 45, 167–256. doi: 10.1137/S003614450342480
Olaoye, O. A. (2016). Antimicrobial activities of five strains of Lactococcus isolated from beef against indicator organisms of public health significance. Turkish J. Agri. Food Sci. Technol. 4, 887–892.
Pasteris, S. E., Pingitore, E. V., Babot, G. R., Otero, M. C., Bühler, M. I., and Nader-Macías, M. E. (2009a). Characterization of the beneficial properties of lactobacilli isolated from bullfrog (Rana catesbeiana) hatchery. Antonie Van Leeuwenhoek 95, 373–385. doi: 10.1007/s10482-009-9329-4
Pasteris, S. E., Roig Babot, G., Otero, M. C., Bühler, M. I., and Nader-Macías, M. E. (2009b). Beneficial properties of lactic acid bacteria isolated from a Rana catesbeiana hatchery. Aquac. Res. 40, 1605–1615. doi: 10.1111/j.1365-2109.2009.02261.x
Payot, T., Chemaly, Z., and Fick, M. (1999). Lactic acid production by Bacillus coagulans—kinetic studies and optimization of culture medium for batch and continuous fermentations. Enzyme Microb. Technol. 24, 191–199. doi: 10.1016/S0141-0229(98)00098-2
Pedrós-Alió, C. (2012). The rare bacterial biosphere. Ann. Rev. Mar. Sci. 4, 449–466. doi: 10.1146/annurev-marine-120710-100948
Perez, T., Balcazar, J. L., Ruiz-Zarzuela, I., Halaihel, N., Vendrell, D., de Blas, I., et al. (2010). Host-microbiota interactions within the fish intestinal ecosystem. Mucosal Immunol. 3, 355–360. doi: 10.1038/mi.2010.12
Peterson, J., Garges, S., Giovanni, M., McInnes, P., Wang, L., Schloss, J. A., et al. (2009). The NIH human microbiome project. Genome Res. 19, 2317–2323. doi: 10.1101/gr.096651.109
Proulx, S. R., Promislow, D. E. L., and Phillips, P. C. (2005). Network thinking in ecology and evolution. Trends Ecol. Evol. 20, 345–353. doi: 10.1016/j.tree.2005.04.004
Qi, Z., and Nie, P. (2008). Comparative study and expression analysis of the interferon gamma gene locus cytokines in Xenopus tropicalis. Immunogenetics 60, 699–710. doi: 10.1007/s00251-008-0326-y
Qi, Z., Nie, P., Secombes, C. J., and Zou, J. (2010). Intron-containing type I and type III IFN coexist in amphibians: refuting the concept that a retroposition event gave rise to type I IFNs. J. Immunol. 184, 5038–5046. doi: 10.4049/jimmunol.0903374
Qi, Z., Zhang, Q., Wang, Z., Zhao, W., and Gao, Q. (2015). Cloning of interleukin-10 from African Clawed Frog (Xenopus tropicalis), with the finding of IL-19/20 homologue in the IL-10 locus. J. Immunol. Res. 2015:462138. doi: 10.1155/2015/462138
Ramos, M. A., Goncalves, J. F., Batista, S., Costas, B., Pires, M. A., Rema, P., et al. (2015). Growth, immune responses and intestinal morphology of rainbow trout (Oncorhynchus mykiss) supplemented with commercial probiotics. Fish Shellfish Immunol. 45, 19–26. doi: 10.1016/j.fsi.2015.04.001
Reid, G., Sanders, M. E., Gaskins, H. R., Gibson, G. R., Mercenier, A., Rastall, R., et al. (2003). New scientific paradigms for probiotics and prebiotics. J. Clin. Gastroenterol. 37, 105–118. doi: 10.1097/00004836-200308000-00004
Ringø, E., Løvmo, L., Kristiansen, M., Bakken, Y., Salinas, I., Myklebust, R., et al. (2010). Lactic acid bacteria vs. pathogens in the gastrointestinal tract of fish: a review. Aquac. Res. 41, 451–467. doi: 10.1111/j.1365-2109.2009.02339.x
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. doi: 10.1038/nri2515
Shafiei, M., Dunn, K. A., Chipman, H., Gu, H., and Bielawski, J. P. (2014). BiomeNet: a Bayesian model for inference of metabolic divergence among microbial communities. PLoS Comput. Biol. 10:e1003918. doi: 10.1371/journal.pcbi.1003918
Shaw, G. T. W., Pao, Y.-Y., and Wang, D. (2016). MetaMIS: a metagenomic microbial interaction simulator based on microbial community profiles. BMC Bioinformatics 17:488. doi: 10.1186/s12859-016-1359-0
Sonoyama, K., Fujiwara, R., Takemura, N., Ogasawara, T., Watanabe, J., Ito, H., et al. (2009). Response of gut microbiota to fasting and hibernation in Syrian hamsters Appl. Environ. Microbiol. 75, 6451–6456. doi: 10.1128/AEM.00692-09
Stein, R. R., Bucci, V., Toussaint, N. C., Buffie, C. G., Rätsch, G., Pamer, E. G., et al. (2013). Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput. Biol. 9:e1003388. doi: 10.1371/journal.pcbi.1003388
Taur, Y., Xavier, J. B., Lipuma, L., Ubeda, C., Goldberg, J., Gobourne, A., et al. (2012). Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 55, 905–914. doi: 10.1093/cid/cis580
Trosvik, P., Stenseth, N. C., and Rudi, K. (2010). Convergent temporal dynamics of the human infant gut microbiota. ISME J. 4, 151–158. doi: 10.1038/ismej.2009.96
Turnbaugh, P. J., and Gordon, J. I. (2009). The core gut microbiome, energy balance and obesity J. Physiol. 587, 4153–4158. doi: 10.1113/jphysiol.2009.174136
Turnbaugh, P. J., Ridaura, V. K., Faith, J. J., Rey, F. E., Knight, R., and Gordon, J. I. (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 1, 6ra14–16ra14. doi: 10.1126/scitranslmed.3000322
Verschuere, L., Rombaut, G., Sorgeloos, P., and Verstraete, W. (2000). Probiotic bacteria as biological control agents in aquaculture. Microbiol. Mol. Biol. Rev. 64, 655–671. doi: 10.1128/MMBR.64.4.655-671.2000
Xu, J., and Gordon, J. I. (2003). Honor thy symbionts. Proc. Natl. Acad. Sci. U.S.A. 100, 10452–10459. doi: 10.1073/pnas.1734063100
Yang, G., Tian, X., Dong, S., Peng, M., and Wang, D. (2015). Effects of dietary Bacillus cereus G19, B. cereus BC-01, and Paracoccus marcusii DB11 supplementation on the growth, immune response, and expression of immune-related genes in coelomocytes and intestine of the sea cucumber (Apostichopus japonicus Selenka). Fish Shellfish Immunol. 45, 800–807. doi: 10.1016/j.fsi.2015.05.032
Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., and Madden, T. L. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:1. doi: 10.1186/1471-2105-13-S6-S1
Keywords: network, gut microbiota, artificial hibernation, probiotics, Polypedates megacephalus
Citation: Weng FC-H, Shaw GT-W, Weng C-Y, Yang Y-J and Wang D (2017) Inferring Microbial Interactions in the Gut of the Hong Kong Whipping Frog (Polypedates megacephalus) and a Validation Using Probiotics. Front. Microbiol. 8:525. doi: 10.3389/fmicb.2017.00525
Received: 20 November 2016; Accepted: 13 March 2017;
Published: 30 March 2017.
Edited by:
Eria Alaide Rebollar, James Madison University, USAReviewed by:
Valerie Mckenzie, University Of Colorado Boulder, USAMiguel Vences, Technische Universitat Braunschweig, Germany
Copyright © 2017 Weng, Shaw, Weng, Yang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daryi Wang, dywang@gate.sinica.edu.tw