 Elena Heusinger
Elena Heusinger Frank Kirchhoff
Frank Kirchhoff
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 14 February 2017
Sec. Virology
Volume 8 - 2017 | https://doi.org/10.3389/fmicb.2017.00198
This article is part of the Research Topic Highly mutable animal RNA viruses: adaptation and evolution View all 37 articles
The transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) plays a complex role during the replication of primate lentiviruses. On the one hand, NF-κB is essential for induction of efficient proviral gene expression. On the other hand, this transcription factor contributes to the innate immune response and induces expression of numerous cellular antiviral genes. Recent data suggest that primate lentiviruses cope with this challenge by boosting NF-κB activity early during the replication cycle to initiate Tat-driven viral transcription and suppressing it at later stages to minimize antiviral gene expression. Human and simian immunodeficiency viruses (HIV and SIV, respectively) initially exploit their accessory Nef protein to increase the responsiveness of infected CD4+ T cells to stimulation. Increased NF-κB activity initiates Tat expression and productive replication. These events happen quickly after infection since Nef is rapidly expressed at high levels. Later during infection, Nef proteins of HIV-2 and most SIVs exert a very different effect: by down-modulating the CD3 receptor, an essential factor for T cell receptor (TCR) signaling, they prevent stimulation of CD4+ T cells via antigen-presenting cells and hence suppress further induction of NF-κB and an effective antiviral immune response. Efficient LTR-driven viral transcription is maintained because it is largely independent of NF-κB in the presence of Tat. In contrast, human immunodeficiency virus type 1 (HIV-1) and its simian precursors have lost the CD3 down-modulation function of Nef and use the late viral protein U (Vpu) to inhibit NF-κB activity by suppressing its nuclear translocation. In this review, we discuss how HIV-1 and other primate lentiviruses might balance viral and antiviral gene expression through a tight temporal regulation of NF-κB activity throughout their replication cycle.
To allow efficient viral gene expression, replication, and spread, viral pathogens need to exploit the cellular transcriptional machinery. In some cases, they hijack exactly those transcription factors that are activated by the host response to infection to initiate antiviral immune responses. The interaction of human immunodeficiency virus type 1 (HIV-1) and related simian immunodeficiency viruses (SIVs) with the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) family of transcription factors provides a prime example for the hostile takeover of a key mediator of the immune response by viral pathogens (Hiscott et al., 2001; Chan and Greene, 2012). NF-κB is ubiquitously expressed and its dysregulation is associated with many pathologies including cancer, cardiovascular, pulmonary, and inflammatory diseases (Panday et al., 2016). NF-κB can be induced by multiple stimuli and regulates the expression of cellular genes involved in numerous processes, such as cell proliferation, DNA repair and cell differentiation or survival (Ghosh and Hayden, 2012; Napetschnig and Wu, 2013). Furthermore, NF-κB plays a key role in inflammation and the induction of innate and adaptive immune responses, including expression of interferon-stimulated genes (ISGs) representing a first line of defense against viral pathogens (Hayden et al., 2006; Vallabhapurapu and Karin, 2009; Pfeffer, 2011).
NF-κB has a complex role in HIV-1 replication and pathogenesis. ISGs exert numerous effector functions and may target almost every step of the retroviral replication cycle (Kluge et al., 2015; Colomer-Lluch et al., 2016). Thus, their induction by NF-κB transcription factors might have beneficial effects for the host by suppressing HIV-1 replication. However, induction of interferon and other cytokines also contributes to HIV-1-induced chronic and systemic inflammation that drives progression to AIDS. Thus, whether induction of interferon responses is beneficial or harmful in HIV-1 infection is a matter of debate (Doyle et al., 2015; Utay and Douek, 2016). Importantly, NF-κB is also critical for potent viral gene expression from the HIV-1 long terminal repeat (LTR) promoter that typically contains two adjacent NF-κB binding sites in its main enhancer region (Chan and Greene, 2012). Combinations of stimulatory drugs including NF-κB inducing agents, such as prostratin, are currently examined for their ability to activate the latent reservoirs of HIV-1 to render them susceptible for elimination (Jiang and Dandekar, 2015; Cary et al., 2016). Thus, NF-κB presents a target for therapeutic intervention both by suppressing its activation to prevent harmful inflammation (Chan and Greene, 2012) or by activating this transcription factor to stimulate the latent reservoirs of HIV-1 (Jiang and Dandekar, 2015).
Numerous studies have examined the effects of HIV-1 infection on NF-κB activity (reviewed in Chan and Greene, 2012). Altogether, the results were puzzling and frequently controversial. Recent evidence suggests that HIV-1 and other primate lentiviruses may have differential effects during the early and late stages of their replication cycle and tightly regulate NF-κB activity (Sauter et al., 2015). In the present review, we first briefly describe some basic aspects of NF-κB signaling and its interaction with the LTR promoter of HIV-1 and other primate lentiviruses. Subsequently, we discuss how these viruses might modulate NF-κB activity throughout their replication cycle to ensure efficient viral gene expression while minimizing the expression of antiviral genes.
NF-κB was initially discovered in the laboratory of David Baltimore because of its ability to bind the enhancer region of the immunoglobulin κ light-chain- in B cells (Sen and Baltimore, 1986). Since then, five mammalian NF-κB members, i.e., NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB and c-Rel, have been identified (Nabel and Verma, 1993). All of them share a highly conserved N-terminal Rel homology domain that is critical for dimerization and DNA binding, while three of them (p65, RelB, and c-Rel) contain an additional C-terminal transactivation domain. The remaining two factors (NF-κB1 and NF-κB2) are synthesized as large inactive precursors (p105 and p100) that need to undergo proteasomal degradation of their ankyrin repeat containing C-terminal region to generate the mature p50 and p52 NF-κB subunits, respectively (Karin and Ben-Neriah, 2000). DNA binding and transcriptional regulation requires dimerization of two subunits, with p50/p65 dimers being the most abundant active form. Many other dimer combinations were observed but not all of them act as transcriptional activators; e.g., p50/p50 and p52/p52 homodimers were reported to suppress NF-κB mediated transcription as they lack a transactivation domain (Zhong et al., 2002).
In unstimulated cells, NF-κB dimers are bound to so-called inhibitors of κB (IκBs) containing ankyrin repeats that mask nuclear localization signals (NLS) and thereby keep the NF-κB proteins sequestered in the cytoplasm. The p105 and p100 precursors also contain ankyrin repeats in their C-terminal parts. Thus, they inhibit NF-κB activity and may also be classified as IκB proteins. A multitude of stimuli including viral antigens, immune cells, mitogens and cytokines, specific cell surface receptors, such as the T cell receptor (TCR)-CD3 complex, IFN receptors, the interleukin 1 receptor (IL-1R), Toll-like receptors (TLRs), or tumor necrosis factor (TNF) receptors induce degradation of IκB proteins (Vallabhapurapu and Karin, 2009). The main mechanism (canonical pathway) involves activation of IκB kinase (IKK), a heterodimer composed of the catalytic IKKα and IKKβ subunits and a regulatory factor termed NEMO (NF-κB essential modulator), and subsequent phosphorylation of serine residues in IκB regulatory domains allowing its ubiquitination and proteasomal degradation (Figure 1). Degradation of IκB allows exposure of the NLS and thus entry of the NF-κB complex into the nucleus for DNA binding and transcriptional induction of genes containing the appropriate binding sites within their promoter regions. As outlined below, NF-κB activation induces immune responses as well as HIV-1 proviral gene expression. Notably, NF-κB also induces expression of IκBα, resulting in a negative autoregulatory feedback loop and thus oscillating levels of NF-κB activity (Nelson et al., 2004). More in depth reviews of NF-κB signaling and its regulation are provided by Oeckinghaus and Ghosh (2009), Skaug et al. (2009) and Vallabhapurapu and Karin (2009).
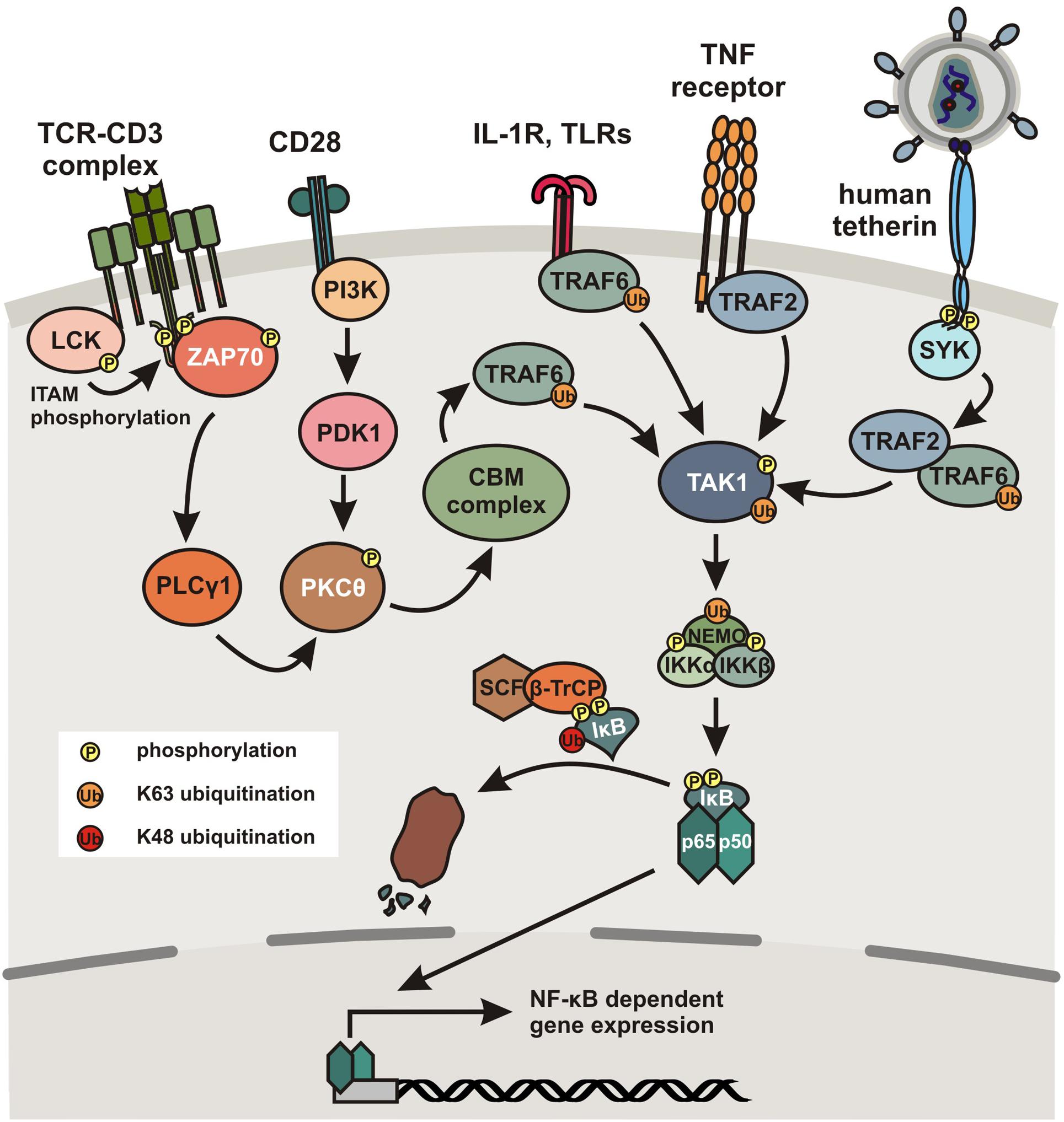
FIGURE 1. Activation of the canonical NF-κB pathway. Simplified schematic presentation of NF-κB activation by different cellular receptors. Binding of the TCR-CD3 complex to antigens presented by MHC molecules induces LCK-dependent phosphorylation of ITAMs in the cytoplasmic tail of CD3. This in turn recruits ZAP70, which induces a signal cascade involving activation of PLCγ1 and PKC𝜃. Alternatively, PKC𝜃 can also be activated via a signal cascade including PI3K following binding of the TCR costimulatory factor CD28 to a B7 ligand. PKC𝜃 subsequently activates the CBM complex consisting of CARD11, BCL10 and MALT1, which activates TAK1 in a TRAF6 dependent manner. Ligand binding to TLRs or TNF receptors also induces TAK1 activation in a TRAF6- or TRAF2-dependent manner, respectively. In humans (and to a much lesser extent in chimpanzees), the cellular restriction factor tetherin also acquired the ability to activate NF-κB. Upon tethering of newly formed viral particles and clustering of tetherin dimers, the cytoplasmic tail of tetherin becomes phosphorylated at a conserved YxY motif by a Scr-family kinase in a RICH2-dependent manner, recruiting SYK, which initiates a signaling cascade involving TRAF2 and/or TRAF6 resulting in the activation of TAK1. TAK1 subsequently phosphorylates IKKβ at two serine residues in the activation loop to activate IKK. Furthermore, TRAFs activate NEMO by poly-ubiquitination, followed by activation of the catalytic subunits IKKα and IKKβ. Subsequently, IκB is phosphorylated and poly-ubiquitinated by the SCF/β-TrCP complex, which results in its proteasomal degradation, thereby releasing p50/p65 heterodimers, which translocate to the nucleus where they bind to specific κB binding sites and initiate NF-κB-dependent gene expression.
It is well established that NF-κB transcription factors regulate many genes involved in immune and inflammatory responses (reviewed in Vallabhapurapu and Karin, 2009). Upon pathogen recognition, a variety of cellular pattern recognition receptors (PRRs), including cGAS [Cyclic-GMP-AMP (cGAMP) synthase], STING (stimulator of interferon genes), IFI16 (interferon γ-inducible protein 16), RIG-I-like helicases, NOD-like receptors (NLRs) and TLRs may activate the NF-κB pathway. Thus, NF-κB signaling is induced by multiple stimuli, including pathogen associated molecular patterns (PAMPs) as well as proinflammatory cytokines, e.g., tumor necrosis factor-α (TNF-α) and interleukin-1. Ligand binding to TLRs induces several downstream effectors and results in the formation of signaling complexes activating the IKK complex (Figure 1). Ubiquitination of TNF-receptor-associated factors (TRAF) and NEMO facilitates activation of the catalytic IKKβ subunit resulting in the phosphorylation and proteasomal degradation of IκB and consequently nuclear translocation of active NF-κB dimers (Skaug et al., 2009). Similarly, signaling via members of the TNF receptor superfamily ultimately leads to activation of the IKK complex, following recruitment of various adaptor proteins and stimulation of TGF-β-activated kinase 1 (TAK1; Wang and Baldwin, 1998). Furthermore, some activated TNFR superfamily members may induce accumulation of mitogen-activated protein kinase 14 (MAP3K14 or NF-κB-inducing kinase, NIK) that activates IKKα to induce processing of p100 and thus activation of e.g., RelB/p52 heterodimers to mediate expression of NF-κB-responsive genes via the non-canonical pathway. This pathway is slower than the canonical NF-κB signaling pathway and can also be activated via the CD40 ligand (CD40L), receptor activator of NF-κB ligand (RANKL) or lymphotoxin β (Cildir et al., 2016). Following activation, RelB/p52 heterodimers translocate to the nucleus for target gene activation.
NF-κB is not only a key factor in the induction of effective innate immune responses but also plays an important role in adaptive immunity. For example, ligand binding to the TCR complex induces recruitment of LCK tyrosine kinase that phosphorylates the ITAMs of the CD3 ζ chains (Figure 1). Subsequent recruitment of ZAP70 mediates activation of a signaling pathway involving PLCγ, protein kinase C (PKC) family members, the CARD11-BCL10-MALT1 (CBM) complex, TRAF6, and ultimately TAK1, inducing NF-κB activity by phosphorylation and activation of IKKs (Cheng et al., 2011; Paul and Schaefer, 2013). NF-κB activation is further enhanced by costimulation via CD28, which triggers distinct signaling cascades involving PI3K and PDK1 (Boomer and Green, 2010). Thus, the signaling pathways induced by CD3/CD28-mediated stimulation of T cells upon interaction with antigen-presenting cells (APCs) allow NF-κB to enter the nucleus to upregulate expression of cytokines and antimicrobial effectors as well as genes involved in T cell survival, proliferation, and differentiation. Consequently, regulation of NF-κB activity plays a key role in innate and adaptive immune function and the defense against bacterial and viral pathogens. In agreement with this important role in inflammatory gene expression, chronic activation of NF-κB signaling is observed in a variety of inflammatory diseases including arthritis, sepsis, gastritis, asthma, atherosclerosis, and inflammatory bowel disease (Panday et al., 2016).
It is long known that HIV-1 transcription can be stimulated by activation of the canonical NF-κB pathway (Nabel and Baltimore, 1987; Tong-Starksen et al., 1987). NF-κB binding sites are found in the enhancer region of all primate lentiviral LTRs, although their numbers may vary between different subtypes of HIV-1 group M and various groups of SIV and HIV. Most subtypes of pandemic HIV-1 group M strains (A, B, D, F, G, H, J, and K) and some SIVs contain two NF-κB binding sites located –104 to –80 bp upstream of the transcriptional start site within the 5′LTR (Figure 2). In contrast, the second human immunodeficiency virus (HIV-2), subtype A/E recombinants of HIV-1 group M and several SIV lineages contain just a single NF-κB binding site. Finally, subtype C strains, which account for almost 50% of HIV-1 infections worldwide, typically contain three binding sites for NF-κB in their enhancer region (Bachu et al., 2012). LTR-mediated transcription is initiated by binding of p50/p65 heterodimers or other members of the NF-κB transcription factor family that recruit p300 to promote acetylation of LTR chromatin and allow access for RNA polymerase II (RNAPII; Figure 2A). NF-κB binding also promotes transcriptional elongation by recruiting the positive transcription elongation factor b (P-TEFb) complex and the transcription factor TFIIH to the carboxyl domain (CTD) of RNAPII, allowing its phosphorylation that results in increased processivity of this enzyme (Barboric et al., 2001). However, this effect is transient since an okadaic acid (OA)-sensitive phosphatase dephosphorylates the CTD of RNAPII to prevent P-TEFb interaction. Thus, short transcripts terminated shortly after the trans-activation response (TAR) RNA element predominate at the early stage. Accumulation of the viral Tat protein that binds to TAR and recruits P-TEFb allows the virus to overcome this bottleneck by allowing hyper-phosphorylation of RNAPII and potent transcriptional elongation and the generation of full-length viral transcripts (Williams et al., 2007) (Figure 2B).
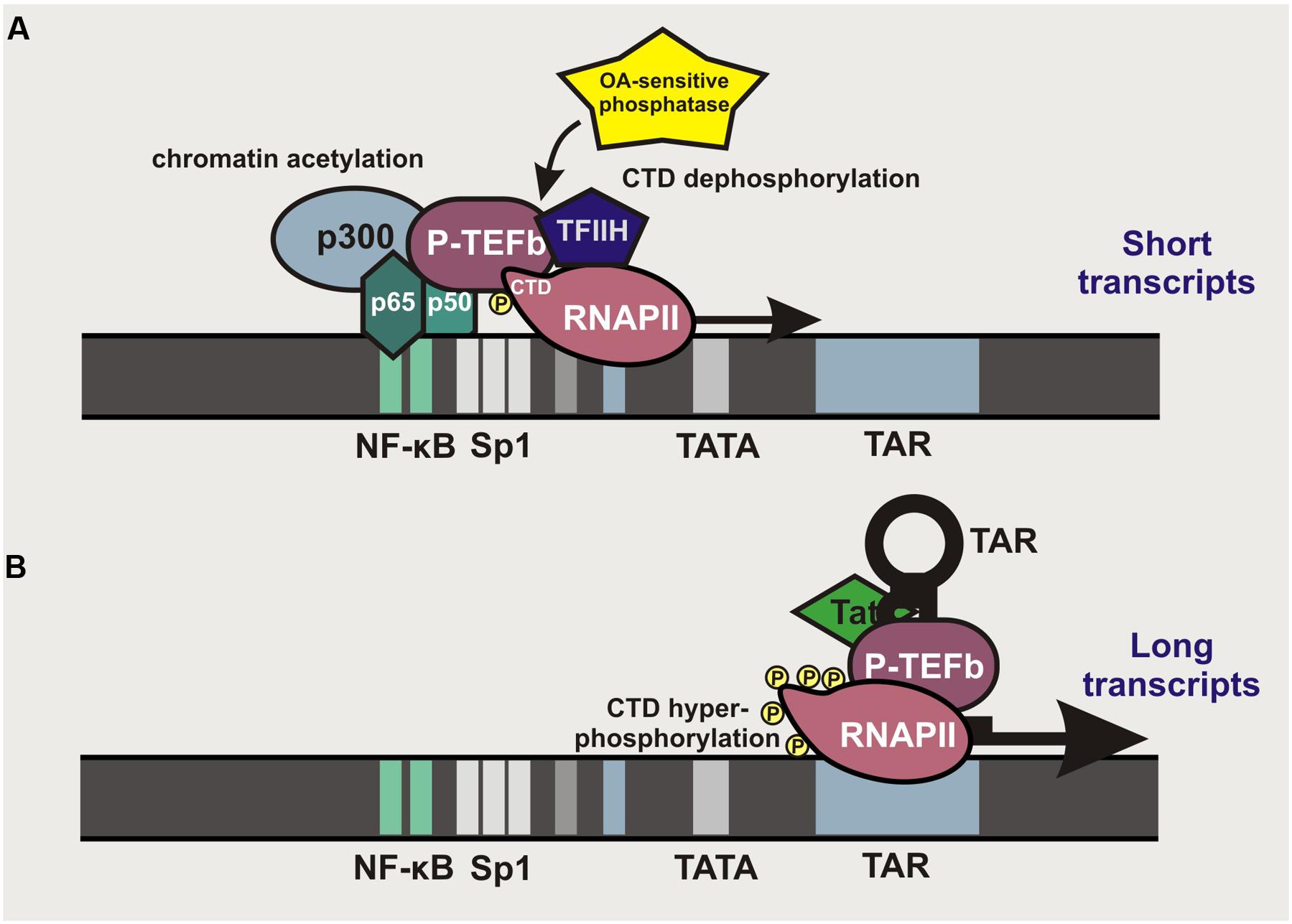
FIGURE 2. Role of κB binding sites in primate lentiviral LTR-mediated transcription. (A) Localization of κB binding sites in primate lentiviral LTRs. NF-κB binding allows recruitment of p300 to initiate chromatin acetylation and to render the LTR better accessible for RNAPII. NF-κB also recruits P-TEFb, which binds to the CTD of RNAPII and strongly enhances its processivity. De-phosphorylation of the CTD by an OA-sensitive phosphatase terminates the elongation, resulting in the production of short TAR-containing transcripts. (B) In the presence of the viral Tat protein, transcription of proviral DNA is maintained independently of NF-κB. Tat binds to the short hairpin loop of TAR and recruits P-TEFb to the RNAPII, thereby allowing efficient elongation and generation of full-length viral transcripts.
Typically, mutation of the NF-κB binding sites in HIV-1 LTRs will prevent efficient proviral transcription. However, one case of a replication-competent pathogenic HIV-1 strain lacking NF-κB binding sites has been reported (Zhang et al., 1997). This unusual HIV-1 strain contained duplications in the TCF-1alpha region that may have compensatory effects. Thus, NF-κB binding sites are important but not essential for HIV-1 replication or pathogenicity in vivo. Since NF-κB activation stimulates HIV-1 transcription, it is also targeted in approaches aiming to eliminate the latent viral reservoirs. Induction of NF-κB activity by T cell activation or treatment with phorbol esters, such as prostratin, potently enhance viral gene expression (Coudronniere et al., 2000; Williams et al., 2004). However, since NF-κB transcription factors are involved in numerous physiological and pathological processes, their induction is prone to adverse effects. Notably, some members of the NF-κB transcription factor family may also promote HIV-1 latency, i.e., it has been reported that in unstimulated T cells, p50 homodimers occupy the NF-κB sites in the proviral LTR to recruit HDAC1, thereby promoting histone hypo-acetylation and hence chromatin condensation rendering the viral LTR poorly accessible to RNAPII binding (Williams et al., 2006).
HIV-1 infection modulates NF-κB signaling by multiple mechanisms. As outlined above, sensing of viral PAMPs by PRRs activates NF-κB to induce an antiviral immune response. HIV-1 sensing is not fully understood as the virus has evolved effective evasion mechanisms. For example, recent evidence suggests that the viral capsid stays largely intact until it docks to the nucleopore (Arhel et al., 2007; Lelek et al., 2012; Dharan et al., 2016) and that it recruits cellular factors, such as PSF6 and cyclophilins, to prevent innate immune activation (Rasaiyaah et al., 2013). Nonetheless, it has become clear that a variety of viral components may trigger immune responses. Viral RNA or DNA intermediates of the reverse transcription (RT) process can be sensed by cytosolic receptors, such as cGAS, IFI16, PQBP1 and RIG-I, particularly under suboptimal conditions for efficient RT (Jakobsen et al., 2015; Sauter and Kirchhoff, 2016). Moreover, antiretroviral restriction factors may also act as immune sensors (Hotter et al., 2013). For example, TRIM5α induces untimely uncoating of the viral capsid and may also act as an activator of the TAK1 kinase complex to stimulate AP-1 and NF-κB signaling (Pertel et al., 2011). Similarly, trapping of HIV-1 particles by the host restriction factor tetherin induces phosphorylation of tyrosine residues in its cytoplasmic tail to mediate recruitment of SYK tyrosine kinase and TRAF2 and/or 6 to activate TAK1 and consequently NF-κB-dependent immune responses (Galão et al., 2012, 2014; Tokarev et al., 2013) (Figure 1). Interestingly, this sensing function of tetherin seems to have an evolutionary recent origin and is only observed for the human and (to a much lesser extent) chimpanzee orthologs (Galão et al., 2012), whereas the ability of tetherin to block virion release has a very ancient origin (Heusinger et al., 2015; Blanco-Melo et al., 2016). Finally, HIV-1 infection induces DNA damage responses since the generation of linear viral RNA/DNA and DNA species, non-integrated circular forms of viral DNA and a double-stand break in the host-genome are inevitable concomitants of the viral replication cycle. DNA damage signaling may activate ATM and mediate phosphorylation and ubiquitination of NEMO to induce IKK and consequently NF-κB activation (Miyamoto, 2011; McCool and Miyamoto, 2012).
HIV-1 not only affects NF-κB activation by triggering immune sensors and inducing DNA damage responses but may also use some of its gene products to manipulate this transcription factor to promote efficient viral gene expression. Many studies investigated the effect of HIV-1 on NF-κB activation but the initial results were often contradictory. One possible reason for this is that NF-κB signaling plays a complex role in the viral replication cycle and that effects may depend on the cell type and state of activation, as well as the stimuli and HIV-1 strains or proteins used in the respective studies. Furthermore, accumulating evidence suggests that the HIV-1 Nef and Tat proteins that are expressed at high levels immediately after initiation of proviral transcription enhance and late viral gene products, such as Vpu, suppress NF-κB activation (Roux et al., 2000; Akari et al., 2001; Bour et al., 2001; Varin et al., 2005; Herbein et al., 2008; Mangino et al., 2011; Fiume et al., 2012; Liu et al., 2013). It has been reported that Tat interacts with IκBα and the p65 subunit of NF-κB to prevent binding of the repressor to the NF-κB complex while promoting p65 binding to DNA (Fiume et al., 2012). It has also been shown that the cytoplasmic domain of the HIV-1 envelope glycoprotein (Env) gp41 interacts with TAK1 to induce NF-κB activation (Postler and Desrosiers, 2012). Whether this mechanism is effective during the earliest stage of infection, i.e., induced by virion fusion with the plasma cell membrane of the target cell, or requires higher quantities of Env achieved during later stages of the replication cycle remains to be determined. Finally, a variety of studies reported that Vpr modulates NF-κB signaling in various cell types. However, the effects are controversial and both stimulatory and inhibitory effects of Vpr have been described (Kogan et al., 2013; Liu et al., 2013, 2014; Liang et al., 2015). Thus, the effects of virion-associated and de novo synthesized Vpr in primary HIV-1 infected T cells need to be further investigated.
The accessory viral Nef protein does not induce NF-κB activity on its own but boosts the responsiveness of HIV-1 infected cells to stimulation (Alexander et al., 1997; Simmons et al., 2001; Fenard et al., 2005) (Figure 3A). Nef-mediated activation of NF-κB, nuclear factor of activated T cells (NFAT), IL-2 and LTR stimulation following TCR-CD3/CD28 costimulation seems to require association with lipid rafts (Schrager and Marsh, 1999; Fortin et al., 2004; Kumar et al., 2016) and may depend on the state of activation of infected T cells (Baur et al., 1994). Interactions of HIV-1 Nef with the CD3 ζ chain (Xu et al., 1999) and downstream effectors of TCR signaling, such as the tyrosine kinase LCK (Baur et al., 1997), serine/threonine p21-activating kinases (Sawai et al., 1994), the DOCK2-ELMO1 complex (Janardhan et al., 2004) and ERK/MAP kinases (Schrager et al., 2002) have been reported. Thus, Nef might affect the catalytic activity of different kinases, induce cytoskeletal changes, and activate a variety of signaling pathways. The relative contribution of these activities and interactions to Nef-mediated enhancement of T cell activation is largely unclear. In either case, Nef promotes nuclear translocation of NF-κB and other transcription factors, such as AP1 and NFAT and activates the viral promoter to induce Tat expression and hence productive viral replication (Kinoshita et al., 1998). Notably, Nef may exert its multiple functions rapidly after viral entry since it is expressed at high levels early during the viral replication cycle and possibly even before proviral integration (Sloan et al., 2011).
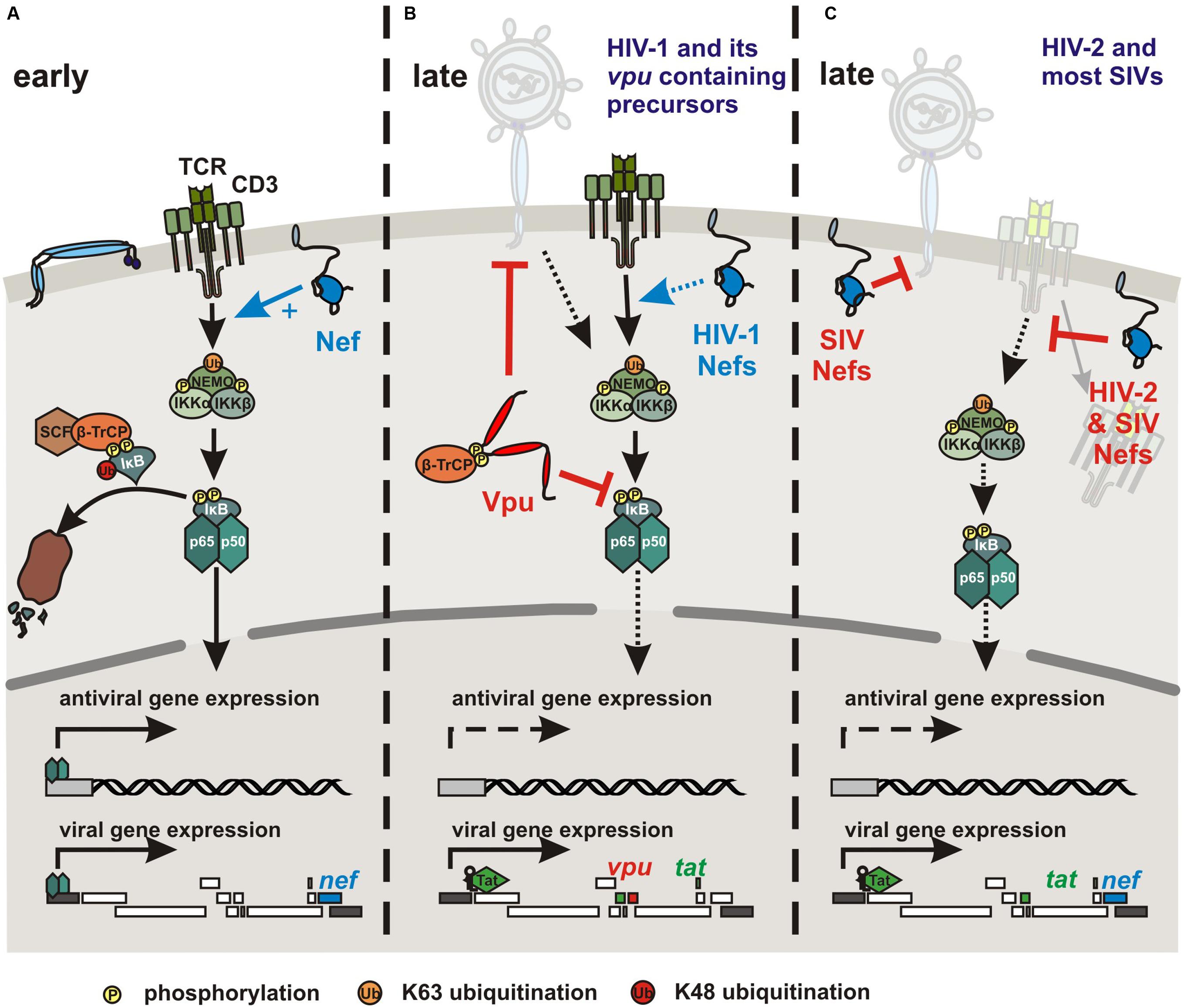
FIGURE 3. Modulation of NF-κB activity by HIV and SIV Nef and Vpu proteins. (A) To initiate early viral gene expression, the viral accessory protein Nef promotes NF-κB activation by boosting TCR-CD3 mediated T cell activation and other yet to be determined mechanisms. (B) HIV-1 and its closest SIV counterparts use their Vpu protein to inhibit NF-κB and thus antiviral gene expression during late stages of the replication cycle. Vpu interferes with IκB degradation by sequestration of β-TrCP and other as-yet-unknown mechanisms. Furthermore, HIV-1 group M and (less effectively) N Vpu proteins counteract the cellular restriction factor tetherin, which traps budding virions at the cell surface and also acts as NF-κB activating immune sensor in the case of the human ortholog. In the presence of the viral transactivator Tat, viral transcription is maintained independently of NF-κB activity. (C) HIV-2 and most SIV strains do not contain a vpu gene but express Nef proteins that efficiently down-modulate CD3 from the cell surface to prevent T cell activation and hence to suppress the induction of NF-κB and other transcription factors. SIV Nefs also counteract tetherin in their respective host. However, although monkey tetherins restrict virus release they are not known to act as NF-κB activating immune sensors.
While Nef-mediated enhancement of NF-κB activity may be important to initiate proviral transcription, it is less critical after accumulation of the viral Tat protein and may even become detrimental to HIV-1 replication because of the induction of antiviral gene expression. Thus, HIV-1 and other primate lentiviruses might tightly regulate NF-κB activity throughout their replication cycle to allow proviral transcription while minimizing antiviral gene expression. Recent studies show that the accessory viral protein U (Vpu) potently suppresses NF-κB activity during later stages of the viral replication cycle (Figure 3B) (Akari et al., 2001; Bour et al., 2001; Sauter et al., 2015). A vpu gene was most likely acquired by the precursor of SIVs infecting Cercopithecus monkeys with subsequent cross-species transmissions and recombination events giving rise to other vpu containing primate lentiviruses (Bailes et al., 2003; Takeuchi et al., 2015). Thus, vpu is found in HIV-1, its chimpanzee and gorilla precursors, SIVcpz and SIVgor, and in SIVgsn, SIVmus, SIVmon, and SIVden, infecting greater spot-nosed, mustached, mona, and Dent’s mona monkeys, respectively (Kirchhoff, 2009). The Vpu proteins of pandemic HIV-1 group M strains counteract tetherin-mediated activation of NF-κB-dependent antiviral immune responses (Cocka and Bates, 2012; Galão et al., 2012, 2014; Tokarev et al., 2013). This is expected since the Vpus of HIV-1 group M and SIVs infecting several Cercopithecus species are potent antagonists of tetherin-mediated inhibition of virion release. More surprisingly, Vpus derived from SIVs infecting chimpanzees and gorillas or HIV-1 group O strains that use their Nef protein to counteract tetherin are also potent inhibitors of NF-κB (Sauter et al., 2015). Indeed, early studies suggested that HIV-1 Vpu prevents NF-κB activation by inhibiting degradation of IκB through sequestration of the adaptor protein β-TrCP (Akari et al., 2001; Bour et al., 2001). More recent data show that the ability of Vpu to prevent NF-κB activation independently of the stimulus is conserved between all lineages of SIV and HIV-1 (except group N) containing this accessory gene and does not correlate with β-TrCP interaction or tetherin antagonism (Sauter et al., 2015). In agreement with the results of early studies (Bour et al., 2001), an intact β-TrCP binding motif and interaction of Vpu with β-TrCP are essential for highly effective inhibition of p65 nuclear translocation by primary Vpu proteins. However, some Vpus failing to recruit β-TrCP still suppressed NF-κB-dependent gene expression (Sauter et al., 2015). Thus, the action of Vpu involves stabilization of IκB and reduced nuclear translocation of p65 but also additional yet-to-be-defined mechanisms. Importantly, the ability of Vpu to inhibit NF-κB is dominant over the stimulatory effect of Nef and associated with reduced induction of IFN and ISGs in HIV-1 infected T cells (Sauter et al., 2015). Accordingly, HIV-1 and its closest simian counterparts seem to utilize Nef to boost NF-κB activation to initiate LTR-driven proviral transcription. At later stages, when viral gene expression is ensured by the presence of Tat, Vpu inhibits NF-κB activity to limit expression of antiviral genes and to attenuate the immune response.
The ability to inhibit NF-κB activity is highly conserved among primate lentiviral Vpu proteins (Sauter et al., 2015) suggesting an important role for viral immune evasion in vivo. As outlined above, however, vpu genes are only found in HIV-1 and its closest simian counterparts, raising the question whether the majority of primate lentiviruses use another mechanism to inhibit NF-κB during late stages of the replication cycle. It is tempting to speculate that originally essentially all primate lentiviruses used the CD3 down-modulation function of Nef to suppress the activity of NF-κB and other transcription factors. This Nef function is conserved among HIV-2 and most lineages of SIVs but was entirely lost in the great majority of vpu containing primate lentiviruses (Schindler et al., 2006). This was most likely not just coincidence. In fact, phylogenetic and functional analyses suggest that the CD3 down-modulation function of Nef may have been lost twice during primate lentiviral evolution when the virus acquired a vpu gene. The first time after acquisition of vpu by a precursor of SIVs nowadays found in various Cercopithecus monkeys and a second time when this virus recombined with the precursor of SIVrcm from Red-capped mangabeys in chimpanzees to give rise to SIVcpz, the precursor of HIV-1 (Kirchhoff, 2010).
It has been suggested that Vpu might diminish the selective advantage of Nef-mediated CD3 down-modulation because it counteracts tetherin and potentially other antiviral factors induced in an inflammatory environment. In fact, lack of CD3 down-modulation by Nef is associated with higher expression levels of early (CD69) and late (CD25) activation markers as well as increased levels of apoptosis, induction of Fas, Fas-L, PD-1, and CTLA-4 cell surface expression and secretion of interferon gamma (IFN-γ) in virally infected cultures of peripheral blood mononuclear cells (Schindler et al., 2006; Schmökel et al., 2011; Yu et al., 2015). The consequences of the presence of vpu and the inability of Nef to block TCR-mediated T cell activation, that distinguish HIV-1 and its simian precursors from other primate lentiviruses, for viral pathogenicity largely remain a matter of speculation. While it is evident that host properties play an important role in the clinical outcome of primate lentiviral infections (Chahroudi et al., 2012), it is conceivable that potent inhibition of T cell activation should help to prevent damagingly high levels of immune activation that drive CD4+ T cell depletion and progression to immunodeficiency (Sodora et al., 2009). Indeed, inefficient Nef-mediated down-modulation of CD3 correlates with low numbers of CD4+ T cells in SIVsmm infected sooty mangabeys (Schindler et al., 2008) and viremic HIV-2 infected individuals (Khalid et al., 2012). Furthermore, HIV-1 is more pathogenic than HIV-2 (Nyamweya et al., 2013) and many SIVs do not cause disease in their natural host species (Chahroudi et al., 2012), whereas SIVcpz causes AIDS in chimpanzees (Keele et al., 2009). It is unknown whether other vpu containing viruses cause disease in the wild. However, the prevalence of SIVgsn/mus/mon in their natural simian hosts seems to be much lower (∼1–4%) than of SIVs capable of CD3 down-modulation (often >40%; Heigele et al., 2016). It will be interesting to further examine whether this is due to differences in the pathogenic outcome of these infections.
Vpu may allow primate lentiviruses to better cope with the antiviral immune response not only by antagonizing innate antiviral factors but also by downregulation of various receptors involved in the activation of natural killer cells (Sugden et al., 2016). However, the recent finding that Vpu suppresses NF-κB activity also suggests a more direct link with the CD3 down-modulation function of Nef. As described above, signaling via the TCR-CD3 complex induces a cascade of events ultimately leading to IKK activation and translocation of NF-κB into the nucleus for target gene expression (Figure 1). It has been established that Nef-mediated down-modulation of CD3 potently blocks the responsiveness of virally infected T cells to TCR-mediated stimulation (Iafrate et al., 1997; Bell et al., 1998; Khalid et al., 2012) and prevents the formation of the immunological synapse between virally infected primary CD4+ T cells and dendritic cells or macrophages (Arhel et al., 2009). It has been shown that CD3 down-modulation potently inhibits the induction of NFAT (Khalid et al., 2012), which also plays an important role in the immune response (Müller and Rao, 2010). While the effect on NF-κB activity is less well investigated, preliminary data clearly indicate that Nef-mediated down-modulation of CD3 would also block activation of this transcription factor (Figure 3C). Thus, our current knowledge suggests that most primate lentiviruses may prevent NF-κB activation by Nef-mediated down-modulation of CD3, whereas HIV-1 and its simian precursors utilize Vpu to inhibit NF-κB signaling further downstream in the cascade. The former mechanism is associated with a more “resting” phenotype of virally infected T cells and disrupts their interaction with and responsiveness to other immune cells (Arhel et al., 2009). The latter may still allow activation of the infected T cells e.g., by APCs but prevent NF-κB-dependent antiviral gene expression during later stages of the viral replication cycle. Notably, most SIVs use their Nef protein to counteract tetherin (Jia et al., 2009; Sauter et al., 2009; Zhang et al., 2009). However, this does most likely not affect NF-κB activity since only the human but not monkey orthologs of tetherin are known to activate this transcription factor (Galão et al., 2012). Finally, HIV-2 uses Env to counteract restriction by human tetherin (Le Tortorec and Neil, 2009) but it has not been reported whether this mechanism also suppresses induction of NF-κB activity.
Importantly, the effects of all primate lentiviral Nefs on T cell activation may differ during the early and late stages of the viral replication cycle. Perhaps most notably, Nef proteins from HIV-2 and SIVs that down-modulate CD3 enhance IKKβ-induced NF-κB activation as efficiently as HIV-1 or SIV Nefs lacking the CD3 down-modulation function entirely (Sauter et al., 2015). Thus, primate lentiviral Nef proteins may generally boost the responsiveness to stimulation during the earliest stage of infection. While down-modulation of CD3 from the cell surface by Nef proteins possessing this function is highly effective, it may take more time than providing an initial boost to NF-κB activity to initiate viral gene expression and productive replication. Consequently, early stimulation and late inhibition of NF-κB may both be achieved by most primate lentiviruses and be mediated by cooperative Nef and Vpu functions in HIV-1 and its precursors. Finally, it is noteworthy that even HIV-1 Nef might have differential effects depending on the stage of infection. It is puzzling that for HIV-1 Nef enhancing (Herbein et al., 2008; Mangino et al., 2011), inhibitory (Niederman et al., 1992; Bandres and Ratner, 1994), and no (Yoon and Kim, 1999; Witte et al., 2008) effects on NF-κB activity have been reported. In part, these differences may depend on the state of activation of infected T cells. However, it will also be interesting to further examine whether the effect of HIV-1 Nef might differ at different stages of the viral replication cycle. In agreement with this possibility, it is known that HIV-1 Nefs down-modulate CD28, an important costimulatory factor of T cell activation, albeit substantially less effectively than HIV-2 and most SIV Nefs (Bell et al., 2001; Swigut et al., 2001). Timing adds another degree of complexity to the already complex role of the multi-functional Nef protein throughout the viral life cycle and clearly warrants further examination.
Numerous studies have investigated how accessory proteins of HIV and SIV, i.e., Vif, Vpr, Nef, Vpu and/or Vpx, counteract antiviral restriction factors, such as APOBEC proteins, tetherin, SAMHD1 and SERINC3/5. Expression of most of these and other antiviral factors is induced by a very limited number of transcription factors that are activated upon viral immune sensing including NF-κB and IRFs. Modulation of the induction and activity of these key regulators of innate and adaptive immunity may have a major benefit for viral replication but has only recently gained significant scientific attention. Specifically, recent data provide evidence that primate lentiviruses tightly regulate the activity of NF-κB to initiate efficient viral transcription while minimizing the expression of antiviral genes. As outlined above, the most common mechanism amongst primate lentiviruses may be initial boosting of NF-κB activity by Nef and prevention of further stimulation of T cell activation by potent down-modulation of TCR-CD3. This strategy seems to be highly successful considering the high prevalence of these viruses and the benign relationship with their natural simian hosts. In contrast, HIV-1 and its SIV precursors seem to use Nef to initially boost and Vpu to later on suppress NF-κB activation. A variety of Vpu functions that might provide a selective advance for viral immune evasion and replication have been reported (Sandberg et al., 2012; Sugden et al., 2016). Nonetheless, the emergence of a vpu containing subset of primate lentiviruses is somewhat surprising as they seem to be less prevalent and potentially more virulent in their natural hosts than other SIVs. Perhaps they have advantages in specific hosts as supported by the more efficient spread of HIV-1 in the human population compared to HIV-2 despite higher virulence. Notably, a few primate lentiviruses lack both vpu and the CD3 down-modulation function of Nef (Schmökel et al., 2011) and our preliminary data suggest that they might use yet another accessory protein (i.e., Vpr) to suppress NF-κB activity during the late stages of infection.
Altogether, recent evidence suggests that primate lentiviruses have evolved several sophisticated mechanisms to tightly regulate the activity of NF-κB and possibly other transcription factors throughout their replication cycle. The conservation of these functions supports an important role for viral replication and immune evasion. Furthermore, modulation of NF-κB activity is most likely also relevant for viral latency and inflammatory responses. NF-κB is targeted for the treatment of inflammatory and proliferative diseases as well as cancer and activation of the latent reservoirs of HIV-1 (Jiang and Dandekar, 2015; Panday et al., 2016). Because of its important role in many physiological processes, however, therapeutic modulation of NF-κB activity is prone to undesired adverse effects. Nevertheless, further studies on the mechanisms used by HIV-1 and other primate lentiviruses to manipulate this central transcription factor may provide important information on how these viruses establish latency and induce inflammation and perhaps even on how to prevent this.
EH and FK both wrote the manuscript and prepared figures.
FK is supported by the Deutsche Forschungsgemeinschaft (DFG) priority program “Innate Sensing and Restriction of Retroviruses” (SPP 1923) and an ERC Advanced grant “Anti-virome.”
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Daniel Sauter, Dré van der Merwe and Dominik Hotter for comments and discussion.
Akari, H., Bour, S., Kao, S., Adachi, A., and Strebel, K. (2001). The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κB-dependent expression of antiapoptotic factors. J. Exp. Med. 194, 1299–1311. doi: 10.1084/jem.194.9.1299
Alexander, L., Du, Z., Rosenzweig, M., Jung, J. U., and Desrosiers, R. C. (1997). A role for natural simian immunodeficiency virus and human immunodeficiency virus type 1 nef alleles in lymphocyte activation. J. Virol. 71, 6094–6099.
Arhel, N., Lehmann, M., Clauss, K., Nienhaus, G. U., Piguet, V., and Kirchhoff, F. (2009). The inability to disrupt the immunological synapse between infected human T cells and APCs distinguishes HIV-1 from most other primate lentiviruses. J. Clin. Invest. 119, 2965–2975. doi: 10.1172/JCI38994
Arhel, N. J., Souquere-Besse, S., Munier, S., Souque, P., Guadagnini, S., Rutherford, S., et al. (2007). HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 26, 3025–3037. doi: 10.1038/sj.emboj.7601740
Bachu, M., Yalla, S., Asokan, M., Verma, A., Neogi, U., Sharma, S., et al. (2012). Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J. Biol. Chem. 287, 44714–44735. doi: 10.1074/jbc.M112.397158
Bailes, E., Gao, F., Bibollet-Ruche, F., Courgnaud, V., Peeters, M., Marx, P. A., et al. (2003). Hybrid origin of SIV in Chimpanzees. Science 300, 1713–1713. doi: 10.1126/science.1080657
Bandres, J. C., and Ratner, L. (1994). Human immunodeficiency virus type 1 Nef protein down-regulates transcription factors NF-kappa B and AP-1 in human T cells in vitro after T-cell receptor stimulation. J. Virol. 68, 3243–3249.
Barboric, M., Nissen, R. M., Kanazawa, S., Jabrane-Ferrat, N., and Peterlin, B. M. (2001). NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8, 327–337. doi: 10.1016/S1097-2765(01)00314-8
Baur, A. S., Sass, G., Laffert, B., Willbold, D., Cheng-Mayer, C., and Peterlin, B. M. (1997). The N-terminus of Nef from HIV-1/SIV associates with a protein complex containing Lck and a serine kinase. Immunity 6, 283–291. doi: 10.1016/S1074-7613(00)80331-3
Baur, A. S., Sawai, E. T., Dazin, P., Fantl, W. J., Cheng-Mayer, C., and Matija Peterlin, B. (1994). HIV-1 nef leads to inhibition or activation of T cells depending on its intracellular localization. Immunity 1, 373–384. doi: 10.1016/1074-7613(94)90068-X
Bell, I., Ashman, C., Maughan, J., Hooker, E., Cook, F., and Reinhart, T. A. (1998). Association of simian immunodeficiency virus Nef with the T-cell receptor (TCR) ζ chain leads to TCR down-modulation. J. Gen. Virol. 79, 2717–2727. doi: 10.1099/0022-1317-79-11-2717
Bell, I., Schaefer, T. M., Trible, R. P., Amedee, A., and Reinhart, T. A. (2001). Down-modulation of the costimulatory molecule, CD28, is a conserved activity of multiple SIV Nefs and is dependent on histidine 196 of Nef. Virology 283, 148–158. doi: 10.1006/viro.2001.0872
Blanco-Melo, D., Venkatesh, S., and Bieniasz, P. D. (2016). Origins and evolution of tetherin, an orphan antiviral gene. Cell Host Microbe 20, 189–201. doi: 10.1016/j.chom.2016.06.007
Boomer, J. S., and Green, J. M. (2010). An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2:a002436. doi: 10.1101/cshperspect.a002436
Bour, S., Perrin, C., Akari, H., and Strebel, K. (2001). The human immunodeficiency virus type 1 Vpu protein inhibits NF-κB activation by interfering with βTrCP-mediated degradation of IκB. J. Biol. Chem. 276, 15920–15928. doi: 10.1074/jbc.M010533200
Cary, D. C., Fujinaga, K., and Peterlin, B. M. (2016). Molecular mechanisms of HIV latency. J. Clin. Invest. 126, 448–454. doi: 10.1172/JCI80565
Chahroudi, A., Bosinger, S. E., Vanderford, T. H., Paiardini, M., and Silvestri, G. (2012). Natural SIV hosts: showing AIDS the door. Science 335, 1188–1193. doi: 10.1126/science.1217550
Chan, J. K., and Greene, W. C. (2012). Dynamic roles for NF-κB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol. Rev. 246, 286–310. doi: 10.1111/j.1600-065X.2012.01094.x
Cheng, J., Montecalvo, A., and Kane, L. P. (2011). Regulation of NF-κB induction by TCR/CD28. Immunol. Res. 50, 113–117. doi: 10.1007/s12026-011-8216-z
Cildir, G., Low, K. C., and Tergaonkar, V. (2016). Noncanonical NF-κB signaling in health and disease. Trends Mol. Med. 22, 414–429. doi: 10.1016/j.molmed.2016.03.002
Cocka, L. J., and Bates, P. (2012). Identification of alternatively translated Tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog. 8:e1002931. doi: 10.1371/journal.ppat.1002931
Colomer-Lluch, M., Gollahon, L. S., and Serra-Moreno, R. (2016). Anti-HIV factors: targeting each step of HIV’s replication cycle. Curr. HIV Res. 14, 175–182. doi: 10.2174/1570162X14999160224094621
Coudronniere, N., Villalba, M., Englund, N., and Altman, A. (2000). NF-κB activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-𝜃. Proc. Natl. Acad. Sci. U.S.A. 97, 3394–3399. doi: 10.1073/pnas.97.7.3394
Dharan, A., Talley, S., Tripathi, A., Mamede, J. I., Majetschak, M., Hope, T. J., et al. (2016). KIF5B and Nup358 cooperatively mediate the nuclear import of HIV-1 during infection. PLOS Pathog. 12:e1005700. doi: 10.1371/journal.ppat.1005700
Doyle, T., Goujon, C., and Malim, M. H. (2015). HIV-1 and interferons: who’s interfering with whom? Nat. Rev. Microbiol. 13, 403–413. doi: 10.1038/nrmicro3449
Fenard, D., Yonemoto, W., de Noronha, C., Cavrois, M., Williams, S. A., and Greene, W. C. (2005). Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J. Immunol. 175, 6050–6057. doi: 10.4049/JIMMUNOL.175.9.6050
Fiume, G., Vecchio, E., De Laurentiis, A., Trimboli, F., Palmieri, C., Pisano, A., et al. (2012). Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α and p65. Nucleic Acids Res. 40, 3548–3562. doi: 10.1093/nar/gkr1224
Fortin, J.-F., Barat, C., Beauséjour, Y., Barbeau, B., and Tremblay, M. J. (2004). Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-κB, and AP-1 induction. J. Biol. Chem. 279, 39520–39531. doi: 10.1074/jbc.M407477200
Galão, R. P., Le Tortorec, A., Pickering, S., Kueck, T., and Neil, S. J. D. (2012). Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 12, 633–644. doi: 10.1016/j.chom.2012.10.007
Galão, R. P., Pickering, S., Curnock, R., and Neil, S. J. D. (2014). Retroviral retention activates a syk-dependent HemITAM in human tetherin. Cell Host Microbe 16, 291–303. doi: 10.1016/j.chom.2014.08.005
Ghosh, S., and Hayden, M. S. (2012). Celebrating 25 years of NF-κB research. Immunol. Rev. 246, 5–13. doi: 10.1111/j.1600-065X.2012.01111.x
Hayden, M. S., West, A. P., and Ghosh, S. (2006). SnapShot: NF-κB signaling pathways. Cell 127, 1286–1287. doi: 10.1016/j.cell.2006.12.005
Heigele, A., Kmiec, D., Regensburger, K., Langer, S., Peiffer, L., Stürzel, C. M., et al. (2016). The potency of nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20, 381–391. doi: 10.1016/j.chom.2016.08.004
Herbein, G., Varin, A., Larbi, A., Fortin, C., Mahlknecht, U., Fulop, T., et al. (2008). Nef and TNFα are coplayers that favor HIV-1 replication in monocytic cells and primary macrophages. Curr. HIV Res. 6, 117–129. doi: 10.2174/157016208783884985
Heusinger, E., Kluge, S. F., Kirchhoff, F., and Sauter, D. (2015). Early vertebrate evolution of the host restriction factor tetherin. J. Virol. 89, 12154–12165. doi: 10.1128/JVI.02149-15
Hiscott, J., Kwon, H., Génin, P., Ghosh, S., May, M., Kopp, E., et al. (2001). Hostile takeovers: viral appropriation of the NF-κB pathway. J. Clin. Invest. 107, 143–151. doi: 10.1172/JCI11918
Hotter, D., Sauter, D., and Kirchhoff, F. (2013). Emerging role of the host restriction factor tetherin in viral immune sensing. J. Mol. Biol. 425, 4956–4964. doi: 10.1016/j.jmb.2013.09.029
Iafrate, A. J., Bronson, S., and Skowronski, J. (1997). Separable functions of Nef disrupt two aspects of T cell receptor machinery: CD4 expression and CD3 signaling. EMBO J. 16, 673–684. doi: 10.1093/emboj/16.4.673
Jakobsen, M. R., Olagnier, D., and Hiscott, J. (2015). Innate immune sensing of HIV-1 infection. Curr. Opin. HIV AIDS 10, 96–102. doi: 10.1097/COH.0000000000000129
Janardhan, A., Swigut, T., Hill, B., Myers, M. P., and Skowronski, J. (2004). HIV-1 Nef binds the DOCK2-ELMO1 complex to activate rac and inhibit lymphocyte chemotaxis. PLoS Biol. 2:E6. doi: 10.1371/journal.pbio.0020006
Jia, B., Serra-Moreno, R., Neidermyer, W., Rahmberg, A., Mackey, J., Fofana, I. B., et al. (2009). Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by Tetherin/BST2. PLoS Pathog. 5:e1000429. doi: 10.1371/journal.ppat.1000429
Jiang, G., and Dandekar, S. (2015). Targeting NF-κB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retroviruses 31, 4–12. doi: 10.1089/AID.2014.0199
Karin, M., and Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18, 621–663. doi: 10.1146/annurev.immunol.18.1.621
Keele, B. F., Jones, J. H., Terio, K. A., Estes, J. D., Rudicell, R. S., Wilson, M. L., et al. (2009). Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460, 515–519. doi: 10.1038/nature08200
Khalid, M., Yu, H., Sauter, D., Usmani, S. M., Schmokel, J., Feldman, J., et al. (2012). Efficient Nef-mediated downmodulation of TCR-CD3 and CD28 is associated with high CD4+ T cell counts in viremic HIV-2 infection. J. Virol. 86, 4906–4920. doi: 10.1128/JVI.06856-11
Kinoshita, S., Chen, B. K., Kaneshima, H., and Nolan, G. P. (1998). Host control of HIV-1 parasitism in T Cells by the nuclear factor of activated T cells. Cell 95, 595–604. doi: 10.1016/S0092-8674(00)81630-X
Kirchhoff, F. (2009). Is the high virulence of HIV-1 an unfortunate coincidence of primate lentiviral evolution? Nat. Rev. Microbiol. 7:467. doi: 10.1038/nrmicro2111
Kirchhoff, F. (2010). Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67. doi: 10.1016/j.chom.2010.06.004
Kluge, S. F., Sauter, D., and Kirchhoff, F. (2015). SnapShot: antiviral restriction factors. Cell 163, 774–774.e1. doi: 10.1016/j.cell.2015.10.019
Kogan, M., Deshmane, S., Sawaya, B. E., Gracely, E. J., Khalili, K., and Rappaport, J. (2013). Inhibition of NF-κB activity by HIV-1 Vpr is dependent on Vpr binding protein. J. Cell. Physiol. 228, 781–790. doi: 10.1002/jcp.24226
Kumar, A., Abbas, W., Colin, L., Khan, K. A., Bouchat, S., Varin, A., et al. (2016). Tuning of AKT-pathway by Nef and its blockade by protease inhibitors results in limited recovery in latently HIV infected T-cell line. Sci. Rep. 6:24090. doi: 10.1038/srep24090
Le Tortorec, A., and Neil, S. J. D. (2009). Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978. doi: 10.1128/JVI.01515-09
Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., and Zimmer, C. (2012). Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. U.S.A. 109, 8564–8569. doi: 10.1073/pnas.1013267109
Liang, Z., Liu, R., Lin, Y., Liang, C., Tan, J., and Qiao, W. (2015). HIV-1 Vpr protein activates the NF-κB pathway to promote G2/M cell cycle arrest. Virol. Sin. 30, 441–448. doi: 10.1007/s12250-015-3654-8
Liu, R., Lin, Y., Jia, R., Geng, Y., Liang, C., Tan, J., et al. (2014). HIV-1 Vpr stimulates NF-κB and AP-1 signaling by activating TAK1. Retrovirology 11:45. doi: 10.1186/1742-4690-11-45
Liu, R., Tan, J., Lin, Y., Jia, R., Yang, W., Liang, C., et al. (2013). HIV-1 Vpr activates both canonical and noncanonical NF-κB pathway by enhancing the phosphorylation of IKKα/β. Virology 439, 47–56. doi: 10.1016/j.virol.2013.01.020
Mangino, G., Percario, Z. A., Fiorucci, G., Vaccari, G., Acconcia, F., Chiarabelli, C., et al. (2011). HIV-1 Nef induces proinflammatory state in macrophages through its acidic cluster domain: involvement of TNF alpha receptor associated factor 2. PLoS ONE 6:e22982. doi: 10.1371/journal.pone.0022982
McCool, K. W., and Miyamoto, S. (2012). DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 246, 311–326. doi: 10.1111/j.1600-065X.2012.01101.x
Miyamoto, S. (2011). Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 21, 116–130. doi: 10.1038/cr.2010.179
Müller, M. R., and Rao, A. (2010). NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10, 645–656. doi: 10.1038/nri2818
Nabel, G., and Baltimore, D. (1987). An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 329, 219–222.
Nabel, G. J., and Verma, I. M. (1993). Proposed NF-κB/IκB family nomenclature. Genes Dev. 7, 2063. doi: 10.1101/gad.7.11.2063
Napetschnig, J., and Wu, H. (2013). Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468. doi: 10.1146/annurev-biophys-083012-130338
Nelson, D. E., Ihekwaba, A. E. C., Elliott, M., Johnson, J. R., Kell, D. B., and White, M. R. H. (2004). Oscillations in NF-κB signaling control the dynamics of gene expression. Science 306, 704–709. doi: 10.1126/science.1099962
Niederman, T. M., Garcia, J. V., Hastings, W. R., Luria, S., and Ratner, L. (1992). Human immunodeficiency virus type 1 Nef protein inhibits NF-κB induction in human T cells. J. Virol. 66, 6213–6219.
Nyamweya, S., Hegedus, A., Jaye, A., Rowland-Jones, S., Flanagan, K. L., and Macallan, D. C. (2013). Comparing HIV-1 and HIV-2 infection: lessons for viral immunopathogenesis. Rev. Med. Virol. 23, 221–240. doi: 10.1002/rmv.1739
Oeckinghaus, A., and Ghosh, S. (2009). The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1:a000034. doi: 10.1101/cshperspect.a000034
Panday, A., Inda, M. E., Bagam, P., Sahoo, M. K., Osorio, D., and Batra, S. (2016). Transcription factor NF-κB: an update on intervention strategies. Arch. Immunol. Ther. Exp. (Warsz) 64, 463–483. doi: 10.1007/s00005-016-0405-y
Paul, S., and Schaefer, B. C. (2013). A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 34, 269–281. doi: 10.1016/j.it.2013.02.002
Pertel, T., Hausmann, S., Morger, D., Züger, S., Guerra, J., Lascano, J., et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. doi: 10.1038/nature09976
Pfeffer, L. M. (2011). The role of nuclear factor κB in the interferon response. J. Interf. cytokine Res. 31, 553–559. doi: 10.1089/jir.2011.0028
Postler, T. S., and Desrosiers, R. C. (2012). The cytoplasmic domain of the HIV-1 glycoprotein gp41 induces NF-κB activation through TGF-β-activated kinase 1. Cell Host Microbe 11, 181–193. doi: 10.1016/j.chom.2011.12.005
Rasaiyaah, J., Tan, C. P., Fletcher, A. J., Price, A. J., Blondeau, C., Hilditch, L., et al. (2013). HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405. doi: 10.1038/nature12769
Roux, P., Alfieri, C., Hrimech, M., Cohen, E. A., and Tanner, J. E. (2000). Activation of transcription factors NF-κB and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces interleukin-8 expression. J. Virol. 74, 4658–4665. doi: 10.1128/JVI.74.10.4658-4665.2000
Sandberg, J. K., Andersson, S. K., Bächle, S. M., Nixon, D. F., and Moll, M. (2012). HIV-1 Vpu interference with innate cell-mediated immune mechanisms. Curr. HIV Res. 10, 327–333. doi: 10.2174/157016212800792513
Sauter, D., Hotter, D., Van Driessche, B., Stürzel, C. M., Kluge, S. F., Wildum, S., et al. (2015). Differential regulation of NF-κB-mediated proviral and antiviral host gene expression by primate lentiviral nef and vpu proteins. Cell Rep. 10, 586–600. doi: 10.1016/j.celrep.2014.12.047
Sauter, D., and Kirchhoff, F. (2016). IFITMs: important factors in trans-mission of HIV-1. Cell Host Microbe 20, 407–408. doi: 10.1016/j.chom.2016.09.009
Sauter, D., Schindler, M., Specht, A., Landford, W. N., Münch, J., Kim, K.-A., et al. (2009). Tetherin-Driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6, 409–421. doi: 10.1016/j.chom.2009.10.004
Sawai, E. T., Baur, A., Struble, H., Peterlin, B. M., Levy, J. A., and Cheng-Mayer, C. (1994). Human immunodeficiency virus type 1 Nef associates with a cellular serine kinase in T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 91, 1539–1543. doi: 10.1073/pnas.91.4.1539
Schindler, M., Münch, J., Kutsch, O., Li, H., Santiago, M. L., Bibollet-Ruche, F., et al. (2006). Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 125, 1055–1067. doi: 10.1016/j.cell.2006.04.033
Schindler, M., Schmökel, J., Specht, A., Li, H., Münch, J., Khalid, M., et al. (2008). Inefficient Nef-mediated downmodulation of CD3 and MHC-I correlates with loss of CD4+T cells in natural SIV infection. PLoS Pathog. 4:e1000107. doi: 10.1371/journal.ppat.1000107
Schmökel, J., Sauter, D., Schindler, M., Leendertz, F. H., Bailes, E., Dazza, M.-C., et al. (2011). The presence of a vpu gene and the lack of Nef-mediated downmodulation of T cell receptor-CD3 are not always linked in primate lentiviruses. J. Virol. 85, 742–752. doi: 10.1128/JVI.02087-10
Schrager, J. A., Der Minassian, V., and Marsh, J. W. (2002). HIV Nef increases T cell ERK MAP kinase activity. J. Biol. Chem. 277, 6137–6142. doi: 10.1074/jbc.M107322200
Schrager, J. A., and Marsh, J. W. (1999). HIV-1 Nef increases T cell activation in a stimulus-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 96, 8167–8172. doi: 10.1073/PNAS.96.14.8167
Sen, R., and Baltimore, D. (1986). Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46, 705–716. doi: 10.1016/0092-8674(86)90346-6
Simmons, A., Aluvihare, V., and McMichael, A. (2001). Nef triggers a transcriptional program in T cells imitating single-signal T cell activation and inducing HIV virulence mediators. Immunity 14, 763–777. doi: 10.1016/S1074-7613(01)00158-3
Skaug, B., Jiang, X., and Chen, Z. J. (2009). The role of ubiquitin in NF-κB regulatory pathways. Annu. Rev. Biochem. 78, 769–796. doi: 10.1146/annurev.biochem.78.070907.102750
Sloan, R. D., Wainberg, M. A., Bar-Magen, T., Sloan, R., Faltenbacher, V., Donahue, D., et al. (2011). The role of unintegrated DNA in HIV infection. Retrovirology 8:52. doi: 10.1186/1742-4690-8-52
Sodora, D. L., Allan, J. S., Apetrei, C., Brenchley, J. M., Douek, D. C., Else, J. G., et al. (2009). Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat. Med. 15, 861–865. doi: 10.1038/nm.2013
Sugden, S. M., Bego, M. G., Pham, T. N. Q., and Cohen, É. A. (2016). Remodeling of the host cell plasma membrane by HIV-1 Nef and Vpu: a strategy to ensure viral fitness and persistence. Viruses 8:67. doi: 10.3390/v8030067
Swigut, T., Shohdy, N., and Skowronski, J. (2001). Mechanism for down-regulation of CD28 by Nef. EMBO J. 20, 1593–1604. doi: 10.1093/emboj/20.7.1593
Takeuchi, J. S., Ren, F., Yoshikawa, R., Yamada, E., Nakano, Y., Kobayashi, T., et al. (2015). Coevolutionary dynamics between tribe Cercopithecini tetherins and their lentiviruses. Sci. Rep. 5:16021. doi: 10.1038/srep16021
Tokarev, A., Suarez, M., Kwan, W., Fitzpatrick, K., Singh, R., and Guatelli, J. (2013). Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 87, 2046–2057. doi: 10.1128/JVI.02272-12
Tong-Starksen, S. E., Luciw, P. A., and Peterlin, B. M. (1987). Human immunodeficiency virus long terminal repeat responds to T-cell activation signals. Proc. Natl. Acad. Sci. U.S.A. 84, 6845–6849. doi: 10.1073/pnas.84.19.6845
Utay, N. S., and Douek, D. C. (2016). Interferons and HIV infection: the good, the bad, and the Ugly. Pathog. Immun. 1, 107–116. doi: 10.20411/pai.v1i1.125
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733. doi: 10.1146/annurev.immunol.021908.132641
Varin, A., Decrion, A.-Z., Sabbah, E., Quivy, V., Sire, J., Van Lint, C., et al. (2005). Synthetic Vpr protein activates activator protein-1, c-Jun N-terminal kinase, and NF-κB and stimulates HIV-1 transcription in promonocytic cells and primary macrophages. J. Biol. Chem. 280, 42557–42567. doi: 10.1074/jbc.M502211200
Wang, D., and Baldwin, A. S. J. (1998). Activation of nuclear factor-κB-dependent transcription by tumor necrosis factor-α is mediated through phosphorylation of RelA/p65 on serine 529. J. Biol. Chem. 273, 29411–29416. doi: 10.1074/jbc.273.45.29411
Williams, S. A., Chen, L.-F., Kwon, H., Fenard, D., Bisgrove, D., Verdin, E., et al. (2004). Prostratin antagonizes HIV latency by activating NF-κB. J. Biol. Chem. 279, 42008–42017. doi: 10.1074/jbc.M402124200
Williams, S. A., Chen, L.-F., Kwon, H., Ruiz-Jarabo, C. M., Verdin, E., and Greene, W. C. (2006). NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 25, 139–149. doi: 10.1038/sj.emboj.7600900
Williams, S. A., Kwon, H., Chen, L.-F., and Greene, W. C. (2007). Sustained induction of NF-κB is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 81, 6043–6056. doi: 10.1128/JVI.02074-06
Witte, V., Laffert, B., Gintschel, P., Krautkrämer, E., Blume, K., Fackler, O. T., et al. (2008). Induction of HIV transcription by Nef involves Lck activation and protein kinase C theta raft recruitment leading to activation of ERK1/2 but not NF kappa B. J. Immunol. 181, 8425–8432. doi: 10.4049/jimmunol.181.12.8425
Xu, X. N., Laffert, B., Screaton, G. R., Kraft, M., Wolf, D., Kolanus, W., et al. (1999). Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor ζ chain. J. Exp. Med. 189, 1489–1496. doi: 10.1084/jem.189.9.1489
Yoon, K., and Kim, S. (1999). Lack of negative influence on the cellular transcription factors NF-κB and AP-1 by the nef protein of human immunodeficiency virus type 1. J. Gen. Virol. 80, 2951–2956. doi: 10.1099/0022-1317-80-11-2951
Yu, H., Khalid, M., Heigele, A., Schmökel, J., Usmani, S. M., van der Merwe, J., et al. (2015). Lentiviral Nef proteins manipulate T cells in a subset-specific manner. J. Virol. 89, 1986–2001. doi: 10.1128/JVI.03104-14
Zhang, F., Wilson, S. J., Landford, W. C., Virgen, B., Gregory, D., Johnson, M. C., et al. (2009). Nef Proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6, 54–67. doi: 10.1016/j.chom.2009.05.008
Zhang, L., Huang, Y., Yuan, H., Chen, B. K., Ip, J., and Ho, D. D. (1997). Identification of a replication-competent pathogenic human immunodeficiency virus type 1 with a duplication in the TCF-1α region but lacking NF-κB binding sites. J. Virol. 71, 1651–1656.
Keywords: HIV, SIV, NF-κB, Nef, Vpu, Tat, LTR
Citation: Heusinger E and Kirchhoff F (2017) Primate Lentiviruses Modulate NF-κB Activity by Multiple Mechanisms to Fine-Tune Viral and Cellular Gene Expression. Front. Microbiol. 8:198. doi: 10.3389/fmicb.2017.00198
Received: 04 January 2017; Accepted: 27 January 2017;
Published: 14 February 2017.
Edited by:
Akio Adachi, Tokushima University, JapanReviewed by:
Mikako Fujita, Kumamoto University, JapanCopyright © 2017 Heusinger and Kirchhoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Kirchhoff, ZnJhbmsua2lyY2hob2ZmQHVuaS11bG0uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.