
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 March 2017
Sec. Food Microbiology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00357
This article is part of the Research Topic Game Changer-Next Generation Sequencing and its Impact on Food Microbiology View all 26 articles
 Christelle F. Iskandar1
Christelle F. Iskandar1 Frédéric Borges1*
Frédéric Borges1* Bernard Taminiau2
Bernard Taminiau2 Georges Daube2
Georges Daube2 Monique Zagorec3
Monique Zagorec3 Benoît Remenant3†
Benoît Remenant3† Jørgen J. Leisner4Martin A. Hansen5
Jørgen J. Leisner4Martin A. Hansen5 Søren J. Sørensen5Cécile Mangavel1
Søren J. Sørensen5Cécile Mangavel1 Catherine Cailliez-Grimal1Anne-Marie Revol-Junelles1
Catherine Cailliez-Grimal1Anne-Marie Revol-Junelles1Lactic acid bacteria (LAB) differ in their ability to colonize food and animal-associated habitats: while some species are specialized and colonize a limited number of habitats, other are generalist and are able to colonize multiple animal-linked habitats. In the current study, Carnobacterium was used as a model genus to elucidate the genetic basis of these colonization differences. Analyses of 16S rRNA gene meta-barcoding data showed that C. maltaromaticum followed by C. divergens are the most prevalent species in foods derived from animals (meat, fish, dairy products), and in the gut. According to phylogenetic analyses, these two animal-adapted species belong to one of two deeply branched lineages. The second lineage contains species isolated from habitats where contact with animal is rare. Genome analyses revealed that members of the animal-adapted lineage harbor a larger secretome than members of the other lineage. The predicted cell-surface proteome is highly diversified in C. maltaromaticum and C. divergens with genes involved in adaptation to the animal milieu such as those encoding biopolymer hydrolytic enzymes, a heme uptake system, and biopolymer-binding adhesins. These species also exhibit genes for gut adaptation and respiration. In contrast, Carnobacterium species belonging to the second lineage encode a poorly diversified cell-surface proteome, lack genes for gut adaptation and are unable to respire. These results shed light on the important genomics traits required for adaptation to animal-linked habitats in generalist Carnobacterium.
Lactic acid bacteria (LAB) include various genera and many species that have been investigated for decades because of their major role in food fermentations and their health benefit potential as probiotics. Understanding how LAB fitness can increase through evolution in these various habitats is critical for exploiting their beneficial properties. LAB are well-known for their ability to colonize animal-derived food, i.e., meat, fish, and dairy products, and for being members of the gastrointestinal (GI) tract and the vagina microbiota (Douglas and Klaenhammer, 2010; Douillard and de Vos, 2014). LAB can be divided into specialized and generalist bacteria. Typically, the specialists that are used as starter cultures for a narrow range of fermented products are characterized by a low genetic diversity (Delorme et al., 2010). Their genomes exhibit signs of massive losses of genes involved in biosynthetic pathways (Douglas and Klaenhammer, 2010). To compensate the loss of these functions, genes encoding transporters for amino acids or carbohydrates were gained to allow growth in nutritional rich fermentation environments (Lorca et al., 2007). These genomic changes were accompanied by a specialization to food matrices, particularly exemplified in dairy strains. Other LAB also exhibit genomes characteristic of their ecological specialization. Lactobacillus iners is described to solely colonize the vaginal cavity and harbors one of the smallest LAB genomes presumably because its niche specialization allowed a substantial genome reduction (Macklaim et al., 2011; Mendes-Soares et al., 2014). Another example is the GI tract symbiont Lactobacillus reuteri, which is characterized by different lineages each one being apparently adapted to one particular vertebrate host: rodent or human (Frese et al., 2011). By contrast, some species are more generalist in their ability to colonize various environments and are therefore ubiquitous. Species from the genus Enterococcus can be found in the GI tract of animals and in a multitude of fermented foods (Santagati et al., 2012). Their adaptation to various environments is strongly linked to the presence in their genome of DNA acquired through horizontal gene transfer (HGT), resulting in a large pan genome (Santagati et al., 2012). Similarly, the genomes of some strains of the ubiquitous species Lactobacillus rhamnosus encode multiple lifestyle traits allowing them to reside in diverse habitats (Douillard et al., 2013). However, the interconnection between the ecology of these bacteria and their genomics is not fully understood and need further investigation. Importantly, the genomic traits responsible for adaptation to multiple animal-associated habitats are not clearly defined.
The LAB genera Enterococcus, Lactobacillus, Lactococcus, and Streptococcus have been intensively investigated, but lately, other genera including Carnobacterium have also drawn attention since 16S meta-barcoding studies have shown their significance in food (Chaillou et al., 2015; Duan et al., 2016; Fougy et al., 2016; Jääskeläinen et al., 2016). The genus Carnobacterium encompasses 11 species that have been isolated from cold and temperate environments, and from the GI tract of animals as well as from foods of animal origin such as seafood, meat, and dairy products. They are mesophilic and some species are psychrotolerant and able to grow down to 0°C. Some are halotolerant and able to grow with 8% NaCl and some are alkaliphilic with growth up to pH 9.5 (Cailliez-Grimal et al., 2014; Pikuta, 2014; Pikuta and Hoover, 2014). These traits could explain their wide distribution. However, it is apparent that some heterogeneity exists within the genus regarding habitat associations. Some species can be isolated from environments where contact with animals is likely rare as exemplified by Carnobacterium inhibens subsp. gilichinskyi WN1359, Carnobacterium sp. 17-4, and Carnobacterium AT7 which were isolated from Siberian permafrost, sea-ice from permanently cold fjords of the Arctic Ocean, and an oceanic trench, respectively (Lauro et al., 2007; Voget et al., 2011; Leonard et al., 2013; Nicholson et al., 2015). Other species, including C. maltaromaticum and C. divergens, have been found in animal-associated habitats. These two species are the most frequently isolated carnobacteria from various sources (Leisner et al., 2007) and belong to the dominant bacterial communities in meat and fish derived food (Chaillou et al., 2015; Duan et al., 2016; Fougy et al., 2016; Jääskeläinen et al., 2016). These bacteria are therefore very interesting models to investigate the genomic traits responsible for adaptation to animal-associated habitats. In the genus Carnobacterium, the genome size ranges from 2.4 Mbp for C. inhibens subsp. gilichinskyi WN1359 (Leonard et al., 2013), to 3.7 Mbp for C. maltaromaticum (Cailliez-Grimal et al., 2013). It has been suggested that the larger genome of the latter species is the basis of its success in colonizing various habitats (Leisner et al., 2007). However, it appears that genome size is not necessarily a predictor of the ability to occupy diverse habitats since the genome of C. divergens strains is relatively small (∼2.7 Mbp), but this species is still able to colonize various habitats (Sun et al., 2015; Remenant et al., 2016).
The aim of this study was to investigate the ecological niches Carnobacterium species can occupy and to identify the genomic traits responsible for the high ecological success of some Carnobacterium species and the possible underlying adaptive mechanisms. For that purpose, we analyzed the relative abundance of Carnobacterium species using 16S rDNA metagenomic data. Subsequently, we compared the genomes of Carnobacterium strains isolated from different environments (mainly from cold aquatic habitats), and animal-associated habitats (live animal and foods).
The data obtained from 681 samples of various ecological origins were analyzed with a focus on Carnobacterium. A database of V1–V3 16S rRNA gene sequence datasets available at the FARAH Institute (University of Liège, Belgium) was used to delineate the species composition of the Carnobacterium genus within four types of matrices: food products, animal samples, human and animal feces and environment. This database was built from 2010 to 2015 by merging the data obtained from several single projects hosted at the FARAH Institute. The datasets were produced as previously described (Rodriguez et al., 2015) by sequencing the V1–V3 16S rDNA hypervariable region with an MiSeq sequencer using v3 reagents (ILLUMINA, USA).
Sequence read processing was employed as previously described (Rodriguez et al., 2015) using the MOTHUR software package v1.35 (Schloss and Handelsman, 2003) and the UCHIME algorithm (Edgar et al., 2011) for alignment and OTU clustering (distance 0.03) and chimera detection, respectively. 16S gene sequence reference alignment and taxonomical assignation were based upon the SILVA database (v1.15) of full-length 16S rDNA sequences.
For each sample, Operational Taxonomic Units (OTUs) belonging to the Carnobacterium genus were extracted. Corresponding reads were further assigned to Carnobacterium species using a local BLASTn algorithm vs. the SILVA database (v1.15). Reads were assigned to a defined species when identical to the best hit (four mismatches were allowed).
Statistical differences of the different species proportion inside each type of matrix were assessed with non-parametric Kruskal–wallis test with Dunn’s post hoc tests using PRISM6 (GraphPad Software).
The Carnobacterium genome sequences available at the start of this study included five C. maltaromaticum, one C. inhibens subsp. gilichinskyi, one C. divergens, and two Carnobacterium sp. genomes among which three were complete (C. maltaromaticum LMA28, Carnobacterium sp. 17.4, and C. inhibens subsp. gilichinskyi WN1359). The strains originated from different ecological habitats, some from animal-derived food such as the C. maltaromaticum strains and C. divergens V41, and others from environmental samples (Table 1).
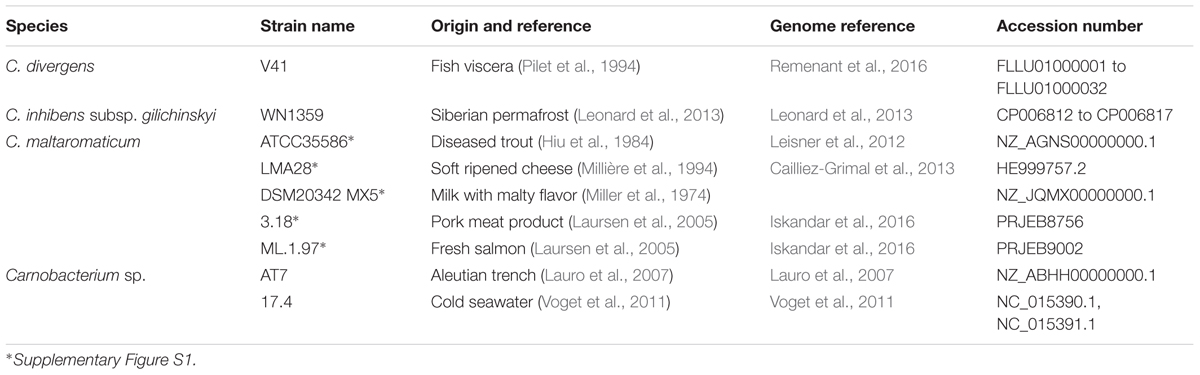
TABLE 1. Characteristics of Carnobacterium strains.
The genome sequences were integrated in the MicroScope platform (Vallenet et al., 2013) to perform automatic and expert annotation of the genes, as well as comparative analysis and secretome prediction by using the integrated SignalP software (Petersen et al., 2011). The gene phyloprofile tool interface was used for searching common or specific genes/regions between a query genome and other genomes or replicons chosen from the ones available in Prokaryotic Genome DataBase (PkGDB; i.e., (re)annotation of bacterial genomes) or complete proteome downloaded from the RefSeq/WGS sections.
The pan/core genome was calculated using two methods. The pan/core genome tool accessible in the comparative genomics section was used with MICFAM parameters of 50 or 80% amino acid (aa) sequence identity and 80% coverage. The Phyloprofile tool from the MicroScope platform was used with a cut-off of 70% aa identity and 80% coverage with the best Bidirectional Best Hit (BBH).
Synteny, defined as an orthologous gene set having the same local organization in species A and B, was determined as sequence similarity by BlastP BBH with at least 30% identity on 80% of the shortest sequence (minLrap 0.8) analyses and co-localization. Metabolic pathways were predicted using the Kyoto Encyclopedia of Genes and the Genomes (KEGG) resources (Kanehisa and Goto, 2000; Kanehisa et al., 2014) and the MetaCyc database (Caspi et al., 2014). A percentage of 70% minimum identity was used to detect the specific and common genes for C. maltaromaticum, excluding genes with 30% minimum identity with the four other Carnobacterium strains.
Neighbor-joining-based phylogenetic reconstruction was based on the nucleic acid sequence of 10 housekeeping genes (dnaK, gyrA, polA, lepA, dnaB, gyrB, secA, ftsZ, recG, ileS) and was performed using MEGA6 by using the Kimura two-parameter model, including transitions and transversions. The candidate tree was tested with 1,000 bootstrap replications (Tamura et al., 2013). The Sequence Type (MLST) was updated using e-BURST analysis from Rahman et al. (2014a). The resulting tree was rooted using the closely related species Enterococcus faecalis as outgroup.
The search for prophages was conducted with the PHAST Search Tool (Zhou et al., 2011).
The annotations were deposited at DDBJ/EMBL/GenBank under the following references: PRJEB9002 for C. maltaromaticum ML.1.97, and PRJEB8756 for C. maltaromaticum 3.18. The annotations are publicly available for consultation in MicroScope1.
Metagenomic data for genes encoding 16S rDNA from 681 samples were analyzed with a focus on the genus Carnobacterium. The samples were categorized as animal-derived food, animal organs, feces, and environment. Overall, the presence of representatives of the species C. divergens, C. iners, C. inhibens, C. jeotgali, C. maltaromaticum, C. mobile, C. viridans, and uncultured Carnobacterium sp was detected and their relative abundance is presented in Figure 1. The most abundant species was C. maltaromaticum accounting for 28–60% of Carnobacterium reads, followed by C. divergens (15–49%) and Carnobacterium spp. from lineages that have not yet been cultured and characterized (14–47% of Carnobacterium reads, depending on the sample origin). C. inhibens, C. mobile, and C. viridans accounted for lesser reads. C. jeotgali was detected only in environmental samples. C. viridans was detected only in food samples whereas C. maltaromaticum reads were observed in the samples from all origins. Other species were detected in the samples from two or three different habitats.
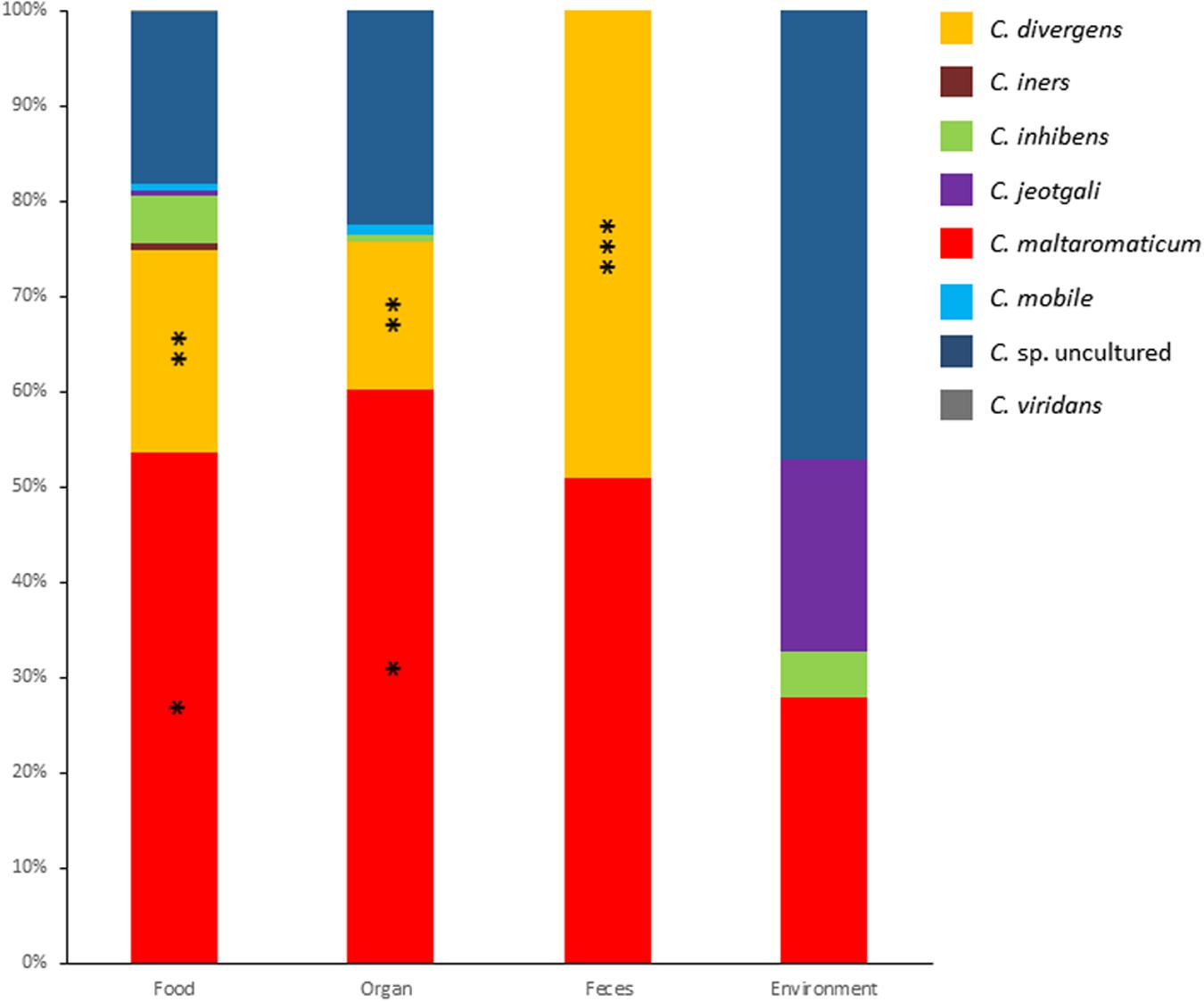
FIGURE 1. Relative abundance of each Carnobacterium species obtained from 16S metagenomic analyses of 681 samples originating from food, organs, feces, and environment. One asterisk indicate significant difference (P < 0.0001) compared to all other Carnobacterium sp, two asterisks indicate significant difference (P < 0.01) compared to C. iners, C. inhibens, C. jeotgali, C. mobile, and C. viridans. Three asterisks indicate significant difference (P < 0.0001) compared to all Carnobacterium sp. except C. maltaromaticum.
The highest species diversity was observed in food and organs (dog lungs, pig nymphal nodes, pig stomach, cattle forestomach, and cattle spleen), with a maximum of eight species in food. The lowest diversity was observed in feces where only two species were recorded. In food and organs, the most abundant species were C. maltaromaticum, C. divergens, and uncultured Carnobacterium sp. The OTUs assigned to the species C. maltaromaticum accounted for 54% and 60% of the reads associated to the genus Carnobacterium in food and organs, respectively (Figure 1). Interestingly, in the feces only C. maltaromaticum and C. divergens sequences were detected, each accounting for about half of the reads. In contrast, C. maltaromaticum sequences represented 27% of the reads, while 47% of the reads were attributed to uncultured Carnobacterium sp. in environmental samples. C. divergens was not found in samples originating from the environment.
These results strongly suggest that C. maltaromaticum and to a lesser extend C. divergens are the most prevalent species of this genus in habitats associated to animals. Although animal-associated habitats differ from each other, they also share common properties: they are nutrient-rich environments, they can induce similar stresses, they are associated with a dense and diversified microbiota and carbon is mainly available in the form of polymeric biomacromolecules. It can therefore be expected that bacteria, such as Carnobacterium species that are associated to animal habitats, share general properties and thereby highly contrast with other Carnobacterium species associated with the external environment.
In order to identify the adaptation factors responsible for the high ability of C. maltaromaticum and C. divergens to colonize multiple ecological niches including animal-associated habitats, a genome based analysis was conducted on nine Carnobacterium genomes (Table 1). The genome sizes of the five C. maltaromaticum strains were the largest, ranging from 3.33 to 3.87 Mbp; the other Carnobacterium genomes were smaller (2.35–2.74 Mbp), with the smallest being that of C. inhibens subsp. gilichinskyi WN1359 (Table 2). Accordingly, the number of predicted CDS in each genome ranged from 3,368 to 3,812 for C. maltaromaticum, and from 2,268 to 2,633 for the other Carnobacterium (Table 2). C. maltaromaticum genomes harbored a lower GC% (34.4–34.5) than other genomes (35.2–35.3).
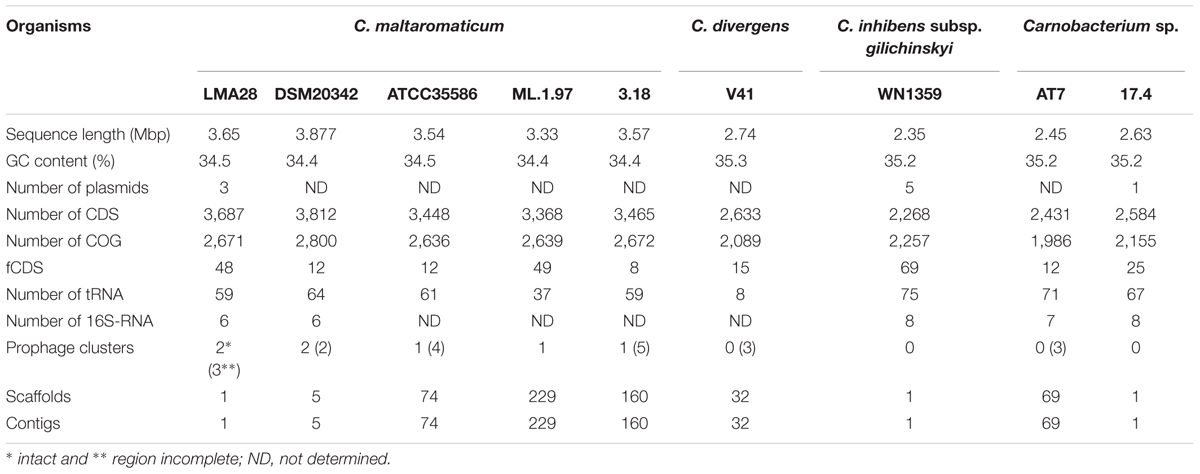
TABLE 2. General features of Carnobacterium genomes.
A phylogenetic tree based on 10 housekeeping genes was constructed. It shows that C. maltaromaticum and C. divergens on one hand, and Carnobacterium sp. 17.4, Carnobacterium sp. AT7, and C. inhibens subp. gilichinskyi WN1359 on the other hand, are closely related. They form two monophyletic taxons sharing a common ancestor, as shown by the outgroup E. faecalis V583 (Figure 2).
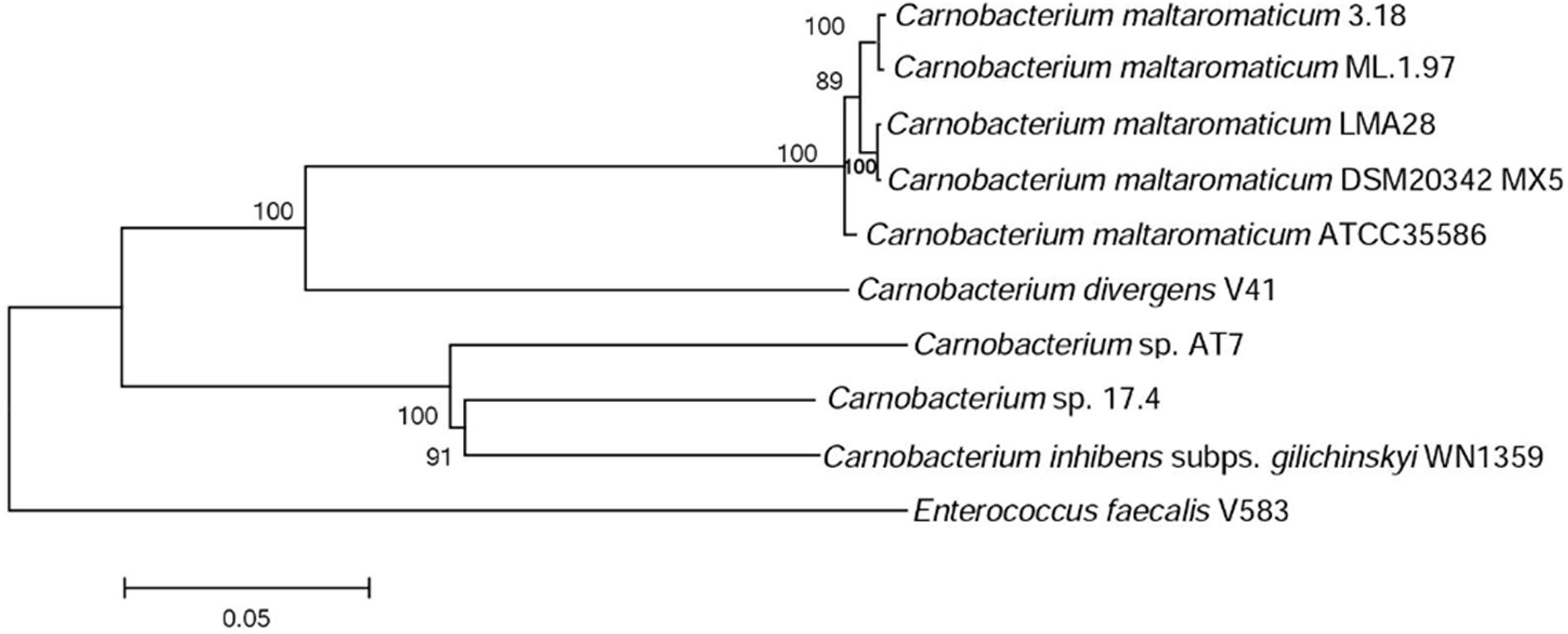
FIGURE 2. Phylogenetic tree of the Carnobacterium strains subjected to comparative genome analysis. The tree is based on the alignment of nucleic acid sequence of 10 housekeeping genes (dnaK, gyrA, polA, lepA, dnaB, gyrB, secA, ftsZ, recG, and ileS).
The pan/core genome analysis of the nine Carnobacterium (cut-off of 50% aa identity and 80% coverage) showed that the core genome represents 1,130 (31%) of the predicted CDS. When comparison was restricted to C. inhibens subsp. gilichinskyi WN1359, Carnobacterium sp. 17-4, and Carnobacterium sp. AT7, the core genome increased up to 1,745–1,785 (67–73%) of the predicted CDS. Similarly, when the comparison was restricted to the 5 C. maltaromaticum strains and C. divergens V41, the core genome increased up to 1,723–1,825 (68–76%) of the predicted CDS. This result is in agreement with the phylogenetic tree showing close phylogenetic proximity of C. maltaromaticum and C. divergens on the one hand, and of C. inhibens subsp. gilichinskyi WN1359, Carnobacterium sp. 17-4, and Carnobacterium sp. AT7 on the other hand.
To be more restrictive, the pan/core genome analysis was performed with a cut-off of 70% aa identity and 80% coverage. It revealed that the strains of C. maltaromaticum share 2,665 CDS, and each of the five strains possesses between 279 and 644 specific genes showing strain-to-strain variations.
The gene repertoire of the different genomes was then compared to search for functions that differ between strains in order to identify the genomic traits that could explain the potential adaptation of C. maltaromaticum and C. divergens to animal-linked habitats.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs; Barrangou et al., 2007), and the CRISPR associated (cas) genes confer resistance to phage infection. The role and mechanism of the CRISPR-cas system in bacterial species have been extensively studied and indicate that the spacer sequences can be considered as a signature of past exposure to exogenous DNA. Among all Carnobacterium genomes, only C. divergens V41 possessed a CRISPR-cas system. More precisely, two loci were predicted in the genome of this strain. The presence of these genes suggests that C. divergens V41 may have systems acting as barriers against HGT, thereby that would limit genome expansion in this taxon.
One to four prophages and/or prophage remnants were detected in all analyzed genomes except in those of C. inhibens subsp. gilichinskyi and Carnobacterium sp. 17.4 (Table 2). Their size, ranged from 11.5 to 74.4 kbp (data not shown). More importantly, complete prophages were found only in the C. maltaromaticum (Table 2).
The genomes of all Carnobacterium species but one -C. maltaromaticum- exhibit a genome of small or relatively small sizes. This suggests that the common ancestor of Carnobacterium exhibited a small size and that the ancestor of the species C. maltaromaticum experienced a massive gain of genes. Compared to C. divergens, no CRISPR-Cas systems were found in the genome of C. maltaromaticum. CRISPR-Cas provide an adaptive immunity against foreign DNA and is considered as a barrier against horizontal transfer (Barrangou et al., 2007). The lack of such a barrier against DNA transfer could have favored the acquisition of genes in the C. maltaromaticum lineage. Consistently, complete prophages were found in the genomes of C. maltaromaticum while only remnants prophages were found in the genome of C. divergens. Similarly, Lactobacillus genomes devoid of CRISPR-Cas systems exhibited the trend of being more abundant in phage sequences (Sun et al., 2015). LAB are mainly described as evolving by massive gene loss. The lineage C. maltaromaticum suggests that LAB may also evolve by a massive gene gain. Consistently, other ubiquitous LAB also exhibited large genomes that could be a result of similar mechanisms (Sun et al., 2015).
Homologs of genes encoding the general secretion route (Sec-pathway) were present in all strains, while no homolog of the Twin-arginine translocation pathway (Tat-pathway) was found (data not shown). The secretome was therefore predicted by identifying genes suspected to encode signal peptide-containing proteins. The five C. maltaromaticum strains contained the largest secretome (319–375 proteins predicted to harbor a signal peptide), whereas C. divergens V41 presented an intermediate number (272 predicted proteins) compared to those predicted in Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and C. inhibens subsp. gilichinskyi WN1359 with only 132–155 proteins containing a peptide signal. Compared to other LAB or genera that can share the same habitats, the sizes of secretomes of C. maltaromaticum and C. divergens are among the largest (Figure 3).
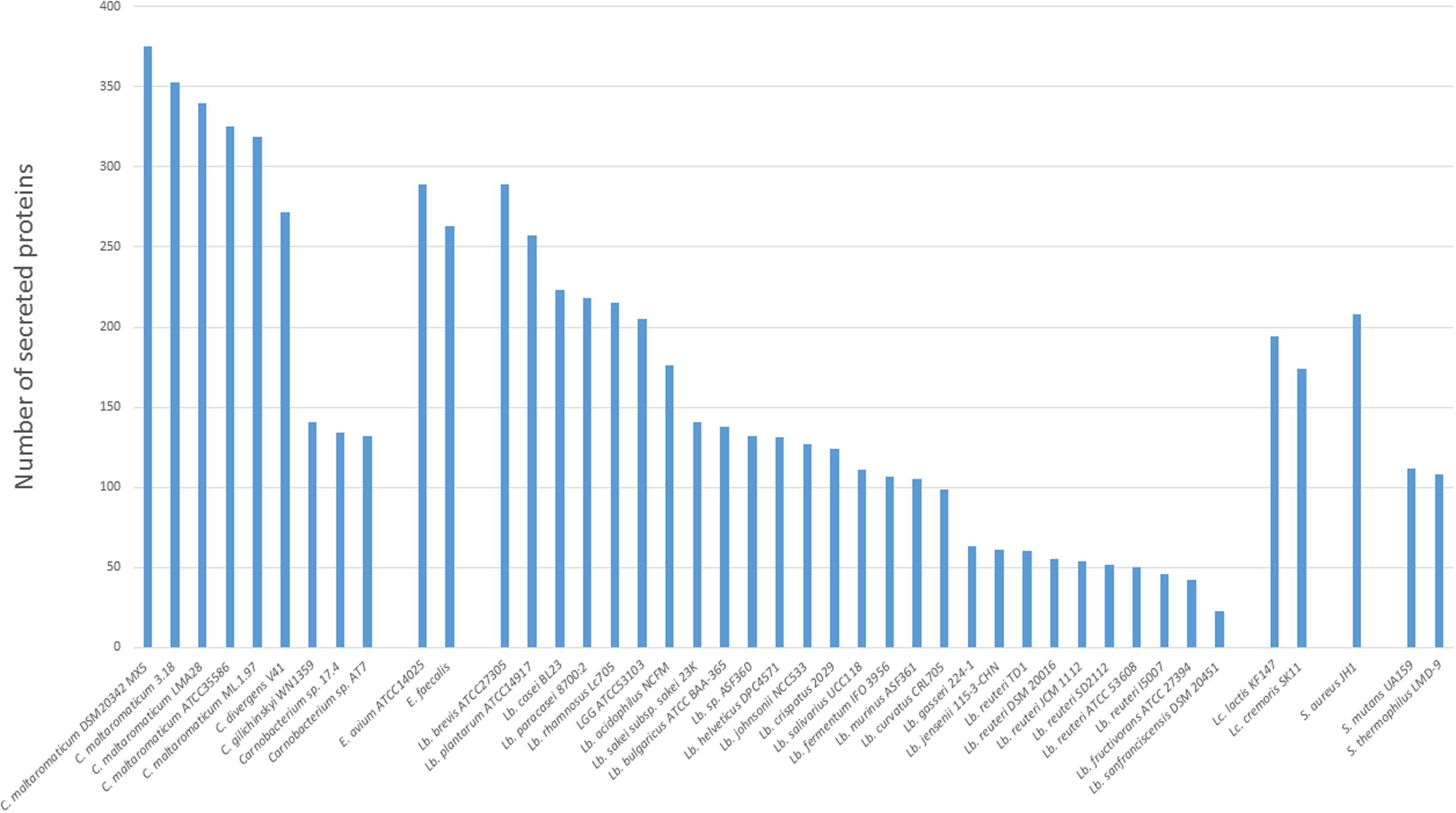
FIGURE 3. Secretome size in the different Carnobacterium genomes. Other genomes belonging to lactic acid bacteria (LAB) (Lactococcus and Lactobacillus) and other Gram-positive species (Staphylococcus, Enterococcus, and Streptococcus) available at Microscope Mage were also included.
Among the COG (cluster of orthologous genes) represented in the secretome, families M (cell wall/membrane/envelop biogenesis), P (inorganic ion transport and metabolism), and R (general functional prediction only) tend to be more represented in C. maltaromaticum and C. divergens compared to the other species (Supplementary Figure S2). This suggests that extracellular functions are more diversified in C. maltaromaticum, and to a lesser extent in C. divergens, than in the other Carnobacterium. For instance, C. maltaromaticum and C. divergens exhibit between 22 and 35 genes in COG R, while the three other strains Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and C. inhibens subsp. gilichinskyi WN1359, would have less than 10 genes encoding a signal peptide within this subclass.
Further, C. maltaromaticum predicted secretomes encompassed a higher number (26–30) of proteins belonging to COG family G (carbohydrate transport and metabolism) than was the case for C. divergens V41 (18) and other species (10–15). This is correlated with a higher content in genes encoding PTS transporters, between 62 and 68, in C. maltaromaticum. By contrast, the other Carnobacterium, including C. divergens, would contain less of such genes (between 27 and 43 depending on the strains). C. maltaromaticum strains, compared to other Carnobacterium species harbor a larger repertoire of PTS transporters. This characteristics is typical of ubiquitous LAB, as these transporters enable the bacteria to exploit a wide range of carbon sources (Douillard and de Vos, 2014).
More strikingly, C. maltaromaticum strains and C. divergens V41 secretomes encompassed between 116 and 155 proteins unclassified in COG families (class X Supplementary Figure S2), while this category was of minor importance (0–23) in other Carnobacterium species. The detailed analysis of those unclassified genes revealed that approximately half of them encode hypothetical proteins or conserved exported proteins of unknown function, and 30% encode secreted proteins associated with the cell wall. These results strongly suggest that C. maltaromaticum and C. divergens cell-surface structures differ significantly from those of Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and C. inhibens subsp. gilichinskyi WN1359. Therefore, we focused on the comparison of the gene repertoire encoding such surface associated proteins.
A larger set of proteins non-covalently bound to the cell wall was predicted for C. maltaromaticum and C. divergens V41 than for the other Carnobacterium. These proteins contain at least one LysM domain, and one WxL domain or SH3 domain (Table 3).
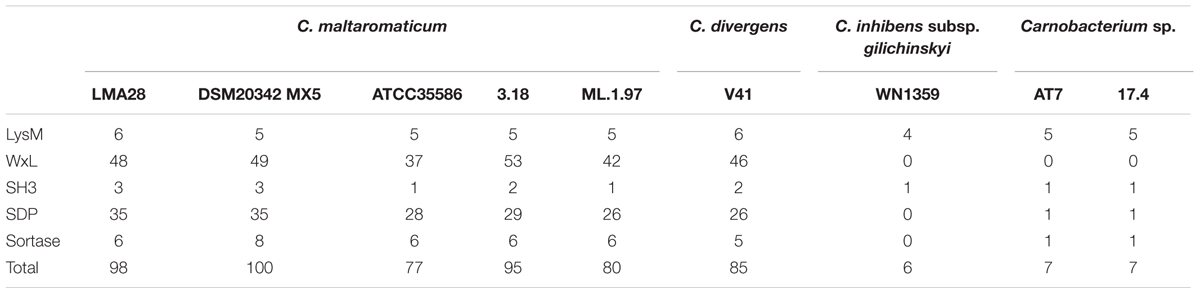
TABLE 3. Number of sortases and surface proteins.
For all strains, at least one protein with an SH3 domain involved in peptidoglycan binding is predicted to be anchored to the cell wall (Table 3 and Supplementary Table S1), and between four and six LysM proteins. Two of them are conserved in all strains, three are found only in C. maltaromaticum and C. divergens V41, and three are variable within Carnobacterium sp. strains (Table 3 and Supplementary Table S1).
The signature of the 160–190 aa long WxL domain is characterized by two WxL motifs. WxL-containing proteins are non-covalently anchored proteins associated with the cell wall (Brinster et al., 2007). Strikingly, the number of WxL-containing proteins is comprised of between 37 and 53 and present only in C. maltaromaticum and C. divergens. Remarkably, no WxL-containing proteins were found to be encoded by the other Carnobacterium genomes. Twenty-nine genes encoding WxL proteins are common to the five C. maltaromaticum strains and C. divergens V41. Overall, 56 genes are variable within the strains (i.e., are absent in at least one strain), and three belong to the C. maltaromaticum core genome (Table 3 and Supplementary Table S1). The WxL proteins belong to the cell-surface complex (Csc) protein family. The Csc protein encoding genes are typically clustered and each cluster contains at least on copy of cscA, cscB, cscC, and cscD. Similarly, C. maltaromaticum and C. divergens contain between 13 and 17 csc clusters. Typically, CscA contains a DUF916 domain with extracellular matrix binding ability (Galloway-Peña et al., 2015) and a C-terminal transmembrane anchor, while CscB and CscC contain WxL domains, and CscD is a small LPXTG protein (Siezen et al., 2006). Similarly, in C. maltaromaticum and C. divergens, all the WxL encoding genes are either cscB or cscC homologs, and are localized in the vicinity of at least one WxL encoding gene. These cscB and cscC homologs can also be clustered with homologs of cscA and cscD. Strikingly, the clusters can encode a high number of WxL proteins, as exemplified by the cluster BN424_324-BN424_330 which is predicted to encode six WxL proteins.
Whereas C. maltaromaticum and C. divergens are predicted to produce a high diversity of non-covalently bound proteins, only a small number of such proteins were found in C. inhibens subsp. gilichinskyi WN1359, Carnobacterium sp. AT7, and Carnobacterium sp 17. Among those, most are predicted as LysM- and SH3-containing proteins and no WxL proteins were detected (Table 3 and Supplementary Table S1).
Sortase-dependent proteins (SDP) are covalently anchored to the cell wall, and possess an LPxTG like motif at their C-terminal end. SDP nomenclature refers to proteins attached to the peptidoglycan by the sortase family of transpeptidases (Schneewind and Missiakas, 2014). Such SDP were found in C. maltaromaticum strains and C. divergens V41 whereas only one LPxTG domain protein was detected in Carnobacterium sp. 17.4 and Carnobacterium sp. AT7, and none in C. inhibens subsp. gilichinskyi WN1359 (Table 3 and Supplementary Table S1).
Sortases decorate the surfaces of Gram-positive bacteria with diverse proteins that enable microbes to interact with their environment (Comfort and Clubb, 2004; Maresso and Schneewind, 2008). The five C. maltaromaticum strains and C. divergens V41 possess many putative sortase A and B genes, while Carnobacterium sp. AT7 and Carnobacterium sp. 17.4 harbor only one and C. inhibens subsp. gilichinskyi WN1359 possesses a pseudogene that might encode a remnant protein with similarities with sortases (Table 3 and Supplementary Table S1).
Depending on the strains, the C. maltaromaticum genomes are predicted to encode 26–35 SDP, and 26 putative SDP could be predicted in C. divergens. Among those, 13 belong to the C. maltaromaticum core genome, including seven also conserved in C. divergens. In addition, 24 C. maltaromaticum SDP-encoding genes are strain specific or shared by only some of the strains. The differences between strains resulted either from the absence of homologs or the presence of 373 predicted pseudogenes. The predicted sortase-encoding gene in the genome of Carnobacterium sp 17.4 is located next to a collagen-binding surface protein encoding gene (ABHHv1_120049, Supplementary Table S1).
As C. maltaromaticum and, to a lesser extent, C. divergens presented a large panel of surface-exposed proteins compared to other Carnobacterium species, we searched for the putative functions of the C. maltaromaticum and C. divergens species specific proteins to illuminate the potential benefits they might provide to these two species.
Homologs of multidomain proteins predicted as nucleotidases/metallophosphatases as well as PrtB homologs are among the proteins conserved in C. maltaromaticum and C. divergens (Figure 4 and Supplementary Table S1). Extracellular 5′-nucleotidase domains that catalyze dephosphorylation of exogenous adenine 5′-nucleotides to adenosine and phosphate (Bengis-Garber and Kushner, 1982) are reported as providing a key function for phosphorous regeneration in aquatic ecosystems (Ammerman and Azam, 1985).
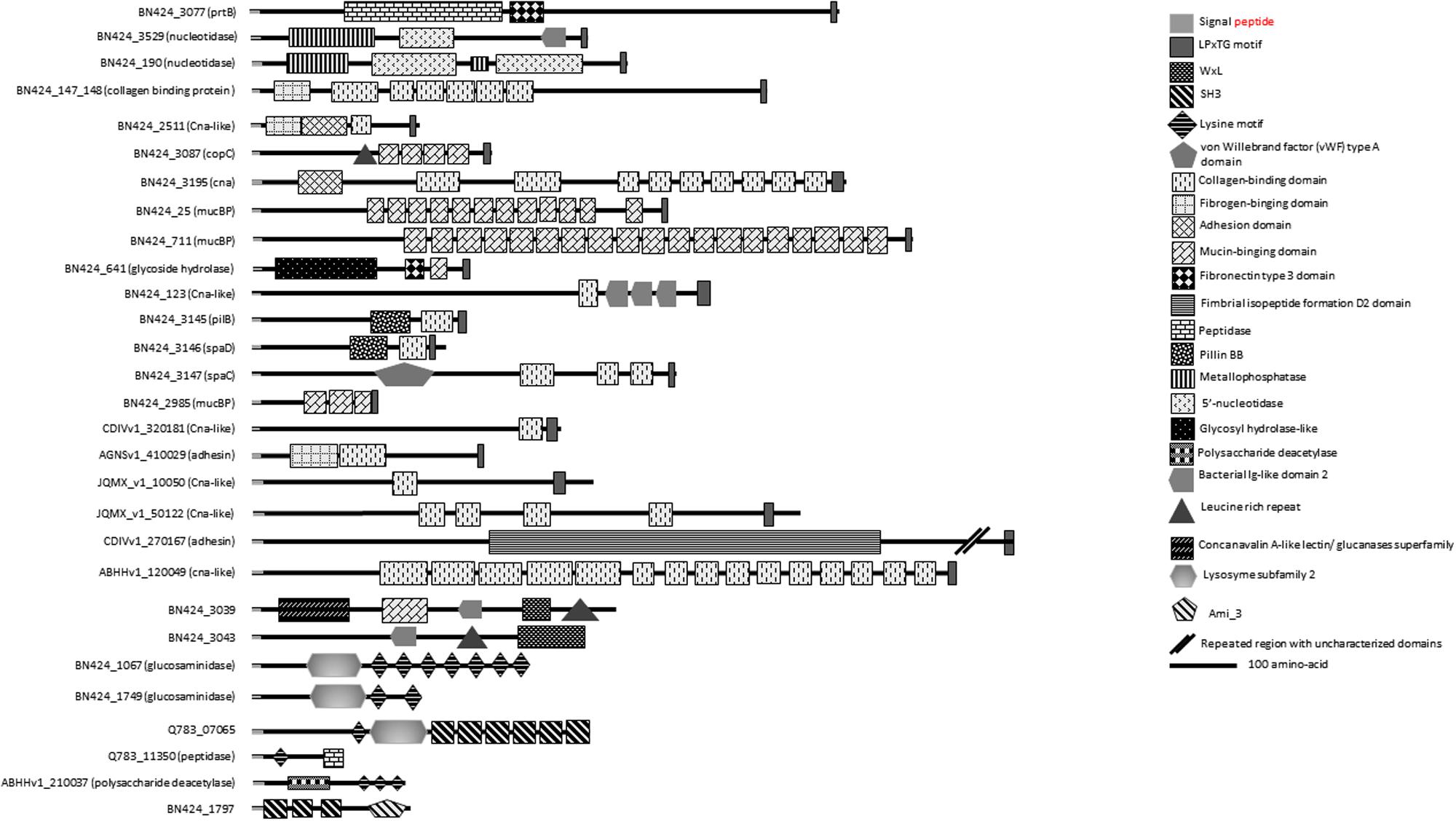
FIGURE 4. Putative surface proteins containing predicted functional domains.
In the dairy LAB Lactobacillus delbrueckii subsp. bulgaricus, PrtB, a cell envelope-associated protease (CEP) has been shown to degrade casein into peptides (Siezen, 1999). Peptides and aas are subsequently internalized and peptides are further hydrolyzed by intracellular peptidases into small peptides and free aa (Savijoki et al., 2006). The analysis of the genomes of C. maltaromaticum and C. divergens revealed the conserved presence of oligopeptides transporter systems OppABCDF and DtpT, as well as intracellular peptidases (Supplementary Table S1). Interestingly, homologs of the Opp system and of intracellular peptidases were also found in Carnobacterium sp. 17.4, Carnobacterium sp. AT7, and C. inhibens subsp. gilichinskyi WN1359 but no homologs of CEP and no DtpT. In the dairy environment, CEP are believed to confer a selective advantage by allowing bacteria to exploit the aa from milk casein (Kunji et al., 1996; Christensen et al., 1999; Doeven et al., 2005; Savijoki et al., 2006). All these data strongly suggest that among Carnobacterium only C. maltaromaticum and C. divergens are indeed able to exploit aa from the proteins present in their environments. The presence of PrtB would be a selective advantage for these two species in protein-rich environments such as food.
Among the SDP proteins present in several C. maltaromaticum strains we noticed a CDS predicted as harboring a glycoside hydrolase activity (BN424_641, Figure 4 and Supplementary Table S1). This multidomain protein also contain a fibronectin type III-like module of unknown function, which is usually associated with glycoside hydrolase domains (Alahuhta et al., 2010). Such enzymatic activity could allow C. maltaromaticum to degrade extracellular carbohydrate polymers. Interestingly, this protein is also predicted to contain a mucin-binding domain. Mucin is the major component of the intestinal mucus. It is tempting to speculate that the putative mucin-binding glycoside hydrolase of C. maltaromaticum would hydrolyze mucin glycan moieties as has been described for some gut bacteria (Tailford et al., 2015).
Two SDP, predicted as heme-binding proteins in C. maltaromaticum and C. divergens, are homologs of IsdA and IsdC (Figure 5 and Supplementary Table S1). The Isd system in Staphylococcus aureus enables to utilize different sources of heme: free heme, heme bound to free hemoglobin, and heme bound to hemoglobin interacting with haptoglobin. The Isd ABC-transport system in S. aureus is encoded by isdABCDEFGH; IsdH, IsdB, and IsdA acting as cell wall anchored receptor proteins: IsdH is the primary haptoglobin-hemoglobin receptor, IsdB the primary hemoglobin receptor, and IsdA can bind free heme or accept heme from IsdB or IsdH. Heme is subsequently transferred from IsdA to IsdC, another cell wall protein and then to a membrane associated transporter, formed by IsdD, IsdE, and IsdF. After internalization, heme is taken up by the heme degrading monooxygenase IsdG that releases the resulting iron in the cytoplasm (Choby and Skaar, 2016). At first glance, among the possible protein candidates able to bind heme from the environment, C. maltaromaticum and C. divergens would only have IsdA as no ortholog of IsdB and IsdH were found. This would suggest that these two Carnobacterium species would primary be able to use free heme as iron source and not heme bound to proteins. However, the IsdA homolog in C. maltaromaticum is predicted to contain four near-iron transporter (NEAT) domains while IsdA from S. aureus contains only two. NEAT domains bind heme compounds or proteins containing heme compound (Gaudin et al., 2011; Balderas et al., 2012). It could therefore be speculated that the presence of two additional NEAT domains in the C. maltaromaticum IsdA homolog might compensate the absence of IsdB.
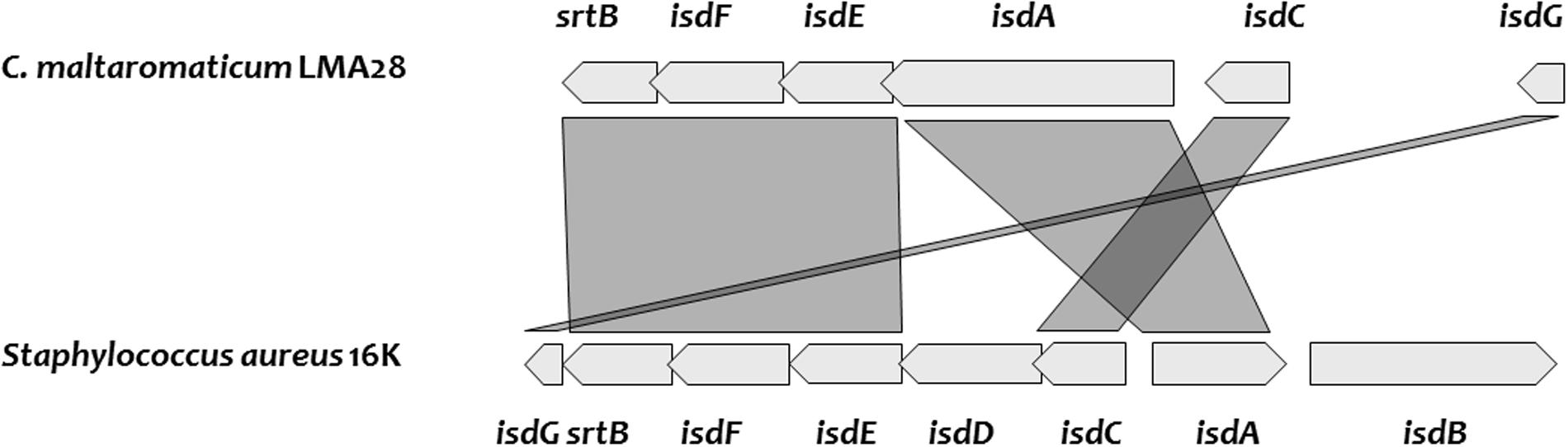
FIGURE 5. Genes encoding heme uptake system, cluster organization, and synteny between C. maltaromaticum LMA28 and Staphylococcus aureus 16K.
Carnobacterium divergens and C. maltaromaticum do not exhibit any IsdD homolog. However, we found two fhuC homologs which encode an ATP-binding component of ABC transporter as well. These FhuC homologs could play the equivalent role of IsdD and thus build a functional ABC transporter with the permease encoded by the IsdF homolog. In C. divergens V41, one fhuC is localized in the vicinity of the putative isd genes, while in the C. maltaromaticum strains, this homolog is localized elsewhere in the genome. In addition, all the analyzed genomes of Carnobacterium contain an isdG homolog and would be able to release iron after heme import in the cytoplasm. The isdA homolog of C. maltaromaticum ML1.97 and 3.18 appear as a pseudogene, indicating that this system is not fully functional in these two strains. Overall, these analyses suggest C. divergens V41 and several strains if not all of C. maltaromaticum would be able to use extracellular heme, for respiration (see below) and/or as an iron source.
The previous analysis of the genome of C. maltaromaticum LMA28 reported the presence of putative cell-surface adhesins (Rahman et al., 2014b). Comparison of Carnobacterium genomes revealed that C. maltaromaticum strains and C. divergens V41 may produce several surface proteins predicted to contain domains reminiscent of adhesion function: collagen-binding, mucBP, Leucine-Rich Repeat (LRR).
Among those, 10 LPxTG proteins are predicted to have a collagen-binding domain (Supplementary Table S1 and Figure 4). Functional domains are annotated collagen-binding protein, Cna, or bacterial adhesin. Collagen-binding proteins found in S. aureus (Elasri et al., 2002) and Listeria monocytogenes (Bierne and Cossart, 2007) are suggested to participate in the infection process. However, proteins putatively involved in adhesion to collagen and mucin have also been reported to be important for the probiotic properties of LAB as shown in Lactobacillus plantarum WCFS1 (Boekhorst et al., 2006b). It is therefore difficult to predict if the binding capacity of such proteins in Carnobacterium can be considered as beneficial or not.
Three LPxTG proteins (Figure 4 and Supplementary Table S2) contain a MucBP (mucin-binding protein) domain. MucBP domains allow adhesion to mucus material (Lukić et al., 2012). Lactobacillales and Listeria species can possess between 1 and 14 MucBP-containing proteins (Boekhorst et al., 2006a; Bierne and Cossart, 2007).
In Enterococcus faecium, the WxL protein SwpA and the protein DufA, which contains a DUF916 domain and is encoded by a gene that belong to a csc cluster, are collagen and fibronectin-binding proteins (Galloway-Peña et al., 2015). Similarly, it can be hypothesized that at least some WxL and DUF916 likely encoded by C. divergens and C. maltaromaticum might exhibit similar adhesion properties. In addition, two LRR domains were found in some strains of C. maltaromaticum holding a WxL anchorage domain (Figure 4 and Supplementary Table S2). These domains are involved in protein–protein interactions. They are described to be associated with domains exhibiting an Ig-like (immunoglobulin) fold, the function of which is thought to facilitate the presentation of the adjacent LRR domain (Bierne and Cossart, 2007). Accordingly, near the LRR domain of the putative C. maltaromaticum LMA28 surface protein BN424_3043, an Ig-like domain was found at the C-terminal end of the LRR region (Figure 4 and Supplementary Table S2). In L. monocytogenes, internalins associated to virulence are LRR-containing proteins. LRR-containing proteins are rather uncommon in Lactobacillus.
The presence of such putative adhesins suggests that C. maltaromaticum and C. divergens exhibit the ability to adhere to intestinal mucosa and extracellular matrices of animals. Overall, 19 putative adhesins were predicted from the genomes of C. maltaromaticum and C. divergens and absent from the other Carnobacterium species. All these proteins are SDP except two LRR-containing proteins which exhibit a WxL binding domain. Of these 19 proteins, two are conserved in all C. maltaromaticum strains and absent in other Carnobacterium: a putative collagen-binding SDP and a mucin-binding protein.
Genes encoding the pili proteins previously described for C. maltaromaticum LMA28 (Rahman et al., 2014b), were found only in one other C. maltaromaticum strain (DSM20342 MX5). Both strains belong to clonal complex CC1, which is suspected to be a lineage well-adapted to the dairy environment. Pili were described as surface components promoting adhesion to dairy matrix in the probiotic strain L. rhamnosus GG and thereby they might contribute to confer an advantage in dairy products (Burgain et al., 2014). In general in Gram-positive bacteria, two or three genes encoding the pilus subunits are organized into an operon, along with at least one sortase gene (Mandlik et al., 2008; Proft and Baker, 2009). The closest homologs of such C. maltaromaticum genes were found in E. faecalis, with also a highly similar genetic organization (Figure 6). However, pilin gene organization is different between C. maltaromaticum and L. rhamnosus GG suggesting that the genetic structures of pili loci in C. maltaromaticum, E. faecalis V583, and L. rhamnosus GG are the result of different gene rearrangements.
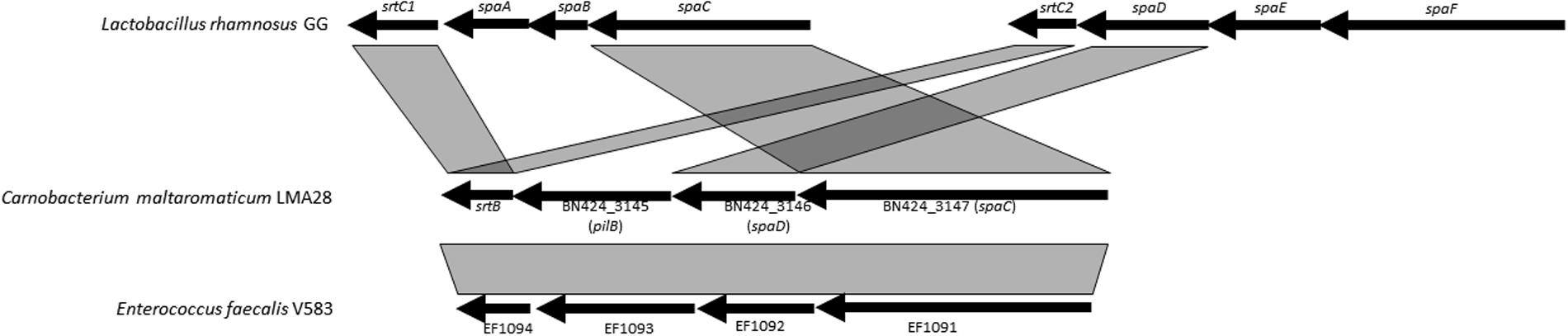
FIGURE 6. Genetic organization and synteny of pili synthesis genes between C. maltaromaticum LMA28, Lactobacillus rhamnosus GG, and Enterococcus faecalis V583.
Overall, these comparative genomic analyses suggest that all C. maltaromaticum strains and C. divergens might have adhesive properties and that strains might exhibit differences in this regard.
Further, these analyses demonstrated striking differences between the group C. maltaromaticum/C. divergens and the other Carnobacterium spp. The secretome and more specifically the cell-surface proteome of C. maltaromaticum and C. divergens are large, the one of C. maltaromaticum being the largest described for LAB as previously noticed by Sun et al. (2015). Conversely the secretome of C. inhibens subsp. gilichinskyi WN1359, Carnobacterium sp. AT7, and Carnobacterium sp. 17.4 is the smallest among LAB. The detailed analysis of the functions provided by such large secretome supports a prediction that the cell-surface proteome would confer the ability to hydrolyze and to adhere to biomacromolecules as well as to capture biomolecules (heme compounds). The cell surface of C. maltaromaticum and C. divergens might closely interact with animal environments and use these nutrient-rich substrates. In addition, C. maltaromaticum would likely exhibit a larger repertoire of hydrolytic enzymes and adhesins that may enable C. maltaromaticum to adapt to multiple habitats. By contrast, the other Carnobacterium lack such a cell-surface proteome and would therefore be expected to be less able to colonize animal-linked habitats. The size of the secretome is highly variable in LAB. Interestingly, all known specialized LAB Streptococcus thermophilus, L. iners, L. reuteri, L. sanfranciscensis, and L. fructivorans are characterized by a small secretome, suggesting that colonizing one specific niche, such as dairy, vagina, sourdough, or the GI tract, respectively, does not require a large secretome. Consistently, the known generalist LAB such as L. rhamnosus, L. plantarum (Martino et al., 2016), and some Enterococcus spp. exhibit a large secretome. We propose that there is an intimate relationship between the secretome size and the ability of LAB to colonize diverse habitats. According to this hypothesis, the bigger a secretome, the higher the capacity to colonize multiple environments.
Carnobacterium maltaromaticum is unable to synthesize heme and exhibits better growth efficiency in the presence of hematin, suggesting that heme is used by C. maltaromaticum to respire oxygen. Consistent with this hypothesis, C. maltaromaticum has been reported to produce cytochrome b and d types when grown aerobically with hematin (Meisel et al., 1994). The gene repertoire of C. maltaromaticum suggests the ability to produce a functional respiratory chain. Indeed, the electron donor-encoding genes, noxB and ndh, and those required for the synthesis of the electron shuttle menaquinone (yhdB, menE, menB, menD, menF, ispA, ispB, and menA) were present. Further, cydABCD, encoding the heme-dependent cytochrome quinol oxidase that performs the final electron transfer to the acceptor oxygen (Lechardeur et al., 2011), were also found. These genes were present in all C. maltaromaticum strains and in C. divergens, except in C. maltaromaticum ML.1.97 whose menF appears to be a pseudogene. Therefore, this last strain might require the presence of quinone in the environment to respire, as reported for Streptococcus agalactiae (Rezaïki et al., 2008). It seems therefore that the components of the cell wall proteome involved in extracellular heme utilization may also contribute to oxygen respiration in C. maltaromaticum and C. divergens.
By contrast, only menA, noxA/noxB, and ispB were found in Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and Carnobacterium inhibens subsp. gilichinskyi WN1359 strongly suggesting that these three bacteria do not possess any functional respiratory chain.
Bacteria have to deal with several stresses in order to survive in the GI tract, including the immune system and bile. All strains of C. maltaromaticum and C. divergens contain genes conferring resistance to some components of the immune system. Indeed, homologs were found for mprF, dltABCD, asnH/asnB, oatA, and pgdA (Supplementary Table S2). The genes mprF and dltABCD are involved in phospholipid lysinylation and teichoic acid D-alanylation, respectively, and thus confer resistance to antimicrobial peptides of the innate immune system by protecting the cell wall. The genes oatA and pgdA confer to peptidoglycan a high resistance to lysozyme, another component of the innate immune system, by introducing O-acetylation and N-deacetylation, respectively. Importantly, no mprF, dltABCD, asnH/asnB, oatA, and pgdA homologs were found in the genomes of Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and Carnobacterium inhibens subsp. gilichinskyi WN1359.
Bile salt hydrolase encoding genes were conserved in all strains of C. maltaromaticum while none were found in other Carnobacteria including C. divergens V41 (Supplementary Table S2).
Carnobacterium maltaromaticum and C. divergens are thus highly contrasting from the three other Carnobacterium taxons by their content of genes described in Lactobacillus as key factor for survival in the GI tract of animals (Kleerebezem et al., 2010). Indeed, while C. maltaromaticum and C. divergens possess genes putatively conferring resistance against the innate immune system, almost none of these homologs where found in Carnobacterium sp. AT7, Carnobacterium sp. 17.4, and C. inhibens subsp. gilichinskyi WN1359. This might explain why the only two species identified in feces samples are C. maltaromaticum and C. divergens. However, surprisingly, genes allowing resistance to bile were only found in C. maltaromaticum and not in C. divergens. Yet the ability to hydrolyze bile is described as a key factor for colonization of the gut (Kleerebezem et al., 2010; Seedorf et al., 2014). Indeed C. maltaromaticum LMA28, a cheese isolate that possesses such genes, is able to survive during the gastrointestinal transit in a mouse model (Rahman et al., 2014b; Sun et al., 2015). However, such ability has not yet been tested in C. divergens V41. Whether these two species differ in their ability to deal with bile and the ecological consequences it has on GI tract survival deserves further investigation.
Performed 16S meta-barcoding: BT and GD. Performed whole genome sequencing and assembly: JL, MH, and SS. Comparative genome analyses: CI, CC-G, FB, A-MR-J, MZ, and BR. Wrote the manuscript: CI, CC-G, BT, MZ, BR, JL, CM, FB, and A-MR-J. Coordinated the study: FB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The LABGeM (CEA/IG/Genoscope & CNRS UMR8030) and the France Génomique National infrastructure (funded as part of Investissement d’avenir program managed by Agence Nationale pour la Recherche, contract ANR-10-INBS-09) are acknowledged for support within the MicroScope annotation platform. They are also thankful to Myriam Michelle, Sylvie Wolff, Arnaud Khemisti, and Camille Collin for their technical support during this study. The authors would like to thank the reviewers for their helpful and constructive comments that greatly contributed to improving the final version of the paper.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00357/full#supplementary-material
Alahuhta, M., Xu, Q., Brunecky, R., Adney, W. S., Ding, S.-Y., Himmel, M. E., et al. (2010). Structure of a fibronectin type III-like module from Clostridium thermocellum. Acta Cryst. F 66, 878–880. doi: 10.1107/S1744309110022529
Ammerman, J. W., and Azam, F. (1985). Bacterial 5-nucleotidase in aquatic ecosystems: a novel mechanism of phosphorus regeneration. Science 227, 1338–1340. doi: 10.1126/science.227.4692.1338
Balderas, M. A., Nobles, C. L., Honsa, E. S., Alicki, E. R., and Maresso, A. W. (2012). Hal is a Bacillus anthracis heme acquisition protein. J. Bacteriol. 194, 5513–5521. doi: 10.1128/JB.00685-12
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Bengis-Garber, C., and Kushner, D. J. (1982). Role of membrane-bound 5′-nucleotidase in nucleotide uptake by the moderate halophile Vibrio costicola. J. Bacteriol. 149, 808–815.
Bierne, H., and Cossart, P. (2007). Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol. Mol. Biol. Rev. 71, 377–397. doi: 10.1128/MMBR.00039-06
Boekhorst, J., Helmer, Q., Kleerebezem, M., and Siezen, R. J. (2006a). Comparative analysis of proteins with a mucus-binding domain found exclusively in lactic acid bacteria. Microbiology 152, 273–280.
Boekhorst, J., Wels, M., Kleerebezem, M., and Siezen, R. J. (2006b). The predicted secretome of Lactobacillus plantarum WCFS1 sheds light on interactions with its environment. Microbiology 152, 3175–3183.
Brinster, S., Furlan, S., and Serror, P. (2007). C-terminal WxL domain mediates cell wall binding in Enterococcus faecalis and other gram-positive bacteria. J. Bacteriol. 189, 1244–1253. doi: 10.1128/JB.00773-06
Burgain, J., Scher, J., Lebeer, S., Vanderleyden, J., Cailliez-Grimal, C., Corgneau, M., et al. (2014). Significance of bacterial surface molecules interactions with milk proteins to enhance microencapsulation of Lactobacillus rhamnosus GG. Food Hydrocoll. 41, 60–70. doi: 10.1016/j.foodhyd.2014.03.029
Cailliez-Grimal, C., Afzal, M. I., and Revol-Junelles, A.-M. (2014). “Carnobacterium,” in Encyclopedia of Food Microbiology, eds C. A. Batt and M. L. Tortorello (Cambridge, MA: Academic Press), 379–383. doi: 10.1016/B978-0-12-384730-0.00381-5
Cailliez-Grimal, C., Chaillou, S., Anba-Mondoloni, J., Loux, V., Afzal, M. I., Rahman, A., et al. (2013). Complete chromosome sequence of Carnobacterium maltaromaticum LMA 28. Genome Announc. 1:e00115-12. doi: 10.1128/genomeA.00115-12
Caspi, R., Altman, T., Billington, R., Dreher, K., Foerster, H., Fulcher, C. A., et al. (2014). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 42, D459–D471. doi: 10.1093/nar/gkt1103
Chaillou, S., Chaulot-Talmon, A., Caekebeke, H., Cardinal, M., Christieans, S., Denis, C., et al. (2015). Origin and ecological selection of core and food-specific bacterial communities associated with meat and seafood spoilage. ISME J. 9, 1105–1118. doi: 10.1038/ismej.2014.202
Choby, J. E., and Skaar, E. P. (2016). Heme synthesis and acquisition in bacterial pathogens. J. Mol. Biol. 428, 3408–3428. doi: 10.1016/j.jmb.2016.03.018
Christensen, J. E., Dudley, E. G., Pederson, J. A., and Steele, J. L. (1999). Peptidases and amino acid catabolism in lactic acid bacteria. Antonie Van Leeuwenhoek 76, 217–246. doi: 10.1023/A:1002001919720
Comfort, D., and Clubb, R. T. (2004). A comparative genome analysis identifies distinct sorting pathways in Gram-positive bacteria. Infect. Immun. 72, 2710–2722. doi: 10.1128/IAI.72.5.2710-2722.2004
Delorme, C., Bartholini, C., Bolotine, A., Ehrlich, S. D., and Renault, P. (2010). Emergence of a cell wall protease in the Streptococcus thermophilus population. Appl. Environ. Microbiol. 76, 451–460. doi: 10.1128/AEM.01018-09
Doeven, M. K., Kok, J., and Poolman, B. (2005). Specificity and selectivity determinants of peptide transport in Lactococcus lactis and other microorganisms: peptide transport in Lactococcus lactis. Mol. Microbiol. 57, 640–649. doi: 10.1111/j.1365-2958.2005.04698.x
Douglas, G. L., and Klaenhammer, T. R. (2010). Genomic evolution of domesticated microorganisms. Annu. Rev. Food Sci. Technol. 1, 397–414. doi: 10.1146/annurev.food.102308.124134
Douillard, F. P., and de Vos, W. M. (2014). Functional genomics of lactic acid bacteria: from food to health. Microb. Cell Fact. 13:S8. doi: 10.1186/1475-2859-13-S1-S8
Douillard, F. P., Ribbera, A., Kant, R., Pietila, T. E., Jarvinen, H. M., Messing, M., et al. (2013). Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 9:e1003683. doi: 10.1371/journal.pgen.1003683
Duan, S., Hu, X., Li, M., Miao, J., Du, J., and Wu, R. (2016). Composition and metabolic activities of the bacterial community in shrimp sauce at the flavor-forming stage of fermentation as revealed by metatranscriptome and 16S rRNA gene sequencing. J. Agric. Food Chem. 64, 2591–2603. doi: 10.1021/acs.jafc.5b05826
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Elasri, M. O., Thomas, J. R., Skinner, R. A., Blevins, J. S., Beenken, K. E., Nelson, C. L., et al. (2002). Staphylococcus aureus collagen adhesin contributes to the pathogenesis of osteomyelitis. Bone 30, 275–280. doi: 10.1016/S8756-3282(01)00632-9
Fougy, L., Desmonts, M.-H., Coeuret, G., Fassel, C., Hamon, E., Hézard, B., et al. (2016). Reducing salt in raw pork sausages increases spoilage and correlates with reduced bacterial diversity. Appl. Environ. Microbiol. 82, 3928–3939. doi: 10.1128/AEM.00323-16
Frese, S. A., Benson, A. K., Tannock, G. W., Loach, D. M., Kim, J., Zhang, M., et al. (2011). The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLOS Genet. 7:e1001314. doi: 10.1371/journal.pgen.1001314
Galloway-Peña, J. R., Liang, X., Singh, K. V., Yadav, P., Chang, C., Rosa, S. L. L., et al. (2015). The identification and functional characterization of WxL proteins from Enterococcus faecium reveal surface proteins involved in extracellular matrix interactions. J. Bacteriol. 197, 882–892. doi: 10.1128/JB.02288-14
Gaudin, C. F. M., Grigg, J. C., Arrieta, A. L., and Murphy, M. E. P. (2011). Unique heme-iron coordination by the hemoglobin receptor IsdB of Staphylococcus aureus. Biochemistry 50, 5443–5452. doi: 10.1021/bi200369p
Hiu, S. F., Holt, R. A., Sriranganathan, N., Seidler, R. J., and Fryer, J. L. (1984). Lactobacillus piscicola, a new species from salmonid fish. Int. J. Syst. Bacteriol. 34, 393–400. doi: 10.1099/00207713-34-4-393
Iskandar, C. F., Cailliez-Grimal, C., Rahman, A., Rondags, E., Remenant, B., Zagorec, M., et al. (2016). Genes associated to lactose metabolism illustrate the high diversity of Carnobacterium maltaromaticum. Food Microbiol. 58, 79–86. doi: 10.1016/j.fm.2016.03.008
Jääskeläinen, E., Hultman, J., Parshintsev, J., Riekkola, M.-L., and Björkroth, J. (2016). Development of spoilage bacterial community and volatile compounds in chilled beef under vacuum or high oxygen atmospheres. Int. J. Food Microbiol. 223, 25–32. doi: 10.1016/j.ijfoodmicro.2016.01.022
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kleerebezem, M., Hols, P., Bernard, E., Rolain, T., Zhou, M., Siezen, R. J., et al. (2010). The extracellular biology of the Lactobacilli. FEMS Microbiol. Rev. 34, 199–230. doi: 10.1111/j.1574-6976.2009.00208.x
Kunji, E. R., Mierau, I., Hagting, A., Poolman, B., and Konings, W. N. (1996). The proteolytic systems of lactic acid bacteria. Antonie Van Leeuwenhoek 70, 187–221. doi: 10.1007/BF00395933
Lauro, F. M., Chastain, R. A., Blankenship, L. E., Yayanos, A. A., and Bartlett, D. H. (2007). The unique 16S rRNA genes of piezophiles reflect both phylogeny and adaptation. Appl. Environ. Microbiol. 73, 838–845. doi: 10.1128/AEM.01726-06
Laursen, B. G., Bay, L., Cleenwerck, I., Vancanneyt, M., Swings, J., Dalgaard, P., et al. (2005). Carnobacterium divergens and Carnobacterium maltaromaticum as spoilers or protective cultures in meat and seafood: phenotypic and genotypic characterization. Syst. Appl. Microbiol. 28, 151–164. doi: 10.1016/j.syapm.2004.12.001
Lechardeur, D., Cesselin, B., Fernandez, A., Lamberet, G., Garrigues, C., Pedersen, M., et al. (2011). Using heme as an energy boost for lactic acid bacteria. Curr. Opin. Biotechnol. 22, 143–149. doi: 10.1016/j.copbio.2010.12.001
Leisner, J. J., Hansen, M. A., Larsen, M. H., Hansen, L., Ingmer, H., and Sørensen, S. J. (2012). The genome sequence of the lactic acid bacterium, Carnobacterium maltaromaticum ATCC 35586 encodes potential virulence factors. Int. J. Food Microbiol. 152, 107–115. doi: 10.1016/j.ijfoodmicro.2011.05.012
Leisner, J. J., Laursen, B. G., Prévost, H., Drider, D., and Dalgaard, P. (2007). Carnobacterium: positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 31, 592–613. doi: 10.1111/j.1574-6976.2007.00080.x
Leonard, M. T., Panayotova, N., Farmerie, W. G., Triplett, E. W., and Nicholson, W. L. (2013). Complete genome sequence of Carnobacterium gilichinskyi strain WN1359T (DSM 27470T). Genome Announc. 1:e00985-13. doi: 10.1128/genomeA.00985-13
Lorca, G. L., Barabote, R. D., Zlotopolski, V., Tran, C., Winnen, B., Hvorup, R. N., et al. (2007). Transport capabilities of eleven gram-positive bacteria: comparative genomic analyses. Biochim. Biophys. Acta 1768, 1342–1366. doi: 10.1016/j.bbamem.2007.02.007
Lukić, J., Strahinić, I., Jovčić, B., Filipić, B., Topisirović, L., Kojić, M., et al. (2012). Different roles for lactococcal aggregation factor and mucin binding protein in adhesion to gastrointestinal mucosa. Appl. Environ. Microbiol. 78, 7993–8000. doi: 10.1128/AEM.02141-12
Macklaim, J. M., Gloor, G. B., Anukam, K. C., Cribby, S., and Reid, G. (2011). At the crossroads of vaginal health and disease, the genome sequence of Lactobacillus iners AB-1. Proc. Natl. Acad. Sci. U.S.A. 108, 4688–4695. doi: 10.1073/pnas.1000086107
Mandlik, A., Das, A., and Ton-That, H. (2008). The molecular switch that activates the cell wall anchoring step of pilus assembly in gram-positive bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 14147–14152. doi: 10.1073/pnas.0806350105
Maresso, A. W., and Schneewind, O. (2008). Sortase as a target of anti-infective therapy. Pharmacol. Rev. 60, 128–141. doi: 10.1124/pr.107.07110
Martino, M. E., Bayjanov, J. R., Caffrey, B. E., Wels, M., Joncour, P., Hughes, S., et al. (2016). Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 18, 4974–4989. doi: 10.1111/1462-2920.13455
Meisel, J., Wolf, G., and Hammes, W. P. (1994). Heme-dependent cytochrome formation in Lactobacillus maltaromicus. Syst. Appl. Microbiol. 17, 20–23. doi: 10.1016/S0723-2020(11)80026-3
Mendes-Soares, H., Suzuki, H., Hickey, R. J., and Forney, L. J. (2014). Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal Lactobacilli to their environment. J. Bacteriol. 196, 1458–1470. doi: 10.1128/JB.01439-13
Miller, A., Morgan, M. E., and Libbey, L. M. (1974). Lactobacillus maltaromicus, a new species producing a malty aroma. Int. J. Syst. Bacteriol. 24, 346–354. doi: 10.1099/00207713-24-3-346
Millière, J. B., Michel, M., Mathieu, F., and Lefebvre, G. (1994). Presence of Carnobacterium spp. in French surface mould-ripened soft-cheese. J. Appl. Bacteriol. 76, 264–269. doi: 10.1111/j.1365-2672.1994.tb01626.x
Nicholson, W. L., Zhalnina, K., de Oliveira, R. R., and Triplett, E. W. (2015). Proposal to rename Carnobacterium inhibens as Carnobacterium inhibens subsp inhibens subsp nov and description of Carnobacterium inhibens subsp gilichinskyi subsp nov., a psychrotolerant bacterium isolated from Siberian permafrost. Int. J. Syst. Evol. Microbiol. 65, 556–561. doi: 10.1099/ijs.0.067983-0
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Meth. 8, 785–786. doi: 10.1038/nmeth.1701
Pikuta, E. V. (2014). The Family Carnobacteriaceae, eds W. H. Holzapfel and B. J. B. Wood (Oxford: Blackwell Science Publishers). doi: 10.1002/9781118655252.part2
Pikuta, E. V., and Hoover, R. B. (2014). The Genus Carnobacterium, eds W. H. Holzapfel and B. J. B. Wood (Oxford: Blackwell Science Publishers). doi: 10.1002/9781118655252.ch10
Pilet, M. F., Dousset, X., Barré, R., Novel, G., Desmazeaud, M., and Piard, J. C. (1994). Evidence for two bacteriocins produced by Carnobacterium piscicola and Carnobacterium divergens isolated from fish and active against Listeria monocytogenes. J. Food Prot. 58, 256–262. doi: 10.4315/0362-028X-58.3.256
Proft, T., and Baker, E. N. (2009). Pili in Gram-negative and Gram-positive bacteria — structure, assembly and their role in disease. Cell. Mol. Life Sci. 66, 613–635. doi: 10.1007/s00018-008-8477-4
Rahman, A., Cailliez-Grimal, C., Bontemps, C., Payot, S., Chaillou, S., Revol-Junelles, A.-M., et al. (2014a). High genetic viversity among strains of the unindustrialized lactic acid bacterium Carnobacterium maltaromaticum in dairy products as revealed by multilocus sequence typing. Appl. Environ. Microbiol. 80, 3920–3929. doi: 10.1128/AEM.00681-14
Rahman, A., Gleinser, M., Lanhers, M.-C., Riedel, C. U., Foligne, B., Hanse, M., et al. (2014b). Adaptation of the lactic acid bacterium Carnobacterium maltaromaticum LMA 28 to the mammalian gastrointestinal tract: from survival in mice to interaction with human cells. Int. Dairy J. 34, 93–99. doi: 10.1016/j.idairyj.2013.07.003
Remenant, B., Borges, F., Cailliez-Grimal, C., Revol-Junelles, A.-M., Marché, L., Lajus, A., et al. (2016). Draft genome sequence of Carnobacterium divergens V41, a bacteriocin-producing strain. Genome Announc. 4:e01109-16. doi: 10.1128/genomeA.01109-16
Rezaïki, L., Lamberet, G., Derré, A., Gruss, A., and Gaudu, P. (2008). Lactococcus lactis produces short-chain quinones that cross-feed Group B Streptococcus to activate respiration growth. Mol. Microbiol. 67, 947–957. doi: 10.1111/j.1365-2958.2007.06083.x
Rodriguez, C., Taminiau, B., Brévers, B., Avesani, V., Van Broeck, J., Leroux, A., et al. (2015). Faecal microbiota characterisation of horses using 16 rdna barcoded pyrosequencing, and carriage rate of Clostridium difficile at hospital admission. BMC Microbiol. 15:181. doi: 10.1186/s12866-015-0514-5
Santagati, M., Campanile, F., and Stefani, S. (2012). Genomic diversification of enterococci in hosts: the role of the mobilome. Front. Microbiol. 3:95. doi: 10.3389/fmicb.2012.00095
Savijoki, K., Ingmer, H., and Varmanen, P. (2006). Proteolytic systems of lactic acid bacteria. Appl. Microbiol. Biotechnol. 71, 394–406. doi: 10.1007/s00253-006-0427-1
Schloss, P. D., and Handelsman, J. (2003). Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 14, 303–310. doi: 10.1016/S0958-1669(03)00067-3
Schneewind, O., and Missiakas, D. (2014). Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta 1843, 1687–1697. doi: 10.1016/j.bbamcr.2013.11.009
Seedorf, H., Griffin, N. W., Ridaura, V. K., Reyes, A., Cheng, J., Rey, F. E., et al. (2014). Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 159, 253–266. doi: 10.1016/j.cell.2014.09.008
Siezen, R., Boekhorst, J., Muscariello, L., Molenaar, D., Renckens, B., and Kleerebezem, M. (2006). Lactobacillus plantarum gene clusters encoding putative cell-surface protein complexes for carbohydrate utilization are conserved in specific gram-positive bacteria. BMC Genomics 7:126. doi: 10.1186/1471-2164-7-126
Siezen, R. J. (1999). Multi-domain, cell-envelope proteinases of lactic acid bacteria. Antonie Van Leeuwenhoek 76, 139–155. doi: 10.1023/A:1002036906922
Sun, C., Fukui, H., Hara, K., Kitayama, Y., Eda, H., Yang, M., et al. (2015). Expression of Reg family genes in the gastrointestinal tract of mice treated with indomethacin. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G736–G744. doi: 10.1152/ajpgi.00362.2014
Tailford, L. E., Crost, E. H., Kavanaugh, D., and Juge, N. (2015). Mucin glycan foraging in the human gut microbiome. Front. Genet. 6:81. doi: 10.3389/fgene.2015.00081
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013). MicroScope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647. doi: 10.1093/nar/gks1194
Voget, S., Klippel, B., Daniel, R., and Antranikian, G. (2011). Complete genome sequence of Carnobacterium sp 17-4. J. Bacteriol. 193, 3403–3404. doi: 10.1128/JB.05113-11
Keywords: lactic acid bacteria, Carnobacterium, 16S meta-barcoding, comparative genomic analyses, ecological differentiation
Citation: Iskandar CF, Borges F, Taminiau B, Daube G, Zagorec M, Remenant B, Leisner JJ, Hansen MA, Sørensen SJ, Mangavel C, Cailliez-Grimal C and Revol-Junelles A-M (2017) Comparative Genomic Analysis Reveals Ecological Differentiation in the Genus Carnobacterium. Front. Microbiol. 8:357. doi: 10.3389/fmicb.2017.00357
Received: 29 November 2016; Accepted: 21 February 2017;
Published: 08 March 2017.
Edited by:
Jennifer Ronholm, McGill University, CanadaReviewed by:
Cristian Botta, University of Turin, ItalyCopyright © 2017 Iskandar, Borges, Taminiau, Daube, Zagorec, Remenant, Leisner, Hansen, Sørensen, Mangavel, Cailliez-Grimal and Revol-Junelles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Borges, ZnJlZGVyaWMuYm9yZ2VzQHVuaXYtbG9ycmFpbmUuZnI=
†Present address: Benoît Remenant, Laboratoire de la Santé des Végétaux, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail, Angers, France
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.